 Micaela Lasser
Micaela Lasser Benjamin Pratt†
Benjamin Pratt† Seung Woo Kim
Seung Woo Kim Laura Anne Lowery
Laura Anne Lowery- Department of Biology, Boston College, Chestnut Hill, MA, United States
Wolf–Hirschhorn syndrome (WHS) is a rare developmental disorder characterized by intellectual disability and various physical malformations including craniofacial, skeletal, and cardiac defects. These phenotypes, as they involve structures that are derived from the cranial neural crest, suggest that WHS may be associated with abnormalities in neural crest cell (NCC) migration. This syndrome is linked with assorted mutations on the short arm of chromosome 4, most notably the microdeletion of a critical genomic region containing several candidate genes. However, the function of these genes during embryonic development, as well as the cellular and molecular mechanisms underlying the disorder, are still unknown. The model organism Xenopus laevis offers a number of advantages for studying WHS. With the Xenopus genome sequenced, genetic manipulation strategies can be readily designed in order to alter the dosage of the WHS candidate genes. Moreover, a variety of assays are available for use in Xenopus to examine how manipulation of WHS genes leads to changes in the development of tissue and organ systems affected in WHS. In this review article, we highlight the benefits of using X. laevis as a model system for studying human genetic disorders of development, with a focus on WHS.
Introduction
Wolf–Hirschhorn syndrome (WHS) is a developmental disorder characterized by craniofacial malformations, intellectual disability, microcephaly, seizures, growth retardation, and developmental delays, though the severity of these symptoms varies from patient to patient (Fisch et al., 2010; Hannes et al., 2010; Sheth et al., 2012; Battaglia et al., 2015; Rutherford and Lowery, 2016). The core phenotype of WHS is the “Greek warrior helmet” appearance, defined by a prominent forehead, widely spaced eyes (hypertelorism), and an unusually wide and protrusive nasal bridge (Engbers et al., 2009). Other craniofacial abnormalities include an undersized jaw (micrognathia), a short philtrum, and cleft lip (Engbers et al., 2009). Most patients present with microcephaly and, less frequently, with skeletal defects, renal and urogenital defects, heart defects, hearing loss, and ocular irregularities (Andersen et al., 2014; Paradowska-Stolarz, 2014).
The chromosomal basis of WHS has been linked to a telomeric heterozygous microdeletion on the short arm of chromosome 4, though the nature and size of this deletion differs between patients (Zollino et al., 2014; Battaglia et al., 2015; Bi et al., 2016). The central phenotypes of WHS are most strongly associated with the deletion of two adjacent, non-overlapping critical regions at 4p16.3, WHS critical region 1 (WHSCR1) and WHS critical region 2 (WHSCR2) (Battaglia et al., 2015). These regions are comprised of the primary candidate genes WHSC1, WHSC2, and LETM1 (Figure 1; Battaglia et al., 2015; Derar et al., 2018). However, most WHS patients harbor mutations that extend beyond these critical regions, affecting several flanking genes such as TACC3, FGFR3, SLBP, CTBP1, CPLX1, PIGG, MSX1, and FGFRL1, demonstrating that deletions encompassing these genes may also contribute to the presentation of WHS symptoms (Hannes et al., 2010; Endele et al., 2011; Battaglia et al., 2015; Ho et al., 2016; Rutherford and Lowery, 2016).
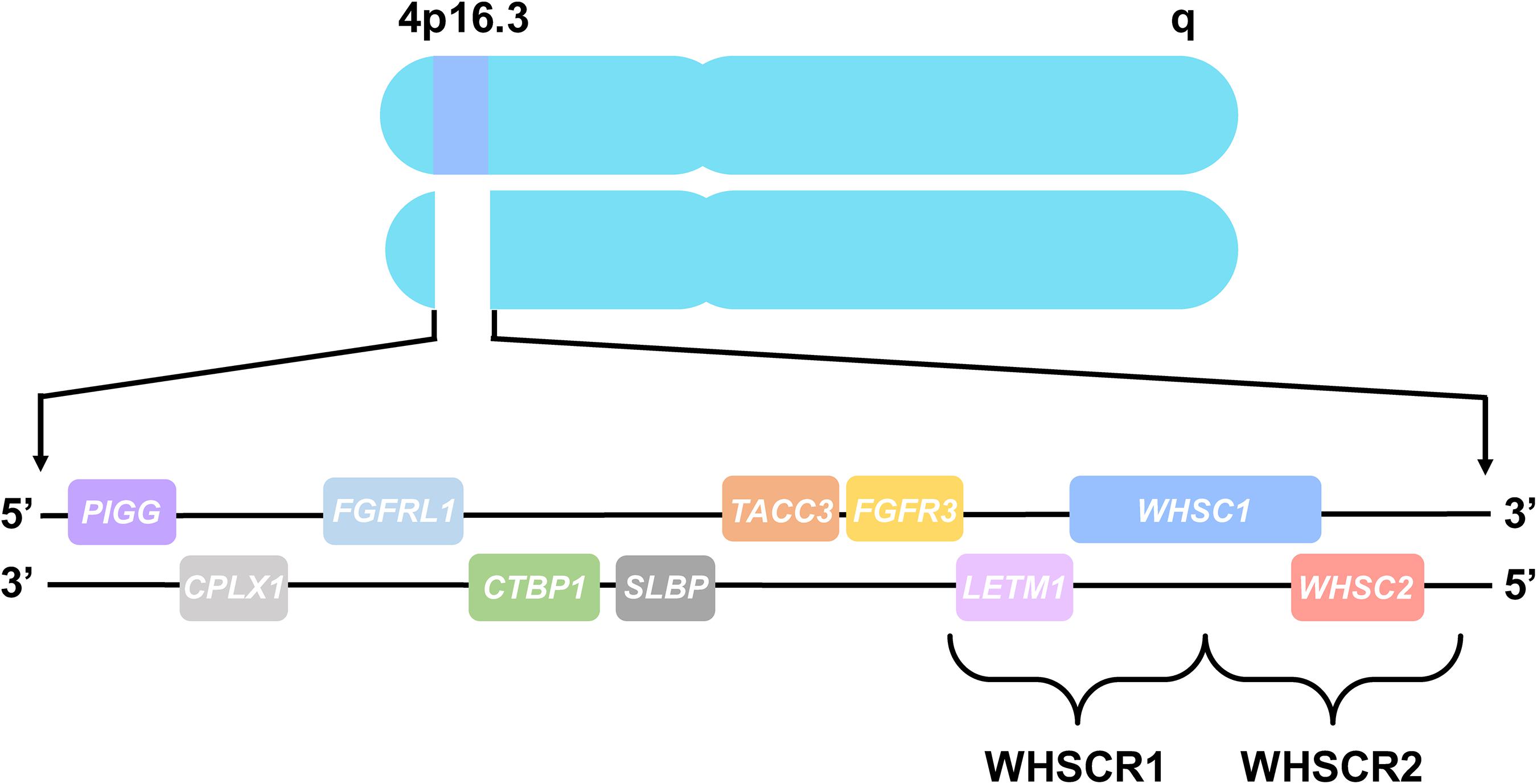
Figure 1. Chromosome 4 and WHS-associated genes. All genes are represented in their order from left to right, telomeric to centromeric orientation, located on the distal arm of chromosome 4p16.3. Brackets delineate WHS critical region 1 (WHSCR1), comprised of LETM1 and WHSC1, and WHS critical region 2 (WHSCR2), comprised of WHSC1 and WHSC2. Genes that are also mutated in WHS patients consist of PIGG, CPLX1, FGFRL1, CTBP1, SLBP, TACC3, and FGFR3, which flank the WHS critical regions.
Due to the clinical and genetic variability of WHS, the pathology underlying the disorder has been difficult to determine. However, the characteristic facial and cardiac phenotypes of WHS suggest a particular cell population may be affected by mutations of WHS-associated genes. Vertebrate facial features are derived from or influenced by neural crest cells (NCCs), which are a group of multipotent stem cells that are born along the neural tube and migrate long distances toward their final destination (Mayor and Theveneau, 2013). Additionally, some WHS-associated genes have defined roles in epigenetic modifications or signaling pathways that are integral for proper embryonic development and cell-motility related processes (Rutherford and Lowery, 2016). Therefore, it is possible that some of the developmental defects associated with WHS may be due to abnormalities in signaling pathways that affect NCC motility or migration.
While some animal models of WHS do exist, most of these studies have focused on the cellular and molecular functions of singular WHS-associated genes, specifically WHSC1 and LETM1 (Battaglia et al., 2015). However, the link between haploinsufficiency of most WHS-associated genes to specific vertebrate developmental processes such as brain, craniofacial, and heart development, has not been carefully examined. Furthermore, clinical studies of WHS patients have provided clear evidence that the syndrome is multigenic (Battaglia et al., 2015); yet, we still lack a mechanistic understanding of how the depletion of WHS-associated genes combinatorially contributes to the phenotypic spectrum of the disorder.
The model organism, Xenopus laevis, has been used extensively in the research community for decades to examine fundamental developmental and cellular biological processes, making it an ideal system for investigating human genetic disorders, such as WHS. X. laevis offers a number of benefits, as they are inexpensive and easy to culture, maintain, manipulate, and image, compared to other vertebrates (Erdogan et al., 2016; Slater et al., 2017). Thus, Xenopus is well-suited to examine the role of WHS-associated genes during vertebrate development, and can be used to test whether abnormal NCC migration may be one mechanism by which mutations of these genes contribute to phenotypes of the disease.
In this review, we provide a brief overview of the known functions of WHS-associated genes and how they may be linked to developmental processes, such as NCC migration. We also highlight how Xenopus is an advantageous system to study WHS as a multigenic disorder, and we discuss ways in which it can be used to investigate how WHS-associated genes individually and combinatorially affect both proper embryonic development and the phenotypes linked to WHS.
WHS Critical Region and Associated Candidate Genes
Wolf–Hirschhorn syndrome is a contiguous gene syndrome associated with small heterozygous deletions of the 4p chromosomal region. Following the initial characterization of the disorder in the 1960s, research has focused on generating a consensus of the core WHS phenotype, which includes intellectual disability, growth delay, seizures, and the typical craniofacial dysmorphisms, as well as defining the critical genomic region that gives rise to this core phenotype (Battaglia et al., 2015). The first critical region to be described, WHSCR1, consisted of a small 165 kb region encompassing two candidate genes, WHS Candidates 1 and 2 (WHSC1, WHSC2), and is deleted in all traditional cases of the disorder (Battaglia et al., 2015).
However, further clinical studies of children with WHS detected an even larger 1.9 Mb deletion that extends distally beyond WHSCR1, sufficient to produce the full WHS core phenotype (Battaglia et al., 2015). This newly described region is referred to as WHSCR2 and encompasses a more telomeric region of WHSC1, as well as a new candidate gene, leucine zipper and EF-hand containing transmembrane protein 1 (LETM1) (Battaglia et al., 2015). Moreover, many WHS patients harbor mutations of genes that flank these critical regions, providing increasing evidence that the manifestation of the disorder is not due to a single gene, but rather due to the haploinsufficiency of several closely linked genes (Battaglia et al., 2015; Rutherford and Lowery, 2016). Thus, WHS is largely considered to be a true multigenic disorder. In the subsequent sections, we describe the current known functions of the primary WHS candidate genes, as well as other WHS-associated genes, their potential roles during embryonic development, and how they may contribute to the spectrum of WHS phenotypes.
