 Paola Romecín
Paola Romecín Esther G. Navarro
Esther G. Navarro David Iyú
David Iyú Joaquín García-Estañ
Joaquín García-Estañ Noemí M. Atucha
Noemí M. Atucha- Department Fisiología, Faculty Medicina, Instituto Murciano de Investigación Biosanitaria, Universidad de Murcia, Murcia, Spain
Previously, we have found that intracellular calcium homeostasis is altered in platelets from an experimental model of liver cirrhosis, the bile-duct ligated (BDL) rat; these alterations are compatible with the existence of a hypercoagulable state and related to an enhanced intracellular calcium release evoked by thrombin and an increased amount of calcium stored in the intracellular organelles. In the present study we have investigated the role of bile acids in those alterations of the BDL cirrhotic model. Cholic acid (CA) or deoxycholic acid (DCA) did not change P-selectin expression or platelet aggregation in any group but elevated baseline platelet calcium levels. Incubation with both bile acids reduced calcium release after stimulation with thrombin in the absence of extracellular calcium. Pretreatment with CA but not with DCA reduced significantly thrombin-induced calcium entry in all three experimental groups. The capacitative calcium entry was also significantly lower in platelets pretreated with both bile acids. The simultaneous addition of thapsigargin and ionomycin to estimate the total amount of calcium in platelet internal stores was decreased by pretreatment with both CA and DCA, although these changes were significantly different in the control rats only with CA and in the BDL platelets with DCA. These results indicate that CA and DCA reduce calcium movements in platelets of control and BDL animals, thus suggesting that bile acids do not participate in the alterations observed in the BDL cirrotic model.
Introduction
Hypercoagulability in patients with primary biliary cirrhosis has been attributed to elevated fibrinogen and hyperactivity of platelets, which is not observed in non-cholestatic liver diseases, such as chronic hepatitis C, and alcoholic cirrhosis. These changes are believed to be the result of a marked systemic inflammatory activity (Ben-Ari et al., 1997; Papadopoulos et al., 2007). The underlying mechanisms for altered platelet function in cholestasis are unclear. Several mechanisms have been suggested (Bowen et al., 1988; Pihusch et al., 2002; Atucha et al., 2007; Witters et al., 2010; Tripodi et al., 2011), including an intrinsic change in platelet function, e.g., increased aggregation resulting from altered intracellular calcium (Ca2+) homeostasis or decreased aggregation due to a storage pool deficiency, the effect of a plasmatic factor such as bile acids or bilirubin or because of the effect of release of ADP-degrading enzymes in the circulation. Regarding platelet activation, the necessary increase in cytoplasmic Ca2+ levels, both by release from internal stores and entry of extracellular Ca2+, has been reported to be defective both in experimental models of liver cirrhosis and patients (Bandi et al., 1997; Atucha et al., 2007; Annie-Jeyachristy et al., 2008). In previous studies from our laboratory, a greater amount of stored Ca2+ together with an increased activity of SERCA, were observed in platelets obtained from a rat model of biliary cirrhosis (Atucha et al., 2007).
The role of bile acids in mediating these alterations in Ca2+ signaling and platelet function is not known. Deoxycholic acid (DCA) has been observed to cause profound Ca2+ release from intracellular stores and entry from the extracellular medium of intact or permeabilized fibroblast cells (Lau et al., 2005). In diets containing cholesterol and cholic acid (CA), increased thrombin and adenosine diphosphate (ADP)-induced platelet aggregation and an acceleration of the platelet-rich plasma clotting time were found (McGregor et al., 1980). Other authors, however, observed that sodium salts of some bile acids, such as cholic acid, showed a clear inhibitory effect on platelet aggregation by ADP, while the sodium salt of chenodeoxycholic acid (CDA) was an inducer of platelet aggregation (Baele et al., 1980). In other experiments, deoxycholic acid inhibited platelet aggregation and ADP secretion in platelets activated with either collagen or thrombin (Tan et al., 2012).
Thus, in the present study we aimed at analyzing the roles of bile acids such as cholic acid and deoxycolic acid on the altered platelet function observed in a model of cholestasis, the bile duct-ligated rat.
