 Alexandre Janin
Alexandre Janin Vincent Gache
Vincent Gache- 1CNRS UMR5310, INSERM U1217, Institut NeuroMyoGène, Université Claude Bernard Lyon 1, Université de Lyon, Lyon, France
- 2Laboratoire de Cardiogénétique Moléculaire, Centre de Biologie et Pathologie Est, Hospices Civils de Lyon, Bron, France
Since the discovery of the inner nuclear transmembrane protein emerin in the early 1990s, nuclear envelope (NE) components and related involvement in nuclei integrity and functionality have been highly investigated. The NE is composed of two distinct lipid bilayers described as the inner (INM) and outer (ONM) nuclear membrane. NE proteins can be specifically “integrated” in the INM (such as emerin and SUN proteins) or in the ONM such as nesprins. Additionally, flanked to the INM, the nuclear lamina, a proteinaceous meshwork mainly composed of lamins A and C completes NE composition. This network of proteins physically interplays to guarantee NE integrity and most importantly, shape the bridge between cytoplasmic cytoskeletons networks (such as microtubules and actin) and the genome, through the anchorage to the heterochromatin. The essential network driving the connection of nucleoskeleton with cytoskeleton takes place in the perinuclear space (the space between ONM and INM) with the contribution of the LINC complex (for Linker of Nucleoskeleton to Cytoskeleton), hosting KASH and SUN proteins interactions. This close interplay between compartments has been related to diverse functions from nuclear integrity, activity and positioning through mechanotransduction pathways. At the same time, mutations in NE components genes coding for proteins such as lamins or nesprins, had been associated with a wide range of congenital diseases including cardiac and muscular diseases. Although most of these NE associated proteins are ubiquitously expressed, a large number of tissue-specific disorders have been associated with diverse pathogenic mutations. Thus, diagnosis and molecular explanation of this group of diseases, commonly called “nuclear envelopathies,” is currently challenging. This review aims, first, to give a better understanding of diverse functions of the LINC complex components, from the point of view of lamins and nesprins. Second, to summarize human congenital diseases with a special focus on muscle and heart abnormalities, caused by mutations in genes coding for these two types of NE associated proteins.
Introduction
The Nuclear Envelope (NE) is composed of two distinct lipid bilayers so called inner (INM) and outer (ONM) nuclear membrane. This particular membrane architecture allows protection of chromatin from the rest of the cell by a physical separation between cytoplasm and nucleoplasm (Osorio and Gomes, 2013). Transmembrane proteins embedded in these two joined-membranes provide an architectural support to the nucleus as well as a physical bridge between nucleoskeleton and cytoskeleton. Physical coupling of these two skeletons is based on the so-called LINC complex (for Linker of the Nucleoskeleton to the Cytoskeleton). It couples, in the perinuclear space, proteins localized at the ONM and containing KASH domains (Klarsicht/Anc-1/Syne homology) with proteins localized at the INM and containing SUN domains (Sad1/UNC-84 homology) (Tapley and Starr, 2013). To date, six KASH domain-containing proteins have been described: nesprins-1, -2, -3, -4, KASH5 and lymphoid-restricted membrane protein (LRMP), respectively, encoded by SYNE1, SYNE2, SYNE3, SYNE4, KASH5, and LRPM genes (Behrens et al., 1994; Zhang et al., 2001; Wilhelmsen et al., 2005; Roux et al., 2009). SUN proteins are embedded in the INM and interact using their N-terminus domain with nuclear pore complex (NPC) and lamins (Padmakumar et al., 2005; Haque et al., 2006). Conversely, C-terminus domain is located in the perinuclear space and interacts with nesprins, which are embedded in the ONM (Padmakumar et al., 2005; Crisp et al., 2006; Haque et al., 2006; Ketema et al., 2007; Horn et al., 2013). This LINC complex play a role in diverse specialized cellular activities such as nuclear morphology maintenance, nuclear positioning, genes expression and cell signaling (Crisp et al., 2006; Lombardi and Lammerding, 2011; Mellad et al., 2011; Stroud et al., 2014). At the nucleoplasmic side, the LINC complex interacts with the nuclear lamina, a network of intermediate filaments just beneath INM and mainly composed of two different groups of lamin: A-type (lamins A and C) and B-type lamins (lamins B1 and B2) (Fisher et al., 1986; McKeon et al., 1986; Peter et al., 1989).
In this review, we will summarize functional diversities of proteins associated to the LINC complex with a particular focus on lamin A/C and nesprins. We will recapitulate known muscular and cardiac abnormalities induced by mutations in those genes and will discuss our recent advances in related pathogenesis.
Nesprins and Lamins as Components of the LINC Complex
In human, full-length nesprin-1 and nesprin-2, which are the ubiquitously expressed giant nesprins isoforms, are respectively, with a molecular weight of 1 MDa (146 exons) and 800 kDa (116 exons), the second and the third largest described proteins, after the “untouchable” titin (4.2 MDa) (Zhang et al., 2001). Nesprins-1 and -2 are ubiquitous proteins, commonly described as ONM components and composed of three major domains: (i) a Calponin Homology (CH) domain, also called Actin-Binding Domain (ABD) located in the N-terminus side that binds to the actin cytoskeleton; (ii) a long central rod domain composed of multiple spectrin repeats (SR) (respectively, 74 and 56 SR in nesprin-1 and -2) and that supports interactions with other proteins such as emerin with the Emerin Binding Domain (EBD) or lamins with the Lamin Binding Domain (LBD) and finally (iii) a C-terminus KASH domain embedded in the ONM (Zhang et al., 2001; Rajgor and Shanahan, 2013; Figure 1A). An additional domain has been described and called “adaptive domain” (AD). This highly conserved domain is located at the C-terminus extremity and is crucial for structural stabilization of SR (Simpson and Roberts, 2008; Zhong et al., 2010).
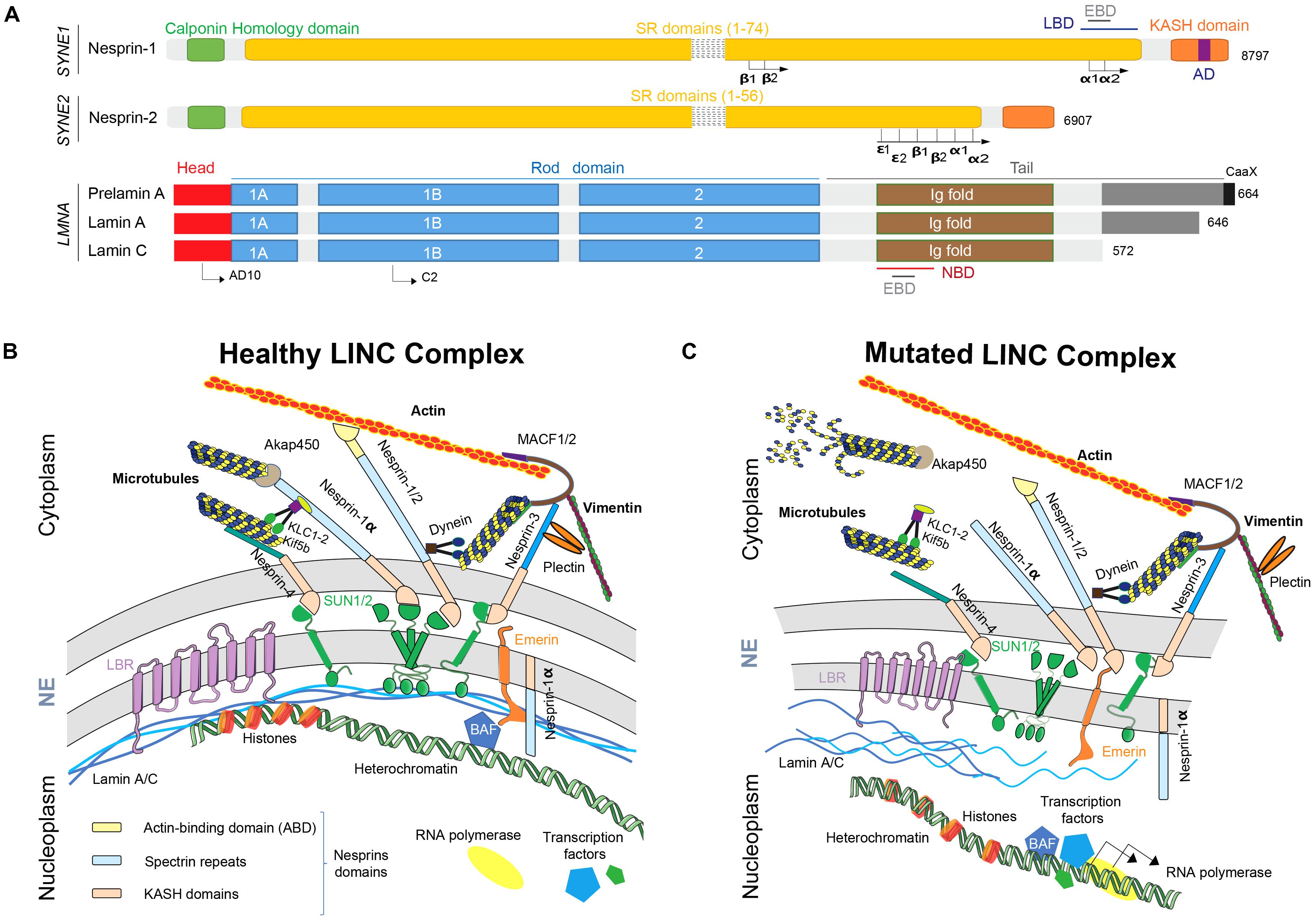
FIGURE 1. Nesprins and lamins as part of the LINC complex. (A) Nesprins and lamins isoforms structures. AD, adaptive domain; EBD, emerin binding domain; KASH domain, Klarsicht/ANC-1/Syne Homology; LBD, lamin binding domain; NBD, nesprin binding domain; SR, spectrin repeats. (B) The healthy LINC complex and its interactors. Nesprins play multiple functions at ONM but also at INM. Full-length nesprins take part at the ONM to the bridge linking the nucleus to the actin cytoskeleton and shorter isoforms, such as nesprin-1a, interact via Akap450 and KLC-1/2 with microtubules. At the same time, smaller isoforms interact with the nucleoskeleton at INM. (C) When a mutation affects LMNA or SYNE genes, the disruption of the LINC complex participates to the physiopathogenesis of the diseases by structural defects, nuclear migration and anchoring defects and cell signaling pathways and gene expression regulation defects. NE, nuclear envelope; LINC, linker of nucleoskeleton to cytoskeleton; ONM, outer nuclear membrane; INM, inner nuclear membrane; KASH domain, Klarsicht/ANC-1/Syne homology; SUN, Sad1p/UNC84; BAF, barrier to auto-integration factor.
