 Elizabeth A. Schroder1,2*†
Elizabeth A. Schroder1,2*† Jennifer L. Wayland1
Jennifer L. Wayland1 Kaitlyn M. Samuels1Syed F. Shah1Don E. Burgess1Tanya Seward1Claude S. Elayi3
Kaitlyn M. Samuels1Syed F. Shah1Don E. Burgess1Tanya Seward1Claude S. Elayi3 Karyn A. Esser4
Karyn A. Esser4 Brian P. Delisle1*†
Brian P. Delisle1*†- 1Department of Physiology, University of Kentucky, Lexington, KY, United States
- 2Internal Medicine and Pulmonary, University of Kentucky, Lexington, KY, United States
- 3CHI Saint Joseph Hospital, Lexington, KY, United States
- 4Department of Physiology and Functional Genomics, University of Florida, Gainesville, FL, United States
Circadian rhythms are generated by cell autonomous circadian clocks that perform a ubiquitous cellular time-keeping function and cell type-specific functions important for normal physiology. Studies show inducing the deletion of the core circadian clock transcription factor Bmal1 in adult mouse cardiomyocytes disrupts cardiac circadian clock function, cardiac ion channel expression, slows heart rate, and prolongs the QT-interval at slow heart rates. This study determined how inducing the deletion of Bmal1 in adult cardiomyocytes impacted the in vivo electrophysiological phenotype of a knock-in mouse model for the arrhythmogenic long QT syndrome (Scn5a+/ΔKPQ). Electrocardiographic telemetry showed inducing the deletion of Bmal1 in the cardiomyocytes of mice with or without the ΔKPQ-Scn5a mutation increased the QT-interval at RR-intervals that were ≥130 ms. Inducing the deletion of Bmal1 in the cardiomyocytes of mice with or without the ΔKPQ-Scn5a mutation also increased the day/night rhythm-adjusted mean in the RR-interval, but it did not change the period, phase or amplitude. Compared to mice without the ΔKPQ-Scn5a mutation, mice with the ΔKPQ-Scn5a mutation had reduced heart rate variability (HRV) during the peak of the day/night rhythm in the RR-interval. Inducing the deletion of Bmal1 in cardiomyocytes did not affect HRV in mice without the ΔKPQ-Scn5a mutation, but it did increase HRV in mice with the ΔKPQ-Scn5a mutation. The data demonstrate that deleting Bmal1 in cardiomyocytes exacerbates QT- and RR-interval prolongation in mice with the ΔKPQ-Scn5a mutation.
Introduction
Circadian rhythms alter physiology in anticipation of predictable changes in the daily environment. They are generated by cell autonomous circadian clocks in most of the cells in the body (Reppert and Weaver, 2002; Hastings et al., 2018; Michel and Meijer, 2020). Circadian clocks are formed by transcription-translation feedback loops that drive rhythmic changes in clock gene and protein expression with a periodicity of ∼24 h (Hastings et al., 2018; Michel and Meijer, 2020). The positive limb of the feedback loop is initiated by the transcription factors BMAL1 and CLOCK, which heterodimerize to activate the transcription of Period (PER) and Cryptochrome (CRY). PER and CRY proteins negatively feedback on BMAL1 and CLOCK activity. In addition to functioning as ubiquitous cellular timekeepers, circadian clocks also contribute to cell- and tissue-specific changes in physiology by regulating the expression of genes outside the timekeeping network. Studies using transgenic mouse models that allow for the selective deletion of Bmal1 in adult cardiomyocytes show the cardiomyocyte circadian clock mechanism contributes to heart rate, ventricular repolarization and the functional expression of several cardiac ion channels (Schroder et al., 2013, 2015; Delisle et al., 2020). In this study, we determined how inducing the deletion of Bmal1 in adult cardiomyocytes impacted cardiac electrophysiology in a genetic mouse model of long QT syndrome (LQTS).
People living with congenital LQTS have a high risk for ventricular arrhythmias that can lead to syncope, seizures, and/or sudden cardiac death (SCD) (Schwartz et al., 1993; Moss, 2003). Most cases of LQTS are caused by mutations in one of three different cardiac ion channel genes (LQT1-LQT3). LQT3 is caused by mutations in the predominant voltage-gated Na+ channel expressed in the heart (SCN5A/Nav1.5). Some LQT3-linked mutations associate with more than one type of arrhythmia syndrome/phenotype in people, including cardiac conduction defects, atrial arrhythmias, and/or Brugada Syndrome (Remme, 2013). People who have the LQT3-linked SCN5A deletion mutation ΔKPQ1505-1507 can suffer LQT3 and cardiac conduction defects (Zareba et al., 2001). Heterozygous knock-in mice with the equivalent ΔKPQ-Scn5a mutation (Scn5a+/ΔKPQ) have abnormally long QT- and RR-intervals (Nuyens et al., 2001; Fabritz et al., 2010; Lemoine et al., 2011; Wu et al., 2012). We tested the hypothesis that inducing the deletion of Bmal1 in Scn5a+/ΔKPQ mice would exacerbate QT- and RR-interval prolongation using electrocardiographic (ECG) telemetry.
Materials and Methods
Animals
All animal procedures were conducted in compliance with the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care and were approved by the Institutional Animal Care and Use Committee at University of Kentucky. Mice for these studies were bred by crossing the floxed Bmal1 (Bmal1f/f) mouse and the cardiac-specific, Myh6-MerCreMer recombinase mouse (iCSΔBmal1) (Schroder et al., 2013). The Scn5a+/ΔKPQ mice (kindly provided by Dr. Peter Carmeliet) were bred with the iCSΔBmal1 mice to generate an B6Cre±;B6Bmal1f/f;129Scn5a+/ΔKPQ (iCSΔBmal1/Scn5a+/ΔKPQ) mouse. The iCSΔBmal1+/+ mice consisted of vehicle injected mice or iCSΔBmal1 mice assessed prior to tamoxifen injection. Cre-recombination was activated by intraperitoneal injections of tamoxifen (2 mg/day) for 5 consecutive days to generate iCSΔBmal1–/– mice. The concentration and duration of the tamoxifen injections used have been shown to cause effective cardiomyocyte-specific recombination without any obvious long-term tamoxifen toxicity as assessed by changes in the structure, function, and the ECG (Eckardt et al., 2004; Andersson et al., 2009; Hall et al., 2011; Schroder et al., 2013). Mice were housed in 12 h light and 12 h dark cycles with ad libitum access to food and water.
Circadian Collections
mRNA collections were done as described previously (Schroder et al., 2013). Male and female iCSΔBmal1 mice at 14–16 weeks of age were housed in light boxes and entrained to a 12:12-h light-dark cycle. Two weeks after the final injection of vehicle (32 mice) or tamoxifen (32 mice), mice were released into constant darkness. After 30 h in darkness (18 h after the beginning of the subjective light phase, Circadian Time or CT = 18 h), mice were euthanized under dim red light (< 5 lux) and hearts were collected every 4 h for 28 h, a total of 8 time points. RNA was prepared for quantitative PCR (qtPCR) using TaqMan (Applied Biosystems) assays to examine transcript expression. The ΔΔCT method was used for the quantification of qtPCR data. Gene expression is shown as the relative value compared with the mean vehicle value.
ECG Telemetry
In vivo ECG telemetry was performed as described previously (Schroder et al., 2013, 2014). Briefly, male mice at 14 weeks of age were anesthetized with isoflurane and transmitter units (DSI, TA11ETA-F10) were implanted in the peritoneal cavity. The two ECG leads were secured near the apex of the heart and the right acromion. Mice were housed singly and allowed to recover for 1 week before beginning recording. Telemetry data were recorded at 1000 Hz before and after the intraperitoneal injection of tamoxifen to activate deletion of Bmal1 by Cre-recombination. After injection, mice were given at least 4 weeks to recover before ECG data were recorded.
Ponemah telemetry software (Data Science International) was used to quantify QT-intervals. QT-intervals were analyzed at RR-intervals ranging from 90 ± 3 ms to 140 ± 3 ms in 10 ms bins. Two independent investigators performed these analyses.
Ponemah was also used to measure RR-intervals and HRV. We plotted 3-days of averaged RR-intervals (15-min averages) and fit the individual data with the following cosine function:
This allowed calculation of the period (T), phase (τ), amplitude of the oscillation (A), and rhythm-adjusted mean (m).
