 Ava Liaghati1,2†
Ava Liaghati1,2† Chantal A. Pileggi1,2†
Chantal A. Pileggi1,2† Gaganvir Parmar1,2
Gaganvir Parmar1,2 David A. Patten1,2Nina Hadzimustafic1,2Alexanne Cuillerier3
David A. Patten1,2Nina Hadzimustafic1,2Alexanne Cuillerier3 Keir J. Menzies1,2,3
Keir J. Menzies1,2,3 Yan Burelle3
Yan Burelle3 Mary-Ellen Harper1,2*
Mary-Ellen Harper1,2*- 1Department of Biochemistry, Microbiology and Immunology, Faculty of Medicine, University of Ottawa, Ottawa, ON, Canada
- 2Ottawa Institute of Systems Biology, University of Ottawa, Ottawa, ON, Canada,
- 3Faculty of Health Science, Interdisciplinary School of Health Sciences, University of Ottawa, Ottawa, ON, Canada
Glutathione is an important antioxidant that regulates cellular redox status and is disordered in many disease states. Glutaredoxin 2 (Grx2) is a glutathione-dependent oxidoreductase that plays a pivotal role in redox control by catalyzing reversible protein deglutathionylation. As oxidized glutathione (GSSG) can stimulate mitochondrial fusion, we hypothesized that Grx2 may contribute to the maintenance of mitochondrial dynamics and ultrastructure. Here, we demonstrate that Grx2 deletion results in decreased GSH:GSSG, with a marked increase of GSSG in primary muscle cells isolated from C57BL/6 Grx2−/− mice. The altered glutathione redox was accompanied by increased mitochondrial length, consistent with a more fused mitochondrial reticulum. Electron microscopy of Grx2−/− skeletal muscle fibers revealed decreased mitochondrial surface area, profoundly disordered ultrastructure, and the appearance of multi-lamellar structures. Immunoblot analysis revealed that autophagic flux was augmented in Grx2−/− muscle as demonstrated by an increase in the ratio of LC3II/I expression. These molecular changes resulted in impaired complex I respiration and complex IV activity, a smaller diameter of tibialis anterior muscle, and decreased body weight in Grx2 deficient mice. Together, these are the first results to show that Grx2 regulates skeletal muscle mitochondrial structure, and autophagy.
Introduction
Disordered mitochondrial glutathione reduction-oxidation reactions (redox) are associated with many disease states, such as type 2 diabetes, aging-associated neurodegenerative diseases, and rare genetic mitochondrial diseases (McMurray et al., 2016; Calabrese et al., 2017; Stockwell et al., 2017; Hoffmann and Griffiths, 2018; Kanaan et al., 2018). As a major non-protein antioxidant in cells, glutathione buffers elevations in hydrogen peroxide (H2O2), a form of reactive oxygen species (ROS) that contributes directly to the pathophysiology of these diseases (Houstis et al., 2006). Cellular ROS production occurs predominantly within the mitochondria of most cell types, making mitochondria the cellular hubs of redox reactions (Murphy, 2009; Diebold and Chandel, 2016; Murphy and Hartley, 2018; Yan et al., 2020). As such, mitochondria employ a variety of mechanisms to facilitate the detoxification of these potentially harmful reactive species. Protein glutathionylation is a rapid and reversible post-translational modification, which functions to protect vulnerable sulfhydryl groups from irreversible oxidation/damage in response to fluctuations in the redox environment (Nikolaienko et al., 2018; Figlia et al., 2020; Mailloux, 2020). In mammalian cells, enzymatic deglutathionylation is catalyzed by glutaredoxins (glutathione dependent reductase and oxidase, GRX; Holmgren, 1976; Beer et al., 2004). Grx2 is the primary mitochondria-specific Grx isoform whose function varies in response to fluctuations in the reduced glutathione (GSH); oxidized glutathione (GSSG) ratio; a high GSH:GSSG ratio promotes protein deglutathionylation, and a low GSH:GSSG ratio promotes Grx2 glutathionylation of target proteins (Beer et al., 2004; Hurd et al., 2008). Glutathionylation reactions can also alter protein activity thereby rendering the targets active or inactive (Adachi et al., 2004; Pimentel et al., 2006), and are particularly abundant in mitochondrial proteins rich in cysteine thiols (Mailloux, 2020).
Mitochondria are dynamic interconnected reticular structures that are continuously undergoing fusion and fission to facilitate metabolic demands and mitochondrial DNA exchange (Chan, 2020; Giacomello et al., 2020; Sabouny and Shutt, 2020; Yu et al., 2020). Fusion is dependent on the activation of large GTPase proteins, mitofusins (MFN)-1 and -2 to facilitate fusion of opposing outer membranes (Santel and Fuller, 2001; Rojo et al., 2002; Chen et al., 2003), and the optic atrophy-1 (OPA1) protein for fusion of the inner mitochondrial membranes (Frezza et al., 2006; Song et al., 2009). Mitochondrial fission requires the recruitment of dynamin-related protein-1 (DRP1), which severs mitochondrial membranes, fragmenting the mitochondrial network (Otsuga et al., 1998). Fission is necessary for mitochondrial quality control to prime damaged mitochondrial fragments for removal via mitophagy (Ashrafi and Schwarz, 2013), whereas fusion can occur as an acute stress-response to protect mitochondria from degradation (Tondera et al., 2009; Gomes et al., 2011). Moreover, it was recently demonstrated that the oligomerization of MFN1 and MFN2 into higher-order complexes is partly controlled by glutathione redox (Mattie et al., 2018). In support of a role for glutathione redox in mitochondrial ultrastructure, Shutt et al. (2012) showed that depletion of GSH, and increased levels of cellular GSSG stimulate mitochondrial hyperfusion. This was later demonstrated to be due in part to a redox-regulated cysteine thiol on MFN2 (Thaher et al., 2018). Importantly, Grx2 is particularly abundant in the mitochondrial intermembrane space (IMS), and plays a central role in regulating the redox state of the IMS import and assembly protein 40 (Mia40; Kojer et al., 2015), suggesting that it may also play a key role in MFN1/2 oligomerization. Moreover, glutathione deficiency has also been implicated in the regulation of autophagy (Filomeni et al., 2010). In retinal cells, depletion of GSH promotes LC3 autophagic flux and the appearance of autophagic vacuoles (Sun et al., 2018), whereas the addition of glutathione mitigates trehalose-induced autophagy (Underwood et al., 2010).
Within skeletal muscle, transient increases in ROS commonly occur in response to fiber contractions, and can result in the glutathionylation of complex I subunits (Gill et al., 2018), contractile proteins, and tricarboxylic acid cycle (TCA) enzymes (Kramer et al., 2018). Moreover, our lab has previously demonstrated that Grx2 plays a key role in skeletal muscle uncoupling protein 3 (UCP3)-dependent leak (Mailloux et al., 2013), and cardiac bioenergetics function and mitochondrial dynamics in mice and humans (Kanaan et al., 2018). However, the role of Grx2 on skeletal muscle mitochondrial ultrastructure and bioenergetic function remains unclear. Therefore, the present study aimed to examine the role of Grx2 on mitochondrial ultrastructure in skeletal muscle. Based on our previous observation of decreased glutathione redox in skeletal muscle mitochondria of Grx2−/− mice (Mailloux et al., 2013), we hypothesized that Grx2 regulates mitochondrial structure, turnover, and function in skeletal muscle.
Materials and Methods
Animals
All experiments involving mice were conducted according to the guidelines and principles of the Canadian Council of Animal Care and approved by the Animal Care Committee of the University of Ottawa. Studies were conducted on male C57BL/6 wild-type (WT) and Grx2−/− (C57BL/6 background) whole body knock-out mice aged 4–6weeks. All mice were genotyped prior to experimentation to confirm knock-out of Grx2. Mice were housed under standard conditions with controlled temperature, humidity, and 06:00–18:00 light cycles. Mice were supplied with ad libitum access to a standard chow diet (44.2% carbohydrate, 6.2% fat, and 18.6% crude protein, 2018 Teklad Global Rodent Diet, Envigo). To evaluate autophagy, mice were injected intraperitoneally with colchicine (0.4mg/kg/day) or an equal volume of saline every 24h for 2days prior to sacrifice (Vainshtein et al., 2015).
Body Composition Analyses
Fat and lean masses were measured by a non-invasive nuclear magnetic resonance imaging whole-body composition analyzer (EchoMRI-700™; Echo Medical Systems).
Indirect Calorimetry
Mouse O2 consumption, CO2 production, respiratory exchange ratio (RER), spontaneous and wheel-running activity as well as food intake were measured in a 12 chamber Comprehensive Lab Animal Monitoring System (CLAMS) instrument (Columbus Instruments) at thermoneutrality (28°C). Mice were individually housed in chambers and acclimated for 24–48h, before collection of data from a 24h period. Mice were provided throughout with ad libitum access to standard chow (as above). Volitional running activity was calculated as the total number of complete wheel rotations; spontaneous activity was assessed by beam breaks (x and z) in the chambers.
Tibialis Anterior Muscle Fiber Diameter
Dissected tibialis anterior muscles were embedded in optimal cutting temperature (OCT, Tissue-Tek) compound and frozen in isopentane cooled in liquid nitrogen. Muscle was cut perpendicular to the muscle fiber axis and sectioned (8μm) at −18°C using a cryotome (Leica Biosystems). Hematoxylin and eosin (H&E) staining was performed according to a standard protocol. Muscle fiber diameter was determined using image analysis using Image J software by determining horizontal muscle fiber diameter length.
Primary Myoblast Isolation
Primary myoblasts from WT and Grx2−/− mice were isolated from the quadriceps, tibialis anterior, soleus, and gastrocnemius muscles. The pooled muscle groups were minced and treated with collagenase/dispase (C/D; Collagenase: 0.1U/ml, Dispase: 0.8U/ml, Sigma Aldrich, 11097113001) solution to liberate myoblasts. The muscles were left in C/D solution for two 15-min incubation periods at 37°C in 5% CO2. After each 15-min incubation period, the suspension was manually triturated 20–25 times using a 10ml serological pipette. Primary cell enrichment was achieved by employing the differential adhesion process to remove fibroblast populations (Pasut et al., 2012). Myoblasts were cultured in Dulbecco’s modified Eagle medium (DMEM) containing 20% bovine growth serum (BGS), 1% Antibiotic-Antimycotic (AA), 30ng/μl basic fibroblast growth factor (bFGF; 13256-029, Life Technologies), and 5μg/ml gentamycin sulfate.
GSH and GSSG Determinations Using High-Performance Liquid Chromatography (HPLC)
Concentrations of GSH and GSSG in myoblasts and tibialis anterior muscle were determined by high-performance liquid chromatography (HPLC) using an Agilent 1100 Series instrument, equipped with a Pursuit 5 C18 column with a flow rate set to 1ml/min with filtered mobile phase [10% methanol, HPLC plus (Sigma, 646377), 90% ddH2O, 0.09% trifluoroacetic acid (Sigma, 302031)]. Serum deprived myoblasts were trypsinized and washed twice in ice-cold 1X phosphate-buffered saline (PBS) solution and counted using a Cell Countess (Thermo Fisher Scientific). Myoblasts were resuspended in 1:1 buffer [1% (v/v) trifluoroacetic acid (Sigma, 302031) and 1% (w/v) meta-phosphoric acid (Sigma, 239275) solution in mobile phase and homogenization buffer (for 25ml, 250mM sucrose, 10mM TRIS, 3mM EDTA dissolved in mobile phase, pH of 7.4) final pH of 1:1 buffer <1.0] and incubated on ice for 20min. For determinations of GSH and GSSG in muscle, frozen tibialis anterior samples were weighed and homogenized in 1:1 buffer with a Teflon pestle drill. The myoblast and homogenized muscle samples were then centrifuged at 14,000g for 20min at 4°C. After centrifugation, the supernatant was collected for analyses. GSH and GSSG were detected using the Agilent UV-visible wavelength detector at 215nm. Retention times were confirmed using standards, which were prepared using 0.1, 0.01, and 0.001mM of GSH (Sigma, G4251) and GSSG (Sigma, G4501) dissolved in 1:1 buffer. Absolute amounts of GSH and GSSG were determined by integrating the area under the respective peaks using a chromatogram, and values were calculated from standard curves.
Immunofluorescence of Primary Myoblasts Mitochondrial Length
Upon 85–90% confluency, cells were trypsinized and plated for imaging. Myoblasts were placed in starvation medium (SM) for an incubation period of 14–16h prior to cell fixation; starvation was used to synchronize cell cycle and reach a quiescent state (G0 state). The SM consisted of DMEM supplemented with 0.1% Bovine Serum Albumin (BSA) and 1% AA. After the starvation period, cells were fixed with 4% paraformaldehyde for 15min. The cells were permeabilized and blocked using a blocking buffer containing 0.1% Triton X-100 and 1% BSA, which also included the primary antibody, TOM20 (Santa Cruz Biotechnology, sc-11415; 1:100 dilution). Oregon green 488 goat anti-rabbit (Life Technologies, O-6381; 1:100 dilution) was diluted in 1x PBS containing Hoechst counter-stain in 1:1 PBS:Glycerol mounting media. Images were obtained using a Zeiss Axiolmager M2 microscope with a Plan-Apochromat 63X/1.4 oil objective. Fifty mitochondria lengths were quantified using ImageJ in five independent experiments for each genotype. The Mitochondrial Network Analysis (MiNA) plugin was used for the semi-automated analysis of mitochondrial network complexity to determine the average number of branches per mitochondrial network (Valente et al., 2017).