WHSC1
WHSC1 is a histone methyltransferase, belonging to a family of nuclear receptor SET domain (NSD) proteins that possess a SET domain encoding lysine melthyltransferase activity (Nimura et al., 2009; Ezponda et al., 2013; Liu et al., 2015). Specifically, WHSC1 is associated with trimethylation of histone H3K36, a mark that is highly correlated with active transcription. WHSC1 is partially or fully deleted in nearly all cases of WHS and is thought to be responsible for many of the core WHS phenotypes, including growth delay and facial irregularities (Andersen et al., 2014; Battaglia et al., 2015; Boczek et al., 2018; Derar et al., 2018); yet, the mechanism behind which its depletion leads to these phenotypes remains to be elucidated.
The spatial and temporal expression of WHSC1, as well as its transcriptional regulation activity, suggests that it plays an important role during development (Rutherford and Lowery, 2016). WHSC1 is a known regulator of TWIST, a key player in both the epithelial-to-mesenchymal transition (EMT) and cell migration (Ezponda et al., 2013). Moreover, WHSC1 is expressed in the developing nervous system, pharyngeal arches, and frontal facial region in both mice and Xenopus laevis, as well as jaw and heart in mice (Stec et al., 1998; Nimura et al., 2009; Yamada-Okabe et al., 2010; Liu et al., 2015; Mills et al., 2019), which is significant, given that many of these structures are both derived from the neural crest and are affected in WHS.
However, until recently, WHSC1 function had not been directly associated with NCC migration. To gain further insight into the role of WHSC1, specifically during processes that govern craniofacial morphogenesis, Mills et al. (2019) sought to observe the effects of reducing WHSC1 dosage levels in Xenopus laevis during embryonic development. They found that WHSC1 KD results in reduced length and area of the pharyngeal arches, as well as reduced NCC migration speed. Depletion of WHSC1 also causes severe craniofacial and brain morphology defects including widening of the facial width and area, and reduction of forebrain size. Together, these results are the first to demonstrate that reduced levels of WHSC1 cause aberrant NCC migration, which may lead to some of the craniofacial abnormalities seen in WHS patients. These results are consistent with other studies showing that WHSC1 depletion leads to irregularities in brain, cartilage, and bone formation, specifically incomplete motor neuron development in zebrafish, deficiencies in midline fusion and cleft palate in mice, as well as delayed bone ossification in both zebrafish and mice (Nimura et al., 2009; Yamada-Okabe et al., 2010; Ezponda et al., 2013; Liu et al., 2015), indicating a conserved developmental role for WHSC1 among various species.
An important question still remains as to which signaling pathways are affected by WHSC1 reduction and how they contribute to abnormal NCC migratory patterns and WHS phenotypes. A promising possibility is the Wnt signaling pathway, which has already been connected to WHSC1 dysregulation in the context of cancer (Toyokawa et al., 2011). Toyokawa et al. (2011) suggests that WHSC1 dosage may impact canonical Wnt signaling by affecting nuclear levels of beta-catenin through transcriptional regulation. Moreover, in Xenopus, canonical Wnt signaling has been shown to be important for NCC specification and migration during development, as well as subsequent craniofacial morphogenesis (Maj et al., 2016; Li et al., 2018). Thus, further experiments are necessary to examine whether alterations in WHSC1 dosage impact the Wnt signaling pathway, potentially leading to abnormal NCC migration and the spectrum of defects observed in WHS.
WHSC2
The WHSC2 gene, also known as NELF-A, encodes a component of the negative elongation factor complex, which negatively regulates RNA polymerase II progression during elongation, subsequently altering the expression of its target genes (Kerzendorfer et al., 2012). Though WHSC2 is commonly deleted in most WHS patients, it has not emerged as a primary contributor to any specific WHS phenotype (Battaglia et al., 2015). However, the cellular and molecular roles of WHSC2 have begun to be illuminated and functionally linked to other WHS-affected genes.
Depletion of WHSC2 in both cell culture and in Drosophila has been shown to reduce cellular proliferation and lead to irregularities in cell cycle progression (Gilchrist et al., 2008; Yung et al., 2009). Interestingly in breast cancer cell lines, reduction of the NELF-E component of the NELF complex is associated with lower levels of trimethylated H3K36 on promoters of NELF-regulated genes (Sun and Li, 2010). As previously mentioned, levels of trimethylated H3K36 are influenced by the activity of WHSC1, highlighting a potential link between WHSC1 and WHSC2 (NELF-A) in altering gene expression, although this remains to be examined.
WHSC2 also functions to recruit stem-loop binding protein (SLBP) to the 3′ ends of histone pre-mRNAs (Narita et al., 2007). SLBP lies telomeric to WHSC2 and mutations of this gene have been found in some WHS patients, though the functional impact of SLBP haploinsufficiency is unclear. SLBP is critical for the correct processing of histone pre-mRNAs and regulating histone degradation. Histone biogenesis is essential for epigenetic gene regulation through histone post-translational modifications and maintenance of chromatin structure (Margueron and Reinberg, 2010). Thus, it is possible that there is a functional connection between WHSC1, WHSC2, and SLBP, and reduced activity of these genes may have impacts on histone levels and transcriptional regulation, which could adversely affect normal development in the context of WHS.
Until recently, WHSC2 function during vertebrate tissue and organ development had not been carefully studied. Mills et al. (2019) found that WHSC2 expression in Xenopus is enriched in the developing nervous system, pharyngeal arches, and frontal facial region. Additionally, altered levels of WHSC2 lead to abnormalities in craniofacial, cartilage, and brain development. Specifically, reduction of WHSC2 causes smaller facial width and area, smaller cartilage elements, as well as smaller brain size (Mills et al., 2019), indicating that WHSC2 may play a larger role regarding WHS phenotypes than what was previously thought. It is possible that these phenotypes arise as a consequence of decreased cellular proliferation, as WHSC2 has already been linked to this process (Sun and Li, 2010), though this connection still needs to be tested. Moreover, it would be interesting to examine whether reduced dosage levels of WHSC2 in combination with WHSC1 or SLBP enhances these phenotypes, providing further evidence that these genes are functionally linked to one another.
LETM1
LETM1 is localized to the inner mitochondrial membrane and is important for maintaining normal mitochondrial function by participating in Ca2+/H+ exchange and regulating Ca2+ homeostasis within the matrix (Dimmer et al., 2008; Jiang et al., 2013). LETM1 has been proposed as the candidate gene for seizures in WHS patients (Endele et al., 1999; McQuibban et al., 2010; Andersen et al., 2014; Battaglia et al., 2015). Heterozygous deletion of LETM1 in mice led to altered glucose metabolism and impaired control of ATP levels in the brain, which could explain the increased susceptibility to seizures observed in these animals (Jiang et al., 2013). However, some WHS patients with LETM1 deletions lack seizures and conversely, some WHS patients have seizures, yet have no detectable mutations in LETM1 (Battaglia et al., 2015). Therefore, it is likely that LETM1 is not solely responsible for the occurrence of seizures and that deletion of additional genes are required for the full expression of the phenotype.
In Xenopus, LETM1 is expressed ubiquitously during early embryonic development. As development progresses, more defined expression is observed in the pharyngeal arches, craniofacial and brain regions (Mills et al., 2019). Reduced dosage of LETM1 in Xenopus revealed that it is critical for proper craniofacial morphology and may contribute to some of the facial defects associated with WHS, which had not been previously demonstrated (Mills et al., 2019). Interestingly, knocking down LETM1 did not have a significant effect on NCC migration or motility, which is surprising, given the robust craniofacial phenotype observed. However, it is possible that LETM1 may play an entirely different role in these cells. Proper mitochondrial function and regulation of energy metabolism is critical for determining cell fate and maintaining cell survival (Khacho et al., 2019). Therefore, while reduced dosage of LETM1 may not directly affect NCC migration, it may greatly impact the metabolic homeostasis of NCCs and lead to downstream effects on differentiation, proliferation, or apoptosis during craniofacial morphogenesis. As LETM1 depletion in fibroblasts derived from WHS patients has already been associated with decreased cellular proliferation (Doonan et al., 2014), this seems a promising avenue of future research to understand the precise molecular mechanisms of LETM1 involvement in the pathogenesis of WHS.
Additional WHS-Associated Genes
While all WHS patients harbor full or partial deletions of the WHSCRs, most have mutations that affect multiple genes flanking these regions. It is widely accepted that haploinsufficiency of the WHS critical region genes alone cannot account for the full spectrum of WHS phenotypes (Battaglia et al., 2015). Thus, it is likely that these flanking genes, including TACC3, FGFR3, FGFRL1, SLBP, CTBP1, CPLX1, and PIGG, and MSX1 also contribute to WHS in some capacity.
Fibroblast growth factor receptor 3 (FGFR3) and fibroblast growth factor receptor like 1 (FGFRL1) are both involved in signaling pathways that influence cell division and differentiation. Both FGFR3 and FGFRL1 have been proposed as candidate genes responsible for the skeletal and craniofacial malformations of WHS patients (Battaglia et al., 2015). Homozygous null mouse lines for Fgfrl1 recapitulate many WHS phenotypes such as hypoplasia of skeletal elements, delayed fusion and bone ossification, abnormal forebrain development, and congenital heart defects (CHD) (Catela et al., 2009). Conversely, Fgfr3 null mice display severe kyphosis, abnormally long tails and femurs, which is indicative of prolonged bone growth (Simon and Bergemann, 2008). With opposing phenotypes, it is possible that these two genes antagonize each other to regulate chondrocyte proliferation and may genetically interact to control bone growth. Moreover, FGFR3 has been shown to directly affect NCC migration in chick, with its loss leading to slower NCC migration velocity and increased neural crest death close to the neural tube (Sato et al., 2011). However, it remains unclear whether the loss of either FGFR3 or FGFRL1 is directly linked to abnormal NCC migration in the WHS phenotypes.
Transforming acidic coiled-coil protein 3 (TACC3) is a microtubule plus-end tracking (+TIP) protein that resides on the end of polymerizing microtubules and has been shown to regulate microtubule dynamics during neural development (Nwagbara et al., 2014; Bearce et al., 2015; Cammarata et al., 2016; Erdogan et al., 2017). The ways in which TACC3 dosage levels affect gross morphological phenotypes reflected in WHS patients was recently examined and provides a strong link to abnormal NCC migration. In Xenopus, TACC3 expression is enriched in the pharyngeal arches, as well as the frontal brain and facial regions (Mills et al., 2019). Knockdown of TACC3 led to overt craniofacial and cartilage defects, including smaller facial width, area and angle, which are likely due to overall smaller cartilage elements (Mills et al., 2019). Furthermore, in vivo migration of NCCs was affected as a consequence of reduced TACC3 levels. This manipulation also severely affected forebrain development, reflecting the microcephaly phenotype commonly associated with WHS. This is interesting given that TACC3 is also known to be an essential centrosome adaptor that is critical for proper cell division, and almost all microcephaly associated genetic defects involve proteins that maintain centrosomes and regulate the mitotic spindle (Peset and Vernos, 2008; Lasser et al., 2018). Thus, it is likely that TACC3 contributes to some WHS phenotypes, possibly by adversely altering the mitotic spindle such that it has downstream effects that influence cell fate and cell migration.
