 Nina Dirkx
Nina Dirkx Francesco Miceli
Francesco Miceli Maurizio Taglialatela
Maurizio Taglialatela Sarah Weckhuysen
Sarah Weckhuysen- 1Applied and Translational Neurogenomics Group, VIB Center for Molecular Neurology, Vlaams Instituut voor Biotechnologie, Antwerp, Belgium
- 2Section of Pharmacology, Department of Neuroscience, University of Naples Federico II, Naples, Italy
- 3Department of Translational Neurosciences, Faculty of Medicine and Health Sciences, University of Antwerp, Antwerp, Belgium
- 4Department of Neurology, Antwerp University Hospital, Antwerp, Belgium
Kv7.2 subunits encoded by the KCNQ2 gene constitute a critical molecular component of the M-current, a subthreshold voltage-gated potassium current controlling neuronal excitability by dampening repetitive action potential firing. Pathogenic loss-of-function variants in KCNQ2 have been linked to epilepsy since 1998, and there is ample functional evidence showing that dysfunction of the channel indeed results in neuronal hyperexcitability. The recent description of individuals with severe developmental delay with or without seizures due to pathogenic variants in KCNQ2 (KCNQ2-encephalopathy) reveals that Kv7.2 channels also have an important role in neurodevelopment. Kv7.2 channels are expressed already very early in the developing brain when key developmental processes such as proliferation, differentiation, and synaptogenesis play a crucial role in brain morphogenesis and maturation. In this review, we will discuss the available evidence for a role of Kv7.2 channels in these neurodevelopmental processes, focusing in particular on insights derived from KCNQ2-related human phenotypes, from the spatio-temporal expression of Kv7.2 and other Kv7 family member, and from cellular and rodent models, highlighting critical gaps and research strategies to be implemented in the future. Lastly, we propose a model which divides the M-current activity in three different developmental stages, correlating with the cell characteristics during these particular periods in neuronal development, and how this can be linked with KCNQ2-related disorders. Understanding these mechanisms can create opportunities for new targeted therapies for KCNQ2-encephalopathy.
Introduction
The KCNQ gene subfamily consist of five members (KCNQ1–5), all encoding voltage-gated potassium (K+) channel subunits (Kv7.1–5). The Kv7.1 subunit is predominantly expressed in the heart tissue, while Kv7.2-5 subunits are expressed most abundantly in the nervous system; Kv7.2 and Kv7.3 subunits expression is mostly restricted to the nervous system, whereas that of Kv7.4 and Kv7.5 subunits are more widespread, also including smooth and skeletal muscles (Sanguinetti et al., 1996; Schroeder et al., 2000; Cooper et al., 2001; Yeung et al., 2007). In adult neurons, Kv7 channels consist most frequently of tetramers containing Kv7.2 and Kv7.3, and more rarely Kv7.5 subunits (Wang et al., 1998; Schroeder et al., 2000). Independently of subunit composition, all Kv7 channels generate voltage-dependent K+ currents and represent the molecular basis of the M-current, a voltage-gated and K+-selective current which derives its name from its suppression upon activation of M1 muscarinic receptors (Brown and Adams, 1980). Muscarinic-dependent M-current suppression is caused by M1 receptor-dependent activation of phospholipase C, which hydrolyses phosphatidylinositol-4,5-bisphosphate (PIP2), a molecule which is necessary for Kv7 channel opening (Suh and Hille, 2002). The M-current is a slowly activating, non-inactivating, time‐ and voltage-dependent potassium current, which regulates the resting membrane potential (RMP) and dampens repetitive neuronal firing, thereby controlling neuronal excitability (Wang et al., 1998; Brown and Passmore, 2009).
During the last decade, an emerging number of studies pointed out that severe disruption of the function of Kv7.2, Kv7.3, and Kv7.5 subunits due to de pathogenic variants in the encoding gene leads to developmental and epileptic encephalopathy (DEE; Weckhuysen et al., 2012; Lehman et al., 2017; Lauritano et al., 2019). DEEs are a heterogeneous group of mostly neonatal-, infantile‐ or childhood-onset disorders characterized by severe, drug-resistant seizures, characteristic electroencephalographic (EEG) signatures, and different levels of developmental delay or regression, often with a poor prognosis. Although anti-seizure drugs can have some effect on seizure control in KCNQ-related DEEs, and several patients become seizure free after a few weeks to months, these patients invariably suffer from severe developmental delay (Pisano et al., 2015). A complex relationship is known to exist between epilepsy and neurodevelopment; before the most recent International League Against Epilepsy classification introducing the DEE term (Fisher et al., 2017), the most-commonly used term of “epileptic encephalopathy” was meant to highlight the classical belief that epileptic activity by itself is the primary cause of cognitive and developmental deterioration. However, some patients can have developmental impairment before seizure onset, or cognitive deterioration may progress despite complete seizure control; given the variable relationship between seizures and cognition, these conditions have been more recently referred to as DEEs. Thus, the possibility that the control exerted over neuronal excitability by Kv7.2, Kv7.3, and Kv7.5 subunits may play a role in neurodevelopment deserves to be investigated. In this review, we will focus on what is currently known about the role of Kv7.2 channels in neurodevelopment.
Human Pathogenic KCNQ2 Variants and Neurodevelopment
The best proof for a role of the M-current in neurodevelopment is the observation that pathogenic variants in KCNQ2 are responsible for a spectrum of neurodevelopmental disorders in humans. Already in 1998, inherited pathogenic variants in KCNQ2 were identified as a cause of benign familial neonatal epilepsy (BFNE; KCNQ2-B; Singh et al., 1998). This disorder is characterized by neonatal seizures, but development in these patients is generally normal (Maljevic and Lerche, 2014). However, the risk of seizure recurrence in BFNE individuals is 10–20 times higher than the general population (Plouin, 1994), and a few BFNE families with individuals affected with various degrees of developmental disability have been described (Dedek et al., 2003; Borgatti et al., 2004). In 2012, some heterozygous pathogenic variants in KCNQ2 were surprisingly shown to result in DEE, with a developmental delay ranging from mild to profound, nowadays referred to as “KCNQ2-encephalopathy” (KCNQ2-E; Weckhuysen et al., 2012). Pathogenic variants in KCNQ2 are shown to be the most common genetic cause of neonatal-onset DEE. A recent prospective cohort study showed that 83% of newborns with a DEE had an identifiable genetic etiology, of whom 40% had a pathogenic variant in KCNQ2 (Shellhaas et al., 2017). The incidence of KCNQ2-E is reported to be 1–3/100.000 births (Lopez-Rivera et al., 2020). By contrast, only a handful of DEE patients with pathogenic variants in KCNQ3 or KCNQ5 have been described (Rauch et al., 2012; Epi4k Consortium et al., 2013; Lehman et al., 2017; Ambrosino et al., 2018; Kothur et al., 2018; Lauritano et al., 2019; Rosti et al., 2019; Sands et al., 2019).
Interestingly, the degree of developmental delay does not completely correlate with the frequency or severity of the epileptic seizures (Weckhuysen et al., 2013). Furthermore, there seems to be a clear distinction between pathogenic variants leading to either KCNQ2-B or KCNQ2-E (Figure 1). Pathogenic variants associated with KCNQ2-B are truncating (splice, nonsense, and frameshift), whole gene deletions, or heterozygous missense variants resulting in haploinsufficiency with a 20–30% reduction of the M-current density when mutant subunits are expressed together with wild-type subunits in heterologous cell systems (Maljevic and Lerche, 2014). KCNQ2-E, on the other hand, is caused by heterozygous missense or in-frame indel mutations shown to have a dominant negative (DN; >50% reduction of the M-current density), or more rarely gain of function effect (GOF; >100% of the M-current density) when co-expressed with wild-type subunits (Miceli et al., 2013, 2015). Interestingly, KCNQ2 GOF variants can lead to distinct neurodevelopmental phenotypes. For example, patients with the mild GOF variant R198Q present infantile spasms without prior neonatal seizure (Millichap et al., 2017), whereas patients with the strong GOF variants R201C/H showed severe neonatal-onset encephalopathy without neonatal seizures but prominent startle-like myoclonus and a burst-suppression EEG pattern (Mulkey et al., 2017). KCNQ2 has also gained interest in the field of autism spectrum disorder (ASD). First of all, many of the reported KCNQ2-E patients show autistic features or have ASD as a behavioral comorbidity (Milh et al., 2013; Millichap et al., 2016). Second, KCNQ2 variants are also commonly identified in genetic studies on ASD cohorts (Jiang et al., 2013; Long et al., 2019). The mechanism by which two opposite effects on the M-channel, either a reduction of more than 50% of the M-current (DN effect) or an increase of the M-current density (GOF effect), both lead to developmental delay, is not known yet. However, this shows that the level of the M-current has to be regulated in a very precise manner to maintain homeostasis of the neuronal network.
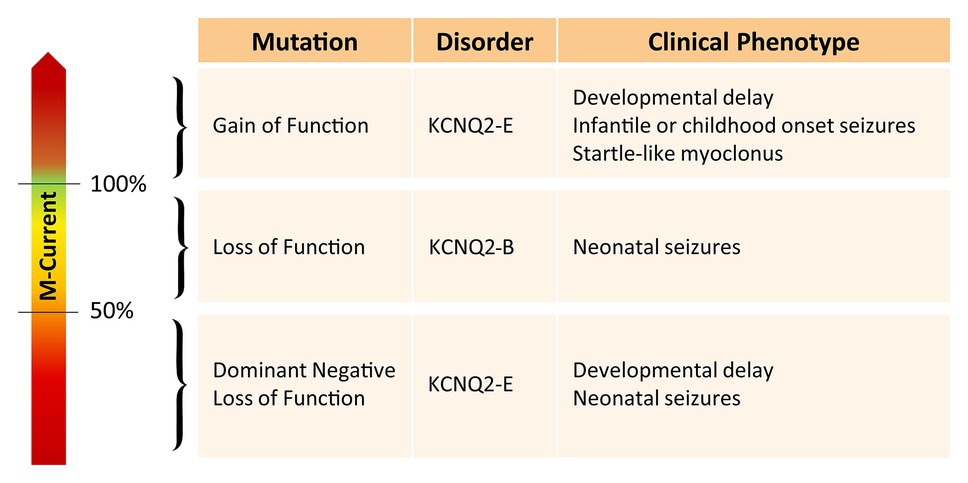
Figure 1. Overview of the effect of different KCNQ2 pathogenic variants on the M-current and their associated clinical phenotype.