Numerous isoforms of nesprin-1 and -2 are produced by alternative initiation/termination of transcription and/or alternative splicing of SYNE1 and SYNE2 genes. These isoforms have a tissue-specific expression and exhibit different subcellular locations, such as actin stress-fibers, focal adhesions or microtubules (Rajgor et al., 2012; Duong et al., 2014). As previously described, full-length isoforms are located at the ONM and, thanks to their KASH domains, interacts in the perinuclear space with SUN1/2. This layout allow the interaction with both actin and microtubule networks through associated molecular motors such as dynein, kinesins light chain (KLC-1/2) and/or microtubule-mediated nucleation factor, such as akap450 (Wilson and Holzbaur, 2015; Gimpel et al., 2017). Interestingly, some isoforms such as full-length nesprin-2 and nesprin-1α, a small isoform, could also localize at the INM leading to the direct interaction with emerin, SUN1/2 and/or lamin A/C (Figure 1A; Zhang, 2005; Holt et al., 2016).
Nesprin-1 and -2, especially small isoforms, nesprin-1α2 and -2α1, are particularly highly expressed in skeletal and cardiac muscles (Duong et al., 2014). Despite the fact that SYNE1 and SYNE2 genes are about 60% homologous, nesprin-1 is more conserved than nesprin-2 (Zhang et al., 2001). However, ABD domains and C-terminus part of SR domains are highly conserved, especially in nesprin-1α2, -2α1 and -2𝜖2, short skeletal muscle-specific and cardiac-specific isoforms. These conserved domains suggest that these isoforms play a similar role in skeletal muscle and cardiac cells physiology (Autore et al., 2013). Nesprin-3, also ubiquitously expressed, was first discovered associated with the ABD of the cytoskeletal linker protein plectin (Wilhelmsen et al., 2005; Ketema et al., 2013). Plectin acts as a mechanical linker between intermediate filaments networks and various cellular structures and is crucial for myofibres integrity. Plectin deficiency has been shown to affect chromatin structure and some genes expression mediating mechanotransduction. Nesprin-3 also interact with ABD of MACF1/2 (Microtubule-Actin Cross Factor 1/2) thanks to its unique spectrin repeat domain and contribute to vimentin accumulation at the vicinity of the nucleus (Wilhelmsen et al., 2005; Ketema et al., 2013; Figure 1B). Nesprin-4 is surprisingly exclusively produced by secretory epithelia and mechanosensory cochlea hair cells (Roux et al., 2009; Horn et al., 2013). KASH5 is only expressed in meiotic cells (Stewart and Burke, 2014). And, finally, LRPM is expressed in some taste cells of mammalian tongues and plays a crucial role in signal transduction by binding IP3 receptors (Shindo et al., 2010). To resume, nesprins-1 and -2 and related isoforms form a strong physical links allowing connection between nucleoskeleton and cytoskeleton including microtubules network, actin and intermediate filaments.
The nuclear lamina is composed of class-V intermediate filaments proteins called “nuclear lamins.” Several isoforms have been described in mammals, divided in two groups: A and B-type lamins (Dechat et al., 2008). A-type lamins are encoded by LMNA gene, located on chromosome 1, producing two major isoforms in somatic cells: lamin-A and lamin-C (Figure 1A). These isoforms are expressed in the majority of differentiated cells, although absent in early stages of embryonic development (Dechat et al., 2010). These two isoforms are produced by an alternative splicing in exon 10. Thus, N-terminus part of lamin-A and -C are similar until the 566th amino-acid and then, lamin-C shows five specific amino-acids in the C-terminus part while lamin A exhibit a specific domain encoded by exons 11 and 12 of LMNA gene (Figure 1A; Furukawa et al., 1994). As the other intermediate filaments, nuclear lamins form dimers by their central rod domain (Stuurman et al., 1998; Herrmann and Aebi, 2016). Additionally, three isoforms of B-type lamins have been described: lamin B1 encoded by LMNB1 gene and lamins B2 and B3, encoded by LMNB2 gene (Dittmer and Misteli, 2011). Nuclear lamin A and C are able to bind both nesprin-1α (Mislow et al., 2002) and nesprin-2 (Yang et al., 2013), heterochromatin (Bruston et al., 2010) and emerin (Sakaki et al., 2001), that itself bind transcription factors and DNA-binding proteins such as barrier to autointegration factor (BAF) (Burke, 1990; Yuan et al., 1991; Furukawa et al., 2003; Segura-Totten and Wilson, 2004; Dialynas et al., 2010; Jamin and Wiebe, 2015; Figure 1B).
This association between nesprins at the ONM through SUN proteins at the INM and lamins at the vicinity of INM is the key place in cells where mechanotransduction occurs. This process allows cytoplasmic mechanical forces, initiated through the dynamics and forces generated by different cytoskeletons (actin, microtubule, and intermediate filaments) to cross the nuclear membrane and influence the transcriptional state of the cell. This transduction can alter transcriptional efficiency through chromatin organization and regulation of epigenetics markers as well as processes linked to DNA replication (Tajik et al., 2016; Kirby and Lammerding, 2018; Wang et al., 2018). The respective role of alternative pathways in mechanical transduction during lifetime of cells remains to be determined. Interestingly, in post-mitotic conditions such as muscle cells, proteins of the LINC complex, especially nesprins and lamins emerged as a fundamental node in the crossroad of muscle pathogenesis.
Roles of Nesprins and Lamins in Muscle Physiology
Cardiac and skeletal muscles are both striated muscles composed of repeated units called sarcomeres. Sarcomeres of both types of muscle are an arrangement of alternating thick and thin filaments resulting in a striated cell pattern. Cardiomyocytes are mononucleated cells with a centrally localized nucleus and form, together with associated cardiomyocytes, a syncytium-like via intercalated disks that link membranes of contiguous cardiomyocytes, providing synchronized contraction. Skeletal muscle is an association of large multinucleated cells with peripheral nuclei called myofibers, resulting from the fusion of specialized cells, myocytes (Tedesco et al., 2010; Chal and Pourquié, 2017). Prior to fusion, myoblasts are highly proliferative, but exit the cell cycle at the onset of myoblasts differentiation and become myocytes, specialized cells possessing the potential to fuse with each other (Yin et al., 2013). As myocytes fuse, they form multinucleated syncytium: myotubes. As myotubes mature into myofibers, contractile units (sarcomeres) are formed. A fully developed muscle is made of an association of myofibers (Sanger et al., 2010; Lindskog et al., 2015). Interestingly, myonuclei positioning is dramatically different between skeletal and cardiac muscles (peripheral vs. central). One could propose that LINC complex composition or arrangement and binding partners will be different between these two tissues. However, evidences about these differences are still lacking and will probably highlight alternative mechanisms in these two systems.
The LINC complex integrity has been shown to be crucial in myonuclei positioning and movements during both fusion of myoblasts and differentiation processes into myotubes/myofibres (Dupin et al., 2011; Gundersen and Worman, 2013; Wilson and Holzbaur, 2015; Stroud et al., 2017). More precisely, nesprin-1 is known to be critical in myonuclear anchoring in skeletal muscle (Zhang et al., 2007b; Puckelwartz et al., 2009) allowed by the bridge between myonuclei and actin cytoskeleton (Padmakumar et al., 2004; Zhang et al., 2007b, 2010; Puckelwartz et al., 2009; Banerjee et al., 2014). Proper nuclear positioning and movements in muscle is mainly mediated by cytoskeletal networks (microtubules and actin) and is known to depend on members of the LINC complex in association with nucleoskeleton components (Metzger et al., 2012; Chang et al., 2015; Wilson and Holzbaur, 2015; Gache et al., 2017; Roman et al., 2017). Both full-length nesprin-1 and nesprin-2 interact with Kif5b molecular motors through a conserved motif located in their C-terminus part of kinesins light chain KLC-1/2. This interaction is crucial for nuclear movement and positioning in differentiated myotubes (Metzger et al., 2012; Wilson and Holzbaur, 2015; Zhou et al., 2017). Moreover, Nesprin-1α is now known to be able to recruit Akap450 to the NE. This recruitment is crucial for nuclear positioning and microtubule organization during myogenesis by switching the Microtubule Organizing Center (MTOC) to the NE (Gimpel et al., 2017). Additionally, differential expression kinetic of alternative nesprins isoforms occurs during muscle differentiation (Randles et al., 2010): in proliferative myoblasts, full-length nesprin-1 isoform is expressed but not nesprin-1α2 while during myogenesis, full-length nesprin-1 and nesprin-1α2 are both up-regulated (Holt et al., 2016; Stroud et al., 2017; Zhou et al., 2017). In human vascular smooth muscle cells (VSMCs), in addition to the NE localization, nesprin-2 is also present in lamellipodia, focal adhesions and filopodia (Zhang, 2005). Moreover, in human skeletal muscle cells, nesprin-2 isoforms had been shown to relocalized at sarcomeres, with a close association with titin (Zhang, 2005). Additionally, in vivo, nesprin-2 can supplant nesprin-1 at the NE (Randles et al., 2010). Finally, nesprins involvements in cardiomyocytes functionality and during cardiac cell differentiation are for now limited mainly because of technical issues (i.e., difficulties to isolate and purify mature cardiomyocytes, absence of cardiac stem cells and regeneration process…). Nonetheless, it has been shown that nesprin-1 knockdown alter differentiation of embryonic stem cells in mature cardiomyocytes in mouse model (Gao et al., 2012) and that nesprins-1 and -2 ablation cause cardiomyopathy in mice with an altered nuclear morphology and altered mechanotransduction (Banerjee et al., 2014).
Plectin, a cytoskeletal protein interacting with nesprin-3, acts as a mechanical linker between intermediate filaments networks and various cellular structures and is crucial for myofibres integrity. Plectin deficiency has been shown to affect chromatin structure, specific genes expression mediating mechanotransduction but also mitochondrial network organization with the upregulation of Mfn-2, the mitochondrial fusion-associated protein mitofusion-2 (Staszewska et al., 2015; Winter et al., 2015).
In the same manner, lamins exert their function thanks to their interaction with numerous lamin-binding partners. Over 100 putative binding proteins are predicted with a high variety of functions: mechanotransduction, cell migration and differentiation (Gerace and Tapia, 2018). Crucial role of lamins reside in the structural support gave to the nuclei in cell, especially in contractile tissues. This role is mediated by a structure and assembly of lamins based on an α-helicoidal coiled coil structure assembled into fibers (Gruenbaum and Foisner, 2015). This typical network pattern provides resistance to contractile cells, such as in cardiomyocytes and muscle fibers, which are subjected to physical forces (Carmosino et al., 2014). For example, lamin A/C, by monitoring nuclei membrane rigidity, is determinant in the control of myonuclear movement to the periphery of myofibers (Roman et al., 2017). As discussed after, this role is the foundation of “mechanical stress hypothesis,” also called “structural hypothesis” about the physiopathological mechanisms underlying LMNA mutations. The second crucial role of lamins is their interactions with chromatin, signaling molecules and transcription factors (Herrmann et al., 2007). This interplay with lamin A/C, but also with other LINC complex components or interactors (signaling molecules or transcription factors) is an important mechanism to explain gene expression regulation by NE proteins (Robson et al., 2016; Kirby and Lammerding, 2018; Wang et al., 2018). Numerous studies have highlighted this regulatory role of lamins in signaling pathways such as MAPK (Muchir et al., 2007), Akt/mTOR (Choi et al., 2012; Ramos et al., 2012), TGFα (Arimura et al., 2005; Cohen et al., 2013), BMP (Janin et al., 2018), YAP (Bertrand et al., 2014), and major apoptosis signaling pathways (Lu et al., 2010). Lamins A/C have also a crucial role for proper skeletal muscle stem cells differentiation: primary Lmna-null myoblasts exhibit altered expression of differentiation regulators such as MyoD1 or desmin (Frock et al., 2006). Moreover, lamin A/C inactivation has been shown to severely impair Serum Response Factor (SRF) pathway, which control genes coding for actin cytoskeleton (Vartiainen et al., 2007; Ho et al., 2013) and for sarcomeric proteins (Balza and Misra, 2006). This characteristic will also contribute to control physiopathological and tissue-specific mechanisms underlying in LMNA mutations. Finally, lamins have been shown also to play a role in autophagy and probably in senescence. In Lmna-/- mice, hyperactivation of mTORC pathway is responsible for autophagy inhibition and, thus, disease progression. This inhibition could lead to energy deficit and accumulation of damaged and toxic proteins and/or organelles (Ramos et al., 2012).
