 Gary Tse1,2*
Gary Tse1,2* Tong Liu3Ka H. C. Li4
Tong Liu3Ka H. C. Li4 Victoria Laxton5
Victoria Laxton5 Yin W. F. Chan6Wendy Keung7Ronald A. Li8Bryan P. Yan1,9*
Yin W. F. Chan6Wendy Keung7Ronald A. Li8Bryan P. Yan1,9*- 1Department of Medicine and Therapeutics, Chinese University of Hong Kong, Hong Kong, Hong Kong
- 2Li Ka Shing Institute of Health Sciences, Chinese University of Hong Kong, Hong Kong, Hong Kong
- 3Tianjin Key Laboratory of Ionic-Molecular Function of Cardiovascular Disease, Department of Cardiology, Tianjin Institute of Cardiology, Second Hospital of Tianjin Medical University, Tianjin, China
- 4Faculty of Medicine, Newcastle University, Newcastle, UK
- 5Intensive Care Department, Royal Brompton and Harefield NHS Trust, London, UK
- 6School of Biological Sciences, University of Cambridge, Cambridge, UK
- 7Stem Cell and Regenerative Medicine Consortium, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Pokfulam, Hong Kong
- 8Ming Wai Lau Centre for Reparative Medicine, Karolinska Institutet, Solna, Sweden
- 9Department of Epidemiology and Preventive Medicine, Monash University, Melbourne, VIC, Australia
Brugada syndrome (BrS), is a primary electrical disorder predisposing affected individuals to sudden cardiac death via the development of ventricular tachycardia and fibrillation (VT/VF). Originally, BrS was linked to mutations in the SCN5A, which encodes for the cardiac Na+ channel. To date, variants in 19 genes have been implicated in this condition, with 11, 5, 3, and 1 genes affecting the Na+, K+, Ca2+, and funny currents, respectively. Diagnosis of BrS is based on ECG criteria of coved- or saddle-shaped ST segment elevation and/or T-wave inversion with or without drug challenge. Three hypotheses based on abnormal depolarization, abnormal repolarization, and current-load-mismatch have been put forward to explain the electrophysiological mechanisms responsible for BrS. Evidence from computational modeling, pre-clinical, and clinical studies illustrates that molecular abnormalities found in BrS lead to alterations in excitation wavelength (λ), which ultimately elevates arrhythmic risk. A major challenge for clinicians in managing this condition is the difficulty in predicting the subset of patients who will suffer from life-threatening VT/VF. Several repolarization risk markers have been used thus far, but these neglect the contributions of conduction abnormalities in the form of slowing and dispersion. Indices incorporating both repolarization and conduction and based on the concept of λ have recently been proposed. These may have better predictive values than the existing markers.
Introduction
The syndrome of right bundle branch block with ST segment elevation was first described by the Italian physicians, Drs Nava, Martini and Thiene, in 1989 (Martini et al., 1989). The term Brugada syndrome (BrS) was later introduced to describe a hereditary condition involving idiopathic ventricular tachycardia, ventricular fibrillation (VT/VF) and sudden cardiac death (SCD) in structurally normal hearts (Brugada and Brugada, 1992). Traditionally, BrS has been linked to loss-of-function mutations in the SCN5A gene, which encodes for the cardiac Na+ channel. Although SCN5A is the commonest affected gene, its mutations are only implicated in approximately a fifth of patients. In some pedigrees, affected members did not have mutations in this gene (Probst et al., 2009). Over the past decades, variants in 19 genes that affect the Na+, K+, Ca2+, and funny currents have been implicated. However, caution must be taken when interpreting such genetic variants, as these do not always contribute significantly to the BrS phenotype (Le Scouarnec et al., 2015). Other than BrS, Scn5a mutations have been associated with sick sinus syndrome (SSS; Benson et al., 2003), progressive cardiac conduction defect (PCCD, or Lenègre disease; Schott et al., 1999; Tan et al., 2001; Probst et al., 2003) and idiopathic VF without BrS findings (Akai et al., 2000). They can also lead to overlap disorders (Remme et al., 2008). For example, the p.Y1449C mutation was associated with conduction disease, Brugada syndrome and atrial flutter (Hothi et al., 2015). The differing disease phenotypes can partly be explained by the biophysical effects of the mutated Scn5a gene product (Liu et al., 2014). The same mutation can affect members of different families, or even members of the same family, differently, suggesting that other factors modify the behavior of the sodium channels.
BrS was initially estimated to account for 12% of the cases of SCD in the general population (Brugada and Brugada, 1992). However, recent epidemiological studies have demonstrated that its prevalence is much lower. Thus, in Southeast Asians, who are more predisposed to BrS than other ethnicities, only 0.1% showed a Brugada pattern (Ng et al., 2012). In Chinese subjects, the overall prevalence of BrS pattern was 3.3%, with 0.08% due to Type 1 BrS, and the remaining contributions from Types 2 and 3 (Juang et al., 2015). In Denmark, a low prevalence of BrS was found, at 0.001% (Holst et al., 2012). BrS has a male preponderance, affecting men four times more frequently than women and also affecting younger adults than infants or children (Nademanee et al., 1997). BrS can present with SCD (aborted or otherwise), syncope, palpitations or agonal breathing, leading them to undergo further investigation. Precipitating factors include increased vagal tone, fever, and other drugs such as tricyclic antidepressants and alcohol (Madeira et al., 2015; Achaiah and Andrews, 2016).
Aside from VT, BrS is associated with pre-excitation syndromes such as Wolff-Parkinson-White syndrome (Eckardt et al., 2001), and supraventricular tachycardia such as atrioventricular nodal reentrant tachycardia, atrial flutter or atrial fibrillation (Bordachar et al., 2004). Sinus node dysfunction in the form of prolonged sinoatrial node recovery time (Morita et al., 2004) has been observed in BrS. Conduction abnormalities such as reduced conduction velocity (CV) from the sinoatrial node to the atria or through the atria, and blocks such as atrial standstill are also observed (Takehara et al., 2004; Tse et al., 2016b).
Diagnosis of BrS is based on a Type 1 electrocardiographic (ECG) pattern of a coved-shaped ST segment elevation (STE) ≥2 mm and negative T-wave in the right precordial leads with or without drug challenge using a class I anti-arrhythmic agent such as flecainide (Priori et al., 2013). BrS can also be diagnosed when a type 2 (saddleback morphology, defined as a J-wave amplitude ≥2 mm but STE ≥1 mm) or type 3 (STE <1 mm with either a coved or saddleback morphology) ECG pattern is converted to type 1 by drug challenge. Clinical findings, such as agonal respiration during sleep, history of ventricular tachycardia or fibrillation (VT/VF), inducible VT/VF observed during an electrophysiological study (EPS) and family history of SCD or Type 1 (coved-type) ECG were part of the original diagnostic criteria, but have now been excluded in the latest consensus criteria (Priori et al., 2013). Some investigators have emphasized that having a Brugada pattern on the ECG should not automatically equate to a clinical diagnosis of BrS, especially in asymptomatic individuals without clinical signs or symptoms (Martini, 2016). Indeed, there are concerns that over-diagnosis may occur if this is based only on ECG criteria (Viskin et al., 2015). However, others pointed out that those with a Type 1 BrS ECG, even if only it is drug-induced, have a low but nevertheless elevated risk of sudden death compared to the general population and therefore patients should be aware of it (Andorin and Probst, 2016). A recent consensus conference report put forward the Shanghai BrS Score, where patients receive points for (i) ECG, (ii) clinical findings, (iii) family history and (iv) genetic results, and are stratified into non-diagnostic, possible or probable/definite BrS (Antzelevitch et al., 2016). The standard placement of ECG leads in the fourth intercostal spaces may not be sufficiently sensitive to detect the presence of BrS ECG patterns, as only mild ST segment elevation was observed (Shimizu et al., 2000). By contrast, the ST segment elevation is more pronounced when the leads are placed in the second intercostal spaces, in keeping with the arrhythmia origins in the right ventricular outflow tract (Curcio et al., 2016; Kumar and Kalman, 2016).
Traditionally, BrS was thought to be a Mendelian disease, having an autosomal dominant inheritance with incomplete penetrance (Sicouri et al., 2012), but this has been refuted (Gourraud et al., 2016). The genotype-phenotype correlation is poor; a recent study examined co-segregation of SCN5A mutations amongst large genotyped families, demonstrating that some affected family members did not carry the familial mutation (Probst et al., 2009). This suggests other mutations in other genes are responsible for BrS (Marian, 2009; Roden, 2010). Moreover, some putatively pathogenic genetic variants do not manifest clinically as abnormal phenotype (Van Driest et al., 2016). Therefore, caution must be taken in notifying patients of these incidental findings, which may not be clinically significant.
Differential Diagnosis: J-Wave Syndromes and Other Causes of Brugada Pattern
J-wave syndrome is a term encompassing both BrS and early repolarization syndrome (ERS; Shinde et al., 2007; Antzelevitch et al., 2011; Wang et al., 2011). The letter “J” refers to the junctional point between the QRS and ST segment, representing the intersection between end of ventricular depolarization and onset of ventricular repolarization. Early repolarization was first described in 1951 (Grant et al., 1951). It has been defined as prominent or elevated J-point with notching or slurring of distal part of R wave in at least two contiguous leads (Mehta et al., 1999; Klatsky et al., 2003). Although traditionally seen as a benign finding (Mehta and Jain, 1995), early repolarization has subsequently been associated with a higher risk of SCD (Haïssaguerre et al., 2008; Rosso et al., 2008, 2012). The estimated prevalence of ERS is around 1 to 13% of the general population and associated with 15 to 70% of idiopathic VF cases (Haïssaguerre et al., 2008; Derval et al., 2011; Haruta et al., 2011). ERS has been reported to have an autosomal dominant origin with incomplete penetrance (Noseworthy et al., 2011; Nunn et al., 2011; Reinhard et al., 2011).
