Abstract
The recognition of invading pathogens and endogenous molecules from damaged tissues by toll-like receptors (TLRs) triggers protective self-defense mechanisms. However, excessive TLR activation disrupts the immune homeostasis by sustained pro-inflammatory cytokines and chemokines production and consequently contributes to the development of many inflammatory and autoimmune diseases, such as systemic lupus erythematosus (SLE), infection-associated sepsis, atherosclerosis, and asthma. Therefore, inhibitors/antagonists targeting TLR signals may be beneficial to treat these disorders. In this article, we first briefly summarize the pathophysiological role of TLRs in the inflammatory diseases. We then focus on reviewing the current knowledge in both preclinical and clinical studies of various TLR antagonists/inhibitors for the prevention and treatment of inflammatory diseases. These compounds range from conventional small molecules to therapeutic biologics and nanodevices. In particular, nanodevices are emerging as a new class of potent TLR inhibitors for their unique properties in desired bio-distribution, sustained circulation, and preferred pharmacodynamic and pharmacokinetic profiles. More interestingly, the inhibitory activity of these nanodevices can be regulated through precise nano-functionalization, making them the next generation therapeutics or “nano-drugs.” Although, significant efforts have been made in developing different kinds of new TLR inhibitors/antagonists, only limited numbers of them have undergone clinical trials, and none have been approved for clinical uses to date. Nevertheless, these findings and continuous studies of TLR inhibition highlight the pharmacological regulation of TLR signaling, especially on multiple TLR pathways, as future promising therapeutic strategy for various inflammatory and autoimmune diseases.
Introduction
Toll-like receptors (TLRs) are a family of pattern recognition receptors (PRRs) that can recognize and respond to a unique repertoire of distinct molecules referred to as pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs; Kawai and Akira, 2010; Moresco et al., 2011). These conserved receptors belong to type I transmembrane glycoproteins and are made of three structurally important components: (1) the leucine-rich repeat motifs for ligand recognition at N-terminus; (2) a single transmembrane helix; and (3) a conserved cytoplasmic Toll/Interleukin-1 (IL-1) receptor (TIR) domain at C-terminus for intracellular signaling transduction (Botos et al., 2011; Bryant et al., 2015). A representative TLR structure is shown in Figure 1. Currently, a total of 13 TLRs have been identified, among which TLRs 1–10 are expressed in human despite the function of TLR10 is still unclear (Moresco et al., 2011). The expression of TLRs can be found in a variety of immune cells (e.g., dendritic cells, monocytes, macrophages, and B lymphocytes) and non-immune cells (e.g., epithelial cells, endothelial cells, and fibroblasts; Takeda et al., 2003). While TLR1, TLR2, and TLR4–6 are mainly found on the cell surface, TLR3 and TLR7–9 are primarily expressed in the endosomes (Gay et al., 2014). Two TLRs form a hetero- or homo-dimer that allow them to recognize and bind to different molecular patterns (Gay et al., 2014).
Figure 1
When engaging with PAMPs or DAMPs, TLRs can transduce the signaling to initiate innate and adaptive immune responses (Akira and Takeda, 2004; Palm and Medzhitov, 2009). The molecular pathways of TLR signal transduction can be simply categorized into two cascades (Akira and Takeda, 2004; Kawasaki and Kawai, 2014). One is through the main adaptor protein, myeloid differentiation factor 88 (MyD88). The ligation of TLR ligands results in the recruitment of MyD88 to the TIR domain and subsequently leads to activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and mitogen-associated protein kinase (MAPK), which drive the expression of pro-inflammatory genes. The other is through the adaptor TIR-domain-containing adaptor-inducing interferon-β (TRIF), also known as the MyD88-independent pathway, to activate interferon (IFN) regulatory factors (IRFs) and NF-κB for the production of type I IFNs. Among all human TLRs, TLR3 exclusively signals through TRIF-dependent pathway while others use MyD88-dependent pathway; (Uematsu and Akira, 2007) notably, TLR4 can signal through both pathways in a time-dependent manner (Sato et al., 2003; Yamamoto et al., 2003).
TLR activation is one of the first defensive mechanisms utilized by the host to mount innate and subsequent adaptive immune responses to fight the invading pathogens and repair the damaged tissues (Takeda and Akira, 2005; Beutler, 2009; Palm and Medzhitov, 2009). However, dysregulated TLR signaling could disrupt the immune homeostasis (i.e., a self-regulating process to remain physiological balance) in the host by sustained pro-inflammatory cytokines and chemokines secretion. This often contributes to the development of many inflammatory and autoimmune diseases, such as systemic lupus erythematosus (SLE), sepsis, atherosclerosis, and asthma (Marshak-Rothstein, 2006; Tsujimoto et al., 2008; Drexler and Foxwell, 2010). Considering the pathological role of TLRs in these inflammatory diseases, inhibitors targeting TLR signaling might be beneficial in treating these disorders (Hennessy et al., 2010).
Despite many efforts have been put in developing (bio)molecular inhibitors targeting TLR signaling pathways, unfortunately, very few compounds are currently available for clinical uses. Therefore, it is important to search for novel TLR inhibitors that can be utilized both as therapeutic agents and as experimental reagents for dissecting innate immune molecular pathways. Manipulation of immune responsiveness using nanodevices provides a new strategy to treat human diseases, because nanotechnology offers many beneficial features for therapeutics development, including cell penetrating and targeting capabilities, desired biodistribution, improved pharmacokinetics, enhanced therapeutic activity, better biocompatibility, and in vivo tracking (bioimaging). Accordingly, novel nanodevices have emerged with bio-activities to potently regulate TLR signaling, aiming for inhibiting disease-associated TLR activation.
In this review, we first briefly summarize the pathophysiological role of TLRs in many diseases of acute and chronic inflammation as well as autoimmunity. These diseases include SLE in autoimmunity, infection-induced sepsis for acute inflammation, vascular system disorders across both acute and chronic inflammation, and other inflammation-associated diseases such as psoriasis, gastrointestinal cancer, asthma, and chronic obstructive pulmonary disease (COPD; Table 1). We then focus on the current knowledge in developing TLR antagonists and inhibitors at various stages from pre-clinical evaluation to clinical trials, including the most recent development of nano-based modulators on inhibition of TLR signaling, and discuss their potential uses in treating various inflammatory diseases. Particularly, we describe the design of these nanodevices and discuss their mechanism(s) of action in TLR inhibition. Due to the unique feature of nanodevices, these nano-inhibitors hold great promises in treating various inflammatory and autoimmune diseases.
Table 1
| Types of TLR | Associated diseases | Evidence | References |
|---|---|---|---|
| TLR2 | SLE | High mRNA expression in patients; TLR2−/− reduces autoantibody production | Komatsuda et al., 2008; Lartigue et al., 2009 |
| Sepsis | Up-regulated expression in patients; TLR2−/− increases survival under microbial infection | Harter et al., 2004; Bergt et al., 2013 | |
| Atherosclerosis | TLR2−/− decreases vasculature inflammation | Mullick et al., 2005, 2008 | |
| Hypertension | Renal IR injury | Khan et al., 2012 | |
| Psoriasis | Respond to HSP60 | Seung et al., 2007 | |
| COPD | Cigarette smoke-induced inflammation; exacerbation under infection | Droemann et al., 2005; Tokairin et al., 2008 | |
| TLR3 | Sepsis | TLR3−/− is protective against microbial challenge | Cavassani et al., 2008 |
| Atherosclerosis | Promote plague instability | Ishibashi et al., 2013 | |
| TLR4 | SLE | High mRNA expression in patients; TLR4−/− reduces autoantibody production; elevated TLR4 presents SLE-like phenotype | Liu et al., 2006; Komatsuda et al., 2008; Lartigue et al., 2009 |
| Sepsis | Up-regulated expression in patients; TLR4−/− is resistant to Gram-negative bacterial induced sepsis | Harter et al., 2004; Roger et al., 2009 | |
| Atherosclerosis | TLR4−/− decreases vasculature inflammation | Michelsen et al., 2004; Higashimori et al., 2011 | |
| Hypertension | Respond to angiotensin II | Hernanz et al., 2015 | |
| Psoriasis | Respond to HSP60 | Seung et al., 2007 | |
| IBD, gastrointestinal malignancies | Respond to resident microbes | Fukata et al., 2007; Fukata and Abreu, 2008 | |
| Asthma | Respond to airway allergens; Th1/Th2 immune homeostasis | Dong et al., 2009 | |
| COPD | Cigarette smoke-induced inflammation; exacerbation under infection | Karimi et al., 2006; Tokairin et al., 2008 | |
| TLR5 | Psoriasis | Respond to TGF-α | Miller et al., 2005 |
| TLR7 | SLE | Up-regulated expression in patients; TLR7−/− reduces autoantibody, IL-6 and IFN-α production | Christensen et al., 2006; Pisitkun et al., 2006; Subramanian et al., 2006; Lee et al., 2008; Kono et al., 2009 |
| Stroke | Dual role: protective in preconditioning, damaging in post-ischemia phase | Brea et al., 2011; Leung et al., 2012 | |
| Psoriasis | Aggravate disease symptoms | Gilliet et al., 2004 | |
| TLR9 | SLE | Regulatory role | Christensen et al., 2006; Nickerson et al., 2010; Chen et al., 2011; Jackson et al., 2014 |
| Sepsis | TLR9−/− is protective against microbial challenge; TLR9 inhibition reduces disease severity | Yasuda et al., 2008; Liu et al., 2012; Hu et al., 2015 | |
| Atherosclerosis | TLR9−/− exacerbates atherosclerosis | Koulis et al., 2014 | |
| Hypertension | Elevate blood pressure | McCarthy et al., 2015 | |
| Psoriasis | Respond to TGF-α | Miller et al., 2005 | |
| COPD | Cigarette smoke-induced inflammation | Mortaz et al., 2010 |
The role of TLR activation in the pathogenesis of inflammatory diseases.
SLE, systemic lupus erythematosus; IR, ischemia-reperfusion; HSP, heat shock protein; COPD, chronic obstructive pulmonary disease; IBD, inflammatory bowel diseases.
Toll-like receptor activation in autoimmune and inflammatory diseases
Systemic lupus erythematosus (SLE)
Systemic lupus erythematosus (SLE) is a complex autoimmune disease characterized by the loss of tolerance to self-nuclear antigens and the production of autoantibodies, leading to the attack of the immune system on the self-components and healthy tissues. Recent evidences have suggested a close relationship between the endosomal TLR activation and the onset of SLE (Celhar and Fairhurst, 2014; Wu et al., 2015). It has been found that in B cells and plasmacytoid dendritic cells (pDCs), endosomal TLRs play an essential role in the generation of anti-nuclear antibodies and type I IFNs (Clancy et al., 2016). Particularly, in lupus-prone mouse models, TLR7 overexpression is associated with the production of autoantibodies and the development of autoimmune phenotypes (Pisitkun et al., 2006; Subramanian et al., 2006); on the other hand, in the situation of TLR7 deletion the levels of circulating autoantibodies and inflammatory cytokines, such as IL-6 and INF-α, are significantly decreased with improved disease symptoms (Christensen et al., 2006; Lee et al., 2008; Kono et al., 2009). In contrast to TLR7, the role of TLR9 in SLE is not well-defined. It has been shown that TLR9 is indispensable in the autoantibody production in B cells from several mouse studies (Christensen et al., 2006; Nickerson et al., 2010; Chen et al., 2011). However, the absence of TLR9 in these lupus-prone models does not help improve disease conditions, but leads to disease exacerbation (Christensen et al., 2006; Nickerson et al., 2010; Jackson et al., 2014). This suggests that TLR9 may have a regulatory role in the progression of SLE. Nevertheless, the expression of both TLR7 and TLR9 is up-regulated in the peripheral blood mononuclear cells (PBMC) from the SLE patients, and their levels are correlated with the production of IFN-α (Lyn-Cook et al., 2014), making them potential therapeutic targets of SLE.
In addition to endosomal TLRs, TLR2 and TLR4 have been found to participate in the pathogenesis of SLE based on the following evidences. First, their mRNA levels in PBMC of SLE patients are much higher than those in healthy controls (Komatsuda et al., 2008; Lee et al., 2016). Second, in a lupus mouse model, the deletion of TLR4 or TLR2 (to a lesser extent) decreases the levels of autoantibodies, and subsequently ameliorates the disease symptoms (Lartigue et al., 2009). Third, the elevation of TLR4 can induce lupus-like autoimmune disease (Liu et al., 2006). All these studies clearly demonstrated the importance of TLR signaling in SLE development and progression.
Infection-associated sepsis
Due to their capability of recognizing a variety of PAMPs, TLRs play a critical role in fighting against pathogen infection (Mogensen, 2009). The first identified TLR4 can detect a wide range of Gram-negative bacteria by recognizing and responding to the coating molecules on the bacteria cell wall—lipopolysaccharide (LPS). TLR2 (in conjunction with either TLR1 or TLR6) primarily recognizes lipoproteins/lipopeptides and glycolipids from various pathogens including microbes and fungi. TLR5 is the only TLR that identifies a protein ligand—flagellin from nearly all flagellated bacteria. On the other hand, the endosomal TLRs (TLR3, and TLR7-TLR9) are mainly responsible for the recognition of nucleic acid ligands, including dsRNA, ssRNA, and CpG-DNA, from viral and intracellular bacterial infection. Therefore, activation of TLRs by engaging with these PAMPs allows the host to combat invading pathogens. In fact, TLR agonists have long been considered as vaccine adjuvants to boost the immune system to eliminate viral infections and most recently fight cancer (O'Neill et al., 2009; Hedayat et al., 2011).