Heterologous cellular systems have been used abundantly to study the functional consequences of human pathogenic KCNQ2 variants, and electrophysiological assays have been used as a read-out to assess genotype-phenotype correlations (Orhan et al., 2014; Abidi et al., 2015; Miceli et al., 2015; Millichap et al., 2017; Kim et al., 2018; Soldovieri et al., 2019). These systems have the advantage of being relatively straightforward, cost-efficient, and relatively high-throughput (Maljevic et al., 2017). The disadvantage of this approach is that the lack of native-neuronal environment may influence channel behavior in such a way that those variant-induced functional changes that affect channel subcellular localization or interaction with neuronally-expressed proteins may not become apparent. This was demonstrated by two recent studies on two different recurrent KCNQ2-E variants, A294V and R560W. Both generated similar currents as seen for KCNQ2-B variants when overexpressed in heterologous systems (Orhan et al., 2014; Abidi et al., 2015). The DN effect could only be observed in transfected neurons; the A294V variant altered distribution of Kv7.2 containing channels to the somato-dendritic compartment, while the R560W variant reduced Kv7.2 axonal surface expression and impaired gating regulation by phosphoinositides (Kim et al., 2018). This data underline the importance of a neuronal environment to fully understand the disease pathomechanism(s) triggered by each pathogenic KCNQ2 variant.
An interesting side note is that the Kv7.2 channel has several accessory proteins, many of which are also associated with neurodevelopmental disorders. SCN1B encodes for NaVβ1, a multifunctional molecule that is involved in modulation of sodium and potassium channels (including Kv7.2 channels), neurite outgrowth, axon pathfinding, and cell migration (Brackenbury and Isom, 2011; Nguyen et al., 2012). Bi-allelic pathogenic variants in the SCN1B gene are linked with DEE with phenotypic properties similar to Dravet syndrome, whereas heterozygous pathogenic variants lead to generalized epilepsy with febrile seizures plus (Scheffer et al., 2007; Aeby et al., 2019). Another important accessory protein of the Kv7.2 channel is Ankyrin-G, which is encoded by the ANK3 gene. Ankyrin-G has the essential role to anchor the Kv7.2 subunit at the axon initial segment (AIS) and nodes of Ranvier (Devaux et al., 2004; Pan et al., 2006). Heterozygous pathogenic missense variants have been linked to intellectual disability, ASD, bipolar disorder, and schizophrenia (Ferreira et al., 2008; Bi et al., 2012; Iqbal et al., 2013). Lastly, syntaxin-1A is a member of the soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptors (SNARE) superfamily. The SNARE protein complex facilitates the docking and fusion of synaptic vesicles at presynaptic membrane to enable neurotransmission (Rizo, 2018). Syntaxin-1A can bind to the C-terminus of Kv7.2 subunit, thereby decreasing open probability via facilitation of the interaction between the C-terminus and the N-terminus (Regev et al., 2009). To date, malfunction of syntaxin-1A itself is not known to be directly associated with neurodevelopmental disorders, however, pathogenic variants in the gene that encodes for the binding partner syntaxin binding protein 1 (STXBP1) are known to cause several DEE forms including a similar phenotype to KCNQ2-encephalopathy (Stamberger et al., 2016). STXBP1-Loss-of-function (LOF) mutations results in an increased binding of syntaxin-1A to the Kv7.2 channel, and thus, a decrease in M-current density (Devaux et al., 2017). Therefore, alterations in M-current density could be a convergence point between STXBP1-E and KCNQ2-E.
Spatiotemporal Expression of Kv7.2 Subunit and its Relation to Neurodevelopment
In this section, we will focus on Kv7.2 channel expression in the central nervous system. However, it should be noted that Kv7 channels are also expressed in the peripheral nervous system (PNS) (Passmore et al., 2003). To date, only few studies are available regarding the Kv7 subunit distribution in the PNS; nonetheless, most of those document a high Kv7.2 and Kv7.3 subunits expression in rat small-diameter dorsal root ganglionic neurons and non-myelinated C fibers (Passmore et al., 2003; King and Scherer, 2012; Huang et al., 2016). In rat peripheral sciatic nerves, most small‐ and medium-sized axons were shown to express Kv7.2/Kv7.3 heteromers, while in the largest sciatic nerve axons, mostly Kv7.2 homomers are observed (Cooper, 2011).
Kv7.2 Subunit Expression in Mature Neurons in the CNS
Kv7.2 channels are expressed widely throughout the brain in both excitatory and inhibitory neurons. In the adult human brain, immunohistochemistry shows a high level of Kv7.2 channels in the cortex, and lower levels in the hippocampus (Cooper et al., 2000). Within the neuron, Kv7.2 channels are located at the AIS, nodes of Ranvier, the soma, and presynaptic terminals (Cooper et al., 2001; Cooper, 2011). Depending on the Kv7.2 channel subcellular localization in the neuron, Kv7.2 subunits may play different roles. In fact, at the AIS and nodes of Ranvier, Kv7.2 has an important role in stabilizing the RMP and increasing the steady state availability of the sodium channels (Nav) (Battefeld et al., 2014). At the soma, it is a key player for the inhibition of repetitive action potential firing by attenuating after depolarization, and at presynaptic terminals of neurons, the Kv7.2 channel has been shown to regulate presynaptic neurotransmitter release (Martire et al., 2004; Luisi et al., 2009; Regev et al., 2009; Battefeld et al., 2014).
Temporal Regulation of KCNQ2 Transcripts and Kv7.2 Subunit Expression During Neurodevelopment
KCNQ2 Transcripts Expression During Neurodevelopment
KCNQ2 transcripts are already significantly expressed at the stem cell stage and during the whole course of neurodevelopment, with different transcripts showing differences in temporal expression. KCNQ2 has six curated transcripts based on the Reference Sequence (RefSeq) database. Transcript a (NM_172107) is the canonical transcript and counts 17 exons. The two most studied transcripts are transcript c (NM_004518; contains a distinct three prime untranslated region and is missing exon 9 and 12) and d (NM_172108; missing exon 12 and has a shorter exon 13), both generating functional channels (Orhan et al., 2014; Abidi et al., 2015; Ambrosino et al., 2015; Devaux et al., 2017). This suggests that exon 9 and 12 are not essential to form a functional channel. In support, no known KCNQ2-E pathogenic variant is located in these exons. Transcript b (NM_172106) and f (NM_001382235), both missing exon 12, are not well studied. Lastly, transcript e (NM_172109) is a shorter KCNQ2 transcript. It consists of exon 1 until exon 8 and has an alternative extension of exon 8 due to alternative splicing, which results in a frameshift. This shorter KCNQ2 transcript is expressed most abundantly in the fetal brain and its expression levels decrease during neuronal development. Therefore, we will refer to this transcript as the “short fetal transcript.” The opposite is seen for the longer KCNQ2 transcripts; they are upregulated during neuronal differentiation and increase with ongoing maturation (Smith et al., 2001); however, the probe used in this study recognizes all transcripts, thus, the specific role of each individual transcript cannot be assessed. Nonetheless, in support of this human brain tissue study, the transcriptome data from the LIBD stem cell browser1 show a similar temporal expression pattern for the different KCNQ2 transcripts in induced pluripotent stem cells (iPSCs) and iPSC-derived neuronal cells. This database shows the expression of all exons over the course of neuronal development, from self-renewal toward neuronal progenitor cells (NPCs) and neurons. Interestingly, the data show that exon 1 until 8 (present in the short fetal transcript) are already significantly expressed during the self-renewal stage, and expression stays relatively stable throughout neuronal development. Exon 10–11 and 13–17, but not exon 9 and 12, show a low expression in the self-renewal and NPC stage, with an increased expression when NPCs differentiate into neurons. Lastly, exon 9 and 12 only show a significant expression in neurons. Because transcript c is the only curated long transcript that is missing exon 9 and 12, this suggests that transcript c is the longer transcript seen in self-renewal and NPC stage. Indeed, the expression of the short fetal transcript and transcript c in iPSCs and embryonic stem cells (ESCs) was confirmed in other studies, where they even demonstrated that KCNQ2 shows the highest expression of all ion channels (Wang et al., 2005; Jiang et al., 2010; Linta et al., 2013). It should, however, be noted that we cannot rule out that additional alternative transcripts without exon 9 and 12 exist that are not included in the curated RefSeq transcripts.
So far, on a protein level, the expression of these different transcript has not been studied during early neurodevelopmental stages. The presence of a shorter transcript of ~40 kDa on western blot has only been observed in adult mouse brain using an antibody targeting the N-terminal of the Kv7.2 subunit (which is conserved among all the Kv7.2 transcripts; Cooper et al., 2001). The size of this transcript is similar to the predicted size of protein encoded by the short fetal transcript. Unfortunately, no subsequent effort has been made in investigating this shorter transcript further.
M-Current Density During Neurodevelopment
Functional Kv7.2-containing channels are expressed early in neurodevelopment, and their expression increases during development, thereby influencing neuronal differentiation by altering the neuronal membrane potential. Not with standing the ample evidence at the transcript level for expression of Kv7.2 transcripts c and e from the stem cell stage, evidence at the protein level is so far lacking. In this context, it is however interesting to note that HEK293T cells transfected with the short fetal transcript do not generate a detectable M-current. Co-transfection with the short fetal transcript and the canonical transcript a suppresses the M-current density compared to transfection with transcript a alone (Smith et al., 2001). At the time of this publication, the necessity of PIP2 binding for the opening of the channel was not known yet (Suh and Hille, 2002). Based on the knowledge we have today, the short fetal transcript misses two important PIP2 binding sites, located at the A-B linker and B-C linker (Hernandez et al., 2008; Kim et al., 2018; Zhang et al., 2020). Therefore, if the short fetal Kv7.2 subunit would reach the plasma membrane and would form tetramers, this would result in a strongly reduced open probability of the channel due to reduced PIP2 binding. However, it is very unlikely that the short fetal Kv7.2 subunit will reach the plasma membrane, as it also lacks the subunit-interaction domain (SID), important for tertramerization, and the calmodulin domain, important for the transport to the plasma membrane (Schwake et al., 2003; Cavaretta et al., 2014).
Kv7.2 subunits are nevertheless definitely present at the plasma membrane of immature neurons, and levels are shown to increase during differentiation (Safiulina et al., 2008; Figueiro-Silva et al., 2015; Telezhkin et al., 2018). A low M-current density and a weak Kv7.2 channel expression was shown in neonatal mouse P0 CA3 neurons using a C-terminal antibody recognizing the long transcripts. After the first postnatal weeks, both the M-current density and Kv7.2 channels markedly increase. The low expression of Kv7.2 channels in neonatal neurons allows intrinsic bursting and neuronal synchronization, leading to generation of giant depolarizing potential, which are important for hippocampal network stabilization (Safiulina et al., 2008). Moreover, during neuronal differentiation of iPSC-derived neurons, hyperpolarization of the RMP was shown to be correlated with the expression levels of Kv7.2/3 (Telezhkin et al., 2018). The RMP is a known regulator of cell fate, as highlighted in several studies (Sontheimer et al., 1989; Ng et al., 2010; Rao et al., 2015). To initiate differentiation, mouse embryonic stem cells (mESC) become more depolarized to enter the G1/G0 phase, after which the cell gradually hyperpolarizes during differentiation (Ng et al., 2010). Therefore, more mature differentiated neurons have a more hyperpolarized RMP compared to immature neurons. It is thus not surprising that forced membrane potential changes can alter the fate of cells. (MacFarlane and Sontheimer, 2000; Sundelacruz et al., 2008). This is in line with the observation that overexpression of Kv7.2/3 channels speeded up the maturation of the iPSC-derived neurons and facilitated spontaneous firing, by forcing the RMP to reach the optimal range for Nav channel activation (Telezhkin et al., 2018).