Protein Extraction
20–30mg of frozen tibialis anterior muscle was homogenized using a bead mill homogenizer (Fisherbrand Bead Mill 24 Homogenizer) in ice-cold modified RIPA buffer [20mM Tris-HCl (pH 7.5), 150mM NaCl, 1mM Na2EDTA, 1mM EGTA, 1% NP-40, 1% Na deoxycholate, 2.5mM Na pyrophosphate, 1mM β-glycerolphosphate, 1mM Na3VO4] supplemented with a protease and phosphatase inhibitor cocktail (Sigma Aldrich; P8340). Homogenates were centrifuged at 14,000g for 10min at 4°C to remove cellular debris. Protein concentration was determined using the BCA method.
Immunoblotting
Sample aliquots containing 10μg of protein were suspended in 1x Laemmli buffer, with or without DTT, boiled at 95°C for 5min and subjected to SDS/PAGE. Proteins were transferred to a nitrocellulose membrane (Bio-Rad, 160112), and incubated with blocking buffer [5% skim milk powder in tris buffered saline with 0.1% tween 20 (TBST)] for 1h at room temperature, followed by overnight incubation at 4°C with primary antibodies in blocking buffer under gentle agitation against p62 (1:2,000, Cell Signaling, 5114s), Parkin (1:2,000, Santa Cruz, sc-32282), LC3I/II (1:2,000, Cell Signaling, 12741s), MFN1/2 (1:2,000, Abcam, ab57602), DRP1 (1:2,000, BD Biosciences 611113), GFER (1:4,000, ProteinTech, 11293-1-AP), COX17 (1:2,000, ProteinTech 11464-1-AP), and Vinculin (1:10,000, Abcam, ab129002). Membranes were washed five times for 5min and probed with an anti-rabbit or anti-mouse IgG conjugated to horseradish peroxidase (HRP) secondary antibody in blocking buffer for 1h at room temperature. Membranes were washed five times for 5min in TBST and protein bands were visualized using the ChemiDoc™ MP Imaging System (Bio-Rad). Densitometry band analysis was performed using Image J software. Abundance of target proteins are presented normalized to Vinculin or Ponceau staining.
Electron Microscopy
Transmission electron microscopy (TEM) was used to examine the mitochondrial structure in WT and Grx2−/− tibialis anterior muscle. In brief, tibialis anterior muscle was fixed in 2.5% glutaraldehyde. The tissue was dehydrated in a graded series of ethanol and then embedded in Spurr’s resin. Resin blocks were sectioned using a Leica EM UC6 ultramicrotome (Leica Microsystems). Sections were mounted on copper grids coated with formvar film, stained with 2% alcoholic uranyl acetate and Reynold’s lead citrate and imaged using the JEOL 1230 Transmission Electron Microscope (JEOL Ltd.). Fifty-five micrographs were examined from each genotype at a magnification of 3,000. Quantitative morphometry was then used to determine mitochondrial volume density, as estimated using the point-counting method using Image J software (Zong et al., 2002). Specifically, 10 × 10 grids were overlaid on each of the 55 micrographs per animal, and the intersection point between horizontal and vertical lines were used to determine if intersection point fell with the mitochondria, the cytoplasm, or extracellularly. The percentage of surface area was calculated, and the mean surface area for each animal was used to determine the mean for each group.
Mitochondrial and Nuclear DNA Quantification
Tibialis anterior muscle was homogenized using a Dounce homogenizer with Tris-based lysis buffer [10mM Tris-HCl (pH 8.0), 1mM EDTA, and 0.1% SDS]. Homogenates were incubated with 15mM proteinase K (Invitrogen) at 55°C overnight (Guo et al., 2009). Lysates were vortexed vigorously and the non-soluble fraction was pelleted by centrifugation at 10,000g for 15min at room temperature. DNA was extracted from the supernatant using phenol/chloroform/isoamyl alcohol (25:4:1; PCIAA) as previously described (Guo et al., 2009). qPCR with SYBR Green FastMix (Quanta Biosciences) was run according to the manufacturer’s protocol. The cytochrome c oxidase subunit I (CO1) gene of the mtDNA (F: 5'-TGC TAG CCG CAG GCA TTA C-3' and R: 5'-GGG TGC CCA AAG AAT CAG AAC-3') and the NDUFV1 nDNA gene (F: 5'-CTT CCC CAC TGG CCT CAA G-3' and R: 5'-CCA AAA CCC AGT GAT CCA GC-3') were amplified by qPCR.
Colocalization Analysis of Mitochondria and Lysosomes
The Mitophagy Detection Kit (Dojindo Molecular Technologies, Rockville, United States) was used according to the manufacturer’s protocol for visualization of mitophagy in primary myoblasts. The kit consists of a Lyso Dye, which stains lysosomes, and a MtPhagy Dye, that accumulates in all mitochondria. Upon acidification due to fusion of the mitochondria to the lysosome, the fluorescent signal of the MtPhagy dye is enhanced to allow for detection of mitophagy (Iwashita et al., 2017). Briefly, 50,000 myoblasts were seeded into four-well Ibidi slides. After 24h, cells were washed with serum-free media, and incubated with the MtPhagy dye for 30min at 37°C. To observe colocalization with lysosomes, cells were washed again with serum-free media and incubated with the Lyso Dye for 30min at 37°C. Following the incubation, cells were washed and imaged with imaging media (phenol red- and serum-free), using a Scientific CMOS Camera using an Olympus 63x/1.4 oil immersion objective with a DeltaVision Elite Microscope (Olympus). Z-stacks were acquired with 1μm spacing and images were deconvolved using the interactive deconvolution in SoftWorx 7.0.
Blinded colocalization analysis between the Lyso dye and MtPhagy dye was performed on images subjected to manual thresholding of the brightest 2% of pixels in each channel images to unbiasedly remove the background (Lorenzen et al., 2010). Images were converted to binary before using the EzColocalization Fiji Plugin (Stauffer et al., 2018). The Mander’s correlation coefficient was calculated to quantify the co-occurrence of signals between the Lyso Dye and MtPhagy Dye, as well as the MtPhagy Dye and Lyso Dye.
High-Resolution Respirometry in Permeabilized Muscle Fibers
Fibers were prepared from tibialis anterior muscle of mice. High-resolution respirometry was conducted using an Oxygraph-2k system (OROBOROS Instruments, Innsbruck, Austria). Dissected tibialis anterior samples were placed in ice-cold relaxation BIOPS medium (5.77mM Na2ATP, 7.23mM K2EGTA, 2.77mMCaK2EGTA, 6.56mM MgCl2-6H2O, 20mM taurine, 60mMK-lactobionate, 15mM phosphocreatine, 20mM imidazole, 0.5mM DTT, and 50mM MES; pH 7.1 at 0°C) immediately after harvesting. Muscle fibers were mechanically separated and permeabilized using saponin (50μg/ml) on ice for 30min. Fibers were then rinsed in mitochondrial respiration medium, MiRO5 (0.5mM EGTA, 3mM MgCl2-6H2O, 20mM taurine, 10mM K2HPO4, 20mM HEPES, 110mM sucrose, and 1g/l BSA; pH 7.1 at 37°C), and weighed muscle fibers were placed into the respirometry chambers. Two separate protocols were used and analyses were performed at 37°C. The first protocol included consecutive additions of 2mM malate, 5mM pyruvate, 10mM glutamate, 5mM ADP (complex I-supported respiration), 10mM succinate (complex I- and complex II-supported respiration), 0.25μM titrations of carbonyl cyanide p-trifluoromethoxyphenyl hydrazine (FCCP; maximal respiration) and 2.5μM antimycin A (AMA). The second protocol included successive additions of 2mM malate, 200μM octanoyl carnitine, 5mM ADP (fatty acid-supported respiration), 5mM pyruvate, 10mM glutamate, 10mM succinate, 2.5μM oligomycin (leak respiration). Values are corrected to non-mitochondrial oxygen consumption (i.e., that in the presence of AMA).
Complex IV Activity
Enzymatic activity of complex IV was determined by quantifying the oxidation of cytochrome C as previously described (Pileggi et al., 2016). In brief, cytochrome C oxidation was quantified at 550nm in 50mM potassium phosphate buffer (pH 7.0), and 50μM of reduced cytochrome C using the BioTek Synergy Mx Microplate Reader (BioTek Instruments, Inc., Winooski, VT, United States). Enzyme activity was calculated using the extinction coefficient ε = 18.5, and expressed per μg of protein.
Identification of Transcript Correlations in the BXD Mouse Tissue Data Sets
Using a BXD mouse genetic reference population (Pirinen et al., 2014), skeletal muscle microarray data (AffymetrixMouseGene1.0 ST) were analyzed for transcript expression correlations between Grx2 and genes associated with the disulfide relay system using the GeneNetwork program.1
Statistical Analysis
Statistical analyses were performed using Prism software (GraphPad). Statistical significance between WT and Grx2−/− mice was determined using a two-tailed t-test. For experiments involving colchicine treatment, statistical significance was determined using two-way ANOVA with genotype and colchicine as factors. Pairwise comparisons were determined using the Holm-Sidak post hoc method. Data are shown as means ± SEM. Statistical significance was accepted at p < 0.05. Prism software (GraphPad) was used to generate graphs.
Results
Grx2 Deficiency Results in Decreased Lean Mass and Skeletal Muscle Cell Cross-Sectional Area
Previously, we demonstrated that Grx2−/− mice aged between 9 and 12 weeks exhibit normal linear growth with no differences in liver, skeletal muscle, and brown adipose tissue weights (Mailloux et al., 2014). We documented hypertension even in young male Grx2−/− mice, despite no evidence of a cardiac phenotype. In the current study, we did not assess blood pressure, but it is possible that hypertension may be contributing overall to our current findings. Therefore, we aimed to further investigate body composition and metabolic phenotyping, and to focus on young male Grx2−/− mice (4–6weeks) when there is no evidence of heart pathology (e.g., hypertrophy), which develops at ~10weeks of age. We first measured body weights and body composition (Echo-MRI) and found that total body weight and lean mass were slightly decreased in the Grx2−/− compared to WT mice (p < 0.01 for body weight and lean mass; Table 1). In contrast, there were no differences in total body fat mass between the two genotypes (Table 1). Ad libitum food intake was measured in the 12 chamber, fully automated CLAMS system and was found not to differ between genotypes (Table 1). VO2 consumption rates normalized to lean body mass were similar between WT and Grx2−/− mice (Table 1). Similarly, the RER (VCO2/VO2), which indicates the relative proportion of fuels being oxidized at the whole-body level (i.e., carbohydrates vs. fats) did not differ between genotypes (Table 1). We next examined the cross-sectional area of H&E stained skeletal muscle sections to determine if differences in skeletal muscle size were contributing to the decrease in lean body mass in Grx2−/− mice. We conducted these and subsequent analyses in tibialis anterior muscles, which are known to be composed of predominantly glycolytic fibers, with a mix of oxidative and intermediate fiber types (Augusto et al., 2004). Tibialis anterior sections from Grx2−/− mice exhibited a smaller myofiber diameter than WT mice (p < 0.05, Figure 1).
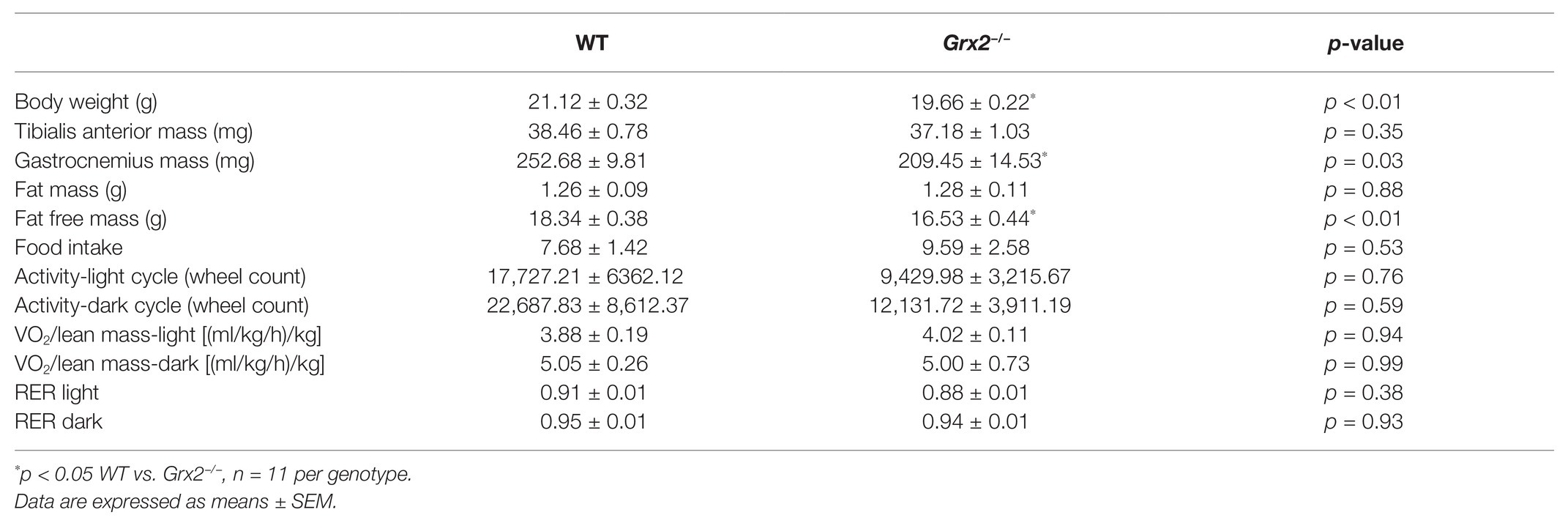
Table 1. Grx2−/− mice weigh less and have decreased lean muscle mass despite equal activity and food intake.