However, excessive and uncontrolled TLR activation can lead to overwhelming systemic inflammation and multiple organ injury, characterized as sepsis. Among all TLRs, TLR2, and TLR4 are the two notable contributors to the pathogenesis of sepsis (Tsujimoto et al., 2008). Evidence came from the observation that the expression of both TLR2 and TLR4 was up-regulated during their ligand stimulation in monocytes from healthy subjects (Wittebole et al., 2005), as well as in PBMC from sepsis patients (Harter et al., 2004). In addition, TLR4 knockout mice are resistant to Gram-negative bacteria-induced septic shock (Roger et al., 2009), and TLR2-deficient mice have increased survival rates compared to wild type mice in a polymicrobial sepsis model (Bergt et al., 2013). Moreover, blockade of TLR2 and TLR4 signaling by antagonistic antibodies successfully decreases disease severity in sepsis models of Gram-positive and Gram-negative bacteria, respectively (Meng et al., 2004; Daubeuf et al., 2007). Interestingly, the protective effect can also be seen in the TLR3- and TLR9-deficient mice against microbial challenge; (Cavassani et al., 2008; Hu et al., 2015) and the pharmacological inhibitors of TLR9 can reduce the mortality of mice in severe sepsis and protect the host from serious complications, such as acute kidney injury (Yasuda et al., 2008; Liu et al., 2012). Since sepsis is one of the leading causes of death in the intensive care units worldwide, much effort has been put to develop therapeutic agents blocking TLR activation, especially TLR2 and TLR4, although none has yet been clinically successful (see Section Toll-Like Receptor Antagonists/Inhibitors and Their Clinical Applications).
Vascular system disorders
Not only can TLRs detect PAMPs in defending pathogen invasion, but they can also recognize circulating host-derived DAMPs released from the dying cells and damaged tissues to trigger the process of self-healing and tissue repair. Till recent years, studies have revealed that sensing the danger signals by TLRs could also contribute to the pathogenesis of many cardiovascular diseases, including atherosclerosis, hypertension, and stroke (Goulopoulou et al., 2016).
Atherosclerosis nowadays is considered as a chronic, progressive inflammatory condition corresponding to lipid accumulation in the vascular system than simply a lipid build-up disease. It has been found that the onset and ongoing of inflammation is mainly caused by the recognition of oxidized lipoproteins (as DAMPs) by TLRs expressed on innate immune cells (e.g., macrophages) and endothelial cells (Miller, 2005); TLR are also involved in the progression of the disease by lesion orientation (Edfeldt et al., 2002), collagen degradation (Monaco et al., 2009), and plaque destruction (Ishibashi et al., 2013). Specifically, it has been shown that deficiency in TLR2 or TLR4 results in diminishing inflammation in the prominent mouse models of atherosclerosis [apolipoprotein E (ApoE)- and LDL receptor-deficient mice; (Michelsen et al., 2004; Mullick et al., 2005, 2008; Higashimori et al., 2011)]. The activation of endosomal TLRs, on the other hand, seems to have controversial effects on atherosclerosis. TLR3 activation has been found to promote atherogenic inflammation, especially in mediating plaque instability (Ishibashi et al., 2013), while the absence of TLR9 exacerbates atherosclerosis in ApoE-deficient mice on a high-fat diet (Koulis et al., 2014).
In the case of hypertension, TLR4 has been well-documented to mediate aberrant immune and inflammation in vasculature, kidneys and autonomic nervous system (McCarthy et al., 2014). This is mainly because TLR4 can respond to angiotensin II, a DAMP, and cause subsequent vascular dysfunction and high blood pressure (Bomfim et al., 2015; Hernanz et al., 2015). Surprisingly, the evidence of TLR2 involved in hypertension is limited to inflammation in the renal system, where TLR2-activated NF-κB signaling is significantly correlated with renal ischemia/reperfusion injury (Khan et al., 2012). Recently, attentions have been drawn to TLR9 for its role in hypertension and blood pressure regulation. Studies have shown that TLR9 activation could elevate blood pressure in normotensive rats (McCarthy et al., 2015), and could also serve as a negative modulator of cardiac autonomic tone and baroreflex function (Rodrigues et al., 2015).
TLR2, TLR4, and endosomal TLRs have all been found to play an important role in stroke and cerebrovascular injury. Interestingly, most of them have a dual function along the time course of the disease: activation of TLRs in post-ischemia state would mediate neuroinflammation and neurodegeneration, but preconditioning with TLR stimulation could be neuroprotective from hypoxia and nutrient deprivation (Wang et al., 2011; Gesuete et al., 2014). For example, TLR7 and TLR8 expression is correlated to the aggravated neuroinflammation and poor outcome in acute ischemic stroke patients (Brea et al., 2011). On the other hand, TLR7 preconditioning [i.e., pre-treatment with a TLR7 agonist, gardiquimod (GDQ), prior to ischemia] has shown protection against subsequent stroke injury through type I IFN-mediated mechanism (Leung et al., 2012).
Other inflammatory diseases
Activation of TLRs is also involved in many other inflammation-associated diseases, such as psoriasis, gastrointestinal malignancies, and inflammatory airway diseases. Their relationships are briefly described hereafter.
Psoriasis is a long-lasting, relapsing inflammatory disease of dermis and epidermis. It has been observed that the expression of transforming growth factor (TGF)-α and heat shock proteins (e.g., HSP60) from keratinocytes can upregulate TLR5 and TLR9 (Miller et al., 2005), and activate TLR2 and TLR4, respectively (Seung et al., 2007). In addition, activation of TLR7/8 by its specific agonist imiquimod could also aggravate the disease symptoms (Gilliet et al., 2004).
It is known nowadays that chronic inflammation is often linked to carcinogenesis. In particular, chronic gastritis due to Helicobacer pylori infection and severe colitis of patients with inflammatory bowel diseases (IBD) greatly increase the risk of having gastrointestinal malignancies (Itzkowitz and Yio, 2004; Houghton and Wang, 2005), and TLR signaling is evidently involved in such inflammatory complications (Fukata and Abreu, 2008). It has been found that TLR4/MyD88 pathway can promote the development of colitis-associated colorectal tumors (Fukata et al., 2007). Since tumor cells also express TLR4, TLR4-induced uncontrolled production of immunosuppressive cytokines has been suggested to contribute to the tumor progression (Sato et al., 2009).
In inflammatory airway diseases, such as asthma and COPD, TLR signaling pathways are closely linked to the disease pathophysiology (Bezemer et al., 2012). For examples, house dust mites, one of the main risk factors for allergic asthma has been suggested to trigger airway neutrophils and monocytes through a TLR4-dependent mechanism (Li et al., 2010). Furthermore, the TLR4 agonist LPS is able to induce either Th1 or Th2 immune response in asthma in a dose dependent manner (Dong et al., 2009). In the case of COPD, TLR2, TLR4, and TLR9 have been shown to participate in cigarette smoke-induced inflammatory responses (Droemann et al., 2005; Karimi et al., 2006; Mortaz et al., 2010). During the exacerbation of the airway diseases, overactive TLR signaling can contribute to excessive inflammatory responses and lung tissue destruction (Tokairin et al., 2008; Stowell et al., 2009).
Toll-like receptor antagonists/inhibitors and their clinical applications
Although, evolutionarily TLRs are essential elements in innate immune system and play a critical role in the host defensive mechanism against invading pathogens, overactivation of TLRs is inevitably involved in the pathogenesis of many inflammatory diseases. Thus, inhibition of TLR signaling pathways has been predicted to be an effective therapeutic strategy to suppress unwanted, disease-associated inflammatory responses. In general, TLR inhibition can be achieved by two major strategies: (1) blocking the binding of TLR ligands to the receptor; (2) interfering the intracellular signaling pathways to stop the signal transduction (Figure 2). Accordingly, various therapeutic agents for inhibiting TLR signaling have been developed to control excessive inflammation; they can be classified as small molecule inhibitors, antibodies, oligonucleotides, lipid-A analogs, microRNAs, and new emerging nano-inhibitors. The current advances of each class are summarized and discussed hereafter (Tables 2–5).
Figure 2
Table 2
| Compound | Target | Indication | Status | Company | References |
|---|---|---|---|---|---|
| TAK-242 | TLR4 | Septic shock | Phase IIIa | Takeda | Rice et al., 2010; Matsunaga et al., 2011 |
| Candesartan1 | ARB, TLR2/4 | Anti-inflammation | Animal study | – | Dasu et al., 2009 |
| Valsartan1 | ARB, TLR4 | Myocardial IR injury | Animal study | Novartis Pharma | Yang et al., 2009 |
| Fluvastatin1 | TLR4 | Chronic heart failure | Experimental | – | Foldes et al., 2008 |
| Simvastatin1 | TLR4 | Atherosclerosis | Experimental | – | Methe et al., 2005 |
| Atorvastatin1 | TLR4 | Atherosclerosis | Animal study | – | Fang et al., 2014 |
| ST2825 | MyD88 | SLE | Experimental | – | Loiarro et al., 2007; Capolunghi et al., 2010 |
| CQ, HCQ | TLR7/8/9 | RA, SLE | Clinical use | Generic | Wozniacka et al., 2006; Lesiak et al., 2010; Lee et al., 2011 |
| CQ, HCQ | TLR7/8/9 | Cerebrovascular ischemia, lupus-associated hypertension, endothelial dysfunction | Animal study | – | Cui et al., 2013; Gomez-Guzman et al., 2014 |
| CQ | TLR7/8/9 | Chikungunya infection | Phase IIIa | Generic | Paton et al., 2011; Borges et al., 2013 |
| Prevention of influenza | Phase IIb | ||||
| Dengue infection | Phase I/IIb | ||||
| CpG-52364 | TLR7/8/9 | RA, SLE | Phase Ic | Pfizer | Lipford et al., 2007 |
| SM934 | TLR7/9 | SLE, autoimmune diseases | IND filed1 | Shanghai Institute of Materia Medica | Hou et al., 2011; Wu et al., 2016 |
The development status of TLR antagonists/inhibitors: class of SMIs.
ARB, angiotensin II receptor blocker; CQ, chloroquine; HCQ, hydroxychloroquine; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; IND, investigational new drug application.
Study terminated.
Unknown status.
Completed with unknown results.
Clinical drugs with new applications.
Small molecule inhibitors (SMIs)
Small molecule inhibitors (SMIs) are synthetic or naturally derived chemical agents with the activity to inhibit TLR signal transduction. Their amphipathic property and small size allow them to cross cell membranes and act on specific intracellular adapter proteins or compartments along the TLR signaling pathways. They usually have good bioavailability but poor specificity and targeting capability. The ease of manufacturing and handling of SMIs makes them popular drug candidates in pharmaceutical industries (Table 2). In the clinical practice, a group of antimalarial drugs, including hydroxychloroquine sulfate (HCQ), chloroquine (CQ), and quinacrine (or mepacrine), have been used to treat autoimmune diseases (arthritis and SLE) after World War II (Fox, 1996; Ruiz-Irastorza et al., 2010). It was not until recent years that their additional mechanisms of action on endosomal TLR signaling (TLR7/8/9) was identified (Kuznik et al., 2011; Lee et al., 2011). These SMIs are all weak bases and tend to accumulate in the acidic intracellular compartments like endosomes and lysosomes, and are able to modulate the pH in these vesicles. The pH modulation can lead to suppression of autoantigen presentation, blockade of endosomal TLR signaling, and decrease in cytokine production (Fox, 1996; Wozniacka et al., 2006; Kuznik et al., 2011). Other mechanisms include inhibition of MAPK signaling and phospholipase A2, antiproliferation, photoprotection as well as reduction of matrix metalloproteinase-9 (MMP-9) activity (Loffler et al., 1985; Weber et al., 2002; Kim et al., 2006; Wozniacka et al., 2008; Lesiak et al., 2010). These mechanisms of action altogether suggest their anti-inflammatory and immuno-suppressive activity.
In addition to autoimmune diseases, these SMIs of TLR7/8/9 are also under investigation in other diseases associated with uncontrolled acute or chronic inflammation. For example, the antimalarial drug CQ has shown its clinical potential in treating severe sepsis, especially in reducing the risk of sepsis-induced acute renal failure (Yasuda et al., 2008). For its capability of blocking endosomal acidification and anti-inflammation, CQ has been applied to prevent and treat various viral infections, such as HIV, influenza, and dengue (Paton et al., 2011; Borges et al., 2013). In pre-clinical studies, CQ pretreatment can significantly improve the cerebral ischemia symptoms in a transient global cerebral ischemia rat model, which suggests that these SMIs of endosomal TLRs could be beneficial for patients with cardiovascular diseases (Cui et al., 2013). Moreover, long-term administration of HCQ has been found to ameliorate hypertension and aortic endothelial dysfunction in a lupus animal model (Gomez-Guzman et al., 2014). These evidences highlight the great potential of the early developed antimalarial drugs and their derivatives as endosomal TLR SMIs in treating a wide range of inflammatory and autoimmune diseases.
Following the success of these early antimalarial drugs, new SMIs have been developed to target endosomal TLR7/8/9 signaling. Among these newly developments, CpG-52364 is a quinacrine derivative, a SMI of TLR7/8/9 (Lipford et al., 2007). Comparing to HCQ, CpG-52364 is more therapeutically effective and has less side effects from animal studies; a phase I clinical trial was completed in 2009 for SLE treatment (Pfizer: NCT00547014), but the results have not been released since. Another exciting advancement is SM934, a novel analog of artemisinin (or “qinghao su,” the new antimalarial drug identified in the early-70's in China), targeting TLR7/9. Studies have shown the therapeutic effects of the orally administered SM934 on lupus-prone MRL/lpr mice by inhibiting Th1 and Th17 cell responses as well as suppressing the B cell activation and plasma cell production (Hou et al., 2011; Wu et al., 2016). ST2825 is a peptidomimetic compound with an inhibitory activity on the MyD88 dimerization and recruitment of interleukin-1 receptor-associated kinase 1 (IRAK1) and IRAK4 (Loiarro et al., 2007); it has been found to suppress TLR9-induced B cell proliferation and differentiation into plasma cells as well as autoantibody production (Capolunghi et al., 2010). Although ST2825 is primarily used as an experimental agent, it provides evidence of inhibiting MyD88 (the universal adaptor used by most TLRs except TLR3) as a potential therapeutic target to treat patients with SLE and possible other autoimmune diseases.