Nesprins and Lamins Mutations in Human Diseases
Mutations in SYNE Genes
Mutations in SYNE1, coding for nesprin-1, have been reported to cause neurodegenerative diseases such as “Autosomal Recessive Cerebellar Ataxia type 1” (ARCA1), also called “Autosomal recessive ataxia, Beauce type” (Gros-Louis et al., 2007). In patients, homogenous phenotype was firstly reported resulting in a relatively pure and slowly progressive cerebellar ataxia with dysarthria and rare extra-neurological features and an onset at adult age. This disease was mostly identified in French and Canadian families (Dupré et al., 2007; Gros-Louis et al., 2007; Noreau et al., 2013). Later, thanks to molecular diagnosis technics improvement, diffuse pure cerebellar ataxia was challenged. Exploration of large SYNE1 mutations cohort in European and non-European patients show that resulting phenotype is wider and encompassed neurological and non-neurological symptoms. To note, in this study, 81% of the patients exhibited additional non-cerebellar features and a multisystemic phenotype (Synofzik et al., 2016). Clinical spectrum encompasses upper and lower motor neuron disease, brainstem abnormalities and musculoskeletal features (kyphosis, scoliosis, pes cavus, or contractures) (Izumi et al., 2013; Synofzik et al., 2016; Algahtani et al., 2017). Similarly, onset age is variable from an early onset for some complex neurodegenerative syndromes to adult-age onset for pure cerebellar ataxia syndromes. In most cases, non-sense or intronic mutations causing premature termination could be highlighted (Synofzik et al., 2016). Thus, different nesprin-1 isoforms could be affected by these mutations, partially explaining the variability in observed phenotypes (Cartwright and Karakesisoglou, 2014; Razafsky and Hodzic, 2015). This observation is consistent with evidences in Syne1 mutant mice model. In Syne1-/- mice, in which the C-terminal SR domains of nesprin-1 were ablated (Zhang et al., 2010), young mice exhibit growth retardation, increased body weight variability and exercise intolerance. In contrast, in Syne1ΔKASH, in which the KASH domain was deleted, mice exhibit normal viability but formation of both synaptic myonuclei clusters and non-synaptic myonuclei organization in skeletal muscle was impaired (Zhang et al., 2007b).
Additionally, SYNE1 polymorphism had been associated, using genome-wide association studies (GWAS) in depression, bipolar disorders and autism and recognized as a risk factor in schizophrenia (Green et al., 2013). Thanks to whole-exome sequencing experiments, a recurrent mutation in SYNE1 (p.Leu3206Met) had been identified as a autism-spectrum disorder candidate gene with a recurrent mutation (Yu et al., 2013).
In musculoskeletal and cardiac diseases, SYNE1 and SYNE2 mutations had been associated with Autosomal-Dominant Emery-Dreifuss Muscular Dystrophy (AD-EDMD) and EDMD-like phenotypes (Zhang et al., 2007a; Koch and Holaska, 2014; Fanin et al., 2015). These myopathies are usually clinically associated with three main symptoms: (i) early contractures of elbows flexors, achilles’ tendons and spine rigidity, (ii) slowly progressive muscle wasting and weakness (initiated at the humeroperoneal stage and diffuse impairment later), and (iii) cardiac misfunctionality characterized by an association of conduction defects and dilated cardiomyopathy (Koch and Holaska, 2014). Cardiac defects occur in late stages of diseases but become the main causes of EDMD patients mortality (Helbling-Leclerc et al., 2002). Mutations in SYNE1 are also associated with pure cardiac phenotype: dilated cardiomyopathy (DCM) with conduction system defects (Puckelwartz et al., 2010; Zhou et al., 2017). Mutations in LMNA and EMD occur in nearly 40% of EDMDs (Bonne et al., 2003). Interestingly, mutations in associated partners of lamins and emerin such as nesprins-1/2 or SUN proteins have been also described in EDMD and EDMD-like phenotypes. In 2007, 190 patients suffering from EDMD or EDMD-like phenotypes without mutations in LMNA and EMD genes were analyzed regarding sequence of SYNE1 and SYNE2 genes. Six variants were identified, absent from control population and showing a segregation pattern compatible with an autosomal dominant inheritance (Zhang et al., 2007a). More than half of identified and characterized SYNE1 and SYNE2 mutants are located in the C-terminus part of full-length nesprins-1 and -2 (Zhang et al., 2007a; Attali et al., 2009; Puckelwartz et al., 2010; Taranum et al., 2012; Fanin et al., 2015; Baumann et al., 2016; Holt et al., 2016; Chen et al., 2017; Haskell et al., 2017; Wu et al., 2017; Zhou et al., 2017). In those contexts, abnormalities in nuclear morphology are frequently observed in mutated cells (commonly fibroblasts or lymphoblasts) such as altered nuclear shape, micronuclei or fragmented nuclei (Zhang et al., 2007a; Haskell et al., 2017). These observations are frequently associated with mislocalization of LINC complex components or interactors such as lamins A/C, SUN 1/2 proteins or kinesin light chain KLC-1/2, bridging cargoes to heavy chain kinesin such as Kif5b and thus to microtubules network (Zhang et al., 2007a; Chen et al., 2017; Zhou et al., 2017). Moreover, impaired fusion between myoblasts in the time course of differentiation process had been described using C2C12 cells infected with DCM-associated nesprin-1 mutations (Zhou et al., 2017). Finally, centralized myonuclei and increased variability in cross-sections area of myofibers (CSA) had been described in muscle biopsies from EDMD patients (Fanin et al., 2015; Chen et al., 2017). Partial genotype-phenotype relationship could be highlighted with SYNE1 and SYNE2 mutations. Most mutations causing muscle phenotype are heterozygous missense located in the C-terminus part of full-length nesprin that also belongs to nesprin-1α2, a muscle specific isoform (Zhou et al., 2017). Mutations affecting CNS development are located all along SYNE1 gene (Synofzik et al., 2016). Most of them are non-sense mutations and induced premature termination codon resulting in truncated proteins or absence of nesprin-1/2 due to nonsense-mediated decay of the mutant mRNA (Zhou et al., 2018). SYNE1 mutations are also involved in Arthrogryposis Multiplex Congenital (AMC) or simply arthrogryposis, a group of diseases with an overall prevalence of one in 3,000 live births, characterized by congenital joint contractures and reduced fetal movements (Baumann et al., 2016). Finally, some emerging role of nesprin-1 in the formation of striated F-actin-based filaments (Packard et al., 2015) or in Drosophila, in the control of glutamate receptors density, specifically at the neuromuscular junction (Morel et al., 2014) suggests that SYNE1 mutations could be highlighted in other neuromuscular junction diseases.
Mutations in LMNA Gene
Laminopathies is the dedicated term to call a group of rare genetic disorders caused by mutations in genes encoding proteins of the nuclear lamina: LMNA, coding for lamins A and C, and LMNB1 and LMNB2, coding for lamins B. Laminopathies caused by LMNA mutations are initially defined on clinical criteria’s and is mostly characterized by an autosomal dominant inheritance (Bertrand et al., 2011). Laminopathies include a group of diseases with a predominant skeletal muscle and/or heart involvement, such as the autosomal dominant form of EDMD (EDMD-2 or AD-EDMD), the autosomal recessive form of EDMD (EDMD-3), a dilated cardiomyopathy phenotype associated with conduction defects (DCM-CD), Congenital Muscular Dystrophy (L-CMD or LMNA-CMD) and Limb-Girdle Muscular Dystrophy 1B (LGMD1B) (Bönnemann et al., 2014). Laminopathies also include diseases with extra-muscular phenotypes such as Dunningan-type Familial Partial Lipodystrophy (FPLD), atypical Werner syndrome, Charcot-Marie-Tooth syndrome 2B1 (CMT2B1), and Hutchinson-Gilford Progeria Syndrome (HGPS) (Shackleton et al., 2000; De Sandre-Giovannoli et al., 2002, 2003; Bonne and Levy, 2003; Bönnemann et al., 2014). Although other tissues are affected in theses diseases, muscle and heart abnormalities are the most frequently observed (Janin et al., 2017).
Skeletal muscle laminopathies share common symptoms: joint contractures affecting predominately elbows (rigidity), ankles and neck and developing toward muscle weakness, life-threatening cardiac conduction defects and dilated cardiomyopathy (Lattanzi et al., 2011). EDMD2, DCM-CD, and L-CMD are overlapping each other and are now considered as a clinical continuum of an unique disease as three phenotypes could be caused by the same mutation and could exist in the same family by variable expressivity (Lattanzi et al., 2011; Janin et al., 2017; Table 1). In laminopathies, serum creatine kinase activity is usually normal or slightly increased (Mercuri et al., 2004). Electromyography and histological analysis usually show unspecific patterns, thus, muscle biopsy is not necessary for diagnosis of phenotype with typical clinical abnormalities (Menezes et al., 2012). Magnetic Resonance Imaging (MRI) of muscle could help differential diagnosis with other myopathies exhibiting fatty infiltration of medial gastrocnemius and vastus muscles in LMNA-related myopathies (also called LMNA-RD) (Carboni et al., 2010, 2013; Mercuri et al., 2010; Menezes et al., 2012).