Based on the currently available studies outlined above, we hypothesize that there are three crucial, temporally-distinct phases in M-current density regulated by KCNQ2 transcript expression that coordinate proper neuronal development. A schematic overview is provided in Figure 2.
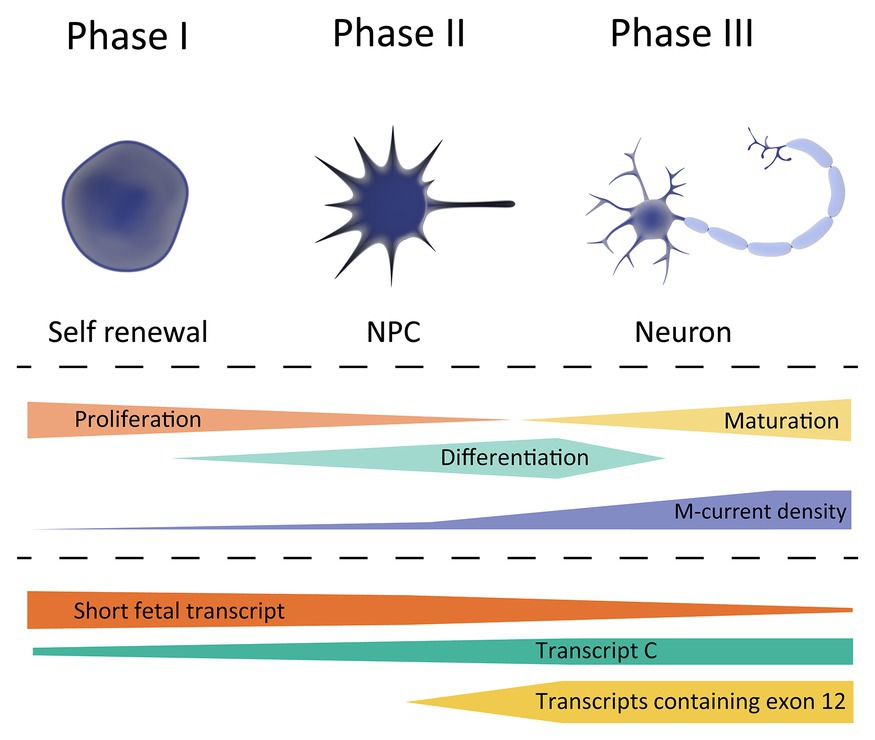
Figure 2. Schematic overview of KCNQ2 transcript expression during the different phases of neuronal development. Top, showing the different cell types during differentiation; stem cell (left), neuronal progenitor cell (NPC, middle), and neuron (right). Middle, showing the cell characteristics during neuronal development, and its correlation with the M-current density. Bottom, expression profile of the different KCNQ2 transcripts based on the data exported from the LIBD stem cell browser.
Phase 1, occurring at very early developmental stages in which a very low M-current density correlates with a strongly depolarized RMP of the cells, a necessary prerequisite to maintain a high proliferation to differentiation ratio. The KCNQ2 expression profile during this period, i.e., a high expression of short fetal KCNQ2 and low expression of long KCNQ2, makes us believe that the short fetal Kv7.2 isoform serves as guard to prevent a large M-current to be generated and prevent hyperpolarization of the cell. Therefore, it may be hypothesized that pathogenic variants in KCNQ2 that reduce the M-current will not lead to large clinical consequence during Phase 1.
Phase 2, in which an enhanced M-current density occurs as a consequence of a gradual increase in Kv7.2 expression, in order to support neuronal differentiation. A tight regulation of fetal and long Kv7.2 isoforms expression is necessary for a gradual increase in M-current density to hyperpolarize the RMP during differentiation. During the second phase, the first clinical consequence of Kv7.2-mediated developmental channelopathy can be expected.
Finally, Phase 3, when a stable M-current density and Kv7.2 expression supports neuronal maturation, corresponding mainly to the postnatal period. During this phase, Kv7.2 channels fulfill their better-known role in neurons; regulating the RMP, dampening repetitive firing, and controlling neurotransmitter release. In this phase, pathogenic variants in KCNQ2 would lead to the different (mostly-neonatal) phenotypes of KCNQ2-B or KCNQ2-E, depending on the extent of M-current functional derangement.
Temporal Regulation of Kv7.2 and Kv7.3 Subunit Expression
In addition to the temporal changes in Kv7.2 subunit expression during development, available data also suggest a time-dependent regulation of the expression of other members of the Kv7 subfamily, the Kv7.3 subunit in particular. In mice, Kv7.2 subunits are already significantly expressed at birth, and its levels are relatively stable or even increase postnatally (Tinel et al., 1998; Safiulina et al., 2008). Expression of Kv7.3 subunit, on the other hand, is very low at birth and gradually increases to eventually become even higher than that of Kv7.3 subunit (Tinel et al., 1998; Zhang et al., 2016). Therefore, the ratio of M-channels consisting of Kv7.2 homomers and Kv7.2/Kv7.3 heteromers will shift during development. A similar pattern of age-dependent expression, with Kv7.2 subunits being already detected in fetal life and Kv7.3 subunits increasing from late fetal life to infancy, has also been shown in human brain tissue (Figure 3; Kanaumi et al., 2008). Interestingly, the M-current density of the Kv7.2/Kv7.3 heteromers is 3.8 times bigger than Kv7.2 homomers (Selyanko et al., 2001). Furthermore, when studying DN KCNQ2-E variants in heterologous cell systems, the incorporation of the Kv7.3 subunit is able to (partially) rescue the mutational effect on the M-current observed in homomeric Kv7.2 channels (Gomis-Perez et al., 2019). Therefore, the differences in the temporal expression of Kv7.2 and Kv7.3 subunits probably contribute to the typical age-dependent characteristics of KCNQ2-related disorders: (i) the neonatal or even prenatal onset, at a time where Kv7.2 homomers are the predominant M-channels and (ii) the transient epileptic phenotype in which the increased expression of Kv7.3 subunits could play a role (Singh et al., 1998; Weckhuysen et al., 2012).
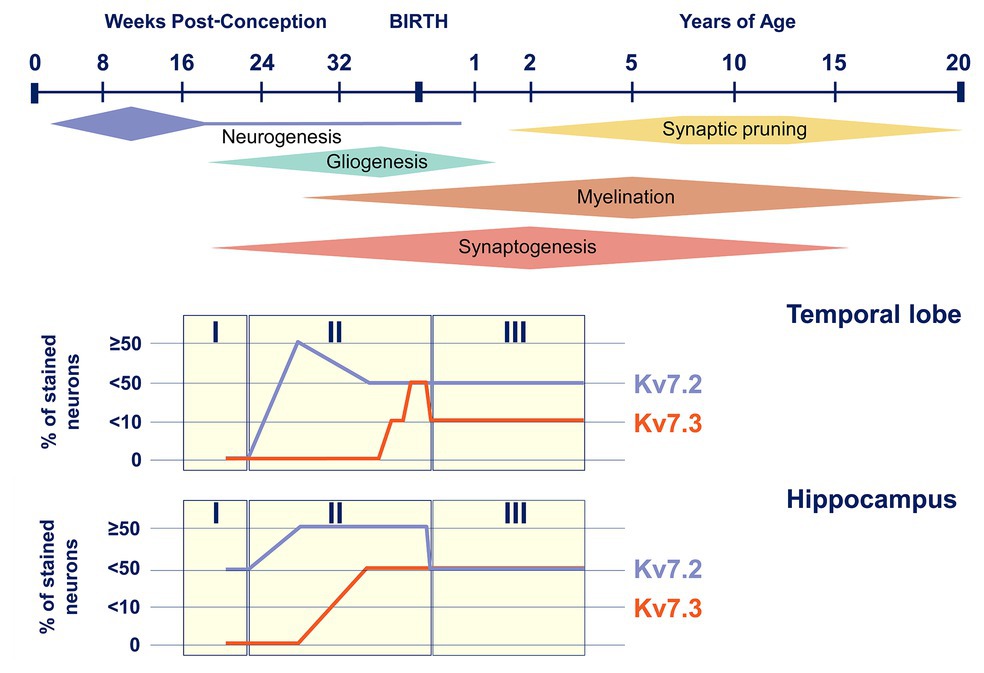
Figure 3. Kv7.2 and Kv7.3 expression during human brain development. Top, timeline of key neural developmental cellular processes in the human brain (adapted from Semple et al., 2013). Bottom, representation of Kv7.2 and Kv7.3 positive neurons in the temporal lobe and the hippocampus during the three neurodevelopmental phases (adapted from Kanaumi et al., 2008).
Kv7.2 Subunit Expression in Fetal Astrocytes
RNA sequencing data show preliminary evidence for early expression of kv7.2 subunits in human astrocytes. In a study on bulk RNA extracted from isolated cell types from mouse and both fetal and adult human cortex, a high expression of KCNQ2 was found in neurons and oligodendrocyte precursor cells of mice, whereas in humans, the highest expression of KCNQ2 was found in fetal astrocytes or astrocyte precursor cells, with rather low expression in neurons (Zhang et al., 2016). Open source, adult human brain, single-nuclei RNAseq databases such as the Allen Brain Map2 and the UCSC cell browser,3 on the other hand, show expression of KCNQ2 in neurons, but no clear evidence of KCNQ2 in astrocytes, suggesting expression in astrocytes is restricted to the early neurodevelopmental period. To date, no proof exists for a functional role of Kv7.2 channels in astrocytes. However, if this proves to be the case, this could open a whole new level of complexity to KCNQ2-related disorders, as astrocytes are known to contribute to the maintenance and formation of neuronal circuits and are known to play a role in the pathogenesis of several neurodevelopmental disorders, including ASD, Rett syndrome, and Fragile X (Molofsky et al., 2012; Sloan and Barres, 2014; Petrelli et al., 2016)
Neuronal Cell Models as a Tool to Study the Role of Kv7.2 Subunits in Neurodevelopment
A common method to study the function of M-channels is the use of pharmacological blockers or openers in in vitro or ex vivo cell models. Retigabine and flupirtine are the most commonly used openers and have a specificity for Kv7.2-5 subunits (Gribkoff, 2003; Yeung et al., 2008). In 2011, retigabine was approved as a treatment for epilepsy by the Food and Drug Administration and the European Medicines Agency; however, it was withdrawn from the market in June 2017 due to side effects including blue discoloration of skin and retina (Tompson et al., 2016). 10,10-bis(pyridin-4-ylmethyl)anthracen-9-one (XE991) and linopirdine are the most commonly used Kv7 channel blockers, both inhibiting all members of the Kv7 family. When interpreting these pharmacological studies, it is, thus, important to realize that none of these drugs are truly Kv7.2 subunit specific.
