 Jiajia Song
Jiajia Song Ruilin Yang
Ruilin Yang Jing Yang
Jing Yang Lufang Zhou
Lufang Zhou- 1Division of Cardiovascular Disease, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL, United States
- 2Key Laboratory of Mechanism Theory and Equipment Design of Ministry of Education, Tianjin University, Tianjin, China
There is increasing evidence that diabetic cardiomyopathy increases the risk of cardiac arrhythmia and sudden cardiac death. While the detailed mechanisms remain incompletely understood, the loss of mitochondrial function, which is often observed in the heart of patients with diabetes, has emerged as a key contributor to the arrhythmogenic substrates. In this mini review, the pathophysiology of mitochondrial dysfunction in diabetes mellitus is explored in detail, followed by descriptions of several mechanisms potentially linking mitochondria to arrhythmogenesis in the context of diabetic cardiomyopathy.
Introduction
Diabetes mellitus (DM) is a group of chronic metabolic diseases that affects around 425 million people worldwide (Tuomilehto and Lindstrom, 2003). It is widely accepted that DM leads to a cascade of long-term severe complications, including cardiovascular diseases, renal failure and blindness (Association, 2000). Importantly, cardiovascular diseases account for nearly 70% of deaths in diabetic patients (Walker and Cubbon, 2015).
Diabetes mellitus can affect cardiac structure and function independently of vascular disease, hypertension or coronary artery disease, leading to the development of a heart disorder termed diabetic cardiomyopathy (DbCM). DbCM is characterized by a series of cardiac structural and functional remodeling, including left ventricular hypertrophy, interstitial fibrosis, lipid deposition, cell death, and decreased systolic dysfunction, which eventually lead to heart failure (Akar et al., 2005; Xie et al., 2009; Singh et al., 2018). In addition to contractile function, DbCM can also disrupt cardiac electrical activity, leading to arrhythmia (Jeong et al., 2016) and sudden cardiac death. It has been reported that patients with diabetes have a significantly increased risk of arrhythmogenesis compared with the general population (Huxley et al., 2011). Indeed, DbCM has become a major contributor to mortality in diabetic patients (Chen C. et al., 2018; D’Souza et al., 2018). Moreover, abnormal mitochondrial morphology (e.g., fragmented organelle, mottled matrix, and damaged membrane) and energetics have been observed in diabetes patients with arrhythmia (Montaigne et al., 2014; Montaigne and Pentiah, 2015), suggesting that mitochondrial dysfunction could contribute to arrhythmogenesis in the setting of DbCM.
In this mini view, we revisit some of the main features of DbCM, focusing on pathophysiological mechanisms associated with cardiac mitochondrial dysfunction, oxidative stress and fibrosis, and their proarrhythmic roles in DbCM.
Mitochondria and Oxidative Stress in Diabetic Cardiomyopathy
Mitochondria are the major powerhouse in cardiomyocytes, producing more than 95% of energy in the form of adenosine triphosphate (ATP). ATP is formed from adenosine diphosphate (ADP) and inorganic phosphate (Pi) by F0F1 ATP synthase (complex V), which uses the electrochemical gradient generated by the electron transport chain (complexes I to IV) driven by NADH produced in the tricarboxylic acid (TCA) cycle (Figure 1). ATP is transported into cytosol via adenine nucleotide translocator (ANT) and hydrolyzed to support cellular mechanical work (excitation and contraction), ion homeostasis, and molecular synthesis (Nicholls and Ferguson, 2002). Importantly, mitochondria are also a major source of reactive oxygen species (ROS) production (Chance et al., 1979), likely at complex I and complex III (Murphy, 2009; Figure 1). While physiological level ROS is essential for cellular redox signaling (Vanden Hoek et al., 1998), excessive ROS are toxic and can affect large molecules such as redox sensitive ion channels [e.g., ryanodine receptors (RyRs), sarcoplasmic reticulum (SR) Ca2+ transport ATPase (SERCA) and L-type Ca2+ channels (LCCs)]. Furthermore, mitochondria can directly regulate cytosolic Ca2+ via mitochondrial Ca2+ uniporter (mCU) and Na+/Ca2+ exchanger (mNCE). Not surprisingly, loss of mitochondrial function has been implicated to be closely associated with a variety of human diseases including DM (Bagheri et al., 2016; Jeong et al., 2016). In particular, studies have revealed that mitochondrial functional and structural alterations, including redox signaling, energy production, biogenesis, dynamics, and quality control, are important contributing factors to the pathogenesis of DbCM (Palikaras and Tavernarakis, 2014).
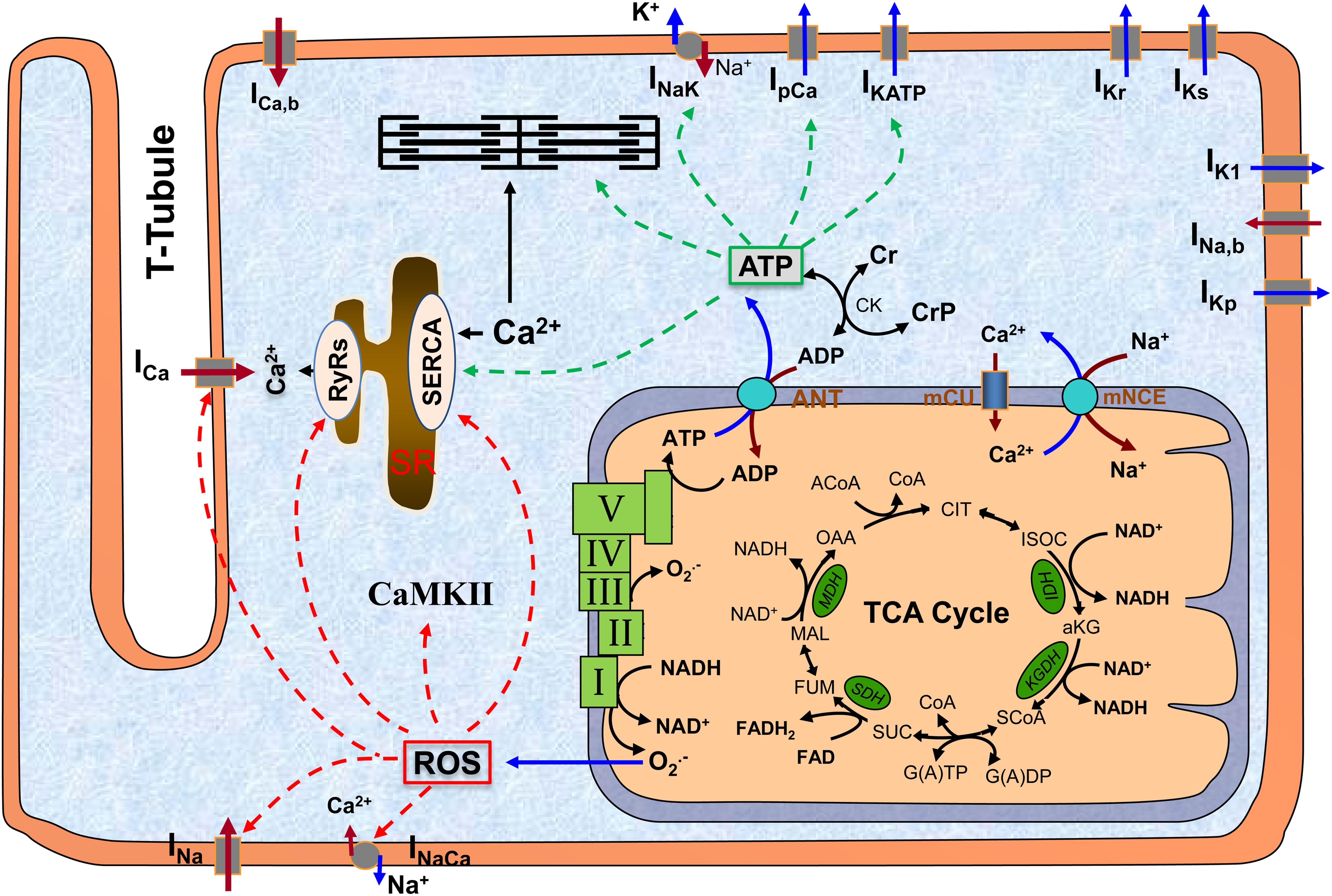
Figure 1. The scheme of mitochondrion and its interaction with other subcellular systems in cardiomyocyte. The major function of mitochondrion is to produce ATP, which occurs at complex V (a.k.a. F0F1 ATPase) using the electrochemical gradient generated by electron transport chain (complex I to IV). A byproduct of ATP production is superoxide (O2.-), which is probably generated at complex I and complex III. ATP is translocated to cytosol via ANT and hydrolyzed to support excitation-contraction and energy-sensitive ion transporters (indicated by green dashed lines). O2.- can freely diffuse to cytosol and form ROS, which can modify a variety of redox sensitive ion transporters (indicated by red dashed lines). Altogether, mitochondrial dysfunction-associated ATP depletion and ROS accumulation can significantly affect cellular action potentials and ion homeostasis. ROS, reactive oxygen species; CaMKII, Ca2+/calmodulin-dependent protein kinase II; SR, sarcoplasmic reticulum; TCA, tricarboxylic acid; ANT, adenine nucleotide translocator; mCU, mitochondrial Ca2+ uniporter; mNCE, mitochondrial Na+/Ca2+ exchanger; I–V, complex I to complex V.