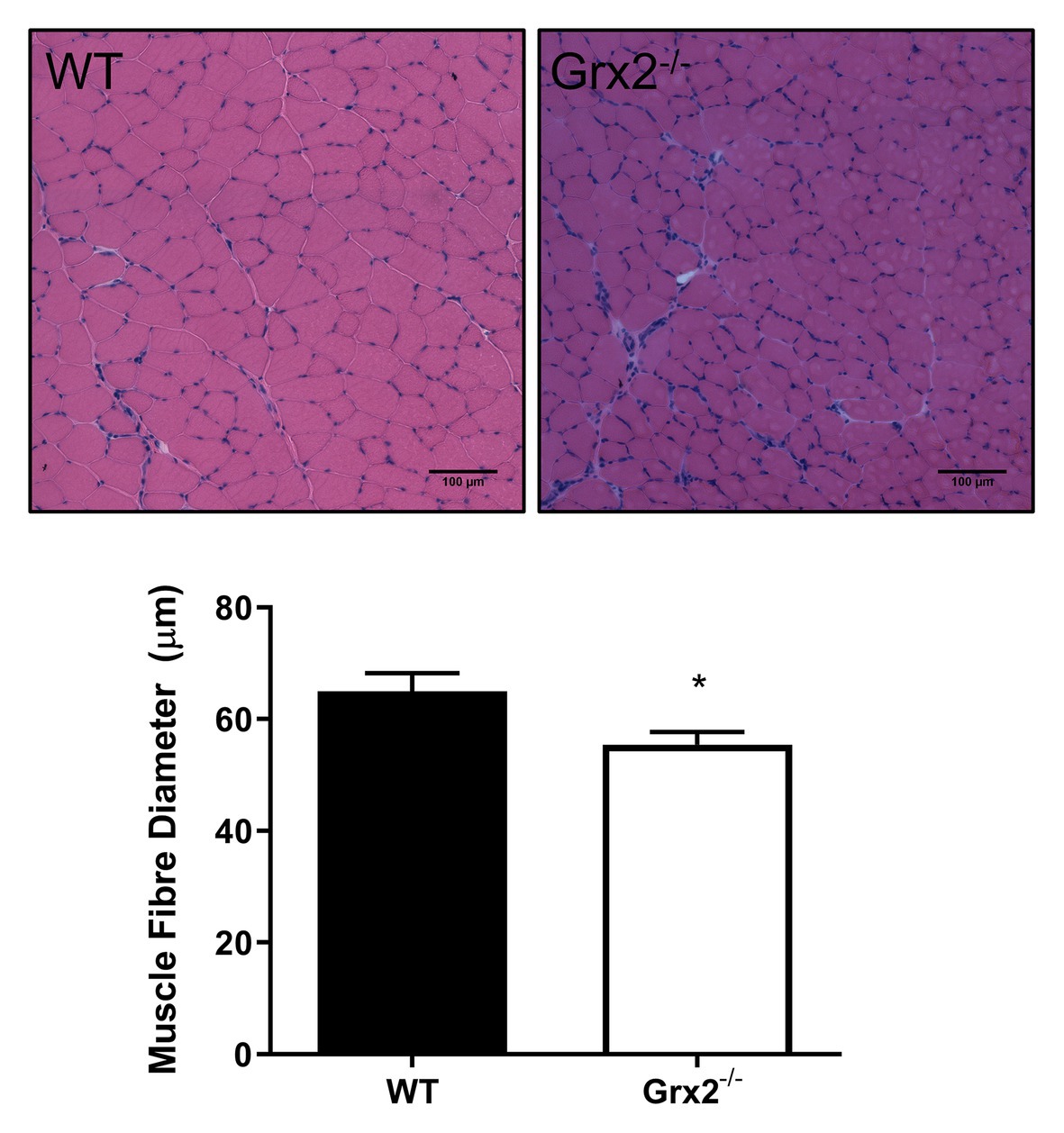
Figure 1. Skeletal muscle fiber diameter is decreased in tibialis anterior muscle in Grx2−/− mice. Muscle fiber diameter was determined in cross-sections of frozen optimal cutting temperature (OCT)-embedded tibialis anterior muscle. Grx2−/− mice displayed decreased muscle fiber diameter compared to wild-type (WT) mice. Representative images are shown above the graph. Data are expressed as mean ± SEM. n = 5, *p < 0.05.
Absence of Grx2 Perturbs Muscle Glutathione Redox
Grx2 plays an important role in regulating protein (de)glutathionylation reactions of mitochondrial target proteins during oxidative stress. Absence of Grx2 results in a decreased GSH:GSSG ratio in isolated mitochondria from skeletal muscle (Mailloux et al., 2013) and the accumulation of GSSG has been linked to molecular adaptions that favor mitochondrial fusion during cellular stress (Shutt et al., 2012). Therefore, we measured GSH in primary myoblasts from within Grx2−/− mice to potentially confirm the finding of GSSG, and to investigate the effects of altered GSH redox on mitochondrial fusion. Our findings revealed that absolute amounts of GSH and GSSG per cell did not differ between genotypes (Figures 2A,B) in primary myoblasts. However, Grx2−/− myoblasts had a significantly decreased glutathione redox ratio when compared to their WT counterparts (p < 0.01, Figure 2C). Next, we aimed to confirm our findings in Grx2−/− tibialis anterior tissue. Our results demonstrate that Grx2−/− muscle tended to have increased concentrations of GSSG (p = 0.0905, Figure 2E), whereas the concentration of GSH did not differ between genotypes (Figure 2D). The increase in GSSG in Grx2−/− muscle was reflected by a trend for decreased glutathione redox ratio in the tibialis anterior from Grx2−/− mice (p = 0.0826, Figure 2F).
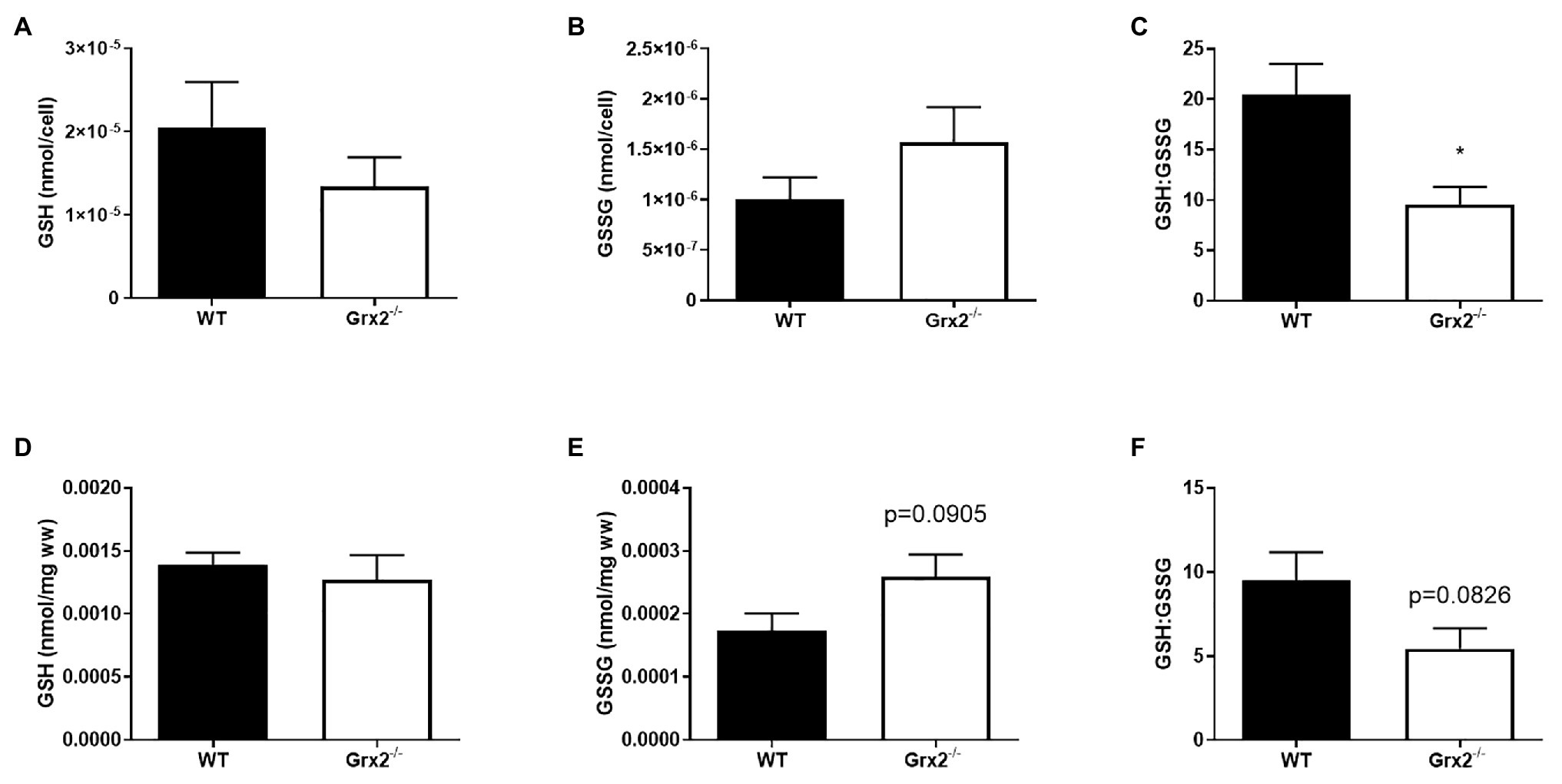
Figure 2. GSH:GSSG is decreased in Grx2−/− myoblasts. High-performance liquid chromatography (HPLC) analyses were used to determine the concentrations of reduced glutathione (GSH), and oxidized glutathione (GSSG) in primary myoblasts and tibialis anterior muscle. (A) GSH and (B) GSSG did not differ between WT and Grx2−/− primary myoblasts. (C) GSH:GSSG is decreased in Grx2−/− myoblasts compared to their WT counterparts. (D) Absolute concentrations of GSH did not differ between genotypes in tibialis anterior muscle. (E) There was a trend for increased GSSG in Grx2−/− muscle. (F) GSH:GSSG tended to decrease in Grx2−/− muscle. Data are mean ± SEM. n = 6–7 for determinations in primary myoblasts (A-C); n = 5 for determinations in in tibialis anterior muscle (D-F); *p < 0.05.
Deletion of Grx2 Promotes Mitochondrial Fusion
Following the confirmation of a decreased GSH:GSSG in Grx2−/− primary myoblasts, we hypothesized that mitochondria in primary muscle cells of Grx2−/− mice would show increased fusion compared to muscle cells of WT mice. Quantitative morphometric analyses of TOM20-stained primary myoblasts from both genotypes showed that mitochondria were indeed more fused, as evidenced by an increased mitochondrial length in Grx2−/− vs. WT myoblasts (p < 0.05, Figure 3A). When assessing mitochondrial network complexity, we did not observe an increase in mitochondrial branching in Grx2−/− primary myoblasts (p = 0.18, Figure 3B). Next, to examine whether the increase in mitochondria length was attributable to redox control of the formation of MFN oligomer complexes, we immunoblotted for MFN1/2 and DRP1 under non-reducing conditions to preserve disulfide bonds. There was a trend for increased content of higher order MFN1/2 oligomers between 160 and 250kDa (p = 0.071, Figure 3C). In contrast, there was no difference in the formation of DRP1 oligomers (Figure 3D). Given that we observed MFN1/2 and DRP1 high molecular weight (MW) oligomers by western blot only under non-reducing conditions, we conclude that these high MW oligomers are similarly disulfide-mediated and would reduce in response to redox potential.