A clear distinction should also be made between studies using acute and chronic interventions. Acutely blocking the M-current in neurons is known to increase the intrinsic excitability, whereas chronic blocking gradually reduces the intrinsic excitability again. The chronic block induces a compensatory distal shift of the AIS and a relocation away from the soma of Nav and Kv7-channels, thus, again stabilizing network excitability (Lezmy et al., 2017). Chronical blocking of the M-current does not only influence excitability properties but has also been shown to alter neurodevelopmental processes, such as neurite outgrowth and synaptogenesis in different neuronal cell models (an overview of the studies is given in Table 1).
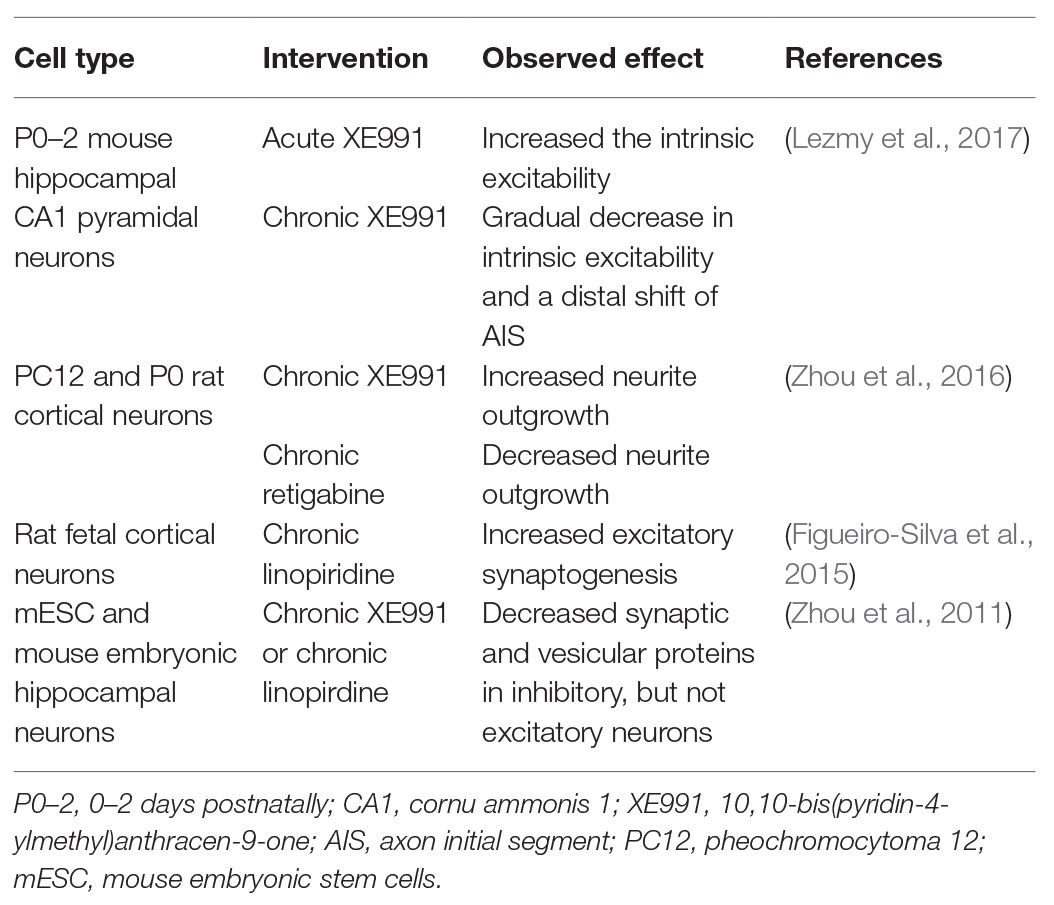
Table 1. Overview of in vitro studies that use pharmacological blockers or openers to chronically block the M-current.
Chronic blocking of the M-current with XE991 in PC12 and P0 rat cortical neurons was shown to result in an increased neurite outgrowth, while retigabine had the opposite effect (Zhou et al., 2016). Additionally, Kv7.2 channels have been described to play a key role in the regulation of synaptic plasticity through its interaction with neuronal pentraxin 1 (NP1) at presynaptic terminals of excitatory synapses and the axonal growth cones of fetal cortical neurons. Chronic blocking of Kv7.2 channels in these neurons increases excitatory synaptogenesis (Figueiro-Silva et al., 2015). Remarkably, another study using chronic XE991 treatment in differentiating mESC and embryonic hippocampal neurons was shown to decrease synaptic and vesicular proteins in inhibitory neurons but not in excitatory neurons (Zhou et al., 2011). A possible explanation for these opposite results could be the difference in studied cell type and cell maturity between the two studies, as Kv7 subunits and transcripts show distinct expression levels, spatial expression, and interactors depending on cell type and maturity stage.
Mouse Models as Tools to Study the Role of Kv7.2 Subunits in Neurodevelopment
To date, animal models have led to many fundamental discoveries of brain function and have given us a better understanding of the regulation of neurodevelopmental processes. Both knockout (KO) or knock-in (KI) mice models have been generated to study the role of Kv7.2 channels in vivo.
KCNQ2 KO Mouse Models
The first mouse models to study the function of Kv7.2 channels were KO models, some generated already 2 decades ago. They have taught us that specific neuronal subtypes are more vulnerable to changes in Kv7.2 expression levels and/or function, and especially that loss of Kv7.2 channels leads to complex homeostatic network changes that generally favor hyperexcitability. Heterozygous deletion of KCNQ2, engineered in attempt to mimic the human BFNE genotype, fails to prompt spontaneous seizures but lowers the seizure threshold to proconvulsants in adult mice (Watanabe et al., 2000; Yang et al., 2003). The focus of these earlier studies was mainly on seizure susceptibility, as Kv7.2 was not linked yet to behavioral abnormalities. With the current evidence that the Kv7.2 channel plays a role in neurodevelopment and that variants in KCNQ2 are enriched in ASD cohorts, the KCNQ2 KO model was also subjected to extensive behavioral analysis; an in-depth behavioral study, indeed, recently showed autism-associated behaviors such as a decrease in social behavior and enhanced repetitive behaviors in KCNQ2 heterozygous KO mice (Kim et al., 2020).
Homozygous KCNQ2 KO mice die within hours after birth, which highlights the importance of Kv7.2 channels for survival. To circumvent lethality, brain region-specific KCNQ2-null mice have been developed to enable studying complete Kv7.2 channel loss in specific cell types (Soh et al., 2014, 2018; Niday et al., 2017). Transgenic mice with conditional deletion of KCNQ2 in cortical pyramidal neurons are characterized by abnormal electrocorticogram activity and early death and showed increased excitability of CA1 pyramidal neurons. Interestingly, KCNQ2-null neurons did not show alterations in the RMP although they presented a decrease by ~85% of the M-current, suggesting that the contribution of K7.2 subunits to the RMP in p15-p20 mice cerebral cortical pyramidal neurons is small and/or that homeostatic adaptations occur to correct for the absence of Kv7.2 channels (Soh et al., 2014). A follow-up study furthermore showed neuronal hyperexcitability in layer 2/3 pyramidal neurons and alterations in action potential properties (AP), including increase in AP amplitude, decrease in medium after hyperpolarization (mAHP) and a (subsequent) increase in AP frequency (Niday et al., 2017). Specific ablation of Kv7.2 in interneurons, on the other hand, led to elevated excitability of parvalbumin positive (PV+), but not somatostatin positive, neurons. This, in turn, resulted in homeostatic potentiation of excitatory transmission in pyramidal neurons and increased seizure susceptibility in PV-KCNQ2 null mice (Soh et al., 2018). Importantly, this study shows that Kv7.2 loss induced an increase in interneuron excitability, which influences the course of excitatory network development. In this respect, it is important to point out that γ-aminobutyric acid (GABA) drives excitatory synapse maturation and formation in pyramidal neurons and that early in neurodevelopment, GABA is known to act as an excitatory, instead of an inhibitory neurotransmitter, due to high Na+ − K+ − Cl- cotransporters 1 (NKCC1) expression (Li and Xu, 2008; Le Magueresse and Monyer, 2013).
KI Mouse Models of Human Pathogenic KCNQ2 Variants
Most KI mouse models published so far express human KCNQ2-B variants and show a decreased seizure threshold similar to the heterozygous KO mice (Singh et al., 2008; Tomonoh et al., 2014). Interestingly, the BFNE KCNQ2-Y284C KI mouse also showed an increase in presynaptic GABA release at brain examination in the first postnatal week (Uchida et al., 2017). Because of the excitatory behavior of GABA early in neurodevelopment, the M-current is one of the most important regulators of neuronal inhibition during this period (Peters et al., 2005). Therefore, the combination of a reduction of the M-current and an increase in GABA release is believed to strongly contribute to the observed neonatal brain hyperexcitability in KCNQ2-B.
Until recently, the only genetic KCNQ2-E mouse model that had been published was a conditional overexpression model of the DN KCNQ2-G279S variant, inserted on the mouse X chromosome (Peters et al., 2005). This variant was modeled based on a DN variant in KCNQ1, and has, in the meantime, been described in a patient with KCNQ2-E (Sharawat et al., 2019). The mice displayed spontaneous seizures, as well as cognitive and behavioral impairments when scored with the Morris water maze and open-field tests. Morphological characterization of the hippocampus showed loss of mossy fiber terminals in the infrapyramidal layer. Remarkably, the observed phenotype was reversed when mutated KCNQ2 expression was turned off, using a Tet-off system, in the first postnatal weeks (Peters et al., 2005). In a follow-up study, the authors showed that bumetanide treatment during this same critical period also resulted in a rescue of the phenotype (Marguet et al., 2015). Bumetanide is a NKCC1 antagonist reducing the GABA-mediated depolarization that is seen in early neurodevelopment (Wang and Kriegstein, 2011). In conclusion, both the model with conditional KCNQ2 KO in PV+ neurons discussed above, the KCNQ2-Y284C KI and the KCNQ2-E overexpression mouse models, point toward an important role for GABA during early neurodevelopment in KCNQ2-related disorders.
Very recently, the first KI mouse model of a recurrent KCNQ2-E variant, KCNQ2-T274M, has been published, reproducing several phenotypic traits of human KCNQ2-E (Weckhuysen et al., 2012; Milh et al., 2020). These mice show learning and memory deficits when tested in the Morris water maze or the Barnes maze, as well as reduced exploratory behavior, suggesting that hippocampal dysfunction is involved in the disease pathology, although no abnormalities were observed upon morphological examination of the hippocampus. Furthermore, these mice present spontaneous clinical seizures starting from P20, which are rarely observed after P100 (Milh et al., 2020). This nicely mimics what is seen in patients, as most of them become seizure free a few months after seizure onset (Weckhuysen et al., 2012). This mouse model will contribute significantly to the understanding of KCNQ2-E phenotypic traits, which are often impossible to fully recapitulate in simplistic cell models and will be a very valuable tool for future drug discovery (Maljevic et al., 2017).
