 Carolina Jaquenod De Giusti
Carolina Jaquenod De Giusti Barbara Roman2
Barbara Roman2 Samarjit Das
Samarjit Das- 1Centro de Investigaciones Cardiovasculares CIC-CONICET, Facultad de Ciencias Médicas, Universidad Nacional de La Plata, La Plata, Argentina
- 2Department of Pathology, Johns Hopkins University, Baltimore, MD, United States
Abnormal mitochondrial calcium ([Ca2+]m) handling and energy deficiency results in cellular dysfunction and cell death. Recent studies suggest that nuclear-encoded microRNAs (miRNA) are able to translocate in to the mitochondrial compartment, and modulate mitochondrial activities, including [Ca2+]m uptake. Apart from this subset of miRNAs, there are several miRNAs that have been reported to target genes that play a role in maintaining [Ca2+]m levels in the cytoplasm. It is imperative to validate miRNAs that alter [Ca2+]m handling, and thereby alter cellular fate. The focus of this review is to highlight the mitochondrial miRNAs (MitomiRs), and other cytosolic miRNAs that target mRNAs which play an important role in [Ca2+]m handling.
Introduction
One of the cell's major sources of energy comes from mitochondria. However, the role of mitochondria is not limited only to ATP generation; mitochondria participate and control numerous metabolic pathways and signaling cascades, redox balance, β-oxidation of fatty acids, the synthesis of amino acids, heme and steroids, and cellular apoptosis. The ability of mitochondria to act as Ca2+ buffer has important consequences on the pattern of the [Ca2+]cyto signals. In addition, mitochondria are also a major storage hub of cellular calcium (Ca2+), and Ca2+ homeostasis is fundamental for a wide range of cellular activities such as control of oxidative phosphorylation (OXPHOS), modulation of cytosolic Ca2+ ([Ca2+]cyto) signals, cell death, secretion, and the production of reactive oxygen species (ROS) (Mammucari et al., 2018). Approximately 50 years ago Engstrom et al., demonstrated that mitochondria were able to take up Ca2+(Deluca and Engstrom, 1961), which validated the observation made by Slater and Cleland as early as 1953. In this report, the authors evaluated the effect of Ca2+ on respiratory activity of heart sarcosomes (previous nomenclature of mitochondria) (Slater and Cleland, 1953). This work is the basis of future research in the area of Ca2+-mediated regulation of mitochondrial bioenergetics and mitochondrial calcium ([Ca2+]m) uptake. Additional studies have demonstrated a clear role for [Ca2+]m as a pleiotropic signal that regulates many essential functions of cellular physiology. [Ca2+]m concentration is about 10−7 M and it is the result of the dynamic equilibrium between two continuous and opposite processes: mitochondrial Ca2+ influx vs. efflux (Granatiero et al., 2017).
The functional relevance of [Ca2+]m in energy processes has been shown in studies that reflect a positive effect on the tricarboxylic acid (TCA) cycle that increases the synthesis of reduced substrates (NADH and FADH2), enhances electron transport chain (ETC) activity, and subsequently increases H+ pumping (Rizzuto et al., 2012). Three mitochondrial matrix dehydrogenases are activated by Ca2+: α-ketoglutarate (α-KGDH) and isocitrate-dehydrogenases (ICDH) are regulated by direct binding of Ca2+, while pyruvate dehydrogenase (PDH) is regulated by a Ca2+-dependent phosphatase (PDP1) (Glancy and Balaban, 2012; Das et al., 2014). Stimulation of these dehydrogenases increases NADH and FADH2 availability, which is not only the central mechanism to provide electron carriers for ATP production, but also important for regenerating the antioxidative capacity of the mitochondrial matrix (Nickel et al., 2014). In addition, Ca2+ activates α-glycerolphosphate dehydrogenase, a component of the glycerol phosphate shuttle that supplies NAD+ for glycolysis (Wernette et al., 1981). Apart from these proteins, [Ca2+]m can also modulate the adenine nucleotide transporter (Mildaziene et al., 1995) and the mitochondrial ATP synthase (complex V) expression (Das and Harris, 1990), harnessing the H+ gradient to upregulate ATP production.
It is generally understood that ROS production occurs through complexes I and III of the electron transport chain, and by NADPH-oxidase (NOX 4) (Ago et al., 2010). Additionally, ROS can also be produced by β-oxidation of fatty acids (Perevoshchikova et al., 2013). It has also been demonstrated that ROS production is dependent on the activity of α-glycerolphosphate dehydrogenase (Zorov et al., 2014). ROS production is directly related to increased [Ca2+]m by the activation of metabolic fluxes through the TCA cycle, and increased oxygen consumption by the ETC (Hempel and Trebak, 2017). In fact, there is reciprocal regulation between ROS and [Ca2+]m signaling, and calcium signaling is essential for ROS production through activation of mitochondrial dehydrogenases in the ETC (Görlach et al., 2015). On the other hand, under physiological conditions, a small amount of H2O2 release from mitochondria acts as a signal to stimulate physiological Ca2+ release from endoplasmisc reticulum (ER). This happens via the ryanodine receptors (RyR) which increases [Ca2+]m uptake to increase ATP production to meet cellular energetic demands (Camara et al., 2010; Görlach et al., 2015).
Finally, [Ca2+]m also stimulates cell death pathways, such as apoptosis and necrosis, via activating the intrinsic pathway. [Ca2+]m overload or mitochondrial depolarization can open the mitochondrial permeability transition pore (mPTP) (Rasola and Bernardi, 2011). The mPTP is formed by interactions between IMM and OMM proteins such as VDAC1, ANT (adenine nucleotide translocase), and Cyp-D (cyclophilin D), however, deletion of each one of these proteins does not block the permeability increase caused by some inducers, indicating that the mPTP may not be formed by specific proteins, but just by the interaction between both membranes (Tomasello et al., 2009). While briefopening and closing of mPTP (more commonly known as “mitochondrial flickering”) can help to transient transport [Ca2+]m, complete mPTP openings can result in release of proapoptotic factors and [Ca2+]m resulting in apoptosis (Bernardi et al., 2015). When the stress signal results in simultaneous mPTP opening from several mitochondria, ATP depletion would result in necrosis (Rasola and Bernardi, 2011). Therefore, it is quite evident that [Ca2+]m plays an important role in both the necrotic and apoptotic cell death pathways. The role of [Ca2+]m and ROS in apoptosis is particularly important considering that a small amount of cytochrome c released by mitochondria can bind and promote the opening of inositol triphosphate (IP3R) channels. Activated IP3R can release Ca2+ from the ER, which increases [Ca2+]cyto. Consequently, this cycle further leads to a massive release of cytochrome c, and activation of caspases and apoptosis (Boehning et al., 2003).
