 Nico Kresin1,2
Nico Kresin1,2 Sabrina Stücker1,2
Sabrina Stücker1,2 Elisabeth Krämer1,2Frederik Flenner1,2
Elisabeth Krämer1,2Frederik Flenner1,2 Giulia Mearini1,2Julia Münch3Monica Patten3Charles Redwood4
Giulia Mearini1,2Julia Münch3Monica Patten3Charles Redwood4 Lucie Carrier1,2*
Lucie Carrier1,2* Felix W. Friedrich1,2*
Felix W. Friedrich1,2*- 1Institute of Experimental Pharmacology and Toxicology, Cardiovascular Research Center, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 2DZHK (German Centre for Cardiovascular Research), Partner Site Hamburg/Kiel/Lübeck, Hamburg, Germany
- 3University Heart Center Hamburg, Hamburg, Germany
- 4Radcliffe Department of Medicine, University of Oxford, John Radcliffe Hospital, Oxford, United Kingdom
Background: Many forms of hypertrophic cardiomyopathy (HCM) show an increased myofilament Ca2+ sensitivity. This observation has been mainly made in HCM mouse models, myofilament systems, and cardiomyocytes. Studies of multicellular tissues from patients with different HCM-associated gene mutations are scarce. We investigated Ca2+ sensitivity in multicellular cardiac muscle strips of HCM patients. We furthermore evaluated the use of epigallocatechin-3-gallate (EGCg), a Ca2+ desensitizer.
Methods: After strip isolation from cardiac tissues with single (MYBPC3, MYH7) or double heterozygous mutations (MYBPC3/FLNC, MYH7/LAMP2, MYBPC3/MYH7) and permeabilization, we performed contractility measurements ±EGCg. We furthermore evaluated gene expression with a customized heart failure gene panel using the NanoString technology.
Results: Fmax tended to be higher in HCM than in non-failing (NF) control strips and in single than in double heterozygous strips. Ca2+ sensitivity was higher by trend in most HCM vs. NF strips and by trend in tissues with double vs. single heterozygous mutations. EGCg desensitized myofilaments to Ca2+ in most of the strips and tended to induce a more pronounced shift in strips with truncating than missense or single than double heterozygous mutations. Gene expression analysis revealed lower ATP2A2, PPP1R1A, and FHL2 and higher NPPA, NPPB, COL1A1, CTGF, and POSTN marker levels in HCM than in NF tissues. NPPA, NPPB, ACTA1, CTGF, COL1A1, and POSTN levels were higher in tissues with missense than truncating mutations.
Conclusion: We report an increased myofilament Ca2+ sensitivity in native multicellular cardiac HCM strips, which by trend was more pronounced in samples with double heterozygous mutations. EGCg could have differential effects depending on the underlying genetic status (single vs. double heterozygous) and type (missense vs. truncating).
Introduction
Hypertrophic cardiomyopathy (HCM) is the most frequent genetic disease of the myocardium and is primarily caused by mutations coding for sarcomeric proteins (Friedrich and Carrier, 2012; Maron et al., 2014; Ho et al., 2015). The most frequently mutated genes are MYBPC3, encoding cardiac myosin-binding protein C, and MYH7, encoding β-myosin heavy chain (Walsh et al., 2017). HCM is characterized by asymmetric left ventricular hypertrophy, diastolic dysfunction, and myocardial disarray (Elliott et al., 2008). Current pharmacological treatment of HCM is mainly based on beta-adrenoceptor (AR, beta blockers) and Ca2+ channel antagonists, which improve clinical symptoms, in part prevent arrhythmias and recover diastolic dysfunction by extending left ventricular (LV) filling time and reducing outflow tract obstruction (Maron et al., 2003; Gersh et al., 2011; Spoladore et al., 2012; Hamada et al., 2014; Tardiff et al., 2015). An attractive concept for many forms of HCM is an increased myofilament Ca2+ sensitivity. This could contribute to compromised diastolic relaxation causing remaining actin-myosin interaction at low diastolic Ca2+ concentrations and arrhythmias (Morimoto et al., 1998; Baudenbacher et al., 2008). Interventions decreasing this myofilament Ca2+ sensitivity may be appealing for the treatment of HCM and improvement in symptoms (Jagatheesan et al., 2007; Alves et al., 2014; Tardiff et al., 2015). These observations have mainly been made in genetically engineered HCM mouse models, reconstituted myofilament systems (Tardiff et al., 1999; Cazorla et al., 2006; Pohlmann et al., 2007; Morimoto, 2008; Vignier et al., 2009; Fraysse et al., 2012; Alves et al., 2014; Barefield et al., 2014; Wijnker et al., 2016), and in isolated cardiac cells of human HCM samples (Jacques et al., 2008; van Dijk et al., 2009, 2012). We previously reported contractile data from cardiac strips of HCM patients with mutations in the most frequently mutated HCM gene MYBPC3, but to the best of our knowledge, studies of multicellular tissues from patients with different HCM-associated gene mutations have not been reported so far. We therefore investigated myofilament Ca2+ sensitivity in native multicellular cardiac muscle strips derived from septal myectomies of patients with different HCM gene mutations. We furthermore evaluated the potential use of epigallocatechin-3-gallate (EGCg), the major catechin in green tea and a known Ca2+ desensitizer.
Materials and Methods
Human Samples
We received cardiac tissue of seven HCM patients carrying single heterozygous mutations in MYBPC3 (3) or MYH7 (1) and double heterozygous mutations [MYBPC3/FLNC (1), MYH7/LAMP2 (1), MYH7/MYBPC3 (1)], who underwent septal myectomy due to outflow tract obstruction (Table 2) in a completely anonymized way. Control donor tissues were from non-failing human heart tissues not suitable for transplantation (n = 1, control for contractile function measurements) or from donors who did not die from cardiac disease but of another cause (n = 7 for gene expression analysis). All samples were immediately frozen and stored in liquid nitrogen. An approval by and with the Standing Ethics Committee is and was not required for our study since all data obtained and used in this study were de-identified. No identifying information regarding our patients or donors’ identities was included in the manuscript nor do we have concerns that the anonymity cannot be maintained in the way the data are presented. We have written informed consent from all our patients and donors regarding the use of acquired tissue or other materials such as but not limited to clinical data or blood samples for scientific research in accordance with state laws and ethics. No material was used or included without that written informed consent from the patients or donors. This study is in accordance with the Code of the Ethics Committee of Hamburg and the Code of Ethics of the World Medical Association (Declaration of Helsinki). In summary, ethics approval was not required as per our institution’s guidelines and national regulations.
Skinned Ventricular Trabeculae Force Measurements
Cardiac strips of 2.42 ± 0.62 mm in length, 0.45 ± 0.08 mm in width, and 0.18 ± 0.06 mm2 in cross-sectional area (CSA), calculated by 2πr2 assuming a circular shape, were isolated from human cardiac tissues (n = 4–25/group) and frozen in liquid nitrogen until further analysis. Before contractile analysis, strips were permeabilized in a pCa 9 EGTA-buffer (Kooij et al., 2010; Stoehr et al., 2014; Friedrich et al., 2016) containing 1% Triton X-100 at 4°C for 18 h. The following day strips were either used directly for measurements or stored at −20°C in a 50% glycerol/relaxing solution containing protease inhibitors (EDTA-free, complete tablets, mini, Roche). A fiber test system (1400A; Aurora Scientific) was used to evaluate the contractile function of cardiac strips. Sarcomere length could not be determined reliably in skinned HCM strips due to myocardial and myofilament disarray. Therefore, after mounting strips between a force transducer and a length controller and stretching until slack length, they were stretched another 10% of length. For maximum force measurements, strips were kept in pCa 9 to achieve full relaxation and were then moved to pCa 4.5 until maximal force development was reached, as reported before (Friedrich et al., 2016; Stucker et al., 2017). For force-Ca2+ curves, strips were exposed to increasing Ca2+ concentrations from pCa 9 to pCa 4.5 in EGTA-buffer, and force development was measured in each pCa solution. Measurements were repeated in the presence of 30 μM epigallocatechin-3-gallate (EGCg, Sigma Life Sciences) after 5 min preincubation in pCa 9 (Flenner et al., 2016; Friedrich et al., 2016; Stucker et al., 2017). To exclude time-dependent loss of force, EGCg was tested first and a control measurement was performed 5 min after EGCg washout in every second measurement. Each strip was measured pair wisely (paired analysis baseline vs. intervention), and the length of the strip was not changed in between. Thereby, each strip served as its own control. Data were analyzed with the Hill equation (Hill et al., 1980), with pCa50 as the free Ca2+ concentration which produces 50% of the maximal force and nH representing the Hill coefficient. The pCa50 represents the measure of myofilament Ca2+ sensitivity.
