 Madeleine Iafrate
Madeleine Iafrate Gilbert O. Fruhwirth
Gilbert O. Fruhwirth- Imaging Therapy and Cancer Group, Department of Imaging Chemistry and Biology, School of Biomedical Engineering & Imaging Sciences, King’s College London, London, United Kingdom
Immunotherapy is a relatively new treatment regimen for cancer, and it is based on the modulation of the immune system to battle cancer. Immunotherapies can be classified as either molecular or cell-based immunotherapies, and both types have demonstrated promising results in a growing number of cancers. Indeed, several immunotherapies representing both classes are already approved for clinical use in oncology. While spectacular treatment successes have been reported, particularly for so-called immune checkpoint inhibitors and certain cell-based immunotherapies, they have also been accompanied by a variety of severe, sometimes life-threatening side effects. Furthermore, not all patients respond to immunotherapy. Hence, there is the need for more research to render these promising therapeutics more efficacious, more widely applicable, and safer to use. Whole-body in vivo imaging technologies that can interrogate cancers and/or immunotherapies are highly beneficial tools for immunotherapy development and translation to the clinic. In this review, we explain how in vivo imaging can aid the development of molecular and cell-based anti-cancer immunotherapies. We describe the principles of imaging host T-cells and adoptively transferred therapeutic T-cells as well as the value of traceable cancer cell models in immunotherapy development. Our emphasis is on in vivo cell tracking methodology, including important aspects and caveats specific to immunotherapies. We discuss a variety of associated experimental design aspects including parameters such as cell type, observation times/intervals, and detection sensitivity. The focus is on non-invasive 3D cell tracking on the whole-body level including aspects relevant for both preclinical experimentation and clinical translatability of the underlying methodologies.
Introduction
Immunotherapy is a relatively new concept that is increasingly applied to a variety of conditions. Most of the currently approved or emerging immunotherapy approaches are in the oncology arena. In some cases, they were curative, which represents a major leap over most previous treatment concepts. Mechanistically, they modulate the immune system to better attack the cancer. There are two types of anti-cancer immunotherapy, molecular and cell-based immunotherapy. Both approaches are already in clinical use, whereby molecular immunotherapies currently are further developed with more applications and more approved therapeutics.
Molecular immunotherapies usually modulate the immune system by targeting immune checkpoints using antibodies or antibody-derived molecules. Examples include ICIs targeted at CTLA-4 (e.g. ipilimumab) or the PD-1/PD-L1 axis (e.g. nivolumab, atezolizumab, and pembrolizumab) (Hodi et al., 2010; Topalian et al., 2012; Larkin et al., 2015; Darvin et al., 2018). These immunotherapeutics were largely developed using similar regulatory approval frameworks to other receptor-targeting drugs. Although in several cases the whole-body distribution of these therapeutics would be accessible through imaging the molecular immunotherapy itself, this is not routinely performed. Only very recently did studies report the whole-body distribution of radiolabeled checkpoint inhibitors in man (e.g. atezolizumab, Bensch et al., 2018; Jauw et al., 2019) to assess whether imaging them might reveal prognostic information. Despite molecular immunotherapies changing the landscape of cancer treatment (Ledford et al., 2018), significant challenges remain. These include non-responding patients (Feng et al., 2013), severe immune-related adverse events (IrAE, i.e., ICI weakening the normal physiological barriers against autoimmunity resulting in various local and systemic autoimmune responses), and the development of resistance (Darvin et al., 2018).
Cell-based immunotherapies consist of live immune cells that are administered to patients. The anti-tumor properties are either intrinsic to these therapeutic cells or conferred to them through genetic engineering. The therapeutic immune cells are either taken from a different human donor (allogeneic) or are isolated from the patient (autologous) before undergoing manipulations that transform the cells into immunotherapeutic cells. A historic lack of clarity surrounding the regulatory aspect of live cell-based therapy resulted in debates on what constitutes manipulations requiring regulatory approval (Anon, 2014), but it is now accepted that any cells that have been cultured with any drugs are subject to regulatory approval. The new paradigm of cell-based immunotherapy has forced regulatory agencies to re-evaluate their approval processes to accommodate for living drugs and to avoid slowing progress; for example, the new ATMP framework accelerates the approval process if there is demonstrable clinical need (Marks and Gottlieb, 2018). The first ever clinically approved cell-based anti-cancer immunotherapies were the chimeric antigen receptor T-cell (CAR-T) therapies tisagenlecleucel and axicabtagene ciloleucel, both of which are autologous CD19-targeted CAR-T immunotherapies for the treatment of certain hematological malignancies (B-cell lymphomas; U.S. Food and Drug Administration, 2017). While spectacular treatment successes have been reported for CAR-T immunotherapies, alike molecular immunotherapeutics, not all patients responded and sometimes the effects were only temporary (Neelapu et al., 2017; Schuster et al., 2017; Maude et al., 2018), and these therapeutics have also been associated with severe side-effects and fatalities during trials (Linette et al., 2013; Saudemont et al., 2018). In addition, CAR-T immunotherapy has generally yielded disappointing results in solid tumors (Martinez and Moon, 2019). Nonetheless, the portfolio of immune cells envisaged for cell-based anti-cancer immunotherapy is increasing and now includes T-cell receptor-modified T-cells (TCR-T), γδ T-cells, NK and dendritic cells (DC). Importantly, there are several unknowns including the in vivo distribution, persistence and survival of cell-based immunotherapies as well as their efficacy at target and non-target sites, and there is a need to investigate these aspects during their development and translation into the clinics.
The Need for Imaging in Immunotherapy Development
During the early stages of drug development, animal models are frequently employed to investigate the efficacies of drug candidates in defined disease settings. For instance, multiple animal tumor models have been used in the development of chemotherapeutics and targeted therapies (Cekanova and Rathore, 2014). Similar experimentation has also been necessary for the development of immunotherapies to establish targeting efficiencies, pharmacokinetics/pharmacodynamics, whether there is spatial heterogeneity to therapy delivery, and whether therapy presence is related to efficacy. Novel and accurate biomarkers are also essential to guide immunotherapy development to ensure optimal benefit for cancer patients. Notably, imaging biomarkers differ from conventional tissue/blood-based biomarkers in several important aspects (O’Connor et al., 2017). Foremost, imaging biomarkers are non-invasive, thus overcoming sampling limitations and associated tissue morbidities of conventional tissue/blood biomarkers, and they provide whole-body information albeit usually for only one target at the time. Furthermore, dynamic imaging can provide pharmacokinetic information. As with other biomarkers, imaging biomarkers should be standardized across multiple centers to unleash their full potential for diagnosis, patient stratification and treatment monitoring. Pathways for the development and standardization of dedicated imaging biomarkers have been structured and excellently described by a large team of cancer researchers (O’Connor et al., 2017), and we refer the reader to this publication for specific details.
Whole-body in vivo imaging technologies (Figure 1) that can interrogate cancers and therapeutics in preclinical models are very valuable tools in this context. They show great potential to provide answers to various challenges central to immunotherapy:
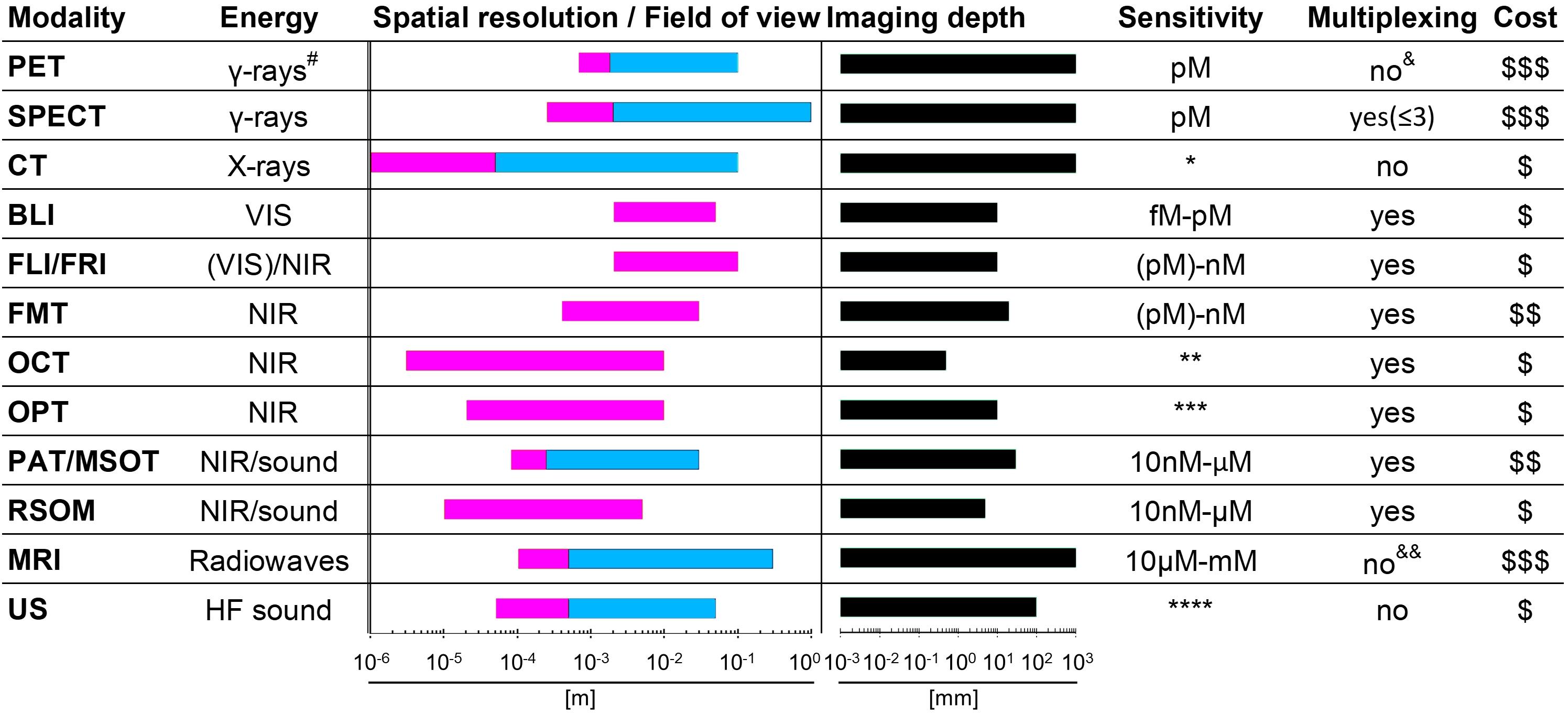
Figure 1. Properties of various whole-body imaging modalities. Imaging modalities are ordered according to the electromagnetic spectrum they exploit for imaging (top, high energy; bottom, low energy). Routinely achievable spatial resolution (left end) and fields of view (right end) are shown in red. Where bars are blue, they overlap red bars and indicate the same parameters but achievable with instruments used routinely in the clinic. Imaging depth is shown in black alongside next to sensitivity ranges. Instrument cost estimations are classified as ($) < 125,000 $, ($$) 125-300,000 $ and ($$$) > 300,000 $. #Generated by positron annihilation (511keV). *Contrast agents sometimes used to obtain different anatomical/functional information. **In “emission mode” comparable to other fluorescence modalities (∼nM). ***Fluorophore detection can suffer from photobleaching by excitation light. ****Highly dependent on contrast agent. & Dual isotope PET is feasible but not routinely in use; it requires two tracers, one with a positron emitter (e.g. 18F and 89Zr) and the other with a positron-gamma emitter (e.g. 124I, 76Br, and 86Y), and is based on recent reconstruction algorithms to differentiate the two isotopes based on the prompt-gamma emission (Andreyev and Celler, 2011; Cal-Gonzalez et al., 2015; Lage et al., 2015). &&Multichannel MRI imaging has been shown to be feasible (Zabow et al., 2008). PET, positron emission tomography; SPECT, single photon emission computed tomography; CT, computed tomography; BLI, bioluminescence imaging; FLI, fluorescent lifetime imaging; FRI, fluorescent reflectance imaging; FMT, fluorescence molecular tomography; OCT, optical coherence tomography; OPT, optical projection tomography; PAT, photoacoustic tomography; MSOT, multispectral optoacoustic tomography; RSOM, raster-scan optoacoustic mesoscopy; MRI, magnetic resonance imaging; US, ultrasound.
(1) Which immune cell classes are present in tumors and are they critical for response?
(2) What role do other components of the tumor microenvironment play?
(3) What are the consequences of heterogeneity within tumors and between lesions?
(4) What are biomarkers of true response and true progression?
(5) What is the relationship between target expression levels, affinity, and response?
(6) Can resistance be detected early or even be predicted?
(7) How can the distribution, fate, persistence and efficacy of cell-based immunotherapies be tracked in vivo?
(8) Can off-target effects and associated toxicities be detected early or be predicted?
(9) How can combination treatments be designed in a rational and effective manner?
Given that metastasis is responsible for >90% of cancer mortality, novel immunotherapy treatments need also to be evaluated for their efficacies against secondary lesions. Metastases can significantly differ from the primary tumor because of tumor evolution, and consequently can show a different therapy response compared to the primary lesion (Caswell and Swanton, 2017; Kim et al., 2019). While anti-metastatic endpoints have long been regarded impractical, it is noteworthy that an anti-metastatic prostate cancer drug, apalutamide, recently received FDA approval on the basis of metastasis-free survival as a new endpoint (measuring the length of time that tumors did not spread to other parts of the body or that death occurred after starting treatment, U.S. Food and Drug Administration, 2018). This raised the prospects for further such research, not least in the context of immunotherapy, and if immunotherapy were to be used as a treatment at earlier stages of cancer. Thus, preclinical models of metastasis employing in vivo traceable cancer cells have also a role to play in the development of immunotherapies (Gomez-Cuadrado et al., 2017).
Imaging Approaches in Immunotherapy Development
Brief Overview of Relevant Imaging Technologies
Medical imaging revolutionized the diagnosis and treatment of human disease by providing anatomical, physiological and molecular information (Mankoff, 2007). Imaging technologies differ in their capabilities and limitations. Figure 1 details the properties of those imaging technologies relevant to this review. Notably, several modalities are already in routine clinical use, for example US, magnetic resonance imaging (MRI), the radionuclide imaging modalities SPECT and PET, and X–ray computed tomography (CT). PAT and MSOT are two closely related relatively new modalities and have recently been translated into the clinical for special applications. PAT/MSOT delivers near infrared laser pulses into biological tissues with the latter absorbing and converting some of the laser pulse energy into heat, leading to transient thermoelastic expansion and thus wideband ultrasonic emission, which is used to compute an image (Ntziachristos et al., 2005; Wang and Yao, 2016). A purely optical imaging approach that is currently used in the clinical setting is OCT with applications in ophthalmology (Jung et al., 2011; Tao et al., 2013) and dermatology (Mogensen et al., 2009; Olsen et al., 2015). In general, the various imaging technologies can be categorized into modalities that subject the patient to a radiation dose (CT, PET, and SPECT) and modalities that are employing non-ionizing radiation (MRI, OCT, PAT/MSOT, and US). Depending on the research/clinical question, CT, MRI, PAT/MSOT, and US can be used with or without a contrast agent. In contrast, PET and SPECT strictly require contrast agents for image formation; these are often termed radiotracers, not least in reference to the very small concentrations required (“tracer levels;” picomolar concentration range) as both PET and SPECT are orders of magnitude more sensitive than the other clinically useable imaging technologies (Figure 1).
In preclinical settings, BLI can compete in sensitivity with radionuclide imaging modalities, but at much reduced experimental complexity and cost (instrument cost and running cost), which renders it a widely used tool. It relies on the presence of luciferase reporter proteins, which convert an administered chemical substrate into light that is then collected by highly sensitive cameras. As luciferase proteins are of non-mammalian origin, BLI is not translatable to the human setting. Another disadvantage is that BLI relies on light emitted within tissues which in turn is subject to absorption and scatter within the tissue matrix, thereby precluding reliable 3D quantification (Li et al., 2013; Dunlap, 2014; Jiang et al., 2016). Fluorescence-based whole-body imaging has also been developed (FLI/FRI), whereby fluorescence light is generated within a thick samples/small animal through excitation light; the approach has the same issues as BLI but is far less sensitive. To obtain true 3D data a tomographic design is required. Among the optical modalities listed in Figure 1, this is provided by optical projection tomography (OPT), which can be considered as the optical analog of CT. OPT operates on the micrometer to millimeter scales (Sharpe et al., 2002; Cheddad et al., 2012) thereby bridging the scale gap between classical whole-body imaging technologies and microscopy. It can either provide tomographic data on light absorption or fluorescence signals, and has been used in live zebrafish (Bassi et al., 2011; McGinty et al., 2011), fruit flies (Vinegoni et al., 2008; Arranz et al., 2014) and for whole organ imaging in mice (Alanentalo et al., 2008; Gleave et al., 2012; Gupta et al., 2013). An alternative approach offering larger fields of view in the centimeter range is diffuse optical tomography or FMT, which exploits photon tissue propagation theory to allow for 3D reconstruction at centimeter depth but its resolution is affected by weak signals and high tissues scattering (Figure 1; Graves et al., 2004; Ntziachristos, 2006; Venugopal et al., 2010; Zacharakis et al., 2011; Wang et al., 2015; Lian et al., 2017). In this review we lay emphasize on methodologies that are providing reliable quantifiable 3D information and have the potential to be clinically translatable.
As imaging modalities differ in their capabilities and limitations (Figure 1), combination technologies have become particularly important. For example, PET offers excellent sensitivity and provides absolute quantitative data (Lajtos et al., 2014) but can only detect signals at millimeter resolution. Hence, PET imaging was combined with other modalities providing higher anatomical resolution, such as CT (Basu et al., 2014) or MRI (Catana, 2017). This resulted in multi-modal whole-body imaging approaches adding anatomical context (from CT, MRI) to molecular imaging information (e.g. from PET or SPECT). Very recently, ultrafast US was combined with PET technology to form a new hybrid technology with the potential to provide molecular, anatomical and functional imaging data (Provost et al., 2018). Multi-modal imaging technologies are extremely useful to obtain maximal information from imaging, whereby the recent work of Bensch et al. provides a very good example for its power in the context of immunotherapy (Bensch et al., 2018).
The Role of Anatomical Imaging
Anatomical imaging methods such as computed tomography (CT) or MRI provide excellent 3D resolution in vivo and enable quantification of tumor size and growth if the tumor differs sufficiently in contrast from surrounding tissues. Importantly, these techniques are non-specific and do not quantify tumor or immune cells specifically, but can account for the entirety of the tumor mass or reveal parameters such as texture (Lambin et al., 2012). This can cause issues for treatment monitoring if tumor size or radiomic features are not correlated to treatment response. If efficacy assessment is based on tumor shrinkage (cf. RECIST criteria in humans, Eisenhauer et al., 2009), then anatomical imaging is not appropriate for the assessment of immunotherapeutics, which initially can cause tumor sizes to increase or plateau before tumor regression occurs. This phenomenon is termed “pseudo-progression” and is evident in both molecular and cell-based immunotherapeutics (Nishino et al., 2017). It is caused by the very mechanisms of immunotherapy, which re-educates the immune system to detect and attack cancer cells, thereby resulting in immune cell infiltration/expansion, and tumors initially enlarging rather than regressing. Pseudo-progression has been recognized and is being accounted for in new criteria relevant for immunotherapy monitoring (Wolchok et al., 2009; Seymour et al., 2017).
Molecular Imaging and Immunotherapy
Molecular imaging differs from anatomical imaging in that it provides specific molecular information on the whole-body level. Molecular imaging can be exploited to visualize and quantify the presence of a target of interest at a given time on the whole-body level. This can be used to diagnose and guide patient stratification and treatment decisions. Via molecular imaging, the heterogeneity of target expression can be assessed, for example, between primary and secondary lesions or within individual tumors (Alizadeh et al., 2015; Kurland et al., 2017; Bensch et al., 2018). Importantly, molecular imaging can support treatment monitoring, for example, inform on target engagement, therapy efficacy, and in certain cases can be used to probe the activity of a therapeutic. Molecular imaging employs a broad variety of different contrast agent classes based on target-specific small molecules as well as a variety of biomolecules. The latter include full-length antibodies, bivalent F(ab’)2 fragments, minibodies, monovalent Fab fragments, diabodies, single-chain variable fragments (scFv), nanobodies, affibodies (listed in order of decreasing molecular weight). Strategies for developing and optimizing such targeted probes for non-invasive imaging using radioactive, optical, magnetic resonance, and ultrasound approaches have been recently summarized by Freise and Wu (Freise and Wu, 2015). The imaging of T-cell effector molecules such as the PD-L1/PD1 axis has been shown to be a successful approach to study T-cell in vivo distribution in preclinical models (Natarajan et al., 2015; Heskamp et al., 2019) and in humans (Jauw et al., 2019). However, a caveat of molecular imaging is its reliance on one chosen molecular target, because its expression might change during tumor progression, and with these changes also the imaging read-outs would change. Recently, the predictive power of molecular imaging for treatment outcome was demonstrated through visualization of the radiolabeled ICI atezolizumab by multimodal PET/CT imaging (combined molecular and anatomical imaging, Bensch et al., 2018; Figure 2). In this study, clinical responses were better correlated with pre-treatment [89Zr]Zr-desferrioxamine (DFO)-atezolizumab PET signals than with immunohistochemistry- or RNA-sequencing-based predictive biomarkers. [89Zr]Zr-DFO-pembrolizumab, which targets PD-1 on T-cells, is currently being tested in clinical trials involving non-small cell lung cancer or metastatic melanoma patients (NCT02760225, NCT03065764). Similarly, the T-cell expressed ICI target CTLA-4 has been imaged in preclinical mouse models of colon cancer to better understand target expression and therapy side effects (Higashikawa et al., 2014). Additionally, 89Zr-labeled ipilimumab targeting CTLA-4 in humans is in phase II trials (NCT03313323) to better comprehend the pharmacodynamics/pharamacokinetics of this antibody-based immunotherapeutic and its IrAEs. Other imaging targets related to T-cell effector functions include interferon-γ and granzyme B, which have both been studied in mice (Larimer et al., 2017, 2019; Gibson et al., 2018).