Oxidative stress is a hall marker of mitochondrial dysfunction, which is known to damage a variety of intracellular macromolecules, leading to lipid peroxidation and detrimental protein modifications (Zhang et al., 2012). Increased ROS levels may also activate multiple redox-sensitive signaling pathways involved in DbCM-relevant cardiac dysfunction, leading to impaired intracellular Ca2+ regulation, fibrosis, and loss of essential trace metal homeostasis. In the cardiomyocytes, the major site of ROS production is mitochondria (Chance et al., 1979; Murphy, 2009). In patients with diabetes, as glucose utilization is greatly impaired, the heart relies almost exclusively on fatty acids oxidation (FAO) to produce ATP. The switching to FAO leads to increased ROS production in DM (Block et al., 2009). Another important source of ROS production in DbCM is the hyperglycemia-induced advanced glycation end-products (AGEs) (Pal et al., 2014). In addition, it has been shown that inhibiting protein kinase C (PKC)-α signaling pathway reduces ROS levels and alleviates oxidative stress damage in DbCM in rats, suggesting that PKC-α is involved in ROS production in DM (Min et al., 2017).
Mitochondrial-Associated Cardiac Arrhythmia
Cardiac arrhythmia refers to conditions in which the heart’s normal rhythm is disrupted, or the electrical activity is abnormal. Cardiac arrhythmias are traditionally considered to occur due to an abnormality in impulse initiation and/or electrical propagation. Abnormal impulse initiation is associated with triggered activity (i.e., trigger) resulting from premature activation of cardiac tissues by afterdepolarizations (e.g., early afterdepolarizations, EADs or delayed afterdepolarizations, DADs) or enhanced automaticity in the pacemaker cells, which may propagate as focal activity. Abnormal impulse propagation is associated with block of conduction or re-entry, which occurs when a single propagating impulse traveling through the heart gives rise to two or more propagated responses through wave-breaks. Reentrant arrhythmias can be attributed to three main cellular mechanisms: (i) loss of cell to cell electrical coupling and communication by closure of gap junctions, (ii) regional uncoupling caused by anatomical barriers such as scar tissue, and (iii) dynamic functional block due to heterogeneity of intrinsic electrophysiological restitution properties. The detailed review of mechanism of cardiac arrhythmogenesis can be found here (Antzelevitch and Burashnikov, 2011; Tse, 2016).
While arrhythmia is a leading cause of sudden cardiac death in patients with diabetes (Cohn et al., 1986; Cohn, 1996), the precise molecular mechanisms underlying cardiac arrhythmogenesis in DbCM are poorly understood, hindering the development of effective therapeutic strategy. Recently, the loss of mitochondrial function, which is often observed in DbCM, has emerged as a key contributor to the arrhythmogenic substrates (Li et al., 2008; Xie et al., 2013), probably via modulating the redox and/or energy sensitive signaling pathways that regulate ion handling channels/transporters (Maack and O’Rourke, 2007; Yan et al., 2008; Zhou et al., 2009, 2011).
Oxidative Stress-Associated Arrhythmic Substrates
Although hyperglycaemia regulates multiple pathways associated with the pathogenesis of DbCM, oxidative stress is considered as a central mechanism underlying the adverse remodeling in diabetic hearts (Liu Q. et al., 2014), including electrophysiological alternations (Li et al., 2008; Karam et al., 2017). Both in vitro and computational studies have shown that ROS cause action potential duration (APD) prolongation and induce EADs or DADs in guinea pig (Liu T. et al., 2014; Li et al., 2015) and rabbit cardiomyocytes (Xie et al., 2009), leading to focal activity. ROS can also promote reentry via heterogeneous APD prolongation (Morita et al., 2011). It is worth noting that both reentry and focal activity account for ROS-mediated arrhythmogenesis.
At the cellular level, the proarrhythmic effect of ROS is attributed to their capability to modulate multiple redox ion channels/transporters underlying ion handling and action potentials (Howe et al., 2004; Zima and Blatter, 2006; Wagner et al., 2013), which include RyRs (Eager et al., 1997; Gen et al., 2001; Yan et al., 2008; Zhou et al., 2011), SERCA (Morris and Sulakhe, 1997; Zima and Blatter, 2006), voltage-gated Na+ channels (Nav) (Liu et al., 2010; Jeong et al., 2012), K+ channels (Kir and Kv) (Zhang et al., 2006), Na+/Ca2+ exchanger (NCX) (Bers and Despa, 2006), and LCCs (Barrington et al., 1988; Coetzee and Opie, 1992; Nakaya et al., 1992; Ward and Giles, 1997; Kourie, 1998; Zima and Blatter, 2006). One of the most well-characterized redox-sensitive ion transporters is RyRs, which have 89 cysteine residues, of which approximately 21 are susceptible to oxidation by free radicals (Xu et al., 1998). Studies have shown that extracellular H2O2 activates RyRs, leading to increased SR Ca2+ release in rat (Gen et al., 2001; Oda et al., 2015) and sheep (Eager et al., 1997) ventricular myocytes. Moreover, recent studies have shown that mitochondrial-derived ROS (mdROS) are closely correlated with enhanced Ca2+ sparks in resting guinea pig cardiomyocytes (Yan et al., 2008; Zhou et al., 2011). In addition to RyRs, ROS can affect SR Ca2+ uptake. It has been shown that ROS inhibit SERCA activity by directly oxidizing its thiol groups (Morris and Sulakhe, 1997; Zima and Blatter, 2006). Thus, ROS, by simultaneously activating RyRs and inhibiting SERCA, may lead to increased Ca2+ transients (Ward and Moffat, 1995; Li et al., 2015). With regards to diabetes, studies have found that free fatty acids-induced mdROS contributes to Ca2+ dysregulation, likely by triggering aberrant endoplasmic reticulum/SR Ca2+ release, leading to increased DADs and probability of focal excitation (Roussel et al., 2015; Ly et al., 2017).
Sarcoplasmic Na+ channels encoded by the SCN5A gene are key to cardiac excitability and rapid impulse propagation. Song et al. (2006) have demonstrated that the late Na+ current is involved in H2O2-induced APD prolongation, EADs and DADs in both guinea pig and rabbit cardiomyocytes. Another study showed that ranolazine, a late Na+ current blocker, suppressed ROS-mediated EAD and arrhythmias (Morita et al., 2011). Importantly, while ROS-induced increase in late Na+ current can elicit EADs and arrhythmia, increased ROS may downregulate the total Na+ channel expression, resulting in reduced Na+ currents and conduction velocity, providing substrates for reentry (Liu et al., 2010).
In addition to directly modulating redox sensitive ion channels/transporters, ROS can also indirectly influence ion homeostasis and consequently action potentials via redox signaling such as Ca2+/calmodulin dependent kinase II (CaMKII) oxidation. CaMKII is a ubiquitously expressed multifunctional protein kinase that can be activated by binding to Ca2+/calmodulin (Zhang and Brown, 2004; Wagner et al., 2013). Recent studies suggest that CaMKII can also be activated by ROS (Erickson et al., 2008, 2011; Huke and Knollmann, 2011; Joiner et al., 2012), leading to phosphorylation of a wide range of ion handling proteins such as Na+ channels (Wagner et al., 2006, 2011), LCCs (Dzhura et al., 2000; Hudmon et al., 2005; Grueter et al., 2006; Blaich et al., 2010), RyRs (Maier et al., 2003; Wehrens et al., 2004; Guo et al., 2006; Kohlhaas et al., 2006; Maier and Bers, 2007; Sag et al., 2013; Ho et al., 2014), and phospholamban (Bassani et al., 1995; Maier and Bers, 2007; Sag et al., 2013). Xie et al. (2009) have showed that H2O2 perfusion-induced oxidative CaMKII activation leads to afterdepolarizations in isolated rabbit cardiomyocytes, likely via phosphorylation of Na+ channels and LCCs. In a computational study, Yang R. et al. (2018) reported that mdROS-mediated oxidative CaMKII activation induces EADs in guinea pig cardiomyocytes by enhancing the late component of Na+ current. The proarrhythmic role of CaMKII oxidation in diabetic hearts has been demonstrated in a recent study showing that selective genetic blocking of CaMKII oxidation prevents the enhanced atrial fibrillation (AF) risk (Mesubi et al., 2017), which has been implicated to be associated with increased mortality in response to myocardial infraction (Luo et al., 2013). Redox signaling can also regulate several pro-arrhythmic transcription factors such as nuclear factor-κB (NF-κB), one of the key transcriptional regulators that mediates gene expressions under stress conditions including diabetes. Importantly, Dudley and colleagues have shown that the promoter region of the SCN5A gene encoding cardiac Na+ channels contains a NF-κB binding domain, suggesting that SCN5A can be regulated by NF-κB in response to oxidative stress (Shang et al., 2008).