RNA Isolation and Expression Analysis With the NanoString nCounter® Elements
Total RNA was extracted from cardiac tissues using the SV Total RNA Isolation kit (Promega). For gene expression analysis, we used a customized NanoString’s nCounter® Elements TagSet panel of 27 genes coding for proteins regulated in hypertrophy/heart failure, including Ca2+ and K+ handling proteins (Prondzynski et al., 2017; Singh et al., 2017; Braumann et al., 2018). About 50 ng of each sample were hybridized to target-specific capture and reporter probes at 67°C overnight (16 h) according to manufacturer’s instructions. Samples were cooled down at 4°C, filled up with 15 μl H2O, and loaded into the NanoString cartridge, and the nCounter Gene Expression Assay was started immediately. Raw data were analyzed with nCounter® Sprint Profiler including background subtraction using negative controls and normalization to five housekeeping genes (ABCF1, CLTC, GAPDH, PGK1, and TUBB). Data represented the mean of normalized counts and were expressed as fold-change. We selected genes that were lower than 0.8-fold and higher than 1.25-fold dysregulated in cardiomyopathy.
Statistical Analysis
Data were expressed as mean ± SEM. Comparisons were performed by paired Student’s t-test (baseline vs. EGCg) and by mixed-effect model analysis followed by Dunnett multiple comparison post-test when analyzing all groups as indicated in the figure legends. For subgroup analyses (single vs. double heterozygous mutations, missense vs. truncating, MYH7 vs. MYBPC3), a qualitative comparison was performed. Concentration response curves were fitted to the data points, and force-pCa relationship comparison was done by using extra sum-of-squares F-test (GraphPad, Prism 8). A value of p < 0.05 was considered statistically significant.
Results
Patients’ Characteristics
HCM patients of the study had undergone septal myectomy due to LV outflow tract obstruction. Echocardiographic and clinical data of the patients are given in Table 1. Patients presented with NYHA states 1.5–3.0 (mean 2.5), preserved ejection fraction (EF > 60%), and increased septum wall thickness (mean 28 ± 7 mm). Most of them had a high LV transaortic pressure gradient (mean 87 ± 34 mm Hg), a systolic anterior motion (SAM) of the mitral valve, differing grades of mitral valve insufficiency, and diastolic dysfunction. Some had encountered ventricular tachycardia and syncope and were equipped with an ICD. Medication consisted of ACE inhibitors, beta blockers, and Ca2+ channel antagonists.
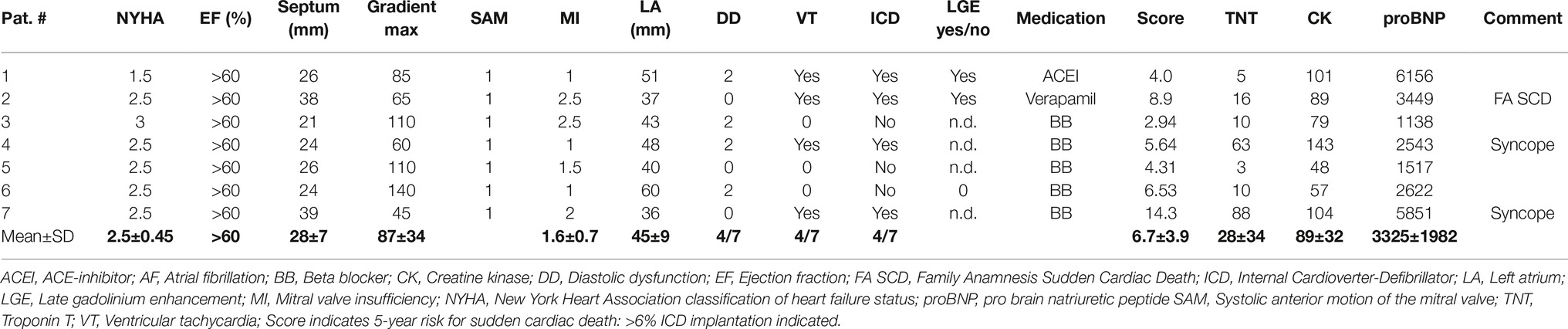
Table 1. Echocardiographic and clinical data of the patients.
Mutation Characteristics
Patients had either single heterozygous mutation in MYBPC3 (n = 3) or MYH7 (n = 1), or double heterozygous mutations [MYBPC3/FLNC (n = 1), MYH7/LAMP2 (n = 1), MYBPC3/MYH7 (n = 1)]. Two mutations are truncating, and the rest are missense mutations. In silico analysis using the prediction programs Mutation Taster1 and PolyPhen-22 classified most of the mutations as disease causing or possibly damaging (Table 2).
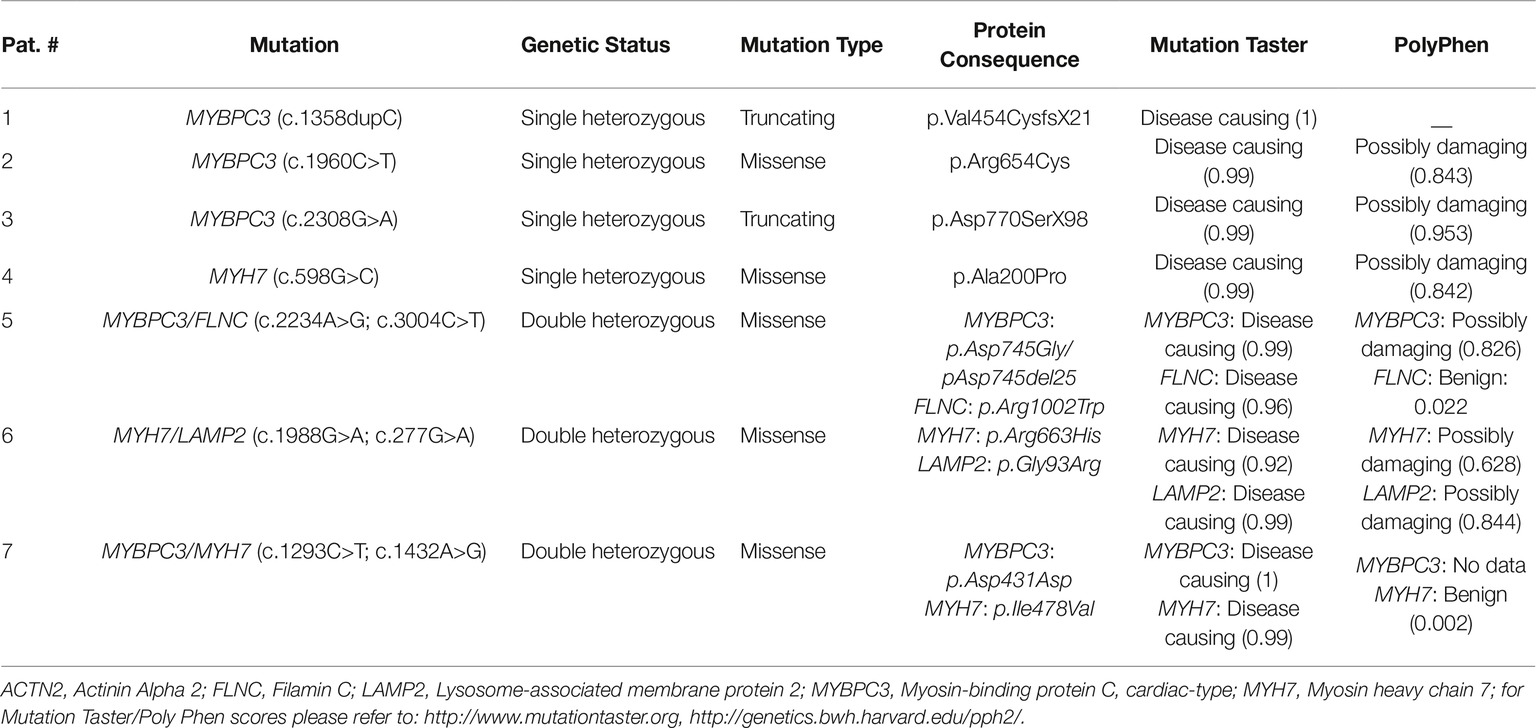
Table 2. Mutation characteristics.