A group of heterogeneous conditions induce a Brugada ECG pattern, including “metabolism conditions, mechanical compression, myocardial ischaemia, pulmonary embolism, myocardial, and pericardial disease, ECG modulations and miscellaneous conditions” (Dendramis, 2016; Enriquez et al., 2016; Gottschalk et al., 2016). These must be distinguished from true BrS as these are potential reversible causes and do not necessitate invasive treatments such as implantable cardioverter-defibrillator (ICD) insertion.
Electrophysiological Mechanisms Underlying Arrhythmogenesis in Brugada Syndrome
The Na+ channel consists of one α subunit (SCN5A) and one or two β subunits (SCN1B, SCN2B, SCN3B). Loss-of-function mutations in Scn5a have been associated with BrS (Chen et al., 1998), sick sinus syndrome (SSS; Benson et al., 2003), progressive cardiac conduction defect (PCCD, or Lenègre disease; Schott et al., 1999; Tan et al., 2001; Probst et al., 2003) and overlap disorders between these conditions (Remme et al., 2008). By contrast, gain-of-function Scn5a mutations are observed in long QT syndrome type 3 (Wang et al., 1995). BrS and LQTS share many similarities, existing in congenital or acquired forms (Havakuk and Viskin, 2016).
BrS has been associated with reduced INa and loss-of-function mutations in Scn5a. The latter can lead to impaired trafficking to the cell membrane, reduced expression and expression of non-functional proteins (Bezzina et al., 1999; Dumaine et al., 1999; Akai et al., 2000; Kyndt et al., 2001; Valdivia et al., 2004; Amin et al., 2005). Gating of Na+ channels can also be altered such as delayed activation, premature inactivation, enhanced slow inactivation and slower recovery from inactivation (Akai et al., 2000; Amin et al., 2005). Although the commonest mutations of BrS have been localized to SCN5a, accounting for 25% of cases (Probst et al., 2010), other genes have also been implicated. These include the different Na+ channel subunits, e.g., SCN5A, SCN1B, SCN2B, or SCN3B (Watanabe et al., 2008; Hu et al., 2009; Riuró et al., 2013; Ricci et al., 2014). Recently, SCN10A, a neuronal sodium channel gene, was proposed to be a putative causative gene in a large fraction of BrS cases (Hu et al., 2014a). Reduced INa can also arise from mutations in genes encoding for glycerol-3-phosphate dehydrogenase 1-like (GPD1-L) protein (London et al., 2007), MOG1 (Kattygnarath et al., 2011), sarcolemmal membrane-associated protein (SLMAP) (Ishikawa et al., 2012), the desmosomal component plakophilin-2 (Cerrone et al., 2014), FGF2 (fibroblast growth factor homologous factor-1) (Hennessey et al., 2013) and the transcriptional factor HEY2 (Bezzina et al., 2013; Boukens et al., 2013).
BrS can involve reduced ICa (Antzelevitch et al., 2007). LTCC consists of 4 protein subunits α1 (CACNA1C), β2 (CACNB2), α2 (CACNA2D), and δ (CACNA2D). Loss-of-function mutations in these genes can lead to abnormal trafficking, reduced expression or function of the LTCC, similar to the Na+ channel mutations (Antzelevitch et al., 2007; Burashnikov et al., 2010). However, a difference is that BrS secondary to loss-of-function of LTCCs are associated with shortened QT intervals, as opposed to classical BrS in which the QT interval is not normally altered. BrS has also been associated with gain-of-function mutations in genes encoding for proteins that constitute or modulate the different K+ channels. These include KCNE3, KCND3, and SEMA3A (semaphorin, an endogenous K+ channel inhibitor) responsible for Ito (Delpón et al., 2008; Giudicessi et al., 2011, 2012; Ohno et al., 2011; Nakajima et al., 2012; Boczek et al., 2014), KCNJ8 and ABCC9 (encoding for SUR2A, the ATP-binding cassette transporter for the KATP channel) determining IK, ATP (Medeiros-Domingo et al., 2010; Hu et al., 2014b) and KCNH2 encoding for IKr (Wang et al., 2014). Most recently, dysfunction in the KCNAB2, which encodes the voltage-gated K+ channel β2-subunit, was associated with increased Ito activity and identified as a putative gene involved in BrS (Portero et al., 2016). Finally, loss-of-function mutations in HCN4 leading to decreased If and gain- or loss-of-function mutations in the transient receptor potential melastatin protein 4 gene (TRPM4) have also been implicated in BrS (Ueda et al., 2009; Liu et al., 2013).
To explain how these molecular changes lead to the BrS phenotype, the depolarization, repolarization, and current-to-load mismatch hypotheses have been proposed, as summarized in Figure 1 (Nishii et al., 2010; Wilde et al., 2010; Tse et al., 2016i). Even some 37 years before Martini and colleagues described their syndrome of right bundle branch block, ST segment elevation and sudden death (Nava et al., 1988; Martini et al., 1989), delayed depolarization was thought to underlie ECG changes in a healthy male patient simulating acute myocardial infarction (Osher and Wolff, 1953). Regarding the electrophysiological abnormalities observed in this case, they wrote: “due to prolongation of the depolarization process by right bundle block or possibly focal block with delayed activation of a portion of the right ventricle: unusually early onset of repolarization may also play a role”. Since then, the relative contributions of depolarization vs. repolarization have been intensively studied in pre-clinical models (Antzelevitch et al., 2005, 2007; Antzelevitch, 2008; Schweizer et al., 2014). More recently, a third hypothesis based on electrotonic current posits that current-to-load mismatch in the RV and RVOT subepicardium is responsible for ST segment elevation in BrS (Hoogendijk et al., 2010).
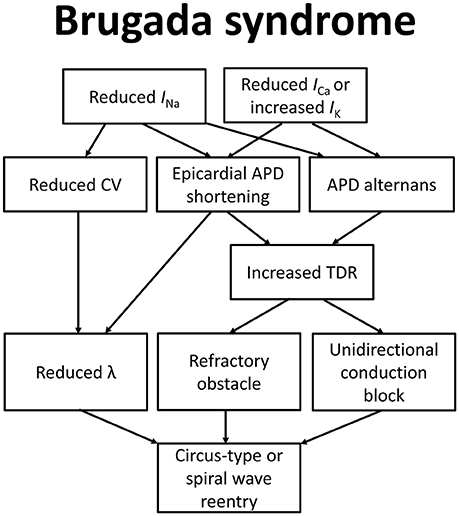
Figure 1. Molecular and electrophysiological mechanisms underlying arrhythmogenesis in Brugada syndrome.
Depolarization Hypothesis
The depolarization theory proposes that slower upstroke of phase 0 and the consequent reduction in conduction velocity (CV) of the APs are responsible for arrhythmogenesis. The seminal work by Martini and colleagues found that patients suffering from RBBB and ST segment elevation also had fibrosis of the right ventricle and the conduction system (Martini et al., 1989). Indeed, targeted disruption of Scn5a (Scn5a+/−), Scn5a1798insD/+ and SCN5aG1408R mice demonstrate reduced CV associated with interstitial fibrosis (Leoni et al., 2010; Boukens et al., 2013; Schweizer et al., 2014). Scn5a+/− mice also show progressive conduction defects that are suggestive of Lenègre disease (Royer et al., 2005). Na+ channels were assumed to be distinct from gap junctions, which are found predominantly at the ends of cardiomyocytes for mediating intercellular conduction. However, they were later shown to co-localize with gap junctions at the intercalated disks (Cohen, 1996). Indeed, the macrostructure connexome has been coined to describe an interacting network of Na+ channels, desomosomal components and gap junctions (Rhett and Gourdie, 2012; Rhett et al., 2013; Agullo-Pascual et al., 2014; Veeraraghavan et al., 2014a,b; Veeraraghavan et al., 2015; George et al., 2015). Autopsy findings support the idea that components of the connexome are not independent of each other, by demonstrating increased myocardial fibrosis from collagen deposition and reduced gap junction expression in the RVOT of hearts from BrS patients (Campuzano and Brugada, 2015; Nademanee et al., 2015).
Mouse hearts with plakophilin-2 missense mutations showed a phenotype that is consistent with arrhythmogenic right ventricular dysplasia (ARVD; Cruz et al., 2015). BrS subjects possessing PKP2 mutations have a reduced number of Na+ channels at the intercalated disk (Agullo-Pascual et al., 2014; Cerrone and Delmar, 2014; Cerrone et al., 2014; Campuzano et al., 2016). Although ARVD and BrS have been considered to be different disorders, both affect primarily the RV and may represent opposite ends of the same disease spectrum, with decreasing degree of structural abnormalities from ARVD toward BrS (Peters, 2014, 2015). Both ARVD and BrS can be therefore considered as diseases of the connexome.
Together, Scn5a models of BrS demonstrate that these conduction disturbances and structural abnormalities are greater in the RV compared to the LV. These are in keeping with clinical findings of delayed depolarization in the RV outflow tract demonstrated using electroanatomical mapping (Nagase et al., 2002; Tukkie et al., 2004; Coronel et al., 2005; Postema et al., 2008, 2010; Lambiase et al., 2009; Nademanee et al., 2011; Ten Sande et al., 2015). Most recently, a panoramic ventricular mapping study in humans showed electrogram prolongation and fractionation, reflecting reduced CV and increased CV dispersion (Zhang et al., 2015). Furthermore, catheter ablation of the RVOT led to resolution of the BrS ECG pattern, and prevention of spontaneous and provoked VT/VF episodes, thereby supporting the depolarization hypothesis (Nademanee et al., 2011; Brugada et al., 2015). However, abnormal depolarization may be less relevant in BrS pathogenic variants in which the INa is not affected. For example, mutations leading to reduced ICa or increased IK would not be expected to influence the AP upstroke but rather shortens the plateau phase of AP repolarization.
