 Yu-Feng Liu1,2†
Yu-Feng Liu1,2† Jia Song
Jia Song Xiao-Lan Zhang
Xiao-Lan Zhang- 1Department of Gastroenterology, The Second Hospital of Hebei Medical University, Shijiazhuang, China
- 2Department of Gastroenterology, Dingzhou People’s Hospital of Hebei Province, Dingzhou, China
Background: The progression of liver disorders is frequently associated with inflammatory bowel disease through the gut-liver axis. However, no direct evidence showed the mechanisms of ulcerative colitis (UC) in the development of liver fibrosis per se. Thus, this study aimed to evaluate the effects of UC on liver fibrosis and its potential mechanism in the experimental model.
Methods: Male C57BL/6 mice were allocated into five groups (n = 10 per group) to receive either drinking water (control), 2% dextran sulfate sodium (DSS), olive oil, carbon tetrachloride (CCl4) or DSS + CCl4 for 4 cycles. Blood was collected for biochemical analysis. Colons were excised for the evaluation of colon length and morphological score. Liver, colon, and mesenteric lymph nodes (MLNs) were collected for histopathological staining, expression analysis, and bacterial translocation assay to evaluate the inflammation, fibrosis, the activation of hepatic stellate cells (HSCs), and gut barrier function.
Results: DSS caused severe colitis in mice treated or treated with CCl4, as evident from the elevation of disease activity index (DAI), histological abnormalities, and increased pro-inflammatory cytokines (TNF-α, IFN-γ, and IL-17A). Histopathological staining revealed that DSS treatment aggravated the CCl4-induced extracellular matrix deposition, liver fibrosis, and inflammation in mice. Additionally, biochemical and expression analysis indicated the DSS treatment caused the increase of hydroxyproline and pro-inflammatory cytokines, as well as the abnormal liver function indexes in CCl4-induced mice. Gut barrier function was impaired in DSS- and DSS + CCl4-treated mice, manifesting as the increase in bacterial translocation and lipopolysaccharide level, and the reduction in tight junction proteins (occluding, claudin-1 and ZO-1) expression. Further, the activations of HSCs and TLR4 signaling pathway were observed after DSS + CCl4 treatment, presenting with the increase in expression of α-SMA, vimentin, TGF-β, collagen type I, collagen type II, TIMP-2, TLR4, TRAF6, and NF-κB p65, and a decrease in GFAP and MMP-2 expression.
Conclusion: The present study verified that UC aggravated CCl4-induced liver injury, inflammation, and fibrosis in mice through the gut-liver axis. Gut barrier dysfunction in UC leads to bacterial translocation and elevated lipopolysaccharide, which may promote the activation of TLR4 signaling and HSCs in the liver.
Introduction
Liver fibrosis is a common disease associated with various liver disorders such as viral hepatitis, and alcoholic liver, which often leads to gradual loss of liver function (Shiha et al., 2017). It has the potential to progress into cirrhosis, even liver cancer, and liver failure if not necessarily prevented (Aydın and Akçalı, 2018). Moreover, the progression of fibrosis, and particularly cirrhosis, is responsible for significant morbidity and mortality (Berumen et al., 2021). However, liver transplantation is still the only available curative treatment for patients with end-stage of liver fibrosis at present (Koyama et al., 2016). In recent years, liver fibrosis is considered a dynamic and potentially bidirectional process that has an inherent capacity for recovery and remodeling (Hintermann and Christen, 2019), which translates new possibilities into the development of anti-fibrotic therapies. Despite numerous therapeutic targets have been identified in liver fibrosis, unfortunately, no clinical trial has so far provided unequivocal evidence for a complete reversal of cirrhosis (Povero et al., 2010; Pellicoro et al., 2014). Accordingly, identifying the critical pathways and mechanisms in liver fibrosis is likely to provide novel insights for clinical translation.
Notably, a considerable number of studies have been indicated that the progression of liver disorders is frequently associated with inflammatory bowel disease through the gut-liver axis (Frasinariu et al., 2013), as the blood supply in the liver mostly comes from the intestine through the portal vein (Seki and Schnabl, 2012). Particularly, liver disorders are typical extra-intestinal manifestations in ulcerative colitis (UC). Several previous studies have demonstrated the dextran sodium sulfate (DSS)-induced colitis promotes liver damage, inflammation, fibrogenesis, and even tumorigenesis in the non-alcoholic steatohepatitis (NASH) model (Gäbele et al., 2011; Trivedi and Jena, 2013; Achiwa et al., 2016). Remarkably, little attention was paid to liver fibrosis per se for many years, even though almost all morbidity of liver diseases can be tied directly to progressive fibrosis (Albanis and Friedman, 2001). Currently, no direct evidence has shown the mechanisms of UC in the development of liver fibrosis per se. Therefore, elucidating the role of DSS-induced colitis in the liver fibrosis model would provide additional evidence for the development of anti-fibrotic therapies.
Gut-liver axis is widely recognized to be implicated in the pathogenesis of liver disorders and is emerging as the focus of clinical research increasingly (Wiest et al., 2017). Early studies identified the increased level of portal lipopolysaccharide (LPS) and intestinal permeability in experimental NASH or alcoholic liver disease (ALD), suggesting the gut barrier dysfunction (Szabo, 2015). More recently, evidence from clinical patients proposed a new viewpoint on the role of the gut-liver axis in the pathogenesis of liver disorders (Haderer et al., 2021). Liver cirrhosis could result in a reduction in the thickness of the colonic mucus layer, which allows bacteria-to-epithelial cell contact, and subsequently bacteria led to a marked reduction of cell-to-cell junctions (Haderer et al., 2021). Furthermore, the destruction of intestinal tight junctions increases intestinal permeability, leading to the transport of intestinal bacterial products to the liver via the portal vein (Matsuda and Seki, 2020). These bacterial products stimulate innate immune receptors, namely Toll-like receptors (TLRs), eventually activating downstream pathways involved in liver inflammation and fibrogenesis (Frasinariu et al., 2013; Hu et al., 2020). However, the evidence to support this pathogenesis mechanism of LPS-activated TLR4 signaling comes largely from experimental NASH, data from carbon tetrachloride (CCl4)-induced liver fibrosis model has not been reported currently. As reported, the development of different liver fibrosis (such as hepatotoxic and cholestatic) depends on the etiology of the underlying liver disease (Kisseleva and Brenner, 2021). Thus, the purpose of this study was to evaluate the effects of DSS-induced colitis on liver inflammation and fibrosis in mice with CCl4-induced liver fibrosis, hoping to provide additional and direct evidence that highlight the mechanisms of gut–liver axis relevant to liver fibrosis.
Materials and Methods
Animals and Experimental Protocol
Male C57BL/6 mice (weight 18–22 g; age 6–8 weeks) were obtained from Laboratory Animal Centre of Hebei Medical University (Shijiazhuang, Hebei, China). All mice were maintained under standard conditions (temperature, 23 ± 2°C; humidity, 70–75%; lighting regimen, 12-h light/dark cycle), and free access to food and water. All experimental protocols were conducted according to the Institutional Animal Care guidelines and approved by the Laboratory Animal Ethics Committee of Hebei Medical University.