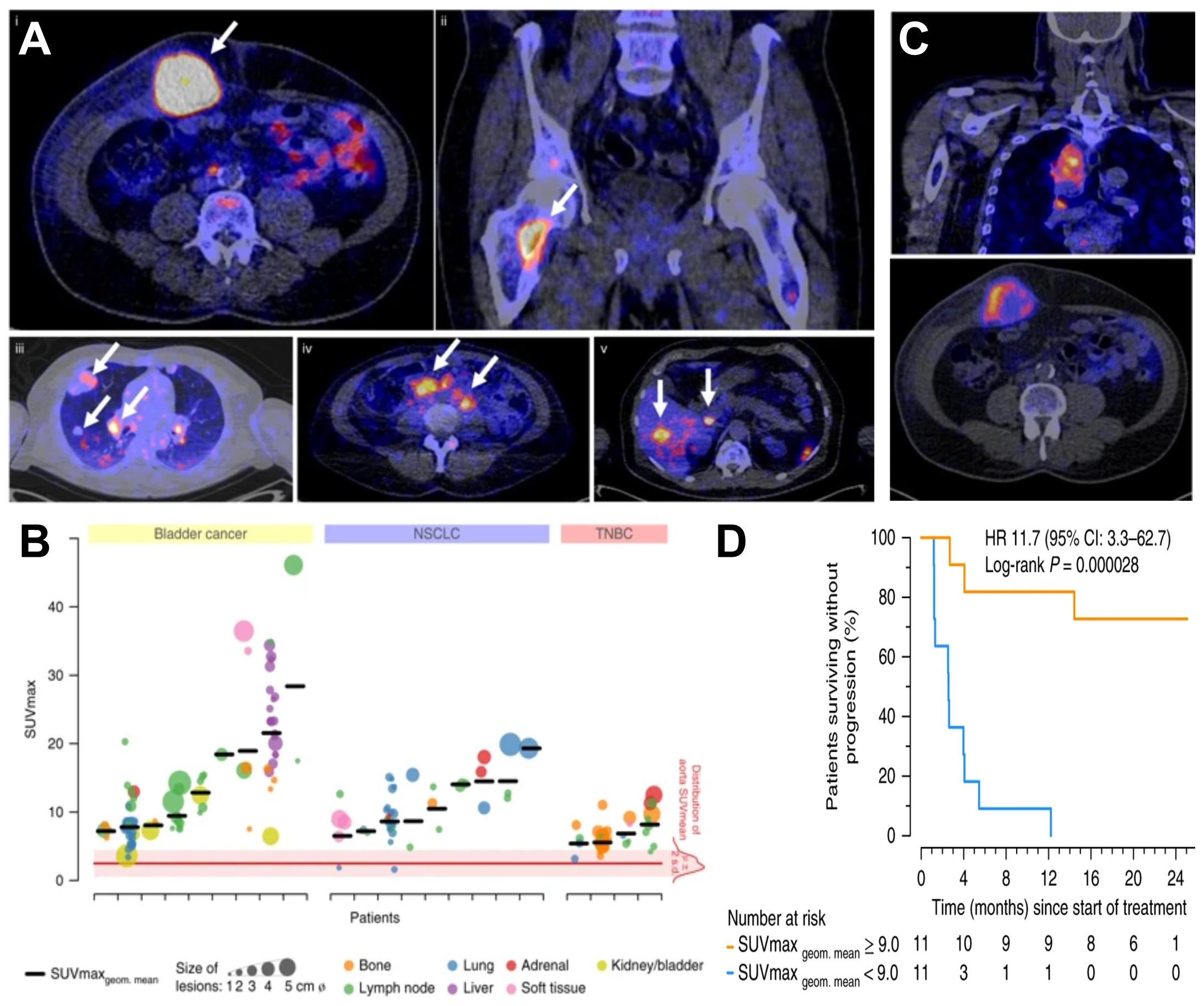
Figure 2. Molecular imaging can be used as a non-invasive tool to predict clinical response of immunotherapy. (A) Examples of PET/CT images of four patients illustrating 89Zr-atezolizumab tumor uptake in five different locations on day 7 post-contrast agent administration (white arrows indicate tumor lesions; PET scans were performed once per patient and time point). Images (i) and (ii) are from the same patient, whereas images (iii), (iv), and (v) are each from a separate patient. (B) Overview of 89Zr-atezolizumab uptake as SUVmax at day 7 post-contrast agent administration in 196 tumor lesions with a diameter >2 cm grouped per tumor type and ordered by increasing geometric mean SUVmax per patient, visualizing tumor size and site, and with the distribution of aorta for blood pool background uptake as reference. Horizontal bars indicate geometric mean SUVmax per patient. (C) PET/CT images of lesions of two patients with heterogeneous intralesional 89Zr-atezolizumab uptake on day 7 post contrast agent administration. (Top) Mediastinal lesion of a NSCLC patient (SUVmax 19.9) and (Bottom) abdominal wall metastases of a bladder cancer patient (SUVmax 36.4). (D) Progression-free survival according to the geometric mean standard uptake value (SUVmax) per patient obtained by non-invasive PET imaging using 89Zr-labeled atezolizumab (orange, above-median geometric mean uptake; blue, below-median geometric mean uptake; N = 22 patients; two-sided log-rank test). For comparison, Hazard Ratios (HR) were only 2.6 and 1.3 for two different PD-L1 antibodies used in histology. For details see Bensch et al. (2018). (Reproduced with modifications from the indicated reference).
In preclinical models, the use of reporter genes to detect cancer cells in vivo (see Section “Non-invasive Whole-Body in vivo Cell Tracking”) can overcome specificity issues of anatomical imaging. Cancer cell tracking by means of reporter gene imaging is frequently performed using bioluminescence technology which is cost-effective and fast but suffers from the limitations of optical imaging, which preclude accurate quantification (Figure 1, see Section “Brief Overview of Relevant Imaging Technologies”). A recent article comparing BLI alone with combined BLI and radionuclide imaging demonstrated such aspects providing real-life examples in the context of cancer cell tracking (Vandergaast et al., 2019). It is noteworthy that other imaging technologies can fare well in some specialized cases. For example, metastasis tracking of melanin-producing murine melanoma cells was achieved in mice at reasonable sensitivity and resolution compared to the study aims by using PAT (Lavaud et al., 2017). Alternatively, radionuclide cancer cell tracking methodologies have been developed but they are more expensive and lower throughput techniques but provide 3D tomographic and fully quantitative information (e.g. Fruhwirth et al., 2014; Diocou et al., 2017; Volpe et al., 2018). The latter is currently being tackled by the development of multi-animal radionuclide imaging beds (e.g. four-mouse hotels, Greenwood et al., 2019).
In vivo Imaging of T-Cell Populations
Specific cell surface markers on T-cells are attractive imaging targets as they enable the in vivo visualization of either all T-cells or distinct T-cell sub-populations. They can also be exploited for the quantification of therapeutic responses affecting T-cell presence (or absence) in cancerous tissues. For example, targeting T-cell receptors (TCR) is attractive because due to their high turnover on the plasma membrane, the bound radiotracers can gradually accumulate within the T-cells. In one preclinical study, TCRs were targeted using a 89Zr-conjugated anti-murine TCR F(ab’)2 fragment selective for the murine TCR beta domain. Using PET/CT imaging, this radiotracer was shown to track the location of adoptively transferred engineered T-cells in vivo; notably, imaging data and ex vivo quantification of transgenic T-cell numbers in tumors correlated well (Yusufi et al., 2017). Additionally, using a 64Cu-labeled anti-chicken OVA-TCR antibody, it was demonstrated that associated TCR internalization neither impaired antigen recognition via the TCR, nor did it diminish T-cell viability or function in mice (Griessinger et al., 2015).
Alternatively, targeting CD3, a T-cell surface glycoprotein and pan-T-cell marker, has been suggested. Therefore, a radiometal-chelated antibody against CD3 ([89Zr]Zr-DFO-CD3) was designed to quantify T-cell infiltration during anti-CTLA-4 treatment in colon cancer xenograft models. As the host species were mice, a murine anti-CD3 antibody was required in this case, and large amounts of infiltrated T-cells were found in the tumor prior to regression (Larimer et al., 2016). The LAG-3 was similarly exploited to image T-cells in xenografts established in transgene mice expressing human LAG-3 as host strains (Kelly et al., 2018). The radiolabelling strategy also utilized the siderophore-based chelator DFO, and this 89Zr-based radiotracer is now in clinical development. Moreover, 89Zr-LAG-3 PET is currently investigated in patients suffering from head and neck cancer or non-small cell lung cancer (NCT03780725). Notably, both anti-CD4 and anti-CD8 cys-diabodies have been radiometal-labeled (89Zr, 64Cu) to track the corresponding T-cell sub-populations in preclinical models. Using these radiotracers, researchers imaged treatment responses of immunotherapies, for example response to checkpoint inhibitors such as anti-PD-1 (Seo et al., 2018) or anti-PD-L1 (alone or in combination with adoptive cell therapy) (Tavare et al., 2016; Freise et al., 2017; Zettlitz et al., 2017). The advantage of incorporating antibody fragments rather than full-length antibodies into the design of in vivo imaging agents is that the end product will reach its target more quickly, and it will be excreted faster (Bates et al., 2019). Such engineered antibody fragments targeting CD8 have already progressed into clinical trials (NCT03107663, NCT03802123, and NCT03610061). To increase specificity and reduce liver toxicity and Fc γ receptor binding, bispecific antibodies targeting both T-cells (e.g. via 4-1BB) and either tumor antigens (e.g. CD19) or tumor stroma (e.g. FAP) have been developed. The bispecific antibodies have also been conjugated to radioisotopes to track their in vivo distribution in rodents by SPECT or PET imaging (Claus et al., 2019).
The mentioned approaches are applicable to the development of various molecular immunotherapies and cell-based immunotherapies. A general limitation is that the obtained imaging signals cannot be used to back-calculate precise T-cell numbers because the precise expression levels of T-cell surface marker molecules are unknown at the point of imaging. All above described methods probe T-cell presence but not their activities. As for cell-based immunotherapies, there is an additional limitation, namely the lack of discrimination between adoptively transferred and resident cells. To overcome this the adoptively transferred cells would need to be labeled to distinguish them from the resident ones (cf. Section “Non-invasive Whole-Body in vivo Cell Tracking”).
Imaging the Activation of T-Cells
Upon antigen-recognition and co-stimulation, naïve T-cells become activated in secondary lymphoid organs, which results in the expression of various cell surface markers of T-cell activation. The latter can be imaged using specific antibodies or antibody-fragments. For example, OX40 (CD134/TNFRSF4) is such a cell surface-expressed marker of T-cell activation and it has been used to image the spatiotemporal dynamics of T-cell activation following in situ vaccination with CpG oligodeoxynucleotide in a dual tumor-bearing mouse model (Alam et al., 2018). Moreover, it was shown that OX40 imaging using 64Cu-DOTA-AbOX40 as a contrast agent for PET predicted tumor responses with greater accuracy than both blood-based measurements for early response (i.e., Luminex analyses including interferon-γ, tumor necrosis factor α, MCP1, MIP1B etc.) and anatomical measurements in this mouse model. Another example is the trimeric IL -2 receptor (CD25/IL-2Ra), which was exploited to visualize activated T-cells in immune-compromised mice by PET imaging using the contrast agent N-(4-[18F]fluorobenzoyl)- IL -2 (Di Gialleonardo et al., 2012). IL-12 has also been implicated as a specific target for T-cell activation. Consequently, 99mTc-labeled IL-12 has been used to detect T-cell activation in vivo in mice, albeit in colitis and not yet in tumor models (Annovazzi et al., 2006). Moreover, bioluminescence and radionuclide imaging tools to assess TCR-specific activation of T-cells have been developed (Ponomarev et al., 2001; Kleinovink et al., 2018), however, these approaches are still in preclinical development and are based on genetic engineering of T-cells and thus constitute specialized variants of cell tracking as described in Section “Non-invasive Whole-Body in vivo Cell Tracking”.
Alternatively, it is possible to exploit the observation that T-cells undergo metabolic changes upon activation in tissues (van der Windt and Pearce, 2012; Buck et al., 2016) resulting in the influx of substrates not normally present in non-activated T-cells. While targeting metabolic pathways with imaging agents can distinguish activated from non-activated T-cells, this approach can suffer from competing signals generated by different cells in close vicinity. A very promising PET tracer in this context is 2′-deoxy-2′-[18F]fluoro-9-β-D-arabinofuranosylguanine ([18F]F-AraG), which accumulates in activated T-cells predominantly via two salvage kinase pathways (Ronald et al., 2017). Notably, the unlabeled compound has previously been used as a T-cell depleting drug in refractory T-cell acute lymphoblastic leukemia. [18F]F-AraG PET imaging in a murine acute graft-vs.-host-disease (GvHD) model enabled visualization of secondary lymphoid organs harboring activated donor T-cells prior to clinical symptoms of GvHD. Notably, the biodistribution of [18F]F-AraG was favorable and it may be useful for imaging activated T-cells in the context of immunooncology, which is currently investigated in several clinical trials (NCT03311672, NCT03142204, and NCT03007719).
Non-Invasive Whole-Body in vivo Cell Tracking
The exploitation of molecular imaging has also enabled spatiotemporal whole-body in vivo tracking of administered cells (Kircher et al., 2011). One form of in vivo cell tracking has long been used to localize occult infections in patients (de Vries et al., 2010; Roca et al., 2010). Technological and methodological advances over the last decade led to a resurgence of cell tracking, this time in conjunction with the emergence of live cell therapeutics. For their development, several important questions remain largely elusive and require attention;
(i) the whole-body distribution of therapeutic cells;
(ii) their potential for re-location during treatment and the kinetics of this process;
(iii) whether on-target off-site toxicities occur;
(iv) how long the administered cells survive; and
(v) which biomarkers are best suited to predict and monitor cell therapy efficacy.
Traditional approaches in preclinical cell therapy development relied on dose escalation with toxicity evaluation, tumorigenicity tests, and qPCR-based persistence determination. Whole-body imaging-based in vivo cell tracking can inform on questions (i)-(iv) of these aspects in a truly non-invasive manner. However, many clinical trials are still performed largely without knowledge of the in vivo distribution and fate of the administered therapeutic cells, making it impossible to adequately monitor and assess their safety, thereby raising ethical questions when considering complications in clinical trials that could have been averted or mitigated if whole-body imaging had been used (Linette et al., 2013; Saudemont et al., 2018). With cell-based anti-cancer immunotherapies currently centered on adoptively transferred T-cells, either subjected to genetic engineering or ex vivo expansion only, there was a need to develop corresponding imaging tools to quantify T-cells in vivo on the whole-body level.
Methods of in vivo Cell Tracking
In vivo cell tracking rests on the principles and mechanisms of molecular imaging to achieve contrast between cells of interest and the other cells of the organism. In some cases, there are intrinsic features of the cells of interest that can be exploited for generating contrast, for example, when cells produce targetable molecules that show low or no expression in other tissues. Under these circumstances, conventional molecular imaging offers cell tracking possibilities both preclinically and clinically. Examples demonstrating this are; tracking thyroid cancer metastases using the NIS (Kogai and Brent, 2012; Portulano et al., 2014), exploiting the PSMA to image prostate cancer and its spread (Perera et al., 2016; Oliveira et al., 2017), carcinoembryonic antigen (CEA) for colorectal cancer imaging (Tiernan et al., 2013), or melanin imaging in melanomas (Tsao et al., 2012). However, in most in vivo cell tracking scenarios, including all reported cases of cell-based immunotherapy, contrast agents or contrast-generating features must be introduced to the cells of interest. Fundamentally, cell labels can be introduced to cells via two different methodologies, direct or indirect cell labeling.
Direct Cell Labeling for Cell-Based Immunotherapies
Direct cell labeling is performed upon cells ex vivo, and the subsequently labeled cells are re-administered into subjects, where they can be tracked using the relevant imaging technology (Figure 3A). Cells can either take up the contrast agents on their own (e.g. through phagocytosis, via internalizing receptors etc.) or are labeled through assisted contrast agent uptake (e.g. using cell permeant contrast agents, transfection etc.). There is a large variety of ready-to-use contrast agents available including chelated radiometals (for PET or SPECT), 19F-fluorinated nanoparticles and iron oxide nanoparticles (for various MRI types), as well as organic fluorophores and fluorescent nanoparticles (for optical imaging); for more details the reader is referred to a recent review by Kircher et al. (2011). One strength of MRI imaging is its excellent whole-body resolution. Consequently, various nanoparticles have been used to label and track adoptively transferred cells in preclinical models by MRI (Qiu et al., 2018). When applied to cell-based immunotherapy in humans, 19F-fluorinated nanoparticles have been proven effective cell-tracking contrast agents for MRI (Srinivas et al., 2013), as 19F is naturally almost absent in tissues. Unfortunately, the detection sensitivity of 19F is very low and requires specialized equipment. Attempts to improve detection sensitivities included the use of molecules and nanoparticles incorporating many 19F atoms (Srinivas et al., 2012). Longitudinal tracking of activated T-cells in vivo was reported for a period of nearly three weeks in mice (Srinivas et al., 2009), but others found only limited utility for in vivo tracking of similarly labeled CD4+/CD8+ T-cells (in a murine diabetes model, Saini et al., 2019). Despite multiple optical contrast agents available for cell labeling, whole-body in vivo cell tracking using optical methodologies is very limited. This is caused by the intrinsic shortcomings of optical imaging including high tissue absorption and scatter precluding accurate in vivo localization and quantification. This includes 3D fluorescence molecular tomography (FMT), which also suffers from poor resolution, limited depth penetration and low sensitivity compared to other modalities (Figure 1). In the following, we focus on direct cell labeling with radioisotopes, because radionuclide imaging is currently the most sensitive tool for in vivo tracking of directly labeled cells in mammals. When co-registered with CT or MRI for additional anatomical detail, SPECT/PET-MRI/CT images are most promising to aid clinical translation of cell-based immunotherapies. Radioisotope can be used for cell labeling by either (i) exposing cells to chelating agents such as radiometal-complexed hydroxyquinolines (oxines) resulting in cellular uptake via diffusion or transport-mediated processes, or by (ii) linking radioisotopes onto cell surfaces, either electrostatically (e.g. using cell insertion peptides) or covalently.
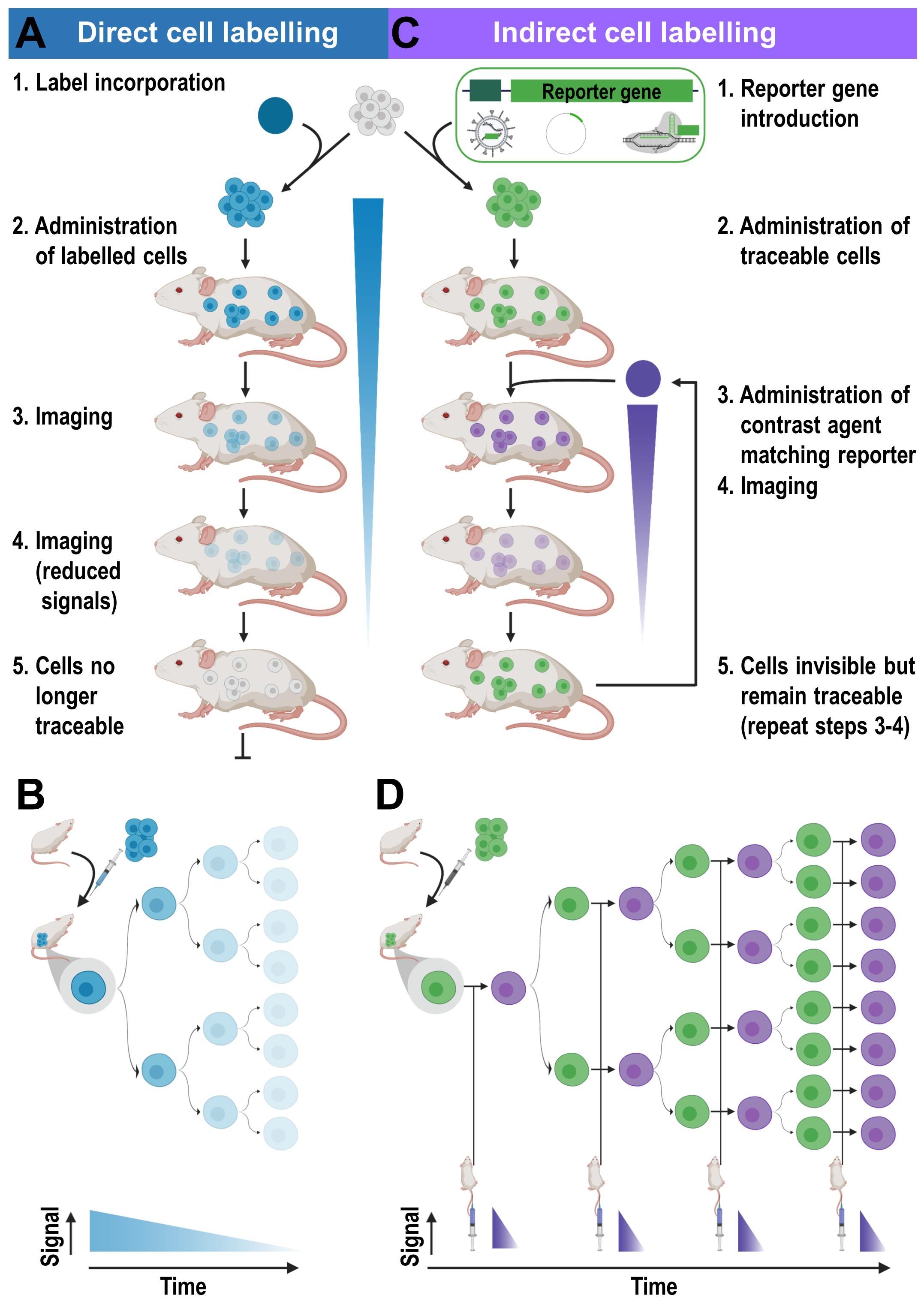
Figure 3. Cell labeling approaches and their consequences for in vivo detectability of cells. (A) Cells are directly labeled by incorporation of a contrast agent (blue) matching the desired imaging technology. Cells can either take up the contrast agent on their own (e.g. through phagocytosis, via internalizing receptors etc.) or are labeled through assisted contrast agent uptake (e.g. cell permeant contrast agents, transfection etc.). The labeled cells (blue) are administered to animals and remain traceable until the contrast agent concentration per cell becomes too dilute to be detectable. Several processes including label efflux, label dilution through cell division, and in the case of radioisotopes also radioactive decay contribute and limit the maximum observation time in vivo. (B) Scheme depicting the effects of label dilution on cell detectability. (C) Indirect cell labeling requires the incorporation of a reporter gene (green) under the control of a suitable promoter (dark green). Reporter genes are frequently introduced using viruses but can also be incorporated via episomal plasmids or gene editing. Engineered cells (green) are administered to animals and can be visualized in vivo via administration of corresponding contrast agents (purple) followed by imaging, which can be repeated to enable long-term tracking. (D) Filial generations of reporter gene expressing cells remain traceable, hence indirectly labeled cells are in vivo traceable indefinitely.
In inflammatory conditions and infectious disease, the radiometal chelators [111In]In-oxine and [99mTc]Tc-HMPAO have been routinely used clinically for tracking ex vivo labeled cells, e.g. white blood cells (Roca et al., 2010; de Vries et al., 2010). This decades-old methodology has more recently been applied to clinical studies of CD4+ T-cells in Hodgkin’s lymphoma (Grimfors et al., 1989), to assess penetrance of tumor-infiltrating lymphocytes in melanoma (Fisher et al., 1989; Griffith et al., 1989) or autologous CD8+ T-cells in early stage non-small cell lung cancer patients who receive anti-PD-L1 immunotherapy in a neo-adjuvant setting (NCT03853187). Both 111In and 99mTc are compatible with SPECT imaging or scintigraphy, an imaging technology which has previously been shown to be insufficiently sensitive in clinical studies (James and Gambhir, 2012). Although technological advances in SPECT instrumentation are improving the situation somewhat, PET imaging remains the method of choice, since it offers absolute quantification and higher sensitivity on clinical instrumentation. The PET isotope equivalent of 111In (τ = 2.8 days) is 89Zr (τ = 3.3 days), which has a similar half-life but different decay properties (89Zr: 23% positron emission, higher energy γ–rays than 111In but lacking Auger electron emission). Like 111In, cell labeling with 89Zr became possible with oxine chelators (Charoenphun et al., 2015; Sato et al., 2015), and it was shown to be better retained inside cells than [111In]In-oxine (Charoenphun et al., 2015). This is a major advantage because the images correspond to the locations of the radioisotope; if labels leak out of cells rapidly they are more likely to give unreliable results. [89Zr]Zr-oxine has been widely applied preclinically for immune cell labeling of cytotoxic T lymphocytes (CTL), γδ T-cells, DC and CAR-T (Sato et al., 2015; Weist et al., 2018; Man et al., 2019). With GMP-compatible protocols now available, [89Zr]Zr-oxine is on a trajectory toward clinical translation and ultimately it will replace [111In]In-oxine, which has become increasingly scarce in the EU due to economic reasons (Dhawan and Peters, 2014). A limitation of PET is its restricted spatial resolution (Figure 1) which is fundamentally limited by the radioisotope-dependent average positron range in matter (Phelps et al., 1975; Cal-Gonzalez et al., 2013). A way to mitigate its low resolution is to combine it with anatomical imaging methods that feature higher resolution (PET/MRI and PET/CT). For cell tracking applications also nanoparticle-based, multimodal PET/MRI probes have been envisaged, for example iron oxide nanoparticles that are cross-linked to radioisotopes (Garcia et al., 2015).
An alternative direct cell labeling methodology is to link contrast agents to the cell surface of cells. For example, [89Zr]Zr-DFO-NCS was used to label human mesenchymal stem cells and while retained on the cell surface for about a week, cell viability appeared to be unaffected (Bansal et al., 2015). This approach is constrained by the availability of cell surface reactive groups that can be exploited, in this case primary amines, and it has the potential to interfere with cell surface proteins and impair cell function. This could restrict its use, particularly if tracer-level concentrations are superseded to achieve high ex vivo cell labeling to expand the cell tracking time (cf. Figures 3A,B). However, no systematic comparative studies between cell uptake and cell surface linking of radiometals in lymphocytes have so far been reported.