Varying Maximal Force Development in HCM Strips
Functional implications of the HCM mutations were evaluated by contractile function measurements of permeabilized cardiac muscle strips. Force development (Fmax) related to cross-sectional area showed a trend to higher Fmax in HCM (black bar, Figure 1A) than in the NF strips. It was higher in one sample carrying a single heterozygous MYBPC3 mutation (Figure 1A). Fmax did not differ between tissues with missense or truncating mutations (Figure 1B), whereas it tended to be higher in tissues with single than double heterozygous mutations or NF (Figure 1C) and in tissues with an MYBPC3 vs. MYH7 genotype (Figure 1D).
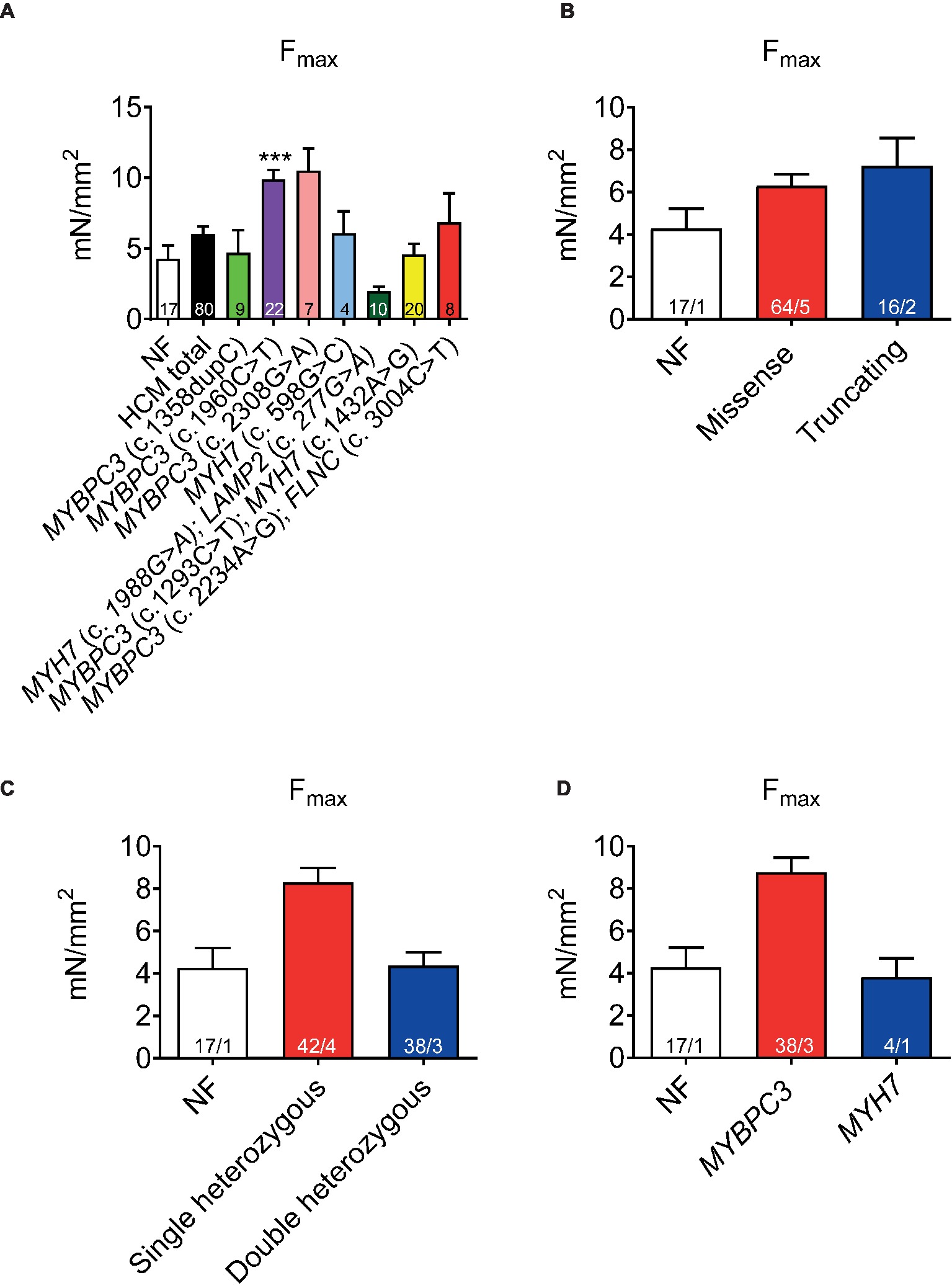
Figure 1. Force development of permeabilized cardiac muscle strips isolated from seven HCM patients with either single heterozygous (MYBPC3, MYH7) or double heterozygous mutations (MYH7/LAMP2, MYBPC3/MYH7, MYBPC3/FLNC). Maximal force development (Fmax) related to cross-sectional area in non-failing control vs. HCM strips (A), grouped in missense or truncating (B), single or double heterozygous mutations (C), and classified according to a pure MYBPC3 or MYH7 genotype (D). Values are related to non-failing control (NF) strips. Data are expressed as mean ± SEM. **p < 0.01 and ***p < 0.001 vs. NF, mixed-effect model analysis followed by Dunnett multiple comparison post-test; for the different subgroup analyses, only qualitative comparisons were performed. n = number of strips; n/N = number of strips/number of patients as depicted in the bars.
Myofilament Ca2+ Sensitivity Is Higher in HCM Than in NF Strips
Force-Ca2+ curves revealed a higher myofilament Ca2+ sensitivity (=higher pCa50) in HCM vs. NF strips (pCa50 NF: 5.57; pCa50 HCMtotal: 5.65; Figures 2A,B). This was independent of the mutation type (Figure 2C), whereas pCa50 was by trend higher in muscle strips with double than single heterozygous mutations (Figure 2D) and in MYBPC3 than MYH7 genotypes (Figure 2E). Hill slope (NH) as the indicator of cooperativity was lower or showed a trend to lower values in almost all HCM in comparison to NF strips except for strips carrying the double MYH7/LAMP2 mutations (Figure 2F).
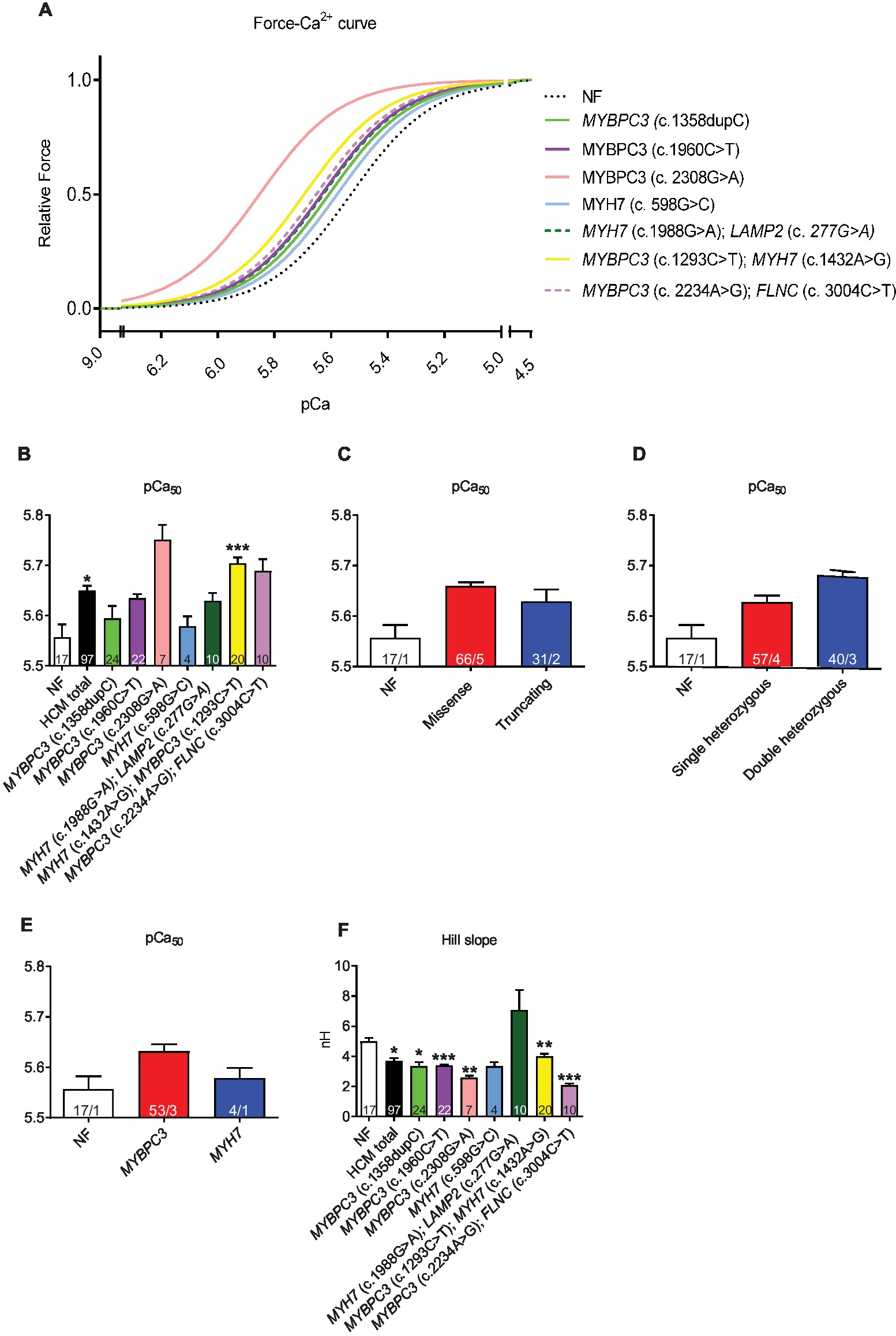
Figure 2. Force-Ca2+ relationship of permeabilized cardiac muscle strips isolated from seven HCM patients with either single heterozygous (MYBPC3, MYH7) or double heterozygous mutations (MYH7/LAMP2, MYBPC3/MYH7, MYBPC3/FLNC). (A) Force-Ca2+ relationship curves in human strips, Hill slope set to 3. (B) pCa50 representing the negative logarithm of the Ca2+ concentration needed for half-maximal activation. (C–E) pCa50 in strips grouped according to mutation type (missense or truncating), to genetic status (single or double heterozygous mutations), and to a pure MYBPC3 or MYH7 genotype. (F) nHill coefficient (Hill slope). Values are related to non-failing control (NF) strips. Data are expressed as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 vs. NF, mixed-effect model analysis followed by Dunnett multiple comparison post-test; for the different subgroup analyses, only qualitative comparisons were performed; concentration response curves were fitted to the data points and curve comparison was done by using extra sum-of-squares F-test. n = number of strips; n/N = number of strips/number of patients as depicted in the bars.