HRV was analyzed using the time domain parameters for the standard deviation of all RR-intervals in sinus rhythm (SDNN, in ms); the root mean square differences between successive RR-intervals (RMSSD, in ms); and the percentage of normal consecutive RR-intervals differing by ≥ 6 ms similar to that previously described (Thireau et al., 2008; Fenske et al., 2016; Moghtadaei et al., 2017). SDNN is an index of total sinus rhythm HRV, whereas RMSSD is a beat to beat index of HRV and reflects abrupt changes in RR-intervals. PNN6 is thought to reflect HRV secondary to parasympathetic tone. Each parameter was measured in 15-min episodes. Individual mouse data sets were not well described using a cosine function, so we compared averaged values that corresponded to 2–5, 8–11, 14–17, or 20–23 h after the start of the light phase (Zeitgeber time or ZT = 2–5, 8–11, 14–17, or 20–23 h, respectively).
Statistical Analysis
The data were analyzed using a two-way ANOVA to identify significant interactions (PRISM, MathWorks). We used the Šidák correction method to correct for multiple comparisons. For gene expression studies, the statistical JTK_CYCLE package was used to identify mRNA transcripts that had circadian expression profiles (Hughes et al., 2010).
Results
Inducing the Deletion of Bmal1 in Adult Cardiomyocyte Decreases the mRNA Transcript Levels for Several Cardiac Ion Channels and Transcription Factors Important for Repolarization and Conduction
Studies suggest that Bmal1 directly and indirectly contributes to the transcription of cardiac ion channel genes Scn5a, Kcnh2, Hcn4, and Kchip2 (Jeyaraj et al., 2012; Schroder et al., 2013, 2015; D’Souza et al., 2020). Bmal1 may regulate cardiac ion channel transcription by binding to enhancer box (E-box) elements in the promoters of ion channel genes, or Bmal1 can modify the expression of other transcription factors that regulate ion channel transcription. We wanted to determine how inducing the deletion of Bmal1 in the adult heart impacts the expression profiles for a large number of candidate ion channel genes that encode proteins important for cardiac depolarization, repolarization, conduction, and ion channel transcription in humans and mice (London, 2001; Eckardt et al., 2004; Nerbonne and Kass, 2005; Arnolds et al., 2012; Herrmann et al., 2012; Jeyaraj et al., 2012; Mao et al., 2012; Cai et al., 2014; Nerbonne, 2014; Wahl-Schott et al., 2014; Tarradas et al., 2017).
In mice and humans, the cardiac Na+ current (INa) generates the rapid upstroke of the action potential in the working myocardium and the funny current (IF) initiates depolarization in the autorhythmic myocardium. Scn5a and Hcn4 encode the major pore-forming proteins that conduct INa and IF (Abriel, 2007; Kozasa et al., 2018; D’Souza et al., 2020). Also similar to humans, the inward rectifier K+ current (IK1) in the mouse working myocardium is responsible for stabilizing the resting membrane potential. Kcnj2 encodes a critical pore-forming subunit that conducts IK1 (Nerbonne and Kass, 2005; Nerbonne, 2014). Unlike humans, the rapid and slowly activating delayed rectifier K+ currents (IKs and IKr, respectively) are not major contributors to ventricular repolarization in mice. However, the genes that encode the pore-forming proteins for IKs and IKr, Kcnq1, and Kcnh2, are expressed in the mouse myocardium (Pond et al., 2000; Knollmann et al., 2007). Ventricular repolarization in mice is regulated by several K+ currents, including the transient outward K+ currents (Ito) and the slow K+ currents (IK,slow). Kcnd2, Kchip2, Kcna5, and Kcnb1 encode important pore-forming and auxiliary ion channel proteins that contribute to Ito and IK,slow (Nerbonne and Kass, 2005; Nerbonne, 2014). Gja1, Gja5, and Gjc1 encode the cardiac gap junction proteins (connexins) that mediate conduction between cardiomyocytes (Jalife et al., 1999). The Scn4b auxiliary Na+ channel subunit is a genetic modifier of cardiac conduction speed in mouse models of Na+ channelopathies (Remme et al., 2009). The transcription factor genes Klf15, Tbx5, Gata4, and Foxo1 regulate the expression of certain cardiac ion channel genes, including Kchip2 or Scn5a, previous studies show that these transcription factors have circadian expression profiles in the mouse heart (Arnolds et al., 2012; Jeyaraj et al., 2012; Mao et al., 2012; Pizarro et al., 2013; Cai et al., 2014; Tarradas et al., 2017).
We previously reported the Scn5a, Kcnh2, and Kcnd2 mRNA transcript expression profiles in iCSΔBmal1+/+ and iCSΔBmal1–/– mouse hearts (Schroder et al., 2013, 2015). We found that Scn5a, Kcnh2 and Kcnd2 had circadian expression profiles in iCSΔBmal1+/+ mouse hearts. The circadian expression profiles for Scn5a and Kcnh2 (but not Kcnd2) were lost after inducing the deletion of Bmal1 (iCSΔBmal1–/–), and, compared to mRNA transcript levels in iCSΔBmal1+/+ mouse hearts, the mRNA transcript levels for Scn5a and Kcnh2 (but not Kcnd2) were reduced in iCSΔBmal1–/– mice.
In this study, Hcn4, Kcnj2, Kcnq1, Kchip2, Kcna5, Kcnb1, Gja1, Gja5, Gjc1, Scn4b, Klf15, Tbx5, Gata4, and Foxo1 transcripts were assessed for both the circadian expression and overall expression level by quantitative PCR. These data demonstrate that Hcn4, Kcnj2, Kcnq1, Scn4b, Tbx5, Gata4, Gja5, and Gjc1 mRNA transcript levels have circadian expression profiles in the hearts of iCSΔBmal1+/+ mice but not in iCSΔBmal1–/– mice (Figures 1A,B). Compared to mRNA transcript levels in iCSΔBmal1+/+ mouse hearts, the mRNA transcript levels in the hearts of iCSΔBmal1–/– mice for Hcn4, Kcnj2, Kcnq1, Scn4b, Gata4, and Tbx5 (but not Gja5 or Gjc1) were reduced.