Indirect Cell Labeling Applied to Immunotherapy Development
Indirect cell labeling is based on genetic engineering of cells to ectopically express a reporter, which serves as an imaging target (Figure 3C). This imaging target is then imaged in vivo after administration of suitable contrast agents, for example short half-life radiotracers, in a process that can be repeated to detect the traceable reporter-expressing cells over time (Figures 3C,D). Introduction of genetically encoded reporters is most frequently performed by viral transduction to ensure genomic integration and long-term expression. In some cases, episomal plasmids have been used (e.g. delivered by transfection or electroporation; Lufino et al., 2008; Ronald et al., 2013). Lately, gene editing approaches have been exploited for reporter insertion as they can be advantageous to viral transduction because they offer precise control over the genomic site of reporter insertion (Bressan et al., 2017). With feasibility having been demonstrated, this approach is likely to receive greater attention in the cell therapy field in future. Contrast formation relies on one of several mechanisms (Figure 4): either
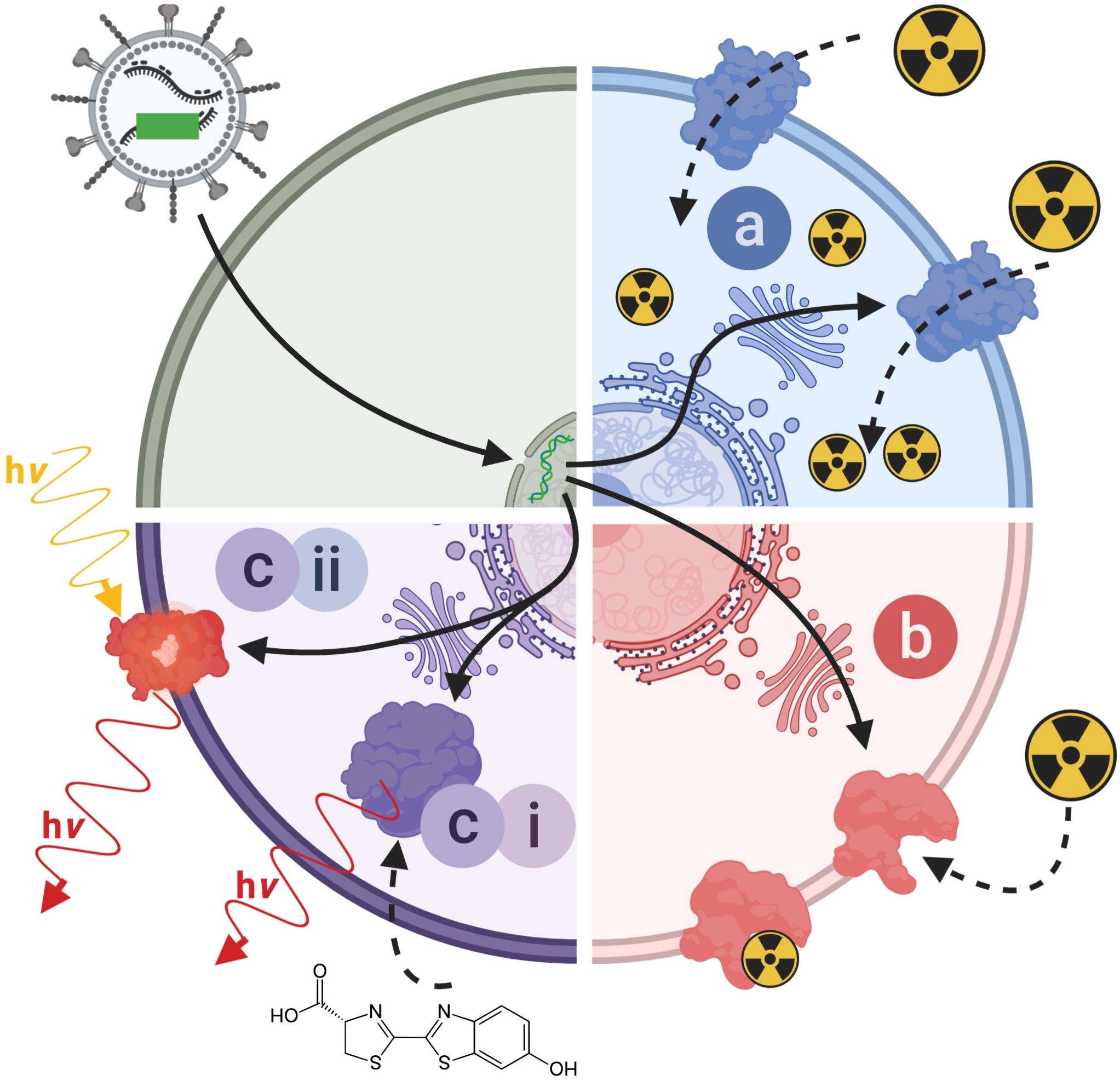
Figure 4. Molecular imaging mechanisms relevant to reporter genes for indirect cell labeling. Cartoon showing the three main molecular imaging mechanism that are exploited for indirect cell labeling. (a) Transport (blue): these reporters are expressed at the plasma membrane of cells and each expressed reporter can transport several contrast agent molecules into the cell, which constitutes a signal amplification mechanism. The radionuclide transporters NIS and NET belong to this class of reporters. (b) Protein binding (red): these reporters are also normally expressed at the plasma membrane of cells and contrast agents bind directly to them; minor levels of signal amplification are theoretically possible if several contrast agents could bind to the reporter, or if several contrast agents could be fused to a reporter binding molecule; however, signal amplification is inferior compared to transporters. Examples for this reporter class are PSMA and SSTR2. (c) Contrast forming reporters (purple) can be sub-divided into two categories; enzymes that can generate contrast, and proteins that act as labels with intrinsic contrast. (ci) Enzymatic contrast formation: such reporters either entrap a molecular probe or generate a contrast agent from a precursor that needs to be either supplied externally or is available within the cell. Thymidine kinases such as HSV1-tk are examples for enzymes that entrap a radiotracer through its phosphorylation, and thereby generate contrast. Firefly luciferases are examples of reporters that convert an externally supplied substrate [shown: luciferin light (hν)]. Tyrosinase is an example of a reporter which converts cell-intrinsic precursors to the contrast agent melanin. (cii) Intrinsic contrast: these reporters produce a signal on their own, normally upon stimulation. Classical examples are all fluorescent proteins, which generate specific light emissions upon excitation with light matching their excitation spectra. For details and literature references to relevant reporter genes see Tables 1, 2. NIS, sodium/iodide symporter; NET, norepinephrine transporter; PSMA, prostate specific membrane antigen; SSTR2, somatostatin receptor 2.
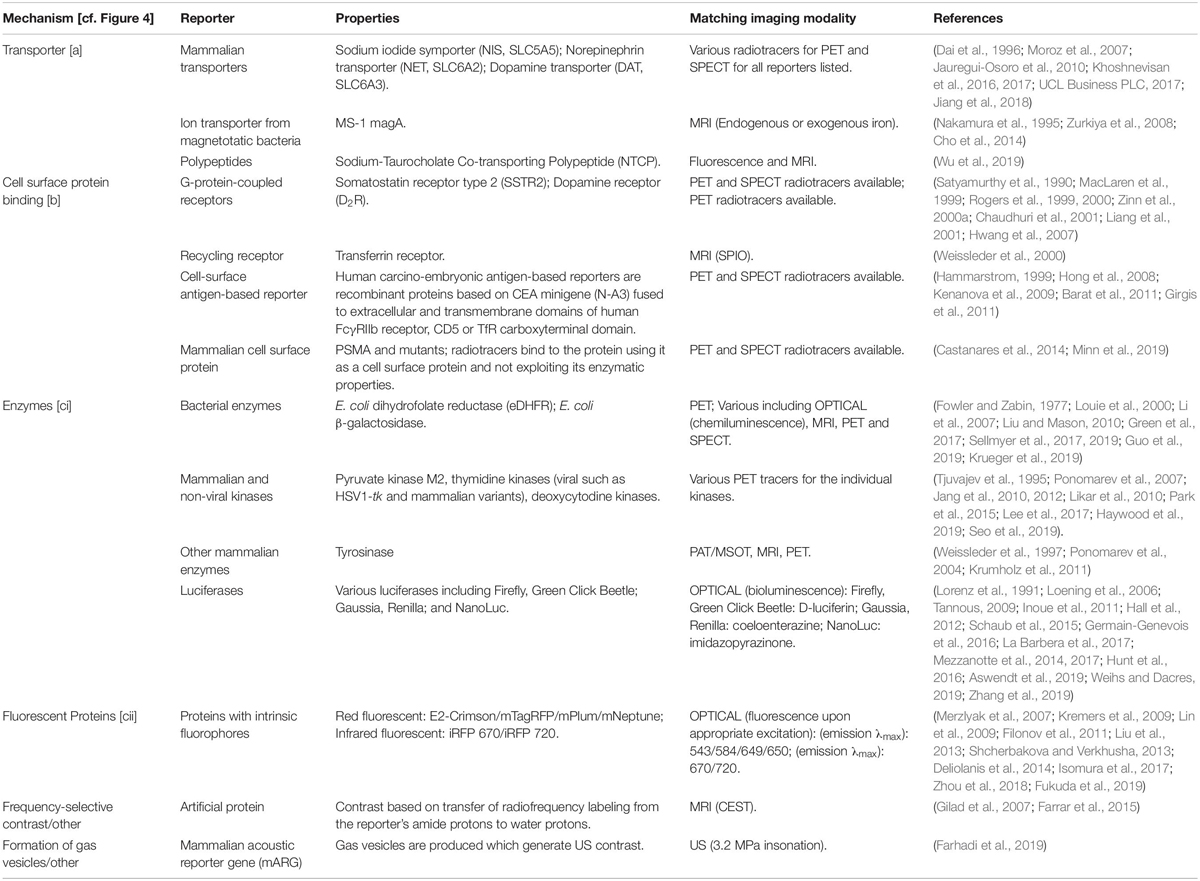
Table 1. Reporter gene classes according to their molecular imaging mechanisms (cf. Figure 4) including selected examples.
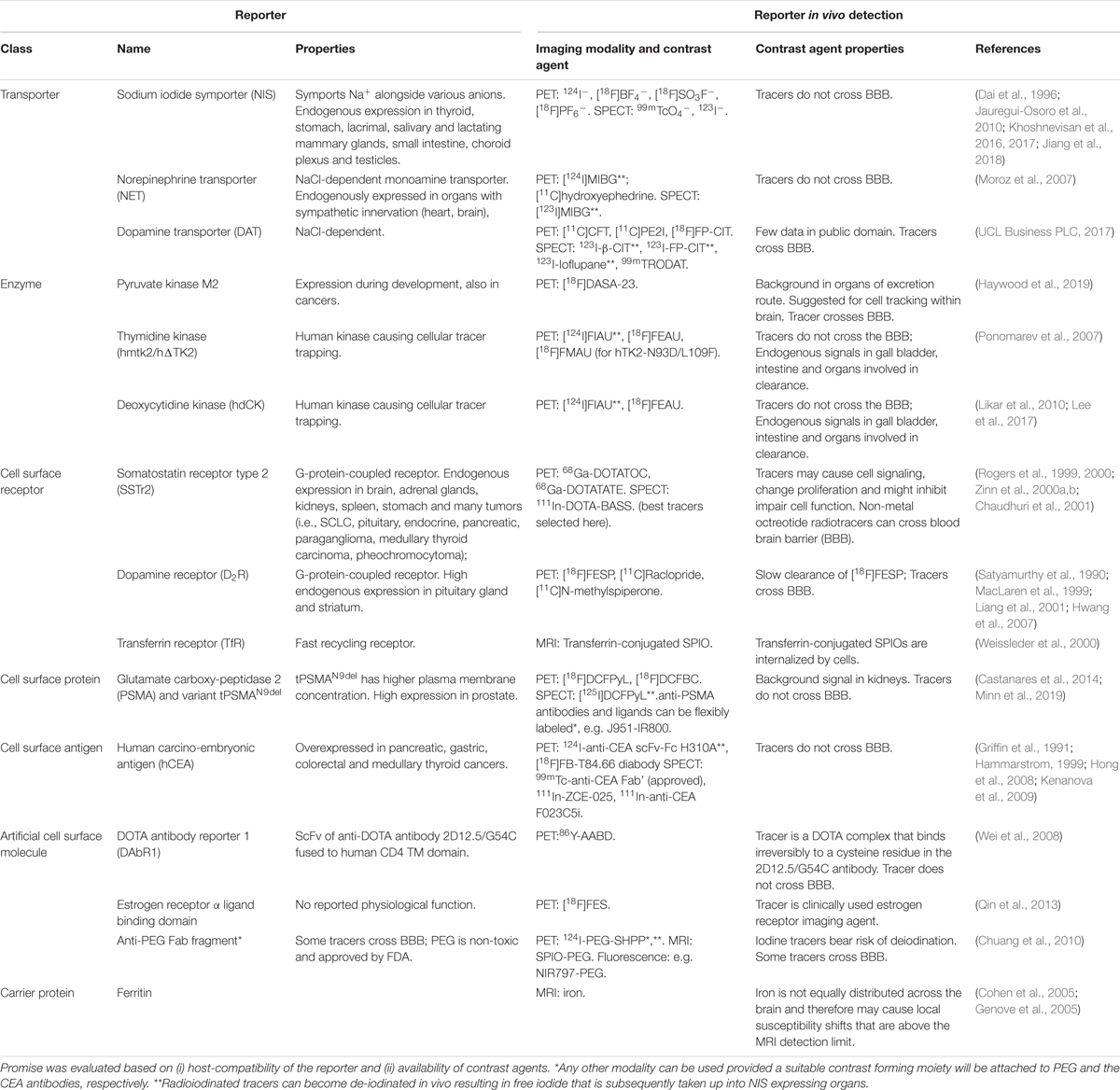
Table 2. Promising host-compatible reporter genes and their corresponding imaging tracers.
(a) label uptake into cells by transporters,
(b) label binding to cell surface-expressed reporters, or
(c) expression of contrast-forming proteins, which either (i) produce a label through enzymatic action (e.g. luciferases, tyrosinase), or (ii) act as labels themselves (e.g. fluorescent proteins).
All these mechanisms can be useful for preclinical cell tracking and a variety of corresponding reporter genes are listed in Table 1. For clinical cell tracking, the emphasis must lies on the mechanisms (a) and (b), because the contrast-forming proteins are either not of human origin or produce toxic products if expressed outside their original context (e.g. tyrosinase; Urabe et al., 1994) and thus not clinically translatable. Alongside improvements of imaging technologies, also the corresponding reporter genes have been developed and optimized. A fundamental drawback of indirect cell labeling is that it requires genetic engineering. However, this is neither a concern for preclinical experimentation nor for cell therapies already reliant on it (e.g. CAR-T) (Saudemont et al., 2018). Several factors require careful consideration when planning reporter gene-afforded in vivo cell tracking experiments, particularly in the context of immunotherapies (see Section “Experimental Design Considerations for in vivo Cell Tracking”).
Multiplex Cell Tracking
It would be highly beneficial to track both primary tumors and metastases alongside the therapeutic in preclinical models. Combining preclinical whole-body cancer cell tracking with imaging of molecular or cell-based immunotherapeutics could enable image-based quantification of the extent a labeled/traceable immunotherapy reaches in vivo traceable cancers, and whether the immunotherapy is delivered to all primary/secondary lesions. Dual-modality approaches would be required for this, ideally both tomographic in nature to enable the 3D quantification of metastasis burden alongside the immunotherapy. While almost every well-performed preclinical immunotherapy imaging study cross-correlates tumor targeting of the traceable therapeutic with either anatomical or molecular imaging in the primary tumor, metastases have rarely been accounted for. One example of such a study evaluating also the metastatic sites involved was performed by Edmonds et al. (Edmonds et al., 2016) in a preclinical breast cancer model. The authors employed dual-radioisotope imaging to co-track cancer metastases and a liposomally encapsulated immunomodulatory drug with the aim to optimize the time between liposome administration and the subsequent adoptive transfer of γδ T-cell immunotherapy involving both primary and secondary lesions. In a subsequent preclinical study, the same authors co-tracked 89Zr-oxine labeled γδ T-cells (direct labeling approach) to NIS reporter expressing breast cancer cells (indirect labeling approach using 99mTcO4– as a NIS radiotracer) employing sequential multi-modal PET-SPECT-CT imaging (Man et al., 2019).
Experimental Design Considerations for in vivo Cell Tracking
To ensure immunotherapy development benefits from cell tracking, it is imperative that the cell labeling approaches for therapeutic and/or cancer cells are chosen with the experimental goals in mind. Considerations must include a variety of different aspects such as cell tracking time, cell tracking interval, experimental setting (preclinical or clinical), use of immunocompetent or immunocompromised host organisms, imaging technology, and contrast agent properties and availability. To detect cells, the employed cell label must match the envisaged imaging technology to be used. The choice of the imaging technology dictates the achievable spatial resolution and imaging depth (Figure 1), impacts on minimal temporal resolution through image acquisition speeds, and contributes majorly to detection sensitivity and cost. Availability of the imaging technology and the necessary label further impact on the feasibility of collaborative across different institutions, which is of particular importance for clinical translation of a methodology and the chances of its subsequent adoption in clinical practice.
Imaging Technology and Its Impact on Cell Detection Sensitivity
Considerations for the Selection of the Imaging Technology
Exquisite detection sensitivity is required for in vivo cell tracking applications. In practice, this means sensitivities should be within or below the picomolar concentration range (Figure 1), which can be achieved best with bioluminescence and radionuclide imaging modalities. Unlike radionuclide imaging technologies, BLI neither provides absolute quantitative data nor true 3D information and is applicable only preclinically. However, despite its shortcomings, BLI has so far been the most frequently used preclinical approach to measure the impact of immunotherapeutics on in vivo traceable bioluminescent tumors; most likely due to BLI being relatively cheap and fast. In special cases, BLI can currently provide unique information relevant to immunotherapy development on the preclinical level. For example, dual-luciferase reporter methodology enabled the quantification of in vivo T-cell activation in specifically engineered transgene mice (Mezzanotte et al., 2011; Kleinovink et al., 2018). While it is fundamentally possible to perform such preclinical experiments with more quantitative 3D radionuclide tomography, it has not been reported so far; most likely due to more complex logistics and higher costs associated with this approach (e.g. two different radiotracers with similar pharmacokinetics/pharmacodynamics would be needed for each individual imaging session).
In many cases, 3D tomographic whole-body imaging data is required in rodents or larger mammals, i.e., non-translucent organisms. This generally limits the use of optical imaging technologies due to their inherent limitations relating to tissue light absorption and scatter. Hence, radionuclide imaging modalities are preferred for such purposes, but they require dedicated reporter genes, which are scarce compared to the plethora of different fluorescent proteins or luciferases that have been developed (Thorn, 2017; Shcherbakova et al., 2018; Mezzanotte et al., 2017). If clinical translation is the main goal for cell tracking applications, then radionuclide imaging is the most suitable approach to address the questions raised in Section “Non-invasive Whole-Body in vivo Cell Tracking.”
Detection Sensitivity and the Duration of Cell Tracking
Detection sensitivity of labeled cells depends on the cellular label concentration and the matched imaging technology. The different labeling methodologies affect the cellular label concentration in different ways (see Section “Non-invasive Whole-Body in vivo Cell Tracking”). Label dilution, label efflux and in the case of radioactive labels dosimetry, can be severe limitations of direct cell labeling methodologies. The impact of label dilution has been discussed above (Section “Direct Cell Labeling for Cell-Based Immunotherapies”). Copper isotopes are the main example wherein label efflux causes issues. 64Cu (τ = 12 h) had been suggested as a shorter half-life PET isotope (Adonai et al., 2002; Li et al., 2009; Bhargava et al., 2009) potentially competing with the SPECT isotope 99mTc (τ = 6.0 h). However, its unfavorably high cellular efflux (>50% per 4-5 h) paired with efficient liver uptake resulted in low signal-to-background ratios in vivo (Adonai et al., 2002; Bhargava et al., 2009; Li et al., 2009; Griessinger et al., 2014). High label efflux also limited the use of the long half-life PET radiometal 52Mn for cell tracking (Gawne et al., 2018). For considerations regarding dosimetry see Section “Impact of Cell Labeling Methodology on Cell Function.”
For indirectly labeled cells, the molecular imaging mechanism of the used reporter gene and the cellular expression level of the reporter gene are crucial, while the label dilution aspect plays no significant role (Figure 3D). Reporter genes which enzymatically entrap radiotracers that are taken up into cells offer high cell detection sensitivities. Examples are thymidine kinases, which phosphorylate and thereby entrap radiotracers in the cells, e.g. HSV1-tk is detected through its corresponding PET radiotracer [18F]FHBG. Transporters (e.g. NET or NIS) provide signal amplification as each reporter protein can transport several radiotracer molecules into the cell. It is noteworthy that ectopic expression of reporters can affect the fate of their substrates. For example, NIS is normally expressed in thyroid follicular cells and its regular substrate, iodide, is metabolized into thyroid hormones after cell import. Upon ectopic expression in non-thyroidal cells, e.g. cancer cells or immune cells, this downstream mechanism affecting the equilibrium of the imported iodide is non-existent resulting in iodide not being accumulated to the same extent compared to thyroid tissues. To apply radioiodide for cell tracking in humans, it would be necessary to counteract high thyroid uptake and radioiodide metabolization there as this could lead to thyroid damage. The latter is possible by prior administration of non-radioactive iodide, but this also impacts on detection sensitivity of the traceable cells of interest. Non-iodide NIS radiotracers, which are not metabolized and wash out of thyroid cells would thus be preferable. As NIS is not very selective regarding its anion substrates (Paroder-Belenitsky et al., 2011) anionic radiotracers that are roughly similar in size and shape were developed for NIS imaging; they include 99mTcO4–, [18F]BF4–, [18F]SO3– or [18F]PF6– (Jauregui-Osoro et al., 2010; Khoshnevisan et al., 2017; Jiang et al., 2018). They are not entrapped in cells, neither in thyroidal tissues nor in cells ectopically expressing NIS. Therefore, it would be advantageous to use non-iodide NIS radiotracers for clinical cell tracking. Another advantage of the new NIS PET radiotracers is that they are based on 18F, which has superior decay properties compared to 124I. 18F decays with a half-life of 109.8 min to 18O with 96.9% positrons (Emean = 0.250 MeV and 0.6 mm average positron range), while 124I decays with a half-life of 4.18 days to 124Te with only 22.7% positrons (11.7% β2+ at Emean = 975 MeV and 4.4 mm mean range, and 10.7% β1+ at Emean = 0.687 MeV and mean 2.8 mm range, plus a minor 0.3% β3+ at 0.367 MeV and 1.1 mm mean range) accompanied by several γ–rays and a high proportion of electron capture (Conti and Eriksson, 2016). Consequently, 18F produces more positrons per decay resulting in better detectability. It is noteworthy that 18F positrons also have a lower mean energy than those of 124I resulting in lower mean positron ranges (until annihilation and emission of detectable γ–rays) and therefore enabling better PET resolution. While free positron range considerations are currently irrelevant for clinical PET imaging (instrument resolution with 3-4 mm larger than most average free positron ranges of relevant PET isotopes), they are of concern for preclinical PET imaging (instruments can provide resolution even below 1 mm with the right isotope) (Deleye et al., 2013; Nagy et al., 2013). Notably, first-in-man clinical studies using [18F]BF4– to image NIS have already been completed (O’Doherty et al., 2017), thereby lowering the hurdles for NIS-afforded PET reporter gene imaging as a means of cell tracking in humans. Such considerations regarding the selection of radioisotopes as part of an individual reporter:contrast agent pair are transferable also to other reporters for which contrast agents with different radioisotopes are available (see Table 2). Selection of the best suited reporter:contrast agent pair is paramount.
The detection sensitivities of NIS-expressing extra-thyroidal cells have been reported preclinically to be as good as hundreds/thousands for cancer cells expressing NIS (Fruhwirth et al., 2014; Diocou et al., 2017) and CAR-T expressing PSMA in vitro (Minn et al., 2019), or tens of thousands for effector T-cells using various different reporter genes in vivo (Moroz et al., 2015) (Figure 5). Comparative studies aiming at the evaluation of how different reporter genes impact on T-cell detectability have been performed in the past (Moroz et al., 2015). Importantly, since this study new reporter gene:contrast agent pairs have become available, for example PSMA paired with its high-affinity PET ligand [18F]DCFPyL (Minn et al., 2019) or NIS paired with its PET radiotracer [18F]BF4– (see above). Consequently, new comparative studies are needed to conclude on relative reporter:contrast agent performance in relevant immune cells; ideally performed such that also reporter expression levels and their intracellular availabilities for interaction with their contrast agents are precisely controlled. As reporter expression levels are cell type-dependent, it is highly recommended to determine detection sensitivities of indirectly labeled cells in each case and also on the available instrumentation before designing in vivo cell tracking experiments.