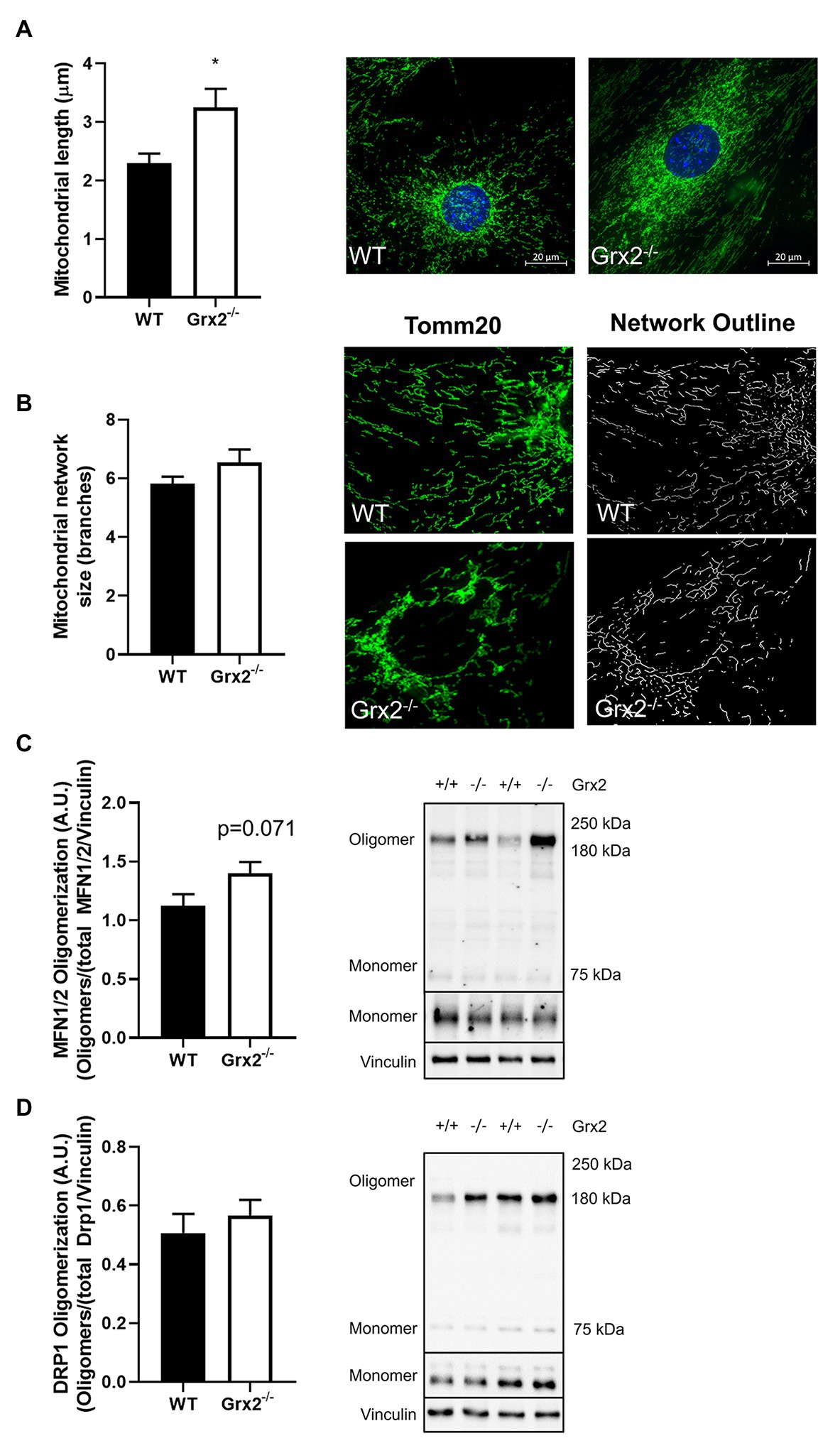
Figure 3. Mitochondrial length is increased in Grx2−/− myoblasts. (A) Mitochondrial length is increased by ~30% in Grx2−/− myoblasts. Representative images of WT and Grx2−/− myoblasts are shown beside the graph; scale bars, 20μm. Quantification of 50 mitochondrial lengths from five independent experiments. (B) Mitochondrial branching does not differ between WT and Grx2−/− myoblasts. Representative images of WT and Grx2−/− myoblasts are shown (C) Oligomerization of MFN1/2 tends to be increased in Grx2−/− skeletal muscle (p = 0.071). (D) Oligomerization of DRP1 in skeletal muscle does not differ between WT and Grx2−/−. Data are expressed as mean ± SEM. n = 6–7 for mitochondrial length determinations in primary myoblasts (A); n = 5 for for mitochondrial branching analysis (B); n = 6 for quantification of MFN1/2 and DRP1 oligomerization in tibialis anterior muscle (C,D); *p < 0.05.
Mitochondrial Ultrastructure and Autophagy Are Altered in Grx2 Deficient Muscle
Following our findings of elevated mitochondrial fusion in primary Grx2−/− myoblasts, we then examined mitochondrial ultrastructure in skeletal muscle tissue using TEM to observe the effects of Grx2 deficiency in vivo. Electron micrographs showed that mitochondrial morphology is markedly abnormal in the Grx2−/− muscle (Figure 4A). The WT mitochondria cristae were found to be well-defined, and tubular. However, micrographs showed that Grx2−/− mitochondria frequently lacked ordered cristae (Figure 4A) and had unusual vacuole-like structures with double membranes that resemble mitochondria engulfed in autophagosomes, consistent with the possibility of impaired mitophagy (Ding and Yin, 2012; Menges et al., 2017; Chakraborty et al., 2020). Furthermore, quantitative morphometric analyses revealed that mitochondrial content in the Grx2−/− muscle was less than half that in the WT skeletal muscle (Figure 4B). We then sought to confirm this by using a common proxy method to measure mitochondrial content, the ratio of mitochondrial DNA to nuclear DNA (mtDNA:nDNA); however, no differences were observed between the WT and Grx2−/− mtDNA:nDNA (Figure 4C).
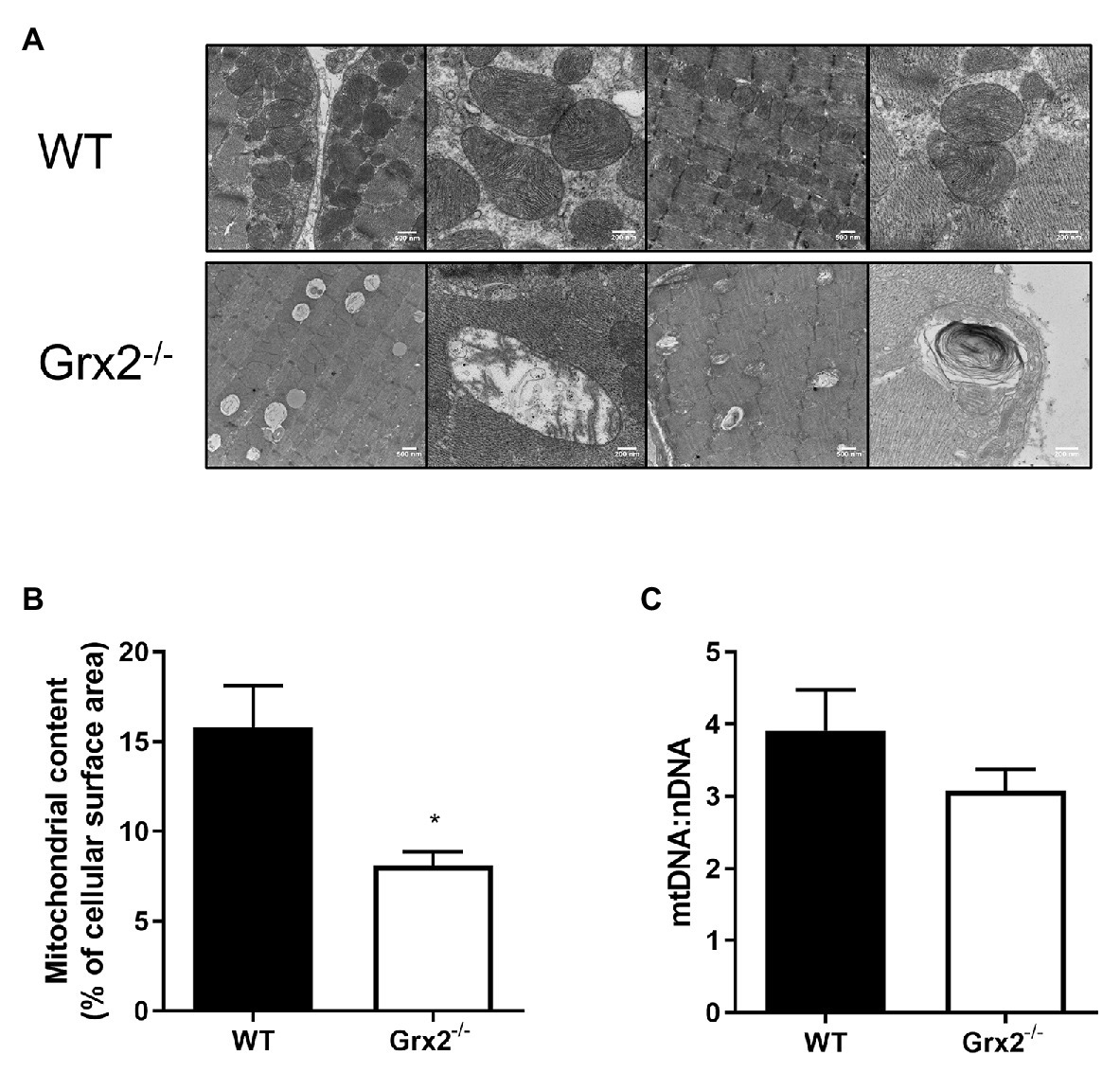
Figure 4. Electron micrographs show disordered mitochondrial morphology and ultrastructure, and a decrease in mitochondrial volume in Grx2−/− skeletal muscle. (A) Representative micrographs of WT and Grx2−/− tibialis anterior muscle. WT mitochondria have well-defined cristae structures with normal mitochondrial shape. In contrast, in Grx2−/− tibialis anterior there are vacuole-like structures with double membranes, and abnormal mitochondrial cristae. (B) Quantitative analyses of images revealed decreased mitochondrial surface area in Grx2−/−. (C) No differences were found in the ratio of mtDNA to nDNA. Data are expressed as mean ± SEM. n = 3 for TEM analysis (A,B); n = 6 for quantification of mtDNA:nDNA (C); *p < 0.05.
We then hypothesized that the irregular ultrastructure of the mitochondria and the multi-lamellar structures in the Grx2−/− skeletal muscle may be indicative of increased autophagy of mitochondria, and that the mtDNA may be retained within autophagosomes. Therefore, we immunoblotted for key proteins involved in autophagy and mitochondrial clearance in skeletal muscle homogenates from mice that were injected intraperitoneally with colchicine or an equal volume of saline for 2days prior to sacrifice (Vainshtein et al., 2015). Autophagic flux is determined by quantifying protein levels in the presence and absence of chemical agents such as colchicine, that destabilize microtubules and block lysosomal degradation (Ju et al., 2010). Immunoblotting for key proteins involved in the priming of autophagic and mitophagic pathways revealed that protein expression of parkin was elevated in Grx2−/− muscle (main effect of genotype p < 0.05; Figure 5A). In contrast, while colchicine treatment promoted increased expression of p62, the expression of p62 did not differ between genotypes (main effect of colchicine p < 0.05; Figure 5B). Expression of the downstream autophagic precursor, LC3I, did not differ between genotypes or with colchicine treatment (Figure 5C), whereas there was a main effect for increased LC3II expression for both colchicine treatment (main effect of colchicine p < 0.05) and absence of Grx2 (main effect of genotype p < 0.05; Figure 5D). The ratio of LC3II/I is used as a quantitative indicator of autophagic flux (Kadowaki and Karim, 2009). LC3II/I was elevated in the tibialis anterior of Grx2−/− mice (main effect of genotype p < 0.05; Figure 5E). Next, to confirm our findings, we used a commercially available mitophagy kit to quantify the colocalization of mitochondria and lysosomes in primary myoblasts. Blinded analysis of acquired images revealed a trend for increased colocalization between mitochondria and lysosomes in Grx2−/− myoblasts compared to WT (p = 0.067, Figure 5F).
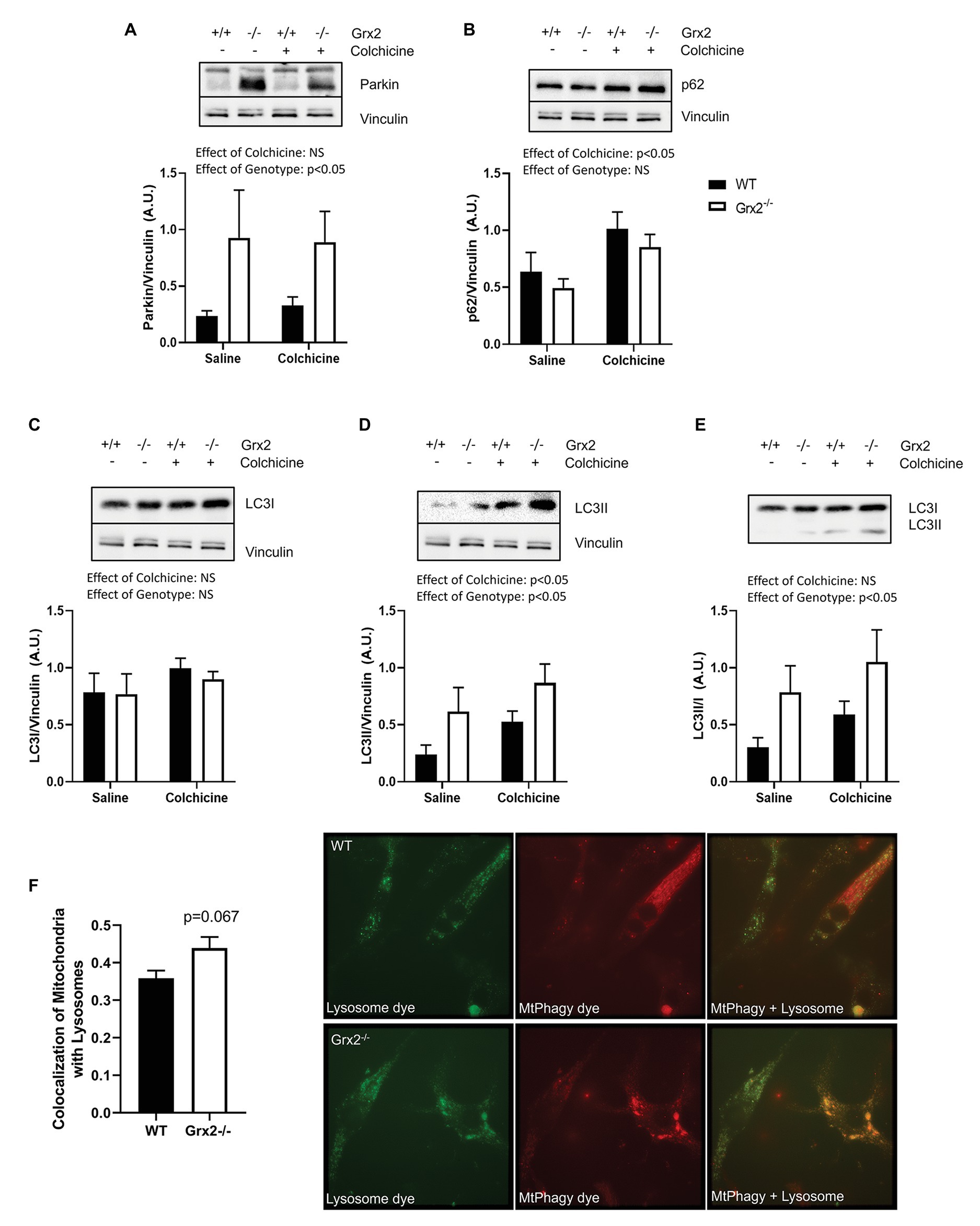
Figure 5. Grx2−/− skeletal muscle has elevated autophagic flux. Immunoblot analyses was performed on tibialis anterior muscle; protein was extracted from colchicine injected mice. (A) Protein expression of parkin is elevated in Grx2−/− muscle. (B) Expression of p62 does not change in the absence of Grx2, but increases with colchicine treatment. (C) LC3I expression does not differ between genotypes or with colchicine treatment. (D) LC3II protein expression is increased with colchicine treatment and in Grx2−/− muscle. (E) Autophagic flux is elevated in Grx2−/− muscle as determined by LC3II/I. Equal protein loading was determined by immunoblotting for vinculin. Data are expressed as mean ± SEM, n = 6–7. Representative blots are shown above the graphs. (F) Grx2−/− primary myoblasts exhibit increased colocalization of mitochondria and lysosomes. Colocalization of mitochondria and lysosomes in primary myoblasts using Mander’s correlation coefficient (0 = no colocalization, 1 = full colocalization). Representative images of WT and Grx2−/− myoblasts. Quantification of 6–10 blinded images per animal. Data expressed as mean ± SEM, n = 4.