Mitochondrial Calcium Transport
The mitochondrion is an organelle formed by two different phospholipid bilayers, an outer mitochondrial membrane (OMM) that allows the entrance of ions and small proteins (MW < 10 kDa) and an inner mitochondrial membrane (IMM), which is ion-impermeable (Mammucari et al., 2018). Calcium enters the mitochondria through voltage dependent anion channels (VDAC) in the OMM (Shoshan-Barmatz et al., 2018) and through the mitochondrial calcium uniporter (MCU) complex from the IMM using the mitochondrial membrane potential (ΔΨm around −150 to −180 mV) as the driving force generated by the electron transport chain (Liu et al., 2017). MCU has high Ca2+ affinity and selectivity, enabling high Ca2+ transport in spite of low [Ca2+]cyto (Kirichok et al., 2004; Baughman et al., 2011; De Stefani et al., 2011). The MCU complex is formed by MCU core proteins and several regulatory proteins such as MCUb, EMRE, MICU1, MICU2, and MCUR1 (Mallilankaraman et al., 2012; Hajnóczky et al., 2014; Tomar et al., 2016). Calcium release is mediated by the Na+/Ca2+ exchanger (NCLX) (Palty et al., 2010) and through the H+/Ca2+ exchanger (HCX) in tissues where mitochondrial NCLX activity is low, such as the liver, kidney, lung, and smooth muscle (Takeuchi et al., 2015). HCX is also known as LETM1 (the leucine zipper-EF hand-containing transmembrane protein 1), but its role as an H+/Ca2+ exchanger or a K+/H+ exchanger is still controversial (Jiang et al., 2009; Austin et al., 2017). NCLX activity can be regulated by post-translational modifications, such as phosphorylation. Alterations in phosphorylation status in NCLX result in its decreased activity, increased [Ca2+]m overload, partial mitochondrial depolarization and increased oxidative stress (Kostic et al., 2015).
The Role of the Endoplasmic Reticulum
The ER is the main Ca2+ storage location in the cell. There are overlapping regions between the ER and mitochondria where highly efficient Ca2+ transfer can occur (Patergnani et al., 2011). Various pathophysiological conditions increase the cross-talk between ER and mitochondria. The VDAC-IP3R interaction is important for [Ca2+]m accumulation (Szabadkai et al., 2006). Increased sarcoplasmic reticulum (SR)/ER calcium leakage through RyR or IP3R channels has been implicated in mitochondrial calcium overload (Santulli et al., 2015; Ivanova et al., 2017). In fact, RyR or IP3R oxidation by mitochondrial ROS increases ER Ca2+ leakage and leads to higher [Ca2+]m entry (Camara et al., 2010). Protein kinase A (PKA) (Banerjee and Ghosh, 2006; Gupta, 2017) and GSK-3β (Das et al., 2008) can regulate VDAC activity by regulating the phosphorylation status of VDAC. Similarly, another OMM translocator protein of 18 kDa size, TSPO, has been shown to alter [Ca2+]m intake. TSPO is capable of complexing with PKA and Acyl-CoA binding domain containing 3 (ACBD3), recruiting them to the OMM, which leads to the phosphorylation of VDAC (Gatliff et al., 2017). Additionally, it has been shown that transient increases in VDAC expression result in increased [Ca2+]m intake (Rapizzi et al., 2002).
The Role of the MCU Complex
The MCU, a 40 kDa protein, has two transmembrane domains that need additional interactions to form a functional channel. It lacks a Ca2+ sensing domain (EF-hand domain), indicating that Ca2+ regulation capacity is not mediated by MCU (Mammucari et al., 2018). It has been shown that phosphorylation by CaMKII can modulate MCU activity (Joiner et al., 2012) as well as other proteins involved in the complex (Mammucari et al., 2018). MCU expression and activity may differ between tissues. The highest MCU mRNA expression levels are seen in skeletal muscle, followed by the liver, heart, spleen, lung, kidney, brain, and white fat (Raffaello et al., 2013). MCU activity measured by patch clamp in isolated mitochondria from mouse hearts, skeletal muscle, liver, kidney, and brown fat showed that heart mitochondria exhibited the lowest MCU current. However, this diminished current could be compensated for by the high numbers of mitochondria in cardiac muscle (Fieni et al., 2012).
MCUb is a 330 amino acid long protein that shares 50% similarity with MCU (Raffaello et al., 2013). By studying the MCU:MCUb interaction using FRET analysis and immunoprecipitation (IP), Raffaello et al., showed that co-expression of MCUb with MCU drastically diminishes MCU activity. (Raffaello et al., 2013).
EMRE (Essential MCU Regulator) is a 10 kDa protein with a mitochondrial target sequence at the 3'-end. Using in vitro models such as HEK293T and HeLa cells, the interaction between EMRE and MCU was identified. Silencing EMRE abolishes [Ca2+]m uptake (Sancak et al., 2013). Furthermore, EMRE also facilitates the interaction between MICU1 and MICU2 with MCU. Cells that lack EMRE have no associations between MCU and MICU1 or MICU2, while the MCU:MCUb or MICU1:MICU2 interactions are not affected (Sancak et al., 2013). Unlike MICU1 or MICU2, EMRE expresses a different Ca2+ sensing domain directed towards the mitochondrial matrix. Therefore, EMRE modulates MCU activity via [Ca2+]m levels (Sancak et al., 2013). In fact, EMRE mutation at the Ca2+ sensing acidic motif does not affect MCU complex formation, but results in increased [Ca2+]m (Kameyama and Gemba, 1991). It is also important to note that the modulatory effect of EMRE also depends on the [Ca2+]cyto binding affinity to the MICU1:MICU2 complex (Kameyama and Gemba, 1991). In summary, EMRE plays an important physical and modulatory role in the formation and activity of the MCU complex.
MICU1 has two Ca2+ sensing EF-hand motifs directed toward the intermembrane space (IMS). It has been demonstrated that MICU1 inhibits [Ca2+]m entry at low resting [Ca2+]cyto concentrations (within the range found in cells at rest), protecting mitochondria from [Ca2+]m overload (Mallilankaraman et al., 2012; Csordás et al., 2013; Liu et al., 2016). On the other hand, MICU1 allows for [Ca2+]m uptake when [Ca2+]cyto concentrations increase to the micromolar range (Csordás et al., 2013). This would help mitochondria to rapidly respond to large [Ca2+]cyto increases (like the one from systolic release). Interestingly, results from human skin fibroblasts with MICU1 mutations confirmed the gatekeeper role of MICU1 at low [Ca2+]cyto, but also showed increased [Ca2+]m uptake at higher [Ca2+]cyto (Logan et al., 2014). In contrast, MICU2 has been shown to form a complex with MCU in the presence of MICU1. MICU2 follows the same pattern of [Ca2+]cyto threshold sensing as MICU1. MICU2 knock-out cells showed higher [Ca2+]m uptake at lower [Ca2+]cyto (Kamer and Mootha, 2014). Similarly, using planar lipid bilayers, Patron et al., concluded that MICU2 plays a gatekeeping role for MCU. MICU2 alters the ratio between MICU1:MICU2, which allows for the formation of MICU1-MICU1 dimers, which is responsible for the [Ca2+]m uptake rate (Patron et al., 2014). Taken together, these MCU regulatory proteins regulate the rate of [Ca2+]m entry in response to different [Ca2+]cyto levels. The relative expression of these MCU regulatory proteins in different tissues, such as the heart, brain, and skeletal muscle, confers different mitochondrial calcium handling capabilities (Paillard et al., 2017).