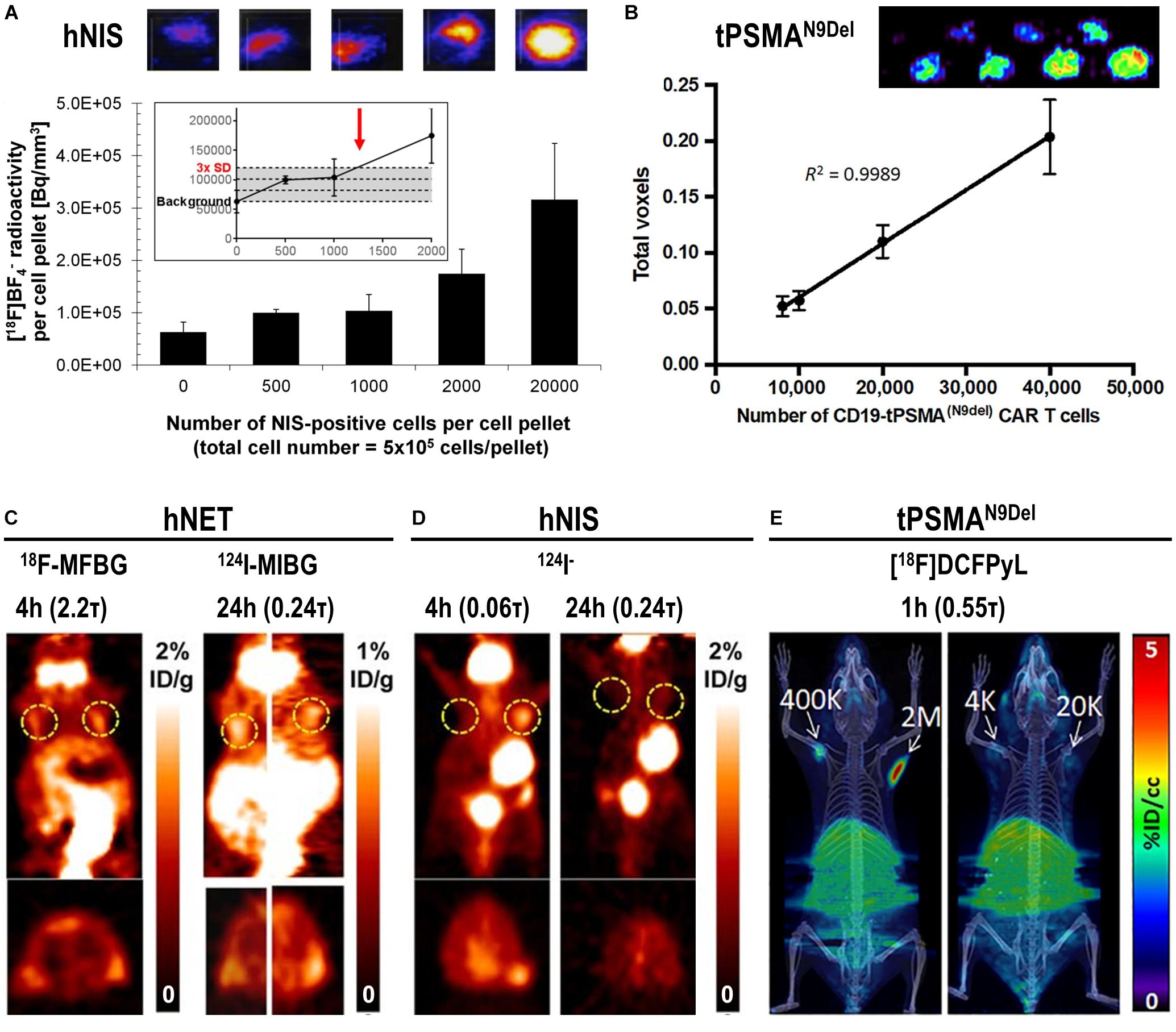
Figure 5. In vitro and in vivo detection sensitivity of reporter gene expressing cells. (A) In vitro determination of the detection limit of NIS-positive cells within a cell pellet of NIS-negative cells for the NIS radio tracer [18F]BF4– using nanoPET/CT equipment from Mediso. For experimental details see Diocou et al. (2017) (top) Typical results of nanoPET/CT imaging of cell pellets and (bottom) quantitative analysis of imaging experiments. The limit of detection was determined to be ∼1,250 NIS-positive cells (inset, red arrow). (B) Standard curve demonstrating a linear relationship between the PET signal and the number of CD19-tPSMAN9del CAR-T. (top) In vitro phantom from which the standard curve was derived. The in vitro phantom used varying numbers of CD19-tPSMA(N9del) CAR T cells incubated with [18F]DCFPyL, a high affinity, positron-emitting ligand targeting PSMA; cell numbers were in the top row 103, 2⋅103, 4⋅103, and 6⋅103, and in the bottom row 8⋅103, 104, 2⋅104, and 4⋅104. Images were acquired using a SuperArgus small-animal PET/CT instrument from Sedecal. The data in the graph show results from the bottom row of images. The detection limit was determined to be around 2,000 cells. For experimental details see Minn et al. (2019). (C,D) PET in vivo imaging of human primary T-cells transduced with hNET (C) or hNIS (D) reporter genes. Different numbers of T-cells were injected subcutaneously, followed by systemic administration of indicated corresponding radiopharmaceuticals. PET imaging at indicated time points after radiotracer administration was performed using a Focus 120 microPET scanner from Siemens. Number of T-cells injected is (left dashed ring) 3⋅105 and (right dashed ring) 106. No potentially interfering signals were thresholded and data are expressed as percentage injected dose per gram (%ID/g). For experimental details see Moroz et al. (2015). (E) NSG mice injected with the indicated number of CD19-tPSMAN9del CAR-T in 50 μL (50% Matrigel) in the shoulders (white arrows). Mice were in vivo imaged on the Sedecal’s SuperArgus small-animal PET/CT at 1 h after administration of the corresponding radiotracer [18F]DCFPyL. PET data are expressed in percentage of injected dose per cubic centimeter of tissue imaged (%ID/cc). To improve the display contrast of the in vivo images, relatively high renal radiotracer uptake was masked using a thresholding method. For experimental details see Minn et al. (2019). (Figure combined from the publications referenced in the legend above; permissions from corresponding publishers obtained).
Proliferation of Traceable Cells and in vivo Tracking Time
Paramount for choosing the cell labeling approach is how rapid the traceable cells divide and how long-term an observer wishes to track them in vivo. Tracking cancer cells in preclinical tumor models normally entails following them over multiple cell divisions, spreading over weeks if not months. To evaluate cell-based anti-cancer immunotherapies, cell engraftment, expansion and survival are of interest, whereby observation times, usually several days up to several weeks, are long-term compared to division/expansion events of therapeutic cells.
For direct cell labeling applications label efflux and label dilution are limiting (Figures 3B,D). If radioisotopes are used for direct cell labeling then their half-lives additionally limit achievable tracking times with 4-5 half-lives being realistic with existing small-animal PET instrumentation; e.g. about two weeks for 89Zr (Khoshnevisan et al., 2017) depending on the amount of radiolabel loading and instrument sensitivity. As the continued presence of the radioisotope in direct cell labeling results in a radiation dose to the cell and consequently radiation damage accumulation, a compromise must be reached between the maximum label concentration, which should not impair cell function (see Section “Impact of Cell Labeling Methodology on Cell Function”), and the maximum achievable tracking time.
In contrast, indirect cell labeling does not suffer from label dilution as the genetically encoded reporter is passed on to filial generations, thereby rendering the observation time theoretically indefinite. Indirect cell labeling relies on repeat administration of contrast agents, if radioactive then short half-live radioisotopes. This adds complexity as for example radiotracers need to be freshly prepared for every imaging session (Figure 3D), but it is certainly advantageous that the overall received doses are smaller than in direct cell labeling when compared over the same tracking periods. Consequently, indirect cell labeling is the preferred method of choice for long-term cell tracking, including cancer cell tracking in spontaneous metastasis models or the long-term evaluation of cell-based immunotherapies.
In vivo Cell Tracking Interval
During tracking of directly labeled cells, the imaging interval is not linked to the imaging technology other than that animal welfare considerations must be considered (e.g. minimum interval of repeat-anesthesia). However, for indirect cell labeling approaches with radionuclide reporter gene and corresponding radiotracers, the choice of radioisotope affects the minimum imaging interval because the radiotracer from an earlier imaging session must have had time to sufficiently decay before a new batch can be administered to enable a subsequent imaging session (Figure 3D). For example, there are various radiotracers for the radionuclide reporter NIS, which have differing radioisotope half-lives; these include 99mTcO4– (τ = 6.01 h) and 123I– (τ = 13.2 h) for SPECT or 124I– (τ = 101 h) and [18F]BF4– (τ = 1.83 h) for PET. Again, ∼4-5 half-lives are needed for sufficient radiotracer decay and this defines the minimum time interval acceptable between imaging sessions. It was previously shown that repeat-imaging of NIS-expressing cells is possible after four half-lives using 99mTcO4–, i.e., after 24 h (Diocou et al., 2017); however, this would not be possible for over two weeks when using 124I–, while [18F]BF4– would allow ∼8 h intervals.
As for radionuclide imaging-afforded cell tracking it is noteworthy that radiotracer concentrations are very low, generally below target saturation, hence presence of prior administered radiotracers is normally negligible for later imaging sessions due to very low picomolar radiotracer concentrations, even if they would stay intact and not be excreted. A special case in this context is 99mTc, which decays to long-lived 99Tc and consequently remains in the same chemical form after its decay. Thus, it could accumulate over repeat imaging sessions unless excreted. For example, in preclinical NIS imaging experiments in mice the radiotracer 99mTcO4– is administered at 15 – 30 MBq per animal. This equates to about 0.5 – 1.0 pmol of the total pertechnetate species (sum of 99mTcO4– and 99TcO4–) taking into account a typical 99mTcO4– generator elution regimen and 99TcO4– carrier presence (Lamson et al., 1975). Even in an unrealistic worst-case scenario excluding its renal excretion, each repeat NIS imaging session would add this amount to the animal. However, the overall pertechnetate concentration would still be far below NIS saturation as its Michaelis-Menten constant for pertechnetate is likely very similar to those reported for ReO4– or ClO4– (Paroder-Belenitsky et al., 2011) and hence in the low micromolar range. Others repeatedly imaged animals with NIS-expressing cancer cells by 99mTcO4– -SPECT and found no impact of earlier imaging sessions on subsequent ones (Diocou et al., 2017). For radiotracers that decompose chemically following radioisotope decay this consideration is irrelevant. Such an example is the NIS PET radiotracer [18F]BF4–, in which 18F decays to 18O resulting in a chemically instable product ultimately generating borate, which is no longer a substrate for NIS (Khoshnevisan et al., 2016). While presented using the example of NIS here, such considerations can also be relevant for other reporter gene:radiotracer pairs.
Cell Viability and Its Impact on Detected Cell Tracking Signals
Signals from directly labeled cells do not report on whether the cells are alive. Moreover, recorded signals might not even stem from the initially labeled cell population (e.g. due to label efflux or cell death and subsequent deposition or uptake into different cells). In contrast, indirect cell labeling is fundamentally linked to cell viability as the reporter is encoded in the DNA of the traceable cells. However, signal loss in reporter expressing cells is also a possibility, for example, when the reporter gene expression cassettes become epigenetically silenced. Notably, so-called ‘safe harbor locations’ have been discovered in mammalian genomes (Pellenz et al., 2019), and reporter genes can be inserted into such locations using gene editing methodologies. The latter has recently be demonstrated in different stem cell types even with large reporter genes such as NIS (Wolfs et al., 2017; Ashmore-Harris et al., 2019).
Stem cell tracking experiments conducted with cells that were both iron oxide nanoparticle-labeled (“direct” cell label) and expressing luciferase and the fluorescent protein GFP (“indirect” reporter genes) elegantly demonstrated the differences between the two different cell labeling methodologies (Figure 6). Even though MRI signals from the iron oxide nanoparticle were detectable for four weeks, these signals were found through ex vivo validation by histology to stem from resident macrophages that had phagocytosed the nanoparticles, which were released from dying stem cells. In contrast, luciferase signals were recorded only from living cells and were validated ex vivo also by histology (Li et al., 2008). This study highlighted comprehensively that reporter gene imaging much better reflects cell viability and that great care must be taken to avoid ascribing signals from directly labeled cells to the wrong cell populations. Consequently, employing direct cell labeling necessitates independent cross-validation, which could include e.g. in vivo co-tracking by reporter gene imaging and ex vivo validation by histology or flow cytometry.
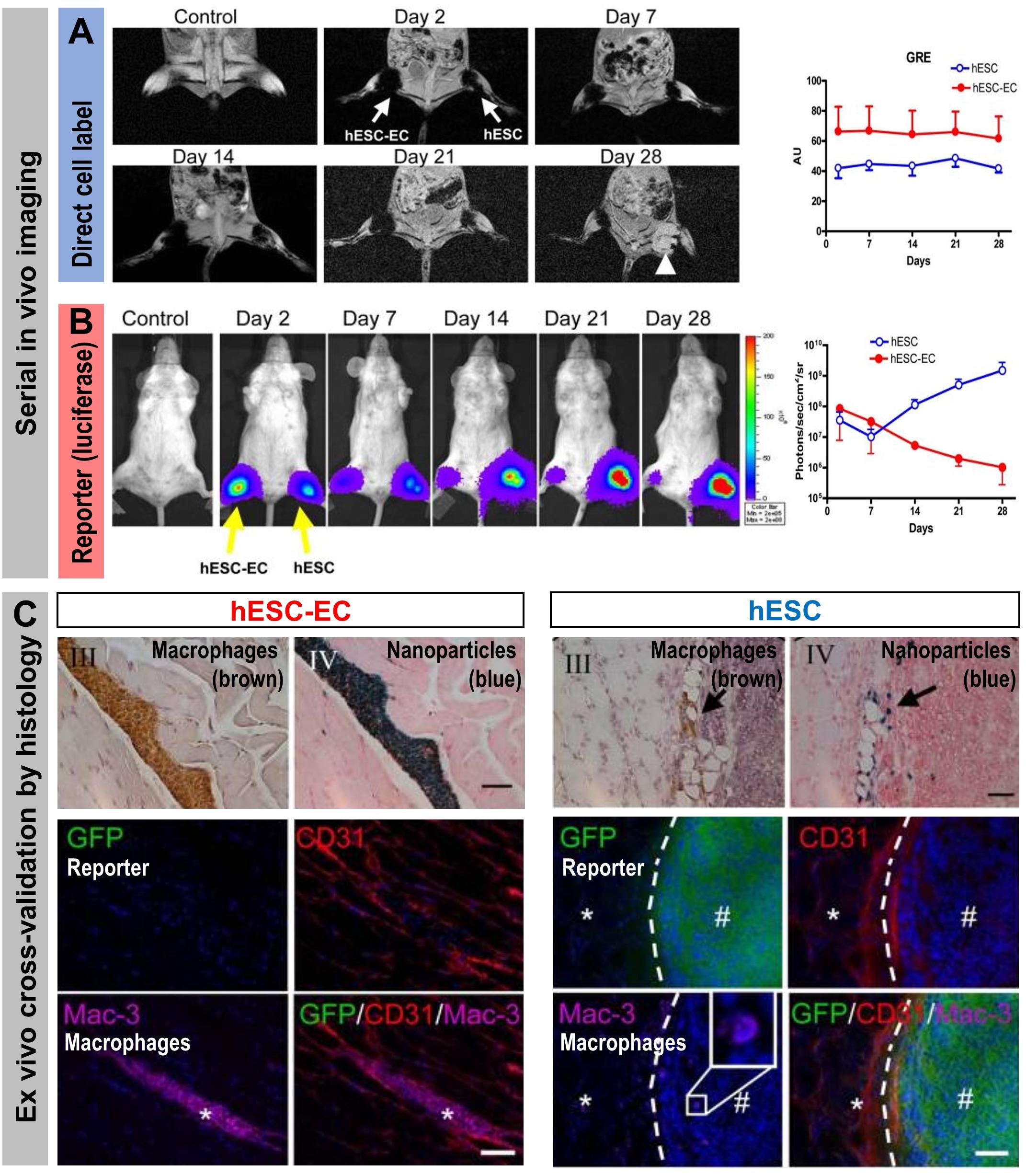
Figure 6. How the type of cell labeling impacts on conclusions drawn from signals obtained from serial imaging. Cell viability can be assessed better from indirectly labeled cells than from directly labeled cells as shown by a cross-validation study using both direct and indirect cell labeling within the same cells. Reporter gene (luciferase and GFP)-expressing human embryonic stem cells (hES) or human embryonic stem cells differentiated to endothelial cells (hESC-EC) were directly labeled using iron oxide nanoparticles. (A) MRI imaging to track the directly loaded nanoparticle cell label (A/left) Serial in vivo MR [gradient-recalled echo (GRE)] images of iron oxide nanoparticles. No hypointense signal was found in control animals injected with unlabeled cells. MR signals showed no significant difference from day 2 to day 28 (the white arrow indicates teratoma formation in the hind limb injected with hES cells). (A/right) Quantitative analysis of GRE signals from all animals transplanted with hES cells and hESC-ECs [signal activity is expressed as authority unit (AU)]. (B) Tracking of the cells by virtue of reporter gene imaging (indirect cell labeling). (B/left) Planar bioluminescence imaging reveals differences in signals obtained from hind limbs that received either hES or hESC-EC cells. After initial similar signal decreases in both limbs, the signals from limbs with hES increased significantly over time, coinciding with teratoma formation in these limbs. (B/right) Quantification of 2D bioluminescence signals from each limb (photons/sec/cm2/sr; note the log10 scale). (C, top) Immunohistochemical (IHC) analysis of initially double labeled hES cells and hESC-ECs clearly reveals iron oxide (by Prussian Blue) co-localizing with a macrophage stain (by specific antibody Mac-3); IHC counterstains were Nuclear Fast Red and Hematoxylin, respectively. Note that macrophages loaded with iron particles can be found in between muscle bundles.(C, bottom) Immunofluorescence staining of GFP for transplanted luciferase co-expressing hESC-ECs (left) or hESC (right). Other panels show respective counterstains for microvasculature (CD31) or macrophages (Mac-3); nuclei were stained with DAPI (blue) in merged images. All images are from four weeks after transplantation. There were no transplanted GFP+ hESC-ECs found nearby macrophages. In tissues that received hES cells, GFP+ hESC were found to form teratoma (#) but no Prussian Blue-stained nanoparticles were found in corresponding IHC regions. The dashed line separates teratoma from normal muscle fibers (*). All scale bars are 20 μm. (Figure modified with permission from Li et al., 2008).
Within indirect cell tracking, there can be variations in how reporter genes reflect cell viability. Differences can arise due to the steady-state concentrations of certain reporter proteins in cells, determined by the production and degradation rate of the reporter. These turnover parameters have not been systematically studied for most reporter genes except some fluorescent proteins (Khmelinskii et al., 2016). In certain conditions, turnover can be manipulated, for example through genetic modification with oxygen degradation domains to speed up reporter degradation in normoxic conditions (Goldman et al., 2011; Misra et al., 2017). In general, both fluorescent proteins and reporters relying on contrast agent binding are likely to produce signals if present. Dying traceable cells or cell debris from them will remain detectable until the reporter proteins are cleared or destroyed. In contrast, reporters with enzyme or transporter functions need to be active to generate contrast in cells, and this requires a form of cellular energy to drive the transport. For example in the case of NIS, the Na+/K+ gradient (Dohan et al., 2003) is critical and its breakdown results in loss of NIS transporter activity, which is the basis for the high sensitivity of NIS to cell death. Consequently, if cell viability is central to the goals of a study, an activity-dependent reporter may yield more reliable data than a reporter which signal relies merely on protein presence.
Impact of Cell Labeling Methodology on Cell Function
It is obvious that there should be no impact of the cell labeling methodology on the function and long-term fate of the labeled cell. Contrast agents for direct cell labeling that are compatible with highly sensitive in vivo detection of labeled cells are often radiotracers. While chemical/biological toxicity is mostly irrelevant due to very low tracer-level concentrations (picomolar), they have the potential to exert radio-damage to the cells depending on their cellular concentration and location, their half-life and type of radioactive decay. For example, despite their short range the Auger electrons emitted by 111In and to a lesser extent by 99mTc have the potential to exert significant DNA damage if they come in close proximity with DNA within the cell nucleus (Sahu et al., 1995). Consequently, cell labeling with agents directly releasing them into the cytosol such as [111In]In-oxine could be more harmful than agents linking them to the cell surface. However, systematic comparative and quantitative studies on such radiobiological effects are not yet available in lymphocytes. Nevertheless, it can be safely assumed that great care must be taken when radiolabelling e.g. T-cells with radiometals, because irradiation is a successful method to deplete the immune system of lymphocytes indicating their distinct sensitivity to radiation (Manda et al., 2012; Piotrowski et al., 2018). Generally, the longer the half-life the larger the dose the labeled cells receive; and this is also valid for their in vivo environment which experiences crossfire from the labeled cells. Consequently, careful consideration of dosimetry is required as well as biological evaluation of radiation effects in radiolabeled cells. For example, [89Zr]Zr-oxine labeled γδ T-cells were tracked to NIS-reporter gene expressing tumors visualized by 99mTcO4– in breast xenograft murine models to determine if the immunostimulatory drug alendronate would result in enhanced tumor targeting by the administered γδ T-cells (Man et al., 2019). To achieve this, the authors first titrated cellular radioactivity amounts delivered into γδ T-cells and validated its impact on γδ T-cell viability/proliferation, occurrence of DNA double strand breaks and retention of tumor cell killing function (Figure 7). This resulted in an optimized radiolabelling regimen that was then used for in vivo cell tracking. Using a similar approach, others determined tumor targeting and tumor retention of [89Zr]Zr-oxine-labeled CAR-T in a glioblastoma and prostate cancer animal model (Weist et al., 2018). The reported tolerated radioactivity levels in labeled cells were 20 mBq/cell for γδ T-cell and 70-80 mBq/cell for effector T-cells/CAR-T in these studies. Fundamentally, the tolerated radioactivity amounts limit the possible tracking time for such labeled cells.
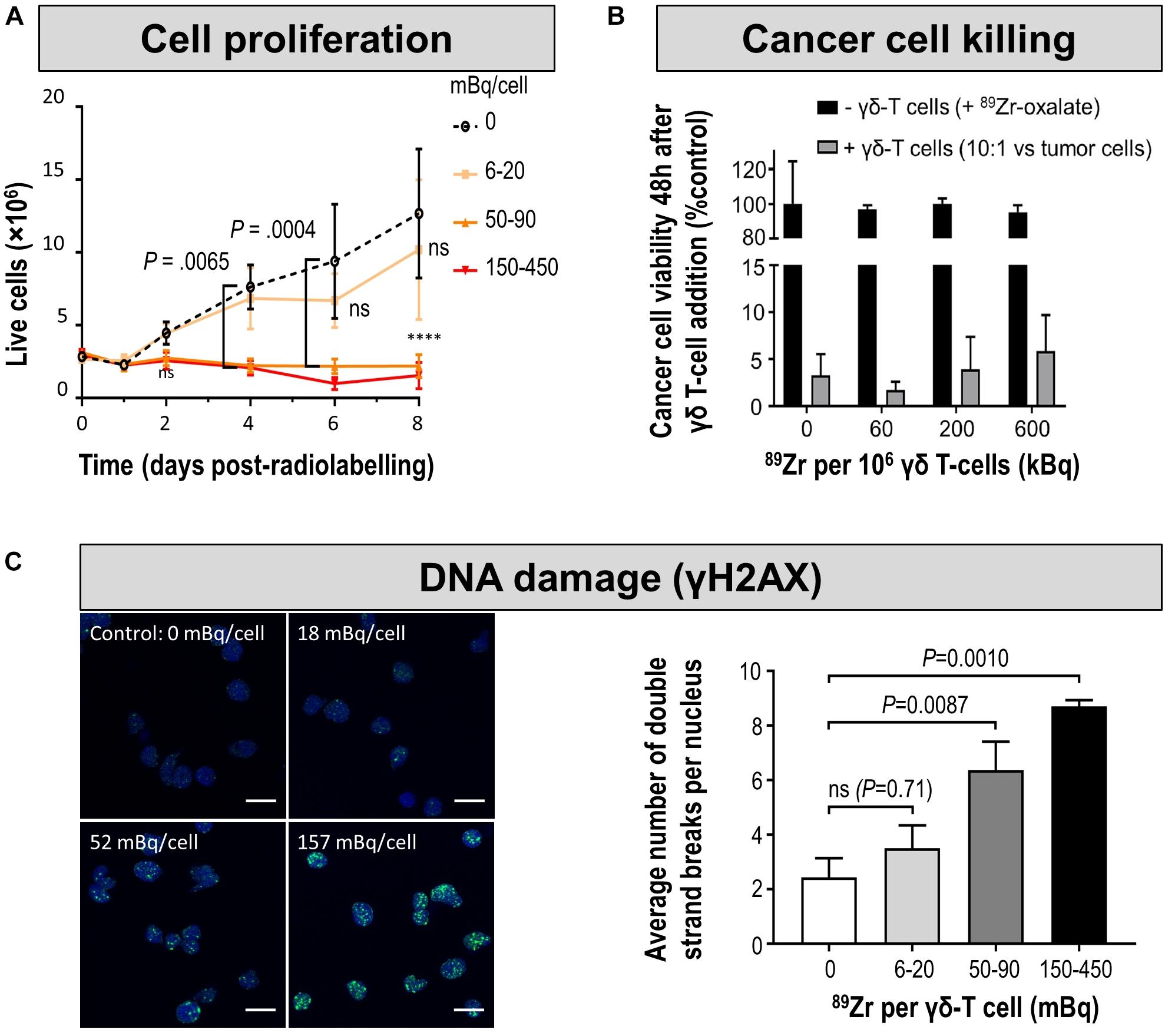
Figure 7. Cell characterization after direct labeling of T-cells with [89Zr]Zr-oxine. (A) In vitro proliferation of differently radiolabeled human γδ T-cells demonstrates that with higher amounts of cell label per cell, the capacity to proliferate diminishes. As expansion capacity is crucial for cell-based immunotherapy applications, it is paramount to perform such proliferation assays for sufficiently long times and quantify any differences even if they happen several days after cell labeling. (B) Tumor cell killing assay demonstrates that even γδ T-cells containing radioactivity levels incompatible with further expansion still retain at least part of their tumor killing function if supplied in sufficiently high amounts. Here, the authors reported this using a triple negative breast cancer cell line in vitro by quantifying tumor cell viability 48 h after immune cell addition. Notably, unchelated 89Zr supplied to tumor cells did not kill them and served as one of the controls. (C) (left) DNA damage analysis in radiolabeled human γδ T-cells. Representative images of γ-H2AX foci (green) and nuclei (blue); scale bars are 10 μm. (right) Cumulative data from the quantification of γ-H2AX foci per nuclei after radiolabelling. For statistical analysis of all data see Man et al. (2019), from where this figure is reproduced with modification and permission.