Materials and Methods
Animals
Male Sprague–Dawley rats born and raised in the Animal House of the Universidad de Murcia were used in the present study. The rats were housed in a temperature controlled environment, with 12:12-h light-dark cycle in the Animal Care Facility of the University of Murcia (REGAES300305440012). The animals were kept and treated according to the guidelines established by the European Union for the protection of animals used in experiments (86/609/EEC). All procedures were approved by the Animal Care and Use Committee of the University of Murcia.
Experimental Groups
Animals weighing ~250 g were subjected to bile-duct ligation (BDL) and excision or sham operation (controls), as described previously (Ortíz et al., 1996; Nadal et al., 2002; Atucha et al., 2003, 2007). Normal rat chow and tap water were offered ad libitum. Experiments were performed 21 days later (BDL non-ascitic group) and at least 28 days after surgery, when ascites was present (BDL ascitic group).
Isolation of Platelets
Isolation of platelets were performed as described previously (Iyú et al., 2004; Atucha et al., 2007). Briefly, platelet-rich plasma (PRP) and platelet-poor plasma (PPP) was obtained by differential centrifugation and the platelet count was adjusted to 3. 105 cels/μl.
Flow Cytometry
Determination of Platelet Activation in PRP Through the Expression of P-Selectin
The expression of P-selectin was performed by flow cytometry in platelets obtained as described above. Briefly, platelets resuspended in PBS were incubated for 30 min with 5 μl of CD61-FITC (for detection of platelet population) and 5 μl CD62P-PE (for detection of P-selectin). The acquisition was set at 50,000 events in the FAC-sort cytometer (Becton-Dickinson). The software used was CELLQuestTM. These experiments were performed in platelets obtained from six animals per group.
Fura-2 Loading and Determination of [Ca2+]i
These methods were carried out as described previously (Grynkiewicz et al., 1985; Atucha et al., 2003, 2007; Iyú et al., 2004) in platelets obtained from six rats per group. After obtaining platelet-rich plasma, platelets were washed and incubated with 2.5 μmol/l fura-2/AM (Molecular Probes) for 45 min at room temperature. Then, after washing out fura-2, platelets were stored at room temperature in the dark until Ca2+ measurements were performed. Platelets were placed in fluorescence-free cuvettes (Sigma) in the optical field of a fluorescence spectrometer (Aminco Bowman 2; Microbeam), and excited alternatively with light at 340 and 380 nm and the light emitted at 510 nm was collected. Changes in cytosolic free Ca2+ concentration ([Ca2+]i) were obtained by using the fura-2 340/380 fluorescence ratio and calibrated as described previously (Atucha et al., 2007). Only one concentration of each drug was tested on every platelet suspension. The calibration procedure was done in every experiment to take into account differences in the number of platelets between animals. After obtaining baseline values for 30 s, the appropriate drug concentration was added and the fluorescence recorded. Three protocols were performed:
(1) The response to thrombin (0.3 U/ml) was studied in the absence (no Ca2+ added plus 0.5 mmol/l EGTA) and presence (1 mmol/l CaCl2) of extracellular Ca2+.
(2) Capacitative Ca2+ entry (CCE) was determined by incubating platelets in a Ca2+-free medium with thapsigargin (1 μmol/l). After 180 s, Ca2+ (1 mmol/l) was added and changes in [Ca2+]i were monitored for another 180 s.
(3) Ca2+ accumulation into intracellular stores was estimated by suspending platelets in a Ca2+-free medium (+100 μmol/l EGTA) and challenging with ionomycin (5 μmol/l) and thapsigargin (1 μmol/l).
In these experiments, to analyze the effect of bile acids, platelets were pre-incubated 10 min at room temperature with CA or DCA at a final concentration of 100 μmol/l). Then, these three protocols were performed again.
Drugs
All the products used were from Sigma, except where indicated. Fura-2 AM (fura 2 acetoxymethyl ester, Molecular Probes) and Thapsigargin (Invitrogen) were dissolved in DMSO. Appropriate dilutions were prepared freshly every day in measurement buffer (Atucha et al., 2007).
Statistical Analysis
Data are expressed as the mean ± S.E.M. In order to compare the Ca2+ responses within the same group, the area under the curve (AUC) of the individual Ca2+ responses were calculated by summation of all experimental values (180 s) corrected by substraction of the averaged baseline (estimated during 30 s). The resulting values as well as baseline values were compared between groups by one-way analysis of variance and a Student-Newman-Keuls as post-hoc test. A probability level of p < 0.05 was considered to be a significant difference.