EGCg Induces a Stronger Myofilament Ca2+ Desensitization in Strips From Patients With Truncating and Single Heterozygous Mutations
We previously reported that EGCg decreased Ca2+ sensitivity of mouse and human cardiac myofilaments with MYBPC3 mutations (Friedrich et al., 2016; Stucker et al., 2017). To assess whether the grade of desensitization is similar between HCM strips, we measured force-Ca2+ relationships in skinned ventricular muscle strips at baseline and after the application of 30 μM EGCg. Incubation (5 min) with EGCg shifted the force-Ca2+ curves to different extent to the right (pCa50 HCMtotal + 30 μM EGCg: 5.54) of all strips (except in strips with double heterozygous MYBPC3/FLNC mutations) suggesting myofilament Ca2+ desensitization (Figure 3A). EGCg application tended to induce a more pronounced shift in strips from patients with truncating than missense (Figure 3B) and in strips with single than heterozygous mutations (Figure 3C). No major difference was observed between strips with MYBPC3 or MYH7 genotypes (Figure 3D). EGCg induced a force-Ca2+ curve right shift also in NF strips.
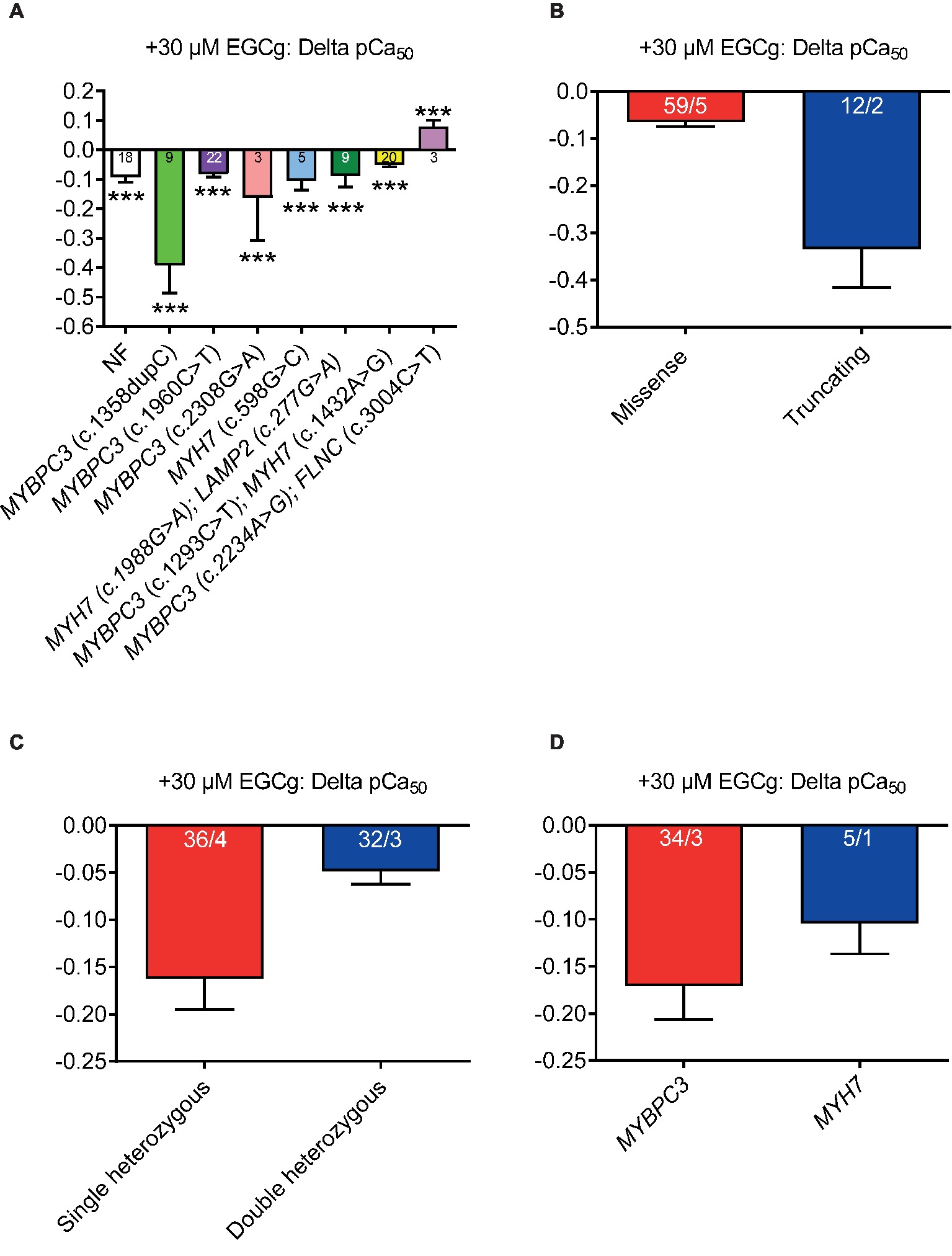
Figure 3. Effect of 30 μM EGCg on pCa50 values in permeabilized cardiac muscle strips isolated from seven HCM patients with either single heterozygous (MYBPC3, MYH7) or double heterozygous mutations (MYH7/LAMP2, MYBPC3/MYH7, MYBPC3/FLNC). (A) Delta of pCa50 ± EGCg (30 μM) in non-failing control vs. HCM permeabilized cardiac muscle strips grouped according to mutation type (missense or truncating, (B), to genetic status (single or double heterozygous mutations, (C), and to a pure MYBPC3, MYH7, or ACTN2 genotype (D). Values are related to baseline. Data are expressed as mean ± SEM. ***p < 0.001 vs. baseline (without EGCg), paired Student’s t-test; for the different subgroup analyses, only qualitative comparisons were performed. n = number of strips; n/N = number of strips/number of patients as depicted in the bars.