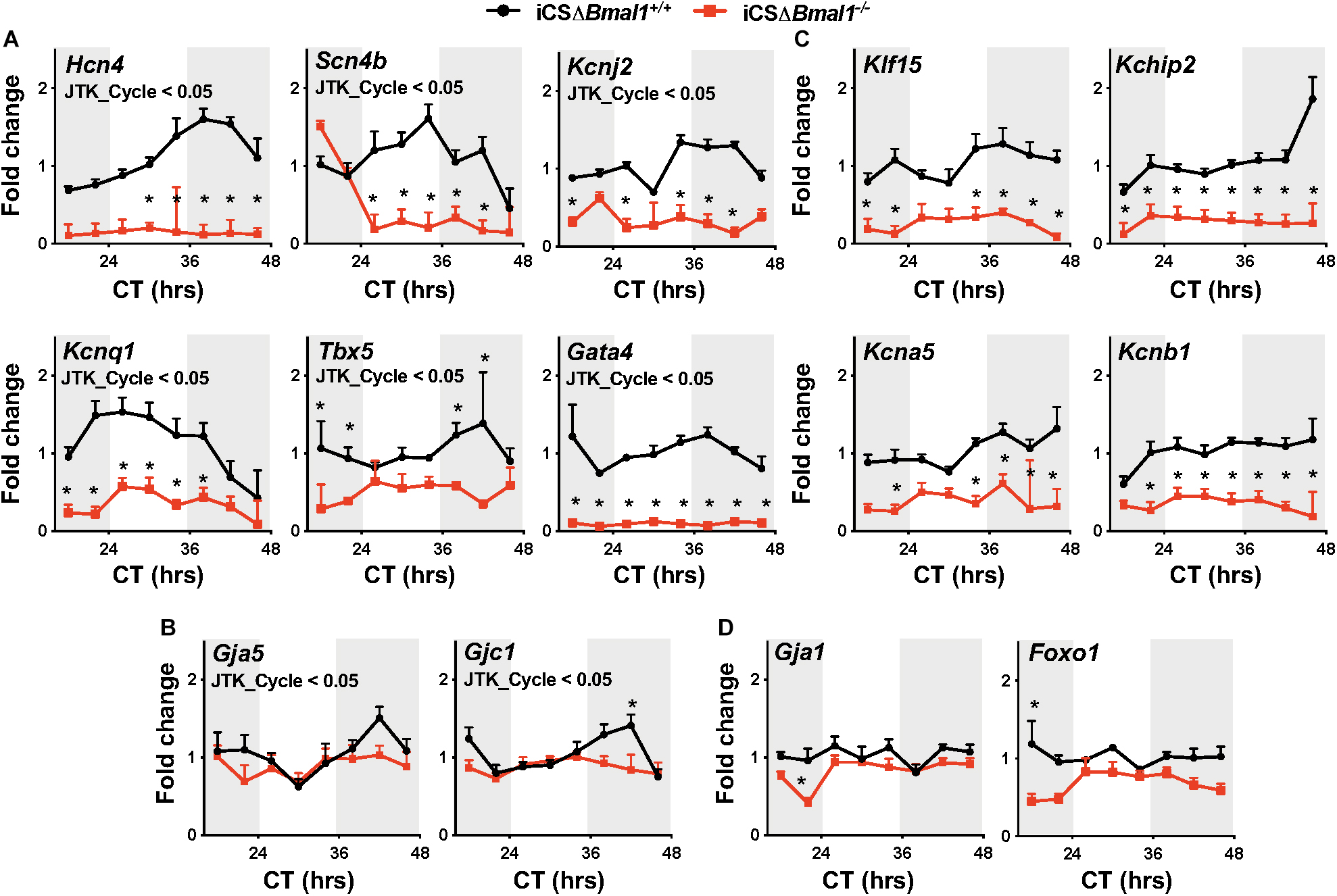
Figure 1. Inducing the deletion of Bmal1 in adult cardiomyocytes differentially impacts the temporal expression of cardiac ion channel transcripts. Shown are qPCR profiles for mRNA transcripts measured from iCSΔBmal1+/+ (black circles) and iCSΔBmal1–/– (red squares) mouse hearts plotted as a function of circadian time (CT). Light and shaded regions on x-axis represent subjective light and dark cycles. (A) Hcn4, Scn4b, Kcnj2, Kcnq1, Tbx5, and Gata4 mRNA transcripts had circadian expression profiles in the hearts of iCSΔBmal1+/+ mice (JTK_Cycle < 0.05) but not iCSΔBmal1–/– mice. mRNA expression was lower at most time points in the hearts of iCSΔBmal1–/– mice when compared to iCSΔBmal1+/+ mouse hearts (n = 3–4/time point; *p < 0.05). (B) Gja5 and Gjc1 mRNA transcripts had circadian expression profiles in iCSΔBmal1+/+ mouse hearts (JTK_Cycle < 0.05) but not iCSΔBmal1–/–mouse hearts. The overall transcript levels were similar in iCSΔBmal1+/+ and iCSΔBmal1–/– mice at most circadian time points (n = 3-4/time point; *p < 0.05). (C) Klf15, Kchip2, Kcna5, and Kcnb1 mRNA transcripts did not have circadian expression profiles in the hearts of iCSΔBmal1+/+ or iCSΔBmal1–/– mice. (JTK_Cycle > 0.05). The overall transcript levels were lower in iCSΔBmal1–/– mouse hearts compared to transcript levels in iCSΔBmal1+/+ mouse hearts at most circadian time points (n = 3–4/time point; *p < 0.05). (D) Gja1 and Foxo1 mRNA transcripts did not have circadian expression profiles in the hearts of iCSΔBmal1+/+ or iCSΔBmal1–/– mice (JTK_Cycle > 0.05) and the overall transcript levels were similar at most circadian time points (n = 3–4/time point; *p < 0.05).
Klf15, Kchip2, Kcna5, Kcnb1, Gja1, and Foxo1 mRNA transcripts did not show a circadian expression profile in the hearts of iCSΔBmal1+/+ or iCSΔBmal1–/– mice (Figures 1C,D). However, compared to mRNA transcript levels in the hearts of iCSΔBmal1+/+ mice, the mRNA transcript levels for Klf15, Kchip2, Kcna5, and Kcnb1 in iCSΔBmal1–/– mice were reduced (Figure 1C). The mRNA transcript levels for Gja1 and Foxo1 in iCSΔBmal1+/+ and iCSΔBmal1–/– mice were not different (Figure 1D). Together, these findings demonstrate that the loss of Bmal1 in adult cardiomyocytes causes a loss of circadian expression and/or a reduction in overall mRNA transcript levels for several cardiac ion channels and transcription factors important for cardiac depolarization, repolarization and conduction.
Inducing the Deletion of Bmal1 Increases the QT-Interval at Slow Heart Rates
The changes in cardiac ion channel expression that occurred after inducing the deletion of Bmal1 in adult cardiomyocytes motivated us to determine how it would impact the cardiac electrophysiological phenotype of mice with the ΔKPQ-Scn5a mutation. We used ECG telemetry to measure QT- and RR-intervals in conscious free moving mice housed in 12-h light and 12-h dark cycles prior to and after inducing Bmal1 deletion in cardiomyocytes (Figure 2A). Previous studies show that the QT-intervals measured from wildtype C57BL/6 mice do not depend on the preceding RR-intervals (Drici et al., 1998; Speerschneider and Thomsen, 2013; Roussel et al., 2016). We confirmed these findings in the iCSΔBmal1+/+ mice by measuring the QT-intervals at RR-intervals ranging from 90 to 140 ms in 10 ms bins (Figure 2B). Deletion of Bmal1 in cardiomyocytes caused the QT-intervals to become longer at slower RR-intervals. Compared to iCSΔBmal1+/+ mice, the iCSΔBmal1–/– mice had longer QT-intervals at RR-intervals that were ≥ 130 ms.
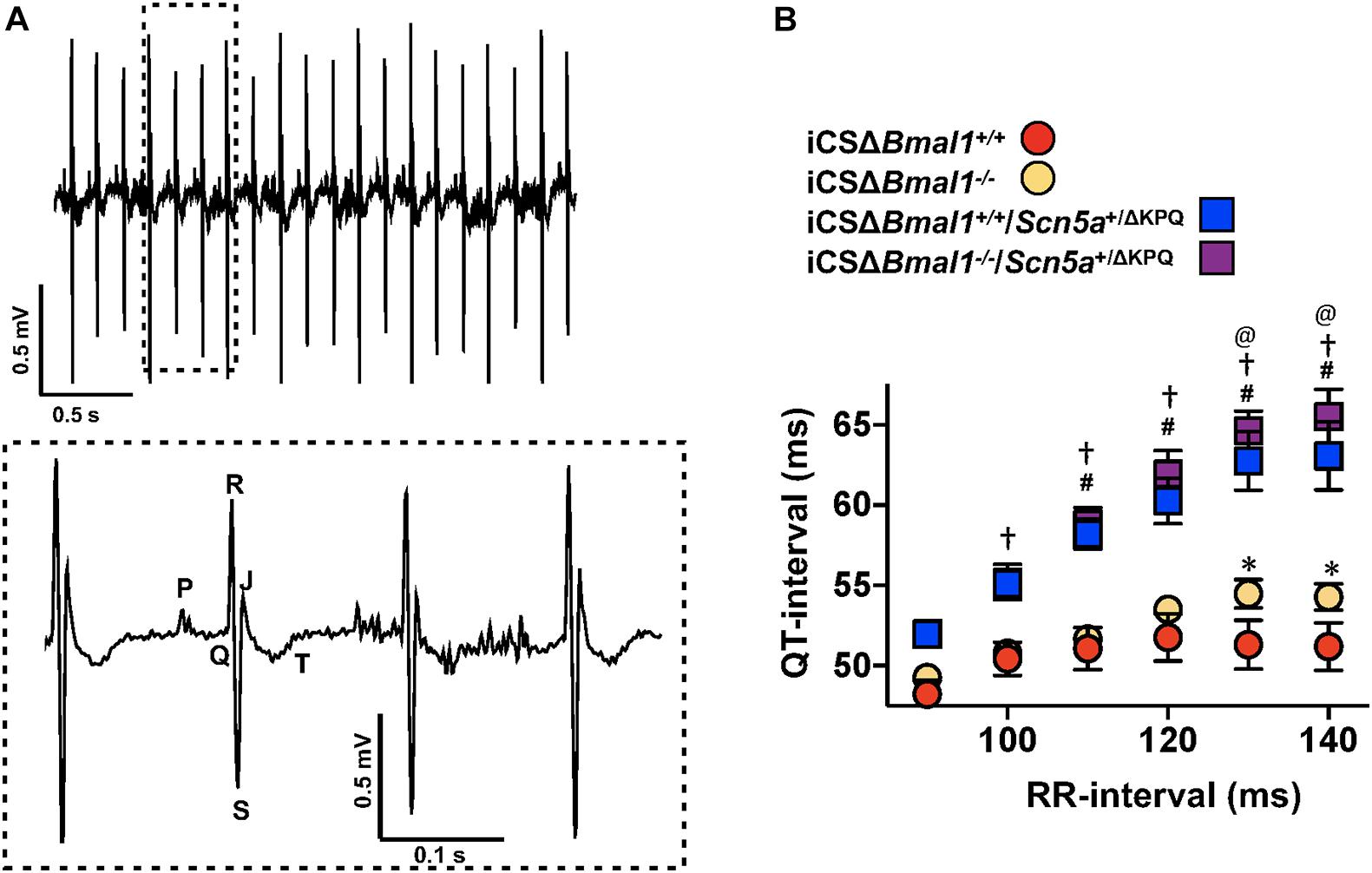
Figure 2. Deletion of Bmal1 in adult cardiomyocytes prolongs the QT-interval at longer RR-intervals. (A) Representative electrocardiogram trace measured from the mice used in this study. The inset shows the dashed box region on the ECG trace in more detail to highlight the P, Q, R, S, J, and T waves. (B) The mean QT-interval plus standard error of the mean measured between 90 and 140 ms in iCSΔBmal1+/+ (red circles), iCSΔBmal1+/+/Scn5a+/ΔKPQ (blue squares), iCSΔBmal1–/– (orange circles) or iCSΔBmal1–/–/Scn5a+/ΔKPQ (purple squares) mice is shown (n = 6 and n = 8, respectively). We compared QT intervals at each RR-interval between iCSΔBmal1+/+ and iCSΔBmal1+/+/Scn5a+/ΔKPQ (#p < 0.05); iCSΔBmal1–/– or iCSΔBmal1–/–/Scn5a+/ΔKPQ (†p < 0.05); iCSΔBmal1+/+ and iCSΔBmal1–/– (*p < 0.05); and iCSΔBmal1+/+/Scn5a+/ΔKPQ and iCSΔBmal1–/–/Scn5a+/ΔKPQ (@p < 0.05).