It has been suggested that C-terminal binding protein 1 (CTBP1), complexin 1 (CPLX1), and phosphatidylinositol glycan anchor biosynthesis class G (PIGG) are possible candidates for developmental delay, intellectual disabilities, and seizures in WHS patients (Zollino et al., 2014). CTBP1 is a transcriptional co-repressor known to mediate E-cadherin repression and play a key role in EMT (Zhang et al., 2013). Increased Ctbp activity has been shown to reduce epileptic behavior in rats following a ketogenic diet by forcing energy production via the TCA cycle and oxidation of NADH (Garriga-Canut et al., 2006). Reduced levels of NADH stimulates Ctbp activity and could be a mechanism by which loss of CTBP1 contributes to seizures. Ctbp1 mutant mice also exhibit defects in skeletal and muscle development (Hildebrand and Soriano, 2002). As CTBP1 is known to be involved in EMT (Zhang et al., 2013), it seems plausible that mutations of this gene may result in these phenotypes by influencing NCC migration.
CPLX1 is a cytosolic protein that is important for synaptic vesicle exocytosis and regulation of neurotransmitter release (Glynn et al., 2005). Mutations of CPLX1 have been associated with epilepsy and intellectual disability, and Cplx1 knockout mice exhibit pronounced motor and social deficits (Glynn et al., 2005; Drew et al., 2007; Redler et al., 2017). Pathogenic variants of PIGG are associated with intellectual disability, hypotonia, and seizures (Makrythanasis et al., 2016). PIGG localizes to the endoplasmic reticulum and is responsible for the biogenesis of GPI anchor proteins in zebrafish, which is critical for expression of voltage-gated sodium channels in a subset of neurons (Nakano et al., 2010). However, it is clear that further research is needed to determine how these genes are distinctly contributing to the development of organs and tissues that are affected in WHS.
MSX1 (Msh homeobox 1) functions as a transcriptional repressor during embryogenesis and has been suggested as a candidate gene for the oral and dental malformations seen in some WHS patients (Nieminen et al., 2003). Mutations of MSX1 have been associated with non-syndromic cleft palate and tooth agenesis in humans, and Msx1-deficient mice exhibit severe craniofacial abnormalities, cleft palate, and absence of teeth (Satokata and Maas, 1994; Zhang et al., 2002; Nieminen et al., 2003). In Xenopus, MSX1 has been shown to be critical for NCC induction and regulates the expression of genes that are necessary for proper NCC migration (Monsoro-Burq et al., 2005; Bonano et al., 2008; Macrì et al., 2016). Thus, it is likely that haploinsufficiency of MSX1 contributes to some craniofacial and dental defects associated with WHS by affecting NCC induction, as well as having downstream regulatory effects on genes that influence their subsequent migration.
Using Xenopus Laevis as a Model to Study WHS
In an effort to answer some outstanding questions regarding the pathogenesis of WHS, it is critical to choose a model organism that is ideal for studying human genetic diseases. Every model system has its benefits and limitations; however, X. laevis has emerged as a particularly advantageous model for studying human genetic diseases, like WHS. While mouse models are an excellent mammalian system due to the similarity between the mouse and human genomes and the large genetic toolkit available, they are very costly to house and maintain. Moreover, litter sizes are small, embryonic development occurs in utero, and creating genetic lines that harbor mutant alleles is time-consuming. While zebrafish produce many offspring and have well-developed genetic manipulation strategies, their genome has lost a great deal of synteny with mammals and many relevant disease-related genes do not perform the same function (Garcia de la Serrana et al., 2014). Zebrafish also lack organ systems, such as limbs, digits, and lungs, that are involved in many human congenital syndromes. Additionally, the zebrafish heart only has one atrium and one ventricle, and cannot fully model developmental heart abnormalities, like those associated with WHS.
However, using Xenopus as a system to investigate human genetic diseases of development has enormous potential, and can complement more established systems, like mouse or zebrafish, to enhance our knowledge about the function of understudied genes and the underlying mechanisms by which developmental abnormalities arise. In the following sections, we highlight why X. laevis is an advantageous model to choose for investigating developmental disorders, with a specific focus on how it can be used to examine multiple aspects of WHS.
Advantages of Xenopus laevis as a Model Organism
Xenopus is an excellent system for relatively high throughput analysis of genetic manipulation on vertebrate embryonic development. Xenopus shares a high degree of synteny with humans and a majority of disease-associated genes are conserved between these species (Hellsten et al., 2010; Session et al., 2016). Despite X. laevis having a duplicated genome, genetic manipulation strategies can still be utilized. However, X. tropicalis may be more suitable, depending on the number of genes being studied, as they are diploid, making genetic manipulation much easier and more rapid. The Xenopus genome is widely available to the research community through the efforts of Xenbase, an online resource that has organized current annotated genetic information, protocols, Xenopus anatomy and development, scientific literature, and provides useful sequencing tools, such as the genome browser and Xenopus specific BLAST (James-Zorn et al., 2018).
A key characteristic of the Xenopus model is the ease of acquiring large amounts of high quality embryos by inducing females to lay eggs via hormone priming (Sive et al., 2007a, b; Erdogan et al., 2016; Slater et al., 2017). Hundreds of embryos can be obtained in a single clutch, enabling numerous embryos to be manipulated and observed in a single experiment. Xenopus development occurs rapidly and externally, with gastrulation and neurulation occurring between 9 and 26 h post fertilization, and organogenesis almost complete by 5 days post fertilization (Nieuwkoop and Faber, 1994; Zahn et al., 2017). Xenopus organ development and morphology have been well-characterized and is comparable to those of mammalian systems, including orofacial (Dickinson, 2016), heart (Hempel and Kühl, 2016), kidney (Getwan and Lienkamp, 2017), and nervous system development (Pratt and Khakhalin, 2013; Lee-Liu et al., 2017), all of which are affected in WHS patients. Moreover, Xenopus is being used extensively as a model to understand a number of different human genetic diseases that lead to defects in these systems, such as congenital heart disorders (Boskovski et al., 2013; Sojka et al., 2014; Duncan and Khokha, 2016; Deniz et al., 2017), kidney disease (Lienkamp, 2016), ciliopathies (Brooks and Wallingford, 2015; Wallmeier et al., 2016), orofacial defects (Tahir et al., 2014; Dickinson, 2016), and neurodevelopmental disorders (Pratt and Khakhalin, 2013; Tandon et al., 2017; Willsey et al., 2018).
In order to characterize WHS-associated gene functions in relation to development and disease, Xenopus embryos can be injected with a variety of materials to manipulate gene expression, such as CRISPR/cas9 or morpholino oligonucleotides (MOs), in either the whole embryo or selected blastomeres (up to the 64-cell stage) (Mimoto and Christian, 2011; Tandon et al., 2012, 2017; Bestman and Cline, 2014; Bhattacharya et al., 2015; Wang et al., 2015; DeLay et al., 2016; Feehan et al., 2017; Moody, 2018a, b). As the lineage of individual cells has been well-documented, injections can be precisely targeted to specific tissues and organs that are affected in WHS, such as the heart, kidney, or brain. A unique feature of Xenopus compared to other models is the ability to perform one-sided embryo injections, wherein only one side of the embryo is experimentally manipulated and the opposite side serves as an internal control (Figure 2; Willsey et al., 2018; Mills et al., 2019). Thus, assessing phenotypic consequences that arise as a result of genetic manipulation can be compared side-by-side to wild-type gene expression.
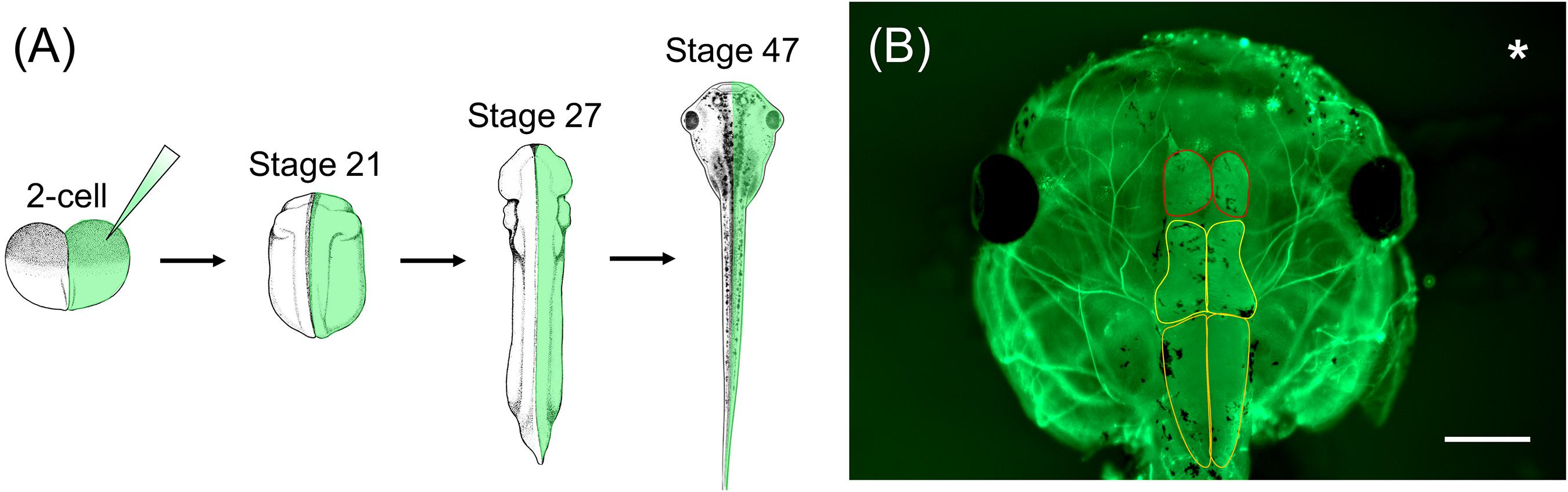
Figure 2. Xenopus one-sided injections allow for side-by-side comparison to wild-type gene expression following genetic manipulation. (A) Xenopus embryos can be injected in one cell at the 2-cell stage with genetic manipulation macromolecules and fluorescent mRNA. At stage 21, embryos can be sorted depending on the side that was injected (left vs. right) and used for various assays throughout development. (B) Representative image of stage 47 X. laevis tadpole immunolabeled for acetylated tubulin. Asterisk represents the side that was injected with MO and fluorescent mRNA. Brain morphology, such as forebrain size (red outline), midbrain size (yellow outline), and hindbrain size (orange outline), of the manipulated side can be compared to the unaltered, wild-type side. Scale bar: 500 μm.