Energy Deficiency-Associated Arrhythmias
Studies have shown that oxygen consumption rate and ATP synthesis are reduced in patients with insulin resistance (Szendroedi et al., 2012) and animals fed with high palmitate (Xu et al., 2015), which can contribute to the development of arrhythmia. A series of studies by O’Rourke’s laboratory have convincingly demonstrated that mitochondrial depolarization-induced ATP depletion elicits reentrant arrhythmias via a mechanism termed “metabolic sink” (Akar et al., 2005; Brown et al., 2008; Aon et al., 2009; Zhou et al., 2014). In particular, they showed that decreased ATP levels, or increased ADP/ATP ratio, caused by mitochondrial depolarization, leads to rapid activation of the sarcoplasmic potassium-sensitive KATP (KATP) channels, causing shortening of APD and reduction of action potential amplitude (APA) (Zhou et al., 2009). When a threshold of opening KATP channels is reached, cardiomyocytes are rendered completely inexcitable. They further showed that mitochondrial oscillations [a phenomena involving local oxidative stress-induced cyclic changes in mitochondrial membrane potential, NADH oxidation and ROS production (Aon et al., 2003; Zhou and O’Rourke, 2012; Zhou et al., 2014)] cause highly correlated, repetitive activations of the KATP channels and fluctuations in both APD and APA (Zhou et al., 2009), indicating that there is a direct link between mitochondrial energetic dysfunction and arrhythmia. At the tissue level, regional mitochondrial depolarization and KATP current activation can create inhomogeneous regions or “metabolic sink,” in which the increased dispersion of repolarization enhances the vulnerability to reentry, as revealed by both experimental studies (Akar et al., 2005) and computer simulations (Zhou et al., 2014).
Other ATP-sensitive ion transporter proteins include SERCA and sarcoplasmic Na+/K+ ATPase. It has been shown that expression of ATP2A2 gene that encodes SERCA2a, an isoform mainly expressed in cardiomyocytes, is reduced in DbCM (Suarez et al., 2008). Studies also showed that reduced mitochondrial ATP production suppresses SERCA activity (Landolfi et al., 1998; De Marchi et al., 2011). The decreased SERCA2a activity and expression present in the diabetic myocardium has been considered as a main contributing factor to impaired Ca2+ cycling and aberrant action potentials. Similarly, ATP depletion can decrease Na+/K+ ATPase activity (Ziegelhoffer et al., 2000), an electrogenic pump on sarcoplasmic membrane, leading to not only dysregulation of ion homeostasis but also alteration of the membrane potential.
In addition to influence cellular ion channels and action potentials, decreased ATP production can alter intercellular coupling via gap junctions. Gap junctions are hemichannels in the heart that form electrical connection between cells, mediating the spread of electrical impulse and coordinated contraction of cardiac chambers (see review Sohl and Willecke, 2004). Connexin 43 (Cx43) is the major component of gap junctions in the ventricular myocytes. In heart failure, the level of Cx43 expression is reduced, which is associated with depressed AP propagation and increased incidence of lethal ventricular arrhythmias. At the single channel level, studies have shown that Cx43 conductance can be inhibited by decreased intracellular ATP concentration in guinea pig (Sugiura et al., 1990) and rat (Duthe et al., 2001) cardiomyocytes, among others (see review Schulz and Heusch, 2004), causing suppression of intercellular electrical conductance. It is worth mentioning that gap junction conductance can also be impaired by oxidative stress (Berthoud and Beyer, 2009; Jeong et al., 2012).
Mitochondrial Dysfunction, Fibrosis and Atrial Fibrillation in Diabetes
Cardiac fibrosis is characterized by fibroblasts accumulation and excessive deposition of extracellular matrix (ECM) that greatly increases the stiffness of the heart wall and reduces the contractility and compliance of cardiac muscle (Fan et al., 2012). Myocardial fibrosis is increasingly recognized as one of the major factors contributing to the pathogenesis of DbCM (Shen et al., 2014). In addition, fibrosis, particularly atrial interstitial fibrosis, increases vulnerability to arrhythmia because of delayed discontinuous and zig-zag conduction and unidirectional conduction block (De Jong et al., 2011; Nguyen et al., 2014).
Mitochondrial Dysfunction and Pathogenesis of Cardiac Fibrosis in DbCM
In the heart, fibrosis can be activated by multiple sources, including increased mechanical stretch (Froese et al., 2016), dysregulated redox signaling (Dai et al., 2011), paracrine cytokines from injured cells (Yue et al., 2013), and infiltration of circulatory immune cells (Ismahil et al., 2014). Among those, the transforming growth factor β1 (TGF-β1) is recognized as a major fibrogenic factor. In diabetes, hyperglycemia changes the levels of microRNAs and long non-coding RNAs expression, which can contribute to TGF-β activation (Yue et al., 2013). TGF-β activity can also be increased by high glucose-induced activation of transcriptional co-regulator p300 (Bugyei-Twum et al., 2014), sustained hyperglycemia (Wu et al., 2016; Xu et al., 2016), and activation of matrix metalloproteinases (MMP), a family of ECM proteolytic enzymes (Dayer and Stamenkovic, 2015; Li et al., 2018). Activated TGF-β1 binds to its membrane receptors phosphorylates transcription factors SMAD2/3, which activates the canonical (SMAD-dependent) fibrogenic pathway (Verrecchia and Mauviel, 2002; Khalil et al., 2017), leading to myofibroblast differentiation and fibrosis (Lijnen et al., 2003). TGF-β1 can also stimulate endothelial-to-mesenchymal transition (EndMT) through convergence of SMAD-dependent and SMAD-independent signaling (Medici et al., 2011). Activated EndMT can increase the expression levels of mesenchymal markers, such as α-smooth muscle actin (α-SMA), fibroblast-specific protein-1, and vimentin (Medici and Kalluri, 2012), promoting cardiac fibrosis in diabetic hearts. Importantly, studies have implicated an interplay between TGF-β1 signaling and mitochondrial dysfunction and associated redox signaling (Cucoranu et al., 2005). On one hand, TGF-β1 can upregulate NADPH oxidase 4 (NOX4) expression, resulting in increased mitochondrial ROS production (Jiang et al., 2014). On the other hand, mdROS have been shown to activate TGF-β1 signaling (Koitabashi et al., 2011). Thus, there is a positive feedback mechanism between TGF-β1 and mitochondrial-associated ROS production, facilitating the amplification of the profibrogenic TGF- β1 signaling in DbCM (Dudley et al., 2005; Anderson et al., 2009).
In addition to TGF-β1, some members of the mitogen-activated protein kinase (MAPK) family and protein kinase B (Akt)/glycogen synthase kinase-3β (GSK-3β) signaling, are also involved in myocardial fibrosis (Ying et al., 2017; Yue et al., 2017). Suppression of the MAPKs signaling or the Akt/GSK-β signaling has been shown to ameliorate myocardial fibrosis in diabetes (Liu et al., 2016; Zhang et al., 2016). The PKC, which is activated under oxidative stress, can also contribute to myocardial fibrosis by activating NF-κB and the sequential collagen accumulation (Bhattacharjee et al., 2017). The inhibition of redox sensitive NF-κB signaling has been shown to attenuate hyperglycemia-induced cardiac injuries (Chen X. et al., 2018).
In summary, mdROS, which is increased in diabetic human atrial tissue (Anderson et al., 2009), could play important roles in the pathophysiology of cardiac fibrosis in DbCM, among many other contributing factors, likely through regulating various redox signaling pathways.
Cardiac Fibrosis and Atrial Fibrillation in DbCM
Atrial fibrillation (AF) is characterized by rapid and uncoordinated atrial activity that causes ineffective atrial contraction. As the most common sustained arrhythmia in clinical practice, AF causes major cardiovascular morbidity and mortality in DM patients (Karam et al., 2017). A recent cross sectional survey in China reported that the prevalence of AF in patients with DM is significantly higher than those without known DM (Hu and Sun, 2008). Another study in Japan showed the similar findings that the incidence of AF in diabetic patients is much higher than in the control group (Iguchi et al., 2008).