In addition to endosomal TLR signaling, TLR2 and TLR4 are other two major targets of therapeutic SMIs. TAK-242 (Resatorvid) is an anti-sepsis SMI that targets TLR4 signaling pathways (Matsunaga et al., 2011). This compound binds to cysteine 747 in the intracellular TIR domain of TLR4, which blocks the interaction between TLR4 and the adaptor proteins-TIR domain containing adaptor protein (TIRAP) and TRIF-related adaptor molecule (TRAM), thereby diminishing LPS-induced TLR4 signaling and inflammation. Based on its pre-clinical success, TAK-242 advanced into clinical investigations. Two phase III clinical trials NCT00143611 for severe sepsis and NCT00633477 for sepsis-induced cardiovascular and respiratory failure were launched. In the first trial, the results were unfortunately not satisfactory due to failure to effectively suppress serum cytokine levels (IL-6, IL-8, and TNFα) comparing to controls, even though the drug was well-tolerated (Rice et al., 2010). The second trial, however, was terminated due to a business decision, and no further clinical development of TAK-242 has been conducted ever since.
Like antimalarial drugs, angiotensin II receptor blockers (ARBs), and statins are among the early developed drugs with newly discovered inhibitory activity on TLR2 and TLR4 signaling. For example, the ARB family valsartan can decrease proinflammatory cytokines release and infarct size by inhibiting TLR4 signaling (Yang et al., 2009) while candesartan can suppress Pam3CSK4 and LPS induced TLR2 and TLR4 activation, respectively (Dasu et al., 2009). Statins, also known as 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase inhibitors, are a class of lipid-lowering drugs. Among the statin family, fluvastatin, simvastatin, and atorvastatin all have shown potent inhibitory activity on TLR4 and subsequent inflammatory pathways to reduce inflammation in vascular systems (Methe et al., 2005; Foldes et al., 2008; Fang et al., 2014).
Antibodies
Due to their high specificity, antibodies have been widely used as biological tools to precisely probe certain molecules. For therapeutic applications, they can be designed to neutralize soluble effectors, block the binding of receptors to their ligands to stop signal transduction, or induce targeted cytotoxicity (Suzuki et al., 2015). The most attractive feature of therapeutic antibodies is their superior specificity to the drug targets comparing to other type of drugs, and hence they are getting great attentions especially for immunotherapy nowadays. However, antibodies still possess a number of limitations including high costs in manufacturing, poor cellular and tissue penetration, and risks of immunogenicity (Chames et al., 2009).
In terms of inhibiting TLR signaling, antibodies are designed (as antagonists) to block the binding of ligands to the specific TLRs (Table 3). One promising advancement is OPN-305, which is the first fully humanized IgG4 monoclonal TLR2-specific antibody. Pre-clinical studies have shown the potency of this compound in blocking TLR2-mediated pro-inflammatory cytokine production in vitro and in ischemia-reperfusion (IR) injury animal models (Ultaigh et al., 2011; Arslan et al., 2012; Farrar et al., 2012). The success of OPN-305 in phase I study suggests that OPN-305 is an effective and well-tolerated treatment for the prevention of IR injury in solid organ transplantation (Reilly et al., 2013). It is currently under a phase II clinical trial (NCT01794663) in renal transplant patients with a high risk of developing delayed graft function.
Table 3
| Compound | Target | Indication | Status | Company | References |
|---|---|---|---|---|---|
| OPN-305 | TLR2 | Delayed graft function | Phase II | Opsona therapeutics | Arslan et al., 2012; Reilly et al., 2013 |
| T2.5 | TLR2 | Septic shock, cardiac fibrosis, cerebrovascular ischemia | Animal study | – | Meng et al., 2004; Spiller et al., 2008; Ziegler et al., 2011; Wang et al., 2014 |
| NI-0101 | TLR4 | RA | Phase I | NovImmune | Monnet et al., 2015, 2017 |
| 1A6 | TLR4/MD2 | Sepsis | Experimental | – | Spiller et al., 2008; Lima et al., 2015 |
The development status of TLR antagonists/inhibitors: class of antibodies.
RA, rheumatoid arthritis.
T2.5 is another anti-TLR2 antibody with therapeutic potential. It was found to increase animal survival and protect against severe shock-like syndrome in a mouse model challenged with Pam3CSK4 (a TLR1/2 ligand) or lethally challenged with Bacillus subtilis (Meng et al., 2004). A combination therapy of T2.5 with 1A6 (an anti-TLR4 antibody, see below) have shown beneficial effects in a sepsis mouse model challenged by S. enterica and E. coli (Spiller et al., 2008). In addition to sepsis models, T2.5 treatment was able to decrease neural death and inflammatory responses in a model of transient brain ischemia (Ziegler et al., 2011). Moreover, T2.5 was found to prevent angiotensin II-induced cardiac fibrosis through suppressing macrophage recruitment and inflammation in the heart (Wang et al., 2014). Altogether, these proof-of-principle studies suggest that blocking TLR2 signaling is therapeutically beneficial to sepsis and vascular system disorders. However, this compound was not moved forward into clinical development but remains as a widely used experimental tool now.
NI-0101 is the first monoclonal anti-TLR4 antibody entering clinical development. It can potently inhibit TLR4 signaling by blocking TLR4 dimerization, which is independent on the ligand type and concentration (Monnet et al., 2015). NI-0101 just completed a phase I clinical trial (NCT01808469) and the results were very encouraging (Monnet et al., 2017). NI-0101 was well-tolerated without safety concerns at various doses; it could successfully block cytokine release ex vivo and in vivo, and prevent flu-like symptoms upon LPS administration. Recent data demonstrated that NI-0101 could also block rheumatoid arthritis (RA) synovial fluids-induced pro-inflammatory cytokine production in monocytes isolated from RA patients (Hatterer et al., 2016). Now, the company is preparing for a phase II clinical trial in RA patients with a TLR4-driven pathotype in 2017.
Another monoclonal antibody of TLR4 is 1A6, which targets the extracellular portion of the TLR4-myeloid differentiation factor 2 (MD2, a co-receptor for TLR4 activation) complex (Spiller et al., 2008). In preclinical study, 1A6 showed protective effect on microbial-induced septic shock in vivo (Spiller et al., 2008; Lima et al., 2015). Interestingly, in a mouse model of dextran sulfate sodium (DSS)-induced colitis, 1A6 could ameliorate inflammation and prevent the progression of the intestine inflammation; however, the blockade of TLR4 signaling by 1A6 could also cause a defect in the mucosal healing during the recovery stage (Ungaro et al., 2009). This study perhaps sent a warning message of blocking early TLR4 signaling as potential therapy for inflammatory bowel diseases (IBD); on the other hand, it provided a strong evidence of multiple roles of TLRs in disease development, progression and remission, guiding the development of next generation of therapeutic TLR inhibitors.
Oligonucleotides
Since endosomal TLRs primarily recognize nucleic acid structures from dsRNA, ssRNA, and CpG-DNA, oligonucleotides with certain sequences are predicted to function as antagonists of endosomal TLRs. They can interfere the binding of endosomal TLRs to their ligands, and hence block the TLR signal transduction. Accordingly, several oligonucleotide-based TLR antagonists have been developed particularly to treat inflammatory diseases associated with endosomal TLR activation, such as SLE; each is discussed hereafter (Table 4).
Table 4
| Compound | Target | Indication | Status | Company | References |
|---|---|---|---|---|---|
| IRS-954 | TLR7/9 | SLE | Preclinicala | Dynayax Technologies | Barrat et al., 2005, 2007; Guiducci et al., 2010 |
| DV-1179 | TLR7/9 | SLE | Phase Ib/IIaa | Dynayax Technologies | Zhu et al., 2011; Suarez-Farinas et al., 2013 |
| IMO-3100 | TLR7/9 | SLE | Preclinical | Idera | Zhu et al., 2011; Suarez-Farinas et al., 2013 |
| Psoriasis | Phase IIb | ||||
| IMO-8400 | TLR7/8/9 | Dermatomyositis | Phase II | Idera | Jiang et al., 2012; Zhu et al., 2012 |
| Plaque psoriasis | Phase II | ||||
| IMO-9200 | TLR7/8/9 | Autoimmune diseases | Phase I | Idera/Vivelix | – |
| IHN-ODN 2088 | TLR9 | Hypertension | Animal study | – | McCarthy et al., 2015 |
| IHN-ODN-24888 | TLR7/9 | SLE | Preclinical | Coley Pharmaceutical GmbH | Rommler et al., 2013, 2015 |
The development status of TLR antagonists/inhibitors: class of oligonucleotides.
SLE, systemic lupus erythematosus.
Study discontinued.
Completed with unknown results.
One of the leading developments of oligonucleotide-based TLR antagonists is named immunoregulatory DNA sequence (IRS; by Dynavax Technologies). Advancing from the earlier encouraging prototypes, IRS-661 (TLR7 specific) and IRS-869 (TLR9 specific), a dual-function targeting both TLR7 and TLR9 antagonist, IRS-954 was developed for the treatment of SLE (Barrat et al., 2005). IRS-954 was found to inhibit IFN-α production by human plasmacytoid predendritic cells in response to DNA or RNA of viruses and immune complexes from SLE patients. In addition, IRS-954 treatment showed a reduction of autoantibodies and improvement of disease symptoms in lupus-prone mice (Barrat et al., 2007). Furthermore, it could also reverse TLR7/9-mediated glucocorticoid resistance of SLE, and thus could be potentially used as corticosteroid-sparing drug (Guiducci et al., 2010). Although IRS-954 has shown promising pre-clinical therapeutic efficacy, there is no further advancement documented in the clinical development to date. On the other hand, DV-1179, another TLR7/9 dual antagonist developed by the same company, entered a phase Ib/IIa study (in partner with GSK) for its safety in both healthy volunteers and patients with active SLE (Dynavax, 2014). Although DV-1179 administration was well tolerated, it failed to achieve the endpoints of reducing the IFN-α-regulated genes1. Such an unfortunate outcome ended the clinical development of this compound ever since.
Another development of oligonucleotide-based antagonists (also termed as immune modulatory oligonucleotide IMO) has made some success into clinical trials (by Idera Pharmaceuticals). The compound IMO-3100 is also a TLR7/9 dual antagonist. In preclinical studies, IMO-3100 could significantly reduce the expression of inflammatory genes, such as IL-17A, β-defensin, CXCL1, and keratin 16 in a mice model of IL-23-induced psoriasis (Suarez-Farinas et al., 2013). Moreover, this compound showed an inhibitory activity on disease progression in a lupus-prone mice model (Zhu et al., 2011). A phase II trial for the treatment of psoriasis was completed in 2012 (NCT01622348), but the results have not yet been disclosed. IMO-8400 is another candidate under current development. Different from IMO-3100, IMO-8400 is capable of inhibiting all TLR7, TLR8, and TLR9. This compound was very effective in preventing inflammation and disease development in both lupus and psoriasis mouse models (Jiang et al., 2012; Zhu et al., 2012; Suarez-Farinas et al., 2013). Currently, two phase II clinical trials are underway for the treatment of plaque psoriasis (NCT01899729) and dermatomyositis (NCT02612857). Similar to IMO-8400, a newly developed IMO-9200 can also target TLR7/8/9. From a phase I clinical trial (in partner with Vivelix), it showed a safe and well-tolerated profile in healthy subjects2.
There are other immunoregulatory oligonucleotides under preclinical developments. The inhibitory oligonucleotide (IHN-ODN) 2088 is a potent TLR9 antagonist. In spontaneously hypertensive rats, IHN-ODN 2088 treatment showed positive effects on systolic blood pressure reduction (McCarthy et al., 2015). Based on IHN-ODN 2088, a guanine-modified inhibitory oligonucleotide, INH-ODN-24888, was designed; this compound demonstrated a promising therapeutic effect in SLE through the suppression of both TLR7 and TLR9 (Rommler et al., 2015). The guanine modification potentiates the inhibitory activity of INH-ODN-24888 and efficiently improves its capacity to reduce TLR7/9-mediated immune signaling in human immune cells (Rommler et al., 2013). These results indicates that IHN-ODN-24888 may be further developed into a promising clinical treatment for SLE.
Lipid A analogs
A special group of TLR antagonists is the lipid A analog (Table 5), specifically targeting TLR4. Lipid A is a lipid component of LPS (or endotoxin) contributing to the toxicity of an endotoxin molecule. Because the structure of lipid A is highly conserved in endotoxins, it becomes an attractive therapeutic target to regulate TLR4 signaling (Rietschel et al., 1994).
Table 5
| Compound | Target | Indication | Status | Company | References |
|---|---|---|---|---|---|
| CLASS OF LIPID A ANALOGS | |||||
| Eritoran (E5564) | TLR4/MD2 | Sepsis | Phase IIIa | Eisai (Boston) | Barochia et al., 2011; Opal et al., 2013 |
| Influenza infection, cardiac hypertrophy, myocardial IR injury, renal IR injury | Animal study | – | Shimamoto et al., 2006; Liu et al., 2010; Ehrentraut et al., 2011; Shirey et al., 2013 | ||
| CLASS OF miRNAs | |||||
| miR-146a | TLR4 | Autoimmune diseases, SLE | Animal study | – | Taganov et al., 2006; Tang et al., 2009; Boldin et al., 2011; Pan et al., 2012 |
| miR-21 | TLR4 | Anti-inflammation | Animal study | – | Sheedy et al., 2010 |
| CLASS OF “NANO-DRUGS” | |||||
| NAHNP | TLR4 | Anti-inflammation | Experimental | – | Babazada et al., 2014a,b |
| HDL-like NP | TLR4 | Anti-inflammation | Experimental | – | Foit and Thaxton, 2016 |
| Bare GNP | TLR4 | Eye inflammation | Animal study | – | Pereira et al., 2012 |
| Glycolipid-coated GNP | TLR4/MD2 | Sepsis | Experimental | – | Rodriguez Lavado et al., 2014 |
| Peptide-GNP hybrid | TLR2/3/4/5 | Anti-inflammation | Experimental | – | Yang et al., 2015, 2016 |
The development status of TLR antagonists/inhibitors: classes of lipid A analogs, miRNAs and “nano-drugs.”
SLE. systemic lupus erythematosus; NAHNP, non-anticoagulant heparin nanoparticle; HDL, high-density lipoprotein; NP, nanoparticle; GNP, gold nanoparticle.
Study discontinued.