QT-intervals in mice with the ΔKPQ-Scn5a mutation were dependent on the duration of preceding RR-intervals (Figure 2B). Compared to the QT-intervals measured in the iCSΔBmal1+/+ mice, the QT-intervals in iCSΔBmal1+/+/Scn5a+/ΔKPQ mice were longer at RR-intervals that were ≥ 110 ms. Compared to the iCSΔBmal1–/– mice, the iCSΔBmal1–/–/Scn5a+/ΔKPQ mice had longer QT-intervals at RR-intervals that were ≥ 100 ms. Inducing the deletion of Bmal1 in mice with the ΔKPQ-Scn5a mutation also increased the QT-interval at RR-intervals that were ≥ 130 ms. These data demonstrate that Bmal1 deletion in adult cardiomyocytes prolongs the QT-interval at slow heart rates in mice with or without the ΔKPQ-Scn5a mutation.
Inducing the Deletion of Bmal1 Increases the Day/Night Rhythm-Adjusted Mean in the RR-Interval in Mice With or Without the ΔKPQ-Scn5a Mutation
We quantified the day/night rhythm in the RR-interval recorded from mice before and after inducing the deletion of Bmal1 in adult cardiomyocytes with or without the ΔKPQ-Scn5a mutation. Mice were housed in 12 h light and 12 h dark cycles. The RR-intervals were averaged every 15-min and plotted as a function of ZT for 3 days (Figure 3A). The individual data from each mouse were fit with a cosine function to calculate the day/night period, acrophase (the ZT at which the rhythms peaked), amplitude, and rhythm adjusted mean (Figure 3B). The day/night rhythm in the RR-intervals had periods that were ∼24 h in mice before and after deletion of Bmal1 in cardiomyocytes with or without the ΔKPQ-Scn5a mutation. The day/night rhythm in the RR-intervals peaked after the beginning of the light cycle and did not change after deletion of Bmal1 in the cardiomyocytes of mice with or without the ΔKPQ-Scn5a mutation. The amplitudes in the day/night rhythm of the RR-intervals were not different before or after deletion of Bmal1 in cardiomyocytes in both groups of mice, however, the iCSΔBmal1–/– mice had larger amplitudes compared to iCSΔBmal1–/–/Scn5a+/ΔKPQ mice. The biggest differences among the groups of mice were in day/night rhythm-adjusted means of the RR-intervals. Compared to iCSΔBmal1+/+ mice, the rhythm-adjusted means for the RR-intervals were longer in iCSΔBmal1+/+/Scn5a+/ΔKPQ mice. Inducing the deletion of Bmal1 in cardiomyocytes increased the rhythm-adjusted means for both groups of mice by similar amounts (Figure 3B). These data demonstrate that deletion of Bmal1 in cardiomyocytes does not impact the period, phase or amplitude in the day/night rhythm in RR-intervals in mice with or without the ΔKPQ-Scn5a mutation. Deletion of Bmal1 in adult cardiomyocytes prolongs the day/night rhythm-adjusted mean in the RR-intervals of mice with or without the ΔKPQ-Scn5a mutation.
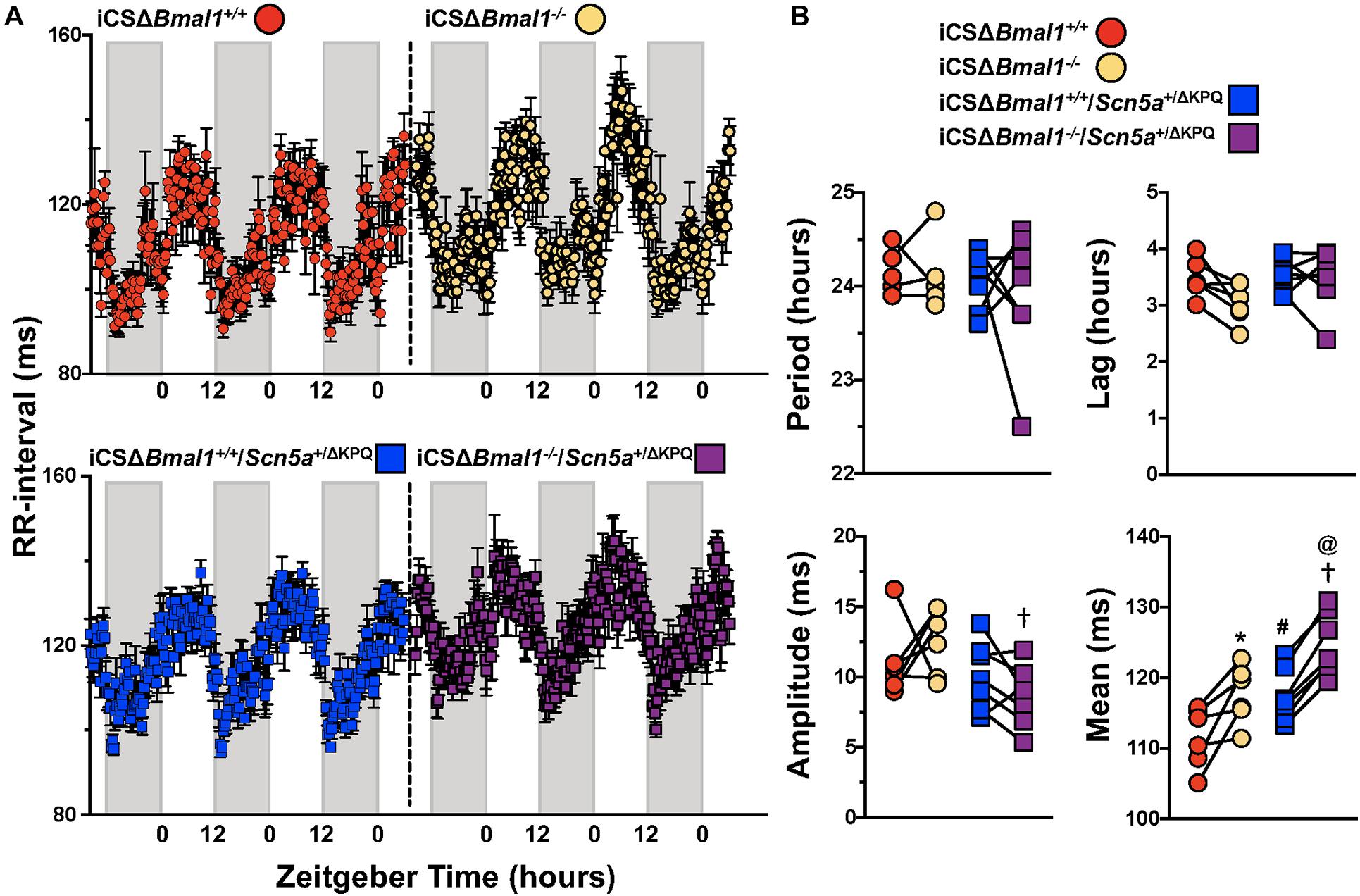