Mitochondrial Calcium Overload in the Pathophysiology of Disease States
[Ca2+]m overload and diminished [Ca2+]m have both been linked to the pathophysiology of different disease states. Particularly, high [Ca2+]m load due to increased SR leakage through RyR channels has been related to ROS production, which ultimately leads to heart failure (Santulli et al., 2015). Human disease conditions, such as proximal myopathy, learning difficulties, and a progressive extrapyramidal movement disorder, have also been linked to [Ca2+]m overload (Logan et al., 2014). Similarly, it has been validated that [Ca2+]m overload is responsible for excessive ROS production and mitochondrial depolarization, which results in neuronal death (Saffari et al., 2017). This mechanism is one of the highlights of Parkinson disease progression (Kostic et al., 2015; Ammal Kaidery and Thomas, 2018; Ludtmann and Abramov, 2018).
Influence of [Ca2+]m in Cancer
In order to meet the energy demand, cancer cells activate mitochondrial metabolism. This increased metabolism is achieved by activation of the TCA cycle dehydrogenases through increased [Ca2+]m entry. Considering the up-regulation of both IP3R and MCU in several types of cancer cells, it seems that ER/mitochondria communication is key for increased mitochondrial metabolism and cancer progression (Bustos et al., 2017). VDAC1 expression is related to poor prognosis in breast, colon, and lung cancers, where it has been demonstrated that VDAC1 promotes cell growth by influencing energy metabolism and inhibiting apoptosis (Mazure, 2017; White, 2017). Additionally, pulmonary arterial cancer cells and colon cancer cells avoid [Ca2+]m overload through downregulation of MCU, which confers resistance to cell death (Liu et al., 2017). Different oncogenes, such as Ras and Akt, have been shown to regulate apoptosis through modulation of [Ca2+]m entry, and thus, inhibition of [Ca2+]m overload (Rimessi et al., 2015). In the same manner, the known tumor suppressor gene, p53, is capable of interacting with sarco/endoplasmic reticulum Ca2+-ATPase (SERCA). p53 has been shown to influence the oxidative state of SERCA, and thus increase SERCA activity in the ER/mitochondria associated membranes (MAM) fraction. This is related to increased ER calcium release that results in [Ca2+]m overload; a priming step for the release of caspase cofactors—and induction of apoptosis (Giorgi et al., 2015; Rimessi et al., 2015). Similarly, the tumor suppressor gene, Fhit, increases MCU affinity toward Ca2+ and induces apoptosis (Rimessi et al., 2009). Based on this, drugs that increase [Ca2+]m entry through ER transfer and/or augmented Ca2+ influx can be used for cancer therapy (Rimessi et al., 2015). However, a strict balance in [Ca2+]m homeostasis is involved in cancer as Ca2+ influx is also needed for cellular energy production and cancer progression (Bustos et al., 2017).
Influence of [Ca2+]m in Cardiovascular Health
Alterations in calcium handling and the processes regulated by Ca2+ have been documented in several cardiovascular diseases (Griffiths, 2009; Kim et al., 2014; Santulli et al., 2015). Santulli et al., showed increased [Ca2+]m in a heart failure model by permanent occlusion of the proximal left anterior descending (LAD) coronary artery (Santulli et al., 2015). In fact, heart failure is characterized by abnormal [Ca2+]m handling and poor energy production, which ultimately leads to contractile dysfunction and myocyte death. This increased [Ca2+]m concentration was related to mitochondrial dysfunction seen as membrane depolarization, reduced ATP production, and ROS generation. In ischemia/reperfusion (IR) injury, both myocytes and whole hearts are characterized by increased [Ca2+]m uptake resulting in excessive ROS production and mPTP opening, which ultimately leads to cell death (Ferrari, 1996; Griffiths, 2009; Kim et al., 2014).
Introduction to Mitochondrial microRNAs
Understanding some of the key cellular abnormalities in various disease conditions and developing specific therapies toward reversing these abnormalities can improve the disease state. microRNAs (miRNA) represent a large set of master regulators of gene expression, which can greatly influence cell death. The binding of miRNAs to their targeted mRNAs can result in translational repression and/or degradation of target genes. These miRNAs are predicted to regulate almost 30–40% of mammalian genes (Kim, 2005; Lewis et al., 2005).
With recent advancements in RNA detecting platforms, we now can detect miRNAs in the mitochondrial compartment of cells. The subset of miRNAs, which translocate into the mitochondria and are functionally active, i.e., RNA-induced silencing complex, or RISC, is inside the mitochondrial compartment, are termed mitochondrial miRNA or MitomiR. The mitochondrial miRNA translocation phenomenon has not been extensively studied (Macgregor-Das and Das, 2018), but MitomiRs can influence various mitochondrial functions, such as the TCA, ETC, lipid metabolism, and amino acid metabolism (Baradan et al., 2017). It has been demonstrated that MitomiRs can post-translationally regulate gene function inside the mitochondrial fraction either by targeting a nuclear-encoded gene present in the mitochondria or by targeting a mitochondrial encoded-gene (Das et al., 2012, 2014, 2017). Interestingly, it has been documented that overexpression of a MitomiR, miR-181c, in the heart can influence [Ca2+]m handling, highlighting the role of miR-181c in heart failure (Das et al., 2014). Thus, in this article, we have focused on the miRNAs that have been found in the mitochondrial compartment, and target either mitochondrial or nuclear genes, which ultimately alters [Ca2+]m handling and cellular fate in different disease conditions.
The Regulation of the MCU Complex Proteins by MicroRNAs
Bargaje et al., showed a role for miR-34a and miR-29a in apoptosis by targeting the 3′-UTR of VDAC1 mRNA in HEK-293 cells. These miRNAs are dowregulated in hepatocellular carcinoma and ovarian cancer. However, [Ca2+]m levels were not documented in this study (Bargaje et al., 2012). Furthermore, miR-7 has been shown to reduce VDAC1 expression in SH-SY5Y neuroblastoma cells and mouse primary cortical neurons. This diminishes efflux, and reduces ROS production, membrane depolarization, cytochrome c release, and apoptosis, all of which are factors involved in the development of Parkinson's disease (PD) (Chaudhuri et al., 2016). Due to its effects on [Ca2+]m, miR-7 could potentially be used as a therapeutic agent in the treatment of Parkinson's disease. Importantly, miR-7 expression has never been measured in PD's patients.
It was proposed from in silico analysis that five miRNAs, miR-15, miR-17, miR-21, miR-25, and miR-137, could target MCU and/or MICU1 in tumor cells (Marchi et al., 2013). However, only miR-25 was able to reduce [Ca2+]m levels in HeLa cells through MCU downregulation (Marchi et al., 2013). In both human colon cancer and prostate cancer, the up-regulation of miR-25 and down-regulation of MCU protein have been observed. Interestingly, the authors performed biochemical assays to demonstrate that miR-25 can directly bind to the 3' UTR of MCU mRNA (Marchi et al., 2013). Treating with antagomiR-25 can increase [Ca2+]m entry and apoptosis (Marchi et al., 2013). In contrast, in breast cancer the downregulation of miR-340 is correlated with increased MCU expression. Targeting MCU, miR-340 increases the rate of glycolysis, a metabolic shift known as the Warburg effect, which promotes cell migration and invasion (Yu et al., 2017). Pulmonary arterial hypertension (PAH) is characterized by excessive pulmonary artery smooth muscle cell proliferation, migration, and apoptosis resistance, resulting in a cancer-like phenotype (Hong et al., 2017). In this model, Hong et al., showed decreased expression of MCU which resulted in elevation of [Ca2+]cyto and reduction in [Ca2+]m. This was associated with increased miR-25 and miR-138 expression. Treatment with antagomiR against both miR-25 and miR-138 can restore MCU expression levels and reverse the pathophysiology of PAH (Hong et al., 2017).