The Role of Kv7.2 Channels in Neurodegeneration
The role of the Kv7.2 channel in neurodegeneration is not the focus of this review; however, it is interesting to note that common genetic variation in KCNQ2 has recently been associated with risk of cognitive decline in healthy elderly (Bonham et al., 2018). Furthermore, some evidence exists for an (indirect) role of Kv7.2 in neurodegeneration disorders. Amyloid beta (Aβ) oligomers, involved in the pathogenesis of Alzheimer’s disease (AD), were reported to reduce Kv7.2 expression, resulting in a decrease of the M-current (Colom et al., 2011; Leao et al., 2012; Duran-Gonzalez et al., 2013; Mayordomo-Cava et al., 2015). In the medial septal neurons in mice, Aβ seems to reduce the M-current only in the glutamatergic neurons, resulting in increased firing and altered medial septal rhythmicity (Leao et al., 2012). As the medial septal nucleus is a key to generate the theta waves in the hippocampus, which is important for memory formation, alterations in the rhythmicity might underlie the cognitive deficits in AD patients (Winson, 1978; Seager et al., 2002). If the hypothesis is correct that hyperexcitability contributes to cognitive decline, specific Kv7.2 channels openers could be of benefit in AD patients. This could also be part of the explanation why no significant improvements in cognition were observed during a clinical trial of linopirdine, a Kv7.2 blocker, in AD patients (Rockwood et al., 1997). Furthermore, Kv7.2 channels openers could also be beneficial for Huntington’s disease (HD), as retigabine has been reported to rescue the electrophysiological and behavioral phenotype of transgenic HD model (Cao et al., 2015). Moreover, retigabine was described to block the hyperexcitability and to improve motor neuron survival of iPSC-derived motor neurons from amyotrophic lateral sclerosis patients (Wainger et al., 2014). Lastly, for Parkinson’s disease, several studies have shown that blocking of Kv7 channels with XE991 increased excitability of the dopaminergic neurons and has a neuroprotective effect toward dopaminergic degeneration in a mouse model of Parkinson’s disease (Shi et al., 2013; Liu et al., 2018). Altogether, those results support the idea that Kv7 subunits, possibly including Kv7.2 subunits, are interesting therapeutic target for different neurodegenerative diseases and cognitive impairment.
Further Perspectives
Since the discovery of the KCNQ2 gene in 1998, KCNQ2-encoded Kv7.2 subunits have been studied extensively (Singh et al., 1998). Although multiple lines of evidence show that Kv7.2 channels are involved in neurodevelopment, most studies have focused on the function of the Kv7.2 channels in a mature neuronal environment. Therefore, in this review, we highlight the importance of Kv7.2 in (early) neurodevelopment. Thanks to the increasing use of RNA-sequencing and the availability of transcriptome databases, a large amount of information on KCNQ2 expression has become available, leading to the consensus that an increasing expression of this gene is consistently observed during neuronal development (Safiulina et al., 2008; Telezhkin et al., 2018). Interestingly, multiple transcriptome datasets show evidence for the expression of a shorter fetal KCNQ2 transcript (transcript e) in iPSCs and neuronal derivatives, in addition to the longer transcript transcript c (Wang et al., 2005; Jiang et al., 2010; Linta et al., 2013). The presence of the short transcript was already reported in 2001 in brain, both at the transcript and protein levels; however, no follow up studies investigating this transcript were performed (Cooper et al., 2001; Smith et al., 2001). Unfortunately, functional evidence for the presence of Kv7.2 protein at stem cell and neuronal progenitor stages is currently lacking. Future studies confirming or negating the presence of Kv7.2 isoforms in stem cells will be needed to clarify this issue. In the meantime, and based on the available data, we introduce a model dividing the M-current density in three phases: (1) very low density important to maintain cell proliferation, (2) increasing M-current density to hyperpolarize the membrane and support neuronal differentiation, and (3) a stable high M-current density present in maturing or mature neurons to dampen repetitive firing and control neurotransmitter release.
Regarding human disease related to Kv7.2 channel dysfunction, more specifically KCNQ2-E, it is clear that pathology goes beyond a simple increased open or closed state of the channel, but includes impact on temporo-spatial expression levels, binding partners, and widespread homeostatic network changes that are difficult to study in simplified models. The recent publication of a KCNQ2-E KI mouse model is certainly promising and will be an important tool for further in-depth studies (Milh et al., 2020). Another promising model that would contribute to our understanding of the role of Kv7.2 channels in neurodevelopment is neuronal cultures derived from human iPSCs, as they provide the neuronal environment which is lacking in heterologous cell systems. Their ability to differentiate in several different brain cell types allows us to look not only at network changes but also to the contribution of different cell types to pathology. They are especially useful in the study of neurodevelopmental disorders, since they largely recapitulate fetal development (Tidball and Parent, 2016; Logan et al., 2019). Compared to monolayer neuronal cultures, brain organoids have the ability to foster even more complex cell interactions that stimulate cellular maturation, better recapitulating potential pathological cascades (Logan et al., 2019). Both 2D iPSC-derived neuronal cultures and 3D brain organoids have already successfully been used for the study of several neurodevelopmental disorders to identify disease mechanisms and potential new therapeutic targets (Fink and Levine, 2018; Russo et al., 2019). To date, no iPSC-derived neuronal model for KCNQ2-E has been reported in the literature yet. Furthermore, the current research performed to understand the role of the Kv7.2 channel in neurodevelopment has focused mainly on LOF of Kv7.2 channels, whereas KCNQ2 GOF variants give rise to an at least equally severe neurodevelopmental phenotype in human patients (Miceli et al., 2015). How both DN and GOF variants lead to neurodevelopmental delay is not understood yet. Future efforts comparing the effects of both DN and GOF variants on the neuronal network in vitro and in vivo are necessary to understand how Kv7.2 regulates neurodevelopment and can potentially open new doors for innovative therapeutic tools. Lastly, the development of Kv7 channel blockers and openers overcoming some of the pharmacokinetic and pharmacodynamic limitations shown by the available drugs (all of which have been synthesized and started to be developed well before the discovery of the KCNQ gene subfamily and the description of their multiple pathophysiological roles) could not only be beneficial for patients suffering from KCNQ2-B and KCNQ2-E but could also positively impact patients affected with multiple neurodegenerative disorders (Wainger et al., 2014; Kumar et al., 2016; Ostacolo et al., 2020).
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication. ND and SW designed the scope and structure of the review. ND, SW, FM, and MT wrote and edited the final manuscript.
Funding
ND receives support from FWO-SB (1S59219N) and SW receives support from FWO-FKM (1861419N) and GSKE. MT receives support from the Italian Ministry for University and Research (PRIN 2017ALCR7C). FM receives support from the Italian Ministry for University and Research (Project Scientific Independence of Researchers 2014 RBSI1444EM; PRIN 2017YH3SXK) and the University of Naples “Federico II” and Compagnia di San Paolo (STAR Program; project number 6-CSP-UNINA-120).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Abidi, A., Devaux, J. J., Molinari, F., Alcaraz, G., Michon, F. X., Sutera-Sardo, J., et al. (2015). A recurrent KCNQ2 pore mutation causing early onset epileptic encephalopathy has a moderate effect on M current but alters subcellular localization of Kv7 channels. Neurobiol. Dis. 80, 80–92. doi: 10.1016/j.nbd.2015.04.017
Aeby, A., Sculier, C., Bouza, A. A., Askar, B., Lederer, D., Schoonjans, A. S., et al. (2019). SCN1B-linked early infantile developmental and epileptic encephalopathy. Ann. Clin. Transl. Neurol. 6, 2354–2367. doi: 10.1002/acn3.50921
Ambrosino, P., Alaimo, A., Bartollino, S., Manocchio, L., De Maria, M., Mosca, I., et al. (2015). Epilepsy-causing mutations in Kv7.2 C-terminus affect binding and functional modulation by calmodulin. Biochim. Biophys. Acta 1852, 1856–1866. doi: 10.1016/j.bbadis.2015.06.012
Ambrosino, P., Freri, E., Castellotti, B., Soldovieri, M. V., Mosca, I., Manocchio, L., et al. (2018). Kv7.3 compound heterozygous variants in early onset encephalopathy reveal additive contribution of C-terminal residues to PIP2-dependent K(+) channel gating. Mol. Neurobiol. 55, 7009–7024. doi: 10.1007/s12035-018-0883-5
Battefeld, A., Tran, B. T., Gavrilis, J., Cooper, E. C., and Kole, M. H. (2014). Heteromeric Kv7.2/7.3 channels differentially regulate action potential initiation and conduction in neocortical myelinated axons. J. Neurosci. 34, 3719–3732. doi: 10.1523/JNEUROSCI.4206-13.2014
Bi, C., Wu, J., Jiang, T., Liu, Q., Cai, W., Yu, P., et al. (2012). Mutations of ANK3 identified by exome sequencing are associated with autism susceptibility. Hum. Mutat. 33, 1635–1638. doi: 10.1002/humu.22174