A synthetic lipid A analog of Rhodobacter sphaeroides, eritoran (E5564), is probably the most exciting but also disappointing clinical development of TLR4 antagonist (by Eisai Research Institute of Boston Inc.) (Barochia et al., 2011). Eritoran is the 2nd generation of its own kind derived from the first developed E5531 (Christ et al., 1995). The design principle of these compounds was to competitively bind to the MD2 pocket by mimicking the lipid A structure, which in turn prevents the LPS binding and the induction of TLR4 signaling (Park et al., 2009). The structural improvement of eritoran makes it superior to E5531 for its higher potency due to extended duration of action, cost-effective manufacturing because of a simpler structure, greater water solubility, and better chemical stability. Preclinically, eritoran could significantly reduce LPS-induced NF-κB activation and pro-inflammatory cytokine production (TNF-α, IL-1β, IL-6, and IL-8) in vitro and in animal models (Mullarkey et al., 2003; Savov et al., 2005). In a phase I clinical study on LPS challenged healthy volunteers, eritoran treatment effectively decreased TNF-α and IL-6 levels in the blood, reduced the white blood cell counts and C-reactive protein levels, and diminished sepsis-associate clinical symptoms; the only observed adverse response was the dose-dependent phlebitis (Lynn et al., 2004; Rossignol et al., 2004, 2008). It then went on a randomized, double-blind, multicenter phase II trial (NCT00046072) to treat critically ill septic patients with high predicted risk of mortality; the results were positive as the intravenous treatment of eritoran at the dose of 105 mg led to a trend toward a lower mortality rate (Tidswell et al., 2010). Sadly, the phase III trial (ACCESS, NCT00334828) of eritoran on severe sepsis patients failed because eritoran did not reduce the patient mortality at 28 days and 1 year from the statistical analysis (Opal et al., 2013). Accordingly, the company decided to give up launching the drug to the market.
Although the development of eritoran led to a disappointing end, valuable lessons can be learned from this failed practice for developing the next generation of TLR inhibitors. First, the patient recruitment is key to objectively evaluate the drug efficacy in a clinical trial. One major reason that eritoran failed in the ACCESS was probably because the recruited patients were not well-assessed according to their disease pathogenesis, particularly the circulating levels of LPS in the patients. This is a critical factor that should be included in the recruiting criteria. Since the mechanism of eritoran is to block LPS binding to TLR4, those patients who had low levels of circulating LPS at the time of studies may not respond well to the treatment, and thus contributed to an overall null efficacy of the drug. Second, the time and route of drug administration need to be optimized. Considering the acute inflammatory responses in sepsis, the timing of eritoran administration could be too late to be therapeutic effective when other inflammatory mediators started to take over. Third, the genetic backgrounds, the infection status, and the disease severity all contributed to the heterogeneity in the patients, which ultimately resulted in different responses toward the treatment. Finally, the inflammatory responses of the septic patients could be triggered by multiple TLR pathways, and blocking one of them (even though TLR4 has been considered the major one) may not be enough to stop the overwhelming inflammation in severe sepsis. To ensure future success in developing new TLR inhibitors for inflammatory diseases like sepsis, perhaps new inhibitory strategy on multiple TLR pathways should be considered; and clearly in the clinical trials, patients should be carefully selected and examined based on appropriate criteria with well-defined biomarkers to obtain the fair evaluation and interpretation of the therapeutic efficacy of the tested drugs.
Apart from treating sepsis, eritoran could still be therapeutically beneficial for other inflammatory diseases. In fact, one study has shown that eritoran could prevent influenza-induced death in a mouse model, where it improved the clinical symptoms, inhibited cytokine production (TNF-α, IL-1β, CXCL1, and IL-6), and reduced viral titers (Shirey et al., 2013). Other studies have shown the therapeutic efficacy of eritoran in several cardiovascular disease models. For example, eritoran could reduce cardiac hypertrophy in an aortic constriction mouse model (Ehrentraut et al., 2011), and attenuate inflammatory cytokine production and myocardial IR injury in a rat model (Shimamoto et al., 2006). In addition, eritoran has also provided protective effects on kidney IR injury (Liu et al., 2010). Unexpectedly, it has also been suggested that eritoran could potentially overcome the blood-brain barrier under certain inflamed conditions (like stoke) and play an advantageous role in treating cerebral infarction (Buchanan et al., 2010).
miRNA inhibitors
MicroRNAs (miRNAs) are small (~22 nucleotides), endogenous non-coding RNAs with post-transcriptional regulatory functions to finely tune gene expression (Bartel, 2009). They bind to a target sequence in the 3′-untranslated region (UTR) of the messenger RNA (mRNA) to facilitate mRNA degradation or inhibit its translation. Since miRNAs are involved in the regulation of diverse biological processes and associated with pathogenesis of many diseases, they have emerged as new therapeutic targets.
Till now, there are about 20 miRNAs identified to be involved in the regulation of TLR signaling pathways (He et al., 2014). Among them, miR-146a, miR-155, and miR-21 are the three miRNAs received extensive attention for their regulatory roles in TLR signaling and autoimmune diseases (Table 5; Quinn and O'Neill, 2011; Shen et al., 2012). MiR-146a was identified from a screen of LPS-responsive genes regulated by NF-κB; it was found to inhibit the translation of the TNF receptor-associated family 6 (TRAF6) and IRAK1, both of which are in the downstream pathway of TLR4 signaling (Taganov et al., 2006). Further studies have shown that miR-146a could play a negative regulatory role in autoimmunity (Boldin et al., 2011), particularly in blocking the type I IFN production in human lupus by targeting TLR-type I IFN associated genes TRAF6, IRAK1, IFN regulatory factor 5 (IRF5), and signal transducer and activator of transcription 1 (STAT1; Tang et al., 2009). Given the fact that miR-146a acts on multiple targets in the signaling cascades of TLR-mediated induction of type I IFN, the integrated effects of negative regulation by miR-146a on the pathway are expected to be beneficial in treating SLE and perhaps RA. To translate miR-146a into clinic uses, one possible way is to apply a delivery vehicle to help miR-146a cross the cell membranes in order to function intracellularly (Pan et al., 2012).
Both miR-21 and miR-155 were originally identified as “onco-miR,” but later discovered to have a new role in regulating TLR signaling and inflammation. They were found to be upregulated in TLR-associated inflammatory conditions (O'Connell et al., 2007; Sheedy et al., 2010). Specifically, miR-21 could enhance the LPS-induced anti-inflammatory cytokine IL-10 production by lowering programed cell death 4 gene (PDCD4) expression, serving as a negative regulator of the inflammatory response (Sheedy et al., 2010). MiR-155, on the other hand, appears to have controversial roles in controlling inflammation. In one study, miR-155 targeted TGF-β activated kinase 1 (TAK1) binding protein 2 (TAB2) and inhibited TAK1 activation, which subsequently blocked the activation of downstream NF-κB and MAPK pathways, and dampened the inflammatory responses (Ceppi et al., 2009). Another study reported that miR-155 expression contributed to the production of the proinflammatory cytokine TNF-α and increased the susceptibility to septic shock (Tili et al., 2007). In addition, miR-155 was found to have a positive effect in the pathogenesis of SLE, as miR-155 knockout mice had phenotypes similar to some human systemic autoimmune disorders, and were highly resistant to experimental autoimmune encephalomyelitis, a SLE mouse models (O'Connell et al., 2010). Accordingly, a miR-155 antagonist, MRG-107, is currently under preclinical development (by miRagen Therapeutics) to treat neuro-inflammation.
New emerging nano-inhibitors
In addition to above described inhibitors/antagonists, nanodevices are emerging as a new class of potent TLR inhibitors that can target a single or multiple TLR pathways (Table 5). Due to their nanoscale sizes, nano-inhibitors are expected to have better bio-distribution and sustained circulation (He et al., 2010; Blanco et al., 2015). They can be functionalized to meet the desired pharmacodynamic and pharmacokinetic profiles (Ernsting et al., 2013; Lin and Tam, 2015). One distinct feature of these new nano-inhibitors is that the bio-activity comes from their self-properties, which can be tailored to different medical complications, rather from a carried therapeutic agent. This characteristic makes them a special class of drug (or “nano-drug”) as the next generation nano-therapeutics. Currently, they are at the preclinical investigation stage. Figure 3 summarizes their targets in the TLR pathways, whereas their structures, physicochemical properties and mechanisms of action are listed in Table 6.
Figure 3
Table 6
| Compound | NAHNP | HDL-like NP | Bare GNP | Glycolipid-coated GNP | Peptide-GNP hybrid |
|---|---|---|---|---|---|
| Structure | 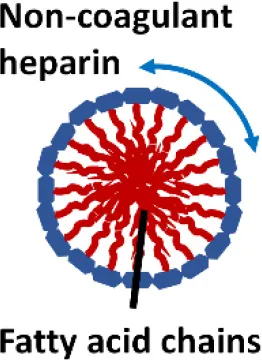 | 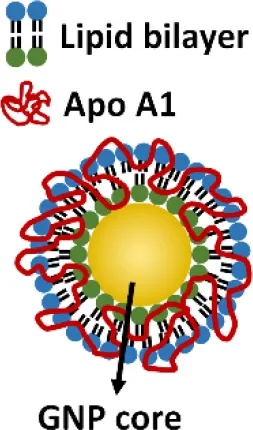 | 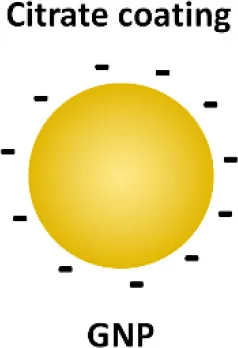 | 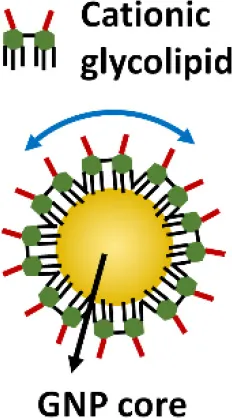 |  |
| Size | 110–160 nm | ~15 nm | ~30 nm | N/A | ~15 nm |
| NP formation | Self-assembling | Surface coating on GNP | GNP only (citrate coated) | Surface coating on GNP | Surface coating on GNP |
| Surface charge | Negative | Negative | Negative | Positive | Negative |
| Biodegradability | Possible | No | No | No | No |
| Mechanisms of action | Interaction with TLR4/MD2 complex | Neutralization of LPS | Decrease in TLR4 and NF-κB expression | Binding to TLR/MD2 complex | Endosomal pH modulation |
| Inhibiting MyD88-dependent NF-κB pathway | Inhibiting LPS induced NF-κB signaling | Inhibiting LPS induced NF-κB signaling | Inhibiting both NF-κB and IRF pathways | ||
| References | Babazada et al., 2014a,b | Foit and Thaxton, 2016 | Pereira et al., 2012 | Rodriguez Lavado et al., 2014 | Yang et al., 2015, 2016 |
The structures, physicochemical properties, and mechanisms of the novel TLR nano-inhibitors.
NAHNP, non-anticoagulant heparin nanoparticle; HDL, high-density lipoprotein; NP, nanoparticle; GNP, gold nanoparticle; Apo A1, apolipoprotein A1; N/A, not available.
One interesting immunomodulatory nanodevice is the lipid-conjugated non-anticoagulant heparin nanoparticle (NAHNP; Babazada et al., 2014a,b). In addition to its well-known anti-coagulant function, heparin is known to have anti-inflammatory activity for its ability to bind and inhibit various cytokines, growth factors and enzymes that are involved in inflammation (Tyrrell et al., 1999); especially, the 6-O sulfation of glucosamine residues of heparin can block the binding of P- and L-selectins for the recruitment of leukocytes at inflammatory sites (Wang et al., 2002). Being intrigued by the intrinsic anti-inflammatory properties of heparin, Babazada et al. synthesized the glycol-split non-anticoagulant heparin with D-erythro-sphingosine grafts, which can self-assemble into NAHNP (Babazada et al., 2014b). These nanoparticles (NPs) were found to suppress LPS-induced MyD88-dependent NF-κB activation and inhibit subsequent cytokines production in mouse macrophages. They further discovered that the length of the alkyl chains of conjugated D-erythro-sphingosine was critical for the anti-inflammatory activity as shortening the alkyl chain of NAHNP resulted in loss of activity. Again, the 6-O-sulfate groups of d-glucosamine residue were important for such an inhibitory activity. By removing the anti-coagulant activity of heparin, NAHNP could serve as new anti-inflammatory nano-therapeutics.
Another novel nanodevice, the high-density lipoprotein (HDL)-like nanoparticle (HDL-like NP), was developed as a TLR4 antagonist by sequestering LPS (Foit and Thaxton, 2016). HDL has been known to naturally bind to and neutralize LPS (Brandenburg et al., 2002), and has been applied to reduce LPS-induced inflammation in vivo (Pajkrt et al., 1996; Guo et al., 2013). The HDL-like NP was designed with a gold NP core and a HDL coating, where the lipid components of HDL can be modified (Foit and Thaxton, 2016). Through screening five variants of the lipid coatings, one HDL-like NP was identified to be able to potently inhibit TLR4 signaling triggered by various sources of LPS and Gram-negative bacteria on human cell lines and PBMC. As expected, the acting mechanism of this HDL-like NP was through scavenging the LPS molecules. It is worth noting that this newly synthesized HDL-like NP exhibits much higher anti-inflammatory activity than the HDL alone, making it a promising candidate as a next generation nano-antagonist of TLR4.
Gold nanoparticles (GNPs) have caught much attention as emerging nanotheranostics in nanomedicine, owing to their ease of fabrication and functionalization, chemical stability, excellent photothermal physical properties, and good biocompatibility (Pedrosa et al., 2015). The fact that a TNF-α-bound GNP agent (CYT-6091 by CytImmune Sciences) showed very-well tolerated profile in a phase I clinical trial warrants the potential clinical uses of GNP-based therapeutics in the near future (Libutti et al., 2010). The exciting advancement in the field is the development of GNP-based “nano-inhibitors” for TLR signaling. In one study, bare GNP was applied as the post-treatment topically to ameliorate LPS-induced eye inflammatory responses and reduce the oxidative damage in irides (Pereira et al., 2012). The possible mechanism was through down-regulating TLR4 expression and the corresponding LPS-induced NF-κB activation. This discovery is quite surprising as the bare GNP is physiologically unstable, and is usually thought to be inert without biological activity. It would be interesting to see the follow-up study and the detailed working mechanisms of bare GNP. In another study, a cationic glycolipid-coated GNP system was developed as a new TLR4 antagonist (Rodriguez Lavado et al., 2014). A new class of glycolipids was synthesized to have a structure mimicking lipid A binding to CD14 and the TLR-MD2 pocket. The biological analysis showed that the GNP coated with a specific glycolipid (11-NP) was able to inhibit the LPS-induced TLR4-MD2 activation in human and murine cells; such an effect was similar to the very efficient TLR4 antagonist eritoran. Further studies on the structure-activity relationship analysis indicated that the presence of fatty ester chains and the facial amphiphilicity of the glycolipids are required to maintain TLR4 antagonistic activity. Preliminary in vivo studies showed that 11-NP was very effective in reducing TNF-α level in the blood in a LPS mouse model.