Results
The BDL rats used in the present study had the typical features of this model, jaundice, enlarged liver and spleen, and mesenteric edema. Ascites was present in variable amounts only in the animals included in this group. Sham-operated animals did not have any of these alterations.
Baseline P-selectin expression was significantly greater in the BDL group as compared to the controls and the BDL-ascitic rats, and CA or DCA did not affect these values in any group (Table 1). Platelet aggregation, which we found significantly elevated in the BDL non-ascitic rats in a previous paper (Romecín et al., 2017) was not modified by pretreatment with those bile acids (data not shown). Basal platelets Ca2+ levels was significantly elevated by incubation with both CA and DCA (Table 2) in platelets of the control animals groups, while in platelets of the BDL rats only DCA elevated basal Ca2+. In platelets of BDL rats with ascites, there was no significant effect of bile acids (BA).
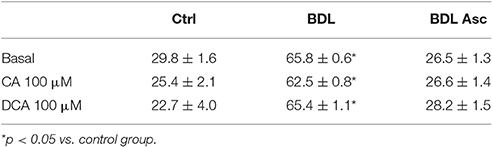
Table 1. P-selectin expression (arbitrary units) in the experimental groups.
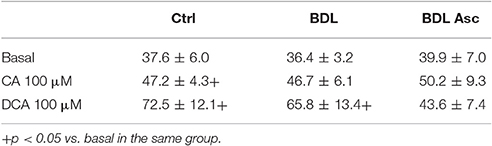
Table 2. Basal calcium levels (nM) in the experimental groups.
In relation with thrombin responses, incubation with both BA reduced the Ca2+ release after stimulation with thrombin (0.3 U/ml) in the absence of extracellular Ca2+ (Figure 1A, top panels), but only CA in the BDL group showed a significant difference as compared to its basal (Figure 1B, top panels). In the presence of extracellular Ca2+ (Figure 1A, bottom panels), thrombin responses were much greater in all three groups and pretreatment with CA but not with DCA, reduced significantly those Ca2+ responses in all three experimental groups (Figure 1B, bottom panels).
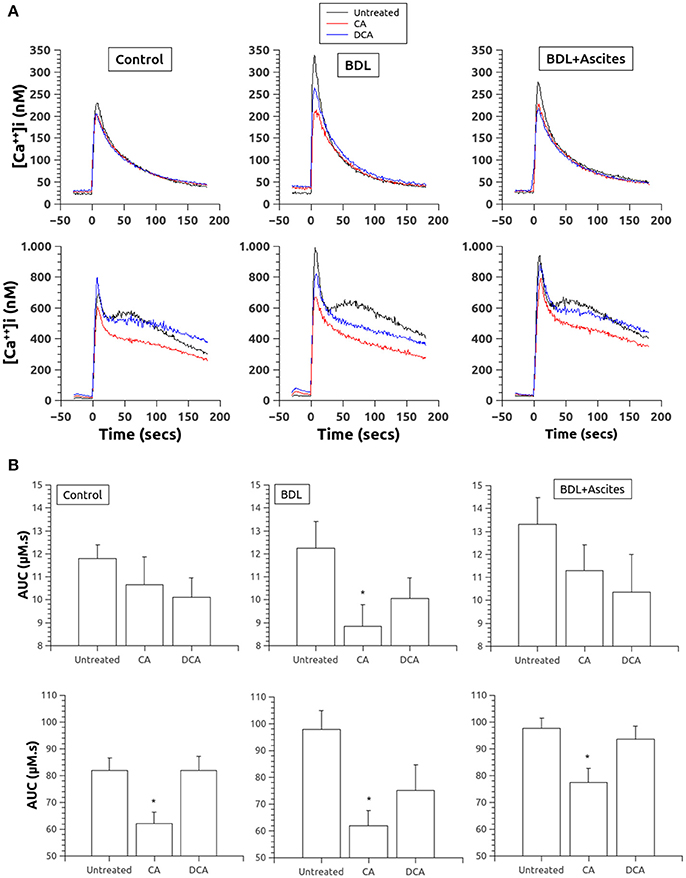
Figure 1. (A) Changes in [Ca2+]i in platelets of control (left column), BDL (middle column) and BDL with ascites rats (right column) in response to thrombin (0.3 U/ml, added at time 0). Top, in the absence of extracellular Ca2+. Bottom, in the presence of extracellular Ca2+. Data were obtained in untreated platelets and after incubation with CA or DCA (100 μM each). (B). Area under the curve of the calcium changes after thrombin administration (0.3 U/ml, added at time 0) in the absence (top panels) and in the presence (bottom panels) of extracellular Ca2+. Data were obtained in untreated platelets and after incubation with CA or DCA (100 μM each). Results are means ± S.E.M. *p < 0.05 vs. Untreated.