Overexpression of miR-25 in the heart protects myocytes from oxidative damage by reducing [Ca2+]m levels through downregulation of MCU (Pan et al., 2015). On the other hand, it has been observed that miR-181c can directly target the mitochondrial encoded-gene, mt-COX1, which leads to increased ΔΨm, which could be related to increased [Ca2+]m influx and ROS production (Das et al., 2014). Zaglia et al., demonstrated a role for muscle-specific miRNAs (myomiR), such as miR-1 and miR-206, in heart tissue. This group successfully showed that miR-1 can directly regulate MCU expression during development in both mouse and human cardiac myocytes (Zaglia et al., 2017). During the neonatal phase, i.e., the first few days after birth, mitochondria are disorganized inside cells. Therefore, in this state the distance between mitochondria and ER or SR is far apart, and any Ca2+ release from the ER and/or SR could not influence [Ca2+]m influx easily. However, by 7–14 days after birth, the membrane system matures, putting the ER, and/or SR closer to the mitochondrial membrane (Griffiths et al., 2010). In this phase, mitochondria are now exposed to ER and/SR high Ca2+ microdomains, i.e., Ca2+ release from the ER and/or SR influences [Ca2+]m influx rapidly. Based on the intracellular location of mitochondria during the neonatal phase, miR-1 upregulation could protect mitochondria from [Ca2+]m overload. This is due to the fact that miR-1 directly targets the mRNA of MCU (Zaglia et al., 2017). In contrast, during physiological and pathological hypertrophy where the ATP consumption rate is higher, downregulation of miR-1, and increased MCU levels were observed (Zaglia et al., 2017). Figure 1 summarizes the role of various miRNAs in the in-take of mitochondrial Ca2+.
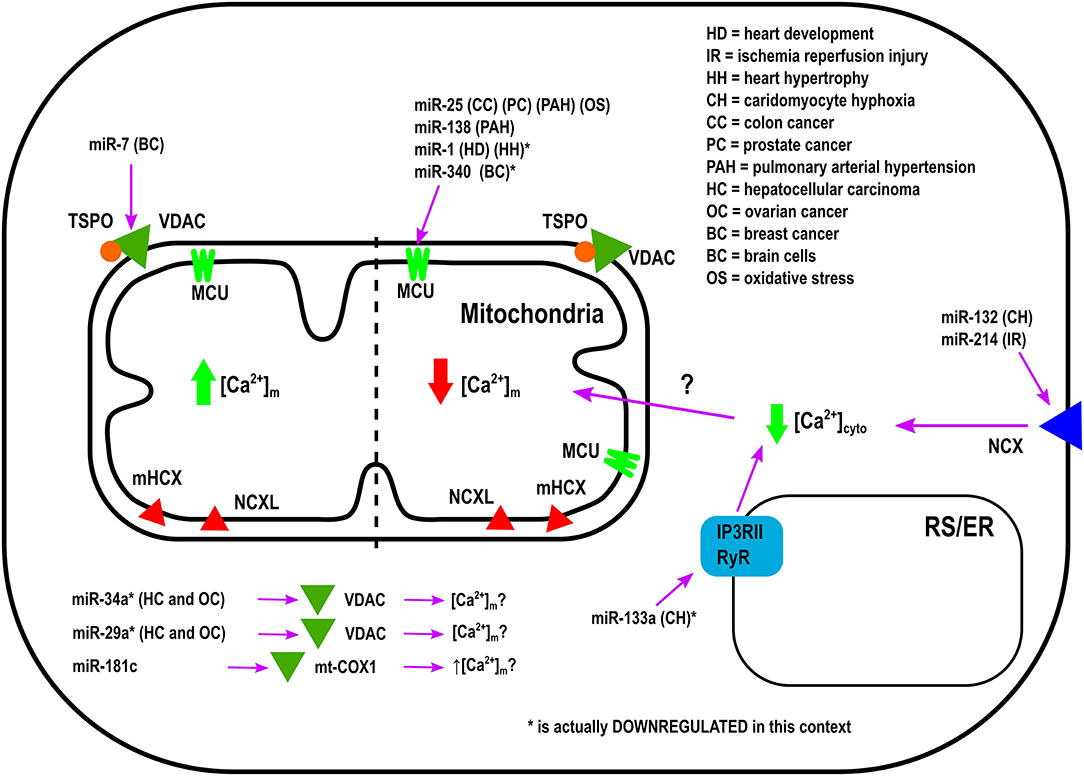
Figure 1. Mechanism of Action by which miRNAs regulate Ca2+-intake into the Mitochondria.
Other MicroRNAs Involved in Calcium Regulation
Other miRNAs have been shown to indirectly regulate [Ca2+]m overload and/or apoptosis. For example, during hypoxic conditions downregulation of miR-132 leads to increased Na+-Ca2+ exchanger 1 (NCX1) expression. Using rat cardiomyocytes it has been documented that miR-132 could influence [Ca2+]m overload by binding to the 3′-UTR of NCX1 mRNA and modulating [Ca2+]cyto (Hong et al., 2015). To release the intracellular H+ accumulation Na+-H+ exchanger (NHE) gets activated. Activated NHE release the H+ from the cell at the cost of Na+ overload. NCX1 plays an important role in releasing the Na+ from the cells to alleviate the intracellular Na+-overload. In cardiac hypertrophy, downregulation of miR-133a has been observed. This leads to higher IP3RII expression and increased [Ca2+]cyto, resulting in left ventricular remodeling and contractile dysfunction (Drawnel et al., 2012). On the contrary, miR-214, which increases its expression during ischemia/reperfusion (I/R) insult, has been shown to protect the heart from Ca2+ overload, heart failure, and apoptosis through targeting NCX1 (Aurora et al., 2012). Multiple pathophysiological conditions, such as hypertrophy and I/R, upregulate NCX1 in the heart (Sipido et al., 2002). Therefore, down-regulation of NCX1 can potentially be a therapeutic intervention from these pathophysiological conditions by reducing [Ca2+]cyto overload and cell death. As it has been shown that miR-214 can directly bind to NCX1 mRNA, increased miR-214 expression can protect the heart from MI and I/R-stress (Aurora et al., 2012). Indeed, it has been observed that genetic deletion of miR-214 in mice results in impaired cardiac function and susceptibility to I/R injury (Aurora et al., 2012). After I/R, miR-214 significantly reduces [Ca2+]cyto overload and protects cardiomyocytes from increased oxidative stress and cell death by reducing NCX1 activity. All of these effects are directly related to [Ca2+]m (Aurora et al., 2012). Additionally, overexpression of miR-145 in rat ventricular cardiomyocytes reduces Ca2+/Calmodulin-Dependent Protein Kinase II (CaMKII) expression (together with other Ca2+ handling proteins), protecting myocytes from apoptosis induced by H2O2 (Cha et al., 2013).