Recently, DbCM-related excessive fibrosis has emerged as a hallmark and independent risk factor for both triggering and sustaining AF (Movahed et al., 2005). Myocardial fibrosis in the atrial tissue can elicit AF by causing abnormal cell coupling, which has been observed in DM patients. Cardiac fibroblasts (CF) are non-excitable cells that obstruct the orderly spread of electrical impulse in the hearts by producing an insulating layer of ECM to physically separate groups of myocytes. Thus, excessive CF forms collagen-rich myocardial tissue that causes reduced inter-cardiomyocyte coupling and increased axial resistance, leading to impaired propagation of cardiac action potentials and formation of reentrant excitations. On the other hand, CF can couple to myocytes via gap junction proteins Cx43 and connexin-45 (Kohl, 2003; Chilton et al., 2007) and studies have shown that the level of Cx43 expression increases significantly in diabetic rat myocardium (Watanabe et al., 2012). As CF have a high cell membrane resistance that makes them excellent long-distance conductors (Kohl, 2003), increased coupling between CF and cardiomyocytes will decrease the conduction velocity and induce synchronous spontaneous activity in distant cardiomyocytes (Viennet et al., 2003; Abdullah et al., 2016). In addition, studies have revealed that atrial effective refractory period (AERP) is shortened in the diabetic rat atrium, which results in increased dispersion of AERP and formation of unidirectional conduction block (Otake et al., 2009), predisposing to reentrant AF.
Mitochondrial-Derived Oxidative Stress and Atrial Electrical Remodeling in DbCM
In addition to promoting arrhythmogenesis through enhancing fibrosis and structural remodeling, mdROS are also involved in the proarrhythmic electrical remodeling in DM [readers are referred to Koektuerk et al. (2016) and Karam et al. (2017) for comprehensive reviews]. For instance, a recent study has shown that dipeptidyl peptidase-4 inhibitor prevents mitochondrial dysfunction (e.g., mdROS production and mitochondrial depolarization) and ameliorates atrial remodeling and arrhythmic substrates in the diabetic rabbits (Zhang et al., 2017). The potential link between mdROS and atrial remodeling in the setting of DM has also been suggested by another study showing that allopurinol, a xanthine oxidase inhibitor, prevents alloxan-induced atrial interstitial fibrosis, abnormal Ca2+ handling and atrial fibrillation inducibility in rabbits (Yang et al., 2018). Furthermore, studies have revealed significant alterations of electrical structure such as effective refractory period (Otake et al., 2009) and gap junction conduction (Joshi et al., 2015) in the atrial of diabetic animals. These studies suggest that mitochondrial dysfunction-induced atrial remodeling may underly the onset and maintaining to AF.
Summary
Mitochondrial dysfunction plays a potential role in the generation and maintenance of cardiac arrhythmias in diabetic hearts. The loss of function of mitochondria is associated with not only reduced ATP production, increased ROS production and acute changes in ion levels, but also dysregulation of multiple signaling pathways regulating transcript expression and fibrosis. All of those could contribute to cardiac arrhythmogenesis (Figure 2), suggesting that mitochondria could be an upstream therapeutic target for treatment of arrhythmias in DbCM.
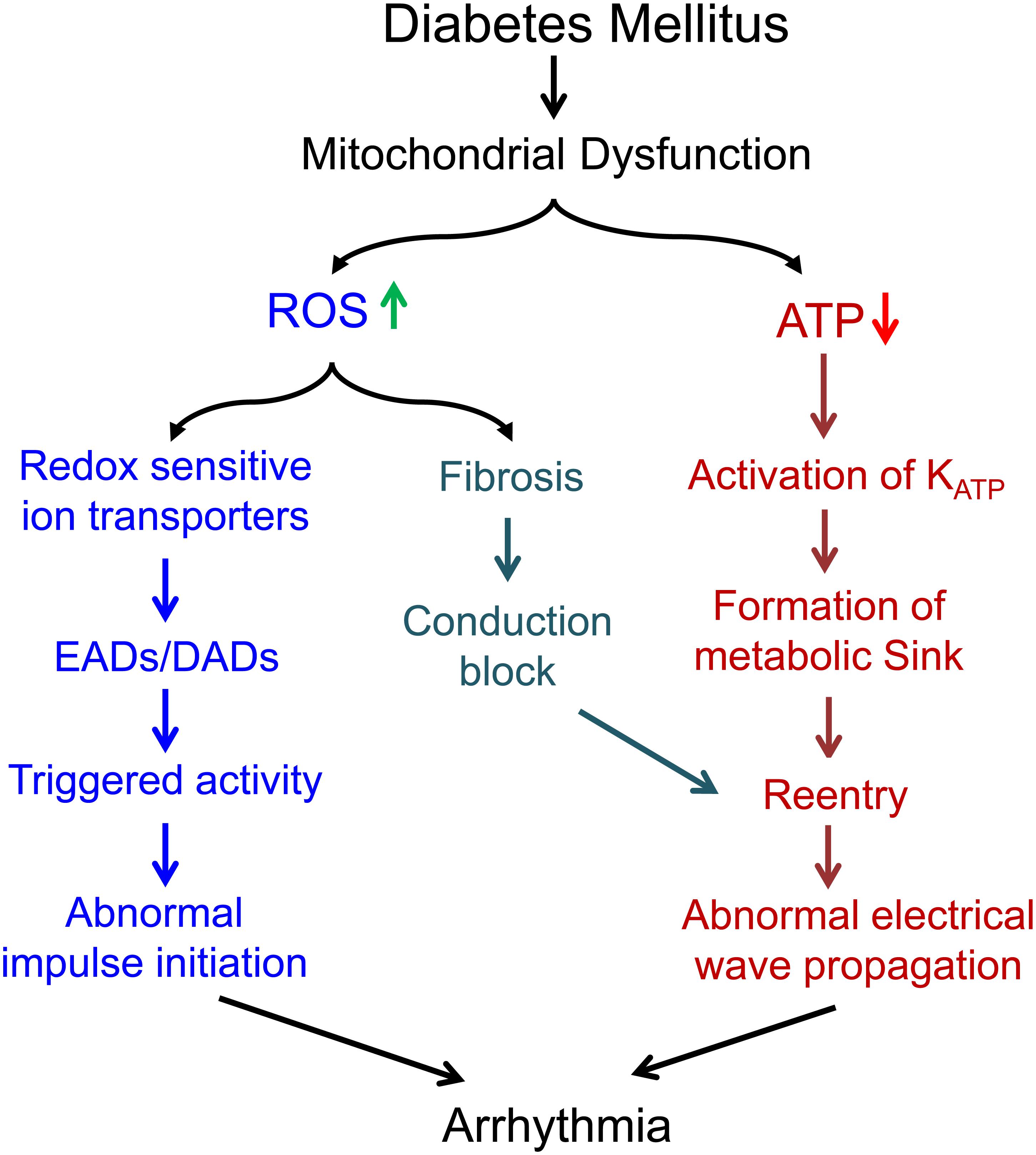
Figure 2. Summary of the potential mechanisms underlying the proarrhythmic role of mitochondrial dysfunction in diabetes mellitus.
Author Contributions
All authors prepared the manuscript. LZ finalized the manuscript.