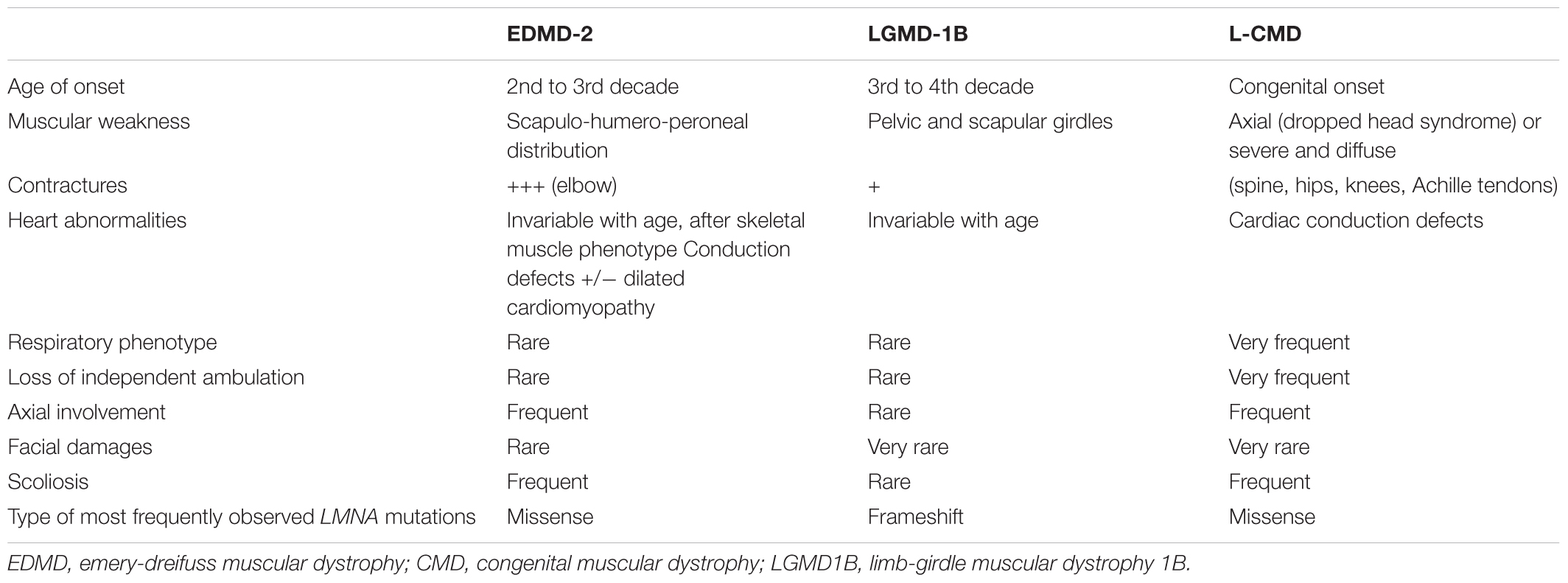
TABLE 1. Clinical and genetics comparison of LMNA-RD myopathies.
The first described muscular phenotype related to LMNA mutations was EDMD-2, so called after the identification of EDMD-1 or X-linked EDMD, a phenotype caused by mutation in EMD coding for emerin, a inner nuclear membrane protein (Bione et al., 1994). EDMD-2 is also clinically characterized by a triad of symptoms: (i) early elbows, ankles and neck contractures with muscle weakness and wasting affecting first the scapulo-humero-peroneal area, (ii) dilated cardiomyopathy, and (iii) heart conduction system defects with high risk of sudden cardiac death (Emery, 2000). Muscle weakness usually appears during the second decade of life, sometimes after contractures occur (Bonne et al., 2000). Main difference between EDMD2 and LGMD1B can be found in wasting and weakness distribution (Maggi et al., 2014). As its name suggests, muscle weakness in LGMD1B is predominant in scapular and pelvic girdle areas in early stages (Muchir et al., 2000). Later, differential diagnosis between EDMD-2 and LGMD1B could be challenging with the progression of muscle weakness to the pelvic girdle in EDMD-2, which mimic LGMD1B phenotype. In these cases, identification of elbow contractures could support EDMD-2 diagnosis (Muchir et al., 2000).
LMNA-CMD phenotype had been described later: this diagnosis concerns children at birth or in their first two years of life (Quijano-Roy et al., 2008). Some of them display a severe congenital form with an absence of motor development. Most common phenotype is an axial muscle weakness with “dropped head syndrome” despite of a former normal acquisition of head carriage, a rigid spine and a scoliosis without locomotion loss (Quijano-Roy et al., 2008). Respiratory abnormalities are very frequent whereas cardiac features are rare, although sudden cardiac death could occur during the first ten years of life (Quijano-Roy et al., 2008). Analysis of an Italian cohort of LMNA-RD patients had confirmed cardiac involvement as it is the main feature in the natural history of patients phenotypes and cardiac follow-up is very important (Maggi et al., 2014). Usually, arrhythmia is the first cardiac symptom with electrocardiogram abnormalities such as low P-wave (atrial depolarization) or elongated PR-interval (period between onset of P-wave and ventricular depolarization) associated with rhythm abnormalities such as sinus bradycardia, sick sinus syndrome, bundle branch block or atrioventricular block or ectopic beats (Fatkin et al., 1999; Nigro et al., 2012). These symptoms appear mainly after the third decade of life (van Berlo et al., 2005).
Mutations Pathogenesis Hypothesis
Nesprins and lamins both localize at NE and are ubiquitously expressed. However, mutations in genes encoding those proteins are responsible of tissue-specific diseases. Thus, several non-exclusive hypotheses have been proposed to explain the physiopathological mechanisms underlying these described diseases (Figure 1C).
Structural Hypothesis
Structural hypothesis is based on “structural function” of the LINC complex and its ability to couple nucleoskeleton with cytoskeleton and associated interactors such as molecular motors and microtubule-associated proteins (MAPs), microtubules and actin network. LINC complex impairments could modulate extracellular transmission forces balance to the nucleus and cells could suffer from this induced mechanical damages (Zhang et al., 2007a; Banerjee et al., 2014; Bertrand et al., 2014). Nesprins have been shown to play a crucial role in myonuclei positioning and migration in muscles. It has been shown in Caenorhabditis elegans (Starr and Fridolfsson, 2010), in Drosophila melanogaster (Yu et al., 2006) but also in higher organisms (Wilson and Holzbaur, 2015; Gimpel et al., 2017; Zhou et al., 2017) that mutated nesprins (or orthologues) are responsible for mislocated nuclei mediated by an impaired recruitment of different partners such as Akap450 or KLC-1/2.
Gene Regulation Hypothesis
Gene regulation hypothesis suggests that impaired LINC complex could alter interaction of NE proteins (mainly lamins) with the chromatin. Thus, mutations in genes encoding LINC complex proteins could alter in a tissue-specific manner transcription factors expression and/or alter expression pattern of tissue-specific genes (Dialynas et al., 2010). For example, such alterations are more and more described for laminopathies with the hyperactivation of the ERK (extracellular signal-regulated kinase) pathway in cardiomyopathy caused by LMNA mutations (Chatzifrangkeskou et al., 2018) or the overexpression of SMAD6, an inhibitor of the BMP signaling pathways, as a cause of accelerated myogenic differentiation of LMNA mutated cells (Janin et al., 2018).
Lamin A/C plays also a crucial role in euchromatin and heterochromatin distribution (Solovei et al., 2013) and in chromatin mobility restriction (Bronshtein et al., 2015). This chromatin interaction pathway exhibits a cell-type specific and differentiation-dependent pattern (Rønningen et al., 2015). Overexpression of wild-type lamin A/C, or mutated lamins related to progeroid syndromes (L647R) (Wang et al., 2016) or to FPLD and L-CMD (R388P) (Barateau et al., 2017) leads to different genes-lamins interactions patterns revealed using ChIP-seq analysis (Paulsen et al., 2017). These modifications are responsible for a more global three dimensional rearrangement of chromatin conformation (Oldenburg et al., 2014), associated with modification of epigenetic state of cells (Cesarini et al., 2015) and thus as a direct modification in gene expressions as it has been shown in FPLD-causing LMNA mutation R482W (Stierlé et al., 2003; Vadrot et al., 2015; Oldenburg et al., 2017).
Evidences support the epigenetic signature modifications hypothesis in cells. First, in patients suffering from HGPS, fibroblasts level of H3K27me3, a histone methylation often associated with the downregulation of nearby genes, is reduced and caused by a decrease level of the H3K27 methyltransferase of the PRC2 complex, called EZH2 (Shumaker et al., 2006). Interestingly, PRC2 complex regulates expression of P16/INK4A and P21, two tumor suppressors genes (Tzatsos et al., 2011), potentially filling the blank in understanding premature senescence observed in laminopathies (Guénantin et al., 2014). Finally, EZH2 plays a crucial role in the regulation of muscle gene expression and skeletal muscle differentiation (Caretti et al., 2004) and requires a functional lamin A/C network (Cesarini et al., 2015). These findings help to better understand how a ubiquitously expressed mutated protein could lead to tissue-specific phenotypes.
Conclusion
The NE not only isolates genetic material from the rest of the cell, but also plays a crucial role in mechanotransduction signaling through the LINC complex that builds a bridge between cytoskeleton and nucleoskeleton. Moreover, interactions between chromatin, inner nuclear membrane and nuclear lamina are important to regulate gene expression. Thus, NE is recognized as a central complex organizing communication between genome and cytoskeleton, and most importantly as a key factor to mediate appropriate responses regarding external mechanical forces applied on cells.
In the past decade, LINC complex components and interactors have been identified either covering inner surface of the nucleus, or located directly inside inner or outer nuclear membrane. In parallel, mutations in genes encoding NE components have been associated with rare human diseases affecting numerous different tissues; from them, nesprin- and lamin-associated muscular diseases have emerged. Despite databases and large-scale studies recapitulating plenty of mutations, no clear correlation between a given genotype and its affected tissues or disease has been established. For example, LMNA mutations can lead to diseases affecting peripheral nerve, adipose tissue, skeletal muscle or heart, making the molecular diagnosis of envelopathies particularly challenging. At the same time, our progressive knowledge of nesprins and lamins roles at the NE, in the LINC complex organization, in regulation of gene expression, especially during myogenesis allows a better understanding of physiopathological mechanisms beneath described muscular and cardiac phenotypes. A complete understanding of different nesprins isoforms functions associated with an overview of proteins interacting network of lamins and nesprins is currently lacking.
Extensive use of Next Generation Sequencing (NGS) tools and of Whole Genome and Exome Sequencing (WGS and WES) in diagnosis laboratories will improve this discovery process. These tools, associated with the democratization of induced pluripotent stem cells (hiPSC) derived from affected patients or from healthy donor coupled with CRISPR-Cas9 technology in research laboratories, will also help to decipher the pathophysiological mechanisms underlying rare diseases without molecular explanation. Finally, taken together, these findings enable development of potential personalized therapeutic targets.
Author Contributions
All authors wrote, read, and approved the manuscript.
Funding
This work was supported by Hospices Civils de Lyon, Université Claude Bernard Lyon 1, INSERM, and MyoNeurAlp Alliance.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer JR and handling editor declared their shared affiliation at the time of the review.