Fifty mice were randomly allocated into five experimental groups: control, DSS, olive oil, CCl4, and DSS + CCl4 (n = 10 per group) and received the assigned treatments. Mice in the control group received normal drinking water throughout the study. Mice in the DSS group received 2% w/v DSS (40000–50000 MW, dissolved in drinking water, Sigma, United States) for 4 cycles (on day 1–5, 8–12, 15–19, and 22–26, respectively) and distilled water (remission period between each cycle) to induce the chronic colitis. Mice in the olive oil group received normal drinking water and intraperitoneal injection of sterile olive oil twice a week and served as a control for CCl4 group. Mice in the CCl4 group were given normal drinking water and the intraperitoneal injection of 5% CCl4 dissolved in sterile olive oil (10 μl/g body weight). CCl4 + DSS group received 4-cycle administration of 2% DSS and intraperitoneal injection of CCl4. DSS was applied in cycles and each cycle consisted of 5 days DSS administration followed by a 2-day interval with normal drinking water. CCl4 was injected twice a week. The cycles were repeated throughout the experimental period of 4 weeks. During the experimental period, the clinical activity of colitis was blindly assessed by an independent observer to determine disease activity index (DAI), including weight loss, diarrhea, and bloody stools and recorded. DAI was calculated using DAI scoring system described by Barrett et al. (2012). Colon myeloperoxidase (MPO) activity was also measured to assess the colonic injury and inflammation using the Myeloperoxidase Activity Assay Kit (Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s protocol.
Sample Preparation
Mice were anesthetized with pentobarbital and sacrificed by cervical dislocation. Blood was collected from arteria femoralis of mice at the time of sacrificed after a 12-h fast. Serum samples were separated from blood by the centrifugation at 3000 rpm for 15 min and stored at –80°C until the biochemical analysis. Colons were excised and dried after washing in phosphate buffer-saline (PBS), then used for the evaluation of colon length and morphological score, as previously described (Zhang et al., 2020). Liver was also resected after sacrificed. Part of liver or colon was fixed in 4% paraformaldehyde, embedded into paraffin, and cut to 4 μm sections for the histological analysis. Remaining liver or colon tissues were stored for the subsequent analysis, such as detection of hydroxyproline in liver and expression analysis of protein.
Biochemical Analysis
Serum lipopolysaccharide (LPS), alanine aminotransferase (ALT), glutamic oxalacetic transaminase (AST), albumin (ALB), total bilirubin (TBIL), direct bilirubin (DBIL), and hydroxyproline were measured using commercially available kits (Jiancheng Bioengineering Institute, Nanjing, China). The analyses were performed in BECKMAN COULTER CX9Automatic Aralyzer (BECKMAN, United States) or UV-2000 spectrophotometer (Unicosh, Shanghai, China).
Histopathological Staining
Paraffin-embedded colon and liver sections were, respectively, stained with hematoxylin and eosin (H&E) for the histological evaluation of inflammation, extent, gland damage, and cell infiltration. Liver sections were stained with Masson’s trichrome (MT) or Sirius red for fibrosis evaluation. The stained sections were examined under a microscope (Olympus BX51, Japan). Histological score was evaluated according to the established scale based on inflammatory and epithelial parameters (Aranda et al., 1997). Fibrotic stage was graded on a 0–4 scale, including no fibrosis, portal, periportal, or bridging fibrosis and cirrhosis, respectively. Fibrotic area were quantified after Sirius red staining.
Bacterial Translocation Assay
After mice were sacrificed, mesenteric lymph nodes (MLN) were resected under aseptic conditions. MLN samples were washed in 200 μl PBS containing gentamicin (50 μg/mL) and homogenized in ice-cold PBS. Tissue homogenates were seeded in Luria-Bertani plate at 37°C for 24 h. The number of colonies was counted based on the dilution ratio and expressed as colony forming unit (CFU). Bacterial translocation were assessed by the amount of bacteria per gram (CFU/g).
Immunohistochemistry (IHC) Staining
IHC staining was conducted according to standard protocol. The paraffin-embedded liver or colon tissues were sequentially deparaffinized, rehydrated and washed. Sections were then treated with pH 6.0 sodium citrate buffer, followed by blocking with 3% H2O2 and goat serum. Next, the following primary antibodies: anti-claudin-1 (Santa Cruz, United States), anti-occludin (Santa Cruz, United States), anti-ZO-1 (Abcam United States), anti-tumor necrosis factor-α (TNF-α, Bio Legend, United States), anti-interferon-γ (IFN-γ, Bio Legend, United States), anti-interleukin-17A (IL-17A, Santa Cruz, United States), anti-TLR4 (Santa Cruz, United States), anti-TNF receptor-associated factor 6 (TRAF6, Santa Cruz, United States), anti-nuclear factor-κB p65 (NF-κB p65, Santa Cruz, United States), anti-α-smooth muscle actin (α-SMA, Abcam, United States), anti-transforming growth factor-β (TGF-β, Santa Cruz, United States), anti-Collagen type I (Col I, Santa Cruz, United States), anti-Collagen type III (Col III, Santa Cruz, United States), anti-matrix metalloproteinase-2 (MMP-2, Proteintech, United States), anti-tissue inhibitor of MMP-2 (TIMP-2, Santa Cruz, United States), and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Santa Cruz, United States) were used to stain the target proteins. Peroxidase-conjugated antibody were used as secondary antibody. Detection was performed using diaminobezidin (Dako, Denmark) as chromogen. Images were acquired under fluorescent microscopy (Olympus, Japan) and analyzed using Image-Pro Plus 6.0 software (Media Cybernetics Inc., United States).
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Analysis
Total RNA was prepared from the liver or colon tissues using Trizol reagent (Invitrogen, Carlsbad, CA, United States) according to the manufacturer’s instructions. Then, cDNA was synthesized from total RNA by iScript select cDNA synthesis kit (Bio-Rad, Hercules, CA, United States). Quantitative real-time PCR was performed with iTaq Universal SYBR Green Supermix (Bio-Rad) by CFX96 Touch real-time PCR detection system (Bio-Rad, Hercules, CA, United States). GAPDH was used as the endogenous control. The relative quantities were normalized to endogenous control values and calculated by using the 2–△△Ct method. Sequences of primers are described in Table 1.