Funding
This work was supported by National Institutes of Health R01HL121206.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdullah, C. S., Li, Z., Wang, X., and Jin, Z.-Q. (2016). Depletion of T lymphocytes ameliorates cardiac fibrosis in streptozotocin-induced diabetic cardiomyopathy. Int. Immunopharmacol. 39, 251–264. doi: 10.1016/j.intimp.2016.07.027
Akar, F. G., Aon, M. A., Tomaselli, G. F., and O’Rourke, B. (2005). The mitochondrial origin of postischemic arrhythmias. J. Clin. Invest. 115, 3527–3535. doi: 10.1172/JCI25371
Anderson, E. J., Kypson, A. P., Rodriguez, E., Anderson, C. A., Lehr, E. J., and Neufer, P. D. (2009). Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 54, 1891–1898. doi: 10.1016/j.jacc.2009.07.031
Antzelevitch, C., and Burashnikov, A. (2011). Overview of basic mechanisms of cardiac arrhythmia. Card Electrophysiol. Clin. 3, 23–45. doi: 10.1016/j.ccep.2010.10.012
Aon, M. A., Cortassa, S., Akar, F. G., Brown, D. A., Zhou, L., and O’Rourke, B. (2009). From mitochondrial dynamics to arrhythmias. Int. J. Biochem. Cell Biol. 41, 1940–1948. doi: 10.1016/j.biocel.2009.02.016
Aon, M. A., Cortassa, S., Marban, E., and O’Rourke, B. (2003). Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J. Biol. Chem. 278, 44735–44744. doi: 10.1074/jbc.M302673200
Association, A. D. (2000). Type 2 diabetes in children and adolescents. Pediatrics 105, 671–680. doi: 10.1542/peds.105.3.671
Bagheri, F., Khori, V., Alizadeh, A. M., Khalighfard, S., Khodayari, S., and Khodayari, H. (2016). Reactive oxygen species-mediated cardiac-reperfusion injury: mechanisms and therapies. Life Sci. 165, 43–55. doi: 10.1016/j.lfs.2016.09.013
Barrington, P. L., Meier, C. F. Jr., and Weglicki, W. B. (1988). Abnormal electrical activity induced by free radical generating systems in isolated cardiocytes. J. Mol. Cell Cardiol. 20, 1163–1178. doi: 10.1016/0022-2828(88)90596-2
Bassani, R. A., Mattiazzi, A., and Bers, D. M. (1995). CaMKII is responsible for activity-dependent acceleration of relaxation in rat ventricular myocytes. Am. J. Physiol. 268(2 Pt 2), H703–H712. doi: 10.1152/ajpheart.1995.268.2.H703
Bers, D. M., and Despa, S. (2006). Cardiac myocytes Ca2+ and Na+ regulation in normal and failing hearts. J. Pharmacol. Sci. 100, 315–322. doi: 10.1254/jphs.CPJ06001X
Berthoud, V. M., and Beyer, E. C. (2009). Oxidative stress, lens gap junctions, and cataracts. Antioxid. Redox Signal. 11, 339–353. doi: 10.1089/ars.2008.2119
Bhattacharjee, N., Dua, T. K., Khanra, R., Joardar, S., Nandy, A., Saha, A., et al. (2017). Protocatechuic acid, a phenolic from Sansevieria roxburghiana leaves, suppresses diabetic cardiomyopathy via stimulating glucose metabolism, ameliorating oxidative stress, and inhibiting inflammation. Front. Pharmacol. 8:251. doi: 10.3389/fphar.2017.00251
Blaich, A., Welling, A., Fischer, S., Wegener, J. W., Kostner, K., Hofmann, F., et al. (2010). Facilitation of murine cardiac L-type Ca(v)1.2 channel is modulated by calmodulin kinase II-dependent phosphorylation of S1512 and S1570. Proc. Natl. Acad. Sci. U.S.A. 107, 10285–10289. doi: 10.1073/pnas.0914287107
Block, K., Gorin, Y., and Abboud, H. E. (2009). Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. U.S.A. 106, 14385–14390. doi: 10.1073/pnas.0906805106
Brown, D., Aon, M. A., Akar, F. G., Liu, T., Sorarrain, N., and O’Rourke, B. (2008). Effect of 4’-chlorodiazepam on cellular excitation-contraction coupling and ischemia-reperfusion injury in rabbit heart. Cardiovasc. Res. 79, 141–149. doi: 10.1093/cvr/cvn053
Bugyei-Twum, A., Advani, A., Advani, S. L., Zhang, Y., Thai, K., Kelly, D. J., et al. (2014). High glucose induces Smad activation via the transcriptional coregulator p300 and contributes to cardiac fibrosis and hypertrophy. Cardiovasc. Diabetol. 13:89. doi: 10.1186/1475-2840-13-89
Chance, B., Sies, H., and Boveris, A. (1979). Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 59, 527–605. doi: 10.1152/physrev.1979.59.3.527
Chen, C., Meng, Z., Zheng, Y., Hu, B., and Shen, E. (2018). Fibroblast growth factor 21 inhibition aggravates cardiac dysfunction in diabetic cardiomyopathy by improving lipid accumulation. Exp. Ther. Med. 15, 75–84. doi: 10.3892/etm.2017.5375
Chen, X., Qian, J., Wang, L., Li, J., Zhao, Y., Han, J., et al. (2018). Kaempferol attenuates hyperglycemia-induced cardiac injuries by inhibiting inflammatory responses and oxidative stress. Endocrine 60, 83–94. doi: 10.1007/s12020-018-1525-4
Chilton, L., Giles, W. R., and Smith, G. L. (2007). Evidence of intercellular coupling between co-cultured adult rabbit ventricular myocytes and myofibroblasts. J. Physiol. 583(Pt 1), 225–236. doi: 10.1113/jphysiol.2007.135038
Coetzee, W. A., and Opie, L. H. (1992). Effects of oxygen free radicals on isolated cardiac myocytes from guinea-pig ventricle: electrophysiological studies. J. Mol. Cell Cardiol. 24, 651–663. doi: 10.1016/0022-2828(92)91049-B
Cohn, J. N. (1996). Prognosis in congestive heart failure. J. Card. Fail. 2(4 Suppl.), S225–S229. doi: 10.1016/S1071-9164(96)80081-9
Cohn, J. N., Archibald, D. G., Ziesche, S., Franciosa, J. A., Harston, W. E., Tristani, F. E., et al. (1986). Effect of vasodilator therapy on mortality in chronic congestive heart failure. Results of a veterans administration cooperative study. N. Engl. J. Med. 314, 1547–1552. doi: 10.1056/NEJM198606123142404
Cucoranu, I., Clempus, R., Dikalova, A., Phelan, P. J., Ariyan, S., Dikalov, S., et al. (2005). NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 97, 900–907. doi: 10.1161/01.RES.0000187457.24338.3D
Dai, D. F., Johnson, S. C., Villarin, J. J., Chin, M. T., Nieves-Cintron, M., Chen, T., et al. (2011). Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ. Res. 108, 837–846. doi: 10.1161/CIRCRESAHA.110.232306
Dayer, C., and Stamenkovic, I. (2015). Recruitment of Matrix Metalloproteinase-9 (MMP-9) to the fibroblast cell surface by Lysyl Hydroxylase-3 (LH3) triggers TGF-β activation and fibroblast differentiation. J. Biol. Chem. 290, 13763–13778. doi: 10.1074/jbc.M114.622274
De Jong, S., van Veen, T. A., van Rijen, H. V., and de Bakker, J. M. (2011). Fibrosis and cardiac arrhythmias. J. Cardiovasc. Pharmacol. 57, 630–638. doi: 10.1097/FJC.0b013e318207a35f
De Marchi, U., Castelbou, C., and Demaurex, N. (2011). Uncoupling protein 3 (UCP3) modulates the activity of Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) by decreasing mitochondrial ATP production. J. Biol. Chem. 286, 32533–32541. doi: 10.1074/jbc.M110.216044
D’Souza, K. M., Oh, C., Zhang, W., and Reaven, P. D. (2018). TXNIP as a mediator of palmitic acid-induced programmed cell-death in cardiomyocytes. FASEB J. 32:719.14.
Dudley, S. C. Jr., Hoch, N. E., McCann, L. A., Honeycutt, C., Diamandopoulos, L., Fukai, T., et al. (2005). Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: role of the NADPH and xanthine oxidases. Circulation 112, 1266–1273. doi: 10.1161/CIRCULATIONAHA.105.538108
Duthe, F., Plaisance, I., Sarrouilhe, D., and Herve, J. C. (2001). Endogenous protein phosphatase 1 runs down gap junctional communication of rat ventricular myocytes. Am. J. Physiol. Cell Physiol. 281, C1648–C1656. doi: 10.1152/ajpcell.2001.281.5.C1648
Dzhura, I., Wu, Y., Colbran, R. J., Balser, J. R., and Anderson, M. E. (2000). Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat. Cell Biol. 2, 173–177. doi: 10.1038/35004052
Eager, K. R., Roden, L. D., and Dulhunty, A. F. (1997). Actions of sulfhydryl reagents on single ryanodine receptor Ca2+-release channels from sheep myocardium. Am. J. Physiol. 272(6 Pt 1), C1908–C1918. doi: 10.1152/ajpcell.1997.272.6.C1908
Erickson, J. R., He, B. J., Grumbach, I. M., and Anderson, M. E. (2011). CaMKII in the cardiovascular system: sensing redox states. Physiol. Rev. 91, 889–915. doi: 10.1152/physrev.00018.2010