HCM Samples Show a Heart Failure Gene Expression Profile
We then evaluated the expression levels of a customized panel of human genes (n = 27) regulated in heart failure using the NanoString nCounter® technology platform. Compared to NF samples (n = 8), gene expression analysis revealed lower MYH6, but higher MYH7 and ACTN2 mRNA levels, lower mRNA levels of Ca2+ handling proteins (ATP2A2, PPP1R1A), lower hypertrophy-associated FHL2, and higher NPPA and NPPB mRNA levels in HCM samples. Furthermore, mRNA levels of fibrosis markers (COL1A1, CTGF, POSTN) were higher (Figure 4A). Gene expression of markers related to hypertrophy (NPPA, NPPB, ACTA1) and fibrosis (CTGF, COL1A1, POSTN) were higher in tissues with missense than with truncating mutations (Figure 4B). mRNA levels of hypertrophy-associated FHL2 were lower and of NPPA and NPPB were higher in tissues with double than with single heterozygous mutations (Figure 4C).
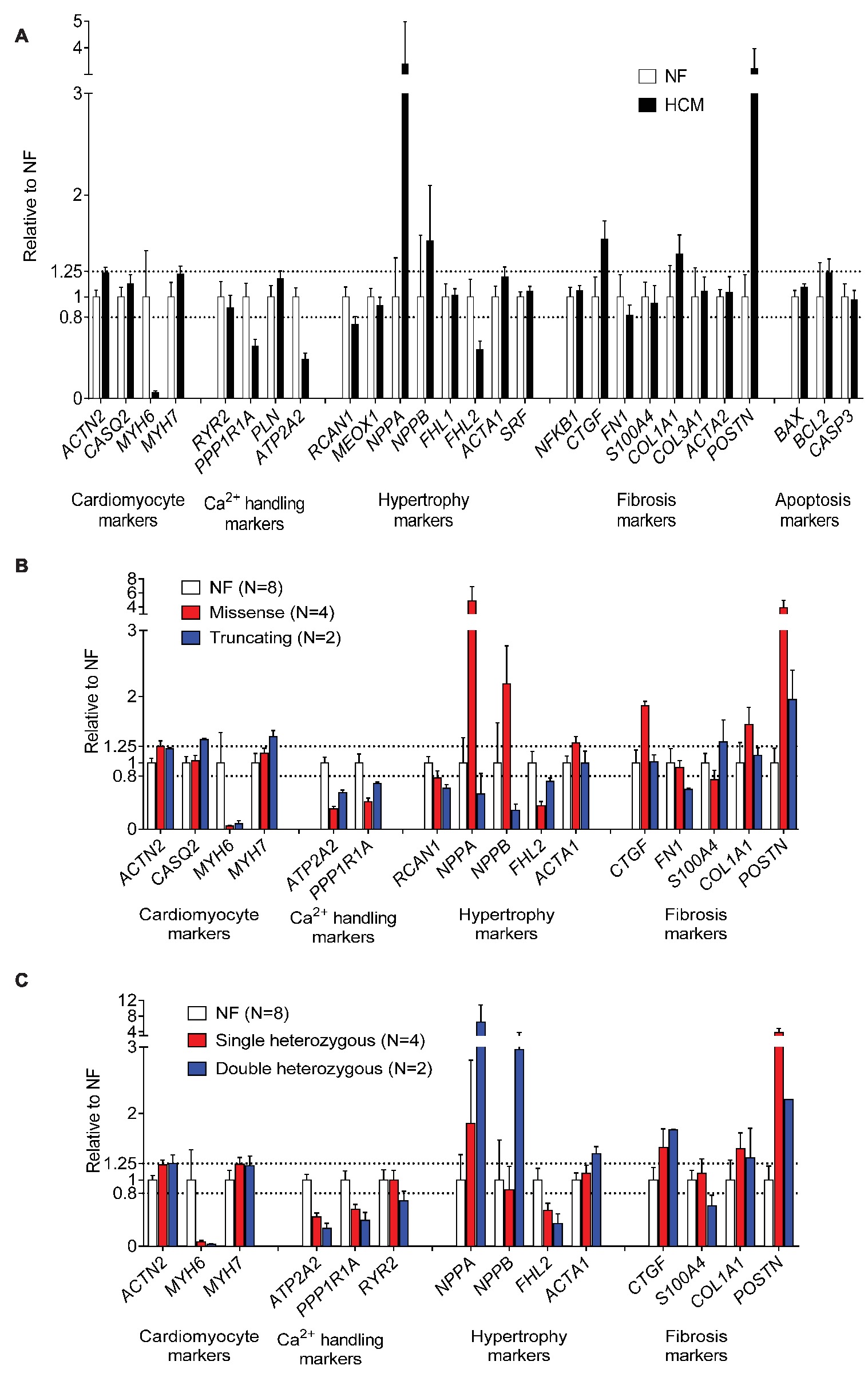
Figure 4. Evaluation of expression of human cardiac genes regulated in heart failure in NF and HCM tissues. Normalized expression levels of selected genes determined with the NanoString nCounter Elements technology comparing gene expression modulating hypertrophy, fibrosis, calcium handling, and apoptosis in NF and HCM (A), (threshold <0.8- or > 1.25-fold change to NF). Control group consisted of RNA isolated from eight different NF control hearts. Gene expression related to (B) mutation type (missense or truncating) or (C) genetic status (single or double heterozygous mutation). Since for patient #7 (Table 1) obtained results repeatedly failed, quality control check data were not included in the analysis.
Discussion
The observation of an increased myofilament Ca2+ sensitivity has mainly been made in HCM transgenic mouse models, reconstituted myofilament systems, and isolated cardiomyocytes, but data from multicellular tissues from HCM patients with mutations in different HCM genes have not been reported before (Morimoto et al., 1998; Tardiff et al., 1999; Cazorla et al., 2006; Pohlmann et al., 2007; Robinson et al., 2007; Jacques et al., 2008; Morimoto, 2008; van Dijk et al., 2009, 2012; Vignier et al., 2009; Huke and Knollmann, 2010; Kimura, 2010; Fraysse et al., 2012; Moore et al., 2012; Barefield et al., 2014; Flenner et al., 2016; Wijnker et al., 2016; Stucker et al., 2017; Ren et al., 2018). We therefore investigated force development and myofilament Ca2+ sensitivity in multicellular cardiac muscle strips derived from septal myectomies of patients with different HCM gene mutations and evaluated the potential use of epigallocatechin-3-gallate (EGCg), a known Ca2+-desensitizer, for myofilament Ca2+ desensitization.
The main findings of this study were as follows: (1) Fmax tended to be higher in permeabilized cardiac muscle strips from patients with single heterozygous than with double heterozygous mutations and NF control, and in MYBPC3 vs. MYH7 strips. (2) Myofilament Ca2+ sensitivity was higher in HCM strips than in NF. (3) Myofilament Ca2+ sensitivity was greater by trend in samples with double than with single heterozygous mutations. (4) EGCg induced myofilament Ca2+ desensitization in almost all strips. EGCg effect was stronger by tendency in strips with truncating or single heterozygous mutations. (5) RNA expression analysis of a cardiomyopathy gene panel showed lower MYH6, but higher MYH7 and ACTN2 levels, lower levels of Ca2+ handling genes (ATP2A2, PPP1R1A), lower hypertrophy-associated FHL2, and higher NPPA and NPPB levels in comparison to NF samples. Furthermore, gene expression level differed between the genetic status (single or double heterozygous mutations) and between the mutation type (missense or truncating).
Force development had a tendency to be higher in cardiac muscle strips from patients with single heterozygous mutations or MYBPC3 mutations, but did not differ to NF in samples carrying double heterozygous or MYH7 mutations. Variable degrees of force development have been reported before. Most of the published data derived from in vitro studies with HCM-associated MYH7 mutations are consistent with increased contractility (Debold et al., 2007; Palmer et al., 2008; Marston, 2011; Moore et al., 2012; Sommese et al., 2013; Spudich et al., 2016). However, a reduced Fmax was also reported despite increased myofilament Ca2+ sensitivity (van Dijk et al., 2009). Force values for NF donor samples are lower than reported by other groups such as by Mamidi et al. (2017). One reason could be different types of measurement setups and another reason could be the difference in dimensions of the measured samples. Thinner preparations are more multicellular, whereas our samples contain also extracellular matrix which does not contribute to force generation.