As illustrated in Table 1, the miRNAs which are not directly involved in [Ca2+]m handling, but they play an important role in buffering [Ca2+]cyto (Shoshan-Barmatz et al., 2018). It has been shown that the alterations in [Ca2+]cyto could influence [Ca2+]m influx, either directly by influencing MCU, or indirectly via other MCU regulatory proteins, such as MICU1. Therefore, one miRNA can influence [Ca2+]m entry by forming the RISC inside the mitochondria or at the cytoplasm. If the RISC is inside the mitochondrial compartment, then that miRNA will be termed as a “MitomiR.”
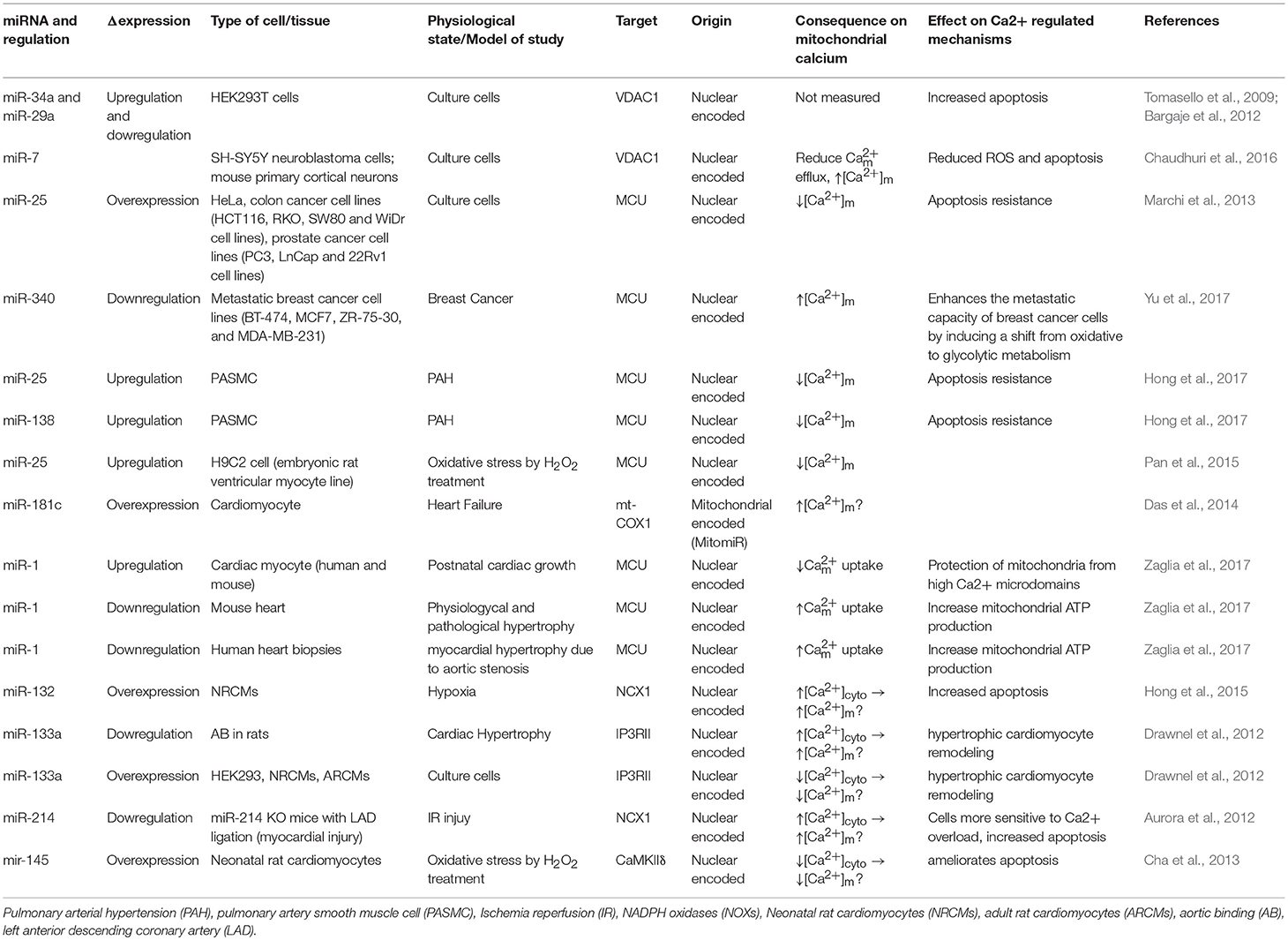
Table 1. List of miRNAs which regulate [Ca2+]m.
Concluding Remarks
[Ca2+]m is regulated by its influx and efflux through VDAC, the MCU complex, NCXL, and NHX. Therefore, the regulation of these Ca2+-sensing proteins can determine the fate of cellular activity. Recent studies have highlighted the importance of MitomiR on [Ca2+]m handling and mitochondrial function, making these MitomiRs attractive as therapeutic targets. While there is still much work to be done with regards to miRNA import into the mitochondria, several groups have documented that MitomiRs can regulate [Ca2+]m entry. The potential for miRNA therapeutics is limitless, and could be applied to any disease, including Alzheimers, Parkinson's, cancer, and heart failure, which are characterized by alterations in their MitomiR profiles. Indeed, it is true that when researchers across the world tried to regulate [Ca2+]m by knocking down MCU protein in a mouse model, there were very conflicting results. In some studies, cardio-specific MCU knock-out proved detrimental against various stresses, such as ischemia/reperfusion injury. On the other hand, global MCU knock-out mice showed a minimal functional effect. MCU knock-out mice are slightly smaller compared to their wild type littermates and the mice have modest defects in skeletal muscle strength and some alterations in metabolic functions. Apart from these minor phenotypical defects, the global MCU knock-out mice are healthy. Therefore, alteration of MCU to regulate [Ca2+]m will require extensive studies to fully uncover the underlying molecular mechanism(s) before proposing that a miRNA for regulating the MCU gene could be an effective strategy to achieve a therapeutic goal. However, the gate-keeper of MCU, MICU1, when altered showed very consistent outcomes in multiple groups. It is known that knocking down MICU1 is detrimental when [Ca2+]cyto is low, whereas knocking-down MICU1 is beneficial when [Ca2+]cyto is high. Studies have clearly shown that it is the ratio between MICU1 and MICU2 that plays a pivotal role in [Ca2+]m influx. Therefore, it is fair to predict that miRNA(s) which can effectively bind to MICU1 and/or MICU2 mRNAs can influence the [Ca2+]m. In particular, there is great potential for treatment of conditions such as diabetes and cardiovascular disease with MitomiR(s) manipulation due to their impact on the mitochondria and overall cardiac function.
Author Contributions
CJDG and BR have performed the literature search. CJDG, BR, and SD wrote the manuscript. All the authors have read and consented to submit this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the grant from the AHA, 14SDG18890049 (SD).
Abbreviations
miRNA, microRNA; ROS, Reactive Oxygen Species; [Ca2+]m, Mitochondrial matrix calcium; mt-COX1, Cytochrome C Oxidase subunit 1; COX IV, Mitochondrial respiratory Complex IV; MCU, Mitochondrial Calcium Uniporter; PDH, Pyruvate Dehydrogenase; RISC, RNA-induced silencing complex; NCX1, Na+-Ca2+ exchanger 1.