Figure 3. Deletion of Bmal1 in adult cardiomyocytes increases the rhythm adjusted mean of the RR-interval. (A) RR intervals were averaged in 15-min bins over 3 days for iCSΔBmal1+/+ (red circles) and iCSΔBmal1–/– (orange circles) (top graph, n = 6) or iCSΔBmal1+/+/Scn5a+/ΔKPQ (blue circles) and iCSΔBmal1–/–/Scn5a+/ΔKPQ (purple circles) mice (bottom graph, n = 7). Gray shaded regions on x-axis represent lights off. (B) The data for each mouse were fit to cosine curves to analyze period, lag, amplitude and median. The graphical representation of the change in period, amplitude and slope for iCSΔBmal1+/+ (red circles) and iCSΔBmal1–/– (orange circles) and iCSΔBmal1+/+/Scn5a+/ΔKPQ (blue squares) and iCSΔBmal1–/–/Scn5a+/ΔKPQ (purple squares) mice are shown. Analysis showed that the rhythm adjusted mean between iCSΔBmal1+/+ and iCSΔBmal1+/+/Scn5a+/ΔKPQ (#p < 0.05) were different; the amplitude and rhythm adjusted mean for iCSΔBmal1–/– or iCSΔBmal1–/–/Scn5a+/ΔKPQ were different (†p < 0.05); the rhythm adjusted mean for iCSΔBmal1+/+ and iCSΔBmal1–/– were different (*p < 0.05); and the rhythm adjusted mean for iCSΔBmal1+/+/Scn5a+/ΔKPQ and iCSΔBmal1–/–/Scn5a+/ΔKPQ were different (@p < 0.05).
Mice With the ΔKPQ-Scn5a Mutation Have Reduced HRV That Is Normalized After Inducing the Deletion of Bmal1
Heart rate variability measures changes in sinoatrial node function secondary to autonomic drive, autonomic sensitivity of the sinoatrial nodal cells, and/or changes in the spontaneous depolarization of SAN cells (Fenske et al., 2016). We tested the hypothesis that the HRV in the iCSΔBmal1+/+/Scn5a+/ΔKPQ mice was different than the iCSΔBmal1+/+ mice. We measured the 3-day average in the SDNN, RMSSD, and pNN6 at ZT = 2-5 h, ZT = 8–11 h, ZT = 14–17 h, or ZT = 20–23 h. We found that when RR-intervals were longest (ZT = 2–5 h), the SDNN, RMSSD and pNN6 were smaller in the iCSΔBmal1+/+/Scn5a+/ΔKPQ mice compared to iCSΔBmal1+/+ mice (Figure 4). Compared to the iCSΔBmal1+/+ mice, the HRV did not change in the iCSΔBmal1–/– mice at any of the timepoints tested. Compared to iCSΔBmal1+/+/Scn5a+/ΔKPQ mice, the iCSΔBmal1–/–/Scn5a+/ΔKPQ mice had SDNN values that increased at all the timepoints tested; RMSSD values that increased at ZT = 2–5 h and ZT = 20–23 h; and pNN6 values that increased at ZT = 2–5 h, ZT = 8–11 h, and ZT = 20–23 h. The data demonstrate that inducing the deletion of Bmal1 in the cardiomyocytes of mice with the ΔKPQ-Scn5a mutation normalize the HRV to levels similar to those seen in iCSΔBmal1+/+ or iCSΔBmal1–/– mice.
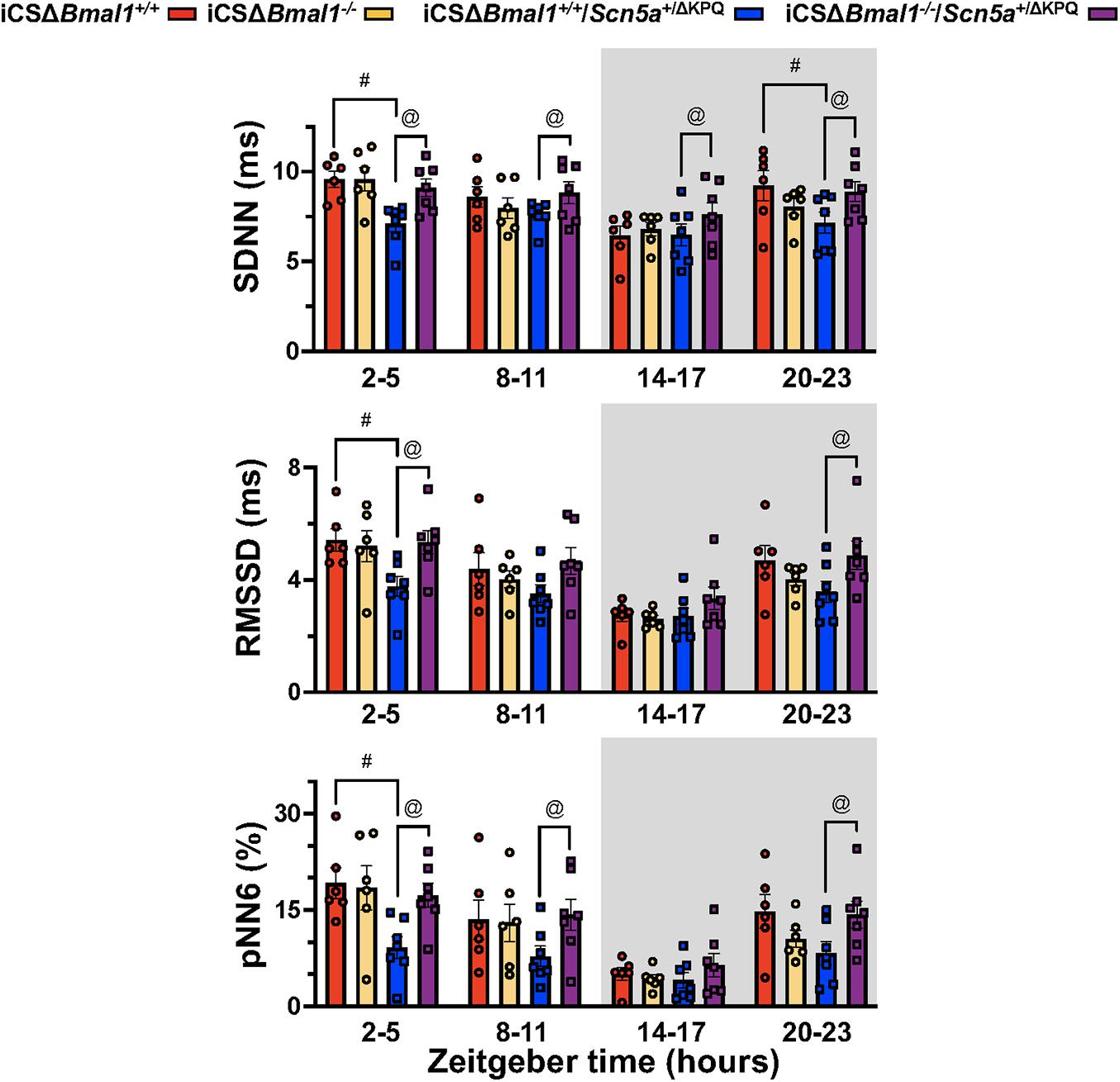
Figure 4. Mice with the ΔKPQ-Scn5a mutation have reduced HRV and this is increased by inducing the deletion of Bmal1 in adult cardiomyocytes. The graphical representation of the change SDNN (top graph), RMSD (middle graph) or pNN6 (bottom graph) for iCSΔBmal1+/+ (red bars, circles), iCSΔBmal1–/– (orange bars, circles), or iCSΔBmal1+/+/Scn5a+/ΔKPQ (blue bars, circles), and iCSΔBmal1–/–/Scn5a+/ΔKPQ (purple bars, circles) mice measured at ZT = 2–5, 8–11, 14–17, and 20–23 h are shown (n = 6 and n = 7, respectively). Analysis detected differences in the SDNN, RMSSD and PNN6 between iCSΔBmal1+/+ and iCSΔBmal1+/+/Scn5a+/ΔKPQ mice (#p < 0.05) at certain timepoints, as well as differences in the SDNN, RMSSD and PNN6 between iCSΔBmal1+/+/Scn5a+/ΔKPQ and iCSΔBmal1–/–/Scn5a+/ΔKPQ mice (@p < 0.05).