As indirect cell tracking is based on repeat administration of short half-life radioisotopes (Figure 3C), total received doses are lower compared to direct cell labeling-afforded cell tracking over equivalent time spans. While radio damage is likely less of concern in this context, there are currently no systematic studies on radio damage of reporter-expressing lymphocytes incubated repeatedly with short half-life radiotracers available.
Another important aspect relates to the question of whether there is any impact of the typically very small administered radiotracer amounts (“tracer levels,” “microdoses”) on the corresponding target biology/physiology; in the context of this article for example whether imaging affects adoptively transferred immunotherapies. Generally accepted is the use of tracer level amounts, whereby a microdose is defined as “less than 1/100 of the dose of a test substance calculated (based on animal data) to yield a pharmacological effect of the test substance with a maximum dose of ≤100 μg or, in the case of biological agents, ≤30 nmol” (European Medicines Agency, 2004; VanBrocklin, 2008). However, there are studies available now, which should serve as a primer to investigate this matter more closely as molecules emerged that have biological effects at administered doses comparable to what is generally accepted as tracer level/microdose amounts. First, in a study aimed at radiotherapeutic evaluation of the human somatostatin receptor (hSSTr2) agonist [90Y]Y-DOTATOC, it was found that the agonist impaired immune function in humans (Barsegian et al., 2015). Hence, it cannot be ruled out at this point that other somatostatin-related imaging agents also have effects on the immune system, and consequently this imaging agent might not be suitable to in vivo track hSSTr2 reporter expressing adoptively transferred T-cells. Moreover, a recent study reported that immunoPET performed to in vivo track adoptively transferred T-cells in tumor-bearing mice impacted on immunotherapy outcome (Mayer et al., 2018). The authors used 89Zr-DFO-conjugated anti-CD7 and anti-CD2 antibody fragments (F(ab’)2), respectively, to quantify adoptively transferred T-cell populations in tumors. While they did not find any impact of both imaging tracers on T-cells in vitro, they found that the anti-CD2 radiotracer caused severe T-cell depletion and abrogated the effects of the adoptively transferred T-cell immunotherapy (the anti-CD7 radiotracer performed as expected and had no impact on adoptively transferred T-cells). The amounts of radiolabeled antibody fragment used in this study were ∼9 μmol/kg [1 mg/kg F(ab’)2], which was, for example, about five times less compared to what was used in the seminal immunoPET study that originally developed the anti-CD8 cis-diabody (∼50 μmol/kg ≈ 3 mg/kg), which is now in clinical development (see Section “In vivo Imaging of T-Cell Populations”). From these two examples follows clearly that great care must be taken with imaging agents in the context of immunology even when used at amounts generally accepted to be in the range of what normally constitutes tracer levels/microdoses, because there can be effects on immune cells and their functions in vivo, depending on the chosen imaging target. Furthermore, this is strong evidence that in vitro experimentation might not be indicative of such effects. Moreover, this is also an argument for the need of careful comprehensive in vivo validation experiments in relevant animal models during the development of imaging agents, irrespective of whether envisaged to aid immunotherapy development or intended for future immunotherapy monitoring in the clinics.
Immunogenicity and Contrast Are Linked in Reporter Gene Applications
For in vivo tracking of cell-based immunotherapies using reporter genes, immunogenicity of the reporter represents another very important aspect. It is linked to the achievable contrast at different body locations, which we explain in the following. For best contrast, a foreign reporter would appear ideal as it is expressed nowhere in the host organism guaranteeing good contrast. In animal disease models, such reporters are, for example, fluorescent proteins, luciferases (Mezzanotte et al., 2017) or the PET reporter herpes simplex virus 1 thymidine kinase (HSV1-tk) (Gambhir et al., 2000; Yaghoubi and Gambhir, 2006; Likar et al., 2009). All of them provide excellent contrast in vivo with varying sensitivities and spatial resolutions depending on the imaging modality used to probe them (cf. Figure 1). However, all of them are proteins foreign to mammals and consequently any cell expressing them in mammals can be detected and cleared by an intact host immune system (Figure 8). While this might represent only a minor issue if heavily immunocompromised animals are used, for example in human tumor xenograft models, it cannot be ignored in syngeneic models or the human clinical setting. While the foreign reporter (HSV1-tk) was used in the first proof-of-principle clinical reporter gene imaging study (Keu et al., 2017), this was performed in the setting of late-stage glioblastoma in heavily pre-treated patients, all of whom died within a year of the study start. In fact, immunogenicity of HSV1-tk has been well documented (Berger et al., 2006) and consequently HSV1-tk has been ruled out for reporter gene-afforded routine cell tracking of adoptive cell-based immunotherapies in humans; it should also not be considered for research in preclinical syngeneic models.
Figure 8. Simplified cartoon illustrating one way of cytotoxic T-cells to recognize foreign reporter antigens. A variety of immune recognition mechanisms exist in mammals as part of their innate and adaptive immune system. Here, as a simplified example, recognition of antigen-presenting MHC class I molecules on target cells by a cytotoxic CD8+ T-cells is visualized. The TCR (orange) of the cytotoxic CD8+ T-cells recognizes foreign antigen presented on host MHC class I molecules (light gray with red foreign antigen) but not host antigen on host MHC class I molecules (green: antigen from host reporter; black: any other host antigen). Foreign MHC class I molecules are also recognized by CD8+ T-cells. The T-cell co-receptor CD8 (dark gray) binds to MHC class I molecules upon TCR binding and the overall process activates CD8+ T-cells. CD8+ T-cell action results in granzyme and perforin release, and consequent killing of the corresponding target cell. Several mechanisms ensure that host antigens are not recognized; they include deletion of self-recognizing T-cells and tolerance conferred by regulatory T-cells. This simplified scheme demonstrates the importance to employ host reporters in experiments involving species with intact adaptive immunity.
Immunogenicity issues can best be overcome by using host reporter proteins (Table 2) that are normally endogenously expressed in the organism of interest. Importantly, these host reporters should be endogenously expressed in only a very limited number of host tissues, only in tissues where signals do not interfere with the experimental goals, and ideally at low levels to ensure favorable contrast in adjacent organs (cf. different background patterns in Figure 5).
Mammalian NIS has been found to be useful as a radionuclide reporter if used together with non-iodine radiotracers, which results in better signal-to-background (Diocou et al., 2017). Generally in mammals, NIS is endogenously expressed at high levels in the thyroid gland and at lower levels in few extrathyroidal tissues (salivary glands, mammary glands, stomach and small intestine, testes) (Portulano et al., 2014). This means that for cell tracking applications in other organs the host reporter gene NIS provides excellent signal-to-background ratios when exogenously expressed in cells of interest. NIS has been used to track many different cell types preclinically (Sieger et al., 2003; Groot-Wassink et al., 2004; Che et al., 2005; Dingli et al., 2006; Merron et al., 2007; Terrovitis et al., 2008; Carlson et al., 2009; Higuchi et al., 2009; Fruhwirth et al., 2014; Diocou et al., 2017) including stem cell and CAR-T-cell therapies (Emami-Shahri et al., 2018; Kurtys et al., 2018; Ashmore-Harris et al., 2019). It has not yet been used in the clinic but due to its favorable properties and the readily available corresponding radiotracers for both PET and SPECT for its detection, it is a promising candidate for use in future clinical trials.
The human somatostatin receptor subtype 2 (hSSTr2) is another reporter with some potential for cell tracking using clinically approved PET tracers based on somatostatin analogs [e.g. [68Ga]Ga-DOTATATE (antagonist) or [68Ga]Ga-DOTATOC (agonist)] and it has been used preclinically for CAR-T tracking (Zhang et al., 2011; Vedvyas et al., 2016). A significant pitfall of hSSTr2 use as a reporter for immunotherapies is that it is expressed endogenously on various immune cell types including T-cells, B-cells and macrophages (Elliott et al., 1999). This negatively affects imaging specificity in immunocompetent models and likely humans. It is also expressed in the cerebrum, kidneys and also the gastrointestinal tract (Yamada et al., 1992). Moreover, it was found that the hSSTr2 agonist [90Y]Y-DOTATOC impaired immune function in humans (Barsegian et al., 2015). Whilst radioactive contrast agent concentrations are very low, it cannot be ruled out without further studies that somatostatin analogs and their derivatives might also impair some immune system functions. Another important caveat of hSSTr2 use as a reporter is that it internalizes upon substrate binding (Oomen et al., 2001; Cescato et al., 2006) and this is likely to affect the detection sensitivity of hSSTr2-expressing cells through reduction of its steady-state concentration on the plasma membrane.
A very promising host reporter gene with very limited endogenous expression is PSMA (Castanares et al., 2014). It has been developed alongside PET radiotracers that were originally intended for molecular imaging of PSMA-expressing prostate cancer. PSMA is a type II plasma membrane protein that can be internalized upon ligand binding. It has a short cytoplasmic N-terminal tail, which is responsible for its internalization (Rajasekaran et al., 2003). N-terminally modified PSMA variants, PSMAW2G and tPSMAN9del, were recently designed to prevent receptor internalization and to increase PSMA surface expression with the authors hypothesizing that would increase PET radiotracer binding and overall imaging sensitivity (Minn et al., 2019). Moreover, the tPSMAN9del variant lacks putative intracellular signaling motifs rendering it less likely to affect normal T-cell function. tPSMAN9del was used as a reporter to track CAR-T cells in a preclinical model of acute lymphoblastic leukemia by PET imaging with the radiotracer [18F]DCFPyL (Minn et al., 2019). [18F]DCFPyL is a radiotracer for PSMA, in fact a high-affinity PSMA ligand that can be produced in good quantities and with high specific activity (Ravert et al., 2016), and it has already been used in humans and is currently also in a phase II clinical trial for the detection of metastatic prostate cancer via PSMA (NCT03173924), another application that requires the detection of small amounts of cells.
Despite some notable advances in recent years, there is still significant room for improvement to optimize host reporter/tracer pairs, for example to improve signal-to-background, tailor them better to application in specific immune cells, and enhance the steady-state concentrations in traceable cells and thereby cell tracking sensitivity.
Conclusion and Outlook
The development of both molecular and cell-based immunotherapies can be greatly assisted by in vivo imaging, which provided valuable insight into spatiotemporal dynamics of immune responses and the complex interactions of the tumor microenvironment. In vivo imaging has earned itself a place among the indispensable tools for immunotherapy development at preclinical stages, and many available molecular imaging technologies can be used for understanding the mechanisms governing immunotherapy function and to improve immunotherapy efficacy and safety. Newly identified relevant targets will require some degree of molecular imaging development to generate the relevant contrast agent, but multiple robust methodologies for turning target-specific biomolecules such as antibodies, antibody fragments/derivatives or peptides into contrast agents are already available. Various molecular imaging techniques aiding immunotherapy are currently at the brink of clinical application, mostly still in explorative studies, some in clinical trials, and they focus on early response monitoring with response prediction representing a major goal. Individual response monitoring at the patient level is particularly important as responses can be heterogeneous between lesions within the same individual and also between patients, rendering this a potential routine clinical application of molecular imaging in the future. A currently somewhat underexplored area is immunotherapy presence and action at secondary lesions. Preclinically, traceable cancer models would be very useful tools in this context, enabling in vivo quantification of therapy arrival and perhaps therapy action at the intended target sites. Clinically, molecular imaging will help inform on lesion heterogeneity as well as potential response heterogeneity in patients.
Cell-based immunotherapies represent an area in need of further development to unleash their full potential and render them more efficacious, safer to use, and more widely applicable. Therefore, it remains highly beneficial to better understand their in vivo distribution, behavior and fate, and to use such non-invasively acquired information to elucidate and tailor their mechanisms of action. Cell-based immunotherapies can be classified into two groups that (a) do not need genetic engineering for efficacy, and those that (b) fundamentally require genetic engineering (e.g. CAR-T, TCR-T).
The first group, which includes immunotherapies based on e.g. TILs and γδ T-cells, the choice between direct and indirect cell labeling depends on the precise research question, practicalities and of course whether clinical translation of the tracking methodology is envisaged and for what purpose. Implementing genetic engineering to enable indirect cell labeling to these therapies adds a significant regulatory burden and it is certainly difficult to justify the additional efforts required for the sole purpose of in vivo cell therapy tracking. Consequently, recently developed direct cell labeling approaches involving cell tracking by PET (e.g. γδ T-cell labeling with [89Zr]Zr-oxine) are promising tools despite their obvious limitations caused by the cell labeling methodology itself (label efflux, label dilution, complex dosimetry, limited observation times). However, the situation is likely to improve through the development of total-body PET, which has been reported to be 40-times more sensitive than conventional PET (Cherry et al., 2018). This sensitivity advantage could either be invested into faster PET scanning or scanning with much less radioactivity. In vivo cell tracking studies using this new technology will reveal to what extent the sensitivity advantage of total-body PET can be used to extend the tracking time of directly labeled cells.
For cell-based immunotherapies that require genetic engineering, an immunocompatible host reporter gene can be implemented without adding to the regulatory burden. Indirect cell labeling is clearly advantageous over direct cell labeling in such cases as it enables longer-term monitoring, reflects cell proliferation/survival, and avoids complex dosimetry considerations during cell labeling. Genetic engineering technologies have been steadily advanced and include now viral as well as non-viral delivery methods as well as site-specific integration via gene editing approaches (Figure 3C). Moreover, the reporter gene can be co-delivered with other relevant components during genetic engineering of the cells as was previously demonstrated rendering CAR-T traceable by SPECT or PET (Emami-Shahri et al., 2018; Kurtys et al., 2018; Minn et al., 2019). If contrast agents can be used that match the reporter and are already clinically approved, this is obviously beneficial. Importantly, it is unlikely that a one-fits-all approach across cancers involving only one immunocompatible host reporter gene is viable. More likely, various cancers at different body locations with varying endogenous host reporter expression levels will be targeted by genetically engineered cell-based immunotherapies in which the targeting moiety as well as the host reporter must be tailored. Undoubtedly, more research into host reporter/contrast agent pairs is warranted to provide the most flexible tools to render these immunotherapies in vivo traceable with best contrast in a quantitative manner.
In summary, we described how in vivo imaging can aid the development of molecular and cell-based anti-cancer immunotherapies and explained a variety of methodological and experimental design aspects. Notably, these concepts can also be extrapolated to immunotherapies intended to treat other conditions, for example, in the fields of regenerative medicine (Naumova et al., 2014), transplantation (Afzali et al., 2013; Safinia et al., 2016), diabetes type I (Alhadj Ali et al., 2017; Smith and Peakman, 2018), multiple sclerosis (Chataway et al., 2018), and infectious diseases (Hotchkiss and Moldawer, 2014).
Author Contributions
GF contributed the article concept. Both authors compiled the figures, wrote the manuscript, and contributed to manuscript revision, read, and approved the submitted version.
Funding
The authors received support from the Cancer Research UK[cpsenter](Multidisciplinary Project Award C48390/A21153 to GF) and EPSRC and GE Healthcare (Ph.D. studentship to MI). Further they are supported by the King’s College London and UCL Comprehensive Cancer Imaging Centre, funded by Cancer Research UK and EPSRC; the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London; and the Wellcome/EPSRC Centre for Medical Engineering at King’s College London [WT 203148/Z/16/Z]. Institutional Open Access funds to support article publication were also received. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the DoH.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Professsor Paul Marsden (King’s College London) for his very helpful suggestions in editing the manuscript. Figures 3, 4, 8 contain graphics that were created using software accessed on BioRender.com via MI’s paid subscription.
Abbreviations
ATMP, advanced therapy medicinal products; BLI, bioluminescence imaging a preclinical imaging technology; CAR, chimeric antigen receptor an artificial molecule; CAR-T, chimeric antigen receptor T-cell therapy; CD2, cluster of differentiation 2 a cell adhesion molecule found on the surface of T-cells and natural killer cells also known as T-cell surface antigen T11/Leu-5, LFA-2 LFA-3 receptor erythrocyte receptor and rosette receptor; CD3, cluster of differentiation 3 a T-cell co-receptor; CD4, cluster of differentiation 4 a glycoprotein found on the surface of immune cells such as T helper cells, monocytes macrophages and dendritic cells; CD7, cluster of differentiation 7 found on thymocytes and mature T-cells; CD8, cluster of differentiation 8 a glycoprotein that serves as a co-receptor for the T-cell receptor predominantly expressed on the surface of cytotoxic T-cells; CD19, cluster of differentiation 19 also known as B-Lymphocyte Surface Antigen B4T-Cell Surface Antigen Leu-12 and CVID3 is a transmembrane protein; CD25, cluster of differentiation 25 also known as IL-2 receptor alpha chaina part of the high-affinity IL receptor; CT, computed tomography a whole-body imaging technology employing X–rays; CTLA-4, cytotoxic T-lymphocyte -associated protein 4 also known as cluster of differentiation 152 a molecule that is part of an immune checkpoint axis; DC, dendritic cell a type of immune cell; DFO, desferrioxamine a chelator for metal ions; FDA, U.S. Food and Drug Administration [18F]FHBG 9-(4-[18F]-Fluoro-3-[hydroxymethyl]butyl)guaninea radiotracer for the reporter gene HSV1-tk; FMT, fluorescence mediated tomography a fluorescence-based whole-body imaging technology; HMPAO, hexamethylene-propyleneamine oxime an agent to label cells directly with 99mTc; HSV1-tk, Herpes Simplex Virus 1 thymidine kinase an enzyme that can be exploited as a reporter gene; ICI, immune checkpoint inhibitor; IL, IL, immune mediators specified by a number to identify the individual molecule (e.g. IL-2 and IL-12) IrAE immune related adverse event; LAG-3, lymphocyte-activation gene 3 an immune checkpoint molecule; MRI, magnetic resonance imaging an imaging technology; MSOT, multispectral optoacoustic tomography an advanced form of PAT imaging; NET, norepinephrine transporter a mammalian protein that can be used as a host reporter gene; NIS, sodium iodide symporter a mammalian protein that can be used as a host reporter gene; NK, natural killer cell a type of immune cell; OCT, optical coherence tomography an optical imaging technology; OPT, optical projection tomography a preclinical optical imaging technology; OVA, ovalbumin main protein found in egg white widely used as a model antigen in T-cell biology Numerous mouse cancer models have been modified to express OVA to aid in enhancing and tracking tumor-specific T-cell responses; PAT, photoacoustic tomography an imaging technology; PD-1, programed cell death protein 1 an immune checkpoint molecule; PD-L1, programed death ligand 1 also known as cluster of differentiation 274 or B7 homolog 1 (B7-H1) constitutes an immune checkpoint axis together with PD-1; PET, positron emission tomography a whole-body imaging technology detecting γ-photons produced by the annihilation of positrons stemming from the decay of certain radioisotopes; PSMA, prostate-specific membrane antigen a glutamate carboxypeptidase 2 also known as folate hydrolase 1; qPCR, quantitative polymerase chain reaction an analysis technique to determine specific DNA amounts in biological samples; RSOM, raster-scanning optoacoustic mesoscopy a preclinical imaging technology exploiting the photoacoustic effect; SPECT, single photon emission computed tomography a whole-body imaging technology detecting radioisotope location in 3D; TCR, T-cell receptor a multi-protein complex responsible for many T-cell activation mechanisms; TCR-T, T-cell receptor-modified T-cell therapy; TIL, tumor infiltrating lymphocyte; US, ultrasound imaging a cheap standard imaging technology.
References
Adonai, N., Adonai, N., Nguyen, K. N., Walsh, J., Iyer, M., Toyokuni, T., et al. (2002). Ex vivo cell labeling with 64Cu-pyruvaldehyde-bis(N4-methylthiosemicarbazone) for imaging cell trafficking in mice with positron-emission tomography. Proc. Natl. Acad. Sci. U.S.A. 99, 3030–3035. doi: 10.1073/pnas.052709599
Afzali, B., Edozie, F. C., Fazekasova, H., Scotta, C., Mitchell, P. J., Canavan, J. B., et al. (2013). Comparison of regulatory T cells in hemodialysis patients and healthy controls, implications for cell therapy in transplantation. Clin. J. Am. Soc. Nephrol. 8, 1396–1405. doi: 10.2215/CJN.12931212
Alam, I. S., Mayer, A. T., Sagiv-Barfi, I., Wang, K., Vermesh, O., Czerwinski, D. K., et al. (2018). Imaging activated T cells predicts response to cancer vaccines. J. Clin. Invest. 128, 2569–2580. doi: 10.1172/JCI98509
Alanentalo, T., Loren, C. E., Larefalk, A., Sharpe, J., Holmberg, D., and Ahlgren, U. (2008). High-resolution three-dimensional imaging of islet-infiltrate interactions based on optical projection tomography assessments of the intact adult mouse pancreas. J. Biomed. Opt. 13:054070. doi: 10.1117/1.3000430
Alhadj Ali, M., Liu, Y. F., Arif, S., Tatovic, D., Shariff, H., Gibson, V. B., et al. (2017). Metabolic and immune effects of immunotherapy with proinsulin peptide in human new-onset type 1 diabetes. Sci. Transl. Med. 9:eaaf7779. doi: 10.1126/scitranslmed.aaf7779
Alizadeh, A. A., Aranda, V., Bardelli, A., Blanpain, C., Bock, C., Borowski, C., et al. (2015). Toward understanding and exploiting tumor heterogeneity. Nat. Med. 21, 846–853. doi: 10.1038/nm.3915
Andreyev, A., and Celler, A. (2011). Dual-isotope PET using positron-gamma emitters. Phys. Med, Biol. 56, 4539–4556. doi: 10.1088/0031-9155/56/14/020
Anon (2014). UNITED STATES of America, Appellee v. REGENERATIVE SCIENCES, LLC, A Corporation, et al., Appellants. Toronto: Thomson Reuters.
Annovazzi, A., D’Alessandria, C., Bonanno, E., Mather, S. J., Cornelissen, B., van de Wiele, C., et al. (2006). Synthesis of 99mTc-HYNIC-interleukin-12, a new specific radiopharmaceutical for imaging T lymphocytes. Eur. J. Nucl. Med. Mol. Imaging 33, 474–482. doi: 10.1007/s00259-005-0001-6
Arranz, A., Dong, D., Zhu, S., Savakis, C., Tian, J., and Ripoll, J. (2014). In-vivo optical tomography of small scattering specimens, time-lapse 3D imaging of the head eversion process in Drosophila melanogaster. Sci. Rep. 4:7325. doi: 10.1038/srep07325
Ashmore-Harris, C., Blackford, S. J., Grimsdell, B., Kurtys, E., Glatz, M. C., Rashid, T. S., et al. (2019). Reporter gene-engineering of human induced pluripotent stem cells during differentiation renders in vivo traceable hepatocyte-like cells accessible. Stem Cell Res. 41:101599. doi: 10.1016/j.scr.2019.101599
Aswendt, M., Vogel, S., Schäfer, C., Jathoul, A., Pule, M., and Hoehn, M. (2019). Quantitative in vivo dual-color bioluminescence imaging in the mouse brain. Neurophotonics 6, 1–11.