As mentioned before, to this extent, data from multicellular tissues from HCM patients with mutations in different HCM genes have not been reported before. In this study, HCM patients’ strips derived from myectomies revealed a higher myofilament Ca2+ sensitivity than the NF control or showed at least a trend. An increased myofilament Ca2+ sensitivity leading to Ca2+ trapping and altered Ca2+ fluxes during excitation-contraction coupling is believed to play a central role in disease presentation (Ashrafian et al., 2011). Increased force development and Ca2+ sensitization can trigger hypertrophy, which leads to HCM phenotypic expression. Hypertrophy in turn can influence Ca2+ transients and other cellular processes such as energy metabolism, myofilament contraction, and electrophysiology. These processes boost hypertrophy and promote HCM disease expression. Clinically, all HCM patients showed an increased LV wall thickness. The increased myofilament Ca2+ sensitivity may also contribute to relaxation deficits and arrhythmias (Morimoto et al., 1998; Baudenbacher et al., 2008). An increased myofilament Ca2+ sensitivity is compatible with the compromised diastolic function and incomplete relaxation observed in HCM patients (Authors/Task Force et al., 2014) and in mice (Tardiff et al., 1999; Cazorla et al., 2006; Pohlmann et al., 2007; Vignier et al., 2009; Fraysse et al., 2012; Barefield et al., 2014). Five of the eight patients of this study presented a marked diastolic dysfunction. As for arrhythmias, four of the patients had episodes of ventricular tachycardia, which made an ICD implantation necessary.
Furthermore, myofilament Ca2+ sensitivity tended to be higher in strips with double than single heterozygous mutations and in strips with MYBPC3 compared to MYH7 mutations. An even higher Ca2+ sensitivity could exaggerate the consequences described above (Ca2+ trapping, increased force development…). Since the number of patients was low, interpretation of these data has to be treated with caution. The difference in Ca2+ sensitivity between single and double heterozygous mutations fits to the often-observed clinical difference in phenotypes. Patients with double mutations generally show an earlier disease presentation, more severe LV hypertrophy, higher prevalence of advanced heart failure, and an increased risk of sudden cardiac death than patients with single heterozygous mutations (Maron et al., 2012; Biagini et al., 2014; Fazeli Dehkordy et al., 2018). A differential Ca2+ sensitivity has also been observed in other studies with different HCM disease genes (Ren et al., 2018; Mamidi et al., 2019). It therefore seems that the direction and magnitude of the change not only depends on the affected gene (MYBPC3 vs. MYH7) but also on the genetic status (single vs. double heterozygous), but not on the mutation type (missense vs. truncating).
Clinical management of HCM is challenging. Guidelines recommend treatment with beta blockers and Ca2+ channel antagonists, which improve clinical symptoms, moderately prevent arrhythmias, improve diastolic dysfunction, and reduce outflow tract obstruction (Tardiff et al., 2015). The idea of myofilament desensitization to reduce diastolic dysfunction and arrhythmias in HCM patients has not been tested clinically because of a lack of clinically applicable Ca2+ desensitizers. We and others reported that EGCg lowered myofilament Ca2+ sensitivity in mouse and human HCM tissues (Warren et al., 2015; Messer et al., 2016; Friedrich et al., 2016; Stucker et al., 2017). Additionally, Sheehan et al. and others reported that EGCg and structurally related compounds restored the coupled relationship between Ca2+-sensitivity and TnI phosphorylation in mutant thin filaments, which was independent of the underlying mutation (Papadaki et al., 2015; Messer et al., 2016; Sheehan et al., 2018). EGCg has been suggested to alter the interaction between cardiac troponin C and troponin I and thereby the sensitivity of the myofilaments to Ca2+ (Robertson et al., 2009). In the current study, we extended the analysis to several HCM muscle strips with differing HCM gene mutations and confirmed that application of EGCg shifted the force-Ca2+ curves of nearly all strips to the right, suggesting myofilament Ca2+ desensitization. EGCg application tended to induce a more prominent shift in strips with truncating than missense mutations or with single than double heterozygous mutations. Similar to the differential effects of EGCg on human HCM strip Ca2+ sensitivity in this study, we previously observed that EGCg had a more prominent effect on cardiac strips of an HCM Mybpc3 knock-in (KI) mouse model than the WT control group (Friedrich et al., 2016). It is also compatible with results of a study in which the Ca2+ desensitizing effect of ranolazine was only present in KI, but not in WT muscle strips (Flenner et al., 2016). In contrast, EGCg induced a force-Ca2+ curve right shift also in NF strips. Thus, these differential EGCg effects could depend on the mutation, genetic status (single vs. double heterozygous), and the mutation type (missense vs. truncating).
Similar to the observations made in cardiomyopathy tissue with a reduced ejection fraction, we observed altered mRNA steady-state concentrations in myosin heavy chain isoforms (MYH6/7), heart-failure-associated Ca2+-handling (ATP2A2, PPP1R1A), hypertrophy (FHL2, NPPA, NPPB), and fibrosis (COL1A1, CTGF, POSTN) markers in comparison to NF samples. This is partially in accordance with previous findings using HCM samples, HCM human-induced pluripotent stem cells (iPSC)-CMs, or human embryonic stem cells carrying MYBPC3 mutations as well as microarray results obtained in cardiac tissues of HCM patients prior to transplantation (Tanaka et al., 2014; Messer et al., 2016; Singh et al., 2017; Braumann et al., 2018; Mohammad et al., 2018). Furthermore, a marked difference in gene expression was detected between mutation type (missense vs. truncating) and genetic status (single vs. double heterozygous mutations). This study does not provide any explanation for this difference. As far as we are aware, such a discrepancy has not been reported before.
Limitations of the Study
Although we were able to study a collection of human HCM samples with different mutations in genes encoding sarcomeric proteins, care must be taken when extrapolating our findings to all patients with HCM. Our HCM population consisted of patients with left ventricular outflow tract obstruction. As all HCM patients received a certain medication regime, we cannot exclude effects of medication on the outcome of our study. Due to the low numbers (each genotype N patient = 1, subgroup analysis N patient = 1–5), the data have to be treated with caution. We used a mixed-effect model analysis when analyzing all groups. To avoid type-II errors, we performed qualitative comparisons for the different subgroup analyses and discussed possible trends. Furthermore, sarcomere length could not be determined reliably in skinned HCM strips due to myocardial and myofilament disarray. Differences in sarcomere length could therefore have an effect on pCa50 values.
Conclusion
We reported an increased myofilament Ca2+-sensitivity in native multicellular cardiac strips of HCM patients. Our findings suggest that myofilament Ca2+ desensitizing approaches might be useful for the treatment of HCM-associated diastolic dysfunction. Our data highlight that mutation-induced changes in myofilament Ca2+ sensitivity and response to EGCg are diverse and depend on the mutation, genetic status, and mutation type.
Author Contributions
NK and SS contributed to isolation and treatment of cardiac muscle strips and execution of experiments. EK contributed to RNA isolation and execution of RNA expression experiments. GM and FF contributed to preservation of human cardiac tissues and database maintenance. JM and MP contributed to patients’ recruitment. CR contributed to genotyping of samples. LC is responsible for interpretation of data and correction of the manuscript. FWF contributed to conception and design of research, execution of experiments, analysis and interpretation of data, figure preparation, and drafting of the manuscript. All authors critically reviewed and approved the manuscript before submission.
Funding
This work was supported by the DZHK (German Centre for Cardiovascular Research) and the Deutsche Stiftung für Herzforschung (F/28/12).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Maksymilian Prondzynski for assistance with the NanoString technology and the patients for donating the cardiac tissue for scientific research.