Erickson, J. R., Joiner, M. L., Guan, X., Kutschke, W., Yang, J., Oddis, C. V., et al. (2008). A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462–474. doi: 10.1016/j.cell.2008.02.048
Fan, D., Takawale, A., Lee, J., and Kassiri, Z. (2012). Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair 5:15. doi: 10.1186/1755-1536-5-15
Froese, A. R., Shimbori, C., Bellaye, P. S., Inman, M., Obex, S., Fatima, S., et al. (2016). Stretch-induced activation of transforming growth Factor-beta1 in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 194, 84–96. doi: 10.1164/rccm.201508-1638OC
Gen, W., Tani, M., Takeshita, J., Ebihara, Y., and Tamaki, K. (2001). Mechanisms of Ca2+ overload induced by extracellular H2O2 in quiescent isolated rat cardiomyocytes. Basic Res. Cardiol. 96, 623–629. doi: 10.1007/s003950170014
Grueter, C. E., Abiria, S. A., Dzhura, I., Wu, Y., Ham, A. J., Mohler, P. J., et al. (2006). L-type Ca2+ channel facilitation mediated by phosphorylation of the beta subunit by CaMKII. Mol. Cell. 23, 641–650. doi: 10.1016/j.molcel.2006.07.006
Guo, T., Zhang, T., Mestril, R., and Bers, D. M. (2006). Ca2+/Calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ. Res. 99, 398–406. doi: 10.1161/01.RES.0000236756.06252.13
Ho, H. T., Liu, B., Snyder, J. S., Lou, Q., Brundage, E. A., Velez-Cortes, F., et al. (2014). Ryanodine receptor phosphorylation by oxidized CaMKII contributes to the cardiotoxic effects of cardiac glycosides. Cardiovasc. Res. 101, 165–174. doi: 10.1093/cvr/cvt233
Howe, C. J., Lahair, M. M., McCubrey, J. A., and Franklin, R. A. (2004). Redox regulation of the calcium/calmodulin-dependent protein kinases. J. Biol. Chem. 279, 44573–44581. doi: 10.1074/jbc.M404175200
Hu, D., and Sun, Y. (2008). Epidemiology, risk factors for stroke, and management of atrial fibrillation in China. J. Am. Coll. Cardiol. 52, 865–868. doi: 10.1016/j.jacc.2008.05.042
Hudmon, A., Schulman, H., Kim, J., Maltez, J. M., Tsien, R. W., and Pitt, G. S. (2005). CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J. Cell Biol. 171, 537–547. doi: 10.1083/jcb.200505155
Huke, S., and Knollmann, B. C. (2011). Oxidized CaMKII: a “heart stopper” for the sinus node? J. Clin. Invest. 121, 2975–2977. doi: 10.1172/JCI58389
Huxley, R. R., Filion, K. B., Konety, S., and Alonso, A. (2011). Meta-analysis of cohort and case-control studies of type 2 diabetes mellitus and risk of atrial fibrillation. Am. J. Cardiol. 108, 56–62. doi: 10.1016/j.amjcard.2011.03.004
Iguchi, Y., Kimura, K., Aoki, J., Kobayashi, K., Terasawa, Y., Sakai, K., et al. (2008). Prevalence of atrial fibrillation in community-dwelling Japanese aged 40 years or older in Japan: analysis of 41,436 non-employee residents in Kurashiki-city. Circ. J. 72, 909–913. doi: 10.1253/circj.72.909
Ismahil, M. A., Hamid, T., Bansal, S. S., Patel, B., Kingery, J. R., and Prabhu, S. D. (2014). Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ. Res. 114, 266–282. doi: 10.1161/CIRCRESAHA.113.301720
Jeong, E. M., Chung, J., Liu, H., Go, Y., Gladstein, S., Farzaneh-Far, A., et al. (2016). Role of mitochondrial oxidative stress in glucose tolerance, insulin resistance, and cardiac diastolic dysfunction. J. Am. Heart Assoc. 5:e003046. doi: 10.1161/JAHA.115.003046
Jeong, E. M., Liu, M., Sturdy, M., Gao, G., Varghese, S. T., Sovari, A. A., et al. (2012). Metabolic stress, reactive oxygen species, and arrhythmia. J. Mol. Cell Cardiol. 52, 454–463. doi: 10.1016/j.yjmcc.2011.09.018
Jiang, F., Liu, G. S., Dusting, G. J., and Chan, E. C. (2014). NADPH oxidase-dependent redox signaling in TGF-beta-mediated fibrotic responses. Redox Biol. 2, 267–272. doi: 10.1016/j.redox.2014.01.012
Joiner, M. L., Koval, O. M., Li, J., He, B. J., Allamargot, C., Gao, Z., et al. (2012). CaMKII determines mitochondrial stress responses in heart. Nature 491, 269–273. doi: 10.1038/nature11444
Joshi, M. S., Mihm, M. J., Cook, A. C., Schanbacher, B. L., and Bauer, J. A. (2015). Alterations in connexin 43 during diabetic cardiomyopathy: competition of tyrosine nitration versus phosphorylation. J. Diabetes 7, 250–259. doi: 10.1111/1753-0407.12164
Karam, B. S., Chavez-Moreno, A., Koh, W., Akar, J. G., and Akar, F. G. (2017). Oxidative stress and inflammation as central mediators of atrial fibrillation in obesity and diabetes. Cardiovasc. Diabetol. 16:120. doi: 10.1186/s12933-017-0604-9
Khalil, H., Kanisicak, O., Prasad, V., Correll, R. N., Fu, X., Schips, T., et al. (2017). Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Invest. 127, 3770–3783. doi: 10.1172/JCI94753
Koektuerk, B., Aksoy, M., Horlitz, M., Bozdag-Turan, I., and Turan, R. G. (2016). Role of diabetes in heart rhythm disorders. World J. Diabetes 7, 45–49. doi: 10.4239/wjd.v7.i3.45
Kohl, P. (2003). Heterogeneous cell coupling in the heart: an electrophysiological role for fibroblasts. Circ. Res. 93, 381–383. doi: 10.1161/01.RES.0000091364.90121.0C
Kohlhaas, M., Zhang, T., Seidler, T., Zibrova, D., Dybkova, N., Steen, A., et al. (2006). Increased sarcoplasmic reticulum calcium leak but unaltered contractility by acute CaMKII overexpression in isolated rabbit cardiac myocytes. Circ. Res. 98, 235–244. doi: 10.1161/01.RES.0000200739.90811.9f
Koitabashi, N., Danner, T., Zaiman, A. L., Pinto, Y. M., Rowell, J., Mankowski, J., et al. (2011). Pivotal role of cardiomyocyte TGF-beta signaling in the murine pathological response to sustained pressure overload. J. Clin. Invest. 121, 2301–2312. doi: 10.1172/JCI44824
Kourie, J. I. (1998). Interaction of reactive oxygen species with ion transport mechanisms. Am. J. Physiol. 275(1 Pt 1), C1–C24. doi: 10.1152/ajpcell.1998.275.1.C1
Landolfi, B., Curci, S., Debellis, L., Pozzan, T., and Hofer, A. M. (1998). Ca2+ homeostasis in the agonist-sensitive internal store: functional interactions between mitochondria and the ER measured In situ in intact cells. J. Cell Biol. 142, 1235–1243. doi: 10.1083/jcb.142.5.1235
Li, G., Xing, W., Zhang, M., Geng, F.-H., Yang, H., Zhang, H., et al. (2018). Anti-fibrotic cardioprotection of berberine via down-regulating myocardial IGF-1 receptor-regulated MMP-2/9 expression in diabetic rats. Am. J. Physiol. Heart Circ. Physiol. 315, H802–H813. doi: 10.1152/ajpheart.00093.2018
Li, N., Frigerio, F., and Maechler, P. (2008). The sensitivity of pancreatic beta-cells to mitochondrial injuries triggered by lipotoxicity and oxidative stress. Biochem. Soc. Trans. 36(Pt 5), 930–934. doi: 10.1042/BST0360930
Li, Q., Su, D., O’Rourke, B., Pogwizd, S. M., and Zhou, L. (2015). Mitochondria-derived ROS bursts disturb Ca(2)(+) cycling and induce abnormal automaticity in guinea pig cardiomyocytes: a theoretical study. Am. J. Physiol. Heart Circ. Physiol. 308, H623–H636. doi: 10.1152/ajpheart.00493.2014
Lijnen, P., Petrov, V., Rumilla, K., and Fagard, R. (2003). Transforming growth factor-beta 1 promotes contraction of collagen gel by cardiac fibroblasts through their differentiation into myofibroblasts. Methods Find. Exp. Clin. Pharmacol. 25, 79–86. doi: 10.1358/mf.2003.25.2.723680
Liu, M., Liu, H., and Dudley, S. C. Jr. (2010). Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ. Res. 107, 967–974. doi: 10.1161/CIRCRESAHA.110.220673
Liu, Q., Wang, S., and Cai, L. (2014). Diabetic cardiomyopathy and its mechanisms: role of oxidative stress and damage. J. Diabetes Invest. 5, 623–634. doi: 10.1111/jdi.12250
Liu, T., Takimoto, E., Dimaano, V. L., DeMazumder, D., Kettlewell, S., Smith, G., et al. (2014). Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ. Res. 115, 44–54. doi: 10.1161/CIRCRESAHA.115.303062
Liu, X., Liu, C., Li, J., Zhang, X., Song, F., and Xu, J. (2016). Urocortin attenuates myocardial fibrosis in diabetic rats via the Akt/GSK-3β signaling pathway. Endocr. Res. 41, 148–157. doi: 10.3109/07435800.2015.1094489