Repolarization Hypothesis
The repolarization theory states that differential APD shortening across the myocardial wall is primarily responsible for the BrS phenotype. Loss-of-function Scn5a mutations can have opposing effects on the fast and slow inactivation of Na+ channels with distinct effects on repolarization (Veldkamp et al., 2000). Evidence in support of the repolarization hypothesis for BrS has been derived from experiments performed in animal models (Di Diego and Antzelevitch, 2003; Antzelevitch and Oliva, 2006; Antzelevitch, 2006; Tsuboi and Antzelevitch, 2006; Sicouri et al., 2012). Data from mouse Scn5a+/− hearts on repolarization durations have been inconsistent (Stokoe et al., 2007; Martin et al., 2010, 2011a,b), which has been discussed by editorials elsewhere (Tse et al., 2016f,h,i). Experiments derived from arterially perfused, canine wedge preparations have provided much insights into the mechanisms by which reduced inward currents contributes to heterogeneities in repolarization and in turn reentry (Yan and Antzelevitch, 1999; Antzelevitch, 2001; Fish and Antzelevitch, 2004, 2008). A greater degree of shortening is observed in the epicardium with high Ito (particularly in the RVOT epicardium) compared to the endocardium with low Ito. Therefore, epicardial APs, but not endocardial APs, lose their dome shape morphology. During phase 1 of the AP, the outward shift of ionic current balance leads to failure of LTCC activation, which also contributes to the loss of the AP dome. Originally, phase 2 reentry was hypothesized to involve electrotonic coupling between these cardiac regions, which permits propagation of the AP dome from endocardial regions where it is maintained to epicardial regions where it is abolished, thereby producing an extrasystole (Yan and Antzelevitch, 1999). Recent work has suggested that such phase 2 reentry is due to heterogeneity in early repolarization in the sub-epicardial layer (Maoz et al., 2014). This can then serve to initiate arrhythmia, and sustain reentry by a circus-type mechanism (Lukas and Antzelevitch, 1996). Steep and reversal of repolarization gradients lead to the ST segment elevation and T-wave inversion, respectively, in the ECG.
However, the above data from wedge preparations on abnormal repolarization must be recognized with some caution, as no significant transmural differences in repolarization durations were found in the clinical setting (Coronel et al., 2005). The ion currents that apparently mediate the propagation of action potential domes across the myocardium can also equilibrate action potential morphology, thereby reducing transmural differences in repolarization time. Nevertheless, increased Tpeak − Tend, a marker of global dispersion of repolarization, have been found in BrS patients compared to healthy subjects (Morita et al., 2008; Sangawa et al., 2009; Karim Talib et al., 2012; Maury et al., 2015). Moreover, using non-invasive ECG imaging, abnormal substrates in the form of conduction slowing, delayed epicardial repolarization and increased spatial gradients of repolarization have been identified in the right ventricular outflow tract (Zhang et al., 2015). The findings of this study suggest that abnormal repolarization causes ST segment elevation, whereas abnormal depolarization is responsible for the arrhythmic phenotype observed in BrS (Zhang et al., 2015). For BrS patients in whom ICa is reduced or IK is increased, abnormal repolarization is likely to be important in generating or sustaining VT/VF. Indeed, subjects with loss-of-function Ca2+ channel mutations have shortened QT intervals, which may support arrhythmias by a reentrant mechanism (Antzelevitch et al., 2007).
Abnormal restitution has long been recognized as a key determinant of arrhythmogenicity in arrhythmic disorders such as long QT and Brugada syndromes (Nishii et al., 2010; Osadchii et al., 2010; Osadchii, 2012a,b, 2013; Tse et al., 2016c,e). Restitution describes the dependence of a parameter on the previous diastolic interval (DI) (Nolasco and Dahlen, 1968; Franz et al., 1983; Tse et al., 2016g). Increased restitution gradients are associated with arrhythmogenesis, likely via the development of APD alternans, which manifest as T-wave alternans on the ECG (Pastore et al., 1999). APD alternans can produce steep gradients in repolarization and refractoriness, unidirectional conduction block and reentry (Tse et al., 2016a).
However, restitution analysis alone underestimates the extent of APD alternans (Matthews et al., 2012) and may not reliably predict arrhythmogenicity (Tse et al., 2016g). This may be due to restitution-independent mechanisms in the generation of alternans (Wu and Patwardhan, 2006; Jing and Patwardhan, 2012), such as divergence of ERP from APD (Tse et al., 2016c,d,e). APD alternans have been demonstrated in a canine model of BrS (Morita et al., 2006). T-wave alternans have also been in Brugada subjects, lending support to the hypothesis that repolarization abnormalities play a role in arrhythmogenesis (Nishizaki et al., 2005; Uchimura-Makita et al., 2014; Sakamoto et al., 2016).
Current-Load-Mismatch, Depolarization-Repolarization Balance and Excitation Wavelength (λ)
A third mechanism involves current-to-load mismatch in the right ventricle (Hoogendijk et al., 2010). This may occur at sites with structural abnormalities, where the myocardial tissue is interspersed with collagen or adipose tissue (Ten Sande et al., 2015). In a preclinical study, ajmaline-mediated sodium channel blockade led to conduction block and excitatory failure, which were associated with ST segment elevation in the pseudo-ECG recordings (Hoogendijk et al., 2011). Computational modeling work demonstrated that the balance between inward and outward currents could affect excitation and the ST segment elevation in concert (Hoogendijk et al., 2011). Thus, either reduced Ito or increased ICa could compensate for the reduced sodium current, in turn reducing the degree of ST segment elevation. Such preclinical findings are consistent with human data (Hoogendijk et al., 2010). In an explanted human heart, only failure of local excitation, but not delayed activation or early repolarization, correlated with ST segment elevation (Hoogendijk et al., 2010).
In patients with Brugada syndrome, structural abnormalities are indeed observed in the RV and RVOT, which would increase current-load mismatch and excitation failure (Coronel et al., 2005; Frustaci et al., 2005; Catalano et al., 2009). In a recent clinical study, a cohort of BrS patients underwent activation mapping procedures, which showed that electrogram fractionation together with conduction delay and electrocardiographic ST segment elevation were likely due to structural abnormalities in the sub-epicardial region of the right ventricle and right ventricular outflow tract (Ten Sande et al., 2015). It was suggested that current-to-load mismatches at discontinuities can cause conduction block. It should be recognized that abnormal depolarization does not act in isolation, but act in concert with discontinuous conduction to produce arrhythmias in BrS (Postema et al., 2008). This notion is consistent with the observations that BrS patients develop ventricular arrhythmias in their thirties, when interstitial fibrosis is more evident (Coronel et al., 2005; Schweizer et al., 2014; Nademanee et al., 2015). It also interacts with action potential repolarization and recovery to determine the excitation (λ) given by CV × ERP. Decreased λ has been associated with increased likelihood of reentrant arrhythmias not only in pre-clinical animal models, but also in BrS patients (Robyns et al., 2016; Tse, in press; Tse and Yan, in press).
Concluding Remarks
In conclusion, an increasing number of genes have been implicated in the pathogenesis of BrS. The genotype-phenotype correlation for many of these variants are poorly characterized. The commonest gene affected, SCN5A, is only implicated in approximately a fifth of patients. In some pedigrees, affected members did not have mutations in this gene (Probst et al., 2009). On genetic diseases, Marian wrote that “by definition the causal mutation cannot be absent in family members with the phenotype” (Marian, 2009). Therefore, other causal genes remain to be discovered. The use of animal models will be crucial in elucidating the electrophysiological mechanisms of arrhythmogenesis in these cases. Stem cell derived cardiomyocytes can be used to construct monolayers or organoid chambers (Kong et al., 2010; Weng et al., 2014), which can serve as useful platforms for disease modeling, high-throughput drug screening and cardiotoxicity testing (Li, 2012; Lui et al., 2012; Chow et al., 2013). Future studies will be needed to improve risk stratification strategies to determine the subset of patients with Brugada syndrome requiring ICD insertion.
Author Contributions
GT: Design of manuscript; interpreted primary research papers; drafted and critically revised the manuscript for important intellectual content; creation of figure. VL: Interpreted primary research papers. Critically revised the manuscript for important intellectual content. KL: Interpreted primary research papers; drafted and critically revised the manuscript for important intellectual content. VL: Critically revised the manuscript for important intellectual content. YC: Drafted and critically revised the manuscript for important intellectual content. WK: Drafted and critically revised the manuscript for important intellectual content. RL: Critically revised the manuscript for important intellectual content. BY: Drafted and critically revised the manuscript for important intellectual content.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
GT received a BBSRC Doctoral CASE Studentship at the University of Cambridge and thanks the Croucher Foundation of Hong Kong for the support of his Clinical Assistant Professorship. YC is supported by the ESRC for her Ph.D. studies at the University of Cambridge. BY received funding from the Research Grant Council of Hong Kong for his research.
References
Achaiah, A., and Andrews, N. (2016). Intoxication with alcohol: an underestimated trigger of Brugada syndrome? JRSM Open 7, 2054270416640153. doi: 10.1177/2054270416640153
Agullo-Pascual, E., Cerrone, M., and Delmar, M. (2014). Arrhythmogenic cardiomyopathy and Brugada syndrome: diseases of the connexome. FEBS Lett. 588, 1322–1330. doi: 10.1016/j.febslet.2014.02.008
Akai, J., Makita, N., Sakurada, H., Shirai, N., Ueda, K., Kitabatake, A., et al. (2000). A novel SCN5A mutation associated with idiopathic ventricular fibrillation without typical ECG findings of Brugada syndrome. FEBS Lett. 479, 29–34. doi: 10.1016/S0014-5793(00)01875-5
Amin, A. S., Verkerk, A. O., Bhuiyan, Z. A., Wilde, A. A., and Tan, H. L. (2005). Novel Brugada syndrome-causing mutation in ion-conducting pore of cardiac Na+ channel does not affect ion selectivity properties. Acta Physiol. Scand. 185, 291–301. doi: 10.1111/j.1365-201X.2005.01496.x
Andorin, A., and Probst, V. (2016). Reply to the Editor—Brugada syndrome is not an ECG. Heart Rhythm. 13, e292. doi: 10.1016/j.hrthm.2016.06.023
Antzelevitch, C. (2001). Transmural dispersion of repolarization and the T wave. Cardiovasc. Res. 50, 426–431.