Footnotes
References
Alves, M. L., Dias, F. A. L., Gaffin, R. D., Simon, J. N., Montminy, E. M., Biesiadecki, B. J., et al. (2014). Desensitization of myofilaments to Ca2+ as a therapeutic target for hypertrophic cardiomyopathy with mutations in thin filament proteins. Circ. Cardiovasc. Genet. 7, 132–143. doi: 10.1161/CIRCGENETICS.113.000324
Ashrafian, H., McKenna, W. J., and Watkins, H. (2011). Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ. Res. 109, 86–96. doi: 10.1161/CIRCRESAHA.111.242974
Authors/Task Force Members Elliott, P. M., Anastasakis, A., Borger, M. A., Borggrefe, M., Cecchi, F., et al. (2014). 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 35, 2733–2779. doi: 10.1093/eurheartj/ehu284
Barefield, D., Kumar, M., de Tombe, P. P., and Sadayappan, S. (2014). Contractile dysfunction in a mouse model expressing a heterozygous MYBPC3 mutation associated with hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 306, H807–H815. doi: 10.1152/ajpheart.00913.2013
Baudenbacher, F., Schober, T., Pinto, J. R., Sidorov, V. Y., Hilliard, F., Solaro, R. J., et al. (2008). Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J. Clin. Invest. 118, 3893–3903. doi: 10.1172/JCI36642
Biagini, E., Olivotto, I., Iascone, M., Parodi, M. I., Girolami, F., Frisso, G., et al. (2014). Significance of sarcomere gene mutations analysis in the end-stage phase of hypertrophic cardiomyopathy. Am. J. Cardiol. 114, 769–776. doi: 10.1016/j.amjcard.2014.05.065
Braumann, S., Thottakara, T., Stucker, S., Reischmann-Dusener, S., Kramer, E., Gross, J., et al. (2018). S100A4 as a target of the E3-ligase Asb2beta and its effect on engineered heart tissue. Front. Physiol. 9:1292. doi: 10.3389/fphys.2018.01292
Cazorla, O., Szilagyi, S., Vignier, N., Salazar, G., Kramer, E., Vassort, G., et al. (2006). Length and protein kinase A modulations of myocytes in cardiac myosin binding protein C-deficient mice. Cardiovasc. Res. 69, 370–380. doi: 10.1016/j.cardiores.2005.11.009
Debold, E. P., Schmitt, J. P., Patlak, J. B., Beck, S. E., Moore, J. R., Seidman, J. G., et al. (2007). Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse alpha-cardiac myosin in the laser trap assay. Am. J. Physiol. Heart Circ. Physiol. 293, H284–H291. doi: 10.1152/ajpheart.00128.2007
Elliott, P., Andersson, B., Arbustini, E., Bilinska, Z., Cecchi, F., Charron, P., et al. (2008). Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 29, 270–276. doi: 10.1093/eurheartj/ehm342
Fazeli Dehkordy, S., Fowler, K. J., Wolfson, T., Igarashi, S., Lamas Constantino, C. P., Hooker, J. C., et al. (2018). Technical report: gadoxetate-disodium-enhanced 2D R2* mapping: a novel approach for assessing bile ducts in living donors. Abdom. Radiol. 43, 1656–1660. doi: 10.1007/s00261-017-1365-3
Flenner, F., Friedrich, F. W., Ungeheuer, N., Christ, T., Geertz, B., Reischmann, S., et al. (2016). Ranolazine antagonizes catecholamine-induced dysfunction in isolated cardiomyocytes, but lacks long-term therapeutic effects in vivo in a mouse model of hypertrophic cardiomyopathy. Cardiovasc. Res. 109, 90–102. doi: 10.1093/cvr/cvv247
Fraysse, B., Weinberger, F., Bardswell, S. C., Cuello, F., Vignier, N., Geertz, B., et al. (2012). Increased myofilament Ca2+ sensitivity and diastolic dysfunction as early consequences of Mybpc3 mutation in heterozygous knock-in mice. J. Mol. Cell. Cardiol. 52, 1299–1307. doi: 10.1016/j.yjmcc.2012.03.009
Friedrich, F. W., and Carrier, L. (2012). Genetics of hypertrophic and dilated cardiomyopathy. Curr. Pharm. Biotechnol. 13, 2467–2476. doi: 10.2174/1389201011208062467
Friedrich, F. W., Flenner, F., Nasib, M., Eschenhagen, T., and Carrier, L. (2016). Epigallocatechin-3-gallate accelerates relaxation and Ca2+ transient decay and desensitizes myofilaments in healthy and Mybpc3-targeted knock-in cardiomyopathic mice. Front. Physiol. 7:607. doi: 10.3389/fphys.2016.00607
Gersh, B. J., Maron, B. J., Bonow, R. O., Dearani, J. A., Fifer, M. A., Link, M. S., et al. (2011). 2011 ACCF/AHA Guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Developed in collaboration with the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J. Am. Coll. Cardiol. 58, e212–e260. doi: 10.1016/j.jacc.2011.06.011
Hamada, M., Ikeda, S., and Shigematsu, Y. (2014). Advances in medical treatment of hypertrophic cardiomyopathy. J. Cardiol. 64, 1–10. doi: 10.1016/j.jjcc.2014.02.022
Hill, T. L., Eisenberg, E., and Greene, L. (1980). Theoretical model for the cooperative equilibrium binding of myosin subfragment 1 to the actin-troponin-tropomyosin complex. Proc. Natl. Acad. Sci. USA 77, 3186–3190.
Ho, C. Y., Charron, P., Richard, P., Girolami, F., Van Spaendonck-Zwarts, K. Y., and Pinto, Y. (2015). Genetic advances in sarcomeric cardiomyopathies: state of the art. Cardiovasc. Res. 105, 397–408. doi: 10.1093/cvr/cvv025
Huke, S., and Knollmann, B. C. (2010). Increased myofilament Ca2+− sensitivity and arrhythmia susceptibility. J. Mol. Cell. Cardiol. 48, 824–833. doi: 10.1016/j.yjmcc.2010.01.011
Jacques, A., Hoskins, A. C., Kentish, J. C., and Marston, S. B. (2008). From genotype to phenotype: a longitudinal study of a patient with hypertrophic cardiomyopathy due to a mutation in the MYBPC3 gene. J. Muscle Res. Cell Motil. 29, 239–246. doi: 10.1007/s10974-009-9174-0
Jagatheesan, G., Rajan, S., Petrashevskaya, N., Schwartz, A., Boivin, G., Arteaga, G. M., et al. (2007). Rescue of tropomyosin-induced familial hypertrophic cardiomyopathy mice by transgenesis. Am. J. Physiol. Heart Circ. Physiol. 293, H949–H958. doi: 10.1152/ajpheart.01341.2006
Kimura, A. (2010). Molecular basis of hereditary cardiomyopathy: abnormalities in calcium sensitivity, stretch response, stress response and beyond. J. Hum. Genet. 55, 81–90. doi: 10.1038/jhg.2009.138
Kooij, V., Boontje, N., Zaremba, R., Jaquet, K., dos Remedios, C., Stienen, G. J., et al. (2010). Protein kinase C alpha and epsilon phosphorylation of troponin and myosin binding protein C reduce Ca2+ sensitivity in human myocardium. Basic Res. Cardiol. 105, 289–300. doi: 10.1007/s00395-009-0053-z
Mamidi, R., Li, J., Doh, C. Y., Holmes, J. B., and Stelzer, J. E. (2019). Lost in translation: interpreting cardiac muscle mechanics data in clinical practice. Arch. Biochem. Biophys. 662, 213–218. doi: 10.1016/j.abb.2018.12.021
Mamidi, R., Li, J., Gresham, K. S., Verma, S., Doh, C. Y., Li, A., et al. (2017). Dose-dependent effects of the myosin activator omecamtiv mecarbil on cross-bridge behavior and force generation in failing human myocardium. Circ. Heart Fail. 10:e004257. doi: 10.1161/CIRCHEARTFAILURE.117.004257
Maron, B. J., Maron, M. S., and Semsarian, C. (2012). Double or compound sarcomere mutations in hypertrophic cardiomyopathy: a potential link to sudden death in the absence of conventional risk factors. Heart Rhythm. 9, 57–63. doi: 10.1016/j.hrthm.2011.08.009
Maron, B. J., McKenna, W. J., Danielson, G. K., Kappenberger, L. J., Kuhn, H. J., Seidman, C. E., et al. (2003). American College of Cardiology/European Society of Cardiology Clinical Expert Consensus Document on Hypertrophic Cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. Eur. Heart J. 24, 1965–1991. doi: 10.1016/S0195-668X(03)00479-2
Maron, B. J., Ommen, S. R., Semsarian, C., Spirito, P., Olivotto, I., and Maron, M. S. (2014). Hypertrophic cardiomyopathy: present and future, with translation into contemporary cardiovascular medicine. J. Am. Coll. Cardiol. 64, 83–99. doi: 10.1016/j.jacc.2014.05.003
Marston, S. B. (2011). How do mutations in contractile proteins cause the primary familial cardiomyopathies? J. Cardiovasc. Transl. Res. 4, 245–255. doi: 10.1007/s12265-011-9266-2
Messer, A. E., Bayliss, C. R., El-Mezgueldi, M., Redwood, C. S., Ward, D. G., Leung, M. C., et al. (2016). Mutations in troponin T associated with hypertrophic cardiomyopathy increase Ca(2+)-sensitivity and suppress the modulation of Ca(2+)-sensitivity by troponin I phosphorylation. Arch. Biochem. Biophys. 601, 113–120. doi: 10.1016/j.abb.2016.03.027
Mohammad, J., Dhillon, H., Chikara, S., Mamidi, S., Sreedasyam, A., Chittem, K., et al. (2018). Piperlongumine potentiates the effects of gemcitabine in in vitro and in vivo human pancreatic cancer models. Oncotarget 9, 10457–10469. doi: 10.18632/oncotarget.23623
Moore, J. R., Leinwand, L., and Warshaw, D. M. (2012). Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ. Res. 111, 375–385. doi: 10.1161/CIRCRESAHA.110.223842
Morimoto, S. (2008). Sarcomeric proteins and inherited cardiomyopathies. Cardiovasc. Res. 77, 659–666. doi: 10.1093/cvr/cvm084
Morimoto, S., Yanaga, F., Minakami, R., and Ohtsuki, I. (1998). Ca2+− sensitizing effects of the mutations at Ile-79 and Arg-92 of troponin T in hypertrophic cardiomyopathy. Am. J. Phys. 275, C200–C207.