Bonham, L. W., Evans, D. S., Liu, Y., Cummings, S. R., Yaffe, K., and Yokoyama, J. S. (2018). Neurotransmitter pathway genes in cognitive decline during aging: evidence for GNG4 and KCNQ2 genes. Am. J. Alzheimers Dis. Other Dement. 33, 153–165. doi: 10.1177/1533317517739384
Borgatti, R., Zucca, C., Cavallini, A., Ferrario, M., Panzeri, C., Castaldo, P., et al. (2004). A novel mutation in KCNQ2 associated with BFNC, drug resistant epilepsy, and mental retardation. Neurology 63, 57–65. doi: 10.1212/01.wnl.0000132979.08394.6d
Brackenbury, W. J., and Isom, L. L. (2011). Na channel beta subunits: overachievers of the ion channel family. Front. Pharmacol. 2:53. doi: 10.3389/fphar.2011.00053
Brown, D. A., and Adams, P. R. (1980). Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature 283, 673–676. doi: 10.1038/283673a0
Brown, D. A., and Passmore, G. M. (2009). Neural KCNQ (Kv7) channels. Br. J. Pharmacol. 156, 1185–1195. doi: 10.1111/j.1476-5381.2009.00111.x
Cao, Y., Bartolome-Martin, D., Rotem, N., Rozas, C., Dellal, S. S., Chacon, M. A., et al. (2015). Rescue of homeostatic regulation of striatal excitability and locomotor activity in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. U. S. A. 112, 2239–2244. doi: 10.1073/pnas.1405748112
Cavaretta, J. P., Sherer, K. R., Lee, K. Y., Kim, E. H., Issema, R. S., and Chung, H. J. (2014). Polarized axonal surface expression of neuronal KCNQ potassium channels is regulated by calmodulin interaction with KCNQ2 subunit. PLoS One 9:e103655. doi: 10.1371/journal.pone.0103655
Colom, L. V., Castaneda, M. T., Hernandez, S., Perry, G., Jaime, S., and Touhami, A. (2011). Intrahippocampal amyloid-beta (1-40) injections injure medial septal neurons in rats. Curr. Alzheimer Res. 8, 832–840. doi: 10.2174/156720511798192763
Cooper, E. C. (2011). Made for “anchorin”: Kv7.2/7.3 (KCNQ2/KCNQ3) channels and the modulation of neuronal excitability in vertebrate axons. Semin. Cell Dev. Biol. 22, 185–192. doi: 10.1016/j.semcdb.2010.10.001
Cooper, E. C., Aldape, K. D., Abosch, A., Barbaro, N. M., Berger, M. S., Peacock, W. S., et al. (2000). Colocalization and coassembly of two human brain M-type potassium channel subunits that are mutated in epilepsy. Proc. Natl. Acad. Sci. U. S. A. 97, 4914–4919. doi: 10.1073/pnas.090092797
Cooper, E. C., Harrington, E., Jan, Y. N., and Jan, L. Y. (2001). M channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J. Neurosci. 21, 9529–9540. doi: 10.1523/JNEUROSCI.21-24-09529.2001
Dedek, K., Fusco, L., Teloy, N., and Steinlein, O. K. (2003). Neonatal convulsions and epileptic encephalopathy in an Italian family with a missense mutation in the fifth transmembrane region of KCNQ2. Epilepsy Res. 54, 21–27. doi: 10.1016/s0920-1211(03)00037-8
Devaux, J., Dhifallah, S., De Maria, M., Stuart-Lopez, G., Becq, H., Milh, M., et al. (2017). A possible link between KCNQ2‐ and STXBP1-related encephalopathies: STXBP1 reduces the inhibitory impact of syntaxin-1A on M current. Epilepsia 58, 2073–2084. doi: 10.1111/epi.13927
Devaux, J. J., Kleopa, K. A., Cooper, E. C., and Scherer, S. S. (2004). KCNQ2 is a nodal K+ channel. J. Neurosci. 24, 1236–1244. doi: 10.1523/JNEUROSCI.4512-03.2004
Duran-Gonzalez, J., Michi, E. D., Elorza, B., Perez-Cordova, M. G., Pacheco-Otalora, L. F., Touhami, A., et al. (2013). Amyloid beta peptides modify the expression of antioxidant repair enzymes and a potassium channel in the septohippocampal system. Neurobiol. Aging 34, 2071–2076. doi: 10.1016/j.neurobiolaging.2013.02.005
Epi4k ConsortiumEpilepsy Phenome/Genome ProjectAllen, A. S., Berkovic, S. F., Cossette, P., Delanty, N., et al. (2013). De novo mutations in epileptic encephalopathies. Nature 501, 217–221. doi: 10.1038/nature12439
Ferreira, M. A., O’Donovan, M. C., Meng, Y. A., Jones, I. R., Ruderfer, D. M., Jones, L., et al. (2008). Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet. 40, 1056–1058. doi: 10.1038/ng.209
Figueiro-Silva, J., Gruart, A., Clayton, K. B., Podlesniy, P., Abad, M. A., Gasull, X., et al. (2015). Neuronal pentraxin 1 negatively regulates excitatory synapse density and synaptic plasticity. J. Neurosci. 35, 5504–5521. doi: 10.1523/JNEUROSCI.2548-14.2015
Fink, J. J., and Levine, E. S. (2018). Uncovering true cellular phenotypes: using induced pluripotent stem cell-derived neurons to study early insults in neurodevelopmental disorders. Front. Neurol. 9:237. doi: 10.3389/fneur.2018.00237
Fisher, R. S., Cross, J. H., French, J. A., Higurashi, N., Hirsch, E., Jansen, F. E., et al. (2017). Operational classification of seizure types by the international league against epilepsy: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 58, 522–530. doi: 10.1111/epi.13670
Gomis-Perez, C., Urrutia, J., Marce-Grau, A., Malo, C., Lopez-Laso, E., Felipe-Rucian, A., et al. (2019). Homomeric Kv7.2 current suppression is a common feature in KCNQ2 epileptic encephalopathy. Epilepsia 60, 139–148. doi: 10.1111/epi.14609
Gribkoff, V. K. (2003). The therapeutic potential of neuronal KCNQ channel modulators. Expert Opin. Ther. Targets 7, 737–748. doi: 10.1517/14728222.7.6.737
Hernandez, C. C., Zaika, O., and Shapiro, M. S. (2008). A carboxy-terminal inter-helix linker as the site of phosphatidylinositol 4,5-bisphosphate action on Kv7 (M-type) K+ channels. J. Gen. Physiol. 132, 361–381. doi: 10.1085/jgp.200810007
Huang, D., Huang, S., Gao, H., Liu, Y., Qi, J., Chen, P., et al. (2016). Redox-dependent modulation of T-type Ca(2+) channels in sensory neurons contributes to acute anti-nociceptive effect of substance P. Antioxid. Redox Signal. 25, 233–251. doi: 10.1089/ars.2015.6560
Iqbal, Z., Vandeweyer, G., van der Voet, M., Waryah, A. M., Zahoor, M. Y., Besseling, J. A., et al. (2013). Homozygous and heterozygous disruptions of ANK3: at the crossroads of neurodevelopmental and psychiatric disorders. Hum. Mol. Genet. 22, 1960–1970. doi: 10.1093/hmg/ddt043
Jiang, P., Rushing, S. N., Kong, C. W., Fu, J., Lieu, D. K., Chan, C. W., et al. (2010). Electrophysiological properties of human induced pluripotent stem cells. Am. J. Phys. Cell Phys. 298, C486–C495. doi: 10.1152/ajpcell.00251.2009
Jiang, Y. H., Yuen, R. K., Jin, X., Wang, M., Chen, N., Wu, X., et al. (2013). Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am. J. Hum. Genet. 93, 249–263. doi: 10.1016/j.ajhg.2013.06.012
Kanaumi, T., Takashima, S., Iwasaki, H., Itoh, M., Mitsudome, A., and Hirose, S. (2008). Developmental changes in KCNQ2 and KCNQ3 expression in human brain: possible contribution to the age-dependent etiology of benign familial neonatal convulsions. Brain Dev. 30, 362–369. doi: 10.1016/j.braindev.2007.11.003
Kim, E. C., Patel, J., Zhang, J., Soh, H., Rhodes, J. S., Tzingounis, A. V., et al. (2020). Heterozygous loss of epilepsy gene KCNQ2 alters social, repetitive and exploratory behaviors. Genes Brain Behav. 19:e12599. doi: 10.1111/gbb.12599
Kim, E. C., Zhang, J., Pang, W., Wang, S., Lee, K. Y., Cavaretta, J. P., et al. (2018). Reduced axonal surface expression and phosphoinositide sensitivity in Kv7 channels disrupts their function to inhibit neuronal excitability in Kcnq2 epileptic encephalopathy. Neurobiol. Dis. 118, 76–93. doi: 10.1016/j.nbd.2018.07.004
King, C. H., and Scherer, S. S. (2012). Kv7.5 is the primary Kv7 subunit expressed in C-fibers. J. Comp. Neurol. 520, 1940–1950. doi: 10.1002/cne.23019
Kothur, K., Holman, K., Farnsworth, E., Ho, G., Lorentzos, M., Troedson, C., et al. (2018). Diagnostic yield of targeted massively parallel sequencing in children with epileptic encephalopathy. Seizure 59, 132–140. doi: 10.1016/j.seizure.2018.05.005
Kumar, M., Reed, N., Liu, R., Aizenman, E., Wipf, P., and Tzounopoulos, T. (2016). Synthesis and evaluation of potent KCNQ2/3-specific channel activators. Mol. Pharmacol. 89, 667–677. doi: 10.1124/mol.115.103200
Lauritano, A., Moutton, S., Longobardi, E., Tran Mau-Them, F., Laudati, G., Nappi, P., et al. (2019). A novel homozygous KCNQ3 loss-of-function variant causes non-syndromic intellectual disability and neonatal-onset pharmacodependent epilepsy. Epilepsia Open 4, 464–475. doi: 10.1002/epi4.12353
Le Magueresse, C., and Monyer, H. (2013). GABAergic interneurons shape the functional maturation of the cortex. Neuron 77, 388–405. doi: 10.1016/j.neuron.2013.01.011
Leao, R. N., Colom, L. V., Borgius, L., Kiehn, O., and Fisahn, A. (2012). Medial septal dysfunction by Abeta-induced KCNQ channel-block in glutamatergic neurons. Neurobiol. Aging 33, 2046–2061. doi: 10.1016/j.neurobiolaging.2011.07.013
Lehman, A., Thouta, S., Mancini, G. M. S., Naidu, S., van Slegtenhorst, M., McWalter, K., et al. (2017). Loss-of-function and gain-of-function mutations in KCNQ5 cause intellectual disability or epileptic encephalopathy. Am. J. Hum. Genet. 101, 65–74. doi: 10.1016/j.ajhg.2017.05.016
Lezmy, J., Lipinsky, M., Khrapunsky, Y., Patrich, E., Shalom, L., Peretz, A., et al. (2017). M-current inhibition rapidly induces a unique CK2-dependent plasticity of the axon initial segment. Proc. Natl. Acad. Sci. U. S. A. 114, E10234–E10243. doi: 10.1073/pnas.1708700114
Li, K., and Xu, E. (2008). The role and the mechanism of gamma-aminobutyric acid during central nervous system development. Neurosci. Bull. 24, 195–200. doi: 10.1007/s12264-008-0109-3
Linta, L., Stockmann, M., Lin, Q., Lechel, A., Proepper, C., Boeckers, T. M., et al. (2013). Microarray-based comparisons of ion channel expression patterns: human keratinocytes to reprogrammed hiPSCs to differentiated neuronal and cardiac progeny. Stem Cells Int. 2013:784629. doi: 10.1155/2013/784629