Morpholino oligonucleotides are particularly useful in modeling genetic diseases, like WHS, as they can be easily titrated to reduce gene dosage levels similar to that in human patients (Tahir et al., 2014; Blum et al., 2015; McCammon and Sive, 2015). Because WHS-associated mutations result in haploinsufficiency, a full knockout of the candidate genes would not appropriately model the disease, and simultaneous knockdown of genes can be achieved by injecting multiple MOs at once, allowing for concurrent knockdown of genes that are often deleted together in WHS (Blum et al., 2015). While it is certainly possible to produce mouse lines with mutations in multiple genes (Simon and Bergemann, 2008), this is a costlier and more time-consuming process than the equivalent in X. laevis. As with all manipulation strategies, the appropriate controls must be used to account for off-target effects, such as generating more than one MO, dose dependency, and rescuing phenotypes by co-injecting mRNA that is not targeted by the MO (Blum et al., 2015; Gentsch et al., 2018).
The CRISPR/cas9 system has also been used as an extremely effective method to knock out target genes in both X. tropicalis and X. laevis (Bhattacharya et al., 2015; Wang et al., 2015; Tandon et al., 2017). While the off-target effects of CRISPR/cas9 are generally minimal, the use of proper controls is critical by carefully designing multiple sgRNAs and performing rescue experiments to confirm that any phenotypes observed are due to the knockout of a particular gene (Nakayama et al., 2013; Wang et al., 2015). The CRISPR/cas9 system can be employed to validate phenotypes that arise as a result of MO-mediated gene knockdown by comparing phenotypes generated by both methods (Bhattacharya et al., 2015; Willsey et al., 2018). Overall, Xenopus is an excellent system for investigating developmental disorders, such as WHS, and for elucidating the mechanism by which manipulation of WHS-associated genes alters proper embryonic development.
Xenopus laevis as a Model for Studying Neural Crest Cell Migration
As the underlying cellular and molecular mechanisms that cause WHS are unknown, it is essential to investigate the basis behind the disorder. One particularly promising avenue of research in this regard is the migration of the NCC population in the developing embryo (Rutherford and Lowery, 2016). As stated previously, NCCs are a multipotent stem cell population that originate along the neural tube, delaminate, and migrate throughout the developing embryo to their final destinations. Once at their proper locations, NCCs differentiate and contribute to various tissues and organ systems, including craniofacial cartilage and bone, smooth muscle of the heart, peripheral and enteric neurons, melanocytes, and glia (Bronner and LeDouarin, 2012). Considering craniofacial abnormalities are one of the defining phenotypes of WHS, the link to potential aberrations in NCC migration is strong. Moreover, almost all WHS-associated genes are connected with NCC migration or signaling pathways that may influence NCC differentiation or proliferation in some capacity. Therefore, research into NCC migration in relation to WHS is of great interest.
Xenopus laevis is an ideal model organism for studying NCC migration, as both in vivo and in vitro NCC migration can be tracked through multiple methods. NCC migration can be observed in vivo by performing transplantation assays, whereby NCCs are dissected from GFP-injected Xenopus embryos and transplanted to wild-type host embryos (Borchers et al., 2000; Cousin, 2018). These embryos can then be imaged using time-lapse confocal microscopy and NCC migration can be analyzed by measuring the number, width, and migration distance of the GFP-marked cranial segments. Additionally, two transgenic X. laevis frog lines, PAX3-GFP and SOX10-GFP, are available through the National Xenopus Resource (NXR) to study the in vivo development and migration of NCCs (Alkobtawi et al., 2018). In vivo NCC migration can also be analyzed through whole-mount in situ hybridization by observing the morphology of the pharyngeal arches using a marker for NCCs, such as TWIST or SOX9 (Figure 3; Devotta et al., 2016; Szabó et al., 2016).
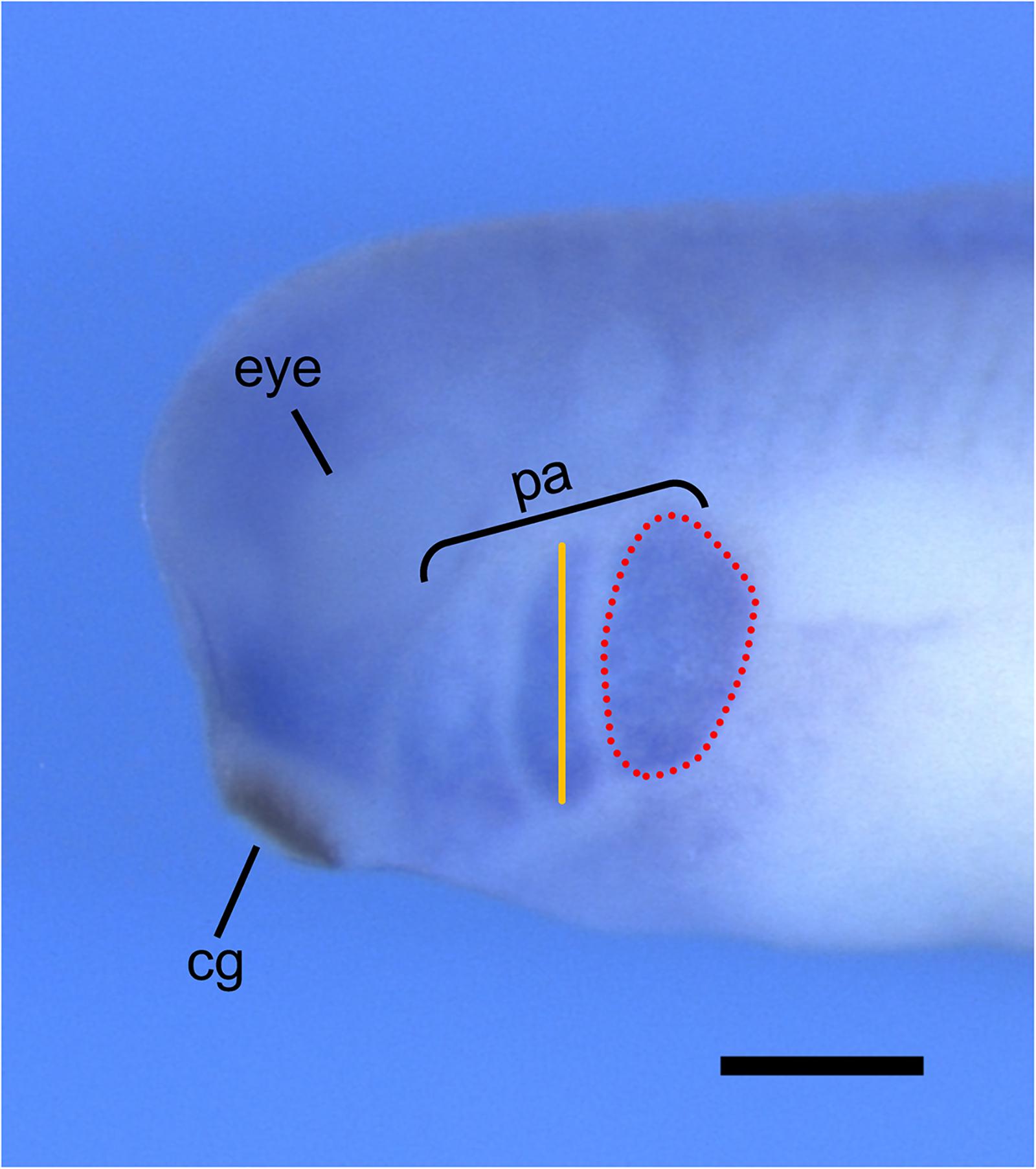
Figure 3. Analysis of in vivo NCC migration using whole-mount in situ hybridization. Representative image of stage 30 X. laevis embryo labeled for xTWIST, a marker for NCCs. Measurements of the length (orange line) and area (red dotted line) of each individual pharyngeal arch can be done using ImageJ. Pharyngeal arch length is measured from the most dorsal point to the most ventral point of each individual arch. Pharyngeal arch area is measured as the area around the periphery of each individual arch. cg, cement gland; pa, pharyngeal arches. Scale bar: 500 μm.
Neural crest cell motility can be examined in vitro by dissecting NCCs from stage 16 Xenopus embryos and culturing explants on fibronectin-coated coverslips (Cousin and Alfandari, 2018). Migratory behavior of NCC explants can be observed using time-lapse confocal microscopy and multiple parameters of migration can be measured including velocity and dispersion (DeSimone et al., 2005; Milet and Monsoro-Burq, 2014; Cousin and Alfandari, 2018). Moreover, chemotaxis assays can be performed in vitro to assess the directional migration of NCCs toward an external, soluble factor by coating beads with a chemoattractant cue, such as SDF-1 (Theveneau and Mayor, 2011; Theveneau et al., 2013; Shellard and Mayor, 2016; Szabó et al., 2016). Recently, these methods were successfully used in X. laevis to test whether knockdown of particular WHS genes led to changes in NCC migration both in vivo and in vitro (Mills et al., 2019), demonstrating how X. laevis can provide a unique and varied approach to study how alterations in NCC migratory behavior may contribute to phenotypes of the disease.
Xenopus laevis as a Model for Studying Other Aspects of WHS
Craniofacial abnormalities are one of the defining phenotypes of WHS and X. laevis has emerged as an excellent system for determining which WHS-associated genes are important for craniofacial morphogenesis. As orofacial development is highly conserved between Xenopus and other mammalian species, craniofacial abnormalities in Xenopus often resemble phenotypes present in human patients (Dickinson and Sive, 2007, 2006; Tahir et al., 2014; Dubey and Saint-Jeannet, 2017). Various techniques to assess changes in Xenopus orofacial development have already been developed and used to study craniofacial defects associated with human genetic disorders (Tahir et al., 2014; Gonzalez Malagon et al., 2018; Mills et al., 2019). Similar to NCC transplant assays, facial transplant assays can be performed in Xenopus (Jacox L. A. et al., 2014; Jacox L. et al., 2014; Jacox et al., 2016; Gonzalez Malagon et al., 2018). This technique allows for the examination of gene or protein function in particular cell types involved in facial development, eliminating non-specific effects in the whole embryo, and can be used to study signaling mechanisms or mechanical forces that are involved in orofacial development (Dickinson, 2016).
More robust quantitative methods to examine alterations of orofacial development have also been adapted for use in Xenopus. Measurements of various craniofacial features such as facial width, height, angle, and area can be done on embryos at different developmental stages using ImageJ (Figure 4; Kennedy and Dickinson, 2014a; Tahir et al., 2014). These measurements can be combined with geometric morphometrics in order to detect subtle differences in face shape and size throughout development (Kennedy and Dickinson, 2014b; Dickinson, 2016). Furthermore, techniques for visualizing cartilage morphology, such as Alcian blue staining, can be used to determine whether craniofacial abnormalities arise as a result of defects in cartilage development (Figure 5; Tahir et al., 2014; Devotta et al., 2016).