One exciting invention is the development of a completely novel peptide-GNP hybrid system as a next generation nano-inhibitor led by Yang et al. (2015, 2016). The system is made of a GNP core coated with a hexapeptide layer to enhance the physiological stability of the GNP, change the surface physicochemical characteristics, and enable the biological activity (Yang et al., 2011, 2013). Through, screening a small library of the established, physiologically stable peptide-GNP hybrids, they discovered a potent multi-TLRs inhibitor, P12, which not only suppressed both arms of TLR4 signaling (i.e., MyD88-dependent NF-κB and TRIF-dependent IRF3 activation), but also inhibited TLR2, TLR3, and TLR5 pathways. The structure-activity relationship analysis revealed that the activity index is strongly dependent on the surface properties of the hybrid, where the hydrophobicity and the aromatic ring-like structure are the two key factors dictating the inhibitory activity (Yang et al., 2015). Further studies conferred the anti-inflammatory activity of P12 in correcting LPS-induced gene expressions, reducing the subsequent pro-inflammatory cytokine production (IL-12p40, MCP-1, and IFN-γ), and increasing the anti-inflammatory cytokine IL-1RA. In searching for the mechanism of action, delicate experiments were performed to show that the potent inhibitory activity of P12 is mainly due to its endosomal pH modulation capability (Yang et al., 2016). Like chloroquine (CQ), a lysotropic agent that can elevate endosome/lysosome pH, P12 was able to prevent the endosomal acidification process, which in turn attenuated downstream signal transduction of endosomal-dependent TLR signaling. More importantly, the inhibitory activity of the “nano-drug” can be well-tuned by selecting different amino acids that are displayed on the GNPs. The in vivo efficacy of P12 was investigated using a murine model of intestinal inflammation; it was found that P12 treatment could reduce the animal weight loss, improve the disease activity index, and ameliorate colonic inflammation.
All the above nanoparticles have minimum acute toxicity in culture cells as well as in the limited animal studies. However, since the gold nanoparticle is not biodegradable, the long-term safety issue of using GNP-based therapeutics is still a concern, even though they are well-tolerated in a phase I study. Therefore, addressing the long-term safety problem of GNP-based TLR inhibitors is critical in order to move this line of research into clinic uses. Alternatively, one could replace the GNP core with a biodegradable NP while maintaining the inhibitory activity, preferably targeting multiple TLRs like peptide-gold nanoparticle hybrids, to accelerate the translation of these novel agents into clinical treatments for inflammatory diseases.
Note that the majority of these developed TLR nano-inhibitors act on TLR4 signaling, except the peptide-gold nanoparticle hybrid P12 which has shown inhibitory activity on multiple TLRs. In the current literatures reviewed, LPS has been used universally as the TLR4 agonist. However, other purinergic receptors, such as P2X7 receptor (encoded by P2RX7 gene), can also recognize LPS and induce the secretion of inflammatory mediators in macrophages (Denlinger et al., 2001). Interestingly, the recognition of LPS by P2X7 requires a co-receptor CD14, which facilitates LPS internalization and binding to the intracellular C-terminal domain of P2X7 (Dagvadorj et al., 2015). Therefore, the specificity of these nanoparticles in inhibition of TLR4 vs. other membrane receptors that also recognize LPS needs to be further addressed. It will also be interesting to see whether these nano-inhibitors can also modulate signaling of other TLRs in addition to TLR4.
Currently, these TLR nano-inhibitors are still at the preclinical investigation stage. Their pharmacokinetics and pharmacodynamics profiles in small and large animals need to be accomplished before launching any clinical trials. However, a few nanoparticles/nano-formulations have already been approved by FDA to deliver water-insoluble drugs or biologics, and have shown enhanced efficacy and safety profiles in cancer treatments. Some well-known examples include the polyethylene glycol (PEG) formulated L-asparaginase (Oncaspar by Sigma-Tau Pharmaceuticals, Inc.) and liposome formulated doxorubicin (Doxil by Jansen Pharmaceutical). Furthermore, several inorganic nanoparticles have been approved by FDA as diagnostic imaging agents. For instance, the iron oxide nanoparticles (Feridex by Bayer HealthCare Pharmaceuticals Inc.) have been used in medical magnetic resonance imaging (MRI). Recent records of clinically used nanoparticles are summarized in two comprehensive reviews in 2016 (Anselmo and Mitragotri, 2016; Bobo et al., 2016). Different from the clinically used nano-therapeutics, the reviewed TLR nano-inhibitors herein would represent a new class of “nano-drug” modulating TLR signaling, and hold great promise for clinical use in the future.
Concluding remarks
TLRs are very important PRRs in the first-line defense system of our bodies. They can recognize both invading pathogens and endogenous danger molecules released from dying cells and damaged tissues, and play a key role in linking innate and adaptive immunity. Despite their well-known function in sensing pathogens and mounting defense mechanisms, overactivation of TLRs can ultimately lead to disruption of immune homeostasis, and thus increase the risk for inflammatory diseases and autoimmune disorders, such as SLE, RA, atherosclerosis, stoke, IR-induced injury, COPD, asthma, gastrointestinal inflammation, and even cancer. Evidences have shown the involvement of TLRs in the development and progression of these diseases. Accordingly, antagonists/inhibitors targeting TLR signaling pathways have emerged as novel therapeutics to treat these diseases. Although, this therapeutic strategy has been well-accepted experimentally, there are only limited numbers of TLR inhibitors/antagonists that have undergone clinical trials to date; many of them failed during the trials, and none have been approved for clinical uses. Lessons have been learned from these failed developments. The efficacy, safety, and stability of the new therapeutic agents, as well as whether these therapies could impair local immune defenses should be considered comprehensively in the clinical development. More importantly, given the facts that pathogen infection or danger signals often activate multiple TLRs, and that the failure of TLR4 specific antagonist/inhibitor in the clinical trials despite its success in preclinical studies, developing more potent antagonists/inhibitors that target multiple TLR signaling pathways perhaps could shine the way to facilitate the translation of such a promising strategy into clinical uses in the future. Certainly, nano-devices will play an important role in the development of next generation TLR inhibitors.
Statements
Author contributions
QL and HY proposed and designed the scope of the review. WG and HY wrote the manuscript. YX and QL critically revised and commented on the manuscript. All the authors read and approved the final manuscript and agreed to be accountable for the accuracy and integrity of any part of the manuscript.
Acknowledgments
QL would like to thank the support from the National Natural Science Foundation of China (Grant No. 81170060/H0111). HY would like to acknowledge the support from the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (TP2016014), the starting fund from Shanghai First People's Hospital, Gaofeng Clinical Medicine Grant support from Shanghai Jiaotong University School of Medicine, and the funding from the Crohn's and Colitis Foundation of Canada (CCFC).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AkiraS.TakedaK. (2004). Toll-like receptor signalling. Nat. Rev. Immunol.4, 499–511. 10.1038/nri1391
2
AnselmoA.MitragotriS. (2016). Nanoparticles in the clinic. Bioeng. Transl. Med.1, 10–29. 10.1002/btm2.10003
3
ArslanF.HoutgraafJ. H.KeoghB.KazemiK.de JongR.McCormackW. J.et al. (2012). Treatment with OPN-305, a humanized anti-Toll-Like receptor-2 antibody, reduces myocardial ischemia/reperfusion injury in pigs. Circ. Cardiovasc. Interv.5, 279–287. 10.1161/CIRCINTERVENTIONS.111.967596
4
BabazadaH.YamashitaF.HashidaM. (2014a). Suppression of experimental arthritis with self-assembling glycol-split heparin nanoparticles via inhibition of TLR4-NF-kappaB signaling. J. Control. Release194, 295–300. 10.1016/j.jconrel.2014.09.015
5
BabazadaH.YamashitaF.YanamotoS.HashidaM. (2014b). Self-assembling lipid modified glycol-split heparin nanoparticles suppress lipopolysaccharide-induced inflammation through TLR4-NF-kappaB signaling. J. Control. Release194, 332–340. 10.1016/j.jconrel.2014.09.011
6
BarochiaA.SolomonS.CuiX.NatansonC.EichackerP. Q. (2011). Eritoran tetrasodium (E5564) treatment for sepsis: review of preclinical and clinical studies. Expert Opin. Drug Metab. Toxicol.7, 479–494. 10.1517/17425255.2011.558190
7
BarratF. J.MeekerT.ChanJ. H.GuiducciC.CoffmanR. L. (2007). Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur. J. Immunol.37, 3582–3586. 10.1002/eji.200737815
8
BarratF. J.MeekerT.GregorioJ.ChanJ. H.UematsuS.AkiraS.et al. (2005). Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med.202, 1131–1139. 10.1084/jem.20050914
9
BartelD. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell136, 215–233. 10.1016/j.cell.2009.01.002
10
BergtS.WagnerN. M.HeidrichM.ButschkauA.Noldge-SchomburgG. E.VollmarB.et al. (2013). Hydrocortisone reduces the beneficial effects of toll-like receptor 2 deficiency on survival in a mouse model of polymicrobial sepsis. Shock40, 414–419. 10.1097/SHK.0000000000000029
11
BeutlerB. A. (2009). TLRs and innate immunity. Blood113, 1399–1407. 10.1182/blood-2008-07-019307
12
BezemerG. F.SagarS.van BergenhenegouwenJ.GeorgiouN. A.GarssenJ.KraneveldA. D.et al. (2012). Dual role of Toll-like receptors in asthma and chronic obstructive pulmonary disease. Pharmacol. Rev.64, 337–358. 10.1124/pr.111.004622
13
BlancoE.ShenH.FerrariM. (2015). Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol.33, 941–951. 10.1038/nbt.3330
14
BoboD.RobinsonK. J.IslamJ.ThurechtK. J.CorrieS. R. (2016). Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm. Res.33, 2373–2387. 10.1007/s11095-016-1958-5
15
BoldinM. P.TaganovK. D.RaoD. S.YangL.ZhaoJ. L.KalwaniM.et al. (2011). miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med.208, 1189–1201. 10.1084/jem.20101823
16
BomfimG. F.EchemC.MartinsC. B.CostaT. J.SartorettoS. M.Dos SantosR. A.et al. (2015). Toll-like receptor 4 inhibition reduces vascular inflammation in spontaneously hypertensive rats. Life Sci.122, 1–7. 10.1016/j.lfs.2014.12.001
17
BorgesM. C.CastroL. A.FonsecaB. A. (2013). Chloroquine use improves dengue-related symptoms. Mem. Inst. Oswaldo Cruz108, 596–599. 10.1590/S0074-02762013000500010
18
BotosI.SegalD. M.DaviesD. R. (2011). The structural biology of Toll-like receptors. Structure19, 447–459. 10.1016/j.str.2011.02.004
19
BrandenburgK.JurgensG.AndraJ.LindnerB.KochM. H.BlumeA.et al. (2002). Biophysical characterization of the interaction of high-density lipoprotein (HDL) with endotoxins. Eur. J. Biochem.269, 5972–5981. 10.1046/j.1432-1033.2002.03333.x
20
BreaD.SobrinoT.Rodriguez-YanezM.Ramos-CabrerP.AgullaJ.Rodriguez-GonzalezR.et al. (2011). Toll-like receptors 7 and 8 expression is associated with poor outcome and greater inflammatory response in acute ischemic stroke. Clin. Immunol.139, 193–198. 10.1016/j.clim.2011.02.001
21
BryantC. E.SymmonsM.GayN. J. (2015). Toll-like receptor signalling through macromolecular protein complexes. Mol. Immunol.63, 162–165. 10.1016/j.molimm.2014.06.033
22
BuchananM. M.HutchinsonM.WatkinsL. R.YinH. (2010). Toll-like receptor 4 in CNS pathologies. J. Neurochem.114, 13–27. 10.1111/j.1471-4159.2010.06736.x
23
CapolunghiF.RosadoM. M.CascioliS.GirolamiE.BordascoS.VivarelliM.et al. (2010). Pharmacological inhibition of TLR9 activation blocks autoantibody production in human B cells from SLE patients. Rheumatology49, 2281–2289. 10.1093/rheumatology/keq226
24
CavassaniK. A.IshiiM.WenH.SchallerM. A.LincolnP. M.LukacsN. W.et al. (2008). TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J. Exp. Med.205, 2609–2621. 10.1084/jem.20081370
25
CelharT.FairhurstA. M. (2014). Toll-like receptors in systemic lupus erythematosus: potential for personalized treatment. Front. Pharmacol.5:265. 10.3389/fphar.2014.00265
26
CeppiM.PereiraP. M.Dunand-SauthierI.BarrasE.ReithW.SantosM. A.et al. (2009). MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc. Natl. Acad. Sci. U.S.A.106, 2735–2740. 10.1073/pnas.0811073106
27
ChamesP.Van RegenmortelM.WeissE.BatyD. (2009). Therapeutic antibodies: successes, limitations and hopes for the future. Br. J. Pharmacol.157, 220–233. 10.1111/j.1476-5381.2009.00190.x
28
ChenM.ZhangW.XuW.ZhangF.XiongS. (2011). Blockade of TLR9 signaling in B cells impaired anti-dsDNA antibody production in mice induced by activated syngenic lymphocyte-derived DNA immunization. Mol. Immunol.48, 1532–1539. 10.1016/j.molimm.2011.04.016
29
ChristW. J.AsanoO.RobidouxA. L.PerezM.WangY.DubucG. R.et al. (1995). E5531, a pure endotoxin antagonist of high potency. Science268, 80–83. 10.1126/science.7701344
30
ChristensenS. R.ShupeJ.NickersonK.KashgarianM.FlavellR. A.ShlomchikM. J. (2006). Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity25, 417–428. 10.1016/j.immuni.2006.07.013
31
ClancyR. M.MarkhamA. J.BuyonJ. P. (2016). Endosomal Toll-like receptors in clinically overt and silent autoimmunity. Immunol. Rev.269, 76–84. 10.1111/imr.12383
32
CuiG.YeX.ZuoT.ZhaoH.ZhaoQ.ChenW.et al. (2013). Chloroquine pretreatment inhibits toll-like receptor 3 signaling after stroke. Neurosci. Lett.548, 101–104. 10.1016/j.neulet.2013.02.072
33
DagvadorjJ.ShimadaK.ChenS.JonesH. D.TumurkhuuG.ZhangW.et al. (2015). Lipopolysaccharide induces Alveolar Macrophage Necrosis via CD14 and the P2X7 receptor leading to interleukin-1α release. Immunity42, 640–653. 10.1016/j.immuni.2015.03.007
34
DasuM. R.RiosvelascoA. C.JialalI. (2009). Candesartan inhibits Toll-like receptor expression and activity both in vitro and in vivo. Atherosclerosis202, 76–83. 10.1016/j.atherosclerosis.2008.04.010
35
DaubeufB.MathisonJ.SpillerS.HuguesS.HerrenS.FerlinW.et al. (2007). TLR4/MD-2 monoclonal antibody therapy affords protection in experimental models of septic shock. J. Immunol.179, 6107–6114. 10.4049/jimmunol.179.9.6107
36
DenlingerL. C.FisetteP. L.SommerJ. A.WattersJ. J.PrabhuU.DubyakG. R.et al. (2001). Cutting edge: the nucleotide receptor P2X7 contains multiple protein- and lipid-interaction motifs including a potential binding site for bacterial lipopolysaccharide. J. Immunol.167, 1871–1876. 10.4049/jimmunol.167.4.1871
37
DongL.LiH.WangS.LiY. (2009). Different doses of lipopolysaccharides regulate the lung inflammation of asthmatic mice via TLR4 pathway in alveolar macrophages. J. Asthma46, 229–233. 10.1080/02770900802610050
38
DrexlerS. K.FoxwellB. M. (2010). The role of toll-like receptors in chronic inflammation. Int. J. Biochem. Cell Biol.42, 506–518. 10.1016/j.biocel.2009.10.009
39
DroemannD.GoldmannT.TiedjeT.ZabelP.DalhoffK.SchaafB. (2005). Toll-like receptor 2 expression is decreased on alveolar macrophages in cigarette smokers and COPD patients. Respir. Res.6:68. 10.1186/1465-9921-6-68
40
Dynavax (2014). Dynavax Regains Full Rights to Investigational TLR 7/9 Inhibitor DV1179 Following Expiration of Collaboration with GSK. Available online at: http://investors.dynavax.com/releasedetail.cfm?releaseid=885172 (Accessed November 28, 2014).