Abbreviations
ABD, actin-binding domain; AD-EDMD, autosomal-dominant emery-dreifuss muscular dystrophy; AD, adaptive domain; AMC, arthrogryposis multiplex congenital; ARCA1, autosomal recessive cerebellar ataxia type 1; BMP, bone morphogenetic proteins; CH, calponin homology; CMD, congenital muscular dystrophy; CMT2B1, charcot-marie-tooth syndrome 2B1; CNS, central nervous system; DCM-CD, dilated cardiomyopathy phenotype with conduction defects; DCM, dilated cardiomyopathy; EDMD, emery-dreifuss muscular dystrophy; ERK, extracellular signal-regulated kinase; EZH2, enhancer of zeste homolog 2; FPLD, dunningan-type familial partial lipodystrophy; GWAS, genome-wide association studies; HGPS, hutchinson-gilford progeria syndrome; INM, inner nuclear membrane; KASH, Klarsicht/Anc-1/Syne Homology; KLC, kinesin light chain (KLC-1/2); LGMD1B, limb-girdle muscular dystrophy 1B; LINC complex, linker of nucleoskeleton to cytoskeleton; LRMP, lymphoid restricted membrane protein; MAP, microtubule-associated proteins; MAPK, mitogen-activated protein kinases; MTOC, microtubule organizing center; NE, nuclear envelope; NGS, next generation sequencing; NPC, nuclear pore complex; ONM, outer nuclear membrane; PRC2, polycomb repressive complex 2; SR, spectrin repeats; SUN, Sad1/UNC-84 homology; TGF, tissue growth factor; VSMC, vascular smooth muscle cells; WES, whole exome sequencing; WGS, whole genome sequencing; YAP, yes-associated protein.
References
Algahtani, H., Marzouk, Y., Algahtani, R., Salman, S., and Shirah, B. (2017). Autosomal recessive cerebellar Ataxia type 1 mimicking multiple sclerosis: a report of two siblings with a novel mutation in SYNE1 gene in a Saudi family. J. Neurol. Sci. 372, 97–100. doi: 10.1016/j.jns.2016.11.036
Arimura, T., Helbling-Leclerc, A., Massart, C., Varnous, S., Niel, F., Lacène, E., et al. (2005). Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet. 14, 155–169. doi: 10.1093/hmg/ddi017
Attali, R., Warwar, N., Israel, A., Gurt, I., McNally, E., Puckelwartz, M., et al. (2009). Mutation of SYNE-1, encoding an essential component of the nuclear lamina, is responsible for autosomal recessive arthrogryposis. Hum. Mol. Genet. 18, 3462–3469. doi: 10.1093/hmg/ddp290
Autore, F., Pfuhl, M., Quan, X., Williams, A., Roberts, R. G., Shanahan, C. M., et al. (2013). Large-scale modelling of the divergent spectrin repeats in nesprins: giant modular proteins. PLoS One 8:e63633. doi: 10.1371/journal.pone.0063633
Balza, R. O., and Misra, R. P. (2006). Role of the serum response factor in regulating contractile apparatus gene expression and sarcomeric integrity in cardiomyocytes. J. Biol. Chem. 281, 6498–6510. doi: 10.1074/jbc.M509487200
Banerjee, I., Zhang, J., Moore-Morris, T., Pfeiffer, E., Buchholz, K. S., Liu, A., et al. (2014). Targeted ablation of nesprin 1 and nesprin 2 from murine myocardium results in cardiomyopathy, altered nuclear morphology and inhibition of the biomechanical gene response. PLoS Genet. 10:e1004114. doi: 10.1371/journal.pgen.1004114
Barateau, A., Vadrot, N., Vicart, P., Ferreiro, A., Mayer, M., Héron, D., et al. (2017). A novel lamin a mutant responsible for congenital muscular dystrophy causes distinct abnormalities of the cell nucleus. PLoS One 12:e0169189. doi: 10.1371/journal.pone.0169189
Baumann, M., Steichen-Gersdorf, E., Krabichler, B., Petersen, B.-S., Weber, U., Schmidt, W. M., et al. (2016). Homozygous SYNE1 mutation causes congenital onset of muscular weakness with distal arthrogryposis: a genotype-phenotype correlation. Eur. J. Hum. Genet. 25, 262–266. doi: 10.1038/ejhg.2016.144
Behrens, T. W., Jagadeesh, J., Scherle, P., Kearns, G., Yewdell, J., and Staudt, L. M. (1994). Jaw1, A lymphoid-restricted membrane protein localized to the endoplasmic reticulum. J. Immunol. Baltim. Med. 1950, 682–690.
Bertrand, A. T., Chikhaoui, K., Yaou, R. B., and Bonne, G. (2011). Clinical and genetic heterogeneity in laminopathies. Biochem. Soc. Trans. 39, 1687–1692. doi: 10.1042/BST20110670
Bertrand, A. T., Ziaei, S., Ehret, C., Duchemin, H., Mamchaoui, K., Bigot, A., et al. (2014). Cellular microenvironments reveal defective mechanosensing responses and elevated YAP signaling in LMNA-mutated muscle precursors. J. Cell Sci. 127, 2873–2884. doi: 10.1242/jcs.144907
Bione, S., Maestrini, E., Rivella, S., Mancini, M., Regis, S., Romeo, G., et al. (1994). Identification of a novel X-linked gene responsible for emery-dreifuss muscular dystrophy. Nat. Genet. 8, 323–327. doi: 10.1038/ng1294-323
Bonne, G., and Levy, N. (2003). LMNA mutations in atypical Werner’s syndrome. Lancet 362, 1585–1586; author reply 1586, doi: 10.1016/S0140-6736(03)14761-7
Bonne, G., Mercuri, E., Muchir, A., Urtizberea, A., Bécane, H. M., Recan, D., et al. (2000). Clinical and molecular genetic spectrum of autosomal dominant emery-dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol. 48, 170–180. doi: 10.1002/1531-8249(200008)48:2<170::AID-ANA6>3.0.CO;2-J
Bonne, G., Yaou, R. B., Béroud, C., Boriani, G., Brown, S., de Visser, M., et al. (2003). 108th ENMC international workshop, 3rd workshop of the MYO-CLUSTER project: EUROMEN, 7th international emery-dreifuss muscular dystrophy (EDMD) workshop, 13–15 september 2002, naarden, the Netherlands. Neuromuscul. Disord. 13, 508–515. doi: 10.1016/S0960-8966(03)00063-4
Bönnemann, C. G., Wang, C. H., Quijano-Roy, S., Deconinck, N., Bertini, E., Ferreiro, A., et al. (2014). Diagnostic approach to the congenital muscular dystrophies. Neuromuscul. Disord. 24, 289–311. doi: 10.1016/j.nmd.2013.12.011
Bronshtein, I., Kepten, E., Kanter, I., Berezin, S., Lindner, M., Redwood, A. B., et al. (2015). Loss of lamin A function increases chromatin dynamics in the nuclear interior. Nat. Commun. 6:8044. doi: 10.1038/ncomms9044
Bruston, F., Delbarre, E., Ostlund, C., Worman, H. J., Buendia, B., and Duband-Goulet, I. (2010). Loss of a DNA binding site within the tail of prelamin A contributes to altered heterochromatin anchorage by progerin. FEBS Lett. 584, 2999–3004. doi: 10.1016/j.febslet.2010.05.032
Burke, B. (1990). On the cell-free association of lamins A and C with metaphase chromosomes. Exp. Cell Res. 186, 169–176. doi: 10.1016/0014-4827(90)90223-W
Carboni, N., Mateddu, A., Marrosu, G., Cocco, E., and Marrosu, M. G. (2013). Genetic and clinical characteristics of skeletal and cardiac muscle in patients with lamin A/C gene mutations. Muscle Nerve 48, 161–170. doi: 10.1002/mus.23827
Carboni, N., Mura, M., Marrosu, G., Cocco, E., Marini, S., Solla, E., et al. (2010). Muscle imaging analogies in a cohort of patients with different clinical phenotypes caused by LMNA gene mutations. Muscle Nerve 41, 458–463. doi: 10.1002/mus.21514
Caretti, G., Di Padova, M., Micales, B., Lyons, G. E., and Sartorelli, V. (2004). The polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev. 18, 2627–2638. doi: 10.1101/gad.1241904
Carmosino, M., Torretta, S., Procino, G., Gerbino, A., Forleo, C., Favale, S., et al. (2014). Role of nuclear lamin A/C in cardiomyocyte functions. Biol. Cell 106, 346–358. doi: 10.1111/boc.201400033
Cartwright, S., and Karakesisoglou, I. (2014). Nesprins in health and disease. Semin. Cell Dev. Biol. 29, 169–179. doi: 10.1016/j.semcdb.2013.12.010
Cesarini, E., Mozzetta, C., Marullo, F., Gregoretti, F., Gargiulo, A., Columbaro, M., et al. (2015). Lamin A/C sustains PcG protein architecture, maintaining transcriptional repression at target genes. J. Cell Biol. 211, 533–551. doi: 10.1083/jcb.201504035
Chal, J., and Pourquié, O. (2017). Making muscle: skeletal myogenesis in vivo and in vitro. Development 144, 2104–2122. doi: 10.1242/dev.151035
Chang, W., Antoku, S., Östlund, C., Worman, H. J., and Gundersen, G. G. (2015). Linker of nucleoskeleton and cytoskeleton (LINC) complex-mediated actin-dependent nuclear positioning orients centrosomes in migrating myoblasts. Nucleus 6, 77–88. doi: 10.1080/19491034.2015.1004947
Chatzifrangkeskou, M., Yadin, D., Marais, T., Chardonnet, S., Cohen-Tannoudji, M., Mougenot, N., et al. (2018). Cofilin-1 phosphorylation catalyzed by ERK1/2 alters cardiac actin dynamics in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. 27, 3060–3078. doi: 10.1093/hmg/ddy215
Chen, Z., Ren, Z., Mei, W., Ma, Q., Shi, Y., Zhang, Y., et al. (2017). A novel SYNE1 gene mutation in a Chinese family of emery-dreifuss muscular dystrophy-like. BMC Med. Genetics 18:63. doi: 10.1186/s12881-017-0424-5
Choi, J. C., Muchir, A., Wu, W., Iwata, S., Homma, S., Morrow, J. P., et al. (2012). Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci. Transl. Med. 4:144ra102. doi: 10.1126/scitranslmed.3003875
Cohen, T. V., Gnocchi, V. F., Cohen, J. E., Phadke, A., Liu, H., Ellis, J. A., et al. (2013). Defective skeletal muscle growth in lamin A/C-deficient mice is rescued by loss of Lap2α. Hum. Mol. Genet. 22, 2852–2869. doi: 10.1093/hmg/ddt135