Complex I Mitochondrial Respiration Is Impaired in Grx2 Deficient Skeletal Muscle
Subsequent to determining that mitochondrial structure was disordered in skeletal muscle, we sought to examine the functional effects of Grx2 deficiency on skeletal muscle mitochondrial bioenergetics in permeabilized muscle fibers. Previously, our investigations into the effects of Grx2 deficiency in skeletal muscle were conducted on mitochondrial isolations from homogenized muscle (Mailloux et al., 2014), in which the isolation process may exclude damaged mitochondria resulting in the preferential selection of healthy mitochondria subpopulations (Piper et al., 1985). Thus, we used published protocols to permeabilize the sarcolemma and examine mitochondrial oxidative phosphorylation (OXPHOS) in situ using high-resolution respirometry. Our results demonstrate impaired complex I (CI) OXPHOS in Grx2−/− permeabilized myofibers (Figure 6A; p < 0.05). Furthermore, maximal respiration, measured by the addition of the uncoupler FCCP (F), was not different between genotypes. Notably, FCCP titrations increased respiration in Grx2−/− muscle, but not WT muscle. Occasionally FCCP titrations do not result in an additional increase in respiratory flux (Christiansen et al., 2015), often due to saturating concentrations of substrates, inhibition of oxygen consumption associated with the use of high FCCP concentrations, or from the use of sub-optimal FCCP concentrations resulting in non-maximal stimulation (Steinlechner-Maran et al., 1996; Pesta and Gnaiger, 2012). To determine if there were OXPHOS defects when respiration was fueled by fatty acid oxidation (FAO), we used a separate protocol using octanoyl carnitine in additional samples of myofibers. FAO+CI OXPHOS was again found to be decreased (Figure 6B).
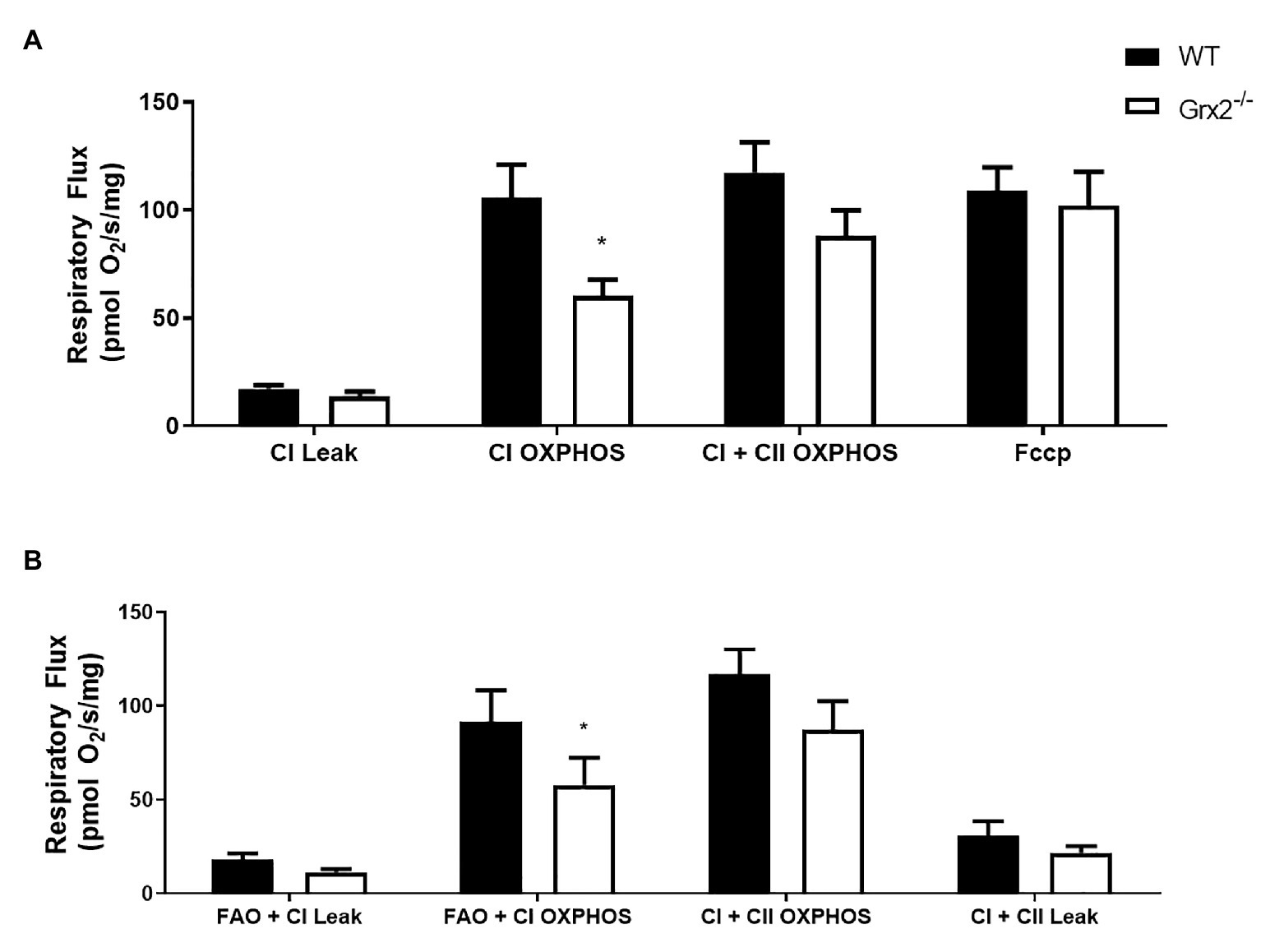
Figure 6. Mitochondrial complex I (CI) oxidative phosphorylation (OXPHOS) is impaired in permeabilized Grx2−/− mouse muscle. (A) Respiratory flux per milligram of saponin-permeabilized tibialis anterior muscle demonstrates that CI OXPHOS is impaired in Grx2−/− mice using the substrates pyruvate, malate, and glutamate. (B) Complex I respiration using octanoylcarnitine, in combination with malate, pyruvate, and glutamate is also decreased in Grx2−/− muscle. Data are expressed as mean ± SEM. n = 6; *p < 0.05.
Grx2 Deletion Is Associated With Dysregulation of the Disulfide Relay System
Lastly, we sought to determine the effects of Grx2 deletion on components of the disulfide relay system. Most nuclear-encoded mitochondrial proteins are synthesized as precursors on cytosolic ribosomes, and subsequently imported via translocases on the mitochondrial outer membrane to the IMS to be directed towards protein folding pathways (Neupert and Herrmann, 2007). A subset of cysteine-rich proteins rely on the disulfide relay system for their retention in the IMS, in which Mia40 (IMS Import and Assembly) binds to cysteine residues on precursor proteins to facilitate the formation of a mixed disulfide bond (Mesecke et al., 2005). The disulfide complex is resolved with the oxidation of the substrate, resulting in the reduction of Mia40. Mia40 is subsequently recycled through oxidation by the growth factor, augmenter of liver regeneration (GFER, ERV1 homolog in Yeast; Mesecke et al., 2005; Hell, 2008; Lionaki et al., 2010; Kojer et al., 2012). GFER is subsequently re-oxidized through electron transfer to cytochrome c, which is recycled through the activity of cytochrome c oxidase (Allen et al., 2005). Grx2 is thought to play a key role in maintaining the redox state of the IMS, as Grx2 is dually localized in the mitochondrial IMS and matrix (Kojer et al., 2015). Moreover, overexpression of Grx2 delays protein folding by maintaining Mia40 in a reduced state (Kojer et al., 2015). As complex IV function is essential to the function of the disulfide relay system, and complex IV assembly proteins COX17 and COX19 are known substrates of this pathway (Mesecke et al., 2005; Gabriel et al., 2007; Hell, 2008), we hypothesized that the deletion of Grx2 would alter complex IV activity. Therefore, we examined the correlation between Grx2 gene expression and key genes involved in the disulfide relay system using BXD mice. Here, we show that Grx2 is positively correlated with Gfer (Figure 7A, Pearson correlation coefficient = 0.484, p < 0.001) and Cox17 gene expression (Figure 7A, Pearson correlation coefficient = 0.461, p < 0.001). Next, we quantified the protein expression of GFER and Cox17 in Grx2−/− skeletal muscle, and determined complex IV activity. Consistent with our hypothesis, GFER expression was decreased in Grx2−/− skeletal muscle (Figure 7B; Main effect of genotype p < 0.05), independent of colchicine treatment. In contrast, COX17 protein expression was increased in Grx2−/− skeletal muscle (Figure 7C; Main effect of genotype p < 0.05), and decreased with colchicine treatment (Figure 7C; Main effect of colchicine p < 0.001). Complex IV activity was decreased in Grx2−/− skeletal muscle (Figure 7D, p < 0.05).
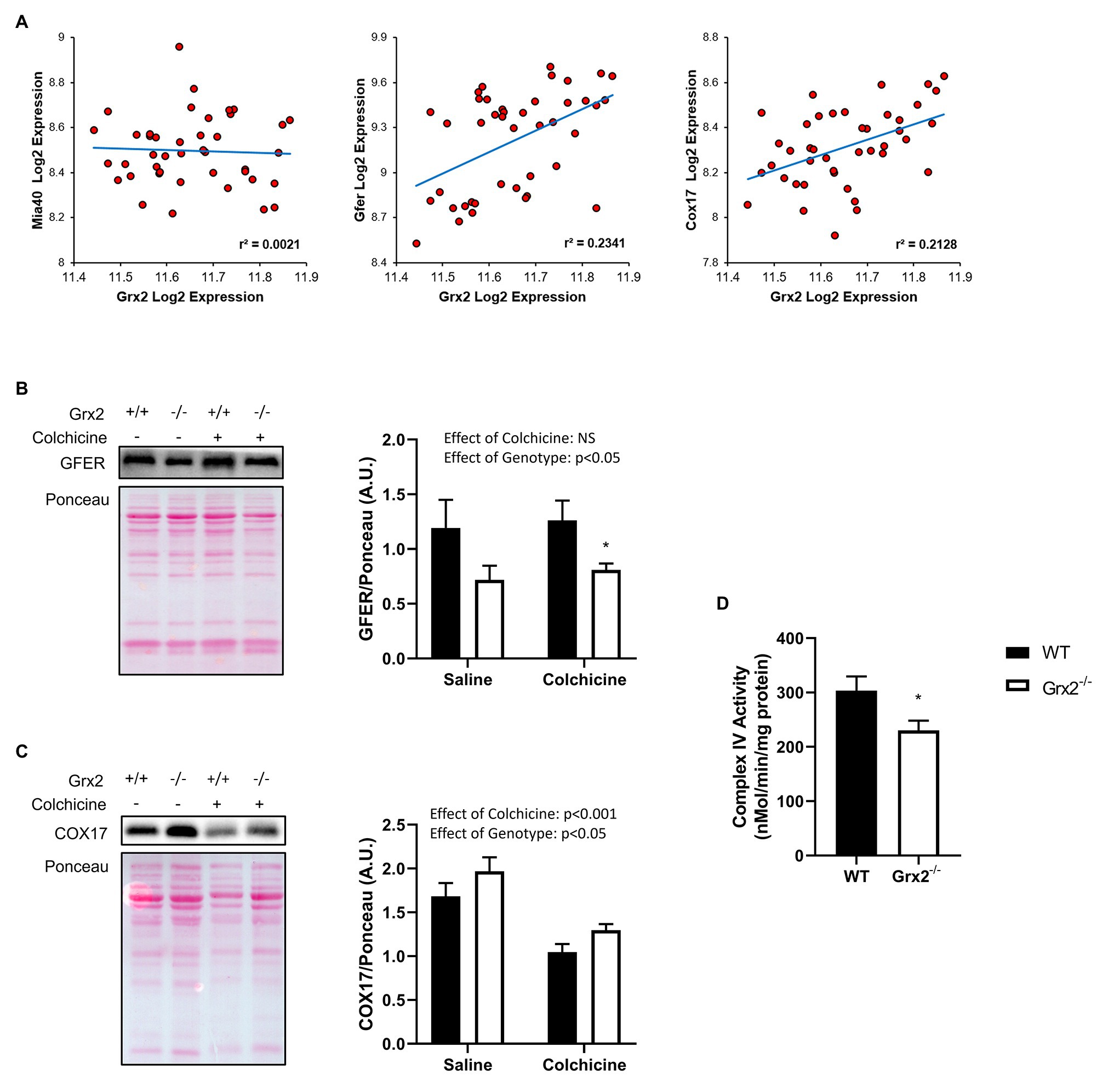
Figure 7. Grx2 deficiency is associated with dysregulation of the disulfide relay system. (A) Genes involved in the disulfide relay system were correlated to Grx2 using 42 BXD mouse strains fed a chow diet. Grx2 is positively correlated with Gfer and Cox17 in skeletal muscle. (B) Tibialis anterior muscle protein was extracted from saline and colchicine injected mice, and protein expression level of GFER was lower in Grx2−/− skeletal muscle, as determined by western blot. (C) Protein expression of COX17 was increased in Grx2−/−, and decreased in skeletal muscle from mice treated with colchicine. Equal protein loading was determined with Ponceau stain. (D) Complex IV activity is decreased in skeletal muscle from Grx2−/− mice. Data are expressed as mean ± SEM. n = 6–7 for GFER and COX17 quantification; n = 6 for complex IV activity. *p < 0.05.