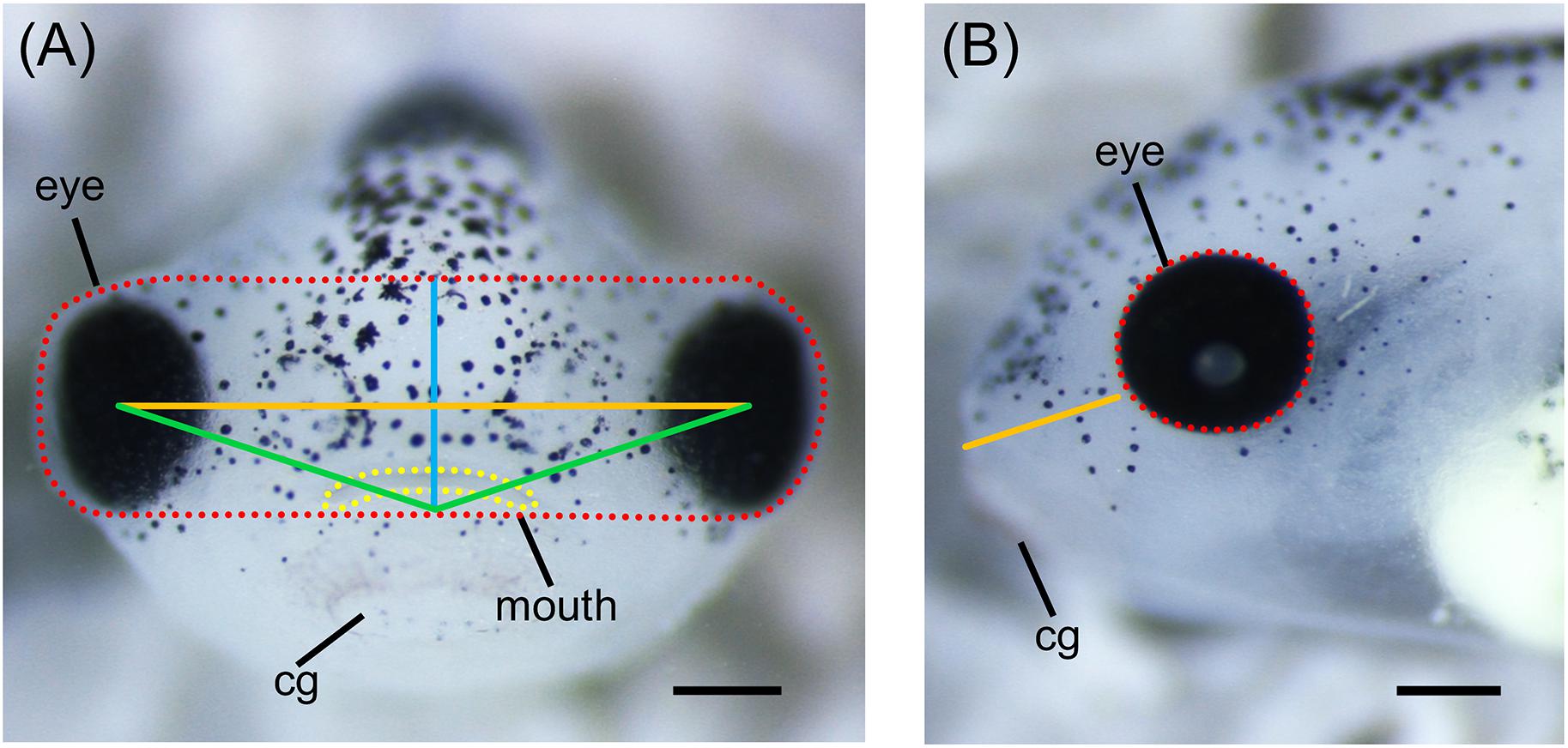
Figure 4. Measurements of Xenopus craniofacial features. (A) Representative frontal view image of stage 42 X. laevis tadpole. Measurements of facial features include facial height (blue line), facial width (orange line), facial area (red dotted line), facial angle (green line), and mouth roundness (yellow dotted line). Facial height is measured from the top of the middle of the eyes to the top of the cement gland. Facial width is measured from the middle of one eye to the middle of the opposite eye. Facial area is measured as the area around the top of the eyes to the top of the cement gland. Facial angle is measured as the angle between the middle of the eyes and the top of the cement gland. Mouth roundness is measured as the area around the periphery of the mouth. (B) Representative lateral view image of stage 42 X. laevis tadpole. Measurements of facial features include eye area (red dotted line) and snout length (orange line). Eye area is measured as the area around the periphery of the eye. Snout length is measured from the most anterior part of the face to the bottom of the eye. All measurements can be performed using ImageJ. cg, cement gland. Scale bar: 100 μm.
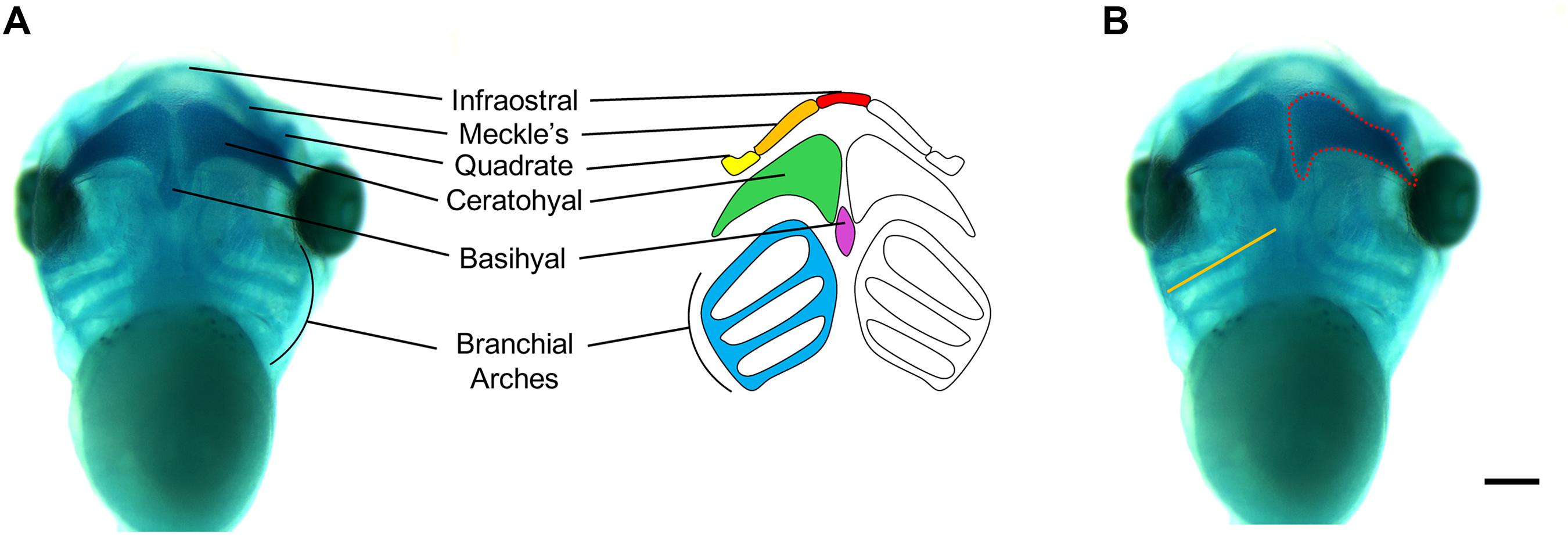
Figure 5. Measurements of Xenopus cartilage elements. (A) Xenopus cartilage anatomy with all major cartilage elements labeled in various colors. (B) Representative ventral view image of stage 45 X. laevis tadpole stained for Alcian blue. Measurements of cartilage elements include branchial arch length (orange line) and ceratohyal area (red dotted line), which can be performed using ImageJ. Scale bar: 300 μm.
It is important to note that there are some craniofacial and cartilage morphological differences between Xenopus and humans that will prevent some direct correlations to disease pathology. For example, Meckel’s cartilage will eventually become a component of the lower jaw in both Xenopus and humans, however in humans, it will also become part of the middle ear skeletal structures. Nevertheless, using these techniques in Xenopus has provided evidence that some WHS-associated genes are required for proper craniofacial development and contribute to the signature craniofacial dysmorphisms seen in WHS patients (Mills et al., 2019).
Individuals with WHS display a variety of other symptoms such as microcephaly, heart defects, and renal defects. Whether WHS-associated genes are required for proper development of these organ and tissue systems in relation to WHS still remains unknown. However, many techniques exist in Xenopus to study brain, heart, and kidney development. Xenopus neurodevelopment has been explored extensively using both in vitro and in vivo methods (Erdogan et al., 2016; Slater et al., 2017). Due to the large size of its growth cones, X. laevis is an excellent model to study cytoskeletal dynamics in axon outgrowth and guidance during early development. Moreover, live imaging can be done on both retinal and spinal axonal tracts in vivo (Erdogan et al., 2016; Slater et al., 2017). Additionally, changes in brain morphology can be quantified simply by measuring the area of the forebrain, midbrain, and hindbrain (Figure 2B), and used as a technique to reflect the microcephaly phenotype associated with WHS (Willsey et al., 2018; Mills et al., 2019).
Congenital heart defects have emerged as the most life-threatening birth defect in newborn patients across different developmental disorders, including WHS (Duncan and Khokha, 2016; Garfinkel and Khokha, 2017). Early heart development is highly conserved between Xenopus and mammalian systems, and assays to analyze CHD candidate genes have been created for use in Xenopus (Sojka et al., 2014; Duncan and Khokha, 2016; Hempel and Kühl, 2016; Deniz et al., 2017; Garfinkel and Khokha, 2017; Tandon et al., 2017). The effect of genetic manipulation on heart development can be explored at early tadpole stages using whole-mount in situ hybridization, immunohistochemistry with anti-tropomyosin (Tmy) antibody, scanning electron microscopy (SEM), transmission electron microscopy (TEM), or optical coherence tomography (OCT) (Sojka et al., 2014; Hempel and Kühl, 2016; Deniz et al., 2017). These assays help to visualize and detect improperly looped hearts, failed chamber formation, and abnormal structure of the adjacent connective tissue, and can be used to elucidate how mutations of WHS genes lead to CHD (Sojka et al., 2014; Duncan and Khokha, 2016).
In vitro explant, transplant, and ablation techniques can be used to derive insight into how mutations of genes associated with renal disease affect the progression of kidney development in Xenopus (Getwan and Lienkamp, 2017). In vivo experiments of kidney development range from optogenetic manipulations of physiological parameters (calcium, pH) to characterizations of electrophysiological recordings (Lienkamp, 2016; Getwan and Lienkamp, 2017). Together, X. laevis can be used as a powerful system to study how mutations of WHS-associated genes affect the development of multiple tissue and organ systems.
Conclusion
Xenopus laevis has emerged as an effective model organism for studying human genetic disorders of development, such as WHS. Xenopus embryos can be collected in large numbers and are easy to maintain, manipulate, and image. Genetic manipulation strategies are straightforward, allow for the concurrent KD or KO of multiple genes at once, and can be titrated to reduce gene dosage levels similar to that in human patients with WHS. Numerous techniques exist in Xenopus to study a variety of tissue and organ systems in order to understand the function of WHS-associated genes during embryonic development. Most importantly, Xenopus provides a system in which phenotypes of WHS, including craniofacial abnormalities, brain defects, heart defects, and kidney defects can be easily examined. Moreover, the mechanism by which mutations of WHS-associated genes affects embryonic development can be studied in Xenopus, such as changes in NCC migration. Thus, X. laevis is an excellent model system for investigating the underlying pathology of WHS, as well as many other human genetic diseases.