Discussion
This study showed that the deletion of Bmal1 in adult cardiomyocytes disrupts the expression profiles for a number of cardiac ion channel and transcription factor transcripts important for normal cardiac repolarization, depolarization, and conduction. We tested the hypothesis that the deletion of Bmal1 in adult cardiomyocytes would modify the cardiac electrophysiological phenotype in mice that harbor the ΔKPQ-Scn5a mutation in vivo. We found that the mice with the ΔKPQ-Scn5a mutation had abnormally long QT-intervals, steeper QT- and RR-interval relations, slower day/night rhythm-adjusted means in RR-intervals and decreased HRV. Inducing the deletion of Bmal1 in the hearts of mice with the ΔKPQ-Scn5a mutation prolonged the QT-intervals measured at slower RR-intervals and increased the day/night rhythm-adjusted mean in RR-interval. The absolute magnitude of these changes was similar in mice without the ΔKPQ-Scn5a mutation, suggesting that the changes caused by deletion of Bmal1 in cardiomyocytes were additive. The deletion of Bmal1 in cardiomyocytes did not alter HRV in mice without the ΔKPQ-Scn5a mutation, but it increased the abnormally low HRV in mice with the ΔKPQ-Scn5a mutation. We conclude that deletion of Bmal1 in cardiomyocytes disrupts the expression for a number of cardiac ion channel genes, prolongs the QT- and RR-intervals in mice with or without the ΔKPQ-Scn5a mutation, and normalizes HRV in mice with the ΔKPQ-Scn5a mutation.
Bmal1 Is Important for Normal Ventricular Repolarization at Slow Heart Rates
Inducing the deletion of Bmal1 prolongs the QT-intervals at slower heart rates in mice with and without the ΔKPQ-Scn5a mutation (Figure 2). This might not have been expected in mice with the ΔKPQ-Scn5a mutation since previous studies show that inducing the deletion of Bmal1 in adult cardiomyocytes decreases the functional expression of Scn5a by ∼30% (Schroder et al., 2013). We now show that Bmal1 directly or indirectly contributes to the expression of many different cardiac transcripts that regulate normal depolarization, repolarization and conduction in mice (Figure 1; Jeyaraj et al., 2012; Schroder et al., 2015; Delisle et al., 2020). Thus, the cardiac electrophysiological properties likely do not reflect the changes in one gene but rather many different genes.
The Day/Night Rhythm in Heart Rate in vivo Does Not Depend on the Cardiomyocyte Circadian Clock
Inducing the deletion of Bmal1 did not change the phase, amplitude or period in the day/night rhythm of heart rate in mice with or without ΔKPQ-Scn5a mutation (Figure 3; Schroder et al., 2013). These data provide additional support that the day/night rhythm in heart rate in vivo does not depend on the functional circadian clock mechanism in cardiomyocytes but rather other factors including the autonomic nervous system, nocturnal behavioral patterns, core body temperature, circadian rhythms in neurohumoral signaling, etc (Sheward et al., 2010; Tong et al., 2013; Schroder et al., 2014). This study confirms previous findings that the deletion of Bmal1 in adult cardiomyocytes slows the day/night rhythm-adjusted mean in heart rate (Schroder et al., 2013). The slowing of the mean heart rate after inducing the deletion of Bmal1 was observed in mice with or without the ΔKPQ-Scn5a mutation.
The Reduction in HRV in Mice With ΔKPQ-Scn5a Is Not Secondary to Slower Heart Rates
Another novel finding in this study is that, compared to mice without the ΔKPQ-Scn5a mutation, mice with the ΔKPQ-Scn5a mutation had a lower HRV when heart rate was slowest. The lower HRV likely reflects sinoatrial node dysfunction, changes in the autonomic sensitivity of the sinoatrial node and/or changes in autonomic tone (Fabritz et al., 2010; Wu et al., 2012). Although studies have not shown changes in the autonomic sensitivity of the sinoatrial node in Scn5a+/ΔKPQ mice, studies do show that, compared to WT mice, the working myocardium of Scn5a+/ΔKPQ mice expresses fewer β-adrenergic receptors (Fabritz et al., 2010). Another possibility is that the reduction in HRV measured in the iCSΔBmal1+/+/Scn5a+/ΔKPQ mice reflects differences in basal heart rate compared to the iCSΔBmal1+/+ mice. However, this seems unlikely because HRV increases as the heart rates become slower (Monfredi et al., 2014). The increase in HRV normally seen at slower heart rates might explain why HRV increased in the iCSΔBmal1–/–/Scn5a+/ΔKPQ mice (Figures 3, 4). Another possible explanation for the effect on HRV is the recent observation that the loss of Bmal1 in the heart alters the intrinsic beating rate of sinoatrial node preparations (D’Souza et al., 2020). The time of day changes in sinoatrial node intrinsic beating rate were suggested to be secondary to a reduction in the functional expression of the Hcn4. Consistent with these studies, we found that cardiac Hcn4 mRNA transcripts in the hearts of iCSΔBmal1+/+ mice had a circadian expression profile that was lost and reduced iCSΔBmal1–/– mice (Figure 1). Whether or not the decreased expression of Hcn4 mRNA transcript levels and/or other cardiac ion channel genes is responsible for the slower rhythm-adjusted mean in heart rate or increased HRV in mice with the ΔKPQ-Scn5a mutation warrants further investigation.
Study Limitations
There are several limitations to this study. Mouse models are widely employed by cardiac electrophysiologists because they are practical for determining how genetic, pharmacological and/or environmental manipulations impact arrhythmogenic triggers and/or pro-arrhythmic changes in the cardiac substrate (Dobrev and Wehrens, 2018; Clauss et al., 2019). However, there are clear species-specific differences in cardiac electrophysiology that limit extrapolation to humans (Sabir et al., 2008; Nerbonne, 2014). Mouse hearts are small and beat about 10 times faster than humans. Although human and mouse ECG waveforms have P, QRS, and T-waves, mouse hearts repolarize quickly to generate a predominant J wave and a small inverted T-wave (Mitchell et al., 1998; Kaese and Verheule, 2012; Boukens et al., 2014). As such, the underlying mechanisms for cardiac excitability and arrhythmias in mouse and human hearts are different (Sabir et al., 2008).
We did not observe any obvious spontaneous ventricular symptoms (e.g., ventricular tachyarrhythmias) in iCSΔBmal1+/+/Scn5a+/ΔKPQ or iCSΔBmal1+/+/Scn5a+/ΔKPQ mice during the ECG recordings (∼6-9 days). Therefore, we cannot conclude that inducing the deletion of Bmal1 in cardiomyocytes increases arrhythmogenicity in mice with the ΔKPQ-Scn5a mutation. However, similar to the Nuyens et al., 2001 study (Nuyens et al., 2001), we did see that mice harboring the ΔKPQ-Scn5a mutation had a higher number of sinus pauses, atrioventricular block and bradyarrhythmias. The absolute number of hourly pauses/blocks during the inactive phase trended higher in the iCSΔBmal1–/–/Scn5a+/ΔKPQ mice compared to the iCSΔBmal1+/+/Scn5a+/ΔKPQ mice but was not significant (iCSΔBmal1+/+/Scn5a+/ΔKPQ = 6.7 ± 2.5 pauses/blocks per hour vs iCSΔBmal1–/–/Scn5a+/ΔKPQ = 20.3 ± 7.8 pauses/blocks per hour, n = 6 mice, p = 0.06). More invasive studies are needed to determine whether the loss of the Bmal1 in mice with the ΔKPQ-Scn5a mutation have higher susceptibility to arrhythmias and/or ventricular tachyarrhythmias.