41
EdfeldtK.SwedenborgJ.HanssonG. K.YanZ. Q. (2002). Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation105, 1158–1161. 10.1161/01.CIR.0000012489.17433.31
42
EhrentrautH.WeberC.EhrentrautS.SchwederskiM.BoehmO.KnuefermannP.et al. (2011). The toll-like receptor 4-antagonist eritoran reduces murine cardiac hypertrophy. Eur. J. Heart Fail.13, 602–610. 10.1093/eurjhf/hfr035
43
ErnstingM. J.MurakamiM.RoyA.LiS. D. (2013). Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J. Control. Release172, 782–794. 10.1016/j.jconrel.2013.09.013
44
FangD.YangS.QuanW.JiaH.QuanZ.QuZ. (2014). Atorvastatin suppresses toll-like receptor 4 expression and NF-κB activation in rabbit atherosclerotic plaques. Eur. Rev. Med. Pharmacol. Sci.18, 242–246.
45
FarrarC. A.KeoghB.McCormackW.O'ShaughnessyA.ParkerA.ReillyM.et al. (2012). Inhibition of TLR2 promotes graft function in a murine model of renal transplant ischemia-reperfusion injury. FASEB J.26, 799–807. 10.1096/fj.11-195396
46
FoitL.ThaxtonC. S. (2016). Synthetic high-density lipoprotein-like nanoparticles potently inhibit cell signaling and production of inflammatory mediators induced by lipopolysaccharide binding Toll-like receptor 4. Biomaterials100, 67–75. 10.1016/j.biomaterials.2016.05.021
47
FoldesG.von HaehlingS.OkonkoD. O.JankowskaE. A.Poole-WilsonP. A.AnkerS. D. (2008). Fluvastatin reduces increased blood monocyte Toll-like receptor 4 expression in whole blood from patients with chronic heart failure. Int. J. Cardiol.124, 80–85. 10.1016/j.ijcard.2006.12.024
48
FoxR. (1996). Anti-malarial drugs: possible mechanisms of action in autoimmune disease and prospects for drug development. Lupus5(Suppl. 1), S4–S10. 10.1177/096120339600500103
49
FukataM.AbreuM. T. (2008). Role of Toll-like receptors in gastrointestinal malignancies. Oncogene27, 234–243. 10.1038/sj.onc.1210908
50
FukataM.ChenA.VamadevanA. S.CohenJ.BreglioK.KrishnareddyS.et al. (2007). Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology133, 1869–1881. 10.1053/j.gastro.2007.09.008
51
GayN. J.SymmonsM. F.GangloffM.BryantC. E. (2014). Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol.14, 546–558. 10.1038/nri3713
52
GesueteR.KohamaS. G.Stenzel-PooreM. P. (2014). Toll-like receptors and ischemic brain injury. J. Neuropathol. Exp. Neurol.73, 378–386. 10.1097/NEN.0000000000000068
53
GillietM.ConradC.GeigesM.CozzioA.ThurlimannW.BurgG.et al. (2004). Psoriasis triggered by toll-like receptor 7 agonist imiquimod in the presence of dermal plasmacytoid dendritic cell precursors. Arch. Dermatol.140, 1490–1495. 10.1001/archderm.140.12.1490
54
Gomez-GuzmanM.JimenezR.RomeroM.SanchezM.ZarzueloM. J.Gomez-MoralesM.et al. (2014). Chronic hydroxychloroquine improves endothelial dysfunction and protects kidney in a mouse model of systemic lupus erythematosus. Hypertension64, 330–337. 10.1161/HYPERTENSIONAHA.114.03587
55
GoulopoulouS.McCarthyC. G.WebbR. C. (2016). Toll-like receptors in the vascular system: sensing the dangers within. Pharmacol. Rev.68, 142–167. 10.1124/pr.114.010090
56
GuiducciC.GongM.XuZ.GillM.ChaussabelD.MeekerT.et al. (2010). TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature465, 937–941. 10.1038/nature09102
57
GuoL.AiJ.ZhengZ.HowattD. A.DaughertyA.HuangB.et al. (2013). High density lipoprotein protects against polymicrobe-induced sepsis in mice. J. Biol. Chem.288, 17947–17953. 10.1074/jbc.M112.442699
58
HarterL.MicaL.StockerR.TrentzO.KeelM. (2004). Increased expression of toll-like receptor-2 and -4 on leukocytes from patients with sepsis. Shock22, 403–409. 10.1097/01.shk.0000142256.23382.5d
59
HattererE.ShangL.SimonetP.HerrenS.DaubeufB.TeixeiraS.et al. (2016). A specific anti-citrullinated protein antibody profile identifies a group of rheumatoid arthritis patients with a toll-like receptor 4-mediated disease. Arthritis Res. Ther.18, 224. 10.1186/s13075-016-1128-5
60
HeC.HuY.YinL.TangC.YinC. (2010). Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials31, 3657–3666. 10.1016/j.biomaterials.2010.01.065
61
HeX.JingZ.ChengG. (2014). MicroRNAs: new regulators of Toll-like receptor signalling pathways. Biomed Res. Int.2014:945169. 10.1155/2014/945169
62
HedayatM.NeteaM. G.RezaeiN. (2011). Targeting of Toll-like receptors: a decade of progress in combating infectious diseases. Lancet Infect. Dis.11, 702–712. 10.1016/S1473-3099(11)70099-8
63
HennessyE. J.ParkerA. E.O'NeillL. A. (2010). Targeting Toll-like receptors: emerging therapeutics?Nat. Rev. Drug Discov.9, 293–307. 10.1038/nrd3203
64
HernanzR.Martinez-RevellesS.PalaciosR.MartinA.CachofeiroV.AguadoA.et al. (2015). Toll-like receptor 4 contributes to vascular remodelling and endothelial dysfunction in angiotensin II-induced hypertension. Br. J. Pharmacol.172, 3159–3176. 10.1111/bph.13117
65
HigashimoriM.TatroJ. B.MooreK. J.MendelsohnM. E.GalperJ. B.BeasleyD. (2011). Role of toll-like receptor 4 in intimal foam cell accumulation in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol.31, 50–57. 10.1161/ATVBAHA.110.210971
66
HouL. F.HeS. J.LiX.YangY.HeP. L.ZhouY.et al. (2011). Oral administration of artemisinin analog SM934 ameliorates lupus syndromes in MRL/lpr mice by inhibiting Th1 and Th17 cell responses. Arthritis Rheum.63, 2445–2455. 10.1002/art.30392
67
HoughtonJ.WangT. C. (2005). Helicobacter pylori and gastric cancer: a new paradigm for inflammation-associated epithelial cancers. Gastroenterology128, 1567–1578. 10.1053/j.gastro.2005.03.037
68
HuD.YangX.XiangY.LiH.YanH.ZhouJ.et al. (2015). Inhibition of Toll-like receptor 9 attenuates sepsis-induced mortality through suppressing excessive inflammatory response. Cell. Immunol.295, 92–98. 10.1016/j.cellimm.2015.03.009
69
IshibashiM.SayersS.D'ArmientoJ. M.TallA. R.WelchC. L. (2013). TLR3 deficiency protects against collagen degradation and medial destruction in murine atherosclerotic plaques. Atherosclerosis229, 52–61. 10.1016/j.atherosclerosis.2013.03.035
70
ItzkowitzS. H.YioX. (2004). Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am. J. Physiol. Gastrointest. Liver Physiol.287, G7–17. 10.1152/ajpgi.00079.2004
71
JacksonS. W.ScharpingN. E.KolhatkarN. S.KhimS.SchwartzM. A.LiQ. Z.et al. (2014). Opposing impact of B cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J. Immunol.192, 4525–4532. 10.4049/jimmunol.1400098
72
JiangW.ZhuF.TangJ.KandimallaE.La MonicaN.AgrawalS. (2012). IMO-8400, a novel TLR7, TLR8 and TLR9 antagonist, inhibits disease development in mouse models of psoriasis. J. Immunol.188 (1 Suppl.), 119.8.
73
KarimiK.SarirH.MortazE.SmitJ. J.HosseiniH.De KimpeS. J.et al. (2006). Toll-like receptor-4 mediates cigarette smoke-induced cytokine production by human macrophages. Respir. Res.7:66. 10.1186/1465-9921-7-66
74
KawaiT.AkiraS. (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol.11, 373–384. 10.1038/ni.1863
75
KawasakiT.KawaiT. (2014). Toll-like receptor signaling pathways. Front. Immunol.5:461. 10.3389/fimmu.2014.00461
76
KhanA. M.LiM.Abdulnour-NakhoulS.MaderdrutJ. L.SimonE. E.BatumanV. (2012). Delayed administration of pituitary adenylate cyclase-activating polypeptide 38 ameliorates renal ischemia/reperfusion injury in mice by modulating Toll-like receptors. Peptides38, 395–403. 10.1016/j.peptides.2012.09.023
77
KimW. U.YooS. A.MinS. Y.ParkS. H.KohH. S.SongS. W.et al. (2006). Hydroxychloroquine potentiates Fas-mediated apoptosis of rheumatoid synoviocytes. Clin. Exp. Immunol.144, 503–511. 10.1111/j.1365-2249.2006.03070.x
78
KomatsudaA.WakuiH.IwamotoK.OzawaM.TogashiM.MasaiR.et al. (2008). Up-regulated expression of Toll-like receptors mRNAs in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Clin. Exp. Immunol.152, 482–487. 10.1111/j.1365-2249.2008.03646.x
79
KonoD. H.HaraldssonM. K.LawsonB. R.PollardK. M.KohY. T.DuX.et al. (2009). Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc. Natl. Acad. Sci. U.S.A.106, 12061–12066. 10.1073/pnas.0905441106
80
KoulisC.ChenY. C.HausdingC.AhrensI.KyawT. S.TayC.et al. (2014). Protective role for Toll-like receptor-9 in the development of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol.34, 516–525. 10.1161/ATVBAHA.113.302407
81
KuznikA.BencinaM.SvajgerU.JerasM.RozmanB.JeralaR. (2011). Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J. Immunol.186, 4794–4804. 10.4049/jimmunol.1000702
82
LartigueA.ColliouN.CalboS.FrancoisA.JacquotS.ArnoultC.et al. (2009). Critical role of TLR2 and TLR4 in autoantibody production and glomerulonephritis in lpr mutation-induced mouse lupus. J. Immunol.183, 6207–6216. 10.4049/jimmunol.0803219
83
LeeP. Y.KumagaiY.LiY.TakeuchiO.YoshidaH.WeinsteinJ.et al. (2008). TLR7-dependent and FcgammaR-independent production of type I interferon in experimental mouse lupus. J. Exp. Med.205, 2995–3006. 10.1084/jem.20080462
84
LeeS. J.SilvermanE.BargmanJ. M. (2011). The role of antimalarial agents in the treatment of SLE and lupus nephritis. Nat. Rev. Nephrol.7, 718–729. 10.1038/nrneph.2011.150
85
LeeY. H.ChoiS. J.JiJ. D.SongG. G. (2016). Association between toll-like receptor polymorphisms and systemic lupus erythematosus: a meta-analysis update. Lupus25, 593–601. 10.1177/0961203315622823
86
LesiakA.NarbuttJ.Sysa-JedrzejowskaA.LukamowiczJ.McCauliffeD. P.WozniackaA. (2010). Effect of chloroquine phosphate treatment on serum MMP-9 and TIMP-1 levels in patients with systemic lupus erythematosus. Lupus19, 683–688. 10.1177/0961203309356455
87
LeungP. Y.StevensS. L.PackardA. E.LessovN. S.YangT.ConradV. K.et al. (2012). Toll-like receptor 7 preconditioning induces robust neuroprotection against stroke by a novel type I interferon-mediated mechanism. Stroke43, 1383–1389. 10.1161/STROKEAHA.111.641522
88
LiN.HarkemaJ. R.LewandowskiR. P.WangM.BrambleL. A.GookinG. R.et al. (2010). Ambient ultrafine particles provide a strong adjuvant effect in the secondary immune response: implication for traffic-related asthma flares. Am. J. Physiol. Lung Cell. Mol. Physiol.299, L374–383. 10.1152/ajplung.00115.2010
89
LibuttiS. K.PaciottiG. F.ByrnesA. A.AlexanderH. R.Jr.GannonW. E.WalkerM.et al. (2010). Phase I and pharmacokinetic studies of CYT-6091, a novel PEGylated colloidal gold-rhTNF nanomedicine. Clin. Cancer Res.16, 6139–6149. 10.1158/1078-0432.CCR-10-0978
90
LimaC. X.SouzaD. G.AmaralF. A.FagundesC. T.RodriguesI. P.Alves-FilhoJ. C.et al. (2015). Therapeutic effects of treatment with anti-TLR2 and anti-TLR4 monoclonal antibodies in polymicrobial sepsis. PLoS ONE10:e0132336. 10.1371/journal.pone.0132336
91
LinP. J.TamY. K. (2015). Enhancing the pharmacokinetic/pharmacodynamic properties of therapeutic nucleotides using lipid nanoparticle systems. Future Med. Chem.7, 1751–1769. 10.4155/fmc.15.108
92
LipfordG.ForsbachA.ZeppC.NguyenT.WeeratnaR.McCluskieM.et al. (2007). Selective toll-like receptor 7/8/9 antagonists for the oral treatment of autoimmune diseases, in American College of Rheumatology 2007 Annual Scientific Meeting (Boston, MA).