Antzelevitch, C. (2006). Brugada syndrome. Pacing Clin. Electrophysiol. 29, 1130–1159. doi: 10.1111/j.1540-8159.2006.00507.x
Antzelevitch, C. (2008). Drug-induced spatial dispersion of repolarization. Cardiol. J. 15, 100–121.
Antzelevitch, C., Brugada, P., Brugada, J., and Brugada, R. (2005). Brugada syndrome: from cell to bedside. Curr. Probl. Cardiol. 30, 9–54. doi: 10.1016/j.cpcardiol.2004.04.005
Antzelevitch, C., and Oliva, A. (2006). Amplification of spatial dispersion of repolarization underlies sudden cardiac death associated with catecholaminergic polymorphic VT, long QT, short QT and Brugada syndromes. J. Intern. Med. 259, 48–58. doi: 10.1111/j.1365-2796.2005.01587.x
Antzelevitch, C., Pollevick, G. D., Cordeiro, J. M., Casis, O., Sanguinetti, M. C., Aizawa, Y., et al. (2007). Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 115, 442–449. doi: 10.1161/CIRCULATIONAHA.106.668392
Antzelevitch, C., Yan, G. X., Ackerman, M. J., Borggrefe, M., Corrado, D., Guo, J., et al. (2016). J-Wave syndromes expert consensus conference report: emerging concepts and gaps in knowledge: endorsed by the Asia Pacific Heart Rhythm Society (APHRS), the European Heart Rhythm Association (EHRA), the Heart Rhythm Society (HRS), and the Latin American Society of Cardiac Pacing and Electrophysiology (Sociedad Latinoamericana de Estimulacifin Cardíaca y Electrofisiología [SOLAECE]). Europace 13, e295–e324. doi: 10.1093/europace/euw235
Antzelevitch, C., Yan, G.-X., and Viskin, S. (2011). Rationale for the use of the terms J-wave syndromes and early repolarization. J. Am. Coll. Cardiol. 57, 1587–1590. doi: 10.1016/j.jacc.2010.11.038
Benson, D. W., Wang, D. W., Dyment, M., Knilans, T. K., Fish, F. A., Strieper, M. J., et al. (2003). Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J. Clin. Invest. 112, 1019–1028. doi: 10.1172/JCI18062
Bezzina, C. R., Barc, J., Mizusawa, Y., Remme, C. A., Gourraud, J. B., Simonet, F., et al. (2013). Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 45, 1044–1049. doi: 10.1038/ng.2712
Bezzina, C., Veldkamp, M. W., van den Berg, M. P., Postma, A. V., Rook, M. B., Viersma, J.-W., et al. (1999). A single Na+ channel mutation causing both long-QT and Brugada syndromes. Circ. Res. 85, 1206–1213. doi: 10.1161/01.res.85.12.1206
Boczek, N. J., Ye, D., Johnson, E. K., Wang, W., Crotti, L., Tester, D. J., et al. (2014). Characterization of SEMA3A-encoded semaphorin as a naturally occurring Kv4.3 protein inhibitor and its contribution to Brugada syndrome. Circ. Res. 115, 460–469. doi: 10.1161/CIRCRESAHA.115.303657
Bordachar, P., Reuter, S., Garrigue, S., Caï, X., Hocini, M., Jaïs, P., et al. (2004). Incidence, clinical implications and prognosis of atrial arrhythmias in Brugada syndrome. Eur. Heart J. 25, 879–884. doi: 10.1016/j.ehj.2004.01.004
Boukens, B. J., Sylva, M., de Gier-de Vries, C., Remme, C. A., Bezzina, C. R., Christoffels, V. M., et al. (2013). Reduced sodium channel function unmasks residual embryonic slow conduction in the adult right ventricular outflow tract. Circ. Res. 113, 137–141. doi: 10.1161/circresaha.113.301565
Brugada, J., Pappone, C., Berruezo, A., Vicedomini, G., Manguso, F., Ciconte, G., et al. (2015). Brugada syndrome phenotype elimination by epicardial substrate ablation. Circ. Arrhythm. Electrophysiol. 8, 1373–1381. doi: 10.1161/CIRCEP.115.003220
Brugada, P., and Brugada, J. (1992). Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J. Am. Coll. Cardiol. 20, 1391–1396.
Burashnikov, E., Pfeiffer, R., Barajas-Martinez, H., Delpón, E., Hu, D., Desai, M., et al. (2010). Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm 7, 1872–1882. doi: 10.1016/j.hrthm.2010.08.026
Campuzano, O., and Brugada, R. (2015). Age, genetics, and fibrosis in the Brugada syndrome. J. Am. Coll. Cardiol. 66, 1987–1989. doi: 10.1016/j.jacc.2015.09.006
Campuzano, O., Fernández-Falgueras, A., Iglesias, A., and Brugada, R. (2016). Brugada syndrome and PKP2: evidences and uncertainties. Int. J. Cardiol. 214, 403–405. doi: 10.1016/j.ijcard.2016.03.194
Catalano, O., Antonaci, S., Moro, G., Mussida, M., Frascaroli, M., Baldi, M., et al. (2009). Magnetic resonance investigations in Brugada syndrome reveal unexpectedly high rate of structural abnormalities. Eur. Heart J. 30, 2241–2248. doi: 10.1093/eurheartj/ehp252
Cerrone, M., and Delmar, M. (2014). Desmosomes and the sodium channel complex: implications for arrhythmogenic cardiomyopathy and Brugada syndrome. Trends Cardiovasc. Med. 24, 184–190. doi: 10.1016/j.tcm.2014.02.001
Cerrone, M., Lin, X., Zhang, M., Agullo-Pascual, E., Pfenniger, A., Chkourko Gusky, H., et al. (2014). Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 129, 1092–1103. doi: 10.1161/CIRCULATIONAHA.113.003077
Chen, Q., Kirsch, G. E., Zhang, D., Brugada, R., Brugada, J., Brugada, P., et al. (1998). Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392, 293–296. doi: 10.1038/32675
Chow, M., Boheler, K. R., and Li, R. A. (2013). Human pluripotent stem cell-derived cardiomyocytes for heart regeneration, drug discovery and disease modeling: from the genetic, epigenetic, and tissue modeling perspectives. Stem Cell Res. Ther. 4:97. doi: 10.1186/scrt308
Cohen, S. A. (1996). Immunocytochemical localization of rH1 sodium channel in adult rat heart atria and ventricle. Presence in terminal intercalated disks. Circulation 94, 3083–3086.
Coronel, R., Casini, S., Koopmann, T. T., Wilms-Schopman, F. J., Verkerk, A. O., de Groot, J. R., et al. (2005). Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation 112, 2769–2777. doi: 10.1161/CIRCULATIONAHA.105.532614
Cruz, F. M., Sanz-Rosa, D., Roche-Molina, M., García-Prieto, J., García-Ruiz, J. M., Pizarro, G., et al. (2015). Exercise triggers ARVC phenotype in mice expressing a disease-causing mutated version of human plakophilin-2. J. Am. Coll. Cardiol. 65, 1438–1450. doi: 10.1016/j.jacc.2015.01.045
Curcio, A., Mazzanti, A., Bloise, R., Monteforte, N., Indolfi, C., Priori, S. G., et al. (2016). Clinical presentation and outcome of Brugada syndrome diagnosed with the new 2013 criteria. J. Cardiovasc. Electrophysiol. 27, 937–943. doi: 10.1111/jce.12997
Delpón, E., Cordeiro, J. M., Núñez, L., Thomsen, P. E., Guerchicoff, A., Pollevick, G. D., et al. (2008). Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ. Arrhythm. Electrophysiol. 1, 209–218. doi: 10.1161/CIRCEP.107.748103
Dendramis, G. (2016). Brugada syndrome and Brugada phenocopy. The importance of a differential diagnosis. Int. J. Cardiol. 210, 25–27. doi: 10.1016/j.ijcard.2016.02.097
Derval, N., Simpson, C. S., Birnie, D. H., Healey, J. S., Chauhan, V., Champagne, J., et al. (2011). Prevalence and characteristics of early repolarization in the CASPER registry: cardiac arrest survivors with preserved ejection fraction registry. J. Am. Coll. Cardiol. 58, 722–728. doi: 10.1016/j.jacc.2011.04.022
Di Diego, J. M., and Antzelevitch, C. (2003). Cellular basis for ST-segment changes observed during ischemia. J. Electrocardiol. 36(Suppl.), 1–5. doi: 10.1016/j.jelectrocard.2003.09.001
Dumaine, R., Towbin, J. A., Brugada, P., Vatta, M., Nesterenko, D. V., Nesterenko, V. V., et al. (1999). Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ. Res. 85, 803–809.
Eckardt, L., Kirchhof, P., Johna, R., Haverkamp, W., Breithardt, G., and Borggrefe, M. (2001). Wolff-Parkinson-White syndrome associated with Brugada syndrome. Pacing Clin. Electrophysiol. 24, 1423–1424. doi: 10.1046/j.1460-9592.2001.01423.x
Enriquez, A., Brugada, J., and Baranchuk, A. (2016). Exercise-induced Brugada phenocopy. J. Cardiovasc. Electrophysiol. 27, 360–361. doi: 10.1111/jce.12838
Fish, J. M., and Antzelevitch, C. (2004). Role of sodium and calcium channel block in unmasking the Brugada syndrome. Heart Rhythm 1, 210–217. doi: 10.1016/j.hrthm.2004.03.061
Fish, J. M., and Antzelevitch, C. (2008). Cellular mechanism and arrhythmogenic potential of T-wave alternans in the Brugada syndrome. J. Cardiovasc. Electrophysiol. 19, 301–308. doi: 10.1111/j.1540-8167.2007.01025.x
Franz, M. R., Schaefer, J., Schöttler, M., Seed, W. A., and Noble, M. I. (1983). Electrical and mechanical restitution of the human heart at different rates of stimulation. Circ. Res. 53, 815–822.