The capacitative Ca2+ response is shown in Figure 2. Pretreatment with thapsigargin to deplete Ca2+ from the intracellular stores slightly elevated Ca2+ levels, being significantly greater in the platelets of the BDL rats. However, regarding the effect of BA, only DCA significantly elevated the thapsigargin effect in platelets of the control rats (Figure 2B, top panels). But, the addition of Ca2+ in those platelets pretreated with thapsigargin to evoke the capacitative Ca2+ entry induced a significantly lower entry of Ca2+ in platelets pretreated with both CA and DCA (Figures 2A,B, bottom panels) in all three experimental groups.
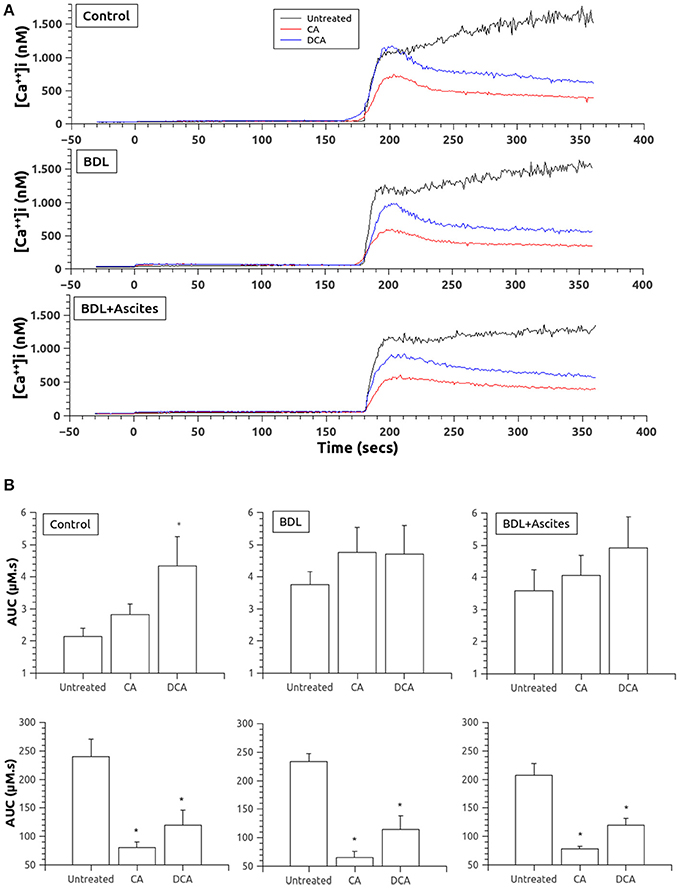
Figure 2. (A) Changes in [Ca2+]i after thapsigargin administration in the absence of extracellular Ca2+ (time 0 seconds) and after addition of calcium to allow for capacitative Ca2+ entry (time 180 s). Data were obtained in untreated platelets and after incubation with CA or DCA (100 μM). (B) Area under the curve of the calcium changes after thapsigargin administration in the absence of extracellular Ca2+ (top panels) and after addition of calcium to allow for capacitative Ca2+ entry (bottom panels). Data were obtained in untreated platelets and after incubation with CA or DCA (100 μM). Results are means ± S.E.M. *p < 0.05 vs. Untreated.
To estimate the amount of Ca2+ accumulated into intracellular stores, we used the simultaneous addition of thapsigargin and ionomycin. As shown in Figures 3A,B, total Ca2+ stored was significantly decreased by pretreatment with both CA and DCA, although these changes were significantly different in the control rats only with CA and in the BDL platelets with DCA.