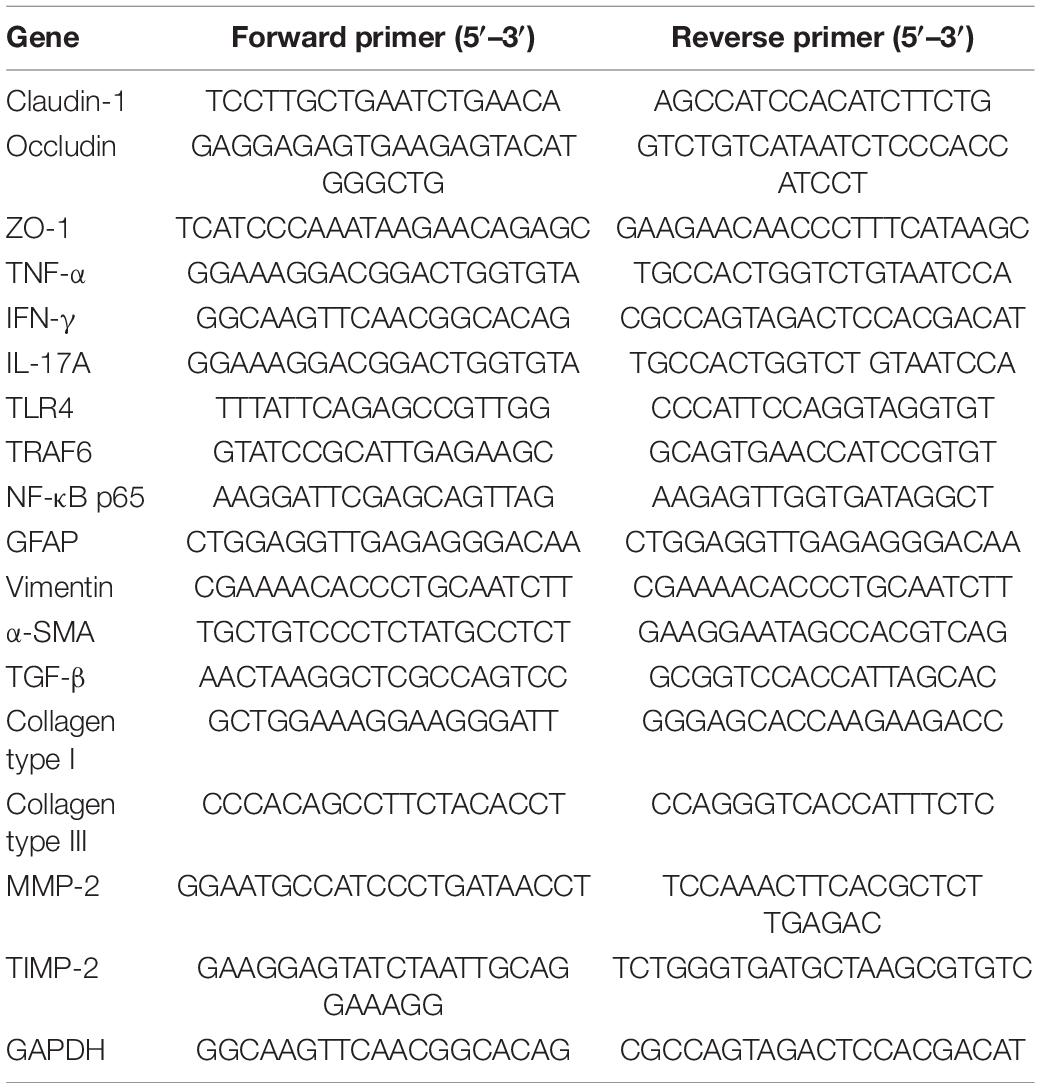
Table 1. Primer sequence for qRT-PCR.
Western Blot Analysis
Samples of liver or colon were homogenized in lysis buffer (Beyotime, Beijing, China) and centrifuged. Protein concentration of the supernatant was determined with coomassie brilliant blue assay. The extracts containing equal quantities of proteins (80 μg) were subjected to 10% polyacrylamide gel electrophoresis and then transferred to a polyvinylidene difluoride (PVDF) membrane (Invitrogen, United States). The membrane was blocked with 5% non-fat milk blocking buffer and blotted with the following specific antibodies: anti-claudin-1 (Santa Cruz, United States), anti-occludin (Santa Cruz, United States), anti-ZO-1 (Abcam United States), anti-TNF-α (Bio Legend, United States), anti-IFN-γ (Bio Legend, United States), anti-IL-17A (Santa Cruz, United States), anti-TLR4 (Santa Cruz, United States), anti-TRAF6 (Santa Cruz, United States), anti-NF-κB p65 (Santa Cruz, United States), anti-glial fibrillary acidic protein (GFAP, Abcam, United States), anti-vimentin (Abcam, United States), anti-α-SMA (Abcam, United States), anti-TGF-β (Santa Cruz, United States), anti-Col I (Santa Cruz, United States), anti-Col III (Santa Cruz, United States), anti-MMP-2 (Proteintech, United States), anti-TIMP-2 (Santa Cruz, United States), and anti-GAPDH (Santa Cruz, United States) overnight at 4°C. Subsequently, they were incubated with goat anti-rabbit IgG (1:2000)/anti-mouse IgG (1:2000)/anti-rat IgG (1:2000) or mouse anti-goat IgG (1:2000) (Zhongshan Goldenbridge, Beijing, China) for 2 h at room temperature. The protein bands on the membranes were developed using enhanced chemiluminescence detection reagents (Santa Cruz, United States) followed by exposure on Kodak Xdmat blue XB-1 film (Kodak, Rochester, NY, United States). GAPDH was used as a loading control.
Statistical Analysis
Statistical analyses were performed with SPSS 13.0 software. Data were expressed as mean ± standard deviation (SD). One-way analysis of variance was used to evaluate the differences among groups and two group comparison was determined by Student-Newman-Keuls test. P value <0.05 was considered statistically significant.
Results
DSS Induced the Intestinal Inflammation in CCl4-Induced Liver Fibrosis Mice
To investigate the mechanism of UC in liver fibrosis, liver fibrosis mouse models with intestinal inflammation were established. After that, the severity of intestinal inflammation was evaluated using the indexes including body weight loss, DAI score, histopathological indicators and the expression of proinflammatory cytokines in colon. As shown in Figure 1A, body weights of mice in DSS and CCl4 groups decreased significantly compared to mice in their corresponding control group (DSS vs. control, p < 0.05; CCl4 vs. Olive oil, p < 0.05). Notably, we found that DSS treatment significantly aggravated the CCl4-induced body weight loss (Figure 1A). Meanwhile, an obvious increase of DAI score were found in DSS and CCl4 groups compared to their controls (Figure 1B). Moreover, the increase of DAI after CCl4 induction were further enhanced by DSS treatment (Figure 1B).