Author Contributions
BP and LL contributed to the conception and design of the manuscript. BP and ML wrote the first draft of the manuscript. CM and SK wrote the sections of the manuscript. LL edited the manuscript. All authors contributed to the revision, read, and approved the final version of the manuscript for submission.
Funding
This work was supported by Charles H. Hood Foundation, NIH R03 DE025824 and NIH R01 MH109651 (to LL), and NIH R01 GM121907 (to ML). American Cancer Society (RSG-16-144-01-CSM) to the funding list for LL.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Lowery Lab for helpful discussions, suggestions, and editing. We also thank the National Xenopus Resource RRID:SCR_013731 and Xenbase RRID:SCR_003280 for their support.
References
Alkobtawi, M., Ray, H., Barriga, E. H., Moreno, M., Kerney, R., Monsoro-Burq, A.-H., et al. (2018). Characterization of Pax3 and Sox10 transgenic Xenopus laevis embryos as tools to study neural crest development. Dev. Biol. 444(Suppl. 1), S202–S208.
Andersen, E. F., Carey, J. C., Earl, D. L., Corzo, D., Suttie, M., Hammond, P., et al. (2014). Deletions involving genes WHSC1 and LETM1 may be necessary, but are not sufficient to cause Wolf-Hirschhorn Syndrome. Eur. J. Hum. Genet. 22, 464–470. doi: 10.1038/ejhg.2013.192
Battaglia, A., Carey, J. C., and South, S. T. (2015). Wolf-Hirschhorn syndrome: a review and update. Am. J. Med. Genet. C Semin. Med. Genet. 169, 216–223. doi: 10.1002/ajmg.c.31449
Bearce, E. A., Erdogan, B., and Lowery, L. A. (2015). TIPsy tour guides: how microtubule plus-end tracking proteins (+TIPs) facilitate axon guidance. Front. Cell. Neurosci. 9:241. doi: 10.3389/fncel.2015.00241
Bestman, J. E., and Cline, H. T. (2014). Morpholino studies in Xenopus brain development. Methods Mol. Biol. 1082, 155–171. doi: 10.1007/978-1-62703-655-9_11
Bhattacharya, D., Marfo, C. A., Li, D., Lane, M., and Khokha, M. K. (2015). CRISPR/Cas9: an inexpensive, efficient loss of function tool to screen human disease genes in Xenopus. Dev. Biol. 408, 196–204. doi: 10.1016/j.ydbio.2015.11.003
Bi, W., Cheung, S. W., Breman, A. M., and Bacino, C. A. (2016). 4p16.3 microdeletions and microduplications detected by chromosomal microarray analysis: new insights into mechanisms and critical regions. Am. J. Med. Genet. A 170, 2540–2550. doi: 10.1002/ajmg.a.37796
Blum, M., De Robertis, E. M., Wallingford, J. B., and Niehrs, C. (2015). Morpholinos: antisense and sensibility. Dev. Cell 35, 145–149. doi: 10.1016/j.devcel.2015.09.017
Boczek, N. J., Lahner, C. A., Nguyen, T.-M., Ferber, M. J., Hasadsri, L., Thorland, E. C., et al. (2018). Developmental delay and failure to thrive associated with a loss-of-function variant in WHSC1 (n.d.). Am. J. Med. Genet. A 176, 2798–2802. doi: 10.1002/ajmg.a.40498
Bonano, M., Tríbulo, C., De Calisto, J., Marchant, L., Sánchez, S. S., Mayor, R., et al. (2008). A new role for the Endothelin-1/Endothelin-A receptor signaling during early neural crest specification. Dev. Biol. 323, 114–129. doi: 10.1016/j.ydbio.2008.08.007
Borchers, A., Epperlein, H. H., and Wedlich, D. (2000). An assay system to study migratory behavior of cranial neural crest cells in Xenopus. Dev. Genes Evol. 210, 217–222. doi: 10.1007/s004270050307
Boskovski, M. T., Yuan, S., Pedersen, N. B., Goth, C. K., Makova, S., Clausen, H., et al. (2013). The heterotaxy gene GALNT11 glycosylates Notch to orchestrate cilia type and laterality. Nature 504, 456–459. doi: 10.1038/nature12723
Bronner, M. E., and LeDouarin, N. M. (2012). Development and evolution of the neural crest: an overview. Dev. Biol. 366, 2–9. doi: 10.1016/j.ydbio.2011.12.042
Brooks, E. R., and Wallingford, J. B. (2015). In vivo investigation of cilia structure and function using Xenopus. Methods Cell Biol. 127, 131–159. doi: 10.1016/bs.mcb.2015.01.018
Cammarata, G. M., Bearce, E. A., and Lowery, L. A. (2016). Cytoskeletal social networking in the growth cone: how +TIPs mediate microtubule-actin cross-linking to drive axon outgrowth and guidance. Cytoskeleton 73, 461–476. doi: 10.1002/cm.21272
Catela, C., Bilbao-Cortes, D., Slonimsky, E., Kratsios, P., Rosenthal, N., and Te Welscher, P. (2009). Multiple congenital malformations of Wolf-Hirschhorn syndrome are recapitulated in Fgfrl1 null mice. Dis. Model. Mech. 2, 283–294. doi: 10.1242/dmm.002287
Cousin, H. (2018). Cranial neural crest transplants. Cold Spring Harb. Protoc. 2018:db.rot097402. doi: 10.1101/pdb.prot097402
Cousin, H., and Alfandari, D. (2018). Cranial neural crest explants. Cold Spring Harb. Protoc. 2018:db.rot097394. doi: 10.1101/pdb.prot097394
DeLay, B. D., Krneta-Stankic, V., and Miller, R. K. (2016). Technique to target microinjection to the developing Xenopus kidney. J. Vis. Exp. 111:e53799. doi: 10.3791/53799
Deniz, E., Jonas, S., Hooper, M., N Griffin, J., Choma, M. A., and Khokha, M. K. (2017). Analysis of craniocardiac malformations in Xenopus using optical coherence tomography. Sci. Rep. 7:42506. doi: 10.1038/srep42506
Derar, N., Al-Hassnan, Z. N., Al-Owain, M., Monies, D., Abouelhoda, M., Meyer, B. F., et al. (2018). De novo truncating variants in WHSC1 recapitulate the Wolf-Hirschhorn (4p16.3 microdeletion) syndrome phenotype. Genet. Med. 21, 185–188. doi: 10.1038/s41436-018-0014-8
DeSimone, D. W., Davidson, L., Marsden, M., and Alfandari, D. (2005). The Xenopus embryo as a model system for studies of cell migration. Methods Mol. Biol. 294, 235–245.
Devotta, A., Juraver-Geslin, H., Gonzalez, J. A., Hong, C.-S., and Saint-Jeannet, J. -P. (2016). Sf3b4-depleted Xenopus embryos: a model to study the pathogenesis of craniofacial defects in Nager syndrome. Dev. Biol. 415, 371–382. doi: 10.1016/j.ydbio.2016.02.010
Dickinson, A., and Sive, H. (2007). Positioning the extreme anterior in Xenopus: cement gland, primary mouth and anterior pituitary. Semin. Cell Dev. Biol. 18, 525–533. doi: 10.1016/j.semcdb.2007.04.002
Dickinson, A. J. G. (2016). Using frogs faces to dissect the mechanisms underlying human orofacial defects. Semin. Cell Dev. Biol. 51, 54–63. doi: 10.1016/j.semcdb.2016.01.016
Dickinson, A. J. G., and Sive, H. (2006). Development of the primary mouth in Xenopus laevis. Dev. Biol. 295, 700–713. doi: 10.1016/j.ydbio.2006.03.054
Dimmer, K. S., Navoni, F., Casarin, A., Trevisson, E., Endele, S., Winterpacht, A., et al. (2008). LETM1, deleted in Wolf-Hirschhorn syndrome is required for normal mitochondrial morphology and cellular viability. Hum. Mol. Genet. 17, 201–214. doi: 10.1093/hmg/ddm297
Doonan, P. J., Chandramoorthy, H. C., Hoffman, N. E., Zhang, X., Cárdenas, C., Shanmughapriya, S., et al. (2014). LETM1-dependent mitochondrial Ca2+ flux modulates cellular bioenergetics and proliferation. FASEB J. 28, 4936–4949. doi: 10.1096/fj.14-256453
Drew, C. J. G., Kyd, R. J., and Morton, A. J. (2007). Complexin 1 knockout mice exhibit marked deficits in social behaviours but appear to be cognitively normal. Hum. Mol. Genet. 16, 2288–2305. doi: 10.1093/hmg/ddm181
Dubey, A., and Saint-Jeannet, J.-P. (2017). Modeling human craniofacial disorders in Xenopus. Curr. Pathobiol. Rep. 5, 79–92. doi: 10.1007/s40139-017-0128-8
Duncan, A. R., and Khokha, M. K. (2016). Xenopus as a model organism for birth defects-Congenital heart disease and heterotaxy. Semin. Cell Dev. Biol. 51, 73–79. doi: 10.1016/j.semcdb.2016.02.022
Endele, S., Fuhry, M., Pak, S. J., Zabel, B. U., and Winterpacht, A. (1999). LETM1, a novel gene encoding a putative EF-hand Ca2+-binding protein, flanks the Wolf-Hirschhorn syndrome (WHS) critical region and is deleted in most WHS patients. Genomics 60, 218–225. doi: 10.1006/geno.1999.5881
Endele, S., Nelkenbrecher, C., Bördlein, A., Schlickum, S., and Winterpacht, A. (2011). C4ORF48, a gene from the Wolf-Hirschhorn syndrome critical region, encodes a putative neuropeptide and is expressed during neocortex and cerebellar development. Neurogenetics 12, 155–163.
Engbers, H., van der Smagt, J. J., van ’t Slot, R., Vermeesch, J. R., Hochstenbach, R., and Poot, M. (2009). Wolf-Hirschhorn syndrome facial dysmorphic features in a patient with a terminal 4p16.3 deletion telomeric to the WHSCR and WHSCR 2 regions. Eur. J. Hum. Genet. 17, 129–132. doi: 10.1038/ejhg.2008.168
Erdogan, B., Cammarata, G. M., Lee, E. J., Pratt, B. C., Francl, A. F., Rutherford, E. L., et al. (2017). The microtubule plus-end-tracking protein TACC3 promotes persistent axon outgrowth and mediates responses to axon guidance signals during development. Neural Dev. 12:3.
Erdogan, B., Ebbert, P. T., and Lowery, L. A. (2016). Using Xenopus laevis retinal and spinal neurons to study mechanisms of axon guidance in vivo and in vitro. Semin. Cell Dev. Biol. 51, 64–72. doi: 10.1016/j.semcdb.2016.02.003
Ezponda, T., Popovic, R., Shah, M. Y., Martinez-Garcia, E., Zheng, Y., Min, D.-J., et al. (2013). The histone methyltransferase MMSET/WHSC1 activates TWIST1 to promote an epithelial-mesenchymal transition and invasive properties of prostate cancer. Oncogene 32, 2882–2890. doi: 10.1038/onc.2012.297
Feehan, J. M., Chiu, C. N., Stanar, P., Tam, B. M., Ahmed, S. N., and Moritz, O. L. (2017). Modeling dominant and recessive forms of retinitis pigmentosa by editing three rhodopsin-encoding genes in Xenopus laevis using Crispr/Cas9. Sci. Rep. 7:6920.