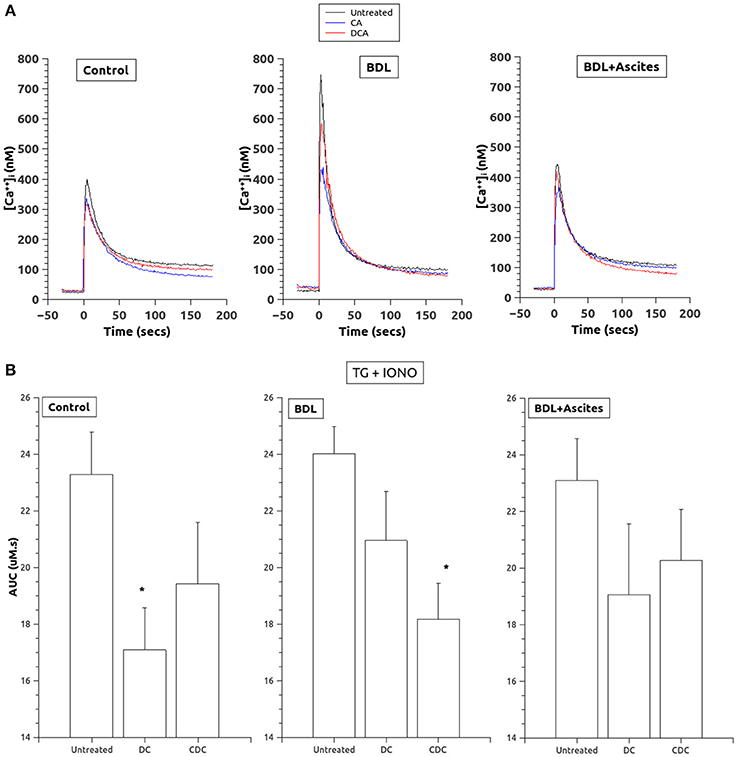
Figure 3. (A) Changes in [Ca2+]i after thapsigargin and ionomycin administration (added at time 0) to allow for estimation of total calcium in intracellular stores. Data were obtained in untreated platelets and after incubation with CA or DCA (100 μM). (B) Area under the curve of the calcium changes after thapsigargin and ionomycin to estimate total calcium in intracellular stores. Data were obtained in untreated platelets and after incubation with CA or DCA (100 μM). Data are means ± S.E.M. *p < 0.05 vs. Untreated.
Discussion
In previous studies (Atucha et al., 2007; Romecín et al., 2017), we showed in a rat model of liver cirrhosis by bile duct ligation, several platelet alterations compatible with the existence of a hyperaggregatory state. These alterations are related to a defective platelet Ca2+ handling, specifically to an enhanced intracellular Ca2+ release evoked by thrombin and an increased amount of Ca2+ stored in the intracellular organelles and are present before the appearance of ascites. In the present study we confirm these alterations and extend them to show their lack of relationship to bile acids. Our results indicate that, if any, bile acids, would tend to reduce Ca2+ movements across platelet membranes which would reduce this hyperaggregatory state characterisitic of the non-ascitic phase of the disease. A similar result would also occur in the ascitic phase, characterized in this case by a lower platelet aggregatory function.
This hyperaggregatory state is related firstly to a higher expression of P-selectin in both groups of cirrhotic rats, indicative of an activated state, which is typical of several types of human cirrhosis (Panasiuk et al., 2001; Vardareli et al., 2007; Xianghong et al., 2013). These data are similar to those found by Watanabe et al. (1998), in cirrhotic patients, who explained it by a change in the fatty acid composition of the platelet membrane. Although bile acids are well known detergents that exhibit membrane-damaging properties against a variety of cells, including blood platelets (Shiao et al., 1993), this does not seem the case, since in our data the expression of P-selectin was unchanged by pretreatment with CA and CDA.
Regarding thrombin-induced intracellular Ca2+ changes, both bile acids clearly decreased both Ca2+ release from internal stores (that observed in the absence of Ca2+) and Ca2+ entry from the extracellular space, although only CA, but not DCA, showed a significantly lower response in all three groups, control and BDL-ligated rats. This result agrees with previous data (Bowen et al., 1988; Shiao et al., 1993) but disagrees with those of McGregor (McGregor et al., 1980) showing that cholic acid increased thrombin and ADP-induced platelet aggregation and accelerated plasma clotting time. A similar result was obtained in fibroblasts, where DCA directly induced both Ca2+ release from internal stores and persistent entry at the plasma membrane (Lau et al., 2005).