Frustaci, A., Priori, S. G., Pieroni, M., Chimenti, C., Napolitano, C., Rivolta, I., et al. (2005). Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation 112, 3680–3687. doi: 10.1161/CIRCULATIONAHA.105.520999
George, S. A., Sciuto, K. J., Lin, J., Salama, M. E., Keener, J. P., Gourdie, R. G., et al. (2015). Extracellular sodium and potassium levels modulate cardiac conduction in mice heterozygous null for the Connexin43 gene. Pflugers Arch. 467, 2287–2297. doi: 10.1007/s00424-015-1698-0
Giudicessi, J. R., Ye, D., Kritzberger, C. J., Nesterenko, V. V., Tester, D. J., Antzelevitch, C., et al. (2012). Novel mutations in the KCND3-encoded Kv4.3 K+ channel associated with autopsy-negative sudden unexplained death. Hum. Mutat. 33, 989–997. doi: 10.1002/humu.22058
Giudicessi, J. R., Ye, D., Tester, D. J., Crotti, L., Mugione, A., Nesterenko, V. V., et al. (2011). Transient outward current (I(to)) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm 8, 1024–1032. doi: 10.1016/j.hrthm.2011.02.021
Gottschalk, B. H., Garcia-Niebla, J., Anselm, D. D., Jaidka, A., De Luna, A. B., and Baranchuk, A. (2016). New methodologies for measuring Brugada ECG patterns cannot differentiate the ECG pattern of Brugada syndrome from Brugada phenocopy. J. Electrocardiol. 49, 187–191. doi: 10.1016/j.jelectrocard.2015.12.011
Gourraud, J. B., Barc, J., Thollet, A., Le Scouarnec, S., Le Marec, H., Schott, J. J., et al. (2016). The Brugada syndrome: a rare arrhythmia disorder with complex inheritance. Front. Cardiovasc. Med. 3:9. doi: 10.3389/fcvm.2016.00009
Grant, R. P., Estes, E. H., and Doyle, J. T. (1951). Spatial vector electrocardiography; the clinical characteristics of S-T and T vectors. Circulation 3, 182–197.
Haïssaguerre, M., Derval, N., Sacher, F., Jesel, L., Deisenhofer, I., de Roy, L., et al. (2008). Sudden cardiac arrest associated with early repolarization. N. Engl. J. Med. 358, 2016–2023. doi: 10.1056/NEJMoa071968
Haruta, D., Matsuo, K., Tsuneto, A., Ichimaru, S., Hida, A., Sera, N., et al. (2011). Incidence and prognostic value of early repolarization pattern in the 12-lead electrocardiogram. Circulation 123, 2931–2937. doi: 10.1161/CIRCULATIONAHA.110.006460
Havakuk, O., and Viskin, S. (2016). A tale of 2 diseases: the history of long-QT syndrome and Brugada syndrome. J. Am. Coll. Cardiol. 67, 100–108. doi: 10.1016/j.jacc.2015.10.020
Hennessey, J. A., Marcou, C. A., Wang, C., Wei, E. Q., Wang, C., Tester, D. J., et al. (2013). FGF12 is a candidate Brugada syndrome locus. Heart Rhythm 10, 1886–1894. doi: 10.1016/j.hrthm.2013.09.064
Holst, A. G., Jensen, H. K., Eschen, O., Henriksen, F. L., Kanters, J., Bundgaard, H., et al. (2012). Low disease prevalence and inappropriate implantable cardioverter defibrillator shock rate in Brugada syndrome: a nationwide study. Europace 14, 1025–1029. doi: 10.1093/europace/eus002
Hoogendijk, M. G., Potse, M., Linnenbank, A. C., Verkerk, A. O., den Ruijter, H. M., van Amersfoorth, S. C., et al. (2010). Mechanism of right precordial ST-segment elevation in structural heart disease: excitation failure by current-to-load mismatch. Heart Rhythm 7, 238–248. doi: 10.1016/j.hrthm.2009.10.007
Hoogendijk, M. G., Potse, M., Vinet, A., de Bakker, J. M., and Coronel, R. (2011). ST segment elevation by current-to-load mismatch: an experimental and computational study. Heart Rhythm 8, 111–118. doi: 10.1016/j.hrthm.2010.09.066
Hothi, S. S., Ara, F., and Timperley, J. (2015). p.Y1449C SCN5A mutation associated with overlap disorder comprising conduction disease, Brugada syndrome, and atrial flutter. J. Cardiovasc. Electrophysiol. 26, 93–97. doi: 10.1111/jce.12470
Hu, D., Barajas-Martinez, H., Burashnikov, E., Springer, M., Wu, Y., Varro, A., et al. (2009). A mutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ. Cardiovasc. Genet. 2, 270–278. doi: 10.1161/CIRCGENETICS.108.829192
Hu, D., Barajas-Martínez, H., Pfeiffer, R., Dezi, F., Pfeiffer, J., Buch, T., et al. (2014a). Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J. Am. Coll. Cardiol. 64, 66–79. doi: 10.1016/j.jacc.2014.04.032
Hu, D., Barajas-Martínez, H., Terzic, A., Park, S., Pfeiffer, R., Burashnikov, E., et al. (2014b). ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int. J. Cardiol. 171, 431–442. doi: 10.1016/j.ijcard.2013.12.084
Ishikawa, T., Sato, A., Marcou, C. A., Tester, D. J., Ackerman, M. J., Crotti, L., et al. (2012). A novel disease gene for Brugada syndrome: sarcolemmal membrane-associated protein gene mutations impair intracellular trafficking of hNav1.5. Circ. Arrhythm. Electrophysiol. 5, 1098–1107. doi: 10.1161/CIRCEP.111.969972
Jing, L., and Patwardhan, A. (2012). Hysteresis in DI independent mechanisms in threshold for transition between 1:1 and 2:2 rhythms in pigs. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2012, 665–668. doi: 10.1109/EMBC.2012.6346019
Juang, J. M., Chen, C. Y., Chen, Y. H., Wu, I. C., Hsu, C. C., Chen, L. N., et al. (2015). Prevalence and prognosis of Brugada electrocardiogram patterns in an elderly Han Chinese population: a nation-wide community-based study (HALST cohort). Europace 17(Suppl. 2), ii54–ii62. doi: 10.1093/europace/euv141
Karim Talib, A., Sato, N., Sakamoto, N., Tanabe, Y., Takeuchi, T., Saijo, Y., et al. (2012). Enhanced transmural dispersion of repolarization in patients with J wave syndromes. J. Cardiovasc. Electrophysiol. 23, 1109–1114. doi: 10.1111/j.1540-8167.2012.02363.x
Kattygnarath, D., Maugenre, S., Neyroud, N., Balse, E., Ichai, C., Denjoy, I., et al. (2011). MOG1: a new susceptibility gene for Brugada syndrome. Circ. Cardiovasc. Genet. 4, 261–268. doi: 10.1161/CIRCGENETICS.110.959130
Klatsky, A. L., Oehm, R., Cooper, R. A., Udaltsova, N., and Armstrong, M. A. (2003). The early repolarization normal variant electrocardiogram: correlates and consequences. Am. J. Med. 115, 171–177. doi: 10.1016/S0002-9343(03)00355-3
Kong, C. W., Akar, F. G., and Li, R. A. (2010). Translational potential of human embryonic and induced pluripotent stem cells for myocardial repair: insights from experimental models. Thromb. Haemost. 104, 30–38. doi: 10.1160/TH10-03-0189
Kumar, S., and Kalman, J. M. (2016). Brugada syndrome diagnosed from the ECG Leads in the high intercostal spaces: searching for answers from a higher source? J. Cardiovasc. Electrophysiol. 27, 944–946. doi: 10.1111/jce.13016
Kyndt, F., Probst, V., Potet, F., Demolombe, S., Chevallier, J.-C., Baro, I., et al. (2001). Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation 104, 3081–3086. doi: 10.1161/hc5001.100834
Lambiase, P. D., Ahmed, A. K., Ciaccio, E. J., Brugada, R., Lizotte, E., Chaubey, S., et al. (2009). High-density substrate mapping in Brugada syndrome: combined role of conduction and repolarization heterogeneities in arrhythmogenesis. Circulation 120, 106–117. doi: 10.1161/CIRCULATIONAHA.108.771401
Leoni, A. L., Gavillet, B., Rougier, J. S., Marionneau, C., Probst, V., Le Scouarnec, S., et al. (2010). Variable Na(v)1.5 protein expression from the wild-type allele correlates with the penetrance of cardiac conduction disease in the Scn5a(+/−) mouse model. PLoS ONE 5:e9298. doi: 10.1371/journal.pone.0009298
Le Scouarnec, S., Karakachoff, M., Gourraud, J. B., Lindenbaum, P., Bonnaud, S., Portero, V., et al. (2015). Testing the burden of rare variation in arrhythmia-susceptibility genes provides new insights into molecular diagnosis for Brugada syndrome. Hum. Mol. Genet. 24, 2757–2763. doi: 10.1093/hmg/ddv036
Li, R. A. (2012). The use of induced pluripotent stem cells for disease modeling: what are the promises and hurdles? Drug Discovery Today: Disease Models 9, e143–e145. doi: 10.1016/j.ddmod.2012.10.001
Lui, K. O., Stachel, M. W., Lieu, D. K., Li, R. A., and Bu, L. (2012). Induced pluripotent stem cells as a disease model for studying inherited arrhythmias: promises and hurdles. Drug Discovery Today: Disease Models 9, e199–e207. doi: 10.1016/j.ddmod.2012.09.001
Liu, M., Yang, K. C., and Dudley, S. C. Jr. (2014). Cardiac sodium channel mutations: why so many phenotypes? Nat. Rev. Cardiol. 11, 607–615. doi: 10.1038/nrcardio.2014.85
Liu, H., Chatel, S., Simard, C., Syam, N., Salle, L., Probst, V., et al. (2013). Molecular genetics and functional anomalies in a series of 248 Brugada cases with 11 mutations in the TRPM4 channel. PLoS ONE 8:e54131. doi: 10.1371/journal.pone.0054131
London, B., Michalec, M., Mehdi, H., Zhu, X., Kerchner, L., Sanyal, S., et al. (2007). Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation 116, 2260–2268. doi: 10.1161/CIRCULATIONAHA.107.703330
Lukas, A., and Antzelevitch, C. (1996). Phase 2 reentry as a mechanism of initiation of circus movement reentry in canine epicardium exposed to simulated ischemia. Cardiovasc. Res. 32, 593–603.