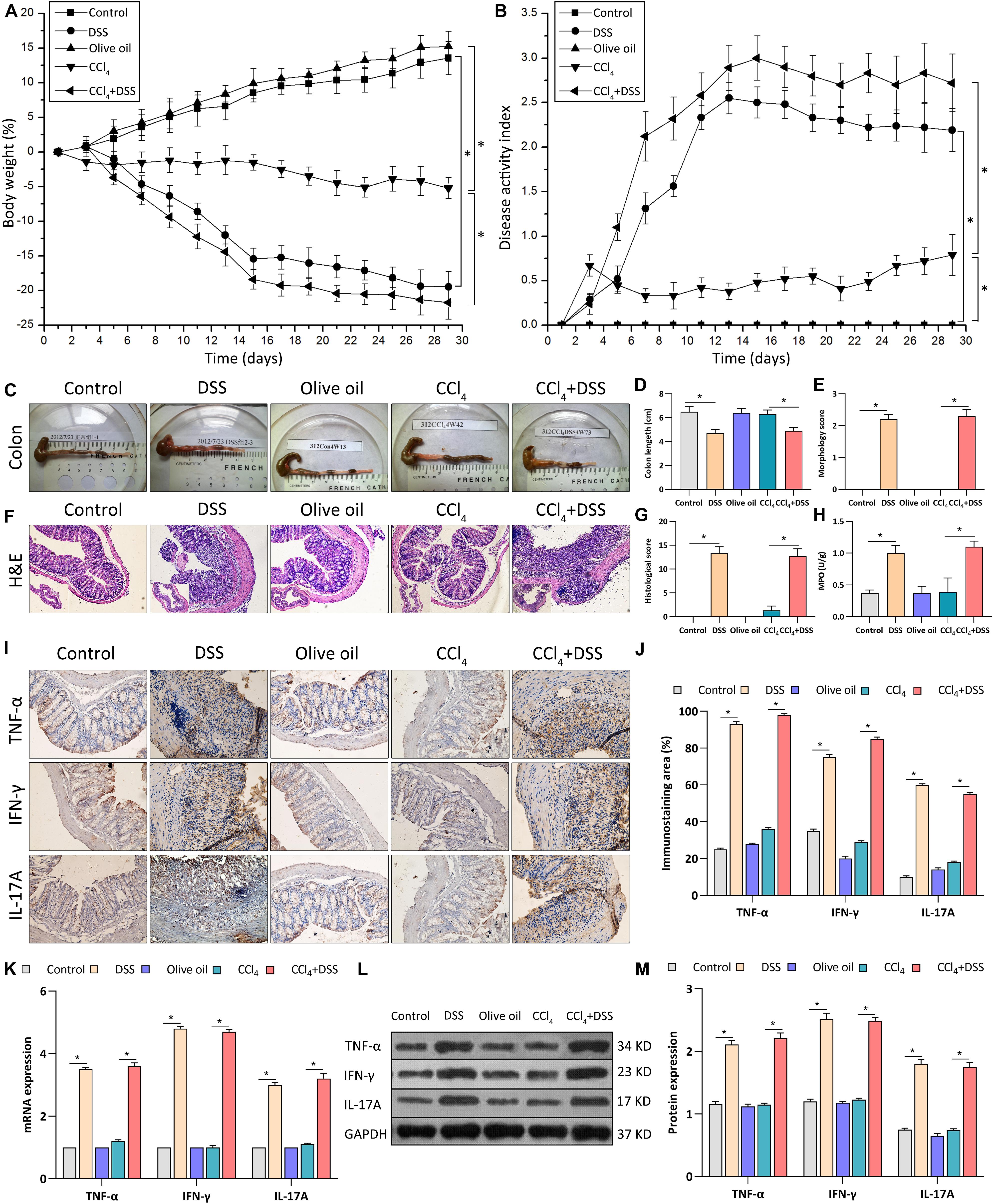
Figure 1. DSS induced the intestinal inflammation in CCl4-induced liver fibrosis mice. (A) Changes of body weight during the experimental period; (B) Disease activity index was evaluated at indicated time points during the experimental period; (C) Representative colon specimen of mice from five groups were shown; (D) Colon length and (E) morphology score were quantified; (F) Representative images of H&E staining colon sections (magnifications ×200) and (G) histological score were shown; (H) Myeloperoxidase activity; (I) Representative images of IHC staining colon sections (magnifications ×400) and (J) staining intensity of pro-inflammatory cytokines were shown. (K–M) Expression levels of pro-inflammatory cytokines in colon were measured by qRT-PCR (K) and western blot (L,M). Data were expressed as mean ± SD. *, p < 0.05.
Through the inspection of colonic specimens, we found that DSS induced the wall thickening, colonic mucosa hyperemia (Figure 1C), decrease of colon length (Figure 1D), and the increase of morphology score (Figure 1E) in CCl4-induced liver fibrosis mice. Pathologically, extensive ulceration of the epithelial layer, edema, crypt damage of bowel wall, infiltration of inflammatory cells into the mucosa were observed in DSS and CCl4 + DSS group (Figure 1F), accompanied by the higher histological score (Figure 1G). In addition, MPO activities in the colonic tissues of DSS and CCl4 + DSS treated mice were significantly increased compared with their control groups (Figure 1H). Furthermore, IHC, qRT-PCR, and western blot analysis all confirmed that the expression levels of proinflammatory cytokines (TNF-α, IFN-γ, and IL-17A) in colon tissues were remarkably increased after DSS treatment (Figures 1I–M). In summary, DSS induced the intestinal inflammation in CCl4-induced liver fibrosis mice, implicating the successful establishment of liver fibrosis mouse models with colitis.
DSS-Induced Colitis Aggravated Liver Damage, Fibrosis and Inflammation in CCl4-Induced Liver Fibrosis Mice
To assess the effects of DSS-induced colitis on liver inflammation and fibrogenesis in CCl4-induced liver fibrosis mice, histopathological changes and expression of proinflammatory cytokines in liver tissues were measured after DSS treatment. Firstly, H&E staining revealed the moderate inflammation in DSS and CCl4 group, while the addition of DSS aggravated the inflammation grade in CCl4-induced liver fibrosis mice (Figures 2A,B). MT and Sirius red staining showed the more obvious deposition of extracellular matrix (ECM), cell vacuolar degeneration, edema and necrosis around central and portal veins in CCl4 + DSS group relative to CCl4 group, along with the increases in fibrotic stage and Sirius red positive area (Figures 2A,C,D). Then, tissue hydroxyproline as objective measure of liver fibrosis were detected. As shown in Figure 2E, the addition of DSS further increased the levels of hydroxyproline in CCl4-induced liver fibrosis mice. By measuring the liver function indexes, we observed the significant elevation of ALT, AST, TBIL, and DBIL levels, but reduction of ALB in CCl4 + DSS group relative to CCl4 group, indicating the aggravation of liver damage (Figure 2F). Moreover, expression analysis using IHC, qRT-PCR, and western blot indicated that CCl4 + DSS treated mice expressed higher levels of proinflammatory cytokines (TNF-α, IFN-γ, and IL-17A) in liver tissues compared to only CCl4 treated mice (Figures 2G–K). Collectively, these results supported the opinion that DSS-induced colitis aggravated liver damage, fibrosis and inflammation in CCl4-induced liver fibrosis mice.