Our results are in keeping with more recent data indicating that bile acids also act as regulatory molecules (Hylemon et al., 2009). Bile acids have been discovered to activate specific nuclear receptors such as farnesoid X receptor (FXR) and others, as well as cell signaling pathways in cells of the liver and gastrointestinal tract. A recent study by Moraes et al. (2016) indicates that FXR ligands inhibited the activation of platelets in response to stimulation of collagen or thrombin receptors, resulting in diminished intracellular Ca2+ mobilization, secretion, fibrinogen binding, and aggregation. However, bile acids also activate a number of cellular Ca2+ related channel receptors (Dopico et al., 2002; Lee et al., 2012; Rainer et al., 2013). Since Ca2+ regulation is critical for multiple platelet functions (Sage, 1997; Rosado and Sage, 2002; Dolan and Diamond, 2014), we suggest that bile acids, acting through platelet receptors, induce inhibition of platelets by reducing Ca2+ signaling. Clearly, more experiments would be necessary in order to prove this hypothesis.
A similar effect of bile acids was found when we studied the capacitative Ca2+ entry, again suggesting an interference with the channels involved in those responses. Interestingly, bile acids did not reduce the intracellular Ca2+ elevation induced by thapsigargin, an inhibitor of SERCA Ca2+ ATP-ase, that although modest compared to the capacitative entry, was significantly elevated with DCA in the platelets of the control animals.
Finally, total amount of Ca2+ in internal stores, as estimated with the use of thapsigargin and ionomycin, was again reduced by the bile acids, suggesting again that bile acids interfere with Ca2+ signaling receptors both at internal and cellular membranes. Clearly, further studies will be necessary to analyze the platelets receptors involved in these alterations.
Conclusions
Patients with liver cirrhosis and experimental models show, in the compensated phase prior to the development of ascites, platelet alterations compatible with a hyperaggregatory state. This is related to enhanced Ca2+ release evoked by agonists in activated platelets, secondary to an increased amount of Ca2+ stored in the intracellular organelles. These alterations are not related to bile acids, since they reduce the amount of internal Ca2+ and the entry from the extracellular space, both necessary to induce platelet aggregation.
Author Contributions
PR was the main performer of most of the experiments, EN is our lab technician and without her nothing can be done, MO and DI played a key role with the flow cytometry technique, JG was the main writer of the manuscript and NA supervised all the experimental protocols and procedure laboratories and directed the research.
Funding
This work has been performed with grants from Instituto de Salud Carlos III (SAF2006-9127) to JG.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Special thanks are given to our colleagues at the Servicio de Apoyo a la Investigación de la Universidad de Murcia for their assistance with Flow Cytometry.
Abbreviations
[Ca2+]i, cytosolic calcium levels; Ca2+, calcium; BDL, bile-duct ligated rat; ADP, adenosine diphosphate; CCE, capacitative calcium entry; SERCA, smooth endoplasmic reticulum calcium ATP-ase.
References
Annie-Jeyachristy, S., Geetha, A., and Surendran, R. (2008). Changes in the level of cytosolic calcium, nitric oxide and nitric oxide synthase activity during platelet aggregation: an in vitro study in platelets from normal subjects and those with cirrhosis. J. Biosci. 33, 45–53. doi: 10.1007/s12038-008-0020-0
Atucha, N. M., Iyú, D., Alcaraz, A., Rosa, V., Martínez-Prieto, C., Ortiz, M. C., et al. (2007). Altered calcium signalling in platelets from bile-duct-ligated rats. Clin. Sci. 112, 167–174. doi: 10.1042/CS20060226
Atucha, N. M., Iyu, D., De Rycker, M., Soler, A., and García-Estañ, J. (2003). Altered calcium regulation in freshly isolated aortic smooth muscle cells from bile duct-ligated rats: Role of nitric oxide. Cell Calcium 33, 129–135. doi: 10.1016/S0143-4160(02)00206-3
Baele, G., Beke, R., and Barbier, F. (1980). In vitro inhibition of platelet aggregation by bile salts. Thromb. Haemost. 44, 62–64.