Discussion
The present study examined the impact of Grx2 deficiency on skeletal muscle mitochondrial ultrastructure and dynamics, as well as on bioenergetics in ex vivo muscle fibers. Our results demonstrate that Grx2 is involved in maintaining mitochondrial ultrastructure by regulating fusion in primary myoblasts. We provide evidence that deletion of Grx2 perturbs muscle glutathione redox status, resulting in increased autophagic flux and the formation of abnormal autophagic vacuoles. These structural abnormalities were accompanied by decreased complex I –supported OXPHOS activity in muscle fibers. Taken together, findings indicate that Grx2 controls skeletal muscle mitochondrial fusion, ultrastructure, and complex I-driven respiration (Figure 8).
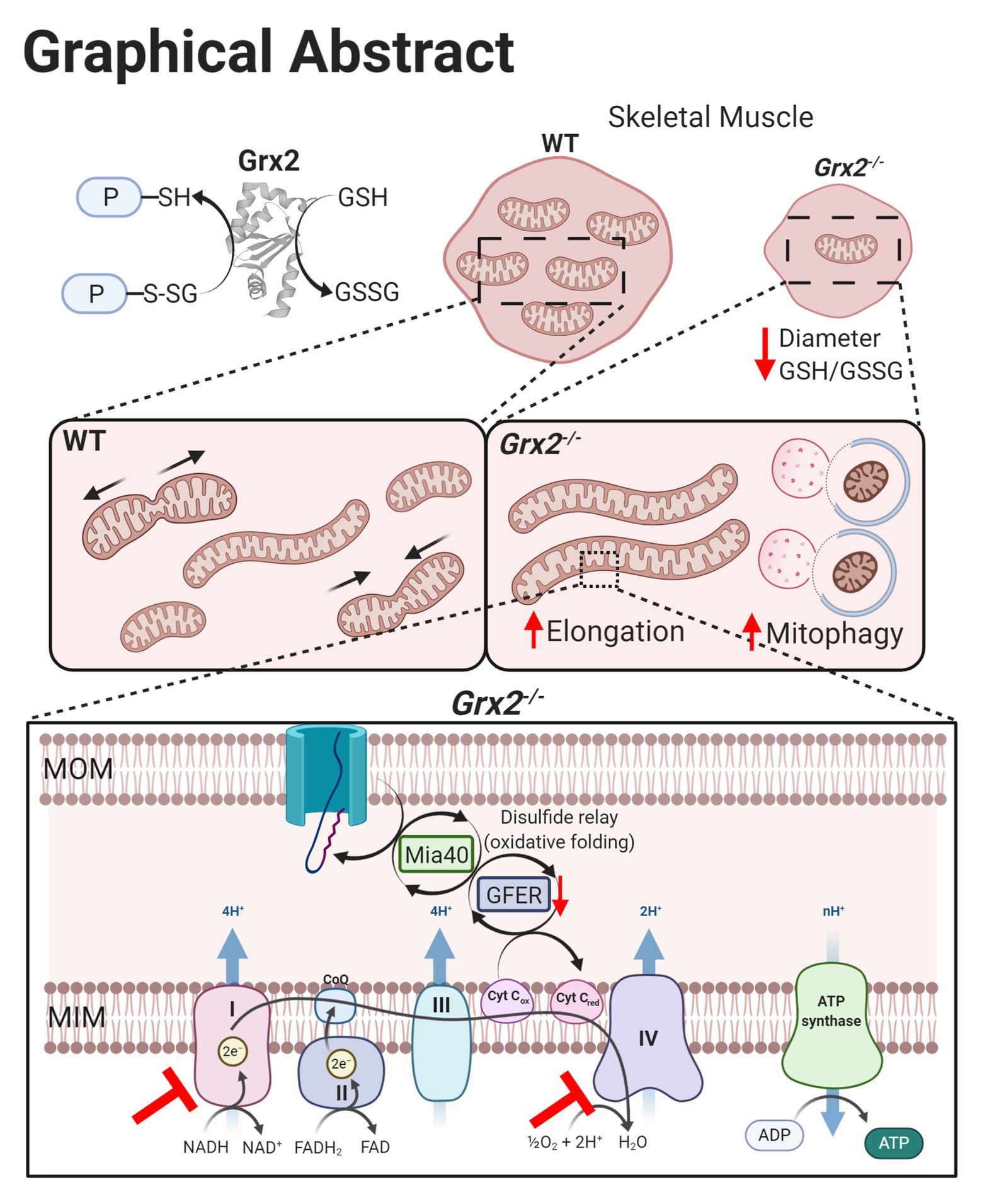
Figure 8. Graphical summary of effects on skeletal muscle following Grx2 deletion. Glutaredoxin-2 (Grx2) is a glutathione-dependent oxidoreductase that reversibly (de)glutathionylates sulfhydryl residues on mitochondrial proteins, thereby directly maintaining the pool of oxidized and GSH and redox homeostasis. Deletion of Grx2 alters skeletal muscle morphology, mitochondrial ultrastructure, and activity. Myofiber diameter of tibialis anterior muscle is smaller following Grx2 deletion, and the ratio of reduced to oxidized glutathione is decreased. The mitochondrial network is elongated in Grx2−/− primary myoblasts and TEM images of Grx2−/− myofibers reveal mitochondria encapsulated in vacuole-like structures resembling autophagosomes. Lastly, the oxidative folding pathway and the electron transfer system are dysregulated as a consequence of Grx2 deletion, consistent with the overall importance of Grx2 in maintaining mitochondrial protein structure and function. Graphical abstract was created with BioRender.com.
Mitochondrial morphology is thought to alter energy transduction, as enhanced fusion is often associated with increased mitochondrial function (Liu et al., 2009; Zick et al., 2009). Mitochondria can also elongate as an acute stress-response to remain protected from autophagic degradation and activation of apoptosis (Tondera et al., 2009; Gomes et al., 2011). Mitochondrial remodeling can occur in response to changes in cellular redox, and the accumulation of GSSG, in particular, has been linked to mitochondrial elongation (Shutt et al., 2012). Specifically, Shutt et al. (2012) demonstrated that a decrease in the GSH:GSSG ratio induces mitochondrial elongation via priming of fusion machinery and generation of MFN disulfide-mediated oligomers. In the context of the current study, our findings of perturbed glutathione redox in Grx2 deficient primary myoblasts, as indicated by a decrease in the ratio of GSH:GSSG, may contribute to the observed mitochondrial elongation in Grx2−/− myoblasts. Furthermore, the trend for increased oligomerization of MFN1/2 in Grx2−/− muscle supports the idea that the coil-coil heptad repeat domains (HR2) domains of MFN1/2 contain cysteine residues located in the intermembrane space that are partially regulated by redox signaling (Mattie et al., 2018). Mitochondrial hyperfusion is generally thought to be a nullifying response to cellular stress; however, prolonged hyperfusion may lead to the accumulation of damaged mitochondrial proteins and activation of autophagy (El Fissi et al., 2018).
Alterations of mitochondrial topology are commonly observed in diseases associated with impaired mitochondrial function (Chan, 2020). Here we show profoundly abnormal mitochondrial ultrastructural characteristics, including decreased mitochondrial surface area and the appearance of vacuole-like double membranous structures. The abnormal mitochondrial ultrastructure observed in the Grx2−/− skeletal muscle is consistent with results from livers dissected from mice treated with ethanol or acetaminophen (Ding and Yin, 2012), and mouse embryonic fibroblasts (MEFS) and SH-SY5Y neuroblasts treated with CCCP (Miyazono et al., 2018; Chakraborty et al., 2020). The decrease in mitochondrial surface area is suggestive of a decrease in mitochondrial density, however analysis of mtDNA:nDNA copy number did not reveal differences between genotypes. Abnormal onion-like structures, similar to the membranous structures detected in the Grx2−/− muscle, have been observed in OPA1 and mitofilin-depleted cells (Olichon et al., 2003; John et al., 2005). The multi-lamellar membranous structures are thought to be the result of lysosomal contact with mitochondria (Wong et al., 2018). Interestingly, similar membranous structures were observed in neurons from Drosophila with a Charcot-Marie-Tooth disease type 2A (CMT2A) MFN2 mutation (R364Wlike) that results in mitochondrial hyperfusion (El Fissi et al., 2018). Similar to our findings in Grx2−/− muscle, of mtDNA copy number did not differ in R364Wlike MFN2 mutant brains, but rather the mtDNA mutation load was elevated (El Fissi et al., 2018). Therefore, it is possible that mtDNA is retained in the membranous onion-like structures, and subjected to damage. Within the context of the current study, the decreased mitochondrial surface area, combined with the presence of abnormal double membranous structures, are consistent with increased autophagy in Grx2−/− muscle.
Autophagy is a highly conserved process that involves the lysosomal degradation of cellular components sequestered in autophagosomes. ROS can induce autophagy as a mechanism to combat oxidative stress and damage, and the activity of autophagy-related protein (ATG)-4 is regulated by the reversible oxidation of Cys81 (Scherz-Shouval et al., 2007; Scherz-Shouval and Elazar, 2011). More recently, glutathione redox has been implicated in the activation of autophagy, with initial evidence demonstrating that the depletion of GSH activates mitophagy in yeast (Deffieu et al., 2009). In mammalian systems, glutathione depletion has been shown to activate autophagy in carcinoma cells (Desideri et al., 2012) and retinal pigment cells (Sun et al., 2018). Thus, our finding of increased autophagic flux in muscle of Grx2−/− mice, indicated by elevated LC3II/I, supports a role for glutathione redox regulation of autophagy. Moreover, the trend for increased colocalization of mitochondria and lysosomes in Grx2−/− primary myoblasts is consistent with increased mitophagy. The removal of mitochondria by mitophagy appears to be occurring through the traditional PINK1/parkin pathway, which relies on parkin to polyubiquitinate outer membrane target proteins to encapsulate mitochondria in the autophagosome, as parkin expression was elevated in Grx2−/− skeletal muscle from mice injected with saline and colchicine. Other proteins in the Bcl-2 family can also interact directly with LC3-II to encapsulate mitochondria independent of parkin-mediated ubiquitination, and may contribute to the induction of mitophagy in Grx2−/− skeletal muscle. Altogether, these findings of increased autophagic flux and appearance of multi-lamellar membranous structures in Grx2−/− skeletal muscle support a role for the redox control of autophagy.
Reversible glutathionylation reactions play important roles in controlling mitochondrial redox, as mitochondria are a major source of superoxide (Turrens, 1997). Protein thiols on specific subunits of complex I (Ndusf1, Ndufv1, and ND3; Beer et al., 2004), are known to be particularly susceptible to regulation by glutathionylation (Cadenas and Davies, 2000). Here, we demonstrate that complex I-supported respiration was decreased in Grx2−/− permeabilized skeletal muscle fibers. In line with these findings, we have previously shown that glutathionylation of complex I subunits, is regulated by Grx2 in cardiac tissue (Mailloux et al., 2014). Modifications to complex I thiols decrease complex I activity in isolated mitochondria from the brain (Balijepalli et al., 1999) and liver (Mailloux et al., 2013). Moreover, deletion of Grx2 in human lens epithelial cells and in primary neonatal cardiomyocytes results in decreased complex I activity and basal mitochondrial function, respectively (Wu et al., 2010; Kanaan et al., 2018). Thus, our current findings, support a role for the redox control of complex I by glutathionylation actions of Grx2.