References
Ago, T., Kuroda, J., Pain, J., Fu, C., Li, H., and Sadoshima, J. (2010). Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ. Res. 106, 1253–1264. doi: 10.1161/CIRCRESAHA.109.213116
Ammal Kaidery, N., and Thomas, B. (2018). Current perspective of mitochondrial biology in Parkinson's disease. Neurochem. Int. 117, 91–113. doi: 10.1016/j.neuint.2018.03.001
Aurora, A. B., Mahmoud, A. I., Luo, X., Johnson, B. A., Van Rooij, E., Matsuzaki, S., et al. (2012). MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca(2)(+) overload and cell death. J. Clin. Invest. 122, 1222–1232. doi: 10.1172/JCI59327
Austin, S., Tavakoli, M., Pfeiffer, C., Seifert, J., Mattarei, A., De Stefani, D., et al. (2017). LETM1-mediated K(+) and Na(+) homeostasis regulates mitochondrial Ca(2+) efflux. Front. Physiol. 8:839. doi: 10.3389/fphys.2017.00839
Banerjee, J., and Ghosh, S. (2006). Phosphorylation of rat brain mitochondrial voltage-dependent anion as a potential tool to control leakage of cytochrome c. J. Neurochem. 98, 670–676. doi: 10.1111/j.1471-4159.2006.03853.x
Baradan, R., Hollander, J. M., and Das, S. (2017). Mitochondrial miRNAs in diabetes: just th e tip of the iceberg. Can. J. Physiol. Pharmacol. 95, 1156–1162. doi: 10.1139/cjpp-2016-0580
Bargaje, R., Gupta, S., Sarkeshik, A., Park, R., Xu, T., Sarkar, M., et al. (2012). Identification of novel targets for miR-29a using miRNA proteomics. PLoS ONE 7:e43243. doi: 10.1371/journal.pone.0043243
Baughman, J. M., Perocchi, F., Girgis, H. S., Plovanich, M., Belcher-Timme, C. A., Sancak, Y., et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. doi: 10.1038/nature10234
Bernardi, P., Rasola, A., Forte, M., and Lippe, G. (2015). The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol. Rev. 95, 1111–1155. doi: 10.1152/physrev.00001.2015
Boehning, D., Patterson, R. L., Sedaghat, L., Glebova, N. O., Kurosaki, T., and Snyder, S. H. (2003). Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat. Cell Biol. 5, 1051–1061. doi: 10.1038/ncb1063
Bustos, G., Cruz, P., Lovy, A., and Cárdenas, C. (2017). Endoplasmic reticulum-mitochondria calcium communication and the regulation of mitochondrial metabolism in cancer: a novel potential target. Front. Oncol. 7:199. doi: 10.3389/fonc.2017.00199
Camara, A. K., Lesnefsky, E. J., and Stowe, D. F. (2010). Potential therapeutic benefits of strategies directed to mitochondria. Antioxid. Redox Signal. 13, 279–347. doi: 10.1089/ars.2009.2788
Cha, M. J., Jang, J. K., Ham, O., Song, B. W., Lee, S. Y., Lee, C. Y., et al. (2013). MicroRNA-145 suppresses ROS-induced Ca2+ overload of cardiomyocytes by targeting CaMKIIdelta. Biochem. Biophys. Res. Commun. 435, 720–726. doi: 10.1016/j.bbrc.2013.05.050
Chaudhuri, A. D., Choi, D. C., Kabaria, S., Tran, A., and Junn, E. (2016). MicroRNA-7 regulates the function of mitochondrial permeability transition pore by targeting VDAC1 expression. J. Biol. Chem. 291, 6483–6493. doi: 10.1074/jbc.M115.691352
Csordás, G., Golenár, T., Seifert, E. L., Kamer, K. J., Sancak, Y., Perocchi, F., et al. (2013). MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab. 17, 976–987. doi: 10.1016/j.cmet.2013.04.020
Das, A. M., and Harris, D. A. (1990). Control of mitochondrial ATP synthase in heart cells: inactive to active transitions caused by beating or positive inotropic agents. Cardiovasc. Res. 24, 411–417. doi: 10.1093/cvr/24.5.411
Das, S., Bedja, D., Campbell, N., Dunkerly, B., Chenna, V., Maitra, A., et al. (2014). miR-181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo. PLoS ONE 9:e96820. doi: 10.1371/journal.pone.0096820
Das, S., Ferlito, M., Kent, O. A., Fox-Talbot, K., Wang, R., Liu, D., et al. (2012). Nuclear miRNA regulates the mitochondrial genome in the heart. Circ. Res. 110, 1596–1603. doi: 10.1161/CIRCRESAHA.112.267732
Das, S., Kohr, M., Dunkerly-Eyring, B., Lee, D. I., Bedja, D., Kent, O. A., et al. (2017). Divergent effects of miR-181 family members on myocardial function through protective cytosolic and detrimental mitochondrial microRNA targets. J. Am. Heart Assoc. 6:e004694. doi: 10.1161/JAHA.116.004694
Das, S., Wong, R., Rajapakse, N., Murphy, E., and Steenbergen, C. (2008). Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation. Circ. Res. 103, 983–991. doi: 10.1161/CIRCRESAHA.108.178970
De Stefani, D., Raffaello, A., Teardo, E., Szabò, I., and Rizzuto, R. (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. doi: 10.1038/nature10230
Deluca, H. F., and Engstrom, G. W. (1961). Calcium uptake by rat kidney mitochondria. Proc. Natl. Acad. Sci. U. S. A. 47, 1744–1750. doi: 10.1073/pnas.47.11.1744
Drawnel, F. M., Wachten, D., Molkentin, J. D., Maillet, M., Aronsen, J. M., Swift, F., et al. (2012). Mutual antagonism between IP(3)RII and miRNA-133a regulates calcium signals and cardiac hypertrophy. J. Cell Biol. 199, 783–798. doi: 10.1083/jcb.201111095
Ferrari, R. (1996). The role of mitochondria in ischemic heart disease. J. Cardiovasc. Pharmacol. 28 (Suppl. 1), S1–S10.