Conclusion
In summary, this study shows inducing the deletion of Bmal1 in mouse hearts exacerbates the prolongation in the QT- and RR-intervals in mice with the ΔKPQ-Scn5a mutation. Specifically, it increases the abnormal prolongation in the QT-intervals at slower heart rates and causes an overall slowing of the heart rate over 24-h. These effects combine to increase the absolute number of abnormal QT-intervals during the 24-h cycle. However, due to the low absolute probability of ventricular symptoms, we cannot conclude it increases the likelihood of ventricular tachyarrhythmias. Future studies that explore the molecular mechanisms with which Bmal1 functions to limit QT-interval prolongation at slow heart rates might lead to the identification of novel therapeutic targets.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by Association for Assessment and Accreditation of Laboratory Animal Care and was approved by the Institutional Animal Care and Use Committee at University of Kentucky.
Author Contributions
ES, KE, CE, and BD contributed and developed the research design. ES, JW, KS, SS, DB, and BD worked on data acquisition and analyses. ES and TS generated the animals and performed the experiments. ES, JW, KS, SS, DB, TS, CE, KE, and BD wrote, edited, and prepared the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Heart Lung and Blood Institute grants R01HL153042, R01HL141343, and the American Heart Association grant 16GRNT30800003.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge Dr. Peter Carmeliet (VIB, Flanders, Belgium) for providing the Scn5a+/ΔKPQ mice.
References
Abriel, H. (2007). Roles and regulation of the cardiac sodium channel Na v 1.5: recent insights from experimental studies. Cardiovasc. Res. 76, 381–389. doi: 10.1016/j.cardiores.2007.07.019
Andersson, K. B., Birkeland, J. A., Finsen, A. V., Louch, W. E., Sjaastad, I., Wang, Y., et al. (2009). Moderate heart dysfunction in mice with inducible cardiomyocyte-specific excision of the Serca2 gene. J. Mol. Cell. Cardiol. 47, 180–187. doi: 10.1016/j.yjmcc.2009.03.013
Arnolds, D. E., Liu, F., Fahrenbach, J. P., Kim, G. H., Schillinger, K. J., Smemo, S., et al. (2012). TBX5 drives Scn5a expression to regulate cardiac conduction system function. J. Clin. Invest. 122, 2509–2518. doi: 10.1172/jci62617
Boukens, B. J., Rivaud, M. R., Rentschler, S., and Coronel, R. (2014). Misinterpretation of the mouse ECG: ‘musing the waves of Mus musculus’. J. Physiol. 592, 4613–4626. doi: 10.1113/jphysiol.2014.279380
Cai, B., Wang, N., Mao, W., You, T., Lu, Y., Li, X., et al. (2014). Deletion of FoxO1 leads to shortening of QRS by increasing Na(+) channel activity through enhanced expression of both cardiac NaV1.5 and beta3 subunit. J. Mol. Cell. Cardiol. 74, 297–306. doi: 10.1016/j.yjmcc.2014.06.006
Clauss, S., Bleyer, C., Schüttler, D., Tomsits, P., Renner, S., Klymiuk, N., et al. (2019). Animal models of arrhythmia: classic electrophysiology to genetically modified large animals. Nat. Rev. Cardiol. 16, 457–475. doi: 10.1038/s41569-019-0179-0
Delisle, B. P., Stumpf, J. L., Wayland, J. L., Johnson, S. R., Ono, M., Hall, D., et al. (2020). Circadian clocks regulate cardiac arrhythmia susceptibility, repolarization, and ion channels. Curr. Opin. Pharmacol. 57, 13–20. doi: 10.1016/j.coph.2020.09.015
Dobrev, D., and Wehrens, X. H. T. (2018). Mouse Models of Cardiac Arrhythmias. Circ. Res. 123, 332–334. doi: 10.1161/circresaha.118.313406
Drici, M. D., Arrighi, I., Chouabe, C., Mann, J. R., Lazdunski, M., Romey, G., et al. (1998). Involvement of IsK-associated K+ channel in heart rate control of repolarization in a murine engineered model of Jervell and Lange-Nielsen syndrome. Circ. Res. 83, 95–102. doi: 10.1161/01.res.83.1.95
D’Souza, A., Wang, Y., Anderson, C., Bucchi, A., Baruscotti, M., Olieslagers, S., et al. (2020). A circadian clock in the sinus node mediates day-night rhythms in Hcn4 and heart rate. Heart Rhythm 18, 801–810. doi: 10.1016/j.hrthm.2020.11.026
Eckardt, D., Theis, M., Degen, J., Ott, T., van Rijen, H. V., Kirchhoff, S., et al. (2004). Functional role of connexin43 gap junction channels in adult mouse heart assessed by inducible gene deletion. J. Mol. Cell Cardiol. 36, 101–110. doi: 10.1016/j.yjmcc.2003.10.006
Fabritz, L., Damke, D., Emmerich, M., Kaufmann, S. G., Theis, K., Blana, A., et al. (2010). Autonomic modulation and antiarrhythmic therapy in a model of long QT syndrome type 3. Cardiovasc. Res. 87, 60–72. doi: 10.1093/cvr/cvq029
Fenske, S., Probstle, R., Auer, F., Hassan, S., Marks, V., Pauza, D. H., et al. (2016). Comprehensive multilevel in vivo and in vitro analysis of heart rate fluctuations in mice by ECG telemetry and electrophysiology. Nat. Protoc. 11, 61–86. doi: 10.1038/nprot.2015.139
Hall, M. E., Smith, G., Hall, J. E., and Stec, D. E. (2011). Systolic dysfunction in cardiac-specific ligand-inducible MerCreMer transgenic mice. Am. J. Physiol. Heart Circ. Physiol. 301, H253–H260.
Hastings, M. H., Maywood, E. S., and Brancaccio, M. (2018). Generation of circadian rhythms in the suprachiasmatic nucleus. Nat. Rev. Neurosci. 19, 453–469.
Hughes, M. E., Hogenesch, J. B., and Kornacker, K. (2010). JTK_CYCLE: An Efficient Nonparametric Algorithm for Detecting Rhythmic Components in Genome-Scale Data Sets. J. Biol. Rhythms 25, 372–380. doi: 10.1177/0748730410379711
Jalife, J., Morley, G. E., and Vaidya, D. (1999). Connexins and impulse propagation in the mouse heart. J. Cardiovasc. Electrophysiol. 10, 1649–1663. doi: 10.1111/j.1540-8167.1999.tb00230.x
Jeyaraj, D., Haldar, S. M., Wan, X., McCauley, M. D., Ripperger, J. A., Hu, K., et al. (2012). Circadian rhythms govern cardiac repolarization and arrhythmogenesis. Nature 483, 96–99. doi: 10.1038/nature10852
Kaese, S., and Verheule, S. (2012). Cardiac electrophysiology in mice: a matter of size. Front. Physiol. 3:345. doi: 10.3389/fphys.2012.00345
Knollmann, B. C., Sirenko, S., Rong, Q., Katchman, A. N., Casimiro, M., Pfeifer, K., et al. (2007). Kcnq1 contributes to an adrenergic-sensitive steady-state K+ current in mouse heart. Biochem. Biophys. Res. Commun. 360, 212–218. doi: 10.1016/j.bbrc.2007.06.038
Kozasa, Y., Nakashima, N., Ito, M., Ishikawa, T., Kimoto, H., Ushijima, K., et al. (2018). HCN4 pacemaker channels attenuate the parasympathetic response and stabilize the spontaneous firing of the sinoatrial node. J. Physiol. 596, 809–825. doi: 10.1113/jp275303
Lemoine, M. D., Duverger, J. E., Naud, P., Chartier, D., Qi, X. Y., Comtois, P., et al. (2011). Arrhythmogenic left atrial cellular electrophysiology in a murine genetic long QT syndrome model. Cardiovasc. Res. 92, 67–74. doi: 10.1093/cvr/cvr166
London, B. (2001). Cardiac arrhythmias: from (transgenic) mice to men. J. Cardiovasc. Electrophysiol. 12, 1089–1091. doi: 10.1046/j.1540-8167.2001.01089.x
Mao, W., You, T., Ye, B., Li, X., Dong, H. H., Hill, J. A., et al. (2012). Reactive oxygen species suppress cardiac NaV1.5 expression through Foxo1. PLoS One. 7:e32738. doi: 10.1371/journal.pone.0032738
Michel, S., and Meijer, J. H. (2020). From clock to functional pacemaker. Eur. J. Neurosci. 51, 482–493. doi: 10.1111/ejn.14388
Mitchell, G. F., Jeron, A., and Koren, G. (1998). Measurement of heart rate and Q-T interval in the conscious mouse. Am. J. Physiol. 274, H747–H751.