Luo, M., Guan, X., Luczak, E. D., Lang, D., Kutschke, W., Gao, Z., et al. (2013). Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J. Clin. Invest. 123, 1262–1274. doi: 10.1172/JCI65268
Ly, L. D., Xu, S., Choi, S. K., Ha, C. M., Thoudam, T., Cha, S. K., et al. (2017). Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 49:e291. doi: 10.1038/emm.2016.157
Maack, C., and O’Rourke, B. (2007). Excitation-contraction coupling and mitochondrial energetics. Basic Res. Cardiol. 102, 369–392. doi: 10.1007/s00395-007-0666-z
Maier, L. S., and Bers, D. M. (2007). Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc. Res. 73, 631–640. doi: 10.1016/j.cardiores.2006.11.005
Maier, L. S., Zhang, T., Chen, L., DeSantiago, J., Brown, J. H., and Bers, D. M. (2003). Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ. Res. 92, 904–911. doi: 10.1161/01.RES.0000069685.20258.F1
Medici, D., and Kalluri, R. (2012). Endothelial–mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin. Cancer Biol. 22, 379–384. doi: 10.1016/j.semcancer.2012.04.004
Medici, D., Potenta, S., and Kalluri, R. (2011). Transforming growth factor-β2 promotes Snail-mediated endothelial–mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem. J. 437, 515–520. doi: 10.1042/BJ20101500
Mesubi, O., Rokita, A., Abrol, N., Wu, Y., Chen, B., Wang, Q., et al. (2017). CaMKII oxidation causes increased atrial fibrillation in diabetic mice. J. Mol. Cell Cardiol. 112, 161–162. doi: 10.1016/j.yjmcc.2017.07.092
Min, Q., Bai, Y., Zhang, Y., Yu, W., Zhang, M., Liu, D., et al. (2017). Hawthorn leaf flavonoids protect against diabetes-induced cardiomyopathy in rats via PKC-α signaling pathway. Evid. Based Complement. Altern. Med. 2017:2071952. doi: 10.1155/2017/2071952
Montaigne, D., Marechal, X., Coisne, A., Debry, N., Modine, T., Fayad, G., et al. (2014). Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 130, 554–564. doi: 10.1161/CIRCULATIONAHA.113.008476
Montaigne, D., and Pentiah, A. D. (2015). Mitochondrial cardiomyopathy and related arrhythmias. Card. Electrophysiol. Clin. 7, 293–301. doi: 10.1016/j.ccep.2015.03.008
Morita, N., Lee, J. H., Xie, Y., Sovari, A., Qu, Z., Weiss, J. N., et al. (2011). Suppression of re-entrant and multifocal ventricular fibrillation by the late sodium current blocker ranolazine. J. Am. Coll. Cardiol. 57, 366–375. doi: 10.1016/j.jacc.2010.07.045
Morris, T. E., and Sulakhe, P. V. (1997). Sarcoplasmic reticulum Ca2+-pump dysfunction in rat cardiomyocytes briefly exposed to hydroxyl radicals. Free Radic. Biol. Med. 22, 37–47. doi: 10.1016/S0891-5849(96)00238-9
Movahed, M. R., Hashemzadeh, M., and Jamal, M. M. (2005). Diabetes mellitus is a strong, independent risk for atrial fibrillation and flutter in addition to other cardiovascular disease. Int. J. Cardiol. 105, 315–318. doi: 10.1016/j.ijcard.2005.02.050
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/bj20081386
Nakaya, H., Takeda, Y., Tohse, N., and Kanno, M. (1992). Mechanism of the membrane depolarization induced by oxidative stress in guinea-pig ventricular cells. J. Mol. Cell Cardiol. 24, 523–534. doi: 10.1016/0022-2828(92)91841-R
Nguyen, T. P., Qu, Z., and Weiss, J. N. (2014). Cardiac fibrosis and arrhythmogenesis: the road to repair is paved with perils. J. Mol. Cell Cardiol. 70, 83–91. doi: 10.1016/j.yjmcc.2013.10.018
Oda, T., Yang, Y., Uchinoumi, H., Thomas, D. D., Chen-Izu, Y., Kato, T., et al. (2015). Oxidation of ryanodine receptor (RyR) and calmodulin enhance Ca release and pathologically alter, RyR structure and calmodulin affinity. J. Mol. Cell Cardiol. 85, 240–248. doi: 10.1016/j.yjmcc.2015.06.009
Otake, H., Suzuki, H., Honda, T., and Maruyama, Y. (2009). Influences of autonomic nervous system on atrial arrhythmogenic substrates and the incidence of atrial fibrillation in diabetic heart. Int. Heart J. 50, 627–641. doi: 10.1536/ihj.50.627
Pal, P. B., Sinha, K., and Sil, P. C. (2014). Mangiferin attenuates diabetic nephropathy by inhibiting oxidative stress mediated signaling cascade, TNFα related and mitochondrial dependent apoptotic pathways in streptozotocin-induced diabetic rats. PLoS One 9:e107220. doi: 10.1371/journal.pone.0107220
Palikaras, K., and Tavernarakis, N. (2014). Mitochondrial homeostasis: the interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 56, 182–188. doi: 10.1016/j.exger.2014.01.021
Roussel, J., Thireau, J., Brenner, C., Saint, N., Scheuermann, V., Lacampagne, A., et al. (2015). Palmitoyl-carnitine increases RyR2 oxidation and sarcoplasmic reticulum Ca2+ leak in cardiomyocytes: Role of adenine nucleotide translocase. Biochim. Biophys. Acta 1852, 749–758. doi: 10.1016/j.bbadis.2015.01.011
Sag, C. M., Wolff, H. A., Neumann, K., Opiela, M. K., Zhang, J., Steuer, F., et al. (2013). Ionizing radiation regulates cardiac Ca handling via increased ROS and activated CaMKII. Basic Res. Cardiol. 108:385. doi: 10.1007/s00395-013-0385-6
Schulz, R., and Heusch, G. (2004). Connexin 43 and ischemic preconditioning. Cardiovasc. Res. 62, 335–344. doi: 10.1016/j.cardiores.2003.12.017
Shang, L. L., Sanyal, S., Pfahnl, A. E., Jiao, Z., Allen, J., Liu, H., et al. (2008). NF-kappaB-dependent transcriptional regulation of the cardiac scn5a sodium channel by angiotensin II. Am. J. Physiol. Cell Physiol. 294, C372–C379. doi: 10.1152/ajpcell.00186.2007
Shen, N., Li, X., Zhou, T., Bilal, M. U., Du, N., Hu, Y., et al. (2014). Shensong Yangxin capsule prevents diabetic myocardial fibrosis by inhibiting TGF-β1/Smad signaling. J. Ethnopharmacol. 157, 161–170. doi: 10.1016/j.jep.2014.09.035
Singh, S., McClung, J., Bellner, L., Cao, J., Waldman, M., Schragenheim, J., et al. (2018). CYP-450 epoxygenase derived epoxyeicosatrienoic acid contribute to reversal of heart failure in obesity-induced diabetic cardiomyopathy via PGC-1 α activation. Cardiovasc. Pharm. Open Access 7:233. doi: 10.4172/2329-6607.1000233
Sohl, G., and Willecke, K. (2004). Gap junctions and the connexin protein family. Cardiovasc. Res. 62, 228–232. doi: 10.1016/j.cardiores.2003.11.013
Song, Y., Shryock, J. C., Wagner, S., Maier, L. S., and Belardinelli, L. (2006). Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J. Pharmacol. Exp. Ther. 318, 214–222. doi: 10.1124/jpet.106.101832
Suarez, J., Scott, B., and Dillmann, W. H. (2008). Conditional increase in SERCA2a protein is able to reverse contractile dysfunction and abnormal calcium flux in established diabetic cardiomyopathy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R1439–R1445. doi: 10.1152/ajpregu.00736.2007
Sugiura, H., Toyama, J., Tsuboi, N., Kamiya, K., and Kodama, I. (1990). ATP directly affects junctional conductance between paired ventricular myocytes isolated from guinea pig heart. Circ. Res. 66, 1095–1102. doi: 10.1161/01.RES.66.4.1095
Szendroedi, J., Frossard, M., Klein, N., Bieglmayer, C., Wagner, O., Pacini, G., et al. (2012). Lipid-induced insulin resistance is not mediated by impaired transcapillary transport of insulin and glucose in humans. Diabetes Metab. Res. Rev. 61, 3176–3180. doi: 10.2337/db12-0108
Tse, G. (2016). Mechanisms of cardiac arrhythmias. J. Arrhythm 32, 75–81. doi: 10.1016/j.joa.2015.11.003
Tuomilehto, J., and Lindstrom, J. (2003). The major diabetes prevention trials. Curr. Diab. Rep. 3, 115–122. doi: 10.1007/s11892-003-0034-9
Vanden Hoek, T. L., Becker, L. B., Shao, Z., Li, C., and Schumacker, P. T. (1998). Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J. Biol. Chem. 273, 18092–18098. doi: 10.1074/jbc.273.29.18092
Verrecchia, F., and Mauviel, A. (2002). Transforming growth factor-beta signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J. Invest. Dermatol. 118, 211–215. doi: 10.1046/j.1523-1747.2002.01641.x