Bansal, A., Pandey, M. K., Demirhan, Y. E., Nesbitt, J. J., Crespo-Diaz, R. J., Terzic, A., et al. (2015). Novel (89)Zr cell labeling approach for PET-based cell trafficking studies. EJNMMI Res. 5:19. doi: 10.1186/s13550-015-0098-y
Barat, B., Kenanova, V. E., Olafsen, T., and Wu, A. M. (2011). Evaluation of two internalizing carcinoembryonic antigen reporter genes for molecular imaging. Mol. Imaging Biol. 13, 526–535. doi: 10.1007/s11307-010-0375-0
Barsegian, V., Hueben, C., Mueller, S. P., Poeppel, T. D., Horn, P. A., Bockisch, A., et al. (2015). Impairment of lymphocyte function following yttrium-90 DOTATOC therapy. Cancer Immunol. Immunother. 64, 755–764. doi: 10.1007/s00262-015-1687-3
Bassi, A., Fieramonti, L., D’Andrea, C., Mione, M., and Valentini, G. (2011). In vivo label-free three-dimensional imaging of zebrafish vasculature with optical projection tomography. J. Biomed. Opt. 16:100502. doi: 10.1117/1.3640808
Basu, S., Hess, S., Nielsen Braad, P. E., Olsen, B. B., Inglev, S., and Hoilund-Carlsen, P. F. (2014). The Basic Principles of FDG-PET/CT Imaging. PET Clin. 9, 355–370. doi: 10.1016/j.cpet.2014.07.006
Bates, A., Power, C. A., and David, V. S. (2019). Goliath, the structure, function, and clinical prospects of antibody fragments. Antibodies 8:28. doi: 10.3390/antib8020028
Bensch, F., van der Veen, E. L., Lub-de Hooge, M. N., Jorritsma-Smit, A., Boellaard, R., Kok, I. C., et al. (2018). (89)Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat. Med. 24, 1852–1858. doi: 10.1038/s41591-018-0255-8
Berger, C., Flowers, M. E., Warren, E. H., and Riddell, S. R. (2006). Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood 107, 2294–2302. doi: 10.1182/blood-2005-08-3503
Bhargava, K. K., Gupta, R. K., Nichols, K. J., and Palestro, C. J. (2009). In vitro human leukocyte labeling with (64)Cu, an intraindividual comparison with (111)In-oxine and (18)F-FDG. Nucl. Med. Biol. 36, 545–549. doi: 10.1016/j.nucmedbio.2009.03.001
Bressan, R. B., Dewari, P. S., Kalantzaki, M., Gangoso, E., Matjusaitis, M., Garcia-Diaz, C., et al. (2017). Efficient CRISPR/Cas9-assisted gene targeting enables rapid and precise genetic manipulation of mammalian neural stem cells. Development 144, 635–648. doi: 10.1242/dev.140855
Buck, M. D., O’Sullivan, D., Klein Geltink, R. I., Curtis, J. D., Chang, C. H., Sanin, D. E., et al. (2016). Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166, 63–76. doi: 10.1016/j.cell.2016.05.035
Cal-Gonzalez, J., Herraiz, J. L., Espana, S., Corzo, P. M., Vaquero, J. J., Desco, M., et al. (2013). Positron range estimations with PeneloPET. Phys. Med. Biol. 58, 5127–5152. doi: 10.1088/0031-9155/58/15/5127
Cal-Gonzalez, J., Lage, E., Herranz, E., Vicente, E., Udias, J. M., Moore, S. C., et al. (2015). Simulation of triple coincidences in PET. Phys. Med. Biol. 60, 117–136. doi: 10.1088/0031-9155/60/1/117
Carlson, S. K., Classic, K. L., Hadac, E. M., Dingli, D., Bender, C. E., Kemp, B. J., et al. (2009). Quantitative molecular imaging of viral therapy for pancreatic cancer using an engineered measles virus expressing the sodium-iodide symporter reporter gene. AJR Am. J. Roentgenol. 192, 279–287. doi: 10.2214/AJR.08.1205
Castanares, M. A., Mukherjee, A., Chowdhury, W. H., Liu, M., Chen, Y., Mease, R. C., et al. (2014). Evaluation of prostate-specific membrane antigen as an imaging reporter. J. Nucl. Med. 55, 805–811. doi: 10.2967/jnumed.113.134031
Caswell, D. R., and Swanton, C. (2017). The role of tumour heterogeneity and clonal cooperativity in metastasis, immune evasion and clinical outcome. BMC Med. 15:133. doi: 10.1186/s12916-017-0900-y
Catana, C. (2017). Principles of Simultaneous PET/MR Imaging. Magn. Reson. Imaging Clin. N. Am. 25, 231–243. doi: 10.1016/j.mric.2017.01.002
Cekanova, M., and Rathore, K. (2014). Animal models and therapeutic molecular targets of cancer, utility and limitations. Drug Des. Devel. Ther. 8, 1911–1921. doi: 10.2147/DDDT.S49584
Cescato, R., Schulz, S., Waser, B., Eltschinger, V., Rivier, J. E., Wester, H. J., et al. (2006). Internalization of sst2, sst3, and sst5 receptors, effects of somatostatin agonists and antagonists. J. Nucl. Med. 47, 502–511.
Charoenphun, P., Meszaros, L. K., Chuamsaamarkkee, K., Sharif-Paghaleh, E., Ballinger, J. R., Ferris, T. J., et al. (2015). [(89)Zr]oxinate4 for long-term in vivo cell tracking by positron emission tomography. Eur. J. Nucl. Med. Mol. Imaging 42, 278–287. doi: 10.1007/s00259-014-2945-x
Chataway, J., Martin, K., Barrell, K., Sharrack, B., Stolt, P., Wraith, D. C., et al. (2018). Effects of ATX-MS-1467 immunotherapy over 16 weeks in relapsing multiple sclerosis. Neurology 90, e955–e962. doi: 10.1212/WNL.0000000000005118
Chaudhuri, T. R., Rogers, B. E., Buchsbaum, D. J., Mountz, J. M., and Zinn, K. R. (2001). A noninvasive reporter system to image adenoviral-mediated gene transfer to ovarian cancer xenografts. Gynecol. Oncol. 83, 432–438. doi: 10.1006/gyno.2001.6333
Che, J., Doubrovin, M., Serganova, I., Ageyeva, L., Zanzonico, P., and Blasberg, R. (2005). hNIS-IRES-eGFP dual reporter gene imaging. Mol. Imaging 4, 128–136.
Cheddad, A., Svensson, C., Sharpe, J., Georgsson, F., and Ahlgren, U. (2012). Image processing assisted algorithms for optical projection tomography. IEEE Trans. Med. Imaging 31, 1–15. doi: 10.1109/TMI.2011.2161590
Cherry, S. R., Jones, T., Karp, J. S., Qi, J., Moses, W. W., and Badawi, R. D. (2018). Total-body PET, maximizing sensitivity to create new opportunities for clinical research and patient care. J. Nucl. Med. 59, 3–12. doi: 10.2967/jnumed.116.184028
Cho, I. K., Moran, S. P., Paudyal, R., Piotrowska-Nitsche, K., Cheng, P.-H., Zhang, X., et al. (2014). Longitudinal monitoring of stem cell grafts in vivo using magnetic resonance imaging with inducible maga as a genetic reporter. Theranostics 4, 972–989. doi: 10.7150/thno.9436
Chuang, K. H., Wang, H. E., Cheng, T. C., Tzou, S. C., Tseng, W. L., Hung, W. C., et al. (2010). Development of a universal anti-polyethylene glycol reporter gene for noninvasive imaging of PEGylated probes. J. Nucl. Med. 51, 933–941. doi: 10.2967/jnumed.109.071977
Claus, C., Ferrara, C., Xu, W., Sam, J., Lang, S., Uhlenbrock, F., et al. (2019). Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci. Transl. Med. 11:eaav5989. doi: 10.1126/scitranslmed.aav5989
Cohen, B., Dafni, H., Meir, G., Harmelin, A., and Neeman, M. (2005). Ferritin as an endogenous MRI reporter for noninvasive imaging of gene expression in C6 glioma tumors. Neoplasia 7, 109–117. doi: 10.1593/neo.04436
Conti, M., and Eriksson, L. (2016). Physics of pure and non-pure positron emitters for PET, a review and a discussion. EJNMMI Phys. 3:8. doi: 10.1186/s40658-016-0144-5
Dai, G., Levy, O., and Carrasco, N. (1996). Cloning and characterization of the thyroid iodide transporter. Nature 379, 458–460. doi: 10.1038/379458a0
Darvin, P., Toor, S. M., Sasidharan Nair, V., and Elkord, E. (2018). Immune checkpoint inhibitors, recent progress and potential biomarkers. Exp. Mol. Med. 50:165. doi: 10.1038/s12276-018-0191-1
de Vries, E. F., Roca, M., Jamar, F., Israel, O., and Signore, A. (2010). Guidelines for the labelling of leucocytes with (99m)Tc-HMPAO. Inflammation/Infection Taskgroup of the European Association of Nuclear Medicine. Eur. J. Nucl. Med. Mol. Imaging 37, 842–848. doi: 10.1007/s00259-010-1394-4
Deleye, S., Van Holen, R., Verhaeghe, J., Vandenberghe, S., Stroobants, S., and Staelens, S. (2013). Performance evaluation of small-animal multipinhole muSPECT scanners for mouse imaging. Eur. J. Nucl. Med Mol Imaging. 40, 744–758. doi: 10.1007/s00259-012-2326-2
Deliolanis, N. C., Ale, A., Morscher, S., Burton, N. C., Schaefer, K., Radrich, K., et al. (2014). Deep-tissue reporter-gene imaging with fluorescence and optoacoustic tomography, a performance overview. Mol. Imaging Biol. 16, 652–660. doi: 10.1007/s11307-014-0728-1
Dhawan, R. T., and Peters, A. M. (2014). Withdrawal of indium-111, implications for white-cell imaging. The nuclear medicine community must act. Nucl. Med. Commun. 35, 789–791. doi: 10.1097/mnm.0000000000000138
Di Gialleonardo, V., Signore, A., Glaudemans, A. W., Dierckx, R. A., and De Vries, E. F. (2012). N-(4-18F-fluorobenzoyl)interleukin-2 for PET of human-activated T lymphocytes. J. Nucl. Med. 53, 679–686. doi: 10.2967/jnumed.111.091306
Dingli, D., Kemp, B. J., O’Connor, M. K., Morris, J. C., Russell, S. J., and Lowe, V. J. (2006). Combined I-124 positron emission tomography/computed tomography imaging of NIS gene expression in animal models of stably transfected and intravenously transfected tumor. Mol. Imaging Biol. 8, 16–23. doi: 10.1007/s11307-005-0025-0
Diocou, S., Volpe, A., Jauregui-Osoro, M., Boudjemeline, M., Chuamsaamarkkee, K., Man, F., et al. (2017). [(18)F]tetrafluoroborate-PET/CT enables sensitive tumor and metastasis in vivo imaging in a sodium iodide symporter-expressing tumor model. Sci. Rep. 7:946. doi: 10.1038/s41598-017-01044-4
Dohan, O., De la Vieja, A., Paroder, V., Riedel, C., Artani, M., Reed, M., et al. (2003). The sodium/iodide Symporter (NIS), characterization, regulation, and medical significance. Endocr. Rev. 24, 48–77. doi: 10.1210/er.2001-0029
Dunlap, P. (2014). Biochemistry and genetics of bacterial bioluminescence. Adv. Biochem. Eng. Biotechnol. 144, 37–64. doi: 10.1007/978-3-662-43385-0_2
Edmonds, S., Volpe, A., Shmeeda, H., Parente-Pereira, A. C., Radia, R., Baguna-Torres, J., et al. (2016). Exploiting the metal-chelating properties of the drug cargo for in vivo positron emission tomography imaging of liposomal nanomedicines. ACS Nano 10, 10294–10307. doi: 10.1021/acsnano.6b05935
Eisenhauer, E. A., Therasse, P., Bogaerts, J., Schwartz, L. H., Sargent, D., Ford, R., et al. (2009). New response evaluation criteria in solid tumours, revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247. doi: 10.1016/j.ejca.2008.10.026
Elliott, D. E., Li, J., Blum, A. M., Metwali, A., Patel, Y. C., and Weinstock, J. V. (1999). SSTR2A is the dominant somatostatin receptor subtype expressed by inflammatory cells, is widely expressed and directly regulates T cell IFN-gamma release. Eur. J. Immunol. 29, 2454–2463. doi: 10.1002/(sici)1521-4141(199908)29:08<2454::aid-immu2454>3.0.co;2-h
Emami-Shahri, N., Foster, J., Kashani, R., Gazinska, P., Cook, C., Sosabowski, J., et al. (2018). Clinically compliant spatial and temporal imaging of chimeric antigen receptor T-cells. Nat. Commun. 9:1081. doi: 10.1038/s41467-018-03524-1
European Medicines Agency. (2004). Position Paper on Non-Clinical Safety Studies to Support Clinical Trials with A Single Microdose. Amsterdam: European Medicines Agency
Farhadi, A., Ho, G. H., Sawyer, D. P., Bourdeau, R. W., and Shapiro, M. G. (2019). Ultrasound imaging of gene expression in mammalian cells. Science 365:1469. doi: 10.1126/science.aax4804
Farrar, C. T., Buhrman, J. S., Liu, G., Kleijn, A., Lamfers, M. L., McMahon, M. T., et al. (2015). Establishing the lysine-rich protein CEST reporter gene as a CEST MR imaging detector for oncolytic virotherapy. Radiology 275, 746–754. doi: 10.1148/radiol.14140251
Feng, Y., Roy, A., Masson, E., Chen, T. T., Humphrey, R., and Weber, J. S. (2013). Exposure-response relationships of the efficacy and safety of ipilimumab in patients with advanced melanoma. Clin. Cancer Res. 19, 3977–3986. doi: 10.1158/1078-0432.CCR-12-3243
Filonov, G. S., Piatkevich, K. D., Ting, L.-M., Zhang, J., Kim, K., and Verkhusha, V. V. (2011). Bright and stable near-infrared fluorescent protein for in vivo imaging. Nat. Biotechnol. 29:757. doi: 10.1038/nbt.1918
Fisher, B., Packard, B. S., Read, E. J., Carrasquillo, J. A., Carter, C. S., Topalian, S. L., et al. (1989). Tumor localization of adoptively transferred indium-111 labeled tumor infiltrating lymphocytes in patients with metastatic melanoma. J. Clin. Oncol. 7, 250–261. doi: 10.1200/jco.1989.7.2.250
Fowler, A. V., and Zabin, I. (1977). The amino acid sequence of beta-galactosidase of Escherichia coli. Proc. Natl Acad. Sci. U. S. A. 74, 1507–1510. doi: 10.1073/pnas.74.4.1507
Freise, A. C., and Wu, A. M. (2015). In vivo imaging with antibodies and engineered fragments. Mol. Immunol. 67, 142–152. doi: 10.1016/j.molimm.2015.04.001
Freise, A. C., Zettlitz, K. A., Salazar, F. B., Lu, X., Tavare, R., and Wu, A. M. (2017). ImmunoPET Imaging of Murine CD4(+) T Cells Using Anti-CD4 Cys-Diabody, Effects of Protein Dose on T Cell Function and Imaging. Mol. Imaging Biol. 19, 599–609. doi: 10.1007/s11307-016-1032-z
Fruhwirth, G. O., Diocou, S., Blower, P. J., Ng, T., and Mullen, G. E. (2014). A whole-body dual-modality radionuclide optical strategy for preclinical imaging of metastasis and heterogeneous treatment response in different microenvironments. J. Nucl. Med. 55, 686–694. doi: 10.2967/jnumed.113.127480
Fukuda, A., Honda, S., Fujioka, N., Sekiguchi, Y., Mizuno, S., Miwa, Y., et al. (2019). Non-invasive in vivo imaging of UCP1 expression in live mice via near-infrared fluorescent protein iRFP720. PLoS One 14:e0225213. doi: 10.1371/journal.pone.0225213
Gambhir, S. S., Bauer, E., Black, M. E., Liang, Q., Kokoris, M. S., Barrio, J. R., et al. (2000). A mutant herpes simplex virus type 1 thymidine kinase reporter gene shows improved sensitivity for imaging reporter gene expression with positron emission tomography. Proc. Natl. Acad. Sci. U.S.A. 97, 2785–2790. doi: 10.1073/pnas.97.6.2785
Garcia, J., Tang, T., and Louie, A. Y. (2015). Nanoparticle-based multimodal PET/MRI probes. Nanomedicine 10, 1343–1359. doi: 10.2217/nnm.14.224
Gawne, P., Man, F., Fonslet, J., Radia, R., Bordoloi, J., Cleveland, M., et al. (2018). Manganese-52, applications in cell radiolabelling and liposomal nanomedicine PET imaging using oxine (8-hydroxyquinoline) as an ionophore. Dalton. Trans. 47, 9283–9293. doi: 10.1039/c8dt00100f
Genove, G., DeMarco, U., Xu, H., Goins, W. F., and Ahrens, E. T. (2005). A new transgene reporter for in vivo magnetic resonance imaging. Nat. Med. 11, 450–454. doi: 10.1038/nm1208
Germain-Genevois, C., Garandeau, O., and Couillaud, F. (2016). Detection of brain tumors and systemic metastases using NanoLuc and fluc for dual reporter imaging. Mol. Imaging Biol. 18, 62–69. doi: 10.1007/s11307-015-0864-2
Gibson, H. M., McKnight, B. N., Malysa, A., Dyson, G., Wiesend, W. N., McCarthy, C. E., et al. (2018). IFNgamma PET imaging as a predictive tool for monitoring response to tumor immunotherapy. Cancer Res. 78, 5706–5717. doi: 10.1158/0008-5472.CAN-18-0253
Gilad, A. A., McMahon, M. T., Walczak, P., Winnard, P. T. Jr., Raman, V., Van Laarhoven, H. W., et al. (2007). Artificial reporter gene providing MRI contrast based on proton exchange. Nat. Biotechnol. 25:217. doi: 10.1038/nbt1277
Girgis, M. D., Olafsen, T., Kenanova, V., McCabe, K. E., Wu, A. M., and Tomlinson, J. S. (2011). Targeting CEA in pancreas cancer xenografts with a mutated scFv-Fc antibody fragment. EJNMMI Res. 1, 24. doi: 10.1186/2191-219X-1-24
Gleave, J. A., Wong, M. D., Dazai, J., Altaf, M., Henkelman, R. M., Lerch, J. P., et al. (2012). Neuroanatomical phenotyping of the mouse brain with three-dimensional autofluorescence imaging. Physiol. Genomics 44, 778–785. doi: 10.1152/physiolgenomics.00055.2012
Goldman, S. J., Chen, E., Taylor, R., Zhang, S., Petrosky, W., Reiss, M., et al. (2011). Use of the ODD-luciferase transgene for the non-invasive imaging of spontaneous tumors in mice. PLoS One 6:e18269. doi: 10.1371/journal.pone.0018269
Gomez-Cuadrado, L., Tracey, N., Ma, R., Qian, B., and Brunton, V. G. (2017). Mouse models of metastasis, progress and prospects. Dis. Model Mech. 10, 1061–1074. doi: 10.1242/dmm.030403
Graves, E. E., Weissleder, R., and Ntziachristos, V. (2004). Fluorescence molecular imaging of small animal tumor models. Curr. Mol. Med. 4, 419–430. doi: 10.2174/1566524043360555
Green, O., Gnaim, S., Blau, R., Eldar-Boock, A., Satchi-Fainaro, R., and Shabat, D. (2017). Near-infrared dioxetane luminophores with direct chemiluminescence emission mode. J. Am. Chem. Soc. 139, 13243–13248. doi: 10.1021/jacs.7b08446
Greenwood, H. E., Nyitrai, Z., Mocsai, G., Hobor, S., and Witney, T. H. (2019). High throughput PET/CT imaging using a multiple mouse imaging system. J. Nucl. Med. 61, 292–297. doi: 10.2967/jnumed.119.228692
Griessinger, C. M., Kehlbach, R., Bukala, D., Wiehr, S., Bantleon, R., Cay, F., et al. (2014). In vivo tracking of Th1 cells by PET reveals quantitative and temporal distribution and specific homing in lymphatic tissue. J. Nucl. Med. 55, 301–307. doi: 10.2967/jnumed.113.126318
Griessinger, C. M., Maurer, A., Kesenheimer, C., Kehlbach, R., Reischl, G., Ehrlichmann, W., et al. (2015). 64Cu antibody-targeting of the T-cell receptor and subsequent internalization enables in vivo tracking of lymphocytes by PET. Proc. Natl. Acad. Sci. U.S.A 112, 1161–1166. doi: 10.1073/pnas.1418391112
Griffin, T. W., Brill, A. B., Stevens, S., Collins, J. A., Bokhari, F., Bushe, H., et al. (1991). Initial clinical study of indium-111-labeled clone 110 anticarcinoembryonic antigen antibody in patients with colorectal cancer. J. Clin. Oncol. 9, 631–640. doi: 10.1200/jco.1991.9.4.631
Griffith, K. D., Read, E. J., Carrasquillo, J. A., Carter, C. S., Yang, J. C., Fisher, B., et al. (1989). In vivo distribution of adoptively transferred indium-111-labeled tumor infiltrating lymphocytes and peripheral blood lymphocytes in patients with metastatic melanoma. J. Natl. Cancer Inst. 81, 1709–1717. doi: 10.1093/jnci/81.22.1709
Grimfors, G., Schnell, P. O., Holm, G., Johansson, B., Mellstedt, H., Pihlstedt, P., et al. (1989). Tumour imaging of indium-111 oxine-labelled autologous lymphocytes as a staging method in Hodgkin’s disease. Eur. J. Haematol. 42, 276–283. doi: 10.1111/j.1600-0609.1989.tb00112.x
Groot-Wassink, T., Aboagye, E. O., Wang, Y., Lemoine, N. R., Keith, W. N., and Vassaux, G. (2004). Noninvasive imaging of the transcriptional activities of human telomerase promoter fragments in mice. Cancer Res. 64, 4906–4911. doi: 10.1158/0008-5472.can-04-0426
Guo, Y., Hui, C.-Y., Liu, L., Zheng, H.-Q., and Wu, H.-M. (2019). Improved Monitoring of Low-Level Transcription in Escherichia coli by a β-Galactosidase α-Complementation System. Front. Microbiol. 10:1454. doi: 10.3389/fmicb.2019.01454
Gupta, S., Utoft, R., Hasseldam, H., Schmidt-Christensen, A., Hannibal, T. D., Hansen, L., et al. (2013). Global and 3D spatial assessment of neuroinflammation in rodent models of Multiple Sclerosis. PLoS One 8:e76330. doi: 10.1371/journal.pone.0076330
Hall, M. P., Unch, J., Binkowski, B. F., Valley, M. P., Butler, B. L., Wood, M. G., et al. (2012). Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 7, 1848–1857. doi: 10.1021/cb3002478
Hammarstrom, S. (1999). The carcinoembryonic antigen (CEA) family, structures, suggested functions and expression in normal and malignant tissues. Semin. Cancer Biol. 9, 67–81. doi: 10.1006/scbi.1998.0119
Haywood, T., Beinat, C., Gowrishankar, G., Patel, C. B., Alam, I. S., Murty, S., et al. (2019). Positron emission tomography reporter gene strategy for use in the central nervous system. Proc. Natl. Acad. Sci. U.S.A. 116, 11402–11407. doi: 10.1073/pnas.1901645116
Heskamp, S., Wierstra, P. J., Molkenboer-Kuenen, J. D. M., Sandker, G. W., Thordardottir, S., Cany, J., et al. (2019). PD-L1 microSPECT/CT imaging for longitudinal monitoring of PD-L1 expression in syngeneic and humanized mouse models for cancer. Cancer Immunol. Res. 7, 150–161. doi: 10.1158/2326-6066.CIR-18-0280
Higashikawa, K., Yagi, K., Watanabe, K., Kamino, S., Ueda, M., Hiromura, M., et al. (2014). 64Cu-DOTA-anti-CTLA-4 mAb enabled PET visualization of CTLA-4 on the T-cell infiltrating tumor tissues. PLoS One 9:e109866. doi: 10.1371/journal.pone.0109866
Higuchi, T., Anton, M., Saraste, A., Dumler, K., Pelisek, J., Nekolla, S. G., et al. (2009). Reporter gene PET for monitoring survival of transplanted endothelial progenitor cells in the rat heart after pretreatment with VEGF and atorvastatin. J. Nucl. Med. 50, 1881–1886. doi: 10.2967/jnumed.109.067801
Hodi, F. S., O’Day, S. J., McDermott, D. F., Weber, R. W., Sosman, J. A., Haanen, J. B., et al. (2010). Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723. doi: 10.1056/NEJMoa1003466
Hong, H., Sun, J., and Cai, W. (2008). Radionuclide-based cancer imaging targeting the carcinoembryonic antigen. Biomark Insights 3, 435–451.