Liu, H., Jia, L., Chen, X., Shi, L., and Xie, J. (2018). The Kv7/KCNQ channel blocker XE991 protects nigral dopaminergic neurons in the 6-hydroxydopamine rat model of Parkinson’s disease. Brain Res. Bull. 137, 132–139. doi: 10.1016/j.brainresbull.2017.11.011
Logan, S., Arzua, T., Canfield, S. G., Seminary, E. R., Sison, S. L., Ebert, A. D., et al. (2019). Studying human neurological disorders using induced pluripotent stem cells: from 2D monolayer to 3d organoid and blood brain barrier models. Compr. Physiol. 9, 565–611. doi: 10.1002/cphy.c180025
Long, S., Zhou, H., Li, S., Wang, T., Ma, Y., Li, C., et al. (2019). The clinical and genetic features of co-occurring epilepsy and autism spectrum disorder in chinese children. Front. Neurol. 10:505. doi: 10.3389/fneur.2019.00505
Lopez-Rivera, J. A., Perez-Palma, E., Symonds, J., Lindy, A. S., McKnight, D. A., Leu, C., et al. (2020). A catalogue of new incidence estimates of monogenic neurodevelopmental disorders caused by de novo variants. Brain 143, 1099–1105. doi: 10.1093/brain/awaa051
Luisi, R., Panza, E., Barrese, V., Iannotti, F. A., Viggiano, D., Secondo, A., et al. (2009). Activation of pre-synaptic M-type K+ channels inhibits [3H]D-aspartate release by reducing Ca2+ entry through P/Q-type voltage-gated Ca2+ channels. J. Neurochem. 109, 168–181. doi: 10.1111/j.1471-4159.2009.05945.x
MacFarlane, S. N., and Sontheimer, H. (2000). Changes in ion channel expression accompany cell cycle progression of spinal cord astrocytes. Glia 30, 39–48. doi: 10.1002/(sici)1098-1136(200003)30:1<39::aid-glia5>3.0.co;2-s
Maljevic, S., and Lerche, H. (2014). Potassium channel genes and benign familial neonatal epilepsy. Prog. Brain Res. 213, 17–53. doi: 10.1016/B978-0-444-63326-2.00002-8
Maljevic, S., Reid, C. A., and Petrou, S. (2017). Models for discovery of targeted therapy in genetic epileptic encephalopathies. J. Neurochem. 143, 30–48. doi: 10.1111/jnc.14134
Marguet, S. L., Le-Schulte, V. T., Merseburg, A., Neu, A., Eichler, R., Jakovcevski, I., et al. (2015). Treatment during a vulnerable developmental period rescues a genetic epilepsy. Nat. Med. 21, 1436–1444. doi: 10.1038/nm.3987
Martire, M., Castaldo, P., D’Amico, M., Preziosi, P., Annunziato, L., and Taglialatela, M. (2004). M channels containing KCNQ2 subunits modulate norepinephrine, aspartate, and GABA release from hippocampal nerve terminals. J. Neurosci. 24, 592–597. doi: 10.1523/JNEUROSCI.3143-03.2004
Mayordomo-Cava, J., Yajeya, J., Navarro-Lopez, J. D., and Jimenez-Diaz, L. (2015). Amyloid-beta(25-35) modulates the expression of GirK and KCNQ channel genes in the hippocampus. PLoS One 10:e0134385. doi: 10.1371/journal.pone.0134385
Miceli, F., Soldovieri, M. V., Ambrosino, P., Barrese, V., Migliore, M., Cilio, M. R., et al. (2013). Genotype-phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of K(v)7.2 potassium channel subunits. Proc. Natl. Acad. Sci. U. S. A. 110, 4386–4391. doi: 10.1073/pnas.1216867110
Miceli, F., Soldovieri, M. V., Ambrosino, P., De Maria, M., Migliore, M., Migliore, R., et al. (2015). Early-onset epileptic encephalopathy caused by gain-of-function mutations in the voltage sensor of Kv7.2 and Kv7.3 potassium channel subunits. J. Neurosci. 35, 3782–3793. doi: 10.1523/JNEUROSCI.4423-14.2015
Milh, M., Boutry-Kryza, N., Sutera-Sardo, J., Mignot, C., Auvin, S., Lacoste, C., et al. (2013). Similar early characteristics but variable neurological outcome of patients with a de novo mutation of KCNQ2. Orphanet J. Rare Dis. 8:80. doi: 10.1186/1750-1172-8-80
Milh, M., Roubertoux, P., Biba, N., Chavany, J., Spiga Ghata, A., Fulachier, C., et al. (2020). A knock-in mouse model for KCNQ2-related epileptic encephalopathy displays spontaneous generalized seizures and cognitive impairment. Epilepsia 61, 868–878. doi: 10.1111/epi.16494
Millichap, J. J., Miceli, F., De Maria, M., Keator, C., Joshi, N., Tran, B., et al. (2017). Infantile spasms and encephalopathy without preceding neonatal seizures caused by KCNQ2 R198Q, a gain-of-function variant. Epilepsia 58, e10–e15. doi: 10.1111/epi.13601
Millichap, J. J., Park, K. L., Tsuchida, T., Ben-Zeev, B., Carmant, L., Flamini, R., et al. (2016). KCNQ2 encephalopathy: features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol. Genet. 2:e96. doi: 10.1212/NXG.0000000000000096
Molofsky, A. V., Krencik, R., Ullian, E. M., Tsai, H. H., Deneen, B., Richardson, W. D., et al. (2012). Astrocytes and disease: a neurodevelopmental perspective. Genes Dev. 26, 891–907. doi: 10.1101/gad.188326.112
Mulkey, S. B., Ben-Zeev, B., Nicolai, J., Carroll, J. L., Grønborg, S., Jiang, Y. H., et al. (2017). Neonatal nonepileptic myoclonus is a prominent clinical feature of KCNQ2 gain-of-function variants R201C and R201H. Epilepsia 58, 436–445. doi: 10.1111/epi.13676
Ng, S. Y., Chin, C. H., Lau, Y. T., Luo, J., Wong, C. K., Bian, Z. X., et al. (2010). Role of voltage-gated potassium channels in the fate determination of embryonic stem cells. J. Cell. Physiol. 224, 165–177. doi: 10.1002/jcp.22113
Nguyen, H. M., Miyazaki, H., Hoshi, N., Smith, B. J., Nukina, N., Goldin, A. L., et al. (2012). Modulation of voltage-gated K+ channels by the sodium channel beta1 subunit. Proc. Natl. Acad. Sci. U. S. A. 109, 18577–18582. doi: 10.1073/pnas.1209142109
Niday, Z., Hawkins, V. E., Soh, H., Mulkey, D. K., and Tzingounis, A. V. (2017). Epilepsy-associated KCNQ2 channels regulate multiple intrinsic properties of layer 2/3 pyramidal neurons. J. Neurosci. 37, 576–586. doi: 10.1523/JNEUROSCI.1425-16.2016
Orhan, G., Bock, M., Schepers, D., Ilina, E. I., Reichel, S. N., Loffler, H., et al. (2014). Dominant-negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann. Neurol. 75, 382–394. doi: 10.1002/ana.24080
Ostacolo, C., Miceli, F., Di Sarno, V., Nappi, P., Iraci, N., Soldovieri, M. V., et al. (2020). Synthesis and pharmacological characterization of conformationally restricted retigabine analogues as novel neuronal Kv7 channel activators. J. Med. Chem. 63, 163–185. doi: 10.1021/acs.jmedchem.9b00796
Pan, Z., Kao, T., Horvath, Z., Lemos, J., Sul, J. Y., Cranstoun, S. D., et al. (2006). A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J. Neurosci. 26, 2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006
Passmore, G. M., Selyanko, A. A., Mistry, M., Al-Qatari, M., Marsh, S. J., Matthews, E. A., et al. (2003). KCNQ/M currents in sensory neurons: significance for pain therapy. J. Neurosci. 23, 7227–7236. doi: 10.1523/JNEUROSCI.23-18-07227.2003
Peters, H. C., Hu, H., Pongs, O., Storm, J. F., and Isbrandt, D. (2005). Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat. Neurosci. 8, 51–60. doi: 10.1038/nn1375
Petrelli, F., Pucci, L., and Bezzi, P. (2016). Astrocytes and microglia and their potential link with autism spectrum disorders. Front. Cell. Neurosci. 10:21. doi: 10.3389/fncel.2016.00021
Pisano, T., Numis, A. L., Heavin, S. B., Weckhuysen, S., Angriman, M., Suls, A., et al. (2015). Early and effective treatment of KCNQ2 encephalopathy. Epilepsia 56, 685–691. doi: 10.1111/epi.12984
Plouin, P. (1994). “Benign familial neonatal convulsions” in Idiopathic generalized epilepsies: Clinical, experimental and genetic aspects. eds. A. Malafosse, E. Hirsch, C. Marescaux, D. Broglin, and R. Bernasconi (London: John Libbey and Co., Ltd.), 39–44.
Rao, V. R., Perez-Neut, M., Kaja, S., and Gentile, S. (2015). Voltage-gated ion channels in cancer cell proliferation. Cancer 7, 849–875. doi: 10.3390/cancers7020813
Rauch, A., Wieczorek, D., Graf, E., Wieland, T., Endele, S., Schwarzmayr, T., et al. (2012). Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380, 1674–1682. doi: 10.1016/S0140-6736(12)61480-9
Regev, N., Degani-Katzav, N., Korngreen, A., Etzioni, A., Siloni, S., Alaimo, A., et al. (2009). Selective interaction of syntaxin 1A with KCNQ2: possible implications for specific modulation of presynaptic activity. PLoS One 4:e6586. doi: 10.1371/journal.pone.0006586
Rizo, J. (2018). Mechanism of neurotransmitter release coming into focus. Protein Sci. 27, 1364–1391. doi: 10.1002/pro.3445
Rockwood, K., Beattie, B. L., Eastwood, M. R., Feldman, H., Mohr, E., Pryse-Phillips, W., et al. (1997). A randomized, controlled trial of linopirdine in the treatment of Alzheimer’s disease. Can. J. Neurol. Sci. 24, 140–145. doi: 10.1017/s031716710002148x
Rosti, G., Tassano, E., Bossi, S., Divizia, M. T., Ronchetto, P., Servetti, M., et al. (2019). Intragenic duplication of KCNQ5 gene results in aberrant splicing leading to a premature termination codon in a patient with intellectual disability. Eur. J. Med. Genet. 62:103555. doi: 10.1016/j.ejmg.2018.10.007
Russo, F. B., Brito, A., de Freitas, A. M., Castanha, A., de Freitas, B. C., and Beltrao-Braga, P. C. B. (2019). The use of iPSC technology for modeling autism spectrum disorders. Neurobiol. Dis. 130:104483. doi: 10.1016/j.nbd.2019.104483
Safiulina, V. F., Zacchi, P., Taglialatela, M., Yaari, Y., and Cherubini, E. (2008). Low expression of Kv7/M channels facilitates intrinsic and network bursting in the developing rat hippocampus. J. Physiol. 586, 5437–5453. doi: 10.1113/jphysiol.2008.156257
Sands, T. T., Miceli, F., Lesca, G., Beck, A. E., Sadleir, L. G., Arrington, D. K., et al. (2019). Autism and developmental disability caused by KCNQ3 gain-of-function variants. Ann. Neurol. 86, 181–192. doi: 10.1002/ana.25522
Sanguinetti, M. C., Curran, M. E., Zou, A., Shen, J., Spector, P. S., Atkinson, D. L., et al. (1996). Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384, 80–83. doi: 10.1038/384080a0
Scheffer, I. E., Harkin, L. A., Grinton, B. E., Dibbens, L. M., Turner, S. J., Zielinski, M. A., et al. (2007). Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B mutations. Brain 130, 100–109. doi: 10.1093/brain/awl272