Viennet, C., Bride, J., Gabiot, A. C., and Humbert, P. (2003). Comparison of different wound dressings on cultured human fibroblasts and collagen lattices. J. Wound Care 12, 385–390. doi: 10.12968/jowc.2003.12.10.26544
Wagner, S., Dybkova, N., Rasenack, E. C., Jacobshagen, C., Fabritz, L., Kirchhof, P., et al. (2006). Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 116, 3127–3138. doi: 10.1172/JCI26620
Wagner, S., Rokita, A. G., Anderson, M. E., and Maier, L. S. (2013). Redox regulation of sodium and calcium handling. Antioxid. Redox. Signal. 18, 1063–1077. doi: 10.1089/ars.2012.4818
Wagner, S., Ruff, H. M., Weber, S. L., Bellmann, S., Sowa, T., Schulte, T., et al. (2011). Reactive oxygen species-activated Ca/calmodulin kinase IIdelta is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ. Res. 108, 555–565. doi: 10.1161/CIRCRESAHA.110.221911
Walker, A. M., and Cubbon, R. M. (2015). Sudden cardiac death in patients with diabetes mellitus and chronic heart failure. Diab. Vasc. Dis. Res. 12, 228–233. doi: 10.1177/1479164115573225
Ward, C. A., and Giles, W. R. (1997). Ionic mechanism of the effects of hydrogen peroxide in rat ventricular myocytes. J. Physiol. 500( Pt 3), 631–642. doi: 10.1113/jphysiol.1997.sp022048
Ward, C. A., and Moffat, M. P. (1995). Role of protein kinase C in mediating effects of hydrogen peroxide in guinea-pig ventricular myocytes. J. Mol. Cell Cardiol. 27, 1089–1097. doi: 10.1016/0022-2828(95)90077-2
Watanabe, M., Yokoshiki, H., Mitsuyama, H., Mizukami, K., Ono, T., and Tsutsui, H. (2012). Conduction and refractory disorders in the diabetic atrium. Am. J. Physiol. Heart Circ. Physiol. 303, H86–H95. doi: 10.1152/ajpheart.00010.2012
Wehrens, X. H., Lehnart, S. E., Reiken, S. R., and Marks, A. R. (2004). Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 94, e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2
Wu, H., Li, G.-N., Xie, J., Li, R., Chen, Q.-H., Chen, J.-Z., et al. (2016). Resveratrol ameliorates myocardial fibrosis by inhibiting ROS/ERK/TGF-β/periostin pathway in STZ-induced diabetic mice. BMC Cardiovasc. Disord. 16:5. doi: 10.1186/s12872-015-0169-z
Xie, C., Biary, N., Tocchetti, C. G., Aon, M. A., Paolocci, N., Kauffman, J., et al. (2013). Glutathione oxidation unmasks proarrhythmic vulnerability of chronically hyperglycemic guinea pigs. Am. J. Physiol. Heart Circ. Physiol. 304, H916–H926. doi: 10.1152/ajpheart.00026.2012
Xie, L. H., Chen, F., Karagueuzian, H. S., and Weiss, J. N. (2009). Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ. Res. 104, 79–86. doi: 10.1161/CIRCRESAHA.108.183475
Xu, L., Eu, J. P., Meissner, G., and Stamler, J. S. (1998). Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279, 234–237. doi: 10.1126/science.279.5348.234
Xu, S., Nam, S. M., Kim, J. H., Das, R., Choi, S. K., Nguyen, T. T., et al. (2015). Palmitate induces ER calcium depletion and apoptosis in mouse podocytes subsequent to mitochondrial oxidative stress. Cell Death Dis. 6:e1976. doi: 10.1038/cddis.2015.331
Xu, Z., Sun, J., Tong, Q., Lin, Q., Qian, L., Park, Y., et al. (2016). The role of ERK1/2 in the development of diabetic cardiomyopathy. Int. J. Mol. Sci. 17:E2001. doi: 10.3390/ijms17122001
Yan, Y., Liu, J., Wei, C., Li, K., Xie, W., Wang, Y., et al. (2008). Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovasc. Res. 77, 432–441. doi: 10.1093/cvr/cvm047
Yang, R., Ernst, P. J., Song, J., Liu, X., Huke, S., Wang, S., et al. (2018). Mitochondrial-mediated oxidative Ca2+/calmodulin-dependent kinase II activation induces early afterdepolarizations in guinea pig cardiomyocytes: an in silico study. J. Am. Heart Assoc. 7:e008939. doi: 10.1161/JAHA.118.008939
Yang, Y., Zhao, J., Qiu, J., Li, J., Liang, X., Zhang, Z., et al. (2018). Xanthine oxidase inhibitor allopurinol prevents oxidative stress-mediated atrial remodeling in alloxan-induced diabetes mellitus rabbits. J. Am. Heart Assoc. 7:e008807. doi: 10.1161/JAHA.118.008807
Ying, C., Liu, T., Ling, H., Cheng, M., Zhou, X., Wang, S., et al. (2017). Glucose variability aggravates cardiac fibrosis by altering AKT signalling path. Diab. Vasc. Dis. Res. 14, 327–335. doi: 10.1177/1479164117698917
Yue, Y., Meng, K., Pu, Y., and Zhang, X. (2017). Transforming growth factor beta (TGF-β) mediates cardiac fibrosis and induces diabetic cardiomyopathy. Diab. Res. Clin. Practice 133, 124–130. doi: 10.1016/j.diabres.2017.08.018
Yue, Z., Zhang, Y., Xie, J., Jiang, J., and Yue, L. (2013). Transient receptor potential (TRP) channels and cardiac fibrosis. Curr. Top. Med. Chem. 13, 270–282. doi: 10.2174/1568026611313030005
Zhang, T., and Brown, J. H. (2004). Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc. Res. 63, 476–486. doi: 10.1016/j.cardiores.2004.04.026
Zhang, X., Zhang, Z., Zhao, Y., Jiang, N., Qiu, J., Yang, Y., et al. (2017). Alogliptin, a dipeptidyl peptidase-4 inhibitor, alleviates atrial remodeling and improves mitochondrial function and biogenesis in diabetic rabbits. J. Am. Heart Assoc. 6:e005945. doi: 10.1161/JAHA.117.005945
Zhang, Y., Tocchetti, C. G., Krieg, T., and Moens, A. L. (2012). Oxidative and nitrosative stress in the maintenance of myocardial function. Free Radic. Biol. Med. 53, 1531–1540. doi: 10.1016/j.freeradbiomed.2012.07.010
Zhang, Y., Xiao, J., Wang, H., Luo, X., Wang, J., Villeneuve, L. R., et al. (2006). Restoring depressed HERG K+ channel function as a mechanism for insulin treatment of abnormal QT prolongation and associated arrhythmias in diabetic rabbits. Am. J. Physiol. Heart Circ. Physiol. 291, H1446–H1455. doi: 10.1152/ajpheart.01356.2005
Zhang, Y., Zhang, L., Zhang, Y., Xu, J.-J., Sun, L.-L., and Li, S.-Z. (2016). The protective role of liquiritin in high fructose-induced myocardial fibrosis via inhibiting NF-κB and MAPK signaling pathway. Biomed. Pharmacother. 84, 1337–1349. doi: 10.1016/j.biopha.2016.10.036
Zhou, L., Aon, M. A., Liu, T., and O’Rourke, B. (2011). Dynamic modulation of Ca2+ sparks by mitochondrial oscillations in isolated guinea pig cardiomyocytes under oxidative stress. J. Mol. Cell Cardiol. 51, 632–639. doi: 10.1016/j.yjmcc.2011.05.007
Zhou, L., Cortassa, S., Wei, A. C., Aon, M. A., Winslow, R. L., and O’Rourke, B. (2009). Modeling cardiac action potential shortening driven by oxidative stress-induced mitochondrial oscillations in guinea pig cardiomyocytes. Biophys. J. 97, 1843–1852. doi: 10.1016/j.bpj.2009.07.029
Zhou, L., and O’Rourke, B. (2012). Cardiac mitochondrial network excitability: insights from computational analysis. Am. J. Physiol. Heart Circ. Physiol. 302, H2178–H2189. doi: 10.1152/ajpheart.01073.2011
Zhou, L., Solhjoo, S., Millare, B., Plank, G., Abraham, M. R., Cortassa, S., et al. (2014). Effects of regional mitochondrial depolarization on electrical propagation: implications for arrhythmogenesis. Circ. Arrhythm Electrophysiol. 7, 143–151. doi: 10.1161/CIRCEP.113.000600
Ziegelhoffer, A., Kjeldsen, K., Bundgaard, H., Breier, A., Vrbjar, N., and Dzurba, A. (2000). Na,K-ATPase in the myocardium: molecular principles, functional and clinical aspects. Gen. Physiol. Biophys. 19, 9–47.
Keywords: mitochondrial dysfunction, arrhythmogenesis, fibrosis, redox signaling, diabetes
Citation: Song J, Yang R, Yang J and Zhou L (2018) Mitochondrial Dysfunction-Associated Arrhythmogenic Substrates in Diabetes Mellitus. Front. Physiol. 9:1670. doi: 10.3389/fphys.2018.01670
Received: 24 August 2018; Accepted: 07 November 2018;
Published: 06 December 2018.
Edited by:
Fadi G. Akar, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Michael Alan Colman, University of Leeds, United KingdomTong Liu, Tianjin Medical University, China
Copyright © 2018 Song, Yang, Yang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lufang Zhou, bGZ6aG91QHVhYi5lZHU=