The mitochondrial disulfide relay system is responsible for the retention and oxidative folding of cysteine-rich proteins in the IMS (Mesecke et al., 2005). Moreover, the disulfide relay system is intricately linked with the mitochondrial electron transport system, as reduced ERV1 (GFER homolog in yeast) can transfer electrons cytochrome C and subsequently cytochrome c oxidase (Complex IV) to induce ERV1 oxidization (Dabir et al., 2007). Grx2 is dually localized in the IMS and matrix of the mitochondria, and is thought to play a regulatory role in the recycling of ERV1/GFER by regulating glutathione redox in the IMS (Kojer et al., 2012, 2015). In yeast, overexpression of Grx2 delays protein folding by disrupting the oxidation of Mia40 by ERV1 (Kojer et al., 2015). The absence of Grx2 also impairs oxidative protein folding in yeast, a consequence attributed to the proofreading actions of glutathione in oxidized protein folding, in which GSH prevents the formation of partially oxidized intermediates to ensure complete and efficient substrate oxidation (Bien et al., 2010). Our findings support the notion that Grx2 may play a proofreading role in the disulfide relay system (Kojer et al., 2012, 2015). Our correlation analyses revealed that Grx2 is positively associated with Gfer and Cox17 and we found that deletion of Grx2 in skeletal muscle results in decreased GFER protein expression, lower complex IV activity, and increased expression of the disulfide relay system substrate COX17, perhaps as a compensatory effect. Interestingly, three patients with a mutation in GFER were identified to have a progressive mitochondrial myopathy that presented with decreased activity of complex IV (Di Fonzo et al., 2009). Electron micrographs of their skeletal muscle revealed enlarged mitochondria, and the appearance of vacuole-like structures (Di Fonzo et al., 2009), similar to our observations in Grx2−/− muscle. These findings indicate that the disulfide relay system may be linked to the activation of mitophagy, as both Grx2 deletion and GFER mutation resulted in abnormal mitochondrial ultrastructure and the appearance of vacuole-like structures (Di Fonzo et al., 2009). This emerging mechanistic link between GFER and autophagy is supported by in vitro evidence demonstrating that genetic depletion of GFER is associated with the activation of autophagy (Todd et al., 2010; Pu et al., 2017), in part through the AMPK/mTOR pathway (Pu et al., 2017).
Our findings demonstrate that Grx2 is involved in the regulation of mitochondrial morphology, autophagy, and energetics in skeletal muscle (Figure 8). The highly irregular ultrastructure of mitochondria and appearance of multi-lamellar structure in TEM micrographs of Grx2−/− muscle, combined with the increase in protein expression of parkin, elevated LC3II/I flux, and the colocalization of mitochondria and lysosomes in Grx2 deficient primary myoblasts are collectively consistent with the conclusion that there is increased autophagic clearance of mitochondria in skeletal muscle of Grx2−/− mice. Deletion of Grx2 decreased the GSH:GSSG ratio, and this was associated with increased mitochondrial length, which also suggests that the absence of Grx2 promotes stress-induced mitochondrial hyperfusion. These structural abnormalities in mitochondria were associated with impaired complex I OXPHOS activity in myofibers in Grx2 deficient muscle. Future studies should further examine the molecular mechanisms that contribute to the glutathione redox control of autophagy. Moreover, studies should investigate the role of glutathione redox and Grx2 in mammalian diseases associated with impaired complex I and IV function and assembly.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by Animal Care Committee of the University of Ottawa.
Author Contributions
Conceived and designed the experiments: M-EH. Performed the experiments: AL, CP, GP, DP, NH, AC. Analyzed data: CP, AL, GP, DP, NH. Drafted the manuscript: CP, GP. Critically evaluated and contributed to the manuscript: CP, GP, DP, NH, AC, YB, M-EH. All authors contributed to the article and approved the submitted version.
Funding
This research was funded through a grant from the Canadian Institutes of Health Research, Institute of Nutrition, Metabolism and Diabetes (CIHR-INMD; FDN-143278, M-EH).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Jian Ying Xuan for excellent technical support, Skye McBride and the Cell Biology and Image Acquisition Core funded by the University of Ottawa, Ottawa, Canada and the Canada Foundation for Innovation. We would also like to thank Arkadiy Reunov for expert assistance with electron microscopy.
Abbreviations
GRX2, Glutaredoxin 2; DRP1, Dynamin related protein-1; GFER, Growth factor, augmenter of liver regeneration; GSH, Reduced glutathione; GSSG, Oxidized glutathione disulfide; IMS, Mitochondrial intermembrane space; MFN, Mitofusin; MFF, Mitochondrial fission factor; Mia40, Mitochondrial intermembrane space import and assembly protein 40; OPA1, Optic atrophy type 1; OXPHOS, Oxidative phosphorylation; RER, Respiratory exchange ratio; ROS, Reactive oxygen species; TEM, Transmission electron microscopy; UCP3, Uncoupling protein 3.
Footnotes
1. Raw microarray data are available at http://www.genenetwork.org.
References
Adachi, T., Pimentel, D. R., Heibeck, T., Hou, X., Lee, Y. J., Jiang, B., et al. (2004). S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J. Biol. Chem. 279, 29857–29862. doi: 10.1074/jbc.M313320200
Allen, S., Balabanidou, V., Sideris, D. P., Lisowsky, T., and Tokatlidis, K. (2005). Erv1 mediates the Mia40-dependent protein import pathway and provides a functional link to the respiratory chain by shuttling electrons to cytochrome c. J. Mol. Biol. 353, 937–944. doi: 10.1016/j.jmb.2005.08.049
Ashrafi, G., and Schwarz, T. L. (2013). The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 20, 31–42. doi: 10.1038/cdd.2012.81
Augusto, V., Padovani, C. R., and Campos, G. E. R. (2004). Skeletal muscle fiber types in C57BL6J mice. Braz. J. Morphol. Sci. 21, 89–94.
Balijepalli, S., Annepu, J., Boyd, M. R., and Ravindranath, V. (1999). Effect of thiol modification on brain mitochondrial complex I activity. Neurosci. Lett. 272, 203–206. doi: 10.1016/s0304-3940(99)00593-5
Beer, S. M., Taylor, E. R., Brown, S. E., Dahm, C. C., Costa, N. J., Runswick, M. J., et al. (2004). Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: implications for mitochondrial redox regulation and antioxidant defence. J. Biol. Chem. 279, 47939–47951. doi: 10.1074/jbc.M408011200
Bien, M., Longen, S., Wagener, N., Chwalla, I., Herrmann, J. M., and Riemer, J. (2010). Mitochondrial disulfide bond formation is driven by intersubunit electron transfer in Erv1 and proofread by glutathione. Mol. Cell 37, 516–528. doi: 10.1016/j.molcel.2010.01.017
Cadenas, E., and Davies, K. J. A. (2000). Mitochondrial free radical generation, oxidative stress, and aging1. Free Radic. Biol. Med. 29, 222–230.
Calabrese, G., Morgan, B., and Riemer, J. (2017). Mitochondrial glutathione: regulation and functions. Antioxid. Redox Signal. 27, 1162–1177. doi: 10.1089/ars.2017.7121
Chakraborty, J., Caicci, F., Roy, M., and Ziviani, E. (2020). Investigating mitochondrial autophagy by routine transmission electron microscopy: seeing is believing? Pharmacol. Res. 160:105097. doi: 10.1016/j.phrs.2020.105097
Chan, D. C. (2020). Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. Mech. Dis. 15, 235–259. doi: 10.1146/annurev-pathmechdis-012419-032711
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200. doi: 10.1083/jcb.200211046
Christiansen, L. B., Dela, F., Koch, J., and Yokota, T. (2015). Tissue-specific and substrate-specific mitochondrial bioenergetics in feline cardiac and skeletal muscles. J. Vet. Med. Sci. 7, 669–675. doi: 10.1292/jvms.14-0573
Dabir, D. V., Leverich, E. P., Kim, S., Tsai, F. D., Hirasawa, M., Knaff, D. B., et al. (2007). A role for cytochrome c and cytochrome c peroxidase in electron shuttling from Erv1. EMBO J. 26, 4801–4811. doi: 10.1038/sj.emboj.7601909
Deffieu, M., Bhatia-Kiššová, I., Salin, B., Galinier, A., Manon, S., and Camougrand, N. (2009). Glutathione participates in the regulation of mitophagy in yeast. J. Biol. Chem. 284, 14828–14837. doi: 10.4161/auto.9065
Desideri, E., Filomeni, G., and Ciriolo, M. R. (2012). Glutathione participates in the modulation of starvation-induced autophagy in carcinoma cells. Autophagy 8, 1769–1781. doi: 10.4161/auto.22037
Diebold, L., and Chandel, N. S. (2016). Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 100, 86–93. doi: 10.1016/j.freeradbiomed.2016.04.198
Di Fonzo, A., Ronchi, D., Lodi, T., Fassone, E., Tigano, M., Lamperti, C., et al. (2009). The mitochondrial disulfide relay system protein GFER is mutated in autosomal-recessive myopathy with cataract and combined respiratory-chain deficiency. Am. J. Hum. Genet. 84, 594–604. doi: 10.1016/j.ajhg.2009.04.004
Ding, W. -X., and Yin, X. -M. (2012). Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol. Chem. 393, 547–564. doi: 10.1515/hsz-2012-0119
El Fissi, N., Rojo, M., Aouane, A., Karatas, E., Poliacikova, G., David, C., et al. (2018). Mitofusin gain and loss of function drive pathogenesis in Drosophila models of CMT 2A neuropathy. EMBO Rep. 19:e45241. doi: 10.15252/embr.201745241
Figlia, G., Willnow, P., and Teleman, A. A. (2020). Metabolites regulate cell signaling and growth via covalent modification of proteins. Dev. Cell 54, 156–170. doi: 10.1016/j.devcel.2020.06.036
Filomeni, G., Desideri, E., Cardaci, S., Rotilio, G., and Ciriolo, M. R. (2010). Under the ROS: thiol network is the principal suspect for autophagy commitment. Autophagy 6, 999–1005. doi: 10.4161/auto.6.7.12754
Frezza, C., Cipolat, S., de Brito, O. M., Micaroni, M., Beznoussenko, G. V., Rudka, T., et al. (2006). OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177–189. doi: 10.1016/j.cell.2006.06.025
Gabriel, K., Milenkovic, D., Chacinska, A., Müller, J., Guiard, B., Pfanner, N., et al. (2007). Novel mitochondrial intermembrane space proteins as substrates of the MIA import pathway. J. Mol. Biol. 365, 612–620. doi: 10.1016/j.jmb.2006.10.038
Giacomello, M., Pyakurel, A., Glytsou, C., and Scorrano, L. (2020). The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 21, 204–224. doi: 10.1038/s41580-020-0210-7
Gill, R. M., O’Brien, M., Young, A., Gardiner, D., and Mailloux, R. J. (2018). Protein S-glutathionylation lowers superoxide/hydrogen peroxide release from skeletal muscle mitochondria through modification of complex I and inhibition of pyruvate uptake. PLoS One 13:e0192801. doi: 10.1371/journal.pone.0192801
Gomes, L. C., Di Benedetto, G., and Scorrano, L. (2011). During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 13, 589–598. doi: 10.1038/ncb2220
Guo, W., Jiang, L., Bhasin, S., Khan, S. M., and Swerdlow, R. H. (2009). DNA extraction procedures meaningfully influence qPCR-based mtDNA copy number determination. Mitochondrion 9, 261–265. doi: 10.1016/j.mito.2009.03.003
Hell, K. (2008). The Erv1–Mia40 disulfide relay system in the intermembrane space of mitochondria. Biochim. Biophys. Acta 1783, 601–609. doi: 10.1016/j.bbamcr.2007.12.005
Hoffmann, M. H., and Griffiths, H. R. (2018). The dual role of reactive oxygen species in autoimmune and inflammatory diseases: evidence from preclinical models. Free Radic. Biol. Med. 125, 62–71. doi: 10.1016/j.freeradbiomed.2018.03.016
Holmgren, A. (1976). Hydrogen donor system for Escherichia coli ribonucleoside-diphosphate reductase dependent upon glutathione. Proc. Natl. Acad. Sci. 73, 2275–2279. doi: 10.1073/pnas.73.7.2275
Houstis, N., Rosen, E. D., and Lander, E. S. (2006). Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 440, 944–948. doi: 10.1038/nature04634
Hurd, T. R., Requejo, R., Filipovska, A., Brown, S., Prime, T. A., Robinson, A. J., et al. (2008). Complex I within Oxidatively stressed bovine heart mitochondria is Glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit POTENTIAL ROLE OF CYS RESIDUES IN DECREASING OXIDATIVE DAMAGE. J. Biol. Chem. 283, 24801–24815. doi: 10.1074/jbc.M803432200