Fieni, F., Lee, S. B., Jan, Y. N., and Kirichok, Y. (2012). Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat. Commun. 3:1317. doi: 10.1038/ncomms2325
Gatliff, J., East, D. A., Singh, A., Alvarez, M. S., Frison, M., Matic, I., et al. (2017). A role for TSPO in mitochondrial Ca(2+) homeostasis and redox stress signaling. Cell Death Dis. 8:e2896. doi: 10.1038/cddis.2017.186
Giorgi, C., Bonora, M., Sorrentino, G., Missiroli, S., Poletti, F., Suski, J. M., et al. (2015). p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. U. S. A. 112, 1779–1784. doi: 10.1073/pnas.1410723112
Glancy, B., and Balaban, R. S. (2012). Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51, 2959–2973. doi: 10.1021/bi2018909
Görlach, A., Bertram, K., Hudecova, S., and Krizanova, O. (2015). Calcium and ROS: a mutual interplay. Redox Biol. 6, 260–271. doi: 10.1016/j.redox.2015.08.010
Granatiero, V., De Stefani, D., and Rizzuto, R. (2017). Mitochondrial calcium handling in physiology and disease. Adv. Exp. Med. Biol. 982, 25–47. doi: 10.1007/978-3-319-55330-6_2
Griffiths, E. J. (2009). Mitochondrial calcium transport in the heart: physiological and pathological roles. J. Mol. Cell. Cardiol. 46, 789–803. doi: 10.1016/j.yjmcc.2009.03.001
Griffiths, E. J., Balaska, D., and Cheng, W. H. (2010). The ups and downs of mitochondrial calcium signalling in the heart. Biochim. Biophys. Acta 1797, 856–864. doi: 10.1016/j.bbabio.2010.02.022
Gupta, R. (2017). Phosphorylation of rat brain purified mitochondrial voltage-dependent anion channel by c-Jun N-terminal kinase-3 modifies open-channel noise. Biochem. Biophys. Res. Commun. 490, 1221–1225. doi: 10.1016/j.bbrc.2017.06.194
Hajnóczky, G., Booth, D., Csordás, G., Debattisti, V., Golenar, T., Naghdi, S., et al. (2014). Reliance of ER-mitochondrial calcium signaling on mitochondrial EF-hand Ca2+ binding proteins: Miros, MICUs, LETM1 and solute carriers. Curr. Opin. Cell Biol. 29, 133–141. doi: 10.1016/j.ceb.2014.06.002
Hempel, N., and Trebak, M. (2017). Crosstalk between calcium and reactive oxygen species signaling in cancer. Cell Calcium 63, 70–96. doi: 10.1016/j.ceca.2017.01.007
Hong, S., Lee, J., Seo, H. H., Lee, C. Y., Yoo, K. J., Kim, S. M., et al. (2015). Na(+)-Ca(2+) exchanger targeting miR-132 prevents apoptosis of cardiomyocytes under hypoxic condition by suppressing Ca(2+) overload. Biochem. Biophys. Res. Commun. 460, 931–937. doi: 10.1016/j.bbrc.2015.03.129
Hong, Z., Chen, K. H., Dasgupta, A., Potus, F., Dunham-Snary, K., Bonnet, S., et al. (2017). MicroRNA-138 and MicroRNA-25 down-regulate mitochondrial calcium uniporter, causing the pulmonary arterial hypertension cancer phenotype. Am. J. Respir. Crit. Care Med. 195, 515–529. doi: 10.1164/rccm.201604-0814OC
Ivanova, H., Kerkhofs, M., La Rovere, R. M., and Bultynck, G. (2017). Endoplasmic reticulum-mitochondrial Ca(2+) fluxes underlying cancer cell survival. Front. Oncol. 7:70. doi: 10.3389/fonc.2017.00070
Jiang, D., Zhao, L., and Clapham, D. E. (2009). Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 326, 144–147. doi: 10.1126/science.1175145
Joiner, M. L., Koval, O. M., Li, J., He, B. J., Allamargot, C., Gao, Z., et al. (2012). CaMKII determines mitochondrial stress responses in heart. Nature 491, 269–273. doi: 10.1038/nature11444
Kamer, K. J., and Mootha, V. K. (2014). MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep. 15, 299–307. doi: 10.1002/embr.201337946
Kameyama, Y., and Gemba, M. (1991). Cisplatin-induced injury to calcium uptake by mitochondria in glutathione-depleted slices of rat kidney cortex. Jpn. J. Pharmacol. 55, 174–176. doi: 10.1254/jjp.55.174
Kim, J. E., Choi, B. K., Choi, J. Y., Ryu, T., Roh, W. S., and Song, S. Y. (2014). Role of calcium channels responsible for phenylephrine-induced contraction in rat aorta 3 days after acute myocardial infarction. Korean J. Anesthesiol. 66, 143–152. doi: 10.4097/kjae.2014.66.2.143
Kim, V. N. (2005). MicroRNA biogenesis: coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol. 6, 376–385. doi: 10.1038/nrm1644
Kirichok, Y., Krapivinsky, G., and Clapham, D. E. (2004). The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364. doi: 10.1038/nature02246
Kostic, M., Ludtmann, M. H., Bading, H., Hershfinkel, M., Steer, E., Chu, C. T., et al. (2015). PKA phosphorylation of NCLX reverses mitochondrial calcium overload and depolarization, promoting survival of PINK1-deficient dopaminergic neurons. Cell Rep. 13, 376–386. doi: 10.1016/j.celrep.2015.08.079
Lewis, B. P., Burge, C. B., and Bartel, D. P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20. doi: 10.1016/j.cell.2004.12.035
Liu, J. C., Liu, J., Holmström, K. M., Menazza, S., Parks, R. J., Fergusson, M. M., et al. (2016). MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep. 16, 1561–1573. doi: 10.1016/j.celrep.2016.07.011
Liu, J. C., Parks, R. J., Liu, J., Stares, J., Rovira, I.i, Murphy, E., et al. (2017). The in vivo biology of the mitochondrial calcium uniporter. Adv. Exp. Med. Biol. 982, 49–63. doi: 10.1007/978-3-319-55330-6_3
Logan, C. V., Szabadkai, G., Sharpe, J. A., Parry, D. A., Torelli, S., Childs, A. M., et al. (2014). Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 46, 188–193. doi: 10.1038/ng.2851
Ludtmann, M. H. R., and Abramov, A. Y. (2018). Mitochondrial calcium imbalance in Parkinson's disease. Neurosci. Lett. 663, 86–90. doi: 10.1016/j.neulet.2017.08.044
Macgregor-Das, A. M., and Das, S. (2018). A microRNA's journey to the center of the mitochondria. Am. J. Physiol. Heart Circ. Physiol. 315, H206–H215. doi: 10.1152/ajpheart.00714.2017
Mallilankaraman, K., Doonan, P., Cárdenas, C., Chandramoorthy, H. C., Müller, M., Miller, R., et al. (2012). MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151, 630–644. doi: 10.1016/j.cell.2012.10.011
Mammucari, C., Raffaello, A., Vecellio Reane, D., Gherardi, G., De Mario, A., and Rizzuto, R. (2018). Mitochondrial calcium uptake in organ physiology: from molecular mechanism to animal models. Pflugers Arch. 470, 1165–1179. doi: 10.1007/s00424-018-2123-2
Marchi, S., Lupini, L., Patergnani, S., Rimessi, A., Missiroli, S., Bonora, M., et al. (2013). Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr. Biol. 23, 58–63. doi: 10.1016/j.cub.2012.11.026
Mazure, N. M. (2017). VDAC in cancer. Biochim. Biophys. Acta 1858, 665–673. doi: 10.1016/j.bbabio.2017.03.002
Mildaziene, V., Baniene, R., Nauciene, Z., Bakker, B. M., Brown, G. C., Westerhoff, H. V., et al. (1995). Calcium indirectly increases the control exerted by the adenine nucleotide translocator over 2-oxoglutarate oxidation in rat heart mitochondria. Arch. Biochem. Biophys. 324, 130–134. doi: 10.1006/abbi.1995.9918