Bandi, J. C., Poch, E., García-Pagán, J. C., Luca, A., Jiménez, W., Escorsell, A., et al. (1997). Platelet cytosolic calcium concentration in patients with liver cirrhosis. Relationship with hepatic and systemic hemodynamics. J. Hepatol. 27, 824–829. doi: 10.1016/S0168-8278(97)80319-4
Ben-Ari, Z., Panagou, M., Patch, D., Bates, S., Osman, E., Pasi, J., et al. (1997). Hypercoagulability in patients with primary biliary cirrhosis and primary sclerosis cholangitis evaluated by thrombelastography. J. Hepatol. 26, 554–559. doi: 10.1016/S0168-8278(97)80420-5
Bowen, D. J., Clemmons, R. M., Meyer, D. J., and Dorsey-Lee, M. R. (1988). Platelet functional changes secondary to hepatocholestasis and elevation of serum bile acids. Thromb. Res. 52, 649–654. doi: 10.1016/0049-3848(88)90137-5
Shiao, Y. J., Chen, J. C., Wang, C. N., and Wang, C. T. (1993). The mode of action of primary bile salts on human platelets. Biochim. Biophys. Acta 1146, 282–293. doi: 10.1016/0005-2736(93)90367-9
Dolan, A. T., and Diamond, S. L. (2014). Systems modeling of Ca(2+) homeostasis and mobilization in platelets mediated by IP3 and store-operated Ca(2+) entry. Biophys. J. 106, 2049–2060. doi: 10.1016/j.bpj.2014.03.028
Dopico, A. M., Walsh, J. V. Jr., and Singer, J. J. (2002). Natural bile acids and synthetic analogues modulate large conductance Ca2+-activated K+ (BKCa) channel activity in smooth muscle cells. J. Gen. Physiol. 119, 251–273. doi: 10.1085/jgp.20028537
Grynkiewicz, G., Poenie, M., and Tsien, R. Y. (1985). A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450.
Hylemon, P. B., Zhou, H., Pandak, W. M., Ren, S., Gil, G., and Dent, P. (2009). Bile acids as regulatory molecules. J. Lipid Res. 50, 1509–1520. doi: 10.1194/jlr.R900007-JLR200
Iyú, D., Atucha, N. M., Martinez-Prieto, C., Ortiz, M. C., and García-Estañ, J. (2004). Altered calcium signaling in platelets from nitric oxide-deficient hypertensive rats. Cell Commun. Signal. 2:1. doi: 10.1186/1478-811X-2-1
Lau, B. W., Colella, M., Ruder, W. C., Ranieri, M., Curci, S., and Hofer, A. M. (2005). Deoxycholic acid activates protein kinase C and phospholipase C via increased Ca2+ entry at plasma membrane. Gastroenterology 128, 695–707. doi: 10.1053/j.gastro.2004.12.046
Lee, H. K., Lee, K. H., and Cho, E. S. (2012). Bile acid inhibition of N-type calcium channel currents from sympathetic ganglion neurons. Korean J. Physiol. Pharmacol. 16, 25–30. doi: 10.4196/kjpp.2012.16.1.25
McGregor, L., Morazain, R., and Renaud, S. (1980). A comparison of the effects of dietary short and long chain saturated fatty acids on platelet functions, platelet phospholipids, and blood coagulation in rats. Lab. Invest. 43, 438–442.
Moraes, L. A., Unsworth, A. J., Vaiyapuri, S., Ali, M. S., Sasikumar, P., Sage, T., et al. (2016). Farnesoid X receptor and its ligands inhibit the function of platelets. Arterioscler. Thromb. Vasc. Biol. 36, 2324–2333. doi: 10.1161/ATVBAHA.116.308093
Nadal, F. J. A., Iyú, D., Atucha, N. M., and García-Estañ, J. (2002). Interaction of nitric oxide with calcium in the mesenteric bed of bile duct-ligated rats. Br. J. Pharmacol. 135, 489–495. doi: 10.1038/sj.bjp.0704504
Ortíz, M. C., Fortepiani, L. A., Martínez, C., Atucha, N. M., and García-Estañ, J. (1996). Vascular hyporesponsiveness in aortic rings from cirrhotic rats: role of nitric oxide and endothelium. Clin. Sci. 91, 733–738.
Panasiuk, A., Prokopowicz, D., Zak, J., Matowicka-Karna, J., Osada, J., and Wysocka, J. (2001). Activation of blood platelets in chronic hepatitis and liver cirrhosis P-selectin expression on blood platelets and secretory activity of beta-thromboglobulin and platelet factor-4. Hepatogastroenterology 48, 818–822.