Crisp, M., Liu, Q., Roux, K., Rattner, J. B., Shanahan, C., Burke, B., et al. (2006). Coupling of the nucleus and cytoplasm: role of the LINC complex. J. Cell Biol. 172, 41–53. doi: 10.1083/jcb.200509124
De Sandre-Giovannoli, A., Bernard, R., Cau, P., Navarro, C., Amiel, J., Boccaccio, I., et al. (2003). Lamin a truncation in hutchinson-gilford progeria. Science 300:2055. doi: 10.1126/science.1084125
De Sandre-Giovannoli, A., Chaouch, M., Kozlov, S., Vallat, J.-M., Tazir, M., Kassouri, N., et al. (2002). Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (charcot-marie-tooth disorder type 2) and mouse. Am. J. Hum. Genet. 70, 726–736. doi: 10.1086/339274
Dechat, T., Adam, S. A., Taimen, P., Shimi, T., and Goldman, R. D. (2010). Nuclear lamins. Cold Spring Harb. Perspect. Biol. 2, a000547. doi: 10.1101/cshperspect.a000547
Dechat, T., Pfleghaar, K., Sengupta, K., Shimi, T., Shumaker, D. K., Solimando, L., et al. (2008). Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 22, 832–853. doi: 10.1101/gad.1652708
Dialynas, G., Speese, S., Budnik, V., Geyer, P. K., and Wallrath, L. L. (2010). The role of drosophila lamin C in muscle function and gene expression. Development 137, 3067–3077. doi: 10.1242/dev.048231
Dittmer, T. A., and Misteli, T. (2011). The lamin protein family. Genome Biol. 12:222. doi: 10.1186/gb-2011-12-5-222
Duong, N. T., Morris, G. E., Lam, L. T., Zhang, Q., Sewry, C. A., Shanahan, C. M., et al. (2014). Nesprins: tissue-specific expression of epsilon and other short isoforms. PLoS One 9:e94380. doi: 10.1371/journal.pone.0094380
Dupin, I., Sakamoto, Y., and Etienne-Manneville, S. (2011). Cytoplasmic intermediate filaments mediate actin-driven positioning of the nucleus. J. Cell Sci. 124, 865–872. doi: 10.1242/jcs.076356
Dupré, N., Gros-Louis, F., Chrestian, N., Verreault, S., Brunet, D., de Verteuil, D., et al. (2007). Clinical and genetic study of autosomal recessive cerebellar ataxia type 1. Ann. Neurol. 62, 93–98. doi: 10.1002/ana.21143
Emery, A. E. (2000). Emery-dreifuss muscular dystrophy - a 40 year retrospective. Neuromuscul. Disord. 10, 228–232. doi: 10.1016/S0960-8966(00)00105-X
Fanin, M., Savarese, M., Nascimbeni, A. C., Di Fruscio, G., Pastorello, E., Tasca, E., et al. (2015). Dominant muscular dystrophy with a novel SYNE1 gene mutation: letter to the editor. Muscle Nerve 51, 145–147. doi: 10.1002/mus.24357
Fatkin, D., MacRae, C., Sasaki, T., Wolff, M. R., Porcu, M., Frenneaux, M., et al. (1999). Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 341, 1715–1724. doi: 10.1056/NEJM199912023412302
Fisher, D. Z., Chaudhary, N., and Blobel, G. (1986). cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl. Acad. Sci. U.S.A. 83, 6450–6454. doi: 10.1073/pnas.83.17.6450
Frock, R. L., Kudlow, B. A., Evans, A. M., Jameson, S. A., Hauschka, S. D., and Kennedy, B. K. (2006). Lamin A/C and emerin are critical for skeletal muscle satellite cell differentiation. Genes Dev. 20, 486–500. doi: 10.1101/gad.1364906
Furukawa, K., Inagaki, H., and Hotta, Y. (1994). Identification and cloning of an mRNA coding for a germ cell-specific A-type lamin in mice. Exp. Cell Res. 212, 426–430. doi: 10.1006/excr.1994.1164
Furukawa, K., Sugiyama, S., Osouda, S., Goto, H., Inagaki, M., Horigome, T., et al. (2003). Barrier-to-autointegration factor plays crucial roles in cell cycle progression and nuclear organization in Drosophila. J. Cell Sci. 116, 3811–3823. doi: 10.1242/jcs.00682
Gache, V., Gomes, E. R., and Cadot, B. (2017). Microtubule motors involved in nuclear movement during skeletal muscle differentiation. Mol. Biol. Cell. 28, 865–874. doi: 10.1091/mbc.E16-06-0405
Gao, F., Yi, Q.-J., Tian, Y.-J., Wang, F.-J., and Liu, G.-X. (2012). Effect of nesprin-1 in the differentiation of mouse embryonic stem cells into cardiomyocytes. Sichuan Da Xue Xue Bao Yi Xue Ban 43, 161–165.
Gerace, L., and Tapia, O. (2018). Messages from the voices within: regulation of signaling by proteins of the nuclear lamina. Curr. Opin. Cell Biol. 52, 14–21. doi: 10.1016/j.ceb.2017.12.009
Gimpel, P., Lee, Y. L., Sobota, R. M., Calvi, A., Koullourou, V., Patel, R., et al. (2017). Nesprin-1α-dependent microtubule nucleation from the nuclear envelope via Akap450 is necessary for nuclear positioning in muscle cells. Curr. Biol. 27, 2999.e9–3009.e9. doi: 10.1016/j.cub.2017.08.031
Green, E. K., Grozeva, D., Forty, L., Gordon-Smith, K., Russell, E., Farmer, A., et al. (2013). Association at SYNE1 in both bipolar disorder and recurrent major depression. Mol. Psychiatry 18, 614–617. doi: 10.1038/mp.2012.48
Gros-Louis, F., Dupré, N., Dion, P., Fox, M. A., Laurent, S., Verreault, S., et al. (2007). Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat. Genet. 39, 80–85. doi: 10.1038/ng1927
Gruenbaum, Y., and Foisner, R. (2015). Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu. Rev. Biochem. 84, 131–164. doi: 10.1146/annurev-biochem-060614-034115
Guénantin, A. C., Briand, N., Bidault, G., Afonso, P., Béréziat, V., Vatier, C., et al. (2014). Nuclear envelope-related lipodystrophies. Semin. Cell Dev. Biol. 29, 148–157. doi: 10.1016/j.semcdb.2013.12.015
Gundersen, G. G., and Worman, H. J. (2013). Nuclear positioning. Cell 152, 1376–1389. doi: 10.1016/j.cell.2013.02.031
Haque, F., Lloyd, D. J., Smallwood, D. T., Dent, C. L., Shanahan, C. M., Fry, A. M., et al. (2006). SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol. Cell. Biol. 26, 3738–3751. doi: 10.1128/MCB.26.10.3738-3751.2006
Haskell, G. T., Jensen, B. C., Samsa, L. A., Marchuk, D., Huang, W., Skrzynia, C., et al. (2017). Whole exome sequencing identifies truncating variants in nuclear envelope genes in patients with cardiovascular disease clinical perspective. Circ. Cardiovasc. Genet. 10:e001443. doi: 10.1161/CIRCGENETICS.116.001443
Helbling-Leclerc, A., Bonne, G., and Schwartz, K. (2002). Emery-dreifuss muscular dystrophy. Eur. J. Hum. Genet. 10, 157–161. doi: 10.1038/sj.ejhg.5200744
Herrmann, H., and Aebi, U. (2016). Intermediate filaments: structure and assembly. Cold Spring Harb. Perspect. Biol. 8:a018242. doi: 10.1101/cshperspect.a018242
Herrmann, H., Bär, H., Kreplak, L., Strelkov, S. V., and Aebi, U. (2007). Intermediate filaments: from cell architecture to nanomechanics. Nat. Rev. Mol. Cell Biol. 8, 562–573. doi: 10.1038/nrm2197
Ho, C. Y., Jaalouk, D. E., Vartiainen, M. K., and Lammerding, J. (2013). Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 497, 507–511. doi: 10.1038/nature12105
Holt, I., Duong, N. T., Zhang, Q., Lam, L. T., Sewry, C. A., Mamchaoui, K., et al. (2016). Specific localization of nesprin-1-α2, the short isoform of nesprin-1 with a KASH domain, in developing, fetal and regenerating muscle, using a new monoclonal antibody. BMC Cell Biol. 17:26. doi: 10.1186/s12860-016-0105-9
Horn, H. F., Brownstein, Z., Lenz, D. R., Shivatzki, S., Dror, A. A., Dagan-Rosenfeld, O., et al. (2013). The LINC complex is essential for hearing. J. Clin. Invest. 123, 740–750. doi: 10.1172/JCI66911
Izumi, Y., Miyamoto, R., Morino, H., Yoshizawa, A., Nishinaka, K., Udaka, F., et al. (2013). Cerebellar ataxia with SYNE1 mutation accompanying motor neuron disease. Neurology 80, 600–601. doi: 10.1212/WNL.0b013e3182815529
Jamin, A., and Wiebe, M. S. (2015). Barrier to autointegration factor (BANF1): interwoven roles in nuclear structure, genome integrity, innate immunity, stress responses and progeria. Curr. Opin. Cell Biol. 34, 61–68. doi: 10.1016/j.ceb.2015.05.006
Janin, A., Bauer, D., Ratti, F., Millat, G., and Méjat, A. (2017). Nuclear envelopathies: a complex LINC between nuclear envelope and pathology. Orphanet J. Rare Dis. 12:147. doi: 10.1186/s13023-017-0698-x
Janin, A., Bauer, D., Ratti, F., Valla, C., Bertrand, A., Christin, E., et al. (2018). SMAD6 overexpression leads to accelerated myogenic differentiation of LMNA mutated cells. Sci. Rep. 8:5618. doi: 10.1038/s41598-018-23918-x
Ketema, M., Kreft, M., Secades, P., Janssen, H., and Sonnenberg, A. (2013). Nesprin-3 connects plectin and vimentin to the nuclear envelope of Sertoli cells but is not required for Sertoli cell function in spermatogenesis. Mol. Biol. Cell 24, 2454–2466. doi: 10.1091/mbc.e13-02-0100
Ketema, M., Wilhelmsen, K., Kuikman, I., Janssen, H., Hodzic, D., and Sonnenberg, A. (2007). Requirements for the localization of nesprin-3 at the nuclear envelope and its interaction with plectin. J. Cell Sci. 120, 3384–3394. doi: 10.1242/jcs.014191
Kirby, T. J., and Lammerding, J. (2018). Emerging views of the nucleus as a cellular mechanosensor. Nat. Cell Biol. 20, 373–381. doi: 10.1038/s41556-018-0038-y
Koch, A. J., and Holaska, J. M. (2014). Emerin in health and disease. Semin. Cell Dev. Biol. 29, 95–106. doi: 10.1016/j.semcdb.2013.12.008
Lattanzi, G., Benedetti, S., Bertini, E., Boriani, G., Mazzanti, L., Novelli, G., et al. (2011). Laminopathies: many diseases, one gene. Report of the first Italian meeting course on laminopathies. Acta Myol. 30, 138–143.