93
LiuB.YangY.DaiJ.MedzhitovR.FreudenbergM. A.ZhangP. L.et al. (2006). TLR4 up-regulation at protein or gene level is pathogenic for lupus-like autoimmune disease. J. Immunol.177, 6880–6888. 10.4049/jimmunol.177.10.6880
94
LiuL.LiY.HuZ.SuJ.HuoY.TanB.et al. (2012). Small interfering RNA targeting Toll-like receptor 9 protects mice against polymicrobial septic acute kidney injury. Nephron Exp. Nephrol.122, 51–61. 10.1159/000346953
95
LiuM.GuM.XuD.LvQ.ZhangW.WuY. (2010). Protective effects of Toll-like receptor 4 inhibitor eritoran on renal ischemia-reperfusion injury. Transplant. Proc.42, 1539–1544. 10.1016/j.transproceed.2010.03.133
96
LofflerB. M.BohnE.HesseB.KunzeH. (1985). Effects of antimalarial drugs on phospholipase A and lysophospholipase activities in plasma membrane, mitochondrial, microsomal and cytosolic subcellular fractions of rat liver. Biochim. Biophys. Acta835, 448–455. 10.1016/0005-2760(85)90114-6
97
LoiarroM.CapolunghiF.FantoN.GalloG.CampoS.ArseniB.et al. (2007). Pivotal advance: inhibition of MyD88 dimerization and recruitment of IRAK1 and IRAK4 by a novel peptidomimetic compound. J. Leukoc. Biol.82, 801–810. 10.1189/jlb.1206746
98
Lyn-CookB. D.XieC.OatesJ.TreadwellE.WordB.HammonsG.et al. (2014). Increased expression of Toll-like receptors (TLRs) 7 and 9 and other cytokines in systemic lupus erythematosus (SLE) patients: ethnic differences and potential new targets for therapeutic drugs. Mol. Immunol.61, 38–43. 10.1016/j.molimm.2014.05.001
99
LynnM.WongY. N.WheelerJ. L.KaoR. J.PerdomoC. A.NoveckR.et al. (2004). Extended in vivo pharmacodynamic activity of E5564 in normal volunteers with experimental endotoxemia [corrected]. J. Pharmacol. Exp. Ther.308, 175–181. 10.1124/jpet.103.056531
100
Marshak-RothsteinA. (2006). Toll-like receptors in systemic autoimmune disease. Nat. Rev. Immunol.6, 823–835. 10.1038/nri1957
101
MatsunagaN.TsuchimoriN.MatsumotoT.IiM. (2011). TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol. Pharmacol.79, 34–41. 10.1124/mol.110.068064
102
McCarthyC. G.GoulopoulouS.WenceslauC. F.SpitlerK.MatsumotoT.WebbR. C. (2014). Toll-like receptors and damage-associated molecular patterns: novel links between inflammation and hypertension. Am. J. Physiol. Heart Circ. Physiol.306, H184–H196. 10.1152/ajpheart.00328.2013
103
McCarthyC. G.WenceslauC. F.GoulopoulouS.OgbiS.BabanB.SullivanJ. C.et al. (2015). Circulating mitochondrial DNA and Toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovasc. Res.107, 119–130. 10.1093/cvr/cvv137
104
MengG.RutzM.SchiemannM.MetzgerJ.GrabiecA.SchwandnerR.et al. (2004). Antagonistic antibody prevents toll-like receptor 2-driven lethal shock-like syndromes. J. Clin. Invest.113, 1473–1481. 10.1172/JCI20762
105
MetheH.KimJ. O.KoflerS.NabauerM.WeisM. (2005). Statins decrease Toll-like receptor 4 expression and downstream signaling in human CD14+ monocytes. Arterioscler. Thromb. Vasc. Biol.25, 1439–1445. 10.1161/01.ATV.0000168410.44722.86
106
MichelsenK. S.WongM. H.ShahP. K.ZhangW.YanoJ.DohertyT. M.et al. (2004). Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc. Natl. Acad. Sci. U.S.A.101, 10679–10684. 10.1073/pnas.0403249101
107
MillerL. S.SorensenO. E.LiuP. T.JalianH. R.EshtiaghpourD.BehmaneshB. E.et al. (2005). TGF-alpha regulates TLR expression and function on epidermal keratinocytes. J. Immunol.174, 6137–6143. 10.4049/jimmunol.174.10.6137
108
MillerY. I. (2005). Toll-like receptors and atherosclerosis: oxidized LDL as an endogenous Toll-like receptor ligand. Future Cardiol.1, 785–792. 10.2217/14796678.1.6.785
109
MogensenT. H. (2009). Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev.22, 240–273. 10.1128/CMR.00046-08
110
MonacoC.GreganS. M.NavinT. J.FoxwellB. M.DaviesA. H.FeldmannM. (2009). Toll-like receptor-2 mediates inflammation and matrix degradation in human atherosclerosis. Circulation120, 2462–2469. 10.1161/CIRCULATIONAHA.109.851881
111
MonnetE.LapeyreG.PoelgeestE. V.JacqminP.GraafK.ReijersJ.et al. (2017). Evidence of NI-0101 pharmacological activity, an anti-TLR4 antibody, in a randomized phase I dose escalation study in healthy volunteers receiving LPS. Clin. Pharmacol. Ther.101, 200–208. 10.1002/cpt.522
112
MonnetE.ShangL.LapeyreG.deGraafK.HattererE.BuatoisV.et al. (2015). NI-0101, a monoclonal antibody targeting toll like receptor 4 (TLR4) being developed for rheumatoid arthritis (RA) treatment with a potential for personalized medicine Ann. Rheum. Dis.74, 1. 10.1136/annrheumdis-2015-eular.3801
113
MorescoE. M.LaVineD.BeutlerB. (2011). Toll-like receptors. Curr. Biol.21, R488–R493. 10.1016/j.cub.2011.05.039
114
MortazE.AdcockI. M.ItoK.KraneveldA. D.NijkampF. P.FolkertsG. (2010). Cigarette smoke induces CXCL8 production by human neutrophils via activation of TLR9 receptor. Eur. Respir. J.36, 1143–1154. 10.1183/09031936.00062209
115
MullarkeyM.RoseJ. R.BristolJ.KawataT.KimuraA.KobayashiS.et al. (2003). Inhibition of endotoxin response by e5564, a novel Toll-like receptor 4-directed endotoxin antagonist. J. Pharmacol. Exp. Ther.304, 1093–1102. 10.1124/jpet.102.044487
116
MullickA. E.SoldauK.KiossesW. B.BellT. A.III.TobiasP. S.CurtissL. K. (2008). Increased endothelial expression of Toll-like receptor 2 at sites of disturbed blood flow exacerbates early atherogenic events. J. Exp. Med.205, 373–383. 10.1084/jem.20071096
117
MullickA. E.TobiasP. S.CurtissL. K. (2005). Modulation of atherosclerosis in mice by Toll-like receptor 2. J. Clin. Invest.115, 3149–3156. 10.1172/JCI25482
118
NickersonK. M.ChristensenS. R.ShupeJ.KashgarianM.KimD.ElkonK.et al. (2010). TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J. Immunol.184, 1840–1848. 10.4049/jimmunol.0902592
119
O'ConnellR. M.KahnD.GibsonW. S.RoundJ. L.ScholzR. L.ChaudhuriA. A.et al. (2010). MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity33, 607–619. 10.1016/j.immuni.2010.09.009
120
O'ConnellR. M.TaganovK. D.BoldinM. P.ChengG.BaltimoreD. (2007). MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. U.S.A.104, 1604–1609. 10.1073/pnas.0610731104
121
O'NeillL. A.BryantC. E.DoyleS. L. (2009). Therapeutic targeting of Toll-like receptors for infectious and inflammatory diseases and cancer. Pharmacol. Rev.61, 177–197. 10.1124/pr.109.001073
122
OpalS. M.LaterreP. F.FrancoisB.LaRosaS. P.AngusD. C.MiraJ. P.et al. (2013). Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA309, 1154–1162. 10.1001/jama.2013.2194
123
PajkrtD.DoranJ. E.KosterF.LerchP. G.ArnetB.van der PollT.et al. (1996). Antiinflammatory effects of reconstituted high-density lipoprotein during human endotoxemia. J. Exp. Med.184, 1601–1608. 10.1084/jem.184.5.1601
124
PalmN. W.MedzhitovR. (2009). Pattern recognition receptors and control of adaptive immunity. Immunol. Rev.227, 221–233. 10.1111/j.1600-065X.2008.00731.x
125
PanY.JiaT.ZhangY.ZhangK.ZhangR.LiJ.et al. (2012). MS2 VLP-based delivery of microRNA-146a inhibits autoantibody production in lupus-prone mice. Int. J. Nanomed.7, 5957–5967. 10.2147/IJN.S37990
126
ParkB. S.SongD. H.KimH. M.ChoiB. S.LeeH.LeeJ. O. (2009). The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature458, 1191–1195. 10.1038/nature07830
127
PatonN. I.LeeL.XuY.OoiE. E.CheungY. B.ArchuletaS.et al. (2011). Chloroquine for influenza prevention: a randomised, double-blind, placebo controlled trial. Lancet Infect. Dis.11, 677–683. 10.1016/S1473-3099(11)70065-2
128
PedrosaP.VinhasR.FernandesA.BaptistaP. V. (2015). Gold nanotheranostics: proof-of-concept or clinical tool?Nanomaterials5, 1853–1879. 10.3390/nano5041853
129
PereiraD. V.PetronilhoF.PereiraH. R.VuoloF.MinaF.PossatoJ. C.et al. (2012). Effects of gold nanoparticles on endotoxin-induced uveitis in rats. Invest. Ophthalmol. Vis. Sci.53, 8036–8041. 10.1167/iovs.12-10743
130
PisitkunP.DeaneJ. A.DifilippantonioM. J.TarasenkoT.SatterthwaiteA. B.BollandS. (2006). Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science312, 1669–1672. 10.1126/science.1124978
131
QuinnS. R.O'NeillL. A. (2011). A trio of microRNAs that control Toll-like receptor signalling. Int. Immunol.23, 421–425. 10.1093/intimm/dxr034
132
ReillyM.MillerR. M.ThomsonM. H.PatrisV.RyleP.McLoughlinL.et al. (2013). Randomized, double-blind, placebo-controlled, dose-escalating phase I, healthy subjects study of intravenous OPN-305, a humanized anti-TLR2 antibody. Clin. Pharmacol. Ther.94, 593–600. 10.1038/clpt.2013.150
133
RiceT. W.WheelerA. P.BernardG. R.VincentJ. L.AngusD. C.AikawaN.et al. (2010). A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit. Care Med.38, 1685–1694. 10.1097/CCM.0b013e3181e7c5c9
134
RietschelE. T.KirikaeT.SchadeF. U.MamatU.SchmidtG.LoppnowH.et al. (1994). Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J.8, 217–225.