Palmer, B. M., Wang, Y., Teekakirikul, P., Hinson, J. T., Fatkin, D., Strouse, S., et al. (2008). Myofilament mechanical performance is enhanced by R403Q myosin in mouse myocardium independent of sex. Am. J. Physiol. Heart Circ. Physiol. 294, H1939–H1947. doi: 10.1152/ajpheart.00644.2007
Papadaki, M., Vikhorev, P. G., Marston, S. B., and Messer, A. E. (2015). Uncoupling of myofilament Ca2+ sensitivity from troponin I phosphorylation by mutations can be reversed by epigallocatechin-3-gallate. Cardiovasc. Res. 108, 99–110. doi: 10.1093/cvr/cvv181
Pohlmann, L., Kroger, I., Vignier, N., Schlossarek, S., Kramer, E., Coirault, C., et al. (2007). Cardiac myosin-binding protein C is required for complete relaxation in intact myocytes. Circ. Res. 101, 928–938. doi: 10.1161/CIRCRESAHA.107.158774
Prondzynski, M., Kramer, E., Laufer, S. D., Shibamiya, A., Pless, O., Flenner, F., et al. (2017). Evaluation of MYBPC3 trans-splicing and gene replacement as therapeutic options in human ipsc-derived cardiomyocytes. Mol. Ther. Nucleic Acids 7, 475–486. doi: 10.1016/j.omtn.2017.05.008
Ren, X., Hensley, N., Brady, M. B., and Gao, W. D. (2018). The genetic and molecular bases for hypertrophic cardiomyopathy: the role for calcium sensitization. J. Cardiothorac. Vasc. Anesth. 32, 478–487. doi: 10.1053/j.jvca.2017.05.035
Robertson, I. M., Li, M. X., and Sykes, B. D. (2009). Solution structure of human cardiac troponin C in complex with the green tea polyphenol, (−)-epigallocatechin 3-gallate. J. Biol. Chem. 284, 23012–23023. doi: 10.1074/jbc.M109.021352
Robinson, P., Griffiths, P. J., Watkins, H., and Redwood, C. S. (2007). Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ. Res. 101, 1266–1273. doi: 10.1161/CIRCRESAHA.107.156380
Sheehan, A., Messer, A. E., Papadaki, M., Choudhry, A., Kren, V., Biedermann, D., et al. (2018). Molecular defects in cardiac myofilament Ca(2+)-regulation due to cardiomyopathy-linked mutations can be reversed by small molecules binding to troponin. Front. Physiol. 9:243. doi: 10.3389/fphys.2018.00243
Singh, S. R., Zech, A. T. L., Geertz, B., Reischmann-Dusener, S., Osinska, H., Prondzynski, M., et al. (2017). Activation of autophagy ameliorates cardiomyopathy in Mybpc3-targeted knockin mice. Circ. Heart Fail. 10. doi: 10.1161/CIRCHEARTFAILURE.117.004140
Sommese, R. F., Sung, J., Nag, S., Sutton, S., Deacon, J. C., Choe, E., et al. (2013). Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human beta-cardiac myosin motor function. Proc. Natl. Acad. Sci. USA 110, 12607–12612. doi: 10.1073/pnas.1309493110
Spoladore, R., Maron, M. S., D’Amato, R., Camici, P. G., and Olivotto, I. (2012). Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur. Heart J. 33, 1724–1733. doi: 10.1093/eurheartj/ehs150
Spudich, J. A., Aksel, T., Bartholomew, S. R., Nag, S., Kawana, M., Yu, E. C., et al. (2016). Effects of hypertrophic and dilated cardiomyopathy mutations on power output by human beta-cardiac myosin. J. Exp. Biol. 219, 161–167. doi: 10.1242/jeb.125930
Stoehr, A., Neuber, C., Baldauf, C., Vollert, I., Friedrich, F. W., Flenner, F., et al. (2014). Automated analysis of contractile force and Ca2+ transients in engineered heart tissue. Am. J. Physiol. Heart Circ. Physiol. 306, H1353–H1363. doi: 10.1152/ajpheart.00705.2013
Stucker, S., Kresin, N., Carrier, L., and Friedrich, F. W. (2017). Nebivolol desensitizes myofilaments of a hypertrophic cardiomyopathy mouse model. Front. Physiol. 8:558. doi: 10.3389/fphys.2017.00558
Tanaka, A., Yuasa, S., Mearini, G., Egashira, T., Seki, T., Kodaira, M., et al. (2014). Endothelin-1 induces myofibrillar disarray and contractile vector variability in hypertrophic cardiomyopathy-induced pluripotent stem cell-derived cardiomyocytes. J. Am. Heart Assoc. 3:e001263. doi: 10.1161/JAHA.114.001263
Tardiff, J. C., Carrier, L., Bers, D. M., Poggesi, C., Ferrantini, C., Coppini, R., et al. (2015). Targets for therapy in sarcomeric cardiomyopathies. Cardiovasc. Res. 105, 457–470. doi: 10.1093/cvr/cvv023
Tardiff, J. C., Hewett, T. E., Palmer, B. M., Olsson, C., Factor, S. M., Moore, R. L., et al. (1999). Cardiac troponin T mutations result in allele-specific phenotypes in a mouse model for hypertrophic cardiomyopathy. J. Clin. Invest. 104, 469–481. doi: 10.1172/JCI6067
van Dijk, S. J., Dooijes, D., dos Remedios, C., Michels, M., Lamers, J. M., Winegrad, S., et al. (2009). Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 119, 1473–1483. doi: 10.1161/CIRCULATIONAHA.108.838672
van Dijk, S. J., Paalberends, E. R., Najafi, A., Michels, M., Sadayappan, S., Carrier, L., et al. (2012). Contractile dysfunction irrespective of the mutant protein in human hypertrophic cardiomyopathy with normal systolic function. Circ. Heart Fail. 5, 36–46. doi: 10.1161/CIRCHEARTFAILURE.111.963702
Vignier, N., Schlossarek, S., Fraysse, B., Mearini, G., Kramer, E., Pointu, H., et al. (2009). Nonsense-mediated mRNA decay and ubiquitin-proteasome system regulate cardiac myosin-binding protein C mutant levels in cardiomyopathic mice. Circ. Res. 105, 239–248. doi: 10.1161/CIRCRESAHA.109.201251
Walsh, R., Thomson, K. L., Ware, J. S., Funke, B. H., Woodley, J., McGuire, K. J., et al. (2017). Reassessment of Mendelian gene pathogenicity using 7, 855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 19, 192–203. doi: 10.1038/gim.2016.90
Warren, C. M., Karam, C. N., Wolska, B. M., Kobayashi, T., de Tombe, P. P., Arteaga, G. M., et al. (2015). Green tea catechin normalizes the enhanced Ca2+ sensitivity of myofilaments regulated by a hypertrophic cardiomyopathy-associated mutation in human cardiac troponin I (K206I). Circ. Cardiovasc. Genet. 8, 765–773. doi: 10.1161/CIRCGENETICS.115.001234
Keywords: myofilament, Ca2+ sensitivity, hypertrophic cardiomyopathy, MYBPC3, epigallocatechin-3-gallate, NanoString nCounter
Citation: Kresin N, Stücker S, Krämer E, Flenner F, Mearini G, Münch J, Patten M, Redwood C, Carrier L and Friedrich FW (2019) Analysis of Contractile Function of Permeabilized Human Hypertrophic Cardiomyopathy Multicellular Heart Tissue. Front. Physiol. 10:239. doi: 10.3389/fphys.2019.00239
Edited by:
Francisco H. Andrade, University of Kentucky, United StatesReviewed by:
Stuart Campbell, Yale University, United StatesKenneth S. Campbell, University of Kentucky, United States
Copyright © 2019 Kresin, Stücker, Krämer, Flenner, Mearini, Münch, Patten, Redwood, Carrier and Friedrich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lucie Carrier, bC5jYXJyaWVyQHVrZS5kZQ==
Felix W. Friedrich, Zi5mcmllZHJpY2hAdWtlLmRl