Lindskog, C., Linné, J., Fagerberg, L., Hallström, B. M., Sundberg, C. J., Lindholm, M., et al. (2015). The human cardiac and skeletal muscle proteomes defined by transcriptomics and antibody-based profiling. BMC Genomics 16:475. doi: 10.1186/s12864-015-1686-y
Lombardi, M. L., and Lammerding, J. (2011). Keeping the LINC: the importance of nucleocytoskeletal coupling in intracellular force transmission and cellular function. Biochem. Soc. Trans. 39, 1729–1734. doi: 10.1042/BST20110686
Lu, D., Lian, H., Zhang, X., Shao, H., Huang, L., Qin, C., et al. (2010). LMNA E82K mutation activates FAS and mitochondrial pathways of apoptosis in heart tissue specific transgenic mice. PLoS One 5:e15167. doi: 10.1371/journal.pone.0015167
Maggi, L., D’Amico, A., Pini, A., Sivo, S., Pane, M., Ricci, G., et al. (2014). LMNA-associated myopathies: the Italian experience in a large cohort of patients. Neurology 83, 1634–1644. doi: 10.1212/WNL.0000000000000934
McKeon, F. D., Kirschner, M. W., and Caput, D. (1986). Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature 319, 463–468. doi: 10.1038/319463a0
Mellad, J. A., Warren, D. T., and Shanahan, C. M. (2011). Nesprins LINC the nucleus and cytoskeleton. Curr. Opin. Cell Biol. 23, 47–54. doi: 10.1016/j.ceb.2010.11.006
Menezes, M. P., Waddell, L. B., Evesson, F. J., Cooper, S., Webster, R., Jones, K., et al. (2012). Importance and challenge of making an early diagnosis in LMNA-related muscular dystrophy. Neurology 78, 1258–1263. doi: 10.1212/WNL.0b013e318250d839
Mercuri, E., Clements, E., Offiah, A., Pichiecchio, A., Vasco, G., Bianco, F., et al. (2010). Muscle magnetic resonance imaging involvement in muscular dystrophies with rigidity of the spine. Ann. Neurol. 67, 201–208. doi: 10.1002/ana.21846
Mercuri, E., Poppe, M., Quinlivan, R., Messina, S., Kinali, M., Demay, L., et al. (2004). Extreme variability of phenotype in patients with an identical missense mutation in the lamin A/C gene: from congenital onset with severe phenotype to milder classic Emery-dreifuss variant. Arch. Neurol. 61, 690–694. doi: 10.1001/archneur.61.5.690
Metzger, T., Gache, V., Xu, M., Cadot, B., Folker, E. S., Richardson, B. E., et al. (2012). MAP and kinesin-dependent nuclear positioning is required for skeletal muscle function. Nature 484, 120–124. doi: 10.1038/nature10914
Mislow, J. M. K., Holaska, J. M., Kim, M. S., Lee, K. K., Segura-Totten, M., Wilson, K. L., et al. (2002). Nesprin-1α self-associates and binds directly to emerin and lamin A in vitro. FEBS Lett. 525, 135–140. doi: 10.1016/S0014-5793(02)03105-8
Morel, V., Lepicard, S., Rey, A. N., Parmentier, M.-L., and Schaeffer, L. (2014). Drosophila nesprin-1 controls glutamate receptor density at neuromuscular junctions. Cell. Mol. Life Sci. 71, 3363–3379. doi: 10.1007/s00018-014-1566-7
Muchir, A., Bonne, G., van der Kooi, A. J., van Meegen, M., Baas, F., Bolhuis, P. A., et al. (2000). Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum. Mol. Genet. 9, 1453–1459. doi: 10.1093/hmg/9.9.1453
Muchir, A., Pavlidis, P., Decostre, V., Herron, A. J., Arimura, T., Bonne, G., et al. (2007). Activation of MAPK pathways links LMNA mutations to cardiomyopathy in emery-dreifuss muscular dystrophy. J. Clin. Invest. 117, 1282–1293. doi: 10.1172/JCI29042
Nigro, G., Russo, V., Rago, A., Papa, A. A., Carbone, N., Marchel, M., et al. (2012). Regional and transmural dispersion of repolarisation in patients with emery-dreifuss muscular dystrophy. Kardiol. Pol. 70, 1154–1159.
Noreau, A., Bourassa, C. V., Szuto, A., Levert, A., Dobrzeniecka, S., Gauthier, J., et al. (2013). SYNE1 mutations in autosomal recessive cerebellar ataxia. JAMA Neurol. 70, 1296–1301. doi: 10.1001/jamaneurol.2013.3268
Oldenburg, A., Briand, N., Sørensen, A. L., Cahyani, I., Shah, A., Moskaug, J. Ø., et al. (2017). A lipodystrophy-causing lamin A mutant alters conformation and epigenetic regulation of the anti-adipogenic MIR335 locus. J. Cell Biol. 216, 2731–2743. doi: 10.1083/jcb.201701043
Oldenburg, A. R., Delbarre, E., Thiede, B., Vigouroux, C., and Collas, P. (2014). Deregulation of Fragile X-related protein 1 by the lipodystrophic lamin A p.R482W mutation elicits a myogenic gene expression program in preadipocytes. Hum. Mol. Genet. 23, 1151–1162. doi: 10.1093/hmg/ddt509
Osorio, D. S., and Gomes, E. R. (2013). The contemporary nucleus: a trip down memory lane. Biol. Cell 105, 430–441. doi: 10.1111/boc.201300009
Packard, M., Jokhi, V., Ding, B., Ruiz-Cañada, C., Ashley, J., and Budnik, V. (2015). Nucleus to synapse nesprin1 railroad tracks direct synapse maturation through RNA localization. Neuron 86, 1015–1028. doi: 10.1016/j.neuron.2015.04.006
Padmakumar, V., Abraham, S., Braune, S., Noegel, A. A., Tunggal, B., Karakesisoglou, I., et al. (2004). Enaptin, a giant actin-binding protein, is an element of the nuclear membrane and the actin cytoskeleton. Exp. Cell Res. 295, 330–339. doi: 10.1016/j.yexcr.2004.01.014
Padmakumar, V. C., Libotte, T., Lu, W., Zaim, H., Abraham, S., Noegel, A. A., et al. (2005). The inner nuclear membrane protein Sun1 mediates the anchorage of Nesprin-2 to the nuclear envelope. J. Cell Sci. 118, 3419–3430. doi: 10.1242/jcs.02471
Paulsen, J., Sekelja, M., Oldenburg, A. R., Barateau, A., Briand, N., Delbarre, E., et al. (2017). Chrom3D: three-dimensional genome modeling from Hi-C and nuclear lamin-genome contacts. Genome Biol. 18:21. doi: 10.1186/s13059-016-1146-2
Peter, M., Kitten, G. T., Lehner, C. F., Vorburger, K., Bailer, S. M., Maridor, G., et al. (1989). Cloning and sequencing of cDNA clones encoding chicken lamins A and B1 and comparison of the primary structures of vertebrate A- and B-type lamins. J. Mol. Biol. 208, 393–404. doi: 10.1016/0022-2836(89)90504-4
Puckelwartz, M. J., Kessler, E., Zhang, Y., Hodzic, D., Randles, K. N., Morris, G., et al. (2009). Disruption of nesprin-1 produces an emery dreifuss muscular dystrophy-like phenotype in mice. Hum. Mol. Genet. 18, 607–620. doi: 10.1093/hmg/ddn386
Puckelwartz, M. J., Kessler, E. J., Kim, G., Dewitt, M. M., Zhang, Y., Earley, J. U., et al. (2010). Nesprin-1 mutations in human and murine cardiomyopathy. J. Mol. Cell. Cardiol. 48, 600–608. doi: 10.1016/j.yjmcc.2009.11.006
Quijano-Roy, S., Mbieleu, B., Bönnemann, C. G., Jeannet, P.-Y., Colomer, J., Clarke, N. F., et al. (2008). De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann. Neurol. 64, 177–186. doi: 10.1002/ana.21417
Rajgor, D., Mellad, J. A., Autore, F., Zhang, Q., and Shanahan, C. M. (2012). Multiple novel nesprin-1 and nesprin-2 variants act as versatile tissue-specific intracellular scaffolds. PLoS One 7:e40098. doi: 10.1371/journal.pone.0040098
Rajgor, D., and Shanahan, C. M. (2013). Nesprins: from the nuclear envelope and beyond. Expert Rev. Mol. Med. 15:e5. doi: 10.1017/erm.2013.6
Ramos, F. J., Chen, S. C., Garelick, M. G., Dai, D.-F., Liao, C.-Y., Schreiber, K. H., et al. (2012). Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med. 4:144ra103. doi: 10.1126/scitranslmed.3003802
Randles, K. N., Lam, L. T., Sewry, C. A., Puckelwartz, M., Furling, D., Wehnert, M., et al. (2010). Nesprins, but not sun proteins, switch isoforms at the nuclear envelope during muscle development. Dev. Dyn. 239, 998–1009. doi: 10.1002/dvdy.22229
Razafsky, D., and Hodzic, D. (2015). A variant of Nesprin1 giant devoid of KASH domain underlies the molecular etiology of autosomal recessive cerebellar ataxia type I. Neurobiol. Dis. 78, 57–67. doi: 10.1016/j.nbd.2015.03.027
Robson, M. I., de Las Heras, J. I., Czapiewski, R., Lê Thành, P., Booth, D. G., Kelly, D. A., et al. (2016). Tissue-specific gene repositioning by muscle nuclear membrane proteins enhances repression of critical developmental genes during myogenesis. Mol. Cell 62, 834–847. doi: 10.1016/j.molcel.2016.04.035
Roman, W., Martins, J. P., Carvalho, F. A., Voituriez, R., Abella, J. V. G., Santos, N. C., et al. (2017). Myofibril contraction and crosslinking drive nuclear movement to the periphery of skeletal muscle. Nat. Cell Biol. 19, 1189–1201. doi: 10.1038/ncb3605
Rønningen, T., Shah, A., Oldenburg, A. R., Vekterud, K., Delbarre, E., Moskaug, J. Ø., et al. (2015). Prepatterning of differentiation-driven nuclear lamin A/C-associated chromatin domains by GlcNAcylated histone H2B. Genome Res. 25, 1825–1835. doi: 10.1101/gr.193748.115
Roux, K. J., Crisp, M. L., Liu, Q., Kim, D., Kozlov, S., Stewart, C. L., et al. (2009). Nesprin 4 is an outer nuclear membrane protein that can induce kinesin-mediated cell polarization. Proc. Natl. Acad. Sci. U.S.A. 106, 2194–2199. doi: 10.1073/pnas.0808602106
Sakaki, M., Koike, H., Takahashi, N., Sasagawa, N., Tomioka, S., Arahata, K., et al. (2001). Interaction between emerin and nuclear lamins. J. Biochem. 129, 321–327. doi: 10.1093/oxfordjournals.jbchem.a002860
Sanger, J. W., Wang, J., Fan, Y., White, J., and Sanger, J. M. (2010). Assembly and dynamics of myofibrils. J. Biomed. Biotechnol. 2010:858606. doi: 10.1155/2010/858606
Segura-Totten, M., and Wilson, K. L. (2004). BAF: roles in chromatin, nuclear structure and retrovirus integration. Trends Cell Biol. 14, 261–266. doi: 10.1016/j.tcb.2004.03.004
Shackleton, S., Lloyd, D. J., Jackson, S. N., Evans, R., Niermeijer, M. F., Singh, B. M., et al. (2000). LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat. Genet. 24, 153–156. doi: 10.1038/72807