Madeira, M., Caetano, F., Providência, R., Almeida, I., Trigo, J., Nascimento, J., et al. (2015). Fever-induced type 1 Brugada pattern. Rev. Port. Cardiol. 34, 287. e281–e287. doi: 10.1016/j.repc.2014.10.002
Maoz, A., Christini, D. J., and Krogh-Madsen, T. (2014). Dependence of phase-2 reentry and repolarization dispersion on epicardial and transmural ionic heterogeneity: a simulation study. Europace 16, 458–465. doi: 10.1093/europace/eut379
Marian, A. J. (2009). Nature's genetic gradients and the clinical phenotype. Circ. Cardiovasc. Genet. 2, 537–539. doi: 10.1161/CIRCGENETICS.109.921940
Martin, C. A., Grace, A. A., and Huang, C. L. (2011a). Refractory dispersion promotes conduction disturbance and arrhythmias in a Scn5a (+/−) mouse model. Pflügers Arch 462, 495–504. doi: 10.1007/s00424-011-0989-3
Martin, C. A., Grace, A. A., and Huang, C. L. (2011b). Spatial and temporal heterogeneities are localized to the right ventricular outflow tract in a heterozygotic Scn5a mouse model. Am. J. Physiol. Heart Circ. Physiol. 300, H605–H616. doi: 10.1152/ajpheart.00824.2010
Martin, C. A., Zhang, Y., Grace, A. A., and Huang, C. L. (2010). Increased right ventricular repolarization gradients promote arrhythmogenesis in a murine model of Brugada syndrome. J. Cardiovasc. Electrophysiol. 21, 1153–1159. doi: 10.1111/j.1540-8167.2010.01767.x
Martini, B. (2016). Brugada syndrome is not an ECG. Heart Rhythm. 13, e292. doi: 10.1016/j.hrthm.2016.06.022
Martini, B., Nava, A., Thiene, G., Buja, G. F., Canciani, B., Scognamiglio, R., et al. (1989). Ventricular fibrillation without apparent heart disease: description of six cases. Am. Heart J. 118, 1203–1209.
Matthews, G. D., Guzadhur, L., Grace, A., and Huang, C. L. (2012). Nonlinearity between action potential alternans and restitution, which both predict ventricular arrhythmic properties in Scn5a+/− and wild-type murine hearts. J. Appl. Physiol. (1985) 112, 1847–1863. doi: 10.1152/japplphysiol.00039.2012
Maury, P., Sacher, F., Gourraud, J. B., Pasquié, J. L., Raczka, F., Bongard, V., et al. (2015). Increased Tpeak-Tend interval is highly and independently related to arrhythmic events in Brugada syndrome. Heart Rhythm 12, 2469–2476. doi: 10.1016/j.hrthm.2015.07.029
Medeiros-Domingo, A., Tan, B. H., Crotti, L., Tester, D. J., Eckhardt, L., Cuoretti, A., et al. (2010). Gain-of-function mutation S422L in the KCNJ8-encoded cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave syndromes. Heart Rhythm 7, 1466–1471. doi: 10.1016/j.hrthm.2010.06.016
Mehta, M. C., and Jain, A. C. (1995). Early repolarization on scalar electrocardiogram. Am. J. Med. Sci. 309, 305–311.
Morita, H., Fukushima-Kusano, K., Nagase, S., Miyaji, K., Hiramatsu, S., Banba, K., et al. (2004). Sinus node function in patients with Brugada-type ECG. Circ. J. 68, 473–476. doi: 10.1253/circj.68.473
Morita, H., Zipes, D. P., Fukushima-Kusano, K., Nagase, S., Nakamura, K., Morita, S. T., et al. (2008). Repolarization heterogeneity in the right ventricular outflow tract: correlation with ventricular arrhythmias in Brugada patients and in an in vitro canine Brugada model. Heart Rhythm 5, 725–733. doi: 10.1016/j.hrthm.2008.02.028
Morita, H., Zipes, D. P., Lopshire, J., Morita, S. T., and Wu, J. (2006). T wave alternans in an in vitro canine tissue model of Brugada syndrome. Am. J. Physiol. Heart Circ. Physiol. 291, H421–H428. doi: 10.1152/ajpheart.01259.2005
Nademanee, K., Raju, H., de Noronha, S. V., Papadakis, M., Robinson, L., Rothery, S., et al. (2015). Fibrosis, Connexin-43, and Conduction Abnormalities in the Brugada Syndrome. J. Am. Coll. Cardiol. 66, 1976–1986. doi: 10.1016/j.jacc.2015.08.862
Nademanee, K., Veerakul, G., Chandanamattha, P., Chaothawee, L., Ariyachaipanich, A., Jirasirirojanakorn, K., et al. (2011). Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation 123, 1270–1279. doi: 10.1161/circulationaha.110.972612
Nademanee, K., Veerakul, G., Nimmannit, S., Chaowakul, V., Bhuripanyo, K., Likittanasombat, K., et al. (1997). Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation 96, 2595–2600.
Nagase, S., Kusano, K. F., Morita, H., Fujimoto, Y., Kakishita, M., Nakamura, K., et al. (2002). Epicardial electrogram of the right ventricular outflow tract in patients with the brugada syndrome: using the epicardial lead. J. Am. Coll. Cardiol. 39, 1992–1995. doi: 10.1016/S0735-1097(02)01888-0
Nakajima, T., Wu, J., Kaneko, Y., Ashihara, T., Ohno, S., Irie, T., et al. (2012). KCNE3 T4A as the genetic basis of Brugada-pattern electrocardiogram. Circ. J. 76, 2763–2772. doi: 10.1253/circj.CJ-12-0551
Nava, A., Canciani, B., Schiavinato, M. L., and Martini, B. (1988). La repolarisation precoce dans le precordiales droites: trouble de la conduction intraventriculaire droite? Correlations de l'electrocardiographie-vectorcardiographie avec l'electro-physiologie. Mises a Jour Cardiologiques 17, 157–159.
Ng, C. T., Ong, H. Y., Cheok, C., Chua, T. S., and Ching, C. K. (2012). Prevalence of electrocardiographic abnormalities in an unselected young male multi-ethnic South-East Asian population undergoing pre-participation cardiovascular screening: results of the Singapore Armed Forces Electrocardiogram and Echocardiogram screening protocol. Europace 14, 1018–1024. doi: 10.1093/europace/eur424
Nishii, N., Nagase, S., Morita, H., Kusano, K. F., Namba, T., Miura, D., et al. (2010). Abnormal restitution property of action potential duration and conduction delay in Brugada syndrome: both repolarization and depolarization abnormalities. Europace 12, 544–552. doi: 10.1093/europace/eup432
Nishizaki, M., Fujii, H., Sakurada, H., Kimura, A., and Hiraoka, M. (2005). Spontaneous T wave alternans in a patient with Brugada syndrome–responses to intravenous administration of class I antiarrhythmic drug, glucose tolerance test, and atrial pacing. J. Cardiovasc. Electrophysiol. 16, 217–220. doi: 10.1046/j.1540-8167.2004.40411.x
Nolasco, J. B., and Dahlen, R. W. (1968). A graphic method for the study of alternation in cardiac action potentials. J. Appl. Physiol. 25, 191–196.
Noseworthy, P. A., Tikkanen, J. T., Porthan, K., Oikarinen, L., Pietilä, A., Harald, K., et al. (2011). The early repolarization pattern in the general population: clinical correlates and heritability. J. Am. Coll. Cardiol. 57, 2284–2289. doi: 10.1016/j.jacc.2011.04.003
Nunn, L. M., Bhar-Amato, J., Lowe, M. D., Macfarlane, P. W., Rogers, P., McKenna, W. J., et al. (2011). Prevalence of J-point elevation in sudden arrhythmic death syndrome families. J. Am. Coll. Cardiol. 58, 286–290. doi: 10.1016/j.jacc.2011.03.028
Ohno, S., Zankov, D. P., Ding, W. G., Itoh, H., Makiyama, T., Doi, T., et al. (2011). KCNE5 (KCNE1L) variants are novel modulators of Brugada syndrome and idiopathic ventricular fibrillation. Circ. Arrhythm. Electrophysiol. 4, 352–361. doi: 10.1161/CIRCEP.110.959619
Osadchii, O. E. (2012a). Dofetilide promotes repolarization abnormalities in perfused Guinea-pig heart. Cardiovasc. Drugs Ther. 26, 489–500. doi: 10.1007/s10557-012-6405-1
Osadchii, O. E. (2012b). Effects of ventricular pacing protocol on electrical restitution assessments in guinea-pig heart. Exp. Physiol. 97, 807–821. doi: 10.1113/expphysiol.2012.065219
Osadchii, O. E. (2013). Quinidine elicits proarrhythmic changes in ventricular repolarization and refractoriness in guinea-pig. Can. J. Physiol. Pharmacol. 91, 306–315. doi: 10.1139/cjpp-2012-0379
Osadchii, O. E., Larsen, A. P., and Olesen, S. P. (2010). Predictive value of electrical restitution in hypokalemia-induced ventricular arrhythmogenicity. Am. J. Physiol. Heart Circ. Physiol. 298, H210–H220. doi: 10.1152/ajpheart.00695.2009
Osher, H. L., and Wolff, L. (1953). Electrocardiographic pattern simulating acute myocardial injury. Am. J. Med. Sci. 226, 541–545.