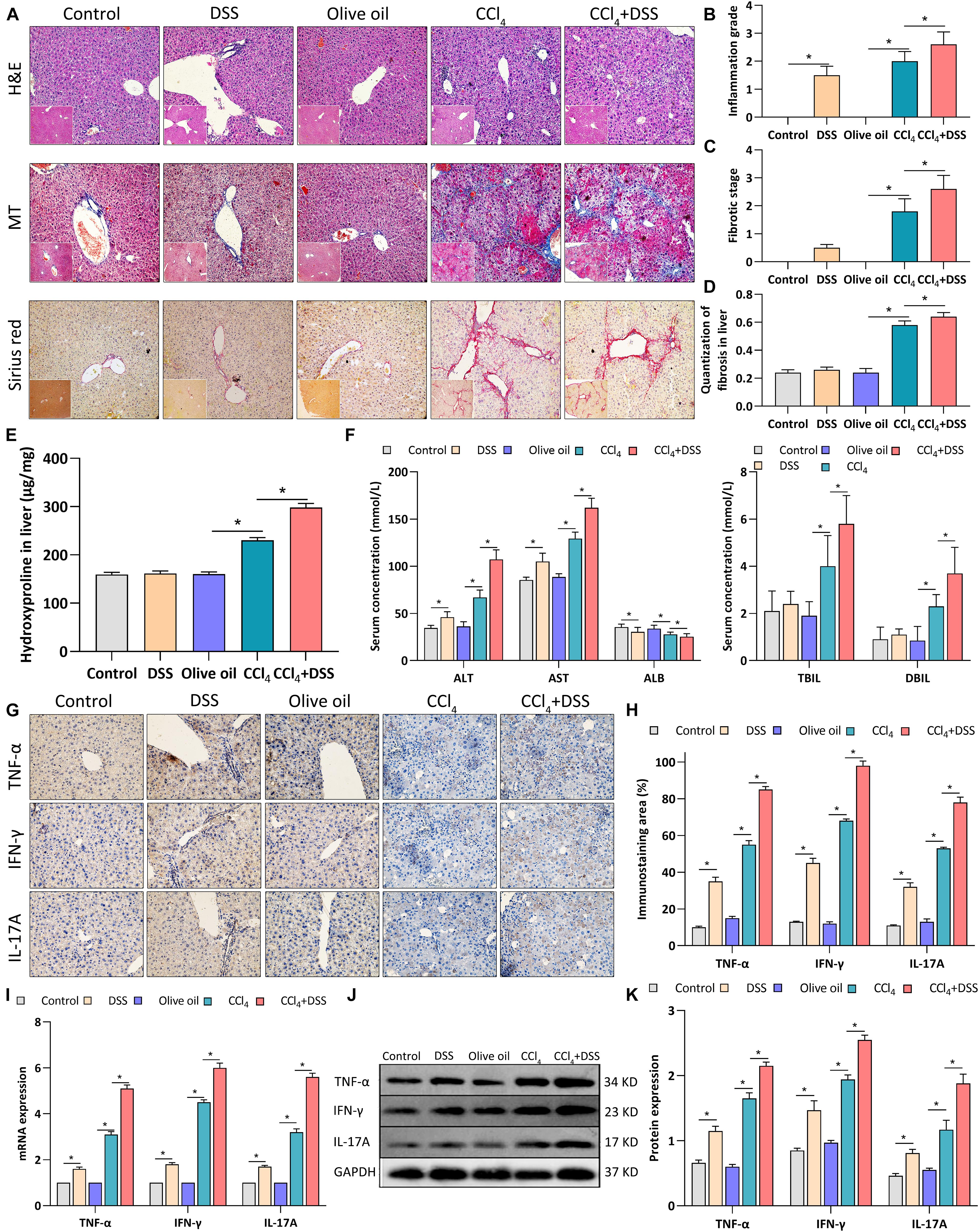
Figure 2. DSS-induced colitis aggravated liver damage, fibrosis and inflammation in CCl4-induced liver fibrosis mice. (A) Representative images of H&E staining, Masson’s trichrome (MT) or Sirius red liver sections (magnifications ×200); (B) Inflammation grade; (C) fibrotic stage and (D) quantitative analysis of fibrous tissue were shown; (E) The content of hydroxyproline in liver tissue; (F) Serum levels of liver function indexes; (G) Representative images of IHC staining liver sections (magnifications ×400) and (H) staining intensity of pro-inflammatory cytokines were shown. (I–K) Expression levels of pro-inflammatory cytokines in liver were measured by qRT-PCR (I) and western blot (J,K). Data were expressed as mean ± SD. *, p < 0.05.
DSS-Induced Colitis Regulated Gut Barrier Function in CCl4-Induced Liver Fibrosis Mice
To assess whether colitis regulated liver fibrosis through gut-liver axis, bacterial translocation, serum LPS level and intestinal epithelial tight junctions were evaluated after DSS treatment. After the bacterial culture of the MLN homogenate, we found that the bacterial translocation to MLN was more frequently present in CCl4 + DSS-treated mice compared to CCl4 treated mice (Figures 3A,B). We next measured the serum level of LPS, a major recognition marker common to gram-negative bacteria, to determine bacterial translocation. As results, both in DSS and CCl4 + DSS groups, serum LPS was significantly higher than that in their controls (Figure 3C). Occludin, claudin-1 and ZO-1 are important tightly linked proteins that play a critical role in maintaining the intestinal epithelial tight junctions and permeability. Therefore, expression levels of these tight junction proteins in colon were detected to evaluate the gut barrier function. Occludin, claudin-1 and ZO-1 staining showed that there were remarkable decrease in the positive staining cells in the livers of DSS and CCl4 + DSS treated mice when compared to their controls (Figures 3D,E). Similarly, the identical results were confirmed by qRT-PCR (Figure 3F) and western blot analysis (Figures 3G,H), that CCl4 + DSS treated mice had lower expression levels of occludin, claudin-1 and ZO-1 compared to controls. In summary, DSS-induced colitis promoted the bacterial translocation and reduced intestinal epithelial tight junctions, leading to the gut barrier dysfunction in a CCl4-induced liver fibrosis mice.
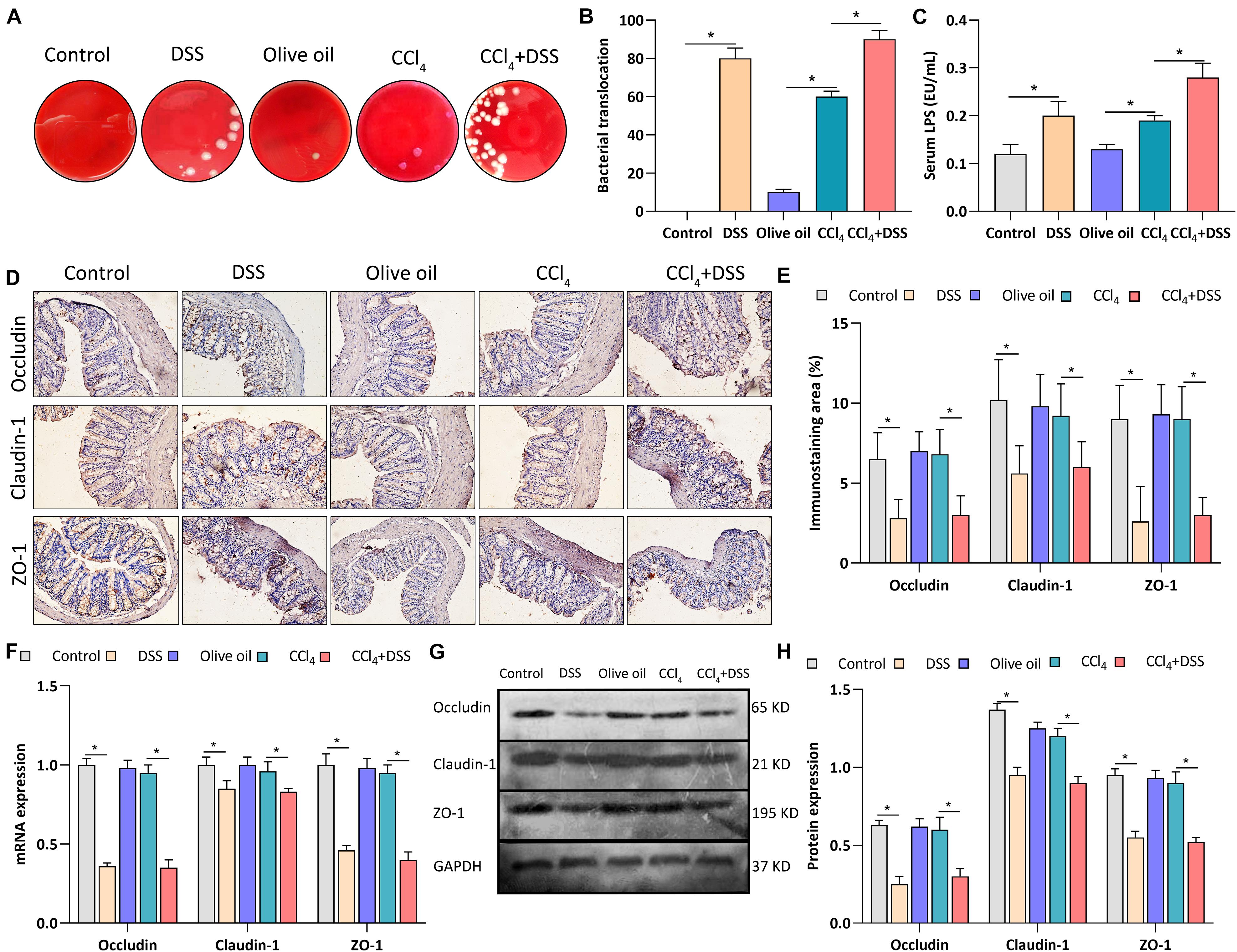
Figure 3. DSS-induced colitis regulated gut barrier function in CCl4-induced liver fibrosis mice. (A) Representative images of bacterial translocation assay and (B) quantitative analysis of the bacterial translocation to MLN were shown; (C) Serum lipopolysaccharide (LPS) levels; (D) Representative images of IHC staining colon sections (magnifications ×400) and (E) staining intensity of tight junction proteins were shown; (F–H) Expression levels of tight junction proteins in colon were measured by qRT-PCR (F) and western blot (G,H). Data were expressed as mean ± SD. *, p < 0.05.