Nickel, A., Kohlhaas, M., and Maack, C. (2014). Mitochondrial reactive oxygen species production and elimination. J. Mol. Cell. Cardiol. 73, 26–33. doi: 10.1016/j.yjmcc.2014.03.011
Paillard, M., Csordas, G., Szanda, G., Golenar, T., Debattisti, V., Bartok, A., et al. (2017). Tissue-specific mitochondrial decoding of cytoplasmic Ca(2+) signals is controlled by the stoichiometry of MICU1/2 and MCU. Cell Rep. 18, 2291–2300. doi: 10.1016/j.celrep.2017.02.032
Palty, R., Silverman, W. F., Hershfinkel, M., Caporale, T., Sensi, S. L., Parnis, J., et al. (2010). NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. U. S. A. 107, 436–441. doi: 10.1073/pnas.0908099107
Pan, L., Huang, B. J., Ma, X. E., Wang, S. Y., Feng, J., Lv, F., et al. (2015). MiR-25 protects cardiomyocytes against oxidative damage by targeting the mitochondrial calcium uniporter. Int. J. Mol. Sci. 16, 5420–5433. doi: 10.3390/ijms16035420
Patergnani, S., Suski, J. M., Agnoletto, C., Bononi, A., Bonora, M., De Marchi, E., et al. (2011). Calcium signaling around Mitochondria Associated Membranes (MAMs). Cell Commun. Signal. 9:19. doi: 10.1186/1478-811X-9-19
Patron, M., Checchetto, V., Raffaello, A., Teardo, E., Vecellio Reane, D., Mantoan, M., et al. (2014). MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 53, 726–737. doi: 10.1016/j.molcel.2014.01.013
Perevoshchikova, I. V., Quinlan, C. L., Orr, A. L., Gerencser, A. A., and Brand, M. D. (2013). Sites of superoxide and hydrogen peroxide production during fatty acid oxidation in rat skeletal muscle mitochondria. Free Radic. Biol. Med. 61, 298–309. doi: 10.1016/j.freeradbiomed.2013.04.006
Raffaello, A., De Stefani, D., Sabbadin, D., Teardo, E., Merli, G., Picard, A., et al. (2013). The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 32, 2362–2376. doi: 10.1038/emboj.2013.157
Rapizzi, E., Pinton, P., Szabadkai, G., Wieckowski, M. R., Vandecasteele, G., Baird, G., et al. (2002). Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 159, 613–624. doi: 10.1083/jcb.200205091
Rasola, A., and Bernardi, P. (2011). Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium 50, 222–233. doi: 10.1016/j.ceca.2011.04.007
Rimessi, A., Marchi, S., Fotino, C., Romagnoli, A., Huebner, K., Croce, C. M., et al. (2009). Intramitochondrial calcium regulation by the FHIT gene product sensitizes to apoptosis. Proc. Natl. Acad. Sci. U. S. A. 106, 12753–12758. doi: 10.1073/pnas.0906484106
Rimessi, A., Patergnani, S., Bonora, M., Wieckowski, M. R., and Pinton, P. (2015). Mitochondrial Ca(2+) remodeling is a prime factor in oncogenic behavior. Front. Oncol. 5:143. doi: 10.3389/fonc.2015.00143
Rizzuto, R., De Stefani, D., Raffaello, A., and Mammucari, C. (2012). Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13, 566–578. doi: 10.1038/nrm3412
Saffari, A., Kölker, S., Hoffmann, G. F., and Ebrahimi-Fakhari, D. (2017). Linking mitochondrial dysfunction to neurodegeneration in lysosomal storage diseases. J. Inherit. Metab. Dis. 40, 631–640. doi: 10.1007/s10545-017-0048-0
Sancak, Y., Markhard, A. L., Kitami, T., Kovács-Bogdán, E., Kamer, K. J., Udeshi, N. D., et al. (2013). EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342, 1379–1382. doi: 10.1126/science.1242993
Santulli, G., Xie, W., Reiken, S. R., and Marks, A. R. (2015). Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. U. S. A. 112, 11389–11394. doi: 10.1073/pnas.1513047112
Shoshan-Barmatz, V., Krelin, Y., and Shteinfer-Kuzmine, A. (2018). VDAC1 functions in Ca(2+) homeostasis and cell life and death in health and disease. Cell Calcium 69, 81–100. doi: 10.1016/j.ceca.2017.06.007
Sipido, K. R., Volders, P. G., Vos, M. A., and Verdonck, F. (2002). Altered Na/Ca exchange activity in cardiac hypertrophy and heart failure: a new target for therapy? Cardiovasc. Res. 53, 782–805. doi: 10.1016/S0008-6363(01)00470-9
Slater, E. C., and Cleland, K. W. (1953). The effect of calcium on the respiratory and phosphorylative activities of heart-muscle sarcosomes. Biochem. J. 55, 566–590. doi: 10.1042/bj0550566
Szabadkai, G., Bianchi, K., Várnai, P., De Stefani, D., Wieckowski, M. R., Cavagna, D., et al. (2006). Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901–911. doi: 10.1083/jcb.200608073
Takeuchi, A., Kim, B., and Matsuoka, S. (2015). The destiny of Ca(2+) released by mitochondria. J. Physiol. Sci. 65, 11–24. doi: 10.1007/s12576-014-0326-7
Tomar, D., Dong, Z., Shanmughapriya, S., Koch, D. A., Thomas, T., Hoffman, N. E., et al. (2016). MCUR1 Is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep. 15, 1673–1685. doi: 10.1016/j.celrep.2016.04.050
Tomasello, F., Messina, A., Lartigue, L., Schembri, L., Medina, C., Reina, S., et al. (2009). Outer membrane VDAC1 controls permeability transition of the inner mitochondrial membrane in cellulo during stress-induced apoptosis. Cell Res. 19, 1363–1376. doi: 10.1038/cr.2009.98
Wernette, M. E., Ochs, R. S., and Lardy, H. A. (1981). Ca2+ stimulation of rat liver mitochondrial glycerophosphate dehydrogenase. J. Biol. Chem. 256, 12767–12771.
White, C. (2017). The regulation of tumor cell invasion and metastasis by endoplasmic reticulum-to-mitochondrial Ca(2+) transfer. Front. Oncol. 7:171. doi: 10.3389/fonc.2017.00171
Yu, C., Wang, Y., Peng, J., Shen, Q., Chen, M., Tang, W., et al. (2017). Mitochondrial calcium uniporter as a target of microRNA-340 and promoter of metastasis via enhancing the Warburg effect. Oncotarget 8, 83831–83844. doi: 10.18632/oncotarget.19747
Zaglia, T., Ceriotti, P., Campo, A., Borile, G., Armani, A., Carullo, P., et al. (2017). Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy. Proc. Natl. Acad. Sci. U. S. A. 114, E9006–E9015. doi: 10.1073/pnas.1708772114
Keywords: microRNA, miRNA, mitochondria, MitomiR, mitochondrial calcium, heart failure
Citation: Jaquenod De Giusti C, Roman B and Das S (2018) The Influence of MicroRNAs on Mitochondrial Calcium. Front. Physiol. 9:1291. doi: 10.3389/fphys.2018.01291
Received: 08 June 2018; Accepted: 27 August 2018;
Published: 21 September 2018.
Edited by:
Gyorgy Szabadkai, University College London, United KingdomReviewed by:
Dhanendra Tomar, Temple University, United StatesAmadou K.S. Camara, Medical College of Wisconsin, United States
Copyright © 2018 Jaquenod De Giusti, Roman and Das. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samarjit Das, c2RhczExQGpobWkuZWR1