Pastore, J. M., Girouard, S. D., Laurita, K. R., Akar, F. G., and Rosenbaum, D. S. (1999). Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation 99, 1385–1394.
Peters, S. (2014). Arrhythmogenic cardiomyopathy and provocable Brugada ECG in a patient caused by missense mutation in plakophilin-2. Int. J. Cardiol. 173, 317–318. doi: 10.1016/j.ijcard.2014.03.071
Peters, S. (2015). Is Brugada syndrome a variant of arrhythmogenic cardiomyopathy? Int. J. Cardiol. 189, 88–90. doi: 10.1016/j.ijcard.2015.03.394
Portero, V., Le Scouarnec, S., Es-Salah-Lamoureux, Z., Burel, S., Gourraud, J.-B., Bonnaud, S., et al. (2016). Dysfunction of the voltage-gated K+ channel β2 subunit in a familial case of Brugada syndrome. J. Am. Heart Assoc. 5:e003122. doi: 10.1161/JAHA.115.003122
Postema, P. G., van Dessel, P. F., de Bakker, J. M., Dekker, L. R., Linnenbank, A. C., Hoogendijk, M. G., et al. (2008). Slow and discontinuous conduction conspire in Brugada syndrome: a right ventricular mapping and stimulation study. Circ. Arrhythm. Electrophysiol. 1, 379–386. doi: 10.1161/CIRCEP.108.790543
Postema, P. G., van Dessel, P. F., Kors, J. A., Linnenbank, A. C., van Herpen, G., Ritsema van Eck, H. J., et al. (2010). Local depolarization abnormalities are the dominant pathophysiologic mechanism for type 1 electrocardiogram in brugada syndrome a study of electrocardiograms, vectorcardiograms, and body surface potential maps during ajmaline provocation. J. Am. Coll. Cardiol. 55, 789–797. doi: 10.1016/j.jacc.2009.11.033
Priori, S. G., Wilde, A. A., Horie, M., Cho, Y., Behr, E. R., Berul, C., et al. (2013). Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace 15, 1389–1406. doi: 10.1093/europace/eut272
Probst, V., Kyndt, F., Potet, F., Trochu, J.-N., Mialet, G., Demolombe, S., et al. (2003). Haploinsufficiency in combination with aging causes SCN5A-linked hereditary Lenégre disease. J. Am. Coll. Cardiol. 41, 643–652. doi: 10.1016/S0735-1097(02)02864-4
Probst, V., Veltmann, C., Eckardt, L., Meregalli, P. G., Gaita, F., Tan, H. L., et al. (2010). Long-term prognosis of patients diagnosed with Brugada syndrome: results from the FINGER Brugada syndrome registry. Circulation 121, 635–643. doi: 10.1161/CIRCULATIONAHA.109.887026
Probst, V., Wilde, A. A., Barc, J., Sacher, F., Babuty, D., Mabo, P., et al. (2009). SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ. Cardiovasc. Genet. 2, 552–557. doi: 10.1161/CIRCGENETICS.109.853374
Reinhard, W., Kaess, B. M., Debiec, R., Nelson, C. P., Stark, K., Tobin, M. D., et al. (2011). Heritability of early repolarization: a population-based study. Circ. Cardiovasc. Genet. 4, 134–138. doi: 10.1161/CIRCGENETICS.110.958298
Remme, C. A., Wilde, A. A., and Bezzina, C. R. (2008). Cardiac sodium channel overlap syndromes: different faces of SCN5A mutations. Trends Cardiovasc. Med. 18, 78–87. doi: 10.1016/j.tcm.2008.01.002
Rhett, J. M., and Gourdie, R. G. (2012). The perinexus: a new feature of Cx43 gap junction organization. Heart Rhythm 9, 619–623. doi: 10.1016/j.hrthm.2011.10.003
Rhett, J. M., Veeraraghavan, R., Poelzing, S., and Gourdie, R. G. (2013). The perinexus: sign-post on the path to a new model of cardiac conduction? Trends Cardiovasc. Med. 23, 222–228. doi: 10.1016/j.tcm.2012.12.005
Ricci, M. T., Menegon, S., Vatrano, S., Mandrile, G., Cerrato, N., Carvalho, P., et al. (2014). SCN1B gene variants in Brugada syndrome: a study of 145 SCN5A-negative patients. Sci. Rep. 4:6470. doi: 10.1038/srep06470
Riuró, H., Beltran-Alvarez, P., Tarradas, A., Selga, E., Campuzano, O., Vergés, M., et al. (2013). A missense mutation in the sodium channel beta2 subunit reveals SCN2B as a new candidate gene for Brugada syndrome. Hum. Mutat. 34, 961–966. doi: 10.1002/humu.22328
Robyns, T., Lu, H. R., Gallacher, D. J., Garweg, C., Ector, J., Willems, R., et al. (2016). Evaluation of index of cardio-electrophysiological balance (iCEB) as a new biomarker for the identification of patients at increased arrhythmic risk. Ann. Noninvasive Electrocardiol. 21, 294–304. doi: 10.1111/anec.12309
Roden, D. M. (2010). Brugada syndrome: lots of questions, some answers. Heart Rhythm 7, 47–49. doi: 10.1016/j.hrthm.2009.10.016
Rosso, R., Glikson, E., Belhassen, B., Katz, A., Halkin, A., Steinvil, A., et al. (2012). Distinguishing “benign” from “malignant early repolarization”: the value of the ST-segment morphology. Heart Rhythm 9, 225–229. doi: 10.1016/j.hrthm.2011.09.012
Rosso, R., Kogan, E., Belhassen, B., Rozovski, U., Scheinman, M. M., Zeltser, D., et al. (2008). J-point elevation in survivors of primary ventricular fibrillation and matched control subjects: incidence and clinical significance. J. Am. Coll. Cardiol. 52, 1231–1238. doi: 10.1016/j.jacc.2008.07.010
Royer, A., van Veen, T. A., Le Bouter, S., Marionneau, C., Griol-Charhbili, V., Léoni, A. L., et al. (2005). Mouse model of SCN5A-linked hereditary Lenegre's disease: age-related conduction slowing and myocardial fibrosis. Circulation 111, 1738–1746. doi: 10.1161/01.CIR.0000160853.19867.61
Sakamoto, S., Takagi, M., Tatsumi, H., Doi, A., Sugioka, K., Hanatani, A., et al. (2016). Utility of T-wave alternans during night time as a predictor for ventricular fibrillation in patients with Brugada syndrome. Heart Vessels 31, 947–956. doi: 10.1007/s00380-015-0692-y
Sangawa, M., Morita, H., Nakatsu, T., Nishii, N., Miura, D., Miura, A., et al. (2009). Abnormal transmural repolarization process in patients with Brugada syndrome. Heart Rhythm 6, 1163–1169. doi: 10.1016/j.hrthm.2009.04.037
Schott, J. J., Alshinawi, C., Kyndt, F., Probst, V., Hoorntje, T. M., Hulsbeek, M., et al. (1999). Cardiac conduction defects associate with mutations in SCN5A. Nat. Genet. 23, 20–21. doi: 10.1038/12618
Schweizer, P. A., Fink, T., Yampolsky, P., Seyler, C., Fabritz, L., Kirchhof, P., et al. (2014). Generation and characterization of SCN5A loss-of-function mutant mice modeling human brugada syndrome. Eur. Heart J. 34:4556. doi: 10.1093/eurheartj/eht310.4556
Shimizu, W., Matsuo, K., Takagi, M., Tanabe, Y., Aiba, T., Taguchi, A., et al. (2000). Body surface distribution and response to drugs of ST segment elevation in Brugada syndrome: clinical implication of eighty-seven-lead body surface potential mapping and its application to twelve-lead electrocardiograms. J. Cardiovasc. Electrophysiol. 11, 396–404. doi: 10.1111/j.1540-8167.2000.tb00334.x
Shinde, R., Shinde, S., Makhale, C., Grant, P., Sathe, S., Durairaj, M., et al. (2007). Occurrence of “J waves” in 12-lead ECG as a marker of acute ischemia and their cellular basis. Pacing Clin. Electrophysiol. 30, 817–819. doi: 10.1111/j.1540-8159.2007.00760.x
Sicouri, S., Blazek, J., Belardinelli, L., and Antzelevitch, C. (2012). Electrophysiological characteristics of canine superior vena cava sleeve preparations: effect of ranolazine. Circ. Arrhythm. Electrophysiol. 5, 371–379. doi: 10.1161/CIRCEP.111.969493
Stokoe, K. S., Balasubramaniam, R., Goddard, C. A., Colledge, W. H., Grace, A. A., and Huang, C. L. (2007). Effects of flecainide and quinidine on arrhythmogenic properties of Scn5a+/− murine hearts modelling the Brugada syndrome. J. Physiol. 581, 255–275. doi: 10.1113/jphysiol.2007.128785
Takehara, N., Makita, N., Kawabe, J., Sato, N., Kawamura, Y., Kitabatake, A., et al. (2004). A cardiac sodium channel mutation identified in Brugada syndrome associated with atrial standstill. J. Intern. Med. 255, 137–142. doi: 10.1046/j.0954-6820.2003.01247.x
Tan, H. L., Bink-Boelkens, M. T., Bezzina, C. R., Viswanathan, P. C., Beaufort-Krol, G. C., van Tintelen, P. J., et al. (2001). A sodium-channel mutation causes isolated cardiac conduction disease. Nature 409, 1043–1047. doi: 10.1038/35059090
Ten Sande, J. N., Coronel, R., Conrath, C. E., Driessen, A. H., de Groot, J. R., Tan, H. L., et al. (2015). ST-segment elevation and fractionated electrograms in Brugada syndrome patients arise from the same structurally abnormal subepicardial RVOT area but have a different mechanism. Circ. Arrhythm. Electrophysiol. 8, 1382–1392. doi: 10.1161/CIRCEP.115.003366
Tse, G. (in press). (Tpeak-Tend)/QRS (Tpeak-Tend)/(QT x QRS): novel markers for predicting arrhythmic risk in the Brugada syndrome. Europace. doi: 10.1093/europace/euw194
Tse, G., Lai, E. T., Lee, A. P., Yan, B. P., and Wong, S. H. (2016a). Electrophysiological mechanisms of gastrointestinal arrhythmogenesis: lessons from the heart. Front. Physiol. 7:230. doi: 10.3389/fphys.2016.00230
Tse, G., Lai, E. T., Yeo, J. M., and Yan, B. P. (2016b). Electrophysiological mechanisms of Bayés syndrome: insights from clinical and mouse studies. Front. Physiol. 7:188. doi: 10.3389/fphys.2016.00188
Tse, G., Tse, V., and Yeo, J. M. (2016c). Ventricular anti-arrhythmic effects of heptanol in hypokalaemic, Langendorff-perfused mouse hearts. Biomed. Rep. 4, 313–324. doi: 10.3892/br.2016.577
Tse, G., Tse, V., Yeo, J. M., and Sun, B. (2016d). Atrial anti-arrhythmic effects of heptanol in Langendorff-perfused mouse hearts. PLoS ONE 11:e0148858. doi: 10.1371/journal.pone.0148858
Tse, G., Wong, S. T., Tse, V., Lee, Y. T., Lin, H. Y., and Yeo, J. M. (2016g). Cardiac dynamics: alternans and arrhythmogenesis. J. Arrhythm. 32, 411–417. doi: 10.1016/j.joa.2016.02.009
Tse, G., Wong, S. T., Tse, V., and Yeo, J. M. (2016h). Determination of action potential wavelength restitution in Scn5a+/− mouse hearts modelling human Brugada syndrome. J. Physiol. Accepted.