Hotchkiss, R. S., and Moldawer, L. L. (2014). Parallels between cancer and infectious disease. N. Engl. J. Med. 371, 380–383. doi: 10.1056/nejmcibr1404664
Hunt, E. A., Moutsiopoulou, A., Ioannou, S., Ahern, K., Woodward, K., Dikici, E., et al. (2016). Truncated variants of gaussia luciferase with tyrosine linker for site-specific bioconjugate applications. Sci. Rep. 6:26814. doi: 10.1038/srep26814
Hwang, D. W., Kang, J. H., Chang, Y. S., Jeong, J. M., Chung, J. K., Lee, M. C., et al. (2007). Development of a dual membrane protein reporter system using sodium iodide symporter and mutant dopamine D2 receptor transgenes. J. Nucl. Med. 48, 588–595. doi: 10.2967/jnumed.106.036533
Inoue, Y., Sheng, F., Kiryu, S., Watanabe, M., Ratanakanit, H., Izawa, K., et al. (2011). Gaussia luciferase for bioluminescence tumor monitoring in comparison with firefly luciferase. Mol. Imaging 10:7290. doi: 10.2310/7290.2010.00057
Isomura, M., Yamada, K., Noguchi, K., and Nishizono, A. (2017). Near-infrared fluorescent protein iRFP720 is optimal for in vivo fluorescence imaging of rabies virus infection. J. Gen. Virol. 98, 2689–2698. doi: 10.1099/jgv.0.000950
James, M. L., and Gambhir, S. S. (2012). A molecular imaging primer, modalities, imaging agents, and applications. Physiol. Rev. 92, 897–965. doi: 10.1152/physrev.00049.2010
Jang, S. J., Kang, J. H., Kim, K. I., Lee, T. S., Lee, Y. J., Lee, K. C., et al. (2010). Application of bioluminescence imaging to therapeutic intervention of herpes simplex virus type I–Thymidine kinase/ganciclovir in glioma. Cancer Lett. 297, 84–90. doi: 10.1016/j.canlet.2010.04.028
Jang, S. J., Lee, Y. J., Lim, S., Kim, K. I., Lee, K. C., An, G. I., et al. (2012). Imaging of a localized bacterial infection with endogenous thymidine kinase using radioisotope-labeled nucleosides. Int. J. Med. Microbio. 302, 101–107. doi: 10.1016/j.ijmm.2011.11.002
Jauregui-Osoro, M., Sunassee, K., Weeks, A. J., Berry, D. J., Paul, R. L., Cleij, M., et al. (2010). Synthesis and biological evaluation of [F-18]tetrafluoroborate, a PET imaging agent for thyroid disease and reporter gene imaging of the sodium/iodide symporter. Eur. J. Nucl. Med. Mol. Imaging 37, 2108–2116. doi: 10.1007/s00259-010-1523-0
Jauw, Y. W. S., Bensch, F., Brouwers, A. H., Hoekstra, O. S., Zijlstra, J. M., Pieplenbosch, S., et al. (2019). Interobserver reproducibility of tumor uptake quantification with (89)Zr-immuno-PET, a multicenter analysis. Eur. J. Nucl. Med. Mol. Imaging 46, 1840–1849. doi: 10.1007/s00259-019-04377-6
Jiang, H., Bansal, A., Goyal, R., Peng, K.-W., Russell, S. J., and DeGrado, T. R. (2018). Synthesis and evaluation of 18F-hexafluorophosphate as a novel PET probe for imaging of sodium/iodide symporter in a murine C6-glioma tumor model. Bioorg. Med. Chem. 26, 225–231. doi: 10.1016/j.bmc.2017.11.034
Jiang, T., Du, L., and Li, M. (2016). Lighting up bioluminescence with coelenterazine, strategies and applications. Photochem. Photobiol. Sci. 15, 466–480. doi: 10.1039/c5pp00456j
Jung, W., Kim, J., Jeon, M., Chaney, E. J., Stewart, C. N., and Boppart, S. A. (2011). Handheld optical coherence tomography scanner for primary care diagnostics. IEEE Trans. Biomed. Eng. 58, 741–744. doi: 10.1109/TBME.2010.2096816
Kelly, M. P., Tavare, R., Giurleo, J., Makonnen, S., Hickey, C., Danton, M., et al. (2018). “Immuno-PET detection of LAG-3 expressing intratumoral lymphocytes using the zirconium-89 radiolabeled fully human anti-LAG-3 antibody REGN3767,” in American Association for Cancer Research Annual Meeting, Chicago.
Kenanova, V., Barat, B., Olafsen, T., Chatziioannou, A., Herschman, H. R., Braun, J., et al. (2009). Recombinant carcinoembryonic antigen as a reporter gene for molecular imaging. Eur. J. Nucl. Med. Mol. Imaging 36, 104–114. doi: 10.1007/s00259-008-0921-z
Keu, K. V., Witney, T. H., Yaghoubi, S., Rosenberg, J., Kurien, A., Magnusson, R., et al. (2017). Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Sci. Transl. Med. 9: eaag2196. doi: 10.1126/scitranslmed.aag2196
Khmelinskii, A., Meurer, M., Ho, C. T., Besenbeck, B., Fuller, J., Lemberg, M. K., et al. (2016). Incomplete proteasomal degradation of green fluorescent proteins in the context of tandem fluorescent protein timers. Mol. Biol. Cell 27, 360–370. doi: 10.1091/mbc.E15-07-0525
Khoshnevisan, A., Chuamsaamarkkee, K., Boudjemeline, M., Jackson, A., Smith, G. E., Gee, A. D., et al. (2017). 18F-Fluorosulfate for PET Imaging of the sodium-iodide symporter, synthesis and biologic evaluation in vitro and in vivo. J. Nucl. Med. 58, 156–161. doi: 10.2967/jnumed.116.177519
Khoshnevisan, A., Jauregui-Osoro, M., Shaw, K., Torres, J. B., Young, J. D., Ramakrishnan, N. K., et al. (2016). [(18)F]tetrafluoroborate as a PET tracer for the sodium/iodide symporter, the importance of specific activity. EJNMMI Res. 6:34. doi: 10.1186/s13550-016-0188-5
Kim, R., Keam, B., Kim, S., Kim, M., Kim, S. H., Kim, J. W., et al. (2019). Differences in tumor microenvironments between primary lung tumors and brain metastases in lung cancer patients, therapeutic implications for immune checkpoint inhibitors. BMC Cancer 19:19. doi: 10.1186/s12885-018-5214-8
Kircher, M. F., Gambhir, S. S., and Grimm, J. (2011). Noninvasive cell-tracking methods. Nat. Rev. Clin. Oncol. 8, 677–688. doi: 10.1038/nrclinonc.2011.141
Kleinovink, J. W., Mezzanotte, L., Zambito, G., Fransen, M. F., Cruz, L. J., Verbeek, J. S., et al. (2018). A dual-color bioluminescence reporter mouse for simultaneous in vivo imaging of t cell localization and function. Front. Immunol. 9:3097. doi: 10.3389/fimmu.2018.03097
Kogai, T., and Brent, G. A. (2012). The sodium iodide symporter (NIS), regulation and approaches to targeting for cancer therapeutics. Pharmacol. Ther. 135, 355–370. doi: 10.1016/j.pharmthera.2012.06.007
Kremers, G.-J., Hazelwood, K. L., Murphy, C. S., Davidson, M. W., and Piston, D. W. (2009). Photoconversion in orange and red fluorescent proteins. Nature Methods 6:355. doi: 10.1038/nmeth.1319
Krueger, M. A., Cotton, J. M., Zhou, B., Wolter, K., Schwenck, J., Kuehn, A., et al. (2019). Abstract 1146, [18F]FPyGal, A novel ß-galactosidase specific PET tracer for in vivo imaging of tumor senescence. Cancer Res. 79, 1118–1146.
Krumholz, A., VanVickle-Chavez, S. J., Yao, J., Fleming, T. P., Gillanders, W. E., and Wang, L. V. (2011). Photoacoustic microscopy of tyrosinase reporter gene in vivo. J. Biomed. Opt. 16:080503. doi: 10.1117/1.3606568
Kurland, B. F., Peterson, L. M., Lee, J. H., Schubert, E. K., Currin, E. R., Link, J. M., et al. (2017). Estrogen receptor binding (18F-FES PET) and glycolytic activity (18F-FDG PET) predict progression-free survival on endocrine therapy in patients with ER+ breast cancer. Clin. Cancer Res. 23, 407–415. doi: 10.1158/1078-0432.CCR-16-0362
Kurtys, E., Lim, L., Man, F., Volpe, A., and Fruhwirth, G. (2018). In vivo tracking of CAR-T by [18f]BF4- PET/CT in human breast cancer xenografts reveals differences in CAR-T tumour retention. Cytotherapy 20:S20.
La Barbera, G., Capriotti, A. L., Michelini, E., Piovesana, S., Calabretta, M. M., Zenezini Chiozzi, R., et al. (2017). Proteomic analysis and bioluminescent reporter gene assays to investigate effects of simulated microgravity on Caco-2 cells. Proteomics 17:1700081. doi: 10.1002/pmic.201700081
Lage, E., Parot, V., Moore, S. C., Sitek, A., Udias, J. M., Dave, S. R., et al. (2015). Recovery and normalization of triple coincidences in PET. Med. Phys. 42, 1398–1410. doi: 10.1118/1.4908226
Lajtos, I., Czernin, J., Dahlbom, M., Daver, F., Emri, M., Farshchi-Heydari, S., et al. (2014). Cold wall effect eliminating method to determine the contrast recovery coefficient for small animal PET scanners using the NEMA NU-4 image quality phantom. Phys. Med. Biol. 59, 2727–2746. doi: 10.1088/0031-9155/59/11/2727
Lambin, P., Rios-Velazquez, E., Leijenaar, R., Carvalho, S., van Stiphout, R. G., Granton, P., et al. (2012). Radiomics, extracting more information from medical images using advanced feature analysis. Eur. J. Cancer 48, 441–446. doi: 10.1016/j.ejca.2011.11.036
Lamson, M. L. III, Kirschner, A. S., Hotte, C. E., Lipsitz, E. L., and Ice, R. D. (1975). Generator-produced 99m TcO4-, carrier free. J. Nucl. Med. 16, 639–641.
Larimer, B. M., Bloch, E., Nesti, S., Austin, E. E., Wehrenberg-Klee, E., Boland, G., et al. (2019). The Effectiveness of Checkpoint Inhibitor Combinations and Administration Timing Can Be Measured by Granzyme B PET Imaging. Clin. Cancer Res. 25, 1196–1205. doi: 10.1158/1078-0432.CCR-18-2407
Larimer, B. M., Wehrenberg-Klee, E., Caraballo, A., and Mahmood, U. (2016). Quantitative CD3 PET imaging predicts tumor growth response to Anti-CTLA-4 therapy. J. Nucl. Med. 57, 1607–1611. doi: 10.2967/jnumed.116.173930
Larimer, B. M., Wehrenberg-Klee, E., Dubois, F., Mehta, A., Kalomeris, T., Flaherty, K., et al. (2017). Granzyme B PET imaging as a predictive biomarker of immunotherapy response. Cancer Res. 77, 2318–2327. doi: 10.1158/0008-5472.CAN-16-3346
Larkin, J., Chiarion-Sileni, V., Gonzalez, R., Grob, J. J., Cowey, C. L., Lao, C. D., et al. (2015). Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 373, 23–34. doi: 10.1056/NEJMoa1504030
Lavaud, J., Henry, M., Coll, J. L., and Josserand, V. (2017). Exploration of melanoma metastases in mice brains using endogenous contrast photoacoustic imaging. Int. J. Pharm. 532, 704–709. doi: 10.1016/j.ijpharm.2017.08.104
Ledford, H., Else, H., and Warren, M. (2018). Cancer immunologists scoop medicine Nobel prize. Nature 562, 20–21. doi: 10.1038/d41586-018-06751-0
Lee, J. T., Zhang, H., Moroz, M. A., Likar, Y., Shenker, L., Sumzin, N., et al. (2017). Comparative analysis of human nucleoside kinase-based reporter systems for PET imaging. Mol. Imaging Biol. 19, 100–108. doi: 10.1007/s11307-016-0981-6
Li, J., Chen, L., Du, L., and Li, M. (2013). Cage the firefly luciferin! - a strategy for developing bioluminescent probes. Chem. Soc. Rev. 42, 662–676. doi: 10.1039/c2cs35249d
Li, L., Zemp, R. J., Lungu, G. F., Stoica, G., and Wang, L. V. (2007). Photoacoustic imaging of lacZ gene expression in vivo. J. Biomed. Opt. 12:020504. doi: 10.1117/1.2717531
Li, Z., Suzuki, Y., Huang, M., Cao, F., Xie, X., Connolly, A. J., et al. (2008). Comparison of reporter gene and iron particle labeling for tracking fate of human embryonic stem cells and differentiated endothelial cells in living subjects. Stem. Cell 26, 864–873. doi: 10.1634/stemcells.2007-0843
Li, Z. B., Chen, K., Wu, Z., Wang, H., Niu, G., and Chen, X. (2009). 64Cu-labeled PEGylated polyethylenimine for cell trafficking and tumor imaging. Mol/Imaging Biol. 11, 415–423. doi: 10.1007/s11307-009-0228-x
Lian, L., Deng, Y., Xie, W., Xu, G., Yang, X., Zhang, Z., et al. (2017). Enhancement of the localization and quantitative performance of fluorescence molecular tomography by using linear nBorn method. Opt. Express 25, 2063–2079. doi: 10.1364/OE.25.002063
Liang, Q., Satyamurthy, N., Barrio, J. R., Toyokuni, T., Phelps, M. P., Gambhir, S. S., et al. (2001). Noninvasive, quantitative imaging in living animals of a mutant dopamine D2 receptor reporter gene in which ligand binding is uncoupled from signal transduction. Gene Ther. 8, 1490–1498. doi: 10.1038/sj.gt.3301542
Likar, Y., Dobrenkov, K., Olszewska, M., Shenker, L., Cai, S., Hricak, H., et al. (2009). PET imaging of HSV1-tk mutants with acquired specificity toward pyrimidine- and acycloguanosine-based radiotracers. Eur. J. Nucl. Med. Mol. Imaging 36, 1273–1282. doi: 10.1007/s00259-009-1089-x
Likar, Y., Zurita, J., Dobrenkov, K., Shenker, L., Cai, S., Neschadim, A., et al. (2010). A new pyrimidine-specific reporter gene, a mutated human deoxycytidine kinase suitable for PET during treatment with acycloguanosine-based cytotoxic drugs. J. Nucl. Med. 51, 1395–1403. doi: 10.2967/jnumed.109.074344
Lin, M. Z., McKeown, M. R., Ng, H.-L., Aguilera, T. A., Shaner, N. C., Campbell, R. E., et al. (2009). Autofluorescent proteins with excitation in the optical window for intravital imaging in mammals. Chem. Biol. 16, 1169–1179. doi: 10.1016/j.chembiol.2009.10.009
Linette, G. P., Stadtmauer, E. A., Maus, M. V., Rapoport, A. P., Levine, B. L., Emery, L., et al. (2013). Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122, 863–871. doi: 10.1182/blood-2013-03-490565
Liu, L., and Mason, R. P. (2010). Imaging β-galactosidase activity in human tumor xenografts and transgenic mice using a chemiluminescent substrate. PLoS One 5:e12024. doi: 10.1371/journal.pone.0012024
Liu, M., Schmitner, N., Sandrian, M. G., Zabihian, B., Hermann, B., Salvenmoser, W., et al. (2013). In vivo three dimensional dual wavelength photoacoustic tomography imaging of the far red fluorescent protein E2-Crimson expressed in adult zebrafish. Biomed. Opt. Express 4, 1846–1855. doi: 10.1364/BOE.4.001846
Loening, A. M., Fenn, T. D., Wu, A. M., and Gambhir, S. S. (2006). Consensus guided mutagenesis of Renilla luciferase yields enhanced stability and light output. Protein Eng., Design Select. 19, 391–400. doi: 10.1093/protein/gzl023
Lorenz, W. W., McCann, R. O., Longiaru, M., and Cormier, M. J. (1991). Isolation and expression of a cDNA encoding Renilla reniformis luciferase. Proc. Natl. Acad. Sci. U.S.A. 88, 4438–4442. doi: 10.1073/pnas.88.10.4438
Louie, A. Y., Hüber, M. M., Ahrens, E. T., Rothbächer, U., Moats, R., Jacobs, R. E., et al. (2000). In vivo visualization of gene expression using magnetic resonance imaging. Nat. Biotechnol. 18:321. doi: 10.1038/73780
Lufino, M. M., Edser, P. A., and Wade-Martins, R. (2008). Advances in high-capacity extrachromosomal vector technology, episomal maintenance, vector delivery, and transgene expression. Mol. Ther. 16, 1525–1538. doi: 10.1038/mt.2008.156
MacLaren, D. C., Gambhir, S. S., Satyamurthy, N., Barrio, J. R., Sharfstein, S., Toyokuni, T., et al. (1999). Repetitive, non-invasive imaging of the dopamine D2 receptor as a reporter gene in living animals. Gene Ther. 6, 785–791. doi: 10.1038/sj.gt.3300877
Man, F., Lim, L., Volpe, A., Gabizon, A., Shmeeda, H., Draper, B., et al. (2019). In Vivo PET Tracking of (89)Zr-Labeled Vgamma9Vdelta2 T cells to mouse xenograft breast tumors activated with liposomal alendronate. Mol. Ther. 27, 219–229. doi: 10.1016/j.ymthe.2018.10.006
Manda, K., Glasow, A., Paape, D., and Hildebrandt, G. (2012). Effects of ionizing radiation on the immune system with special emphasis on the interaction of dendritic and T cells. Front. Oncol. 2:102. doi: 10.3389/fonc.2012.00102
Marks, P., and Gottlieb, S. (2018). Balancing safety and innovation for cell-based regenerative medicine. N. Engl. J. Med. 378, 954–959. doi: 10.1056/nejmsr1715626
Martinez, M., and Moon, E. K. (2019). CAR T cells for solid tumors, new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 10:128. doi: 10.3389/fimmu.2019.00128
Maude, S. L., Laetsch, T. W., Buechner, J., Rives, S., Boyer, M., Bittencourt, H., et al. (2018). Tisagenlecleucel in CHILDREN AND YOUNG ADUlts with B-Cell LYMPHOBLASTIC LEUKEMIA. N. Engl. J. Med. 378, 439–448. doi: 10.1056/NEJMoa1709866
Mayer, K. E., Mall, S., Yusufi, N., Gosmann, D., Steiger, K., Russelli, L., et al. (2018). T-cell functionality testing is highly relevant to developing novel immuno-tracers monitoring T cells in the context of immunotherapies and revealed CD7 as an attractive target. Theranostics 8, 6070–6087. doi: 10.7150/thno.27275
McGinty, J., Taylor, H. B., Chen, L., Bugeon, L., Lamb, J. R., Dallman, M. J., et al. (2011). In vivo fluorescence lifetime optical projection tomography. Biomed. Opt. Express 2, 1340–1350. doi: 10.1364/BOE.2.001340
Merron, A., Peerlinck, I., Martin-Duque, P., Burnet, J., Quintanilla, M., Mather, S., et al. (2007). SPECT/CT imaging of oncolytic adenovirus propagation in tumours in vivo using the Na/I symporter as a reporter gene. Gene Ther. 14, 1731–1738. doi: 10.1038/sj.gt.3303043
Merzlyak, E. M., Goedhart, J., Shcherbo, D., Bulina, M. E., Shcheglov, A. S., Fradkov, A. F., et al. (2007). Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nature Methods 4:555. doi: 10.1038/nmeth1062
Mezzanotte, L., An, N., Mol, I. M., Löwik, C. W., and Kaijzel, E. L. (2014). A new multicolor bioluminescence imaging platform to investigate NF-κB activity and apoptosis in human breast cancer cells. PloS One 9:e85550. doi: 10.1371/journal.pone.0085550.<PMID>PMID:NOPMID</PMID<
Mezzanotte, L., Que, I., Kaijzel, E., Branchini, B., Roda, A., and Lowik, C. (2011). Sensitive dual color in vivo bioluminescence imaging using a new red codon optimized firefly luciferase and a green click beetle luciferase. PLoS One 6:e19277. doi: 10.1371/journal.pone.0019277
Mezzanotte, L., van ’t Root, M., Karatas, H., Goun, E. A., and Lowik, C. (2017). In vivo molecular bioluminescence imaging, new tools and applications. Trends Biotechnol 35, 640–652. doi: 10.1016/j.tibtech.2017.03.012
Minn, I., Huss, D. J., Ahn, H. H., Chinn, T. M., Park, A., Jones, J., et al. (2019). Imaging CAR T cell therapy with PSMA-targeted positron emission tomography. Sci. Adv. 5:eaaw5096. doi: 10.1126/sciadv.aaw5096
Misra, T., Baccino-Calace, M., Meyenhofer, F., Rodriguez-Crespo, D., Akarsu, H., Armenta-Calderon, R., et al. (2017). A genetically encoded biosensor for visualising hypoxia responses in vivo. Biol. Open 6, 296–304. doi: 10.1242/bio.018226
Mogensen, M., Thrane, L., Jorgensen, T. M., Andersen, P. E., and Jemec, G. B. (2009). OCT imaging of skin cancer and other dermatological diseases. J. Biophotonics 2, 442–451. doi: 10.1002/jbio.200910020
Moroz, M. A., Serganova, I., Zanzonico, P., Ageyeva, L., Beresten, T., Dyomina, E., et al. (2007). Imaging hNET reporter gene expression with 124I-MIBG. J. Nucl. Med. 48, 827–836. doi: 10.2967/jnumed.106.037812
Moroz, M. A., Zhang, H., Lee, J., Moroz, E., Zurita, J., Shenker, L., et al. (2015). Comparative analysis of T cell imaging with human nuclear reporter genes. J. Nucl. Med. 56, 1055–1060. doi: 10.2967/jnumed.115.159855
Nagy, K., Toth, M., Major, P., Patay, G., Egri, G., Haggkvist, J., et al. (2013). Performance evaluation of the small-animal nanoScan PET/MRI system. J. Nucl. Med. 54, 1825–1832. doi: 10.2967/jnumed.112.119065
Nakamura, C., Burgess, J. G., Sode, K., and Matsunaga, T. (1995). An iron-regulated gene, magA, encoding an iron transport protein of Magnetospirillum sp. strain AMB-1. J. Biol. Chem. 270, 28392–28396. doi: 10.1074/jbc.270.47.28392
Natarajan, A., Mayer, A. T., Xu, L., Reeves, R. E., Gano, J., and Gambhir, S. S. (2015). Novel radiotracer for ImmunoPET imaging of PD-1 checkpoint expression on tumor infiltrating lymphocytes. Bioconjug Chem. 26, 2062–2069. doi: 10.1021/acs.bioconjchem.5b00318
Naumova, A. V., Modo, M., Moore, A., Murry, C. E., and Frank, J. A. (2014). Clinical imaging in regenerative medicine. Nat. Biotechnol. 32, 804–818. doi: 10.1038/nbt.2993
Neelapu, S. S., Locke, F. L., Bartlett, N. L., Lekakis, L. J., Miklos, D. B., Jacobson, C. A., et al. (2017). Axicabtagene ciloleucel CAR T-Cell therapy in refractory large B-Cell lymphoma. N. Engl. J. Med. 377, 2531–2544. doi: 10.1056/NEJMoa1707447
Nishino, M., Ramaiya, N. H., Hatabu, H., and Hodi, F. S. (2017). Monitoring immune-checkpoint blockade, response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 14, 655–668. doi: 10.1038/nrclinonc.2017.88
Ntziachristos, V., Ripoll, J., Wang, L. V., and Weissleder, R. (2005). Looking and listening to light, the evolution of whole-body photonic imaging. Nat,. Biotechnol. 23, 313–320. doi: 10.1038/nbt1074
O’Connor, J. P., Aboagye, E. O., Adams, J. E., Aerts, H. J., Barrington, S. F., Beer, A. J., et al. (2017). Imaging biomarker roadmap for cancer studies. Nat. Rev. Clin. Oncol. 14, 169–186. doi: 10.1038/nrclinonc.2016.162
O’Doherty, J., Jauregui-Osoro, M., Brothwood, T., Szyszko, T., Marsden, P. K., O’Doherty, M. J., et al. (2017). (18)F-Tetrafluoroborate, a PET probe for imaging sodium/iodide symporter expression, whole-body biodistribution, safety, and radiation dosimetry in thyroid cancer patients. J. Nucl. Med. 58, 1666–1671. doi: 10.2967/jnumed.117.192252
Oliveira, J. M., Gomes, C., Faria, D. B., Vieira, T. S., Silva, F. A., Vale, J., et al. (2017). (68)Ga-prostate-specific membrane antigen positron emission tomography/computed tomography for prostate cancer imaging, a narrative literature review. World J. Nucl. Med. 16, 3–7. doi: 10.4103/1450-1147.198237
Olsen, J., Themstrup, L., and Jemec, G. B. (2015). Optical coherence tomography in dermatology. G Ital Dermatol Venereol 150, 603–615.