Shindo, Y., Kim, M.-R., Miura, H., Yuuki, T., Kanda, T., Hino, A., et al. (2010). Lrmp/Jaw1 is expressed in sweet, bitter, and umami receptor-expressing cells. Chem. Senses 35, 171–177. doi: 10.1093/chemse/bjp097
Shumaker, D. K., Dechat, T., Kohlmaier, A., Adam, S. A., Bozovsky, M. R., Erdos, M. R., et al. (2006). Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. U.S.A. 103, 8703–8708. doi: 10.1073/pnas.0602569103
Simpson, J. G., and Roberts, R. G. (2008). Patterns of evolutionary conservation in the nesprin genes highlight probable functionally important protein domains and isoforms. Biochem. Soc. Trans. 36, 1359–1367. doi: 10.1042/BST0361359
Solovei, I., Wang, A. S., Thanisch, K., Schmidt, C. S., Krebs, S., Zwerger, M., et al. (2013). LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 152, 584–598. doi: 10.1016/j.cell.2013.01.009
Starr, D. A., and Fridolfsson, H. N. (2010). Interactions between nuclei and the cytoskeleton are mediated by SUN-KASH nuclear-envelope bridges. Annu. Rev. Cell Dev. Biol. 26, 421–444. doi: 10.1146/annurev-cellbio-100109-104037
Staszewska, I., Fischer, I., and Wiche, G. (2015). Plectin isoform 1-dependent nuclear docking of desmin networks affects myonuclear architecture and expression of mechanotransducers. Hum. Mol. Genet. 24, 7373–7389. doi: 10.1093/hmg/ddv438
Stewart, C. L., and Burke, B. (2014). The missing LINC: a mammalian KASH-domain protein coupling meiotic chromosomes to the cytoskeleton. Nucleus 5, 3–10. doi: 10.4161/nucl.27819
Stierlé, V., Couprie, J., Ostlund, C., Krimm, I., Zinn-Justin, S., Hossenlopp, P., et al. (2003). The carboxyl-terminal region common to lamins A and C contains a DNA binding domain. Biochemistry 42, 4819–4828. doi: 10.1021/bi020704g
Stroud, M. J., Banerjee, I., Veevers, J., and Chen, J. (2014). Linker of nucleoskeleton and cytoskeleton complex proteins in cardiac structure, function, and disease. Circ. Res. 114, 538–548. doi: 10.1161/CIRCRESAHA.114.301236
Stroud, M. J., Feng, W., Zhang, J., Veevers, J., Fang, X., Gerace, L., et al. (2017). Nesprin 1α2 is essential for mouse postnatal viability and nuclear positioning in skeletal muscle. J. Cell Biol. 216, 1915–1924. doi: 10.1083/jcb.201612128
Stuurman, N., Heins, S., and Aebi, U. (1998). Nuclear lamins: their structure, assembly, and interactions. J. Struct. Biol. 122, 42–66. doi: 10.1006/jsbi.1998.3987
Synofzik, M., Smets, K., Mallaret, M., Di Bella, D., Gallenmüller, C., Baets, J., et al. (2016). SYNE1 ataxia is a common recessive ataxia with major non-cerebellar features: a large multi-centre study. Brain 139, 1378–1393. doi: 10.1093/brain/aww079
Tajik, A., Zhang, Y., Wei, F., Sun, J., Jia, Q., Zhou, W., et al. (2016). Transcription upregulation via force-induced direct stretching of chromatin. Nat. Mater. 15, 1287–1296. doi: 10.1038/nmat4729
Tapley, E. C., and Starr, D. A. (2013). Connecting the nucleus to the cytoskeleton by SUN-KASH bridges across the nuclear envelope. Curr. Opin. Cell Biol. 25, 57–62. doi: 10.1016/j.ceb.2012.10.014
Taranum, S., Vaylann, E., Meinke, P., Abraham, S., Yang, L., Neumann, S., et al. (2012). LINC complex alterations in DMD and EDMD/CMT fibroblasts. Eur. J. Cell Biol. 91, 614–628. doi: 10.1016/j.ejcb.2012.03.003
Tedesco, F. S., Dellavalle, A., Diaz-Manera, J., Messina, G., and Cossu, G. (2010). Repairing skeletal muscle: regenerative potential of skeletal muscle stem cells. J. Clin. Invest. 120, 11–19. doi: 10.1172/JCI40373
Tzatsos, A., Paskaleva, P., Lymperi, S., Contino, G., Stoykova, S., Chen, Z., et al. (2011). Lysine-specific demethylase 2B (KDM2B)-let-7-enhancer of zester homolog 2 (EZH2) pathway regulates cell cycle progression and senescence in primary cells. J. Biol. Chem. 286, 33061–33069. doi: 10.1074/jbc.M111.257667
Vadrot, N., Duband-Goulet, I., Cabet, E., Attanda, W., Barateau, A., Vicart, P., et al. (2015). The p.R482W substitution in A-type lamins deregulates SREBP1 activity in Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 24, 2096–2109. doi: 10.1093/hmg/ddu728
van Berlo, J. H., de Voogt, W. G., van der Kooi, A. J., van Tintelen, J. P., Bonne, G., Yaou, R. B., et al. (2005). Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J. Mol. Med. 83, 79–83. doi: 10.1007/s00109-004-0589-1
Vartiainen, M. K., Guettler, S., Larijani, B., and Treisman, R. (2007). Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science 316, 1749–1752. doi: 10.1126/science.1141084
Wang, S., Stoops, E., Cp, U., Markus, B., Reuveny, A., Ordan, E., et al. (2018). Mechanotransduction via the LINC complex regulates DNA replication in myonuclei. J. Cell Biol. 217, 2005–2018. doi: 10.1083/jcb.201708137
Wang, Y., Lichter-Konecki, U., Anyane-Yeboa, K., Shaw, J. E., Lu, J. T., Östlund, C., et al. (2016). A mutation abolishing the ZMPSTE24 cleavage site in prelamin A causes a progeroid disorder. J. Cell Sci. 129, 1975–1980. doi: 10.1242/jcs.187302
Wilhelmsen, K., Litjens, S. H. M., Kuikman, I., Tshimbalanga, N., Janssen, H., van den Bout, I., et al. (2005). Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J. Cell Biol. 171, 799–810. doi: 10.1083/jcb.200506083
Wilson, M. H., and Holzbaur, E. L. F. (2015). Nesprins anchor kinesin-1 motors to the nucleus to drive nuclear distribution in muscle cells. Dev. Camb. Engl. 142, 218–228. doi: 10.1242/dev.114769
Winter, L., Kuznetsov, A. V., Grimm, M., Zeöld, A., Fischer, I., and Wiche, G. (2015). Plectin isoform P1b and P1d deficiencies differentially affect mitochondrial morphology and function in skeletal muscle. Hum. Mol. Genet. 24, 4530–4544. doi: 10.1093/hmg/ddv184
Wu, L., Xiang, B., Zhang, H., He, X., Shih, C., Chen, X., et al. (2017). Three novel recessive mutations in LAMA2, SYNE1, and TTN are identified in a single case with congenital muscular dystrophy. Neuromuscul. Disord. 27, 1018–1022. doi: 10.1016/j.nmd.2017.06.558
Yang, L., Munck, M., Swaminathan, K., Kapinos, L. E., Noegel, A. A., and Neumann, S. (2013). Mutations in LMNA modulate the lamin A - nesprin-2 interaction and cause LINC complex alterations. PLoS One 8:e71850. doi: 10.1371/journal.pone.0071850
Yin, H., Price, F., and Rudnicki, M. A. (2013). Satellite cells and the muscle stem cell niche. Physiol. Rev. 93, 23–67. doi: 10.1152/physrev.00043.2011
Yu, J., Starr, D. A., Wu, X., Parkhurst, S. M., Zhuang, Y., Xu, T., et al. (2006). The KASH domain protein MSP-300 plays an essential role in nuclear anchoring during drosophila oogenesis. Dev. Biol. 289, 336–345. doi: 10.1016/j.ydbio.2005.10.027
Yu, T. W., Chahrour, M. H., Coulter, M. E., Jiralerspong, S., Okamura-Ikeda, K., Ataman, B., et al. (2013). Using whole-exome sequencing to identify inherited causes of autism. Neuron 77, 259–273. doi: 10.1016/j.neuron.2012.11.002
Yuan, J., Simos, G., Blobel, G., and Georgatos, S. D. (1991). Binding of lamin A to polynucleosomes. J. Biol. Chem. 266, 9211–9215.
Zhang, J., Felder, A., Liu, Y., Guo, L. T., Lange, S., Dalton, N. D., et al. (2010). Nesprin 1 is critical for nuclear positioning and anchorage. Hum. Mol. Genet. 19, 329–341. doi: 10.1093/hmg/ddp499
Zhang, Q. (2005). Nesprin-2 is a multi-isomeric protein that binds lamin and emerin at the nuclear envelope and forms a subcellular network in skeletal muscle. J. Cell Sci. 118, 673–687. doi: 10.1242/jcs.01642
Zhang, Q., Bethmann, C., Worth, N. F., Davies, J. D., Wasner, C., Feuer, A., et al. (2007a). Nesprin-1 and -2 are involved in the pathogenesis of emery dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum. Mol. Genet. 16, 2816–2833. doi: 10.1093/hmg/ddm238
Zhang, Q., Skepper, J. N., Yang, F., Davies, J. D., Hegyi, L., Roberts, R. G., et al. (2001). Nesprins: a novel family of spectrin-repeat-containing proteins that localize to the nuclear membrane in multiple tissues. J. Cell Sci. 114, 4485–4498.
Zhang, X., Xu, R., Zhu, B., Yang, X., Ding, X., Duan, S., et al. (2007b). Syne-1 and Syne-2 play crucial roles in myonuclear anchorage and motor neuron innervation. Development 134, 901–908. doi: 10.1242/dev.02783
Zhong, Z., Chang, S. A., Kalinowski, A., Wilson, K. L., and Dahl, K. N. (2010). Stabilization of the spectrin-like domains of nesprin-1α by the evolutionarily conserved “Adaptive” domain. Cell. Mol. Bioeng. 3, 139–150. doi: 10.1007/s12195-010-0121-3
Zhou, C., Li, C., Zhou, B., Sun, H., Koullourou, V., Holt, I., et al. (2017). Novel nesprin-1 mutations associated with dilated cardiomyopathy cause nuclear envelope disruption and defects in myogenesis. Hum. Mol. Genet. 26, 2258–2276. doi: 10.1093/hmg/ddx116
Keywords: LINC complex, nuclear envelope, lamins A/C, LMNA, nesprins, human diseases
Citation: Janin A and Gache V (2018) Nesprins and Lamins in Health and Diseases of Cardiac and Skeletal Muscles. Front. Physiol. 9:1277. doi: 10.3389/fphys.2018.01277
Received: 29 June 2018; Accepted: 22 August 2018;
Published: 07 September 2018.
Edited by:
Julien Ochala, King’s College London, United KingdomReviewed by:
Tyler John Kirby, Cornell University, United StatesJacob Alexander Ross, King’s College London, United Kingdom
Copyright © 2018 Janin and Gache. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandre Janin, YWxleGFuZHJlLmphbmluMDFAY2h1LWx5b24uZnI= Vincent Gache, dmluY2VudC5nYWNoZUBpbnNlcm0uZnI=