Tse, G., Wong, S. T., Tse, V., and Yeo, J. M. (2016i). Depolarization vs. repolarization: what is the mechanism of ventricular arrhythmogenesis underlying sodium channel haploinsufficiency in mouse hearts? Acta Physiol. (Oxf). doi: 10.1111/apha.12694. [Epub ahead of print].
Tse, G., Wong, S. T., Tse, V., and Yeo, J. M. (2016e). Restitution analysis of alternans using dynamic pacing and its comparison with S1S2 restitution in heptanol-treated, hypokalaemic Langendorff-perfused mouse hearts. Biomed Rep. 4, 673–680. doi: 10.3892/br.2016.659
Tse, G., Wong, S. T., Tse, V., and Yeo, J. M. (2016f). Variability in local action potential durations, dispersion of repolarization and wavelength restitution in aged wild-type and Scn5a+/− mouse hearts modelling human Brugada syndrome. J. Geriatr. Cardiol.
Tse, G., and Yan, B. P. (in press). Traditional novel electrocardiographic conduction repolarization markers of sudden cardiac death. Europace. doi: 10.1093/europace/euw280
Tsuboi, M., and Antzelevitch, C. (2006). Cellular basis for electrocardiographic and arrhythmic manifestations of Andersen-Tawil syndrome (LQT7). Heart Rhythm 3, 328–335. doi: 10.1016/j.hrthm.2005.11.026
Tukkie, R., Sogaard, P., Vleugels, J., de Groot, I. K., Wilde, A. A., and Tan, H. L. (2004). Delay in right ventricular activation contributes to Brugada syndrome. Circulation 109, 1272–1277. doi: 10.1161/01.CIR.0000118467.53182.D1
Uchimura-Makita, Y., Nakano, Y., Tokuyama, T., Fujiwara, M., Watanabe, Y., Sairaku, A., et al. (2014). Time-domain T-wave alternans is strongly associated with a history of ventricular fibrillation in patients with Brugada syndrome. J. Cardiovasc. Electrophysiol. 25, 1021–1027. doi: 10.1111/jce.12441
Ueda, K., Hirano, Y., Higashiuesato, Y., Aizawa, Y., Hayashi, T., Inagaki, N., et al. (2009). Role of HCN4 channel in preventing ventricular arrhythmia. J. Hum. Genet. 54, 115–121. doi: 10.1038/jhg.2008.16
Valdivia, C. R., Tester, D. J., Rok, B. A., Porter, C.-B., Munger, T. M., Jahangir, A., et al. (2004). A trafficking defective, Brugada syndrome-causing SCN5A mutation rescued by drugs. Cardiovasc. Res. 62, 53–62. doi: 10.1016/j.cardiores.2004.01.022
Van Driest, S. L., Wells, Q. S., Stallings, S., Bush, W. S., Gordon, A., Nickerson, D. A., et al. (2016). ASsociation of arrhythmia-related genetic variants with phenotypes documented in electronic medical records. JAMA 315, 47–57. doi: 10.1001/jama.2015.17701
Veeraraghavan, R., Gourdie, R. G., and Poelzing, S. (2014a). Mechanisms of cardiac conduction: a history of revisions. Am. J. Physiol. Heart Circ. Physiol. 306, H619–H627. doi: 10.1152/ajpheart.00760.2013
Veeraraghavan, R., Lin, J., Hoeker, G. S., Keener, J. P., Gourdie, R. G., and Poelzing, S. (2015). Sodium channels in the Cx43 gap junction perinexus may constitute a cardiac ephapse: an experimental and modeling study. Pflugers Arch. 467, 2093–2105. doi: 10.1007/s00424-014-1675-z
Veeraraghavan, R., Poelzing, S., and Gourdie, R. G. (2014b). Old cogs, new tricks: a scaffolding role for connexin43 and a junctional role for sodium channels? FEBS Lett. 588, 1244–1248. doi: 10.1016/j.febslet.2014.01.026
Veldkamp, M. W., Viswanathan, P. C., Bezzina, C., Baartscheer, A., Wilde, A. M. A., and Balser, J. R. (2000). Two distinct congenital arrhythmias evoked by a multidysfunctional Na(+) channel. Circ. Res. 86, E91–E97. doi: 10.1161/01.RES.86.9.e91
Viskin, S., Rosso, R., Friedensohn, L., Havakuk, O., and Wilde, A. A. (2015). Everybody has Brugada syndrome until proven otherwise? Heart Rhythm 12, 1595–1598. doi: 10.1016/j.hrthm.2015.04.017
Wang, D., Yan, G. X., and Antzelevitch, C. (2011). The J wave syndromes and their role in sudden cardiac death. Card. Electrophysiol. Clin. 3, 47–56. doi: 10.1016/j.ccep.2010.10.014
Wang, Q., Ohno, S., Ding, W. G., Fukuyama, M., Miyamoto, A., Itoh, H., et al. (2014). Gain-of-function KCNH2 mutations in patients with Brugada syndrome. J. Cardiovasc. Electrophysiol. 25, 522–530. doi: 10.1111/jce.12361
Wang, Q., Shen, J., Li, Z., Timothy, K., Vincent, G. M., Priori, S. G., et al. (1995). Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum. Mol. Genet. 4, 1603–1607.
Watanabe, H., Koopmann, T. T., Le Scouarnec, S., Yang, T., Ingram, C. R., Schott, J. J., et al. (2008). Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J. Clin. Invest. 118, 2260–2268. doi: 10.1172/JCI33891
Weng, Z., Kong, C. W., Ren, L., Karakikes, I., Geng, L., He, J., et al. (2014). A simple, cost-effective but highly efficient system for deriving ventricular cardiomyocytes from human pluripotent stem cells. Stem Cells Dev. 23, 1704–1716. doi: 10.1089/scd.2013.0509
Wilde, A. A., Postema, P. G., Di Diego, J. M., Viskin, S., Morita, H., Fish, J. M., et al. (2010). The pathophysiological mechanism underlying Brugada syndrome: depolarization versus repolarization. J. Mol. Cell. Cardiol. 49, 543–553. doi: 10.1016/j.yjmcc.2010.07.012
Wu, R., and Patwardhan, A. (2006). Mechanism of repolarization alternans has restitution of action potential duration dependent and independent components. J. Cardiovasc. Electrophysiol. 17, 87–93. doi: 10.1111/j.1540-8167.2005.00319.x
Yan, G. X., and Antzelevitch, C. (1999). Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation 100, 1660–1666.
Keywords: arrhythmia, Brugada syndrome, sodium channel, repolarization, depolarization, risk stratification
Citation: Tse G, Liu T, Li KHC, Laxton V, Chan YWF, Keung W, Li RA and Yan BP (2016) Electrophysiological Mechanisms of Brugada Syndrome: Insights from Pre-clinical and Clinical Studies. Front. Physiol. 7:467. doi: 10.3389/fphys.2016.00467
Received: 01 August 2016; Accepted: 27 September 2016;
Published: 18 October 2016.
Edited by:
Olivier Bernus, Université de Bordeaux, FranceReviewed by:
Flavien Charpentier, Inserm, FranceRichard David Walton, Université de Bordeaux, France
Ruben Coronel, University of Amsterdam, Netherlands
Copyright © 2016 Tse, Liu, Li, Laxton, Chan, Keung, Li and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gary Tse, dHNlZ0BjdWhrLmVkdS5oaw==
Bryan P. Yan, YnJ5YW4ueWFuQGN1aGsuZWR1Lmhr