Oomen, S. P., Hofland, L. J., Lamberts, S. W., Lowenberg, B., and Touw, I. P. (2001). Internalization-defective mutants of somatostatin receptor subtype 2 exert normal signaling functions in hematopoietic cells. FEBS Lett. 503, 163–167. doi: 10.1016/s0014-5793(01)02729-6
Park, J. H., Kim, K. I., Lee, K. C., Lee, Y. J., Lee, T. S., Chung, W. S., et al. (2015). Assessment of α-fetoprotein targeted HSV1-tk expression in hepatocellular carcinoma with in vivo imaging. Cancer Biother. Radiopharm. 30, 8–15. doi: 10.1089/cbr.2014.1716
Paroder-Belenitsky, M., Maestas, M. J., Dohan, O., Nicola, J. P., Reyna-Neyra, A., Follenzi, A., et al. (2011). Mechanism of anion selectivity and stoichiometry of the Na+/I- symporter (NIS). Proc. Natl. Acad. Sci. U.S.A. 108, 17933–17938. doi: 10.1073/pnas.1108278108
Pellenz, S., Phelps, M., Tang, W., Hovde, B. T., Sinit, R. B., Fu, W., et al. (2019). New human chromosomal sites with “Safe Harbor” potential for targeted transgene insertion. Hum. Gene Ther. 30, 814–828. doi: 10.1089/hum.2018.169
Perera, M., Papa, N., Christidis, D., Wetherell, D., Hofman, M. S., Murphy, D. G., et al. (2016). Sensitivity, specificity, and predictors of positive (68)Ga-Prostate-specific membrane antigen positron emission tomography in advanced prostate cancer, a systematic review and meta-analysis. Eur. Urol. 70, 926–937. doi: 10.1016/j.eururo.2016.06.021
Phelps, M. E., Hoffman, E. J., Huang, S. C., and Ter-Pogossian, M. M. (1975). Effect of positron range on spatial resolution. J. Nucl. Med. 16, 649–652.
Piotrowski, A. F., Nirschl, T. R., Velarde, E., Blosser, L., Ganguly, S., Burns, K. H., et al. (2018). Systemic depletion of lymphocytes following focal radiation to the brain in a murine model. Oncoimmunology 7:e1445951. doi: 10.1080/2162402X.2018.1445951
Ponomarev, V., Doubrovin, M., Lyddane, C., Beresten, T., Balatoni, J., Bornman, W., et al. (2001). Imaging TCR-dependent NFAT-mediated T-cell activation with positron emission tomography in vivo. Neoplasia 3, 480–488. doi: 10.1038/sj.neo.7900204
Ponomarev, V., Doubrovin, M., Serganova, I., Vider, J., Shavrin, A., Beresten, T., et al. (2004). A novel triple-modality reporter gene for whole-body fluorescent, bioluminescent, and nuclear noninvasive imaging. Eur. J. Nucl. Med. Mol. Imaging 31, 740–751. doi: 10.1007/s00259-003-1441-5
Ponomarev, V., Doubrovin, M., Shavrin, A., Serganova, I., Beresten, T., Ageyeva, L., et al. (2007). A human-derived reporter gene for noninvasive imaging in humans, mitochondrial thymidine kinase type 2. J. Nucl. Med. 48, 819–826. doi: 10.2967/jnumed.106.036962
Portulano, C., Paroder-Belenitsky, M., and Carrasco, N. (2014). The Na+/I- symporter (NIS), mechanism and medical impact. Endocr. Rev. 35, 106–149. doi: 10.1210/er.2012-1036
Provost, J., Garofalakis, A., Sourdon, J., Bouda, D., Berthon, B., Viel, T., et al. (2018). Simultaneous positron emission tomography and ultrafast ultrasound for hybrid molecular, anatomical and functional imaging. Nat. Biomed. Eng. 2, 85–94. doi: 10.1038/s41551-018-0188-z
Qin, C., Lan, X., He, J., Xia, X., Tian, Y., Pei, Z., et al. (2013). An in vitro and in vivo evaluation of a reporter gene/probe system hERL/(18)F-FES. PLoS One 8:e61911. doi: 10.1371/journal.pone.0061911
Qiu, L., Valente, M., Dolen, Y., Jager, E., Beest, M. T., Zheng, L., et al. (2018). Endolysosomal-escape nanovaccines through adjuvant-induced tumor antigen assembly for enhanced effector CD8(+) T cell activation. Small 14:e1703539. doi: 10.1002/smll.201703539
Rajasekaran, S. A., Anilkumar, G., Oshima, E., Bowie, J. U., Liu, H., Heston, W., et al. (2003). A novel cytoplasmic tail MXXXL motif mediates the internalization of prostate-specific membrane antigen. Mol. Biol. Cell 14, 4835–4845. doi: 10.1091/mbc.e02-11-0731
Ravert, H. T., Holt, D. P., Chen, Y., Mease, R. C., Fan, H., Pomper, M. G., et al. (2016). An improved synthesis of the radiolabeled prostate-specific membrane antigen inhibitor, [(18) F]DCFPyL. J. Labelled Comp. Radiopharm. 59, 439–450. doi: 10.1002/jlcr.3430
Roca, M., de Vries, E. F., Jamar, F., Israel, O., and Signore, A. (2010). Guidelines for the labelling of leucocytes with (111)In-oxine. Inflammation/Infection Taskgroup of the European Association of Nuclear Medicine. Eur. J. Nucl. Med. Mol. Imaging 37, 835–841. doi: 10.1007/s00259-010-1393-5
Rogers, B. E., McLean, S. F., Kirkman, R. L., Della Manna, D., Bright, S. J., Olsen, C. C., et al. (1999). In vivo localization of [(111)In]-DTPA-D-Phe1-octreotide to human ovarian tumor xenografts induced to express the somatostatin receptor subtype 2 using an adenoviral vector. Clin. Cancer Res. 5, 383–393.
Rogers, B. E., Zinn, K. R., and Buchsbaum, D. J. (2000). Gene transfer strategies for improving radiolabeled peptide imaging and therapy. Q. J. Nucl. Med. 44, 208–223.
Ronald, J. A., Cusso, L., Chuang, H. Y., Yan, X., Dragulescu-Andrasi, A., and Gambhir, S. S. (2013). Development and validation of non-integrative, self-limited, and replicating minicircles for safe reporter gene imaging of cell-based therapies. PLoS One 8:e73138. doi: 10.1371/journal.pone.0073138
Ronald, J. A., Kim, B. S., Gowrishankar, G., Namavari, M., Alam, I. S., D’Souza, A., et al. (2017). A PET imaging strategy to visualize activated T Cells in acute graft-versus-host disease elicited by allogenic hematopoietic cell transplant. Cancer Res. 77, 2893–2902. doi: 10.1158/0008-5472.CAN-16-2953
Safinia, N., Vaikunthanathan, T., Fraser, H., Thirkell, S., Lowe, K., Blackmore, L., et al. (2016). Successful expansion of functional and stable regulatory T cells for immunotherapy in liver transplantation. Oncotarget 7, 7563–7577. doi: 10.18632/oncotarget.6927
Sahu, S. K., Kassis, A. I., Makrigiorgos, G. M., Baranowska-Kortylewicz, J., and Adelstein, S. J. (1995). The effects of indium-111 decay on pBR322 DNA. Radiat. Res. 141, 193–198.
Saini, S., Korf, H., Liang, S., Verbeke, R., Manshian, B., Raemdonck, K., et al. (2019). Challenges for labeling and longitudinal tracking of adoptively transferred autoreactive T lymphocytes in an experimental type-1 diabetes model. MAGMA 32, 295–305. doi: 10.1007/s10334-018-0720-x
Sato, N., Wu, H., Asiedu, K. O., Szajek, L. P., Griffiths, G. L., and Choyke, P. L. (2015). (89)Zr-oxine complex PET cell imaging in monitoring cell-based therapies. Radiology 275, 490–500. doi: 10.1148/radiol.15142849
Satyamurthy, N., Barrio, J. R., Bida, G. T., Huang, S. C., Mazziotta, J. C., and Phelps, M. E. (1990). 3-(2’-[18F]fluoroethyl)spiperone, a potent dopamine antagonist, synthesis, structural analysis and in-vivo utilization in humans. Int. J. Rad. Appl. Instrum. A 41, 113–129. doi: 10.1016/0883-2889(90)90096-y
Saudemont, A., Jespers, L., and Clay, T. (2018). Current status of gene engineering cell therapeutics. Front. Immunol. 9:153. doi: 10.3389/fimmu.2018.00153
Schaub, F. X., Reza, M. S., Flaveny, C. A., Li, W., Musicant, A. M., Hoxha, S., et al. (2015). Fluorophore-NanoLuc BRET reporters enable sensitive <em>In Vivo</em> optical imaging and flow cytometry for monitoring tumorigenesis. Cancer Res. 75, 5023–5033. doi: 10.1158/0008-5472.can-14-3538
Schuster, S. J., Svoboda, J., Chong, E. A., Nasta, S. D., Mato, A. R., Anak, O., et al. (2017). Chimeric antigen receptor T Cells in refractory B-Cell lymphomas. N. Engl. J. Med. 377, 2545–2554. doi: 10.1056/NEJMoa1708566
Sellmyer, M. A., Lee, I., Hou, C., Lieberman, B. P., Zeng, C., Mankoff, D. A., et al. (2017). Quantitative PET reporter gene imaging with [11C] trimethoprim. Mol. Ther. 25, 120–126. doi: 10.1016/j.ymthe.2016.10.018
Sellmyer, M. A., Richman, S. A., Lohith, K., Hou, C., Weng, C.-C., Mach, R. H., et al. (2019). Imaging CAR T cell trafficking with eDHFR as a PET reporter gene. Mol. Ther. 28, 42–51. doi: 10.1016/j.ymthe.2019.10.007
Seo, J. W., Tavare, R., Mahakian, L. M., Silvestrini, M. T., Tam, S., Ingham, E. S., et al. (2018). CD8(+) T-Cell density imaging with (64)Cu-Labeled Cys-Diabody informs immunotherapy protocols. Clin. Cancer Res. 24, 4976–4987.
Seo, M.-J., Park, J. H., Lee, K. C., Lee, Y. J., Lee, T. S., Choi, T. H., et al. (2019). Small animal PET imaging of hTERT RNA-Targeted HSV1-tk gene expression with trans-splicing ribozyme. Cancer Biother. Radiopharm. 35, 26–32. doi: 10.1089/cbr.2019.2839
Seymour, L., Bogaerts, J., Perrone, A., Ford, R., Schwartz, L. H., Mandrekar, S., et al. (2017). iRECIST, guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 18, e143–e152. doi: 10.1016/S1470-2045(17)30074-8
Sharpe, J., Ahlgren, U., Perry, P., Hill, B., Ross, A., Hecksher-Sorensen, J., et al. (2002). Optical projection tomography as a tool for 3D microscopy and gene expression studies. Science 296, 541–545. doi: 10.1126/science.1068206
Shcherbakova, D. M., Stepanenko, O. V., Turoverov, K. K., and Verkhusha, V. V. (2018). Near-infrared fluorescent proteins, multiplexing and optogenetics across scales. Trends Biotechnol. 36, 1230–1243. doi: 10.1016/j.tibtech.2018.06.011
Shcherbakova, D. M., and Verkhusha, V. V. (2013). Near-infrared fluorescent proteins for multicolor in vivo imaging. Nature Methods. 10:751. doi: 10.1038/nmeth.2521
Sieger, S., Jiang, S., Schonsiegel, F., Eskerski, H., Kubler, W., Altmann, A., et al. (2003). Tumour-specific activation of the sodium/iodide symporter gene under control of the glucose transporter gene 1 promoter (GTI-1.3). Eur. J. Nucl. Med. Mol. Imaging 30, 748–756. doi: 10.1007/s00259-002-1099-4
Smith, E. L., and Peakman, M. (2018). Peptide immunotherapy for type 1 diabetes-clinical advances. Front. Immunol. 9:392. doi: 10.3389/fimmu.2018.00392
Srinivas, M., Boehm-Sturm, P., Figdor, C. G., de Vries, I. J., and Hoehn, M. (2012). Labeling cells for in vivo tracking using (19)F MRI. Biomaterials 33, 8830–8840. doi: 10.1016/j.biomaterials.2012.08.048
Srinivas, M., Turner, M. S., Janjic, J. M., Morel, P. A., Laidlaw, D. H., and Ahrens, E. T. (2009). In vivo cytometry of antigen-specific t cells using 19F MRI. Magn. Reson. Med. 62, 747–753. doi: 10.1002/mrm.22063
Srinivas, M., Boehm-Sturm, P., Aswendt, M., Pracht, E. D., Figdor, C. G., de Vries, I. J., et al. (2013). In vivo 19F MRI for cell tracking. J. Vis. Exp. e50802. doi: 10.3791/50802
Tannous, B. A. (2009). Gaussia luciferase reporter assay for monitoring biological processes in culture and in vivo. Nature Protoc. 4:582. doi: 10.1038/nprot.2009.28
Tao, A., Shao, Y., Zhong, J., Jiang, H., Shen, M., and Wang, J. (2013). Versatile optical coherence tomography for imaging the human eye. Biomed. Opt. Express 4, 1031–1044. doi: 10.1364/BOE.4.001031
Tavare, R., Escuin-Ordinas, H., Mok, S., McCracken, M. N., Zettlitz, K. A., Salazar, F. B., et al. (2016). An effective immuno-PET imaging method to monitor CD8-dependent responses to immunotherapy. Cancer Res. 76, 73–82. doi: 10.1158/0008-5472.CAN-15-1707
Terrovitis, J., Kwok, K. F., Lautamaki, R., Engles, J. M., Barth, A. S., Kizana, E., et al. (2008). Ectopic expression of the sodium-iodide symporter enables imaging of transplanted cardiac stem cells in vivo by single-photon emission computed tomography or positron emission tomography. J. Am. Coll Cardiol. 52, 1652–1660. doi: 10.1016/j.jacc.2008.06.051
Thorn, K. (2017). Genetically encoded fluorescent tags. Mol. Biol. Cell 28, 848–857. doi: 10.1091/mbc.E16-07-0504
Tiernan, J. P., Perry, S. L., Verghese, E. T., West, N. P., Yeluri, S., Jayne, D. G., et al. (2013). Carcinoembryonic antigen is the preferred biomarker for in vivo colorectal cancer targeting. Br. J. Cancer 108, 662–667. doi: 10.1038/bjc.2012.605
Tjuvajev, J. G., Stockhammer, G., Desai, R., Uehara, H., Watanabe, K., Gansbacher, B., et al. (1995). Imaging the expression of transfected genes in vivo. Cancer Res. 55, 6126–6132.
Topalian, S. L., Hodi, F. S., Brahmer, J. R., Gettinger, S. N., Smith, D. C., McDermott, D. F., et al. (2012). Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454. doi: 10.1056/NEJMoa1200690
Tsao, H., Chin, L., Garraway, L. A., and Fisher, D. E. (2012). Melanoma, from mutations to medicine. Genes Dev. 26, 1131–1155. doi: 10.1101/gad.191999.112
UCL Business PLC (2017). Detecting a Therapeutic Cell. Patent No 20170056534. Mountain View, CA: JUSTIA.
U.S. Food and Drug Administration, (2018). FDA Approves new Treatment for a Certain type of Prostate Cancer Using Novel Clinical Trial Endpoint. FDA News Release.
Urabe, K., Aroca, P., Tsukamoto, K., Mascagna, D., Palumbo, A., Prota, G., et al. (1994). The inherent cytotoxicity of melanin precursors, a revision. Biochim. BiophysActa 1221, 272–278. doi: 10.1016/0167-4889(94)90250-x
U.S. Food and Drug Administration, (2017). FDA Approval Brings First Gene Therapy to the United States. Silver Spring, MA: US Food and Drug Administration.
van der Windt, G. J., and Pearce, E. L. (2012). Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol. Rev. 249, 27–42. doi: 10.1111/j.1600-065X.2012.01150.x
VanBrocklin, H. F. (2008). Radiopharmaceuticals for drug development. United States regulatory perspective. Curr. Radiopharm. 1, 2–6. doi: 10.2174/1874471010801010002
Vandergaast, R., Khongwichit, S., Jiang, H., DeGrado, T. R., Peng, K. W., Smith, D. R., et al. (2019). Enhanced noninvasive imaging of oncology models using the NIS reporter gene and bioluminescence imaging. Cancer Gene Ther.
Vedvyas, Y., Shevlin, E., Zaman, M., Min, I. M., Amor-Coarasa, A., Park, S., et al. (2016). Longitudinal PET imaging demonstrates biphasic CAR T cell responses in survivors. JCI Insight 1, e90064. doi: 10.1172/jci.insight.90064
Venugopal, V., Chen, J., Lesage, F., and Intes, X. (2010). Full-field time-resolved fluorescence tomography of small animals. Opt. Lett. 35, 3189–3191. doi: 10.1364/OL.35.003189
Vinegoni, C., Pitsouli, C., Razansky, D., Perrimon, N., and Ntziachristos, V. (2008). In vivo imaging of Drosophila melanogaster pupae with mesoscopic fluorescence tomography. Nat. Methods 5, 45–47. doi: 10.1038/nmeth1149
Volpe, A., Man, F., Lim, L., Khoshnevisan, A., Blower, J., Blower, P. J., et al. (2018). Radionuclide-fluorescence reporter gene imaging to track tumor progression in rodent tumor models. J. Vis. Exp. 133:e57088. doi: 10.3791/57088
Wang, K., Wang, Q., Luo, Q., and Yang, X. (2015). Fluorescence molecular tomography in the second near-infrared window. Opt. Express 23, 12669–12679.
Wang, L. V., and Yao, J. (2016). A practical guide to photoacoustic tomography in the life sciences. Nat. Methods 13, 627–638. doi: 10.1038/nmeth.3925
Wei, L. H., Olafsen, T., Radu, C., Hildebrandt, I. J., McCoy, M. R., Phelps, M. E., et al. (2008). Engineered antibody fragments with infinite affinity as reporter genes for PET imaging. J. Nucl. Med. 49, 1828–1835. doi: 10.2967/jnumed.108.054452
Weihs, F., and Dacres, H. (2019). Red-shifted bioluminescence resonance energy transfer, improved tools and materials for analytical in vivo approaches. Trac Trends Anal. Chem. 116, 61–73. doi: 10.1016/j.trac.2019.04.011
Weissleder, R., Moore, A., Mahmood, U., Bhorade, R., Benveniste, H., Chiocca, E. A., et al. (2000). In vivo magnetic resonance imaging of transgene expression. Nat. Med. 6, 351–355.
Weissleder, R., Simonova, M., Bogdanova, A., Bredow, S., Enochs, W. S., and Bogdanov, A. Jr. (1997). MR imaging and scintigraphy of gene expression through melanin induction. Radiology 204, 425–429. doi: 10.1148/radiology.204.2.9240530
Weist, M. R., Starr, R., Aguilar, B., Chea, J., Miles, J., Poku, E., et al. (2018). Positron emission tomography of adoptively transferred chimeric antigen receptor T cells with Zirconium-89 oxine. J. Nucl. Med. 59, 1531–1537. doi: 10.2967/jnumed.117.206714
Wolchok, J. D., Hoos, A., O’Day, S., Weber, J. S., Hamid, O., Lebbe, C., et al. (2009). Guidelines for the evaluation of immune therapy activity in solid tumors, immune-related response criteria. Clin. Cancer Res. 15, 7412–7420. doi: 10.1158/1078-0432.CCR-09-1624
Wolfs, E., Holvoet, B., Ordovas, L., Breuls, N., Helsen, N., Schonberger, M., et al. (2017). Molecular imaging of human embryonic stem cells stably expressing human PET reporter genes after zinc finger nuclease-mediated genome editing. J. Nucl. Med. 58, 1659–1665. doi: 10.2967/jnumed.117.189779
Wu, M.-R., Huang, Y.-Y., and Hsiao, J.-K. (2019). Use of indocyanine green (ICG), a medical near infrared dye, for enhanced fluorescent imaging—comparison of organic anion transporting polypeptide 1B3 (OATP1B3) and sodium-taurocholate cotransporting polypeptide (NTCP) reporter genes. Molecules 24:2295. doi: 10.3390/molecules24122295
Yaghoubi, S. S., and Gambhir, S. S. (2006). PET imaging of herpes simplex virus type 1 thymidine kinase (HSV1-tk) or mutant HSV1-sr39tk reporter gene expression in mice and humans using [18F]FHBG. Nat. Protoc. 1, 3069–3075.
Yamada, Y., Post, S. R., Wang, K., Tager, H. S., Bell, G. I., and Seino, S. (1992). Cloning and functional characterization of a family of human and mouse somatostatin receptors expressed in brain, gastrointestinal tract, and kidney. Proc. Natl. Acad. Sci. U.S.A. 89, 251–255. doi: 10.1073/pnas.89.1.251
Yusufi, N., Mall, S., Bianchi, H. O., Steiger, K., Reder, S., Klar, R., et al. (2017). In-depth characterization of a TCR-specific tracer for sensitive detection of tumor-directed transgenic T cells by immuno-PET. Theranostics 7, 2402–2416. doi: 10.7150/thno.17994
Zabow, G., Dodd, S., Moreland, J., and Koretsky, A. (2008). Micro-engineered local field control for high-sensitivity multispectral MRI. Nature 453:1058. doi: 10.1038/nature07048
Zacharakis, G., Favicchio, R., Simantiraki, M., and Ripoll, J. (2011). Spectroscopic detection improves multi-color quantification in fluorescence tomography. Biomed. Opt. Express 2, 431–439. doi: 10.1364/BOE.2.000431
Zettlitz, K. A., Tavare, R., Knowles, S. M., Steward, K. K., Timmerman, J. M., and Wu, A. M. (2017). ImmunoPET of malignant and normal B cells with (89)Zr- and (124)I-Labeled obinutuzumab antibody fragments reveals differential CD20 internalization in vivo. Clin. Cancer Res. 23, 7242–7252. doi: 10.1158/1078-0432.CCR-17-0855
Zhang, H., Moroz, M. A., Serganova, I., Ku, T., Huang, R., Vider, J., et al. (2011). Imaging expression of the human somatostatin receptor subtype-2 reporter gene with 68Ga-DOTATOC. J. Nucl. Med. 52, 123–131. doi: 10.2967/jnumed.110.079004
Zhang, Y., Wang, C., Gao, N., Zhang, X., Yu, X., Xu, J., et al. (2019). Determination of neutralization activities by a new versatile assay using an HIV-1 genome carrying the Gaussia luciferase gene. J. Virol. Methods 267, 22–28. doi: 10.1016/j.jviromet.2019.02.009
Zhou, J., Sharkey, J., Shukla, R., Plagge, A., and Murray, P. (2018). Assessing the effectiveness of a far-red fluorescent reporter for tracking stem cells in vivo. Int. J. Mol. Sci. 19:19. doi: 10.3390/ijms19010019
Zinn, K. R., Buchsbaum, D. J., Chaudhuri, T. R., Mountz, J. M., Grizzle, W. E., and Rogers, B. E. (2000a). Noninvasive monitoring of gene transfer using a reporter receptor imaged with a high-affinity peptide radiolabeled with 99mTc or 188Re. J. Nucl. Med. 41, 887–895.
Zinn, K. R., Buchsbaum, D. J., Chaudhuri, T. R., Mountz, J. M., Grizzle, W. E., and Rogers, B. E. (2000b). Simultaneous in vivo imaging of thymidine kinase and somatostatin receptor expression after gene transfer with an adenoviral vector encoding both genes. Mol. Ther. 2000:S44.
Keywords: adoptive cell therapy, cell tracking, drug development, molecular imaging, multi-modal whole-body imaging, positron emission tomography, reporter gene
Citation: Iafrate M and Fruhwirth GO (2020) How Non-invasive in vivo Cell Tracking Supports the Development and Translation of Cancer Immunotherapies. Front. Physiol. 11:154. doi: 10.3389/fphys.2020.00154
Received: 26 November 2019; Accepted: 12 February 2020;
Published: 03 April 2020.
Edited by:
Claudia Kuntner, Austrian Institute of Technology, AustriaReviewed by:
Ann Ager, Cardiff University, United KingdomShu-Chi A. Yeh, Wellman Center for Photomedicine, Massachusetts General Hospital, United States
Copyright © 2020 Iafrate and Fruhwirth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gilbert O. Fruhwirth, Z2lsYmVydC5mcnVod2lydGhAa2NsLmFjLnVr