Moghtadaei, M., Langille, E., Rafferty, S. A., Bogachev, O., and Rose, R. A. (2017). Altered heart rate regulation by the autonomic nervous system in mice lacking natriuretic peptide receptor C (NPR-C). Sci. Rep. 7:17564.
Monfredi, O., Lyashkov, A. E., Johnsen, A. B., Inada, S., Schneider, H., Wang, R., et al. (2014). Biophysical characterization of the underappreciated and important relationship between heart rate variability and heart rate. Hypertension 64, 1334–1343. doi: 10.1161/hypertensionaha.114.03782
Nerbonne, J. M. (2014). Mouse models of arrhythmogenic cardiovascular disease: challenges and opportunities. Curr. Opin. Pharmacol. 15, 107–114. doi: 10.1016/j.coph.2014.02.003
Nerbonne, J. M., and Kass, R. S. (2005). Molecular physiology of cardiac repolarization. Physiol. Rev. 85, 1205–1253. doi: 10.1152/physrev.00002.2005
Nuyens, D., Stengl, M., Dugarmaa, S., Rossenbacker, T., Compernolle, V., Rudy, Y., et al. (2001). Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat. Med. 7, 1021–1027. doi: 10.1038/nm0901-1021
Pizarro, A., Hayer, K., Lahens, N. F., and Hogenesch, J. B. (2013). CircaDB: a database of mammalian circadian gene expression profiles. Nucleic. Acids. Res. 41, D1009–D1013.
Pond, A. L., Scheve, B. K., Benedict, A. T., Petrecca, K., Van Wagoner, D. R., Shrier, A., et al. (2000). Expression of distinct ERG proteins in rat, mouse, and human heart. Relation to functional I(Kr) channels. J. Biol. Chem. 275, 5997–6006. doi: 10.1074/jbc.275.8.5997
Remme, C. A. (2013). Cardiac sodium channelopathy associated with SCN5A mutations: electrophysiological, molecular and genetic aspects. J. Physiol. 591, 4099–4116. doi: 10.1113/jphysiol.2013.256461
Remme, C. A., Scicluna, B. P., Verkerk, A. O., Amin, A. S., van Brunschot, S., Beekman, L., et al. (2009). Genetically determined differences in sodium current characteristics modulate conduction disease severity in mice with cardiac sodium channelopathy. Circ. Res. 104, 1283–1292. doi: 10.1161/circresaha.109.194423
Reppert, S. M., and Weaver, D. R. (2002). Coordination of circadian timing in mammals. Nature 418, 935–941. doi: 10.1038/nature00965
Roussel, J., Champeroux, P., Roy, J., Richard, S., Fauconnier, J., Le Guennec, J. Y., et al. (2016). The Complex QT/RR Relationship in Mice. Sci. Rep. 6:25388.
Sabir, I. N., Killeen, M. J., Grace, A. A., and Huang, C. L. (2008). Ventricular arrhythmogenesis: insights from murine models. Prog. Biophys. Mol. Biol. 98, 208–218. doi: 10.1016/j.pbiomolbio.2008.10.011
Schroder, E. A., Burgess, D. E., Manning, C. L., Zhao, Y., Moss, A. J., Patwardhan, A., et al. (2014). Light phase-restricted feeding slows basal heart rate to exaggerate the type-3 long QT syndrome phenotype in mice. Am. J. Physiol. Heart Circ. Physiol. 307, H1777–H1785.
Schroder, E. A., Burgess, D. E., Zhang, X., Lefta, M., Smith, J. L., Patwardhan, A., et al. (2015). The cardiomyocyte molecular clock regulates the circadian expression of Kcnh2 and contributes to ventricular repolarization. Heart Rhythm 12, 1306–1314. doi: 10.1016/j.hrthm.2015.02.019
Schroder, E. A., Lefta, M., Zhang, X., Bartos, D. C., Feng, H. Z., Zhao, Y., et al. (2013). The cardiomyocyte molecular clock, regulation of Scn5a, and arrhythmia susceptibility. Am. J. Physiol. Cell Physiol. 304, C954–C965.
Schwartz, P. J., Moss, A. J., Vincent, G. M., and Crampton, R. S. (1993). Diagnostic criteria for the long QT syndrome. An update. Circulation 88, 782–784. doi: 10.1161/01.cir.88.2.782
Sheward, W. J., Naylor, E., Knowles-Barley, S., Armstrong, J. D., Brooker, G. A., Seckl, J. R., et al. (2010). Circadian control of mouse heart rate and blood pressure by the suprachiasmatic nuclei: behavioral effects are more significant than direct outputs. PLoS One. 5:e9783. doi: 10.1371/journal.pone.0009783
Speerschneider, T., and Thomsen, M. B. (2013). Physiology and analysis of the electrocardiographic T wave in mice. Acta. Physiol. 209, 262–271. doi: 10.1111/apha.12172
Tarradas, A., Pinsach-Abuin, M. L., Mackintosh, C., Llora-Batlle, O., Perez-Serra, A., Batlle, M., et al. (2017). Transcriptional regulation of the sodium channel gene (SCN5A) by GATA4 in human heart. J. Mol. Cell. Cardiol. 102, 74–82. doi: 10.1016/j.yjmcc.2016.10.013
Thireau, J., Zhang, B. L., Poisson, D., and Babuty, D. (2008). Heart rate variability in mice: a theoretical and practical guide. Exp. Physiol. 93, 83–94. doi: 10.1113/expphysiol.2007.040733
Tong, M., Watanabe, E., Yamamoto, N., Nagahata-Ishiguro, M., Maemura, K., Takeda, N., et al. (2013). Circadian expressions of cardiac ion channel genes in mouse might be associated with the central clock in the SCN but not the peripheral clock in the heart. Biol. Rhythm. Res. 44, 519–530. doi: 10.1080/09291016.2012.704801
Wahl-Schott, C., Fenske, S., and Biel, M. (2014). HCN channels: new roles in sinoatrial node function. Curr. Opin. Pharmacol. 15, 83–90. doi: 10.1016/j.coph.2013.12.005
Wu, J., Zhang, Y., Zhang, X., Cheng, L., Lammers, W. J., Grace, A. A., et al. (2012). Altered sinoatrial node function and intra-atrial conduction in murine gain-of-function Scn5a+/DeltaKPQ hearts suggest an overlap syndrome. Am. J. Physiol. Heart Circ. Physiol. 302, H1510–H1523.
Zareba, W., Sattari, M. N., Rosero, S., Couderc, J. P., and Moss, A. J. (2001). Altered atrial, atrioventricular, and ventricular conduction in patients with the long QT syndrome caused by the DeltaKPQ SCN5A sodium channel gene mutation. Am. J. Cardiol. 88, 1311–1314. doi: 10.1016/s0002-9149(01)02097-5
Keywords: heart, electrophysiology, ion channel, SCN5A, long QT syndrome, Bmal1, circadian
Citation: Schroder EA, Wayland JL, Samuels KM, Shah SF, Burgess DE, Seward T, Elayi CS, Esser KA and Delisle BP (2021) Cardiomyocyte Deletion of Bmal1 Exacerbates QT- and RR-Interval Prolongation in Scn5a+/ΔKPQ Mice. Front. Physiol. 12:681011. doi: 10.3389/fphys.2021.681011
Received: 15 March 2021; Accepted: 18 May 2021;
Published: 24 June 2021.
Edited by:
Carol Ann Remme, University of Amsterdam, NetherlandsReviewed by:
Bastiaan J. Boukens, University of Amsterdam, NetherlandsPatrick Schweizer, Heidelberg University, Germany
Copyright © 2021 Schroder, Wayland, Samuels, Shah, Burgess, Seward, Elayi, Esser and Delisle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elizabeth A. Schroder, ZXNjaHIwQHVreS5lZHU=; Brian P. Delisle, YnJpYW4uZGVsaXNsZUB1a3kuZWR1
†These authors have contributed equally to this work