Schroeder, B. C., Hechenberger, M., Weinreich, F., Kubisch, C., and Jentsch, T. J. (2000). KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J. Biol. Chem. 275, 24089–24095. doi: 10.1074/jbc.M003245200
Schwake, M., Jentsch, T. J., and Friedrich, T. (2003). A carboxy-terminal domain determines the subunit specificity of KCNQ K+ channel assembly. EMBO Rep. 4, 76–81. doi: 10.1038/sj.embor.embor715
Seager, M. A., Johnson, L. D., Chabot, E. S., Asaka, Y., and Berry, S. D. (2002). Oscillatory brain states and learning: impact of hippocampal theta-contingent training. Proc. Natl. Acad. Sci. U. S. A. 99, 1616–1620. doi: 10.1073/pnas.032662099
Selyanko, A. A., Hadley, J. K., and Brown, D. A. (2001). Properties of single M-type KCNQ2/KCNQ3 potassium channels expressed in mammalian cells. J. Physiol. 534, 15–24. doi: 10.1111/j.1469-7793.2001.00015.x
Semple, B. D., Blomgren, K., Gimlin, K., Ferriero, D. M., and Noble-Haeusslein, L. J. (2013). Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 106-107, 1–16. doi: 10.1016/j.pneurobio.2013.04.001
Sharawat, I. K., Kasinathan, A., Sahu, J. K., and Sankhyan, N. (2019). Response to carbamazepine in KCNQ2 related early infantile epileptic encephalopathy. Indian J. Pediatr. 86, 301–302. doi: 10.1007/s12098-018-2796-8
Shellhaas, R. A., Wusthoff, C. J., Tsuchida, T. N., Glass, H. C., Chu, C. J., Massey, S. L., et al. (2017). Profile of neonatal epilepsies: characteristics of a prospective US cohort. Neurology 89, 893–899. doi: 10.1212/WNL.0000000000004284
Shi, L., Bian, X., Qu, Z., Ma, Z., Zhou, Y., Wang, K., et al. (2013). Peptide hormone ghrelin enhances neuronal excitability by inhibition of Kv7/KCNQ channels. Nat. Commun. 4:1435. doi: 10.1038/ncomms2439
Singh, N. A., Charlier, C., Stauffer, D., DuPont, B. R., Leach, R. J., Melis, R., et al. (1998). A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 18, 25–29. doi: 10.1038/ng0198-25
Singh, N. A., Otto, J. F., Dahle, E. J., Pappas, C., Leslie, J. D., Vilaythong, A., et al. (2008). Mouse models of human KCNQ2 and KCNQ3 mutations for benign familial neonatal convulsions show seizures and neuronal plasticity without synaptic reorganization. J. Physiol. 586, 3405–3423. doi: 10.1113/jphysiol.2008.154971
Sloan, S. A., and Barres, B. A. (2014). Mechanisms of astrocyte development and their contributions to neurodevelopmental disorders. Curr. Opin. Neurobiol. 27, 75–81. doi: 10.1016/j.conb.2014.03.005
Smith, J. S., Iannotti, C. A., Dargis, P., Christian, E. P., and Aiyar, J. (2001). Differential expression of kcnq2 splice variants: implications to m current function during neuronal development. J. Neurosci. 21, 1096–1103. doi: 10.1523/JNEUROSCI.21-04-01096.2001
Soh, H., Pant, R., LoTurco, J. J., and Tzingounis, A. V. (2014). Conditional deletions of epilepsy-associated KCNQ2 and KCNQ3 channels from cerebral cortex cause differential effects on neuronal excitability. J. Neurosci. 34, 5311–5321. doi: 10.1523/JNEUROSCI.3919-13.2014
Soh, H., Park, S., Ryan, K., Springer, K., Maheshwari, A., and Tzingounis, A. V. (2018). Deletion of KCNQ2/3 potassium channels from PV+ interneurons leads to homeostatic potentiation of excitatory transmission. elife 7:e38617. doi: 10.7554/eLife.38617
Soldovieri, M. V., Ambrosino, P., Mosca, I., Miceli, F., Franco, C., Canzoniero, L. M. T., et al. (2019). Epileptic encephalopathy in a patient with a novel variant in the Kv7.2 S2 transmembrane segment: clinical, genetic, and functional features. Int. J. Mol. Sci. 20:3382. doi: 10.3390/ijms20143382
Sontheimer, H., Trotter, J., Schachner, M., and Kettenmann, H. (1989). Channel expression correlates with differentiation stage during the development of oligodendrocytes from their precursor cells in culture. Neuron 2, 1135–1145. doi: 10.1016/0896-6273(89)90180-3
Stamberger, H., Nikanorova, M., Willemsen, M. H., Accorsi, P., Angriman, M., Baier, H., et al. (2016). STXBP1 encephalopathy: a neurodevelopmental disorder including epilepsy. Neurology 86, 954–962. doi: 10.1212/WNL.0000000000002457
Suh, B. C., and Hille, B. (2002). Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron 35, 507–520. doi: 10.1016/s0896-6273(02)00790-0
Sundelacruz, S., Levin, M., and Kaplan, D. L. (2008). Membrane potential controls adipogenic and osteogenic differentiation of mesenchymal stem cells. PLoS One 3:e3737. doi: 10.1371/journal.pone.0003737
Telezhkin, V., Straccia, M., Yarova, P., Pardo, M., Yung, S., Vinh, N. N., et al. (2018). Kv7 channels are upregulated during striatal neuron development and promote maturation of human iPSC-derived neurons. Pflugers Arch. 470, 1359–1376. doi: 10.1007/s00424-018-2155-7
Tidball, A. M., and Parent, J. M. (2016). Concise review: exciting cells: modeling genetic epilepsies with patient-derived induced pluripotent stem cells. Stem Cells 34, 27–33. doi: 10.1002/stem.2203
Tinel, N., Lauritzen, I., Chouabe, C., Lazdunski, M., and Borsotto, M. (1998). The KCNQ2 potassium channel: splice variants, functional and developmental expression. Brain localization and comparison with KCNQ3. FEBS Lett. 438, 171–176. doi: 10.1016/s0014-5793(98)01296-4
Tomonoh, Y., Deshimaru, M., Araki, K., Miyazaki, Y., Arasaki, T., Tanaka, Y., et al. (2014). The kick-in system: a novel rapid knock-in strategy. PLoS One 9:e88549. doi: 10.1371/journal.pone.0088549
Tompson, D. J., Buraglio, M., Andrews, S. M., and Wheless, J. W. (2016). Adolescent clinical development of ezogabine/retigabine as adjunctive therapy for partial-onset seizures: pharmacokinetics and tolerability. J. Pediatr. Pharmacol. Ther. 21, 404–412. doi: 10.5863/1551-6776-21.5.404
Uchida, T., Lossin, C., Ihara, Y., Deshimaru, M., Yanagawa, Y., Koyama, S., et al. (2017). Abnormal gamma-aminobutyric acid neurotransmission in a Kcnq2 model of early onset epilepsy. Epilepsia 58, 1430–1439. doi: 10.1111/epi.13807
Wainger, B. J., Kiskinis, E., Mellin, C., Wiskow, O., Han, S. S., Sandoe, J., et al. (2014). Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 7, 1–11. doi: 10.1016/j.celrep.2014.03.019
Wang, D. D., and Kriegstein, A. R. (2011). Blocking early GABA depolarization with bumetanide results in permanent alterations in cortical circuits and sensorimotor gating deficits. Cereb. Cortex 21, 574–587. doi: 10.1093/cercor/bhq124
Wang, H. S., Pan, Z., Shi, W., Brown, B. S., Wymore, R. S., Cohen, I. S., et al. (1998). KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282, 1890–1893. doi: 10.1126/science.282.5395.1890
Wang, K., Xue, T., Tsang, S. Y., Van Huizen, R., Wong, C. W., Lai, K. W., et al. (2005). Electrophysiological properties of pluripotent human and mouse embryonic stem cells. Stem Cells 23, 1526–1534. doi: 10.1634/stemcells.2004-0299
Watanabe, H., Nagata, E., Kosakai, A., Nakamura, M., Yokoyama, M., Tanaka, K., et al. (2000). Disruption of the epilepsy KCNQ2 gene results in neural hyperexcitability. J. Neurochem. 75, 28–33. doi: 10.1046/j.1471-4159.2000.0750028.x
Weckhuysen, S., Ivanovic, V., Hendrickx, R., Van Coster, R., Hjalgrim, H., Moller, R. S., et al. (2013). Extending the KCNQ2 encephalopathy spectrum: clinical and neuroimaging findings in 17 patients. Neurology 81, 1697–1703. doi: 10.1212/01.wnl.0000435296.72400.a1
Weckhuysen, S., Mandelstam, S., Suls, A., Audenaert, D., Deconinck, T., Claes, L. R., et al. (2012). KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 71, 15–25. doi: 10.1002/ana.22644
Winson, J. (1978). Loss of hippocampal theta rhythm results in spatial memory deficit in the rat. Science 201, 160–163. doi: 10.1126/science.663646
Yang, Y., Beyer, B. J., Otto, J. F., O’Brien, T. P., Letts, V. A., White, H. S., et al. (2003). Spontaneous deletion of epilepsy gene orthologs in a mutant mouse with a low electroconvulsive threshold. Hum. Mol. Genet. 12, 975–984. doi: 10.1093/hmg/ddg118
Yeung, S. Y., Pucovsky, V., Moffatt, J. D., Saldanha, L., Schwake, M., Ohya, S., et al. (2007). Molecular expression and pharmacological identification of a role for K(v)7 channels in murine vascular reactivity. Br. J. Pharmacol. 151, 758–770. doi: 10.1038/sj.bjp.0707284
Yeung, S., Schwake, M., Pucovsky, V., and Greenwood, I. (2008). Bimodal effects of the Kv7 channel activator retigabine on vascular K+ currents. Br. J. Pharmacol. 155, 62–72. doi: 10.1038/bjp.2008.231
Zhang, J., Kim, E. C., Chen, C., Procko, E., Pant, S., Lam, K., et al. (2020). Identifying mutation hotspots reveals pathogenetic mechanisms of KCNQ2 epileptic encephalopathy. Sci. Rep. 10:4756. doi: 10.1038/s41598-020-61697-6
Zhang, Y., Sloan, S. A., Clarke, L. E., Caneda, C., Plaza, C. A., Blumenthal, P. D., et al. (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53. doi: 10.1016/j.neuron.2015.11.013
Zhou, N., Huang, S., Li, L., Huang, D., Yan, Y., Du, X., et al. (2016). Suppression of KV7/KCNQ potassium channel enhances neuronal differentiation of PC12 cells. Neuroscience 333, 356–367. doi: 10.1016/j.neuroscience.2016.07.024
Keywords: KCNQ2, M-current, neurodevelopment, KCNQ2-encephalopathy, Kv7.2
Citation: Dirkx N, Miceli F, Taglialatela M and Weckhuysen S (2020) The Role of Kv7.2 in Neurodevelopment: Insights and Gaps in Our Understanding. Front. Physiol. 11:570588. doi: 10.3389/fphys.2020.570588
Edited by:
Walter Sandtner, Medical University of Vienna, AustriaReviewed by:
Jerome J. Lacroix, Western University of Health Sciences, United StatesIsabella Salzer, Medical University of Vienna, Austria
Copyright © 2020 Dirkx, Miceli, Taglialatela and Weckhuysen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sarah Weckhuysen, c2FyYWgud2Vja2h1eXNlbkB1YW50d2VycGVuLnZpYi5iZQ==