Fisch, G. S., Grossfeld, P., Falk, R., Battaglia, A., Youngblom, J., and Simensen, R. (2010). Cognitive-behavioral features of Wolf-Hirschhorn syndrome and other subtelomeric microdeletions. Am. J. Med. Genet. C Semin. Med. Genet. 154C, 417–426. doi: 10.1002/ajmg.c.30279
Garcia de la Serrana, D., Mareco, E. A., and Johnston, I. A. (2014). Systematic variation in the pattern of gene paralog retention between the teleost superorders Ostariophysi and Acanthopterygii. Genome Biol. Evol. 6, 981–987. doi: 10.1093/gbe/evu074
Garfinkel, A. M., and Khokha, M. K. (2017). An interspecies heart-to-heart: using Xenopus to uncover the genetic basis of congenital heart disease. Curr. Pathobiol. Rep. 5, 187–196. doi: 10.1007/s40139-017-0142-x
Garriga-Canut, M., Schoenike, B., Qazi, R., Bergendahl, K., Daley, T. J., Pfender, R. M., et al. (2006). 2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat. Neurosci. 9, 1382–1387. doi: 10.1038/nn1791
Gentsch, G. E., Spruce, T., Monteiro, R. S., Owens, N. D. L., Martin, S. R., and Smith, J. C. (2018). Innate immune response and off-target mis-splicing are common morpholino-induced side effects in Xenopus. Dev. Cell 44, 597–610.e10. doi: 10.1016/j.devcel.2018.01.022
Getwan, M., and Lienkamp, S. S. (2017). Toolbox in a tadpole: Xenopus for kidney research. Cell Tissue Res. 369, 143–157. doi: 10.1007/s00441-017-2611-2
Gilchrist, D. A., Nechaev, S., Lee, C., Ghosh, S. K. B., Collins, J. B., Li, L., et al. (2008). NELF-mediated stalling of Pol II can enhance gene expression by blocking promoter-proximal nucleosome assembly. Genes Dev. 22, 1921–1933. doi: 10.1101/gad.1643208
Glynn, D., Drew, C. J., Reim, K., Brose, N., and Morton, A. J. (2005). Profound ataxia in complexin I knockout mice masks a complex phenotype that includes exploratory and habituation deficits. Hum. Mol. Genet. 14, 2369–2385. doi: 10.1093/hmg/ddi239
Gonzalez Malagon, S. G., Lopez Muñoz, A. M., Doro, D., Bolger, T. G., Poon, E., Tucker, E. R., et al. (2018). Glycogen synthase kinase 3 controls migration of the neural crest lineage in mouse and Xenopus. Nat. Commun. 9:1126.
Hannes, F., Drozniewska, M., Vermeesch, J. R., and Haus, O. (2010). Duplication of the Wolf-Hirschhorn syndrome critical region causes neurodevelopmental delay. Eur. J. Med. Genet. 53, 136–140. doi: 10.1016/j.ejmg.2010.02.004
Hellsten, U., Harland, R. M., Gilchrist, M. J., Hendrix, D., Jurka, J., Kapitonov, V., et al. (2010). The genome of the Western clawed frog Xenopus tropicalis. Science 328, 633–636. doi: 10.1126/science.1183670
Hempel, A., and Kühl, M. (2016). A matter of the heart: the African clawed frog Xenopus as a model for studying vertebrate cardiogenesis and congenital heart defects. J. Cardiovasc. Dev. Dis. 3:21. doi: 10.3390/jcdd3020021
Hildebrand, J. D., and Soriano, P. (2002). Overlapping and unique roles for C-terminal binding protein 1 (CtBP1) and CtBP2 during mouse development. Mol. Cell. Biol. 22, 5296–5307. doi: 10.1128/MCB.22.15.5296-5307.2002
Ho, K. S., South, S. T., Lortz, A., Hensel, C. H., Sdano, M. R., Vanzo, R. J., et al. (2016). Chromosomal microarray testing identifies a 4p terminal region associated with seizures in Wolf-Hirschhorn syndrome. J. Med. Genet. 53, 256–263. doi: 10.1136/jmedgenet-2015-103626
Jacox, L., Chen, J., Rothman, A., Lathrop-Marshall, H., and Sive, H. (2016). Formation of a “Pre-mouth Array” from the extreme anterior domain is directed by neural crest and Wnt/PCP signaling. Cell Rep. 16, 1445–1455. doi: 10.1016/j.celrep.2016.06.073
Jacox, L., Sindelka, R., Chen, J., Rothman, A., Dickinson, A., and Sive, H. (2014). The extreme anterior domain is an essential craniofacial organizer acting through Kinin-Kallikrein signaling. Cell Rep. 8, 596–609. doi: 10.1016/j.celrep.2014.06.026
Jacox, L. A., Dickinson, A. J., and Sive, H. (2014). Facial transplants in Xenopus laevis embryos. J. Vis. Exp. 85:e50697. doi: 10.3791/50697
James-Zorn, C., Ponferrada, V., Fisher, M. E., Burns, K., Fortriede, J., Segerdell, E., et al. (2018). Navigating xenbase: an integrated Xenopus genomics and gene expression database. Methods Mol. Biol. 1757, 251–305. doi: 10.1007/978-1-4939-7737-6_10
Jiang, D., Zhao, L., Clish, C. B., and Clapham, D. E. (2013). Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf-Hirschhorn syndrome. Proc. Natl. Acad. Sci. U.S.A. 110, E2249–E2254. doi: 10.1073/pnas.1308558110
Kennedy, A. E., and Dickinson, A. J. (2014a). Quantification of orofacial phenotypes in Xenopus. J. Vis. Exp. 93:e52062. doi: 10.3791/52062
Kennedy, A. E., and Dickinson, A. J. (2014b). Quantitative analysis of orofacial development and median clefts in Xenopus laevis. Anat. Rec. 297, 834–855. doi: 10.1002/ar.22864
Kerzendorfer, C., Hannes, F., Colnaghi, R., Abramowicz, I., Carpenter, G., Vermeesch, J. R., et al. (2012). Characterizing the functional consequences of haploinsufficiency of NELF-A (WHSC2) and SLBP identifies novel cellular phenotypes in Wolf-Hirschhorn syndrome. Hum. Mol. Genet. 21, 2181–2193. doi: 10.1093/hmg/dds033
Khacho, M., Harris, R., and Slack, R. S. (2019). Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat. Rev. Neurosci. 20, 34–48. doi: 10.1038/s41583-018-0091-3
Lasser, M., Tiber, J., and Lowery, L. A. (2018). The role of the microtubule cytoskeleton in neurodevelopmental disorders. Front. Cell. Neurosci. 12:165. doi: 10.3389/fncel.2018.00165
Lee-Liu, D., Méndez-Olivos, E. E., Muñoz, R., and Larraín, J. (2017). The African clawed frog Xenopus laevis: a model organism to study regeneration of the central nervous system. Neurosci. Lett. 652, 82–93. doi: 10.1016/j.neulet.2016.09.054
Li, J., Perfetto, M., Neuner, R., Bahudhanapati, H., Christian, L., Mathavan, K., et al. (2018). Xenopus ADAM19 regulates Wnt signaling and neural crest specification by stabilizing ADAM13. Development 145:dev158154. doi: 10.1242/dev.158154
Lienkamp, S. S. (2016). Using Xenopus to study genetic kidney diseases. Semin. Cell Dev. Biol. 51, 117–124. doi: 10.1016/j.semcdb.2016.02.002
Liu, S., Higashihori, N., Yahiro, K., and Moriyama, K. (2015). Retinoic acid inhibits histone methyltransferase Whsc1 during palatogenesis. Biochem. Biophys. Res. Commun. 458, 525–530. doi: 10.1016/j.bbrc.2015.01.148
Macrì, S., Simula, L., Pellarin, I., Pegoraro, S., Onorati, M., Sgarra, R., et al. (2016). Hmga2 is required for neural crest cell specification in Xenopus laevis. Dev. Biol. 411, 25–37. doi: 10.1016/j.ydbio.2016.01.014
Maj, E., Künneke, L., Loresch, E., Grund, A., Melchert, J., Pieler, T., et al. (2016). Controlled levels of canonical Wnt signaling are required for neural crest migration. Dev. Biol. 417, 77–90. doi: 10.1016/j.ydbio.2016.06.022
Makrythanasis, P., Kato, M., Zaki, M. S., Saitsu, H., Nakamura, K., Santoni, F. A., et al. (2016). Pathogenic variants in PIGG cause intellectual disability with seizures and hypotonia. Am. J. Hum. Genet. 98, 615–626. doi: 10.1016/j.ajhg.2016.02.007
Margueron, R., and Reinberg, D. (2010). Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 11, 285–296. doi: 10.1038/nrg2752
Mayor, R., and Theveneau, E. (2013). The neural crest. Development 140, 2247–2251. doi: 10.1242/dev.091751
McCammon, J. M., and Sive, H. (2015). Addressing the genetics of human mental health disorders in model organisms. Annu. Rev. Genomics Hum. Genet. 16, 173–197.
McQuibban, A. G., Joza, N., Megighian, A., Scorzeto, M., Zanini, D., Reipert, S., et al. (2010). A Drosophila mutant of LETM1, a candidate gene for seizures in Wolf-Hirschhorn syndrome. Hum. Mol. Genet. 19, 987–1000. doi: 10.1093/hmg/ddp563
Milet, C., and Monsoro-Burq, A.-H. (2014). Dissection of Xenopus laevis neural crest for in vitro explant culture or in vivo transplantation. J. Vis. Exp. 85:e51118. doi: 10.3791/51118
Mills, A., Bearce, E., Cella, R., Kim, S. W., Selig, M., Lee, S., et al. (2019). Wolf-Hirschhorn Syndrome-associated genes are enriched in motile neural crest and affect craniofacial development in Xenopus laevis. Front. Physiol. 10:431. doi: 10.3389/fphys.2019.00431
Mimoto, M. S., and Christian, J. L. (2011). Manipulation of gene function in Xenopus laevis. Methods Mol. Biol. 770, 55–75. doi: 10.1007/978-1-61779-210-6_3
Monsoro-Burq, A. -H., Wang, E., and Harland, R. (2005). Msx1 and Pax3 cooperate to mediate FGF8 and WNT signals during Xenopus neural crest induction. Dev. Cell 8, 167–178. doi: 10.1016/j.devcel.2004.12.017
Moody, S. A. (2018a). Lineage tracing and fate mapping in Xenopus embryos. Cold Spring Harb. Protoc. 2018:db.rot097253.
Moody, S. A. (2018b). Microinjection of mRNAs and oligonucleotides. Cold Spring Harb. Protoc. 2018:db.rot097261.