DSS-Induced Colitis Promoted the Activation of HSCs Through TLR4 Signaling in CCl4-Induced Liver Fibrosis Mice
As previously reported, the expression of α-SMA, GFAP, vimentin, TGF-β1, collagen type I (Col I), collagen type III (Col III), MMP-2, and TIMP-2 are associated with the activation of HSCs. Thus, the expression levels of these proteins were measured after various treatments. From the IHC staining (Figures 4A,B), qRT-PCR (Figure 4C), and western blot (Figures 4D,E), the activation markers, including α-SMA, vimentin, Col I, and Col III, were up-regulated in the livers of CCl4-induced liver fibrosis mice than that in the control, which were further enhanced by the DSS exposure. On the contrary, the inactivation marker GFAP was down-regulated in the CCl4 group, and was further inhibited in the CCl4 + DSS group. In addition, the staining of other HSCs activation related proteins TGF-β1, MMP-2, and TIMP-2 were more pronounced in the livers of CCl4-induced liver fibrosis mice than that in the control. Furthermore, the DSS exposure further increased the CCl4-induced up-regulation of TGF-β1 and TIMP-2, while decreased the MMP-2 expression (Figures 4A,B). There results were further confirmed by qRT-PCR (Figure 4C) and western blot (Figures 4D,E).
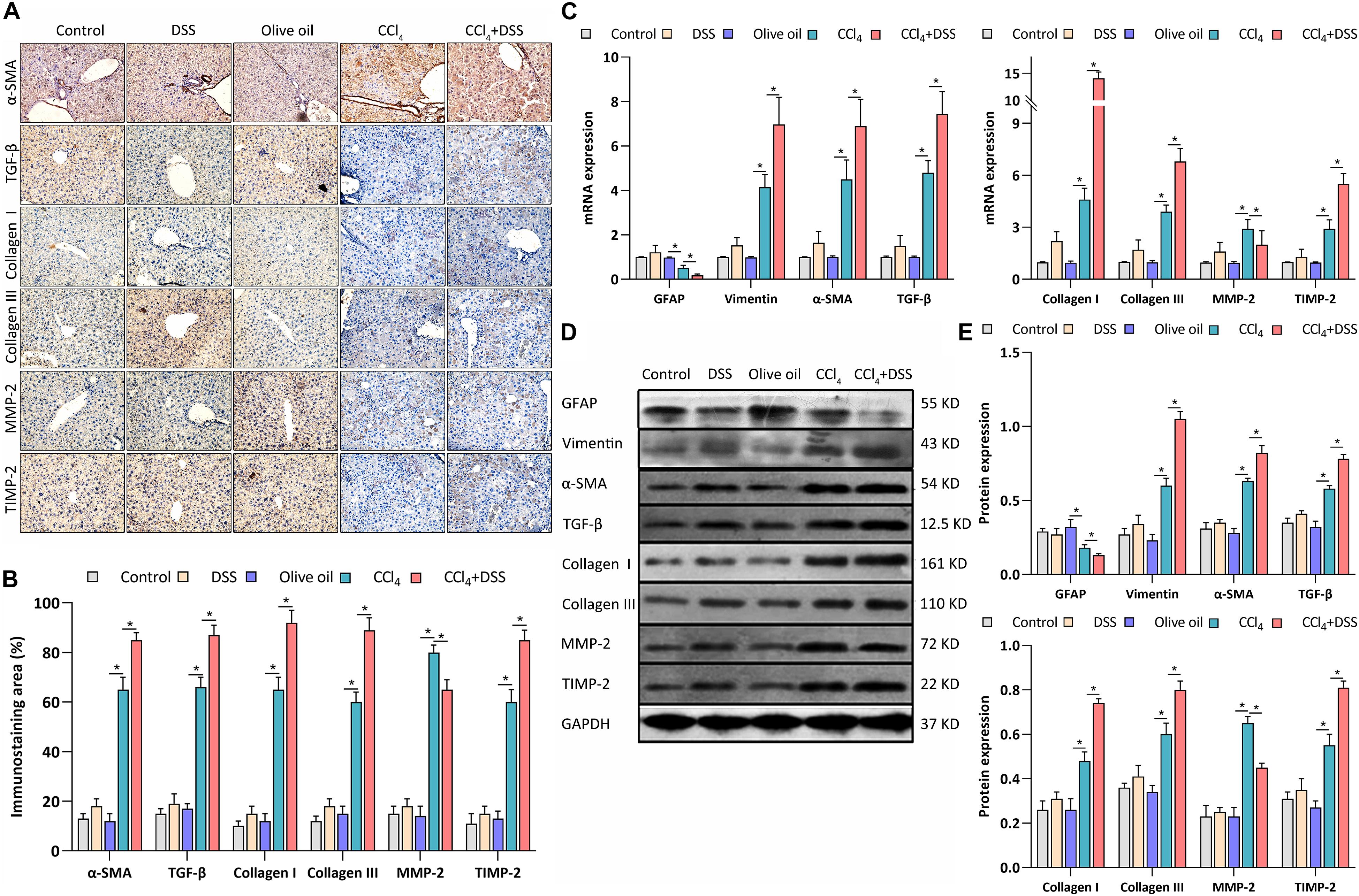
Figure 4. DSS promoted the activation of HSCs in CCl4-induced liver fibrosis mice. (A) Representative images of IHC staining liver sections (magnifications ×400) and (B) staining intensity of proteins-related to HSCs activation were shown; (C–E) Expression levels of proteins-related to HSCs activation in liver were measured by qRT-PCR (C) and western blot (D,E). Data were expressed as mean ± SD. *, p < 0.05.
TLR4 signaling was identified in HSCs, and mediates the inflammatory and fibrogenesis. We therefore detected the key proteins related to TLR4 signaling. IHC staining showed a significant enhancement of the TLR4, TRAF6, and NF-κB p65 expression in the livers of mice in DSS group and CCl4 group relative to their controls (Figures 5A,B). Meanwhile, the CCl4-induced up-regulation of TLR4, TRAF6, and NF-κB p65 were further increased by DSS treatment in the CCl4 + DSS group (Figures 5A,B). In addition, similar results were observed from the analysis by qRT-PCR (Figure 5C) and western blot (Figures 5D,E). Given these above results, we speculated that DSS promoted the activation of HSCs by activating the TLR4 signaling pathway in CCl4-induced liver fibrosis mice.