135
RodriguesF. L.SilvaL. E.HottS. C.BomfimG. F.da SilvaC. A.FazanR.Jr.et al. (2015). Toll-like receptor 9 plays a key role in the autonomic cardiac and baroreflex control of arterial pressure. Am. J. Physiol. Regul. Integr. Comp. Physiol.308, R714–R723. 10.1152/ajpregu.00150.2014
136
Rodriguez LavadoJ.SestitoS. E.CighettiR.Aguilar MoncayoE. M.OblakA.LainscekD.et al. (2014). Trehalose- and glucose-derived glycoamphiphiles: small-molecule and nanoparticle Toll-like receptor 4 (TLR4) modulators. J. Med. Chem.57, 9105–9123. 10.1021/jm501182w
137
RogerT.FroidevauxC.Le RoyD.ReymondM. K.ChansonA. L.MauriD.et al. (2009). Protection from lethal gram-negative bacterial sepsis by targeting Toll-like receptor 4. Proc. Natl. Acad. Sci. U.S.A.106, 2348–2352. 10.1073/pnas.0808146106
138
RommlerF.HammelM.WaldhuberA.MullerT.JurkM.UhlmannE.et al. (2015). Guanine-modified inhibitory oligonucleotides efficiently impair TLR7- and TLR9-mediated immune responses of human immune cells. PLoS ONE10:e0116703. 10.1371/journal.pone.0116703
139
RommlerF.JurkM.UhlmannE.HammelM.WaldhuberA.PfeifferL.et al. (2013). Guanine modification of inhibitory oligonucleotides potentiates their suppressive function. J. Immunol.191, 3240–3253. 10.4049/jimmunol.1300706
140
RossignolD. P.WasanK. M.ChooE.YauE.WongN.RoseJ.et al. (2004). Safety, pharmacokinetics, pharmacodynamics, and plasma lipoprotein distribution of eritoran (E5564) during continuous intravenous infusion into healthy volunteers. Antimicrob. Agents Chemother.48, 3233–3240. 10.1128/AAC.48.9.3233-3240.2004
141
RossignolD. P.WongN.NoveckR.LynnM. (2008). Continuous pharmacodynamic activity of eritoran tetrasodium, a TLR4 antagonist, during intermittent intravenous infusion into normal volunteers. Innate Immun.14, 383–394. 10.1177/1753425908099173
142
Ruiz-IrastorzaG.Ramos-CasalsM.Brito-ZeronP.KhamashtaM. A. (2010). Clinical efficacy and side effects of antimalarials in systemic lupus erythematosus: a systematic review. Ann. Rheum. Dis.69, 20–28. 10.1136/ard.2008.101766
143
SatoS.SugiyamaM.YamamotoM.WatanabeY.KawaiT.TakedaK.et al. (2003). Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-κB and IFN-regulatory factor-3, in the toll-like receptor signaling. J. Immunol.171, 4304–4310. 10.4049/jimmunol.171.8.4304
144
SatoY.GotoY.NaritaN.HoonD. S. (2009). Cancer cells expressing toll-like receptors and the tumor microenvironment. Cancer Microenviron.2(Suppl. 1), 205–214. 10.1007/s12307-009-0022-y
145
SavovJ. D.BrassD. M.LawsonB. L.McElvania-TekippeE.WalkerJ. K.SchwartzD. A. (2005). Toll-like receptor 4 antagonist (E5564) prevents the chronic airway response to inhaled lipopolysaccharide. Am. J. Physiol. Lung Cell. Mol. Physiol.289, L329–L337. 10.1152/ajplung.00014.2005
146
SeungN. R.ParkE. J.KimC. W.KimK. H.KimK. J.ChoH. J.et al. (2007). Comparison of expression of heat-shock protein 60, Toll-like receptors 2 and 4, and T-cell receptor gammadelta in plaque and guttate psoriasis. J. Cutan. Pathol.34, 903–911. 10.1111/j.1600-0560.2007.00756.x
147
SheedyF. J.Palsson-McDermottE.HennessyE. J.MartinC.O'LearyJ. J.RuanQ.et al. (2010). Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat. Immunol.11, 141–147. 10.1038/ni.1828
148
ShenN.LiangD.TangY.de VriesN.TakP. P. (2012). MicroRNAs–novel regulators of ystemic lupus erythematosus pathogenesis. Nat. Rev. Rheumatol.8, 701–709. 10.1038/nrrheum.2012.142
149
ShimamotoA.ChongA. J.YadaM.ShomuraS.TakayamaH.FleisigA. J.et al. (2006). Inhibition of Toll-like receptor 4 with eritoran attenuates myocardial ischemia-reperfusion injury. Circulation114, I270–274. 10.1161/CIRCULATIONAHA.105.000901
150
ShireyK. A.LaiW.ScottA. J.LipskyM.MistryP.PletnevaL. M.et al. (2013). The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature497, 498–502. 10.1038/nature12118
151
SpillerS.ElsonG.FerstlR.DreherS.MuellerT.FreudenbergM.et al. (2008). TLR4-induced IFN-gamma production increases TLR2 sensitivity and drives Gram-negative sepsis in mice. J. Exp. Med.205, 1747–1754. 10.1084/jem.20071990
152
StowellN. C.SeidemanJ.RaymondH. A.SmalleyK. A.LambR. J.EgenolfD. D.et al. (2009). Long-term activation of TLR3 by poly(I:C) induces inflammation and impairs lung function in mice. Respir. Res.10:43. 10.1186/1465-9921-10-43
153
Suarez-FarinasM.ArbeitR.JiangW.OrtenzioF. S.SullivanT.KruegerJ. G. (2013). Suppression of molecular inflammatory pathways by Toll-like receptor 7, 8, and 9 antagonists in a model of IL-23-induced skin inflammation. PLoS ONE8:e84634. 10.1371/journal.pone.0084634
154
SubramanianS.TusK.LiQ. Z.WangA.TianX. H.ZhouJ.et al. (2006). A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc. Natl. Acad. Sci. U.S.A.103, 9970–9975. 10.1073/pnas.0603912103
155
SuzukiM.KatoC.KatoA. (2015). Therapeutic antibodies: their mechanisms of action and the pathological findings they induce in toxicity studies. J. Toxicol. Pathol.28, 133–139. 10.1293/tox.2015-0031
156
TaganovK. D.BoldinM. P.ChangK. J.BaltimoreD. (2006). NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. U.S.A.103, 12481–12486. 10.1073/pnas.0605298103
157
TakedaK.AkiraS. (2005). Toll-like receptors in innate immunity. Int. Immunol.17, 1–14. 10.1093/intimm/dxh186
158
TakedaK.KaishoT.AkiraS. (2003). Toll-like receptors. Annu. Rev. Immunol.21, 335–376. 10.1146/annurev.immunol.21.120601.141126
159
TangY.LuoX.CuiH.NiX.YuanM.GuoY.et al. (2009). MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum.60, 1065–1075. 10.1002/art.24436
160
TidswellM.TillisW.LarosaS. P.LynnM.WittekA. E.KaoR.et al. (2010). Phase 2 trial of eritoran tetrasodium (E5564), a toll-like receptor 4 antagonist, in patients with severe sepsis. Crit. Care Med.38, 72–83. 10.1097/CCM.0b013e3181b07b78
161
TiliE.MichailleJ. J.CiminoA.CostineanS.DumitruC. D.AdairB.et al. (2007). Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol.179, 5082–5089. 10.4049/jimmunol.179.8.5082
162
TokairinY.ShibataY.SataM.AbeS.TakabatakeN.IgarashiA.et al. (2008). Enhanced immediate inflammatory response to Streptococcus pneumoniae in the lungs of mice with pulmonary emphysema. Respirology13, 324–332. 10.1111/j.1440-1843.2007.01229.x
163
TsujimotoH.OnoS.EfronP. A.ScumpiaP. O.MoldawerL. L.MochizukiH. (2008). Role of Toll-like receptors in the development of sepsis. Shock29, 315–321. 10.1097/SHK.0b013e318157ee55
164
TyrrellD. J.HorneA. P.HolmeK. R.PreussJ. M.PageC. P. (1999). Heparin in inflammation: potential therapeutic applications beyond anticoagulation. Adv. Pharmacol.46, 151–208. 10.1016/S1054-3589(08)60471-8
165
UematsuS.AkiraS. (2007). Toll-like receptors and Type I interferons. J. Biol. Chem.282, 15319–15323. 10.1074/jbc.R700009200
166
UltaighS. N.SaberT. P.McCormickJ.ConnollyM.DellacasagrandeJ.KeoghB.et al. (2011). Blockade of Toll-like receptor 2 prevents spontaneous cytokine release from rheumatoid arthritis ex vivo synovial explant cultures. Arthritis Res. Ther.13, R33. 10.1186/ar3261
167
UngaroR.FukataM.HsuD.HernandezY.BreglioK.ChenA.et al. (2009). A novel Toll-like receptor 4 antagonist antibody ameliorates inflammation but impairs mucosal healing in murine colitis. Am. J. Physiol. Gastrointest. Liver Physiol.296, G1167–1179. 10.1152/ajpgi.90496.2008
168
WangL.BrownJ. R.VarkiA.EskoJ. D. (2002). Heparin's anti-inflammatory effects require glucosamine 6-O-sulfation and are mediated by blockade of L- and P-selectins. J. Clin. Invest.110, 127–136. 10.1172/JCI0214996
169
WangL.LiY. L.ZhangC. C.CuiW.WangX.XiaY.et al. (2014). Inhibition of Toll-like receptor 2 reduces cardiac fibrosis by attenuating macrophage-mediated inflammation. Cardiovasc. Res.101, 383–392. 10.1093/cvr/cvt258
170
WangY. C.LinS.YangQ. W. (2011). Toll-like receptors in cerebral ischemic inflammatory injury. J. Neuroinflammation8:134. 10.1186/1742-2094-8-134
171
WeberS. M.ChenJ. M.LevitzS. M. (2002). Inhibition of mitogen-activated protein kinase signaling by chloroquine. J. Immunol.168, 5303–5309. 10.4049/jimmunol.168.10.5303
172
WitteboleX.CoyleS. M.KumarA.GoshimaM.LowryS. F.CalvanoS. E. (2005). Expression of tumour necrosis factor receptor and Toll-like receptor 2 and 4 on peripheral blood leucocytes of human volunteers after endotoxin challenge: a comparison of flow cytometric light scatter and immunofluorescence gating. Clin. Exp. Immunol.141, 99–106. 10.1111/j.1365-2249.2005.02831.x
173
WozniackaA.LesiakA.BoncelaJ.SmolarczykK.McCauliffeD. P.Sysa-JedrzejowskaA. (2008). The influence of antimalarial treatment on IL-1beta, IL-6 and TNF-alpha mRNA expression on UVB-irradiated skin in systemic lupus erythematosus. Br. J. Dermatol.159, 1124–1130. 10.1111/j.1365-2133.2008.08804.x
174
WozniackaA.LesiakA.NarbuttJ.McCauliffeD. P.Sysa-JedrzejowskaA. (2006). Chloroquine treatment influences proinflammatory cytokine levels in systemic lupus erythematosus patients. Lupus15, 268–275. 10.1191/0961203306lu2299oa
175
WuY. W.TangW.ZuoJ. P. (2015). Toll-like receptors: potential targets for lupus treatment. Acta Pharmacol. Sin.36, 1395–1407. 10.1038/aps.2015.91
176
WuY.HeS.BaiB.ZhangL.XueL.LinZ.et al. (2016). Therapeutic effects of the artemisinin analog SM934 on lupus-prone MRL/lpr mice via inhibition of TLR-triggered B-cell activation and plasma cell formation. Cell. Mol. Immunol.13, 379–390. 10.1038/cmi.2015.13
177
YamamotoM.SatoS.HemmiH.HoshinoK.KaishoT.SanjoH.et al. (2003). Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science301, 640–643. 10.1126/science.1087262
178
YangH.FungS. Y.LiuM. (2011). Programming the cellular uptake of physiologically stable peptide-gold nanoparticle hybrids with single amino acids. Angew. Chem. Int. Ed. Engl.50, 9643–9646. 10.1002/anie.201102911
179
YangH.FungS. Y.XuS.SutherlandD. P.KollmannT. R.LiuM.et al. (2015). Amino acid-dependent attenuation of toll-like receptor signaling by peptide-gold nanoparticle hybrids. ACS Nano9, 6774–6784. 10.1021/nn505634h
180
YangH.KozickyL.SaferaliA.FungS. Y.AfacanN.CaiB.et al. (2016). Endosomal pH modulation by peptide-gold nanoparticle hybrids enables potent anti-inflammatory activity in phagocytic immune cells. Biomaterials111, 90–102. 10.1016/j.biomaterials.2016.09.032
181
YangH.ZhouY. Z.FungS. Y.WuL. C.TsaiK.TanR. S.et al. (2013). Amino acid structure determines the immune responses generated by peptide-gold nanoparticle hybrids. Part. Part. Syst. Char.30, 1039–1043. 10.1002/ppsc.201300213
182
YangJ.JiangH.DingJ. W.ChenL. H.LiS.ZhangX. D. (2009). Valsartan preconditioning protects against myocardial ischemia-reperfusion injury through TLR4/NF-kappaB signaling pathway. Mol. Cell. Biochem.330, 39–46. 10.1007/s11010-009-0098-1
183
YasudaH.LeelahavanichkulA.TsunodaS.DearJ. W.TakahashiY.ItoS.et al. (2008). Chloroquine and inhibition of Toll-like receptor 9 protect from sepsis-induced acute kidney injury. Am. J. Physiol. Renal Physiol.294, F1050–F1058. 10.1152/ajprenal.00461.2007
184
ZhuF.JiangW.DongY.KandimallaE.La MonicaN.AgrawalS. (2012). IMO-8400, a novel TLR7, TLR8 and TLR9 antagonist, inhibits disease development in lupus-prone NZBW/F1 mice. J. Immunol.188(1 Suppl.), 119.112.
185
ZhuF.YuD.KandimallaE.La MonicaN.AgrawalS. (2011). Treatment with IMO-3100, a novel TLR7 and TLR9 dual antagonist, inhibits disease development in lupus prone NZBW/F1 mice, in Keystone Symposia: Dendritic Cells and the Initiation of Adaptive Immunity (Santa Fe, NM).
186
ZieglerG.FreyerD.HarhausenD.KhojastehU.NietfeldW.TrendelenburgG. (2011). Blocking TLR2 in vivo protects against accumulation of inflammatory cells and neuronal injury in experimental stroke. J. Cereb. Blood Flow Metab.31, 757–766. 10.1038/jcbfm.2010.161
Summary
Keywords
toll-like receptor (TLR), autoimmune diseases, inflammatory diseases, inflammation, TLR antagonist, TLR inhibitor, nanotherapeutics
Citation
Gao W, Xiong Y, Li Q and Yang H (2017) Inhibition of Toll-Like Receptor Signaling as a Promising Therapy for Inflammatory Diseases: A Journey from Molecular to Nano Therapeutics. Front. Physiol. 8:508. doi: 10.3389/fphys.2017.00508
Received
23 April 2017
Accepted
03 July 2017
Published
19 July 2017
Volume
8 - 2017
Edited by
Enrique Hernandez-Lemus, National Institute of Genomic Medicine, Mexico
Reviewed by
Olalekan Michael Ogundele, Louisiana State University, United States; Yasser Mohamed El-Wazir, Suez Canal University, Egypt
Updates
Copyright
© 2017 Gao, Xiong, Li and Yang.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiang Li liqressh@hotmail.com
This article was submitted to Integrative Physiology, a section of the journal Frontiers in Physiology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.