Nakano, Y., Fujita, M., Ogino, K., Saint-Amant, L., Kinoshita, T., Oda, Y., et al. (2010). Biogenesis of GPI-anchored proteins is essential for surface expression of sodium channels in zebrafish Rohon-Beard neurons to respond to mechanosensory stimulation. Development 137, 1689–1698. doi: 10.1242/dev.047464
Nakayama, T., Fish, M. B., Fisher, M., Oomen-Hajagos, J., Thomsen, G. H., and Grainger, R. M. (2013). Simple and efficient CRISPR/Cas9-mediated targeted mutagenesis in Xenopus tropicalis. Genesis 51, 835–843. doi: 10.1002/dvg.22720
Narita, T., Yung, T. M. C., Yamamoto, J., Tsuboi, Y., Tanabe, H., Tanaka, K., et al. (2007). NELF interacts with CBC and participates in 3’ end processing of replication-dependent histone mRNAs. Mol. Cell 26, 349–365. doi: 10.1016/j.molcel.2007.04.011
Nieminen, P., Kotilainen, J., Aalto, Y., Knuutila, S., Pirinen, S., and Thesleff, I. (2003). MSX1 gene is deleted in Wolf-Hirschhorn syndrome patients with oligodontia. J. Dent. Res. 82, 1013–1017. doi: 10.1177/154405910308201215
Nieuwkoop, P. D., and Faber, J. (1994). Normal Table of Xenopus Laevis (Daudin): A Systematical and Chronological Survey of the Development From the Fertilized Egg Till the End of Metamorphosis. (New York, NY: Garland Pub).
Nimura, K., Ura, K., Shiratori, H., Ikawa, M., Okabe, M., Schwartz, R. J., et al. (2009). A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature 460, 287–291. doi: 10.1038/nature08086
Nwagbara, B. U., Faris, A. E., Bearce, E. A., Erdogan, B., Ebbert, P. T., Evans, M. F., et al. (2014). TACC3 is a microtubule plus end-tracking protein that promotes axon elongation and also regulates microtubule plus end dynamics in multiple embryonic cell types. Mol. Biol. Cell 25, 3350–3362. doi: 10.1091/mbc.E14-06-1121
Paradowska-Stolarz, A. M. (2014). Wolf-Hirschhorn syndrome (WHS) - literature review on the features of the syndrome. Adv. Clin. Exp. Med. 23, 485–489. doi: 10.17219/acem/24111
Peset, I., and Vernos, I. (2008). The TACC proteins: TACC-ling microtubule dynamics and centrosome function. Trends Cell Biol. 18, 379–388. doi: 10.1016/j.tcb.2008.06.005
Pratt, K. G., and Khakhalin, A. S. (2013). Modeling human neurodevelopmental disorders in the Xenopus tadpole: from mechanisms to therapeutic targets. Dis. Model. Mech. 6, 1057–1065. doi: 10.1242/dmm.012138
Redler, S., Strom, T. M., Wieland, T., Cremer, K., Engels, H., Distelmaier, F., et al. (2017). Variants in CPLX1 in two families with autosomal-recessive severe infantile myoclonic epilepsy and ID. Eur. J. Hum. Genet. 25, 889–893. doi: 10.1038/ejhg.2017.52
Rutherford, E. L., and Lowery, L. A. (2016). Exploring the developmental mechanisms underlying Wolf-Hirschhorn Syndrome: evidence for defects in neural crest cell migration. Dev. Biol. 420, 1–10. doi: 10.1016/j.ydbio.2016.10.012
Sato, A., Scholl, A. M., Kuhn, E. N., Kuhn, E. B., Stadt, H. A., Decker, J. R., et al. (2011). FGF8 signaling is chemotactic for cardiac neural crest cells. Dev. Biol. 354, 18–30. doi: 10.1016/j.ydbio.2011.03.010
Satokata, I., and Maas, R. (1994). Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat. Genet. 6, 348–356. doi: 10.1038/ng0494-348
Session, A. M., Uno, Y., Kwon, T., Chapman, J. A., Toyoda, A., Takahashi, S., et al. (2016). Genome evolution in the allotetraploid frog Xenopus laevis. Nature 538, 336–343. doi: 10.1038/nature19840
Shellard, A., and Mayor, R. (2016). Chemotaxis during neural crest migration. Semin. Cell Dev. Biol. 55, 111–118. doi: 10.1016/j.semcdb.2016.01.031
Sheth, F., Akinde, O. R., Datar, C., Adeteye, O. V., and Sheth, J. (2012). Genotype-phenotype characterization of Wolf-Hirschhorn syndrome confirmed by FISH: case reports. Case Rep. Genet. 2012:878796. doi: 10.1155/2012/878796
Simon, R., and Bergemann, A. D. (2008). Mouse models of Wolf-Hirschhorn syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 148C, 275–280. doi: 10.1002/ajmg.c.30184
Sive, H. L., Grainger, R. M., and Harland, R. M. (2007a). Inducing ovulation in Xenopus laevis. CSH Protoc. 2007:db.rot4734.
Sive, H. L., Grainger, R. M., and Harland, R. M. (2007b). Xenopus laevis in vitro fertilization and natural mating methods. CSH Protoc. 2007:db.rot4737. doi: 10.1101/pdb.prot4737
Slater, P. G., Hayrapetian, L., and Lowery, L. A. (2017). Xenopus laevis as a model system to study cytoskeletal dynamics during axon pathfinding. Genesis 55:e22994. doi: 10.1002/dvg.22994
Sojka, S., Amin, N. M., Gibbs, D., Christine, K. S., Charpentier, M. S., and Conlon, F. L. (2014). Congenital heart disease protein 5 associates with CASZ1 to maintain myocardial tissue integrity. Development 141, 3040–3049. doi: 10.1242/dev.106518
Stec, I., Wright, T. J., van Ommen, G. J., de Boer, P. A., van Haeringen, A., Moorman, A. F., et al. (1998). WHSC1, a 90 kb SET domain-containing gene, expressed in early development and homologous to a Drosophila dysmorphy gene maps in the Wolf-Hirschhorn syndrome critical region and is fused to IgH in t(4;14) multiple myeloma. Hum. Mol. Genet. 7, 1071–1082. doi: 10.1093/hmg/7.7.1071
Sun, J., and Li, R. (2010). Human negative elongation factor activates transcription and regulates alternative transcription initiation. J. Biol. Chem. 285, 6443–6452. doi: 10.1074/jbc.M109.084285
Szabó, A., Melchionda, M., Nastasi, G., Woods, M. L., Campo, S., Perris, R., et al. (2016). In vivo confinement promotes collective migration of neural crest cells. J. Cell Biol. 213, 543–555. doi: 10.1083/jcb.201602083
Tahir, R., Kennedy, A., Elsea, S. H., and Dickinson, A. J. (2014). Retinoic acid induced-1 (Rai1) regulates craniofacial and brain development in Xenopus. Mech. Dev. 133, 91–104. doi: 10.1016/j.mod.2014.05.004
Tandon, P., Conlon, F., Furlow, J. D., and Horb, M. E. (2017). Expanding the genetic toolkit in Xenopus: approaches and opportunities for human disease modeling. Dev. Biol. 426, 325–335. doi: 10.1016/j.ydbio.2016.04.009
Tandon, P., Showell, C., Christine, K., and Conlon, F. L. (2012). Morpholino injection in Xenopus. Methods Mol. Biol. 843, 29–46. doi: 10.1007/978-1-61779-523-7_4
Theveneau, E., and Mayor, R. (2011). Beads on the run: beads as alternative tools for chemotaxis assays. Methods Mol. Biol. 769, 449–460. doi: 10.1007/978-1-61779-207-6_30
Theveneau, E., Steventon, B., Scarpa, E., Garcia, S., Trepat, X., Streit, A., et al. (2013). Chase-and-run between adjacent cell populations promotes directional collective migration. Nat. Cell Biol. 15, 763–772. doi: 10.1038/ncb2772
Toyokawa, G., Cho, H.-S., Masuda, K., Yamane, Y., Yoshimatsu, M., Hayami, S., et al. (2011). Histone lysine methyltransferase Wolf-Hirschhorn syndrome candidate 1 is involved in human carcinogenesis through regulation of the Wnt pathway. Neoplasia 13, 887–898.
Wallmeier, J., Shiratori, H., Dougherty, G. W., Edelbusch, C., Hjeij, R., Loges, N. T., et al. (2016). TTC25 deficiency results in defects of the outer dynein arm docking machinery and primary ciliary dyskinesia with left-right body asymmetry randomization. Am. J. Hum. Genet. 99, 460–469. doi: 10.1016/j.ajhg.2016.06.014
Wang, F., Shi, Z., Cui, Y., Guo, X., Shi, Y.-B., and Chen, Y. (2015). Targeted gene disruption in Xenopus laevis using CRISPR/Cas9. Cell Biosci. 5:15.
Willsey, H. R., Walentek, P., Exner, C. R. T., Xu, Y., Lane, A. B., Harland, R. M., et al. (2018). Katanin-like protein Katnal2 is required for ciliogenesis and brain development in Xenopus embryos. Dev. Biol. 442, 276–287. doi: 10.1016/j.ydbio.2018.08.002
Yamada-Okabe, T., Imamura, K., Kawaguchi, N., Sakai, H., Yamashita, M., and Matsumoto, N. (2010). Functional characterization of the zebrafish WHSC1-related gene, a homolog of human NSD2. Biochem. Biophys. Res. Commun. 402, 335–339. doi: 10.1016/j.bbrc.2010.10.027
Yung, T. M. C., Narita, T., Komori, T., Yamaguchi, Y., and Handa, H. (2009). Cellular dynamics of the negative transcription elongation factor NELF. Exp. Cell Res. 315, 1693–1705. doi: 10.1016/j.yexcr.2009.02.013
Zahn, N., Levin, M., and Adams, D. S. (2017). The Zahn drawings: new illustrations of Xenopus embryo and tadpole stages for studies of craniofacial development. Development 144, 2708–2713. doi: 10.1242/dev.151308
Zhang, X.-L., Huang, C.-X., Zhang, J., Inoue, A., Zeng, S.-E., and Xiao, S.-J. (2013). CtBP1 is involved in epithelial-mesenchymal transition and is a potential therapeutic target for hepatocellular carcinoma. Oncol. Rep. 30, 809–814. doi: 10.3892/or.2013.2537
Zhang, Z., Song, Y., Zhao, X., Zhang, X., Fermin, C., and Chen, Y. (2002). Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development 129, 4135–4146.
Keywords: Wolf–Hirschhorn syndrome, Xenopus, development, craniofacial, neural crest cells
Citation: Lasser M, Pratt B, Monahan C, Kim SW and Lowery LA (2019) The Many Faces of Xenopus: Xenopus laevis as a Model System to Study Wolf–Hirschhorn Syndrome. Front. Physiol. 10:817. doi: 10.3389/fphys.2019.00817
Received: 10 October 2018; Accepted: 11 June 2019;
Published: 26 June 2019.
Edited by:
John Noel Griffin, Duke University, United StatesReviewed by:
Engin Deniz, Yale University, United StatesSandra Guadalupe González Malagón, Foundation for Research and Technology Hellas, Greece
Copyright © 2019 Lasser, Pratt, Monahan, Kim and Lowery. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Anne Lowery, TGF1cmEubG93ZXJ5QGJjLmVkdQ==
†These authors have contributed equally to this work