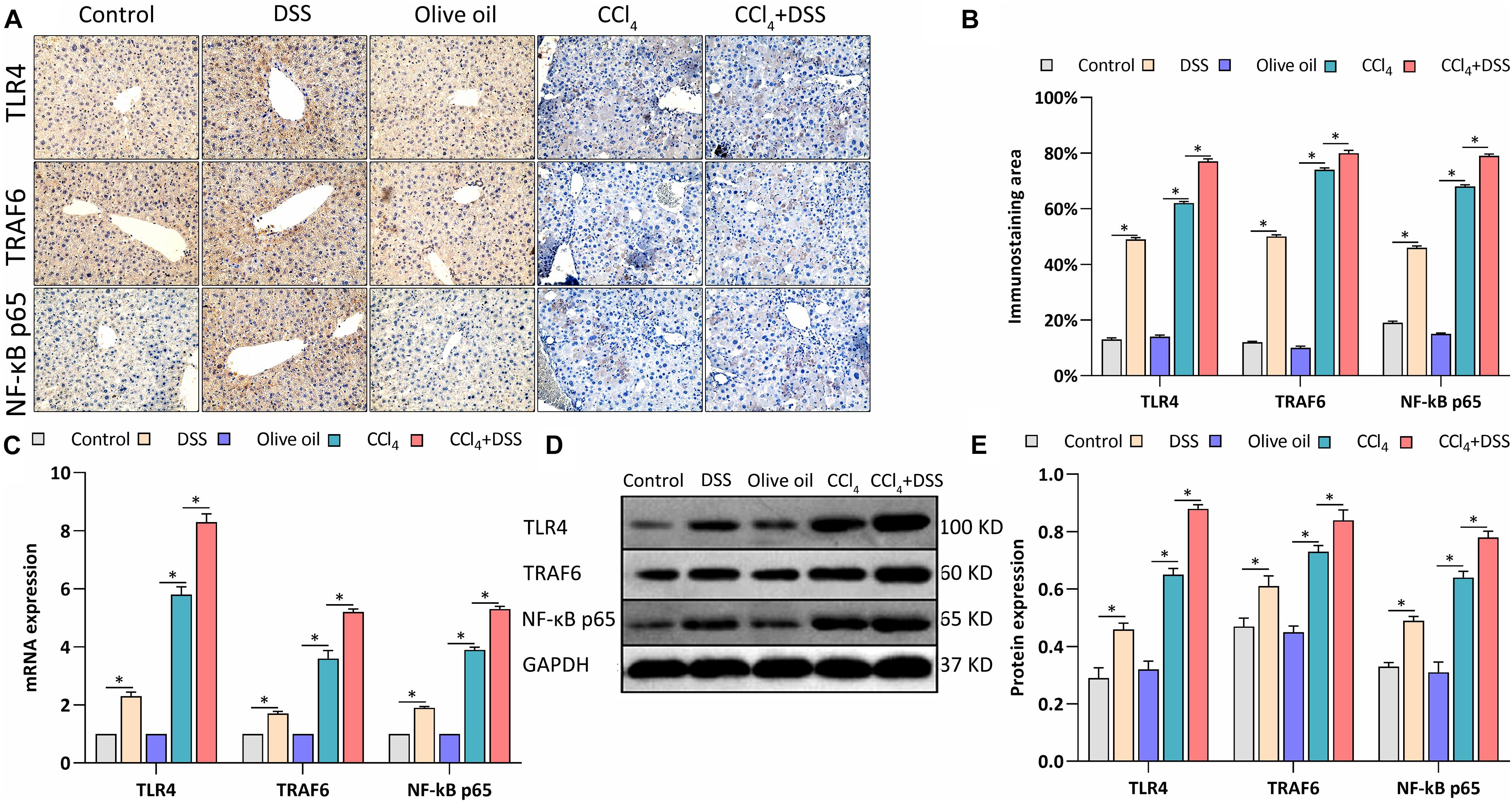
Figure 5. DSS promoted the activation of TLR4 signaling in CCl4-induced liver fibrosis mice. (A) Representative images of IHC staining liver sections (magnifications ×400) and (B) staining intensity of proteins in TLR4 signaling were shown; (C–E) Expression levels of proteins in TLR4 signaling in liver were measured by qRT-PCR (C) and western blot (D,E). Data were expressed as mean ± SD. ∗, p < 0.05.
Discussion
Despite there is increasing evidence for a correlation between the gut-liver axis and the pathogenesis of liver diseases, the majority of studies focused on the fibrosis associated with different etiologies, such as ALD, non-alcoholic fatty liver disease (NAFLD), and viral hepatitis (Elpek, 2014). The intuitive illustration of the pathogenesis of liver fibrosis would contribute to the development of anti-fibrotic therapies. To the best of our knowledge, this is the first direct evidence to support the role of DSS-induced colitis in the pathogenesis of liver fibrosis per se. The present study highlighted that DSS-induced colitis aggravated the liver fibrosis caused by CCl4 through the gut-liver axis. The study of the mechanism indicated that altered intestinal permeability presented as down-regulation of tight junction proteins (occluding, claudin-1) in colitis mice may favor the passage of bacterial products such bacterial LPS into the liver through the portal vein, and then increased LPS activated TLR4 signaling and HSCs activation which involved in liver inflammation and fibrogenesis (Figure 6). These findings provided direct evidence for the role of the gut-liver axis in CCl4-induced liver fibrosis.