Papadopoulos, V., Filippou, D., Manolis, E., and Mimidis, K. (2007). Haemostasis Impairment in Patients with Obstructive Jaundice. J. Gastrointestin Liver Dis. 16, 177–186.
Pihusch, R., Rank, A., Gohring, P., Pihusch, M., Hiller, E., and Beuers, U. (2002). Platelet function rather than plasmatic coagulation explains hypercoagulable state in cholestatic liver disease. J. Hepatol. 37, 548–555. doi: 10.1016/S0168-8278(02)00239-8
Rainer, P. P., Primessnig, U., Harenkamp, S., Doleschal, B., and Wallner, M. (2013). Bile acids induce arrhythmias in human atrial myocardium–implications for altered serum bile acid composition in patients with atrial fibrillation. Heart 99, 1685–1692. doi: 10.1136/heartjnl-2013-304163
Romecín, P., Atucha, N. M., Navarro, E. G., Clara Ortiz, M., Iyú, D., Rosado, J. A., et al. (2017). Role of homocysteine and folic acid on the altered calcium homeostasis of platelets from rats with biliary cirrhosis. Platelets 2, 1–8. doi: 10.1080/09537104.2016.1265920
Rosado, J. A., and Sage, S. O. (2002). “Platelet signalling: calcium,” in Platelets in Thrombotic and Non-Thrombotic Disorders: Pathophysiology, Pharmacology and Therapeutics, eds P. Gresele, C. P. Page, V. Fuster, and J. Vermylen (Cambridge: Cambridge University Press), 260–271.
Sage, S. O. (1997). Calcium entry mechanisms in human platelets (The Wellcome Prize Lecture). Exp. Physiol. 82, 807–823. doi: 10.1113/expphysiol.1997.sp004066
Tan, J., Reddy, E., Murphy, D., Keely, S., and O'Neill, S. (2012). Bile acids differentially impact on platelet activation. BMC Proc. 6(Suppl. 4):P22. doi: 10.1186/1753-6561-6-S4-P22
Tripodi, A., Anstee, Q. M., Sogaard, K. K., Primignani, M., and Valla, D. C. (2011). Hypercoagulability in cirrhosis: causes and consequences. J. Thromb. Haemost. 9, 1713–1723. doi: 10.1111/j.1538-7836.2011.04429.x
Vardareli, E., Saricam, T., Demirustu, C., and Gulbas, Z. (2007). Soluble P selectin levels in chronic liver disease: relationship to disease severity. Hepatogastroenterology 54, 466–469.
Watanabe, M., Shiraishi, K., Itakura, M., and Matsuzaki, S. (1998). Relationship between platelet membrane lipid compositions and platelet aggregability in alcoholic liver disease. Alcohol Clin. Exp. Res. 22, 97S–102S. doi: 10.1097/00000374-199803001-00004
Witters, P., Hoylaerts, M., Freson, K., De Vos, R., van Pelt, J., Nevens, F., et al. (2010). ADP-degrading enzymes inhibit platelet activation in bile duct-ligated rats. J. Thromb. Haemost. 8, 360–368. doi: 10.1111/j.1538-7836.2009.03684.x
Keywords: calcium signaling, bile-duct ligation, capacitative calcium entry, cholestasis, liver cirrhosis, fura-2, thapsigargin, thrombin
Citation: Romecín P, Navarro EG, Ortiz MC, Iyú D, García-Estañ J and Atucha NM (2017) Bile Acids Do Not Contribute to the Altered Calcium Homeostasis of Platelets from Rats with Biliary Cirrhosis. Front. Physiol. 8:384. doi: 10.3389/fphys.2017.00384
Received: 02 March 2017; Accepted: 22 May 2017;
Published: 07 June 2017.
Edited by:
Ovidiu Constantin Baltatu, Anhembi Morumbi University, BrazilReviewed by:
Sulayma A. Albarwani, Sultan Qaboos University, OmanBruno Vogt, University of Bern, Switzerland
Copyright © 2017 Romecín, Navarro, Ortiz, Iyú, García-Estañ and Atucha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Noemí M. Atucha, bnRtYUB1bS5lcw==