Iwashita, H., Torii, S., Nagahora, N., Ishiyama, M., Shioji, K., Sasamoto, K., et al. (2017). Live cell imaging of mitochondrial autophagy with a novel fluorescent small molecule. ACS Chem. Biol. 12, 2546–2551. doi: 10.1021/acschembio.7b00647
John, G. B., Shang, Y., Li, L., Renken, C., Mannella, C. A., Selker, J. M. L., et al. (2005). The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol. Biol. Cell 16, 1543–1554. doi: 10.1091/mbc.e04-08-0697
Ju, J. -S., Varadhachary, A. S., Miller, S. E., and Weihl, C. C. (2010). Quantitation of "autophagic flux" in mature skeletal muscle. Autophagy 6, 929–935. doi: 10.4161/auto.6.7.12785
Kadowaki, M., and Karim, M. R. (2009). Cytosolic LC3 ratio as a quantitative index of macroautophagy. Methods Enzymol. 452, 199–213. doi: 10.1016/S0076-6879(08)03613-6
Kanaan, G. N., Ichim, B., Gharibeh, L., Maharsy, W., Patten, D. A., Xuan, J. Y., et al. (2018). Glutaredoxin-2 controls cardiac mitochondrial dynamics and energetics in mice, and protects against human cardiac pathologies. Redox Biol. 14, 509–521. doi: 10.1016/j.redox.2017.10.019
Kojer, K., Bien, M., Gangel, H., Morgan, B., Dick, T. P., and Riemer, J. (2012). Glutathione redox potential in the mitochondrial intermembrane space is linked to the cytosol and impacts the Mia40 redox state. EMBO J. 31, 3169–3182. doi: 10.1038/emboj.2012.165
Kojer, K., Peleh, V., Calabrese, G., Herrmann, J. M., and Riemer, J. (2015). Kinetic control by limiting glutaredoxin amounts enables thiol oxidation in the reducing mitochondrial intermembrane space. Mol. Biol. Cell 26, 195–204. doi: 10.1091/mbc.E14-10-1422
Kramer, P. A., Duan, J., Gaffrey, M. J., Shukla, A. K., Wang, L., Bammler, T. K., et al. (2018). Fatiguing contractions increase protein S-glutathionylation occupancy in mouse skeletal muscle. Redox Biol. 17, 367–376. doi: 10.1016/j.redox.2018.05.011
Lionaki, E., Aivaliotis, M., Pozidis, C., and Tokatlidis, K. (2010). The N-terminal shuttle domain of Erv1 determines the affinity for Mia40 and mediates electron transfer to the catalytic Erv1 core in yeast mitochondria. Antioxid. Redox Signal. 13, 1327–1339. doi: 10.1089/ars.2010.3200
Liu, X., Weaver, D., Shirihai, O., and Hajnoczky, G. (2009). Mitochondrial “kiss-and-run”: interplay between mitochondrial motility and fusion-fission dynamics. EMBO J. 28, 3074–3089. doi: 10.1038/emboj.2009.255
Lorenzen, A., Samosh, J., Vandewark, K., Anborgh, P. H., Seah, C., Magalhaes, A. C., et al. (2010). Rapid and direct transport of cell surface APP to the lysosome defines a novel selective pathway. Mol. Brain 3:11. doi: 10.1186/1756-6606-3-11
Mailloux, R. J. (2020). Protein S-glutathionylation reactions as a global inhibitor of cell metabolism for the desensitization of hydrogen peroxide signals. Redox Biol. 32:101472. doi: 10.1016/j.redox.2020.101472
Mailloux, R. J., Xuan, J. Y., Beauchamp, B., Jui, L., Lou, M., and Harper, M. E. (2013). Glutaredoxin-2 is required to control proton leak through uncoupling protein-3. J. Biol. Chem. 288, 8365–8379. doi: 10.1074/jbc.M112.442905
Mailloux, R. J., Xuan, J. Y., McBride, S., Maharsy, W., Thorn, S., Holterman, C. E., et al. (2014). Glutaredoxin-2 is required to control oxidative phosphorylation in cardiac muscle by mediating deglutathionylation reactions. J. Biol. Chem. 289, 14812–14828. doi: 10.1074/jbc.M114.550574
Mattie, S., Riemer, J., Wideman, J. G., and McBride, H. M. (2018). A new mitofusin topology places the redox-regulated C terminus in the mitochondrial intermembrane space. J. Cell Biol. 217, 507–515. doi: 10.1083/jcb.201611194
McMurray, F., Patten, D. A., and Harper, M. (2016). Reactive oxygen species and oxidative stress in obesity—recent findings and empirical approaches. Obesity 24, 2301–2310. doi: 10.1002/oby.21654
Menges, S., Minakaki, G., Schaefer, P. M., Meixner, H., Prots, I., Schlötzer-Schrehardt, U., et al. (2017). Alpha-synuclein prevents the formation of spherical mitochondria and apoptosis under oxidative stress. Sci. Rep. 7:42942. doi: 10.1038/srep42942
Mesecke, N., Terziyska, N., Kozany, C., Baumann, F., Neupert, W., Hell, K., et al. (2005). A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell 121, 1059–1069. doi: 10.1016/j.cell.2005.04.011
Miyazono, Y., Hirashima, S., Ishihara, N., Kusukawa, J., Nakamura, K., and Ohta, K. (2018). Uncoupled mitochondria quickly shorten along their long axis to form indented spheroids, instead of rings, in a fission-independent manner. Sci. Rep. 8:350. doi: 10.1038/s41598-017-18582-6
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/BJ20081386
Murphy, M. P., and Hartley, R. C. (2018). Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 17, 865–886. doi: 10.1038/nrd.2018.174
Neupert, W., and Herrmann, J. M. (2007). Translocation of proteins into mitochondria. Annu. Rev. Biochem. 76, 723–749. doi: 10.1146/annurev.biochem.76.052705.163409
Nikolaienko, R., Bovo, E., and Zima, A. V. (2018). Redox dependent modifications of ryanodine receptor: basic mechanisms and implications in heart diseases. Front. Physiol. 9:1775. doi: 10.3389/fphys.2018.01775
Olichon, A., Baricault, L., Gas, N., Guillou, E., Valette, A., Belenguer, P., et al. (2003). Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 278, 7743–7746. doi: 10.1074/jbc.C200677200
Otsuga, D., Keegan, B. R., Brisch, E., Thatcher, J. W., Hermann, G. J., Bleazard, W., et al. (1998). The dynamin-related GTPase, Dnm1p, controls mitochondrial morphology in yeast. J. Cell Biol. 143, 333–349. doi: 10.1083/jcb.143.2.333
Pasut, A., Oleynik, P., and Rudnicki, M. A. (2012). Isolation of muscle stem cells by fluorescence activated cell sorting cytometry. Methods Mol. Biol. 798, 53–64. doi: 10.1007/978-1-61779-343-1_3
Pesta, D., and Gnaiger, E. (2012). High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol. Biol. 810, 25–58. doi: 10.1007/978-1-61779-382-0_3
Pileggi, C. A., Hedges, C. P., Segovia, S. A., Markworth, J. F., Durainayagam, B. R., Gray, C., et al. (2016). Maternal high fat diet alters skeletal muscle mitochondrial catalytic activity in adult male rat offspring. Front. Physiol. 7:546. doi: 10.3389/fphys.2016.00546
Pimentel, D. R., Adachi, T., Ido, Y., Heibeck, T., Jiang, B., Lee, Y., et al. (2006). Strain-stimulated hypertrophy in cardiac myocytes is mediated by reactive oxygen species-dependent Ras S-glutathiolation. J. Mol. Cell. Cardiol. 41, 613–622. doi: 10.1016/j.yjmcc.2006.05.009
Piper, H. M., Sezer, O., Schleyer, M., Schwartz, P., Hütter, J. F., and Spieckermann, P. G. (1985). Development of ischemia-induced damage in defined mitochondrial subpopulations. J. Mol. Cell. Cardiol. 17, 885–896. doi: 10.1016/s0022-2828(85)80102-4
Pirinen, E., Canto, C., Jo, Y. S., Morato, L., Zhang, H., Menzies, K. J., et al. (2014). Pharmacological inhibition of poly (ADP-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 19, 1034–1041. doi: 10.1016/j.cmet.2014.04.002
Pu, T., Liao, X., Sun, H., Guo, H., Jiang, X., Peng, J., et al. (2017). Augmenter of liver regeneration regulates autophagy in renal ischemia–reperfusion injury via the AMPK/mTOR pathway. Apoptosis 22, 955–969. doi: 10.1007/s10495-017-1370-6
Rojo, M., Legros, F., Chateau, D., and Lombès, A. (2002). Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J. Cell Sci. 115, 1663–1674.
Sabouny, R., and Shutt, T. E. (2020). Reciprocal regulation of mitochondrial fission and fusion. Trends Biochem. Sci. 45, 564–577. doi: 10.1016/j.tibs.2020.03.009
Santel, A., and Fuller, M. T. (2001). Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 114, 867–874.
Scherz-Shouval, R., and Elazar, Z. (2011). Regulation of autophagy by ROS: physiology and pathology. Trends Biochem. Sci. 36, 30–38. doi: 10.1016/j.tibs.2010.07.007
Scherz-Shouval, R., Shvets, E., Fass, E., Shorer, H., Gil, L., and Elazar, Z. (2007). Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 26, 1749–1760. doi: 10.1038/sj.emboj.7601623
Shutt, T., Geoffrion, M., Milne, R., and McBride, H. M. (2012). The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 13, 909–915. doi: 10.1038/embor.2012.128
Song, Z., Ghochani, M., McCaffery, J. M., Frey, T. G., and Chan, D. C. (2009). Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 20, 3525–3532. doi: 10.1091/mbc.e09-03-0252
Stauffer, W., Sheng, H., and Lim, H. N. (2018). EzColocalization: an ImageJ plugin for visualizing and measuring colocalization in cells and organisms. Sci. Rep. 8:15764. doi: 10.1038/s41598-018-33592-8
Steinlechner-Maran, R., Eberl, T., Kunc, M., Margreiter, R., and Gnaiger, E. (1996). Oxygen dependence of respiration in coupled and uncoupled endothelial cells. Am. J. Physiol. 271, C2053–C2061. doi: 10.1152/ajpcell.1996.271.6.C2053
Stockwell, B. R., Angeli, J. P. F., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. doi: 10.1016/j.cell.2017.09.021
Sun, Y., Zheng, Y., Wang, C., and Liu, Y. (2018). Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 9:753. doi: 10.1038/s41419-018-0794-4
Thaher, O., Wolf, C., Dey, P. N., Pouya, A., Wüllner, V., Tenzer, S., et al. (2018). The thiol switch C684 in Mitofusin-2 mediates redox-induced alterations of mitochondrial shape and respiration. Neurochem. Int. 117, 167–173. doi: 10.1016/j.neuint.2017.05.009
Todd, L. R., Damin, M. N., Gomathinayagam, R., Horn, S. R., Means, A. R., and Sankar, U. (2010). Growth factor erv1-like modulates Drp1 to preserve mitochondrial dynamics and function in mouse embryonic stem cells. Mol. Biol. Cell 21, 1225–1236. doi: 10.1091/mbc.e09-11-0937
Tondera, D., Grandemange, S., Jourdain, A., Karbowski, M., Mattenberger, Y., Herzig, S., et al. (2009). SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 28, 1589–1600. doi: 10.1038/emboj.2009.89
Turrens, J. (1997). Superoxide production by the mitochondrial respiratory chain. Biosci. Rep. 17, 3–8. doi: 10.1023/a:1027374931887
Underwood, B. R., Imarisio, S., Fleming, A., Rose, C., Krishna, G., Heard, P., et al. (2010). Antioxidants can inhibit basal autophagy and enhance neurodegeneration in models of polyglutamine disease. Hum. Mol. Genet. 19, 3413–3429. doi: 10.1093/hmg/ddq253
Vainshtein, A., Tryon, L. D., Pauly, M., and Hood, D. A. (2015). Role of PGC-1α during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am. J. Physiol. 308, C710–C719. doi: 10.1152/ajpcell.00380.2014
Valente, A. J., Maddalena, L. A., Robb, E. L., Moradi, F., and Stuart, J. A. (2017). A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 119, 315–326. doi: 10.1016/j.acthis.2017.03.001
Wong, Y. C., Ysselstein, D., and Krainc, D. (2018). Mitochondria–lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554, 382–386. doi: 10.1038/nature25486
Wu, H., Xing, K., and Lou, M. F. (2010). Glutaredoxin 2 prevents H2O2-induced cell apoptosis by protecting complex I activity in the mitochondria. Biochim. Biophys. Acta 1797, 1705–1715. doi: 10.1016/j.bbabio.2010.06.003
Yan, J., Jiang, J., He, L., and Chen, L. (2020). Mitochondrial superoxide/hydrogen peroxide: an emerging therapeutic target for metabolic diseases. Free Radic. Biol. Med. 152, 33–42. doi: 10.1016/j.freeradbiomed.2020.02.029
Yu, R., Lendahl, U., Nistér, M., and Zhao, J. (2020). Regulation of mammalian mitochondrial dynamics: opportunities and challenges. Front. Endocrinol. 11:374. doi: 10.3389/fendo.2020.00374
Zick, M., Rabl, R., and Reichert, A. S. (2009). Cristae formation-linking ultrastructure and function of mitochondria. Biochim. Biophys. Acta, Mol. Cell Res. 1793, 5–19. doi: 10.1016/j.bbamcr.2008.06.013
Keywords: mitochondria, glutathione, mitochondrial dynamics, disulfide relay system, glutaredoxin 2, autophagy
Citation: Liaghati A, Pileggi CA, Parmar G, Patten DA, Hadzimustafic N, Cuillerier A, Menzies KJ, Burelle Y and Harper M-E (2021) Grx2 Regulates Skeletal Muscle Mitochondrial Structure and Autophagy. Front. Physiol. 12:604210. doi: 10.3389/fphys.2021.604210
Edited by:
Gareth Davison, Ulster University, United KingdomReviewed by:
Leonardo Yuji Tanaka, University of São Paulo, BrazilVikas Anathy, University of Vermont Larner College of Medicine, United States
Copyright © 2021 Liaghati, Pileggi, Parmar, Patten, Hadzimustafic, Cuillerier, Menzies, Burelle and Harper. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mary-Ellen Harper, bWhhcnBlckB1b3R0YXdhLmNh
†These authors share first authorship