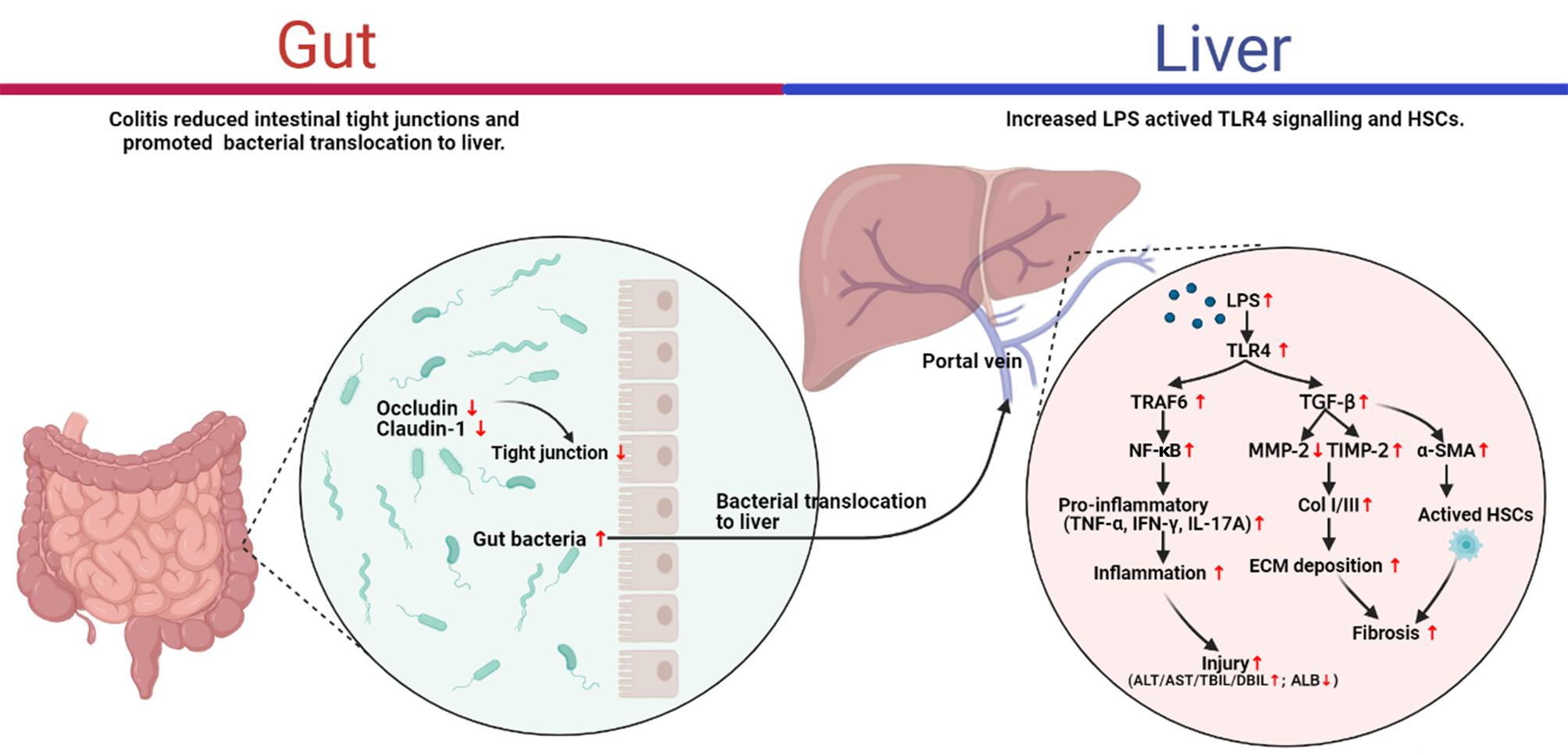
Figure 6. Proposed mechanism for the ulcerative colitis-aggravated liver fibrosis.
CCl4 is considered to be a direct hepatotoxin that causes central necrosis and steatosis in the liver lobule, ultimately leading to liver fibrosis (Ma et al., 2014). Therefore, in the present study, CCl4 was used to establish the experimental liver fibrosis in mice. On the basis of CCl4-induced liver fibrosis, we induced chronic colitis by the repeated administration of DSS. As expected, the combination treatment with DSS and CCl4 successfully induced intestinal inflammation in mice with liver fibrosis, as evident from the elevated DAI score, bodyweight loss, and enhanced inflammatory response. The intestinal pathology caused by DSS treatment was consistent with the previous reports (Chassaing et al., 2014). Subsequently, various histological and biochemical analyses were conducted on this chronic colitis model with liver fibrosis to verify our assumption.
A considerable number of studies have reported that liver abnormalities including inflammation and fibrosis are typical extra-intestinal manifestations of colitis and are frequently observed (Kummen et al., 2013; Marotto et al., 2020). Here, H&E staining revealed the obvious inflammatory cell infiltration, cytoplasm relaxation, and vacuolar degeneration of hepatocytes in DSS-induced colitis mice, which was confirmed by the enhancement of pro-inflammatory cytokines (TNF-α, IFN-γ, and IL-17A) using IHC, qRT-PCR, and western blot analysis. Therefore, the finding from our study was consistent with the above opinion, that DSS-induced colitis mice presented mild liver inflammation and dysfunction. Serum levels of ALT, AST, and ALB, as markers that assess liver injury (Kasarala and Tillmann, 2016), were also found to be abnormal in DSS-induced colitis mice, further supporting that the liver injury is the extra-intestinal manifestation of colitis. Nevertheless, our results were opposite to the previous studies, which demonstrated that the DSS-induced inflammatory process was mainly restricted to the colon and DSS alone did not induce liver pro-inflammatory cytokines, in turn, affect the liver directly (Karlsson et al., 2008; Gäbele et al., 2011). A possible explanation might be that the high dose and long exposure duration of DSS treatment used in our study caused the immune response, which was completely different from previous studies. In fact, 7-day administration of 3% DSS was identified to induce liver inflammation (Trivedi and Jena, 2013). Additionally, a recent study has also reported that the administration of DSS changed the expression of pro-and anti-inflammatory cytokines such as IL-1β, IL-10, and TGFb-2, suggesting that intestinal inflammation led to liver inflammation (Nii et al., 2020), thus, these results supported our finding again. Besides, we found that DSS alone did not increase the fibrotic stage, Sirius red positive area, and hydroxyproline content compared to control, indicating that DSS alone cannot induce liver fibrosis. These results were consistent with previous studies (Gäbele et al., 2011; Ji et al., 2018); thus, we speculated that DSS alone mainly induced liver injury by mediating the inflammation. More importantly, the liver injury, inflammation, and fibrosis that was originally present in CCl4-treated mice were aggravated by the DSS treatment. Actually, this finding was also similar to the data from experimental NASH (Gäbele et al., 2011). The histological abnormalities including ECM deposition, fibrogenesis, inflammatory cell infiltration, and the enhanced inflammatory response caused by colitis were in line with the manifestations in experimental NASH mice (Achiwa et al., 2016). In summary, our study confirmed that DSS-induced colitis aggravated liver damage, fibrosis, and inflammation in CCl4-induced liver fibrosis mice.
In the last few years, a growing particular interest has been devoted to the gut-liver axis in liver disease. Gut-liver axis is a complex structure and its alteration seems to play an important role in the progression of liver disease, in particular, of two of its components (gut barrier and microbiota) (Poeta et al., 2017). Theoretically, bacterial translocation is defined as the passage of intestinal bacteria or bacterial products from the gastrointestinal tract to MLN or other extraintestinal sites, which is correlated with the gut barrier function (Porras et al., 2006). In the present study, more intestinal bacteria were found to translocate into MLN in DSS + CCl4-treated mice compared to CCl4-treated mice, implying that DSS-induced colitis may cause gut barrier dysfunction. Additionally, LPS is a marker for bacterial translocation when detected in the systemic circulation (Alexopoulou et al., 2017). Although LPS in the intestinal cavity is usually unable to penetrate into the healthy intestinal epithelium, it may pass through the intestinal mucosa when the intestinal permeability is disturbed (Stevens et al., 2018). Consistently, the elevated LPS levels in the DSS + CCl4-treated mice further supported the bacterial translocation and gut barrier dysfunction. Unfortunately, our present study did not elucidate which type or what bacteria translocate. However, as previously reported, intestinal bacteria that are able to translocate into extraintestinal sites mainly include Escherichia coli (E. coli), Klebsiella pneumoniae, and other Enterobacteriaceae (Okasha et al., 2000). According to the appearance observation of bacterial colonies, colonial morphology is similar to that of Enterobacteriaceae. Analysis of strains using automatic microbial identification systems identified that the stains is mainly E. coli. Nevertheless, these attempts are preliminary and the definite type of bacteria translocation remains further investigation.
Moreover, increased intestinal permeability is one of the major mechanisms postulated to promote bacterial translocation (Porras et al., 2006). Meanwhile, tight junctions formed by claudins and occludin play a significant role in the formation and maintenance of intestinal epithelial barrier integrity (Vandenbroucke et al., 2013); thus, the tight junction proteins were measured. Here, occluding, claudin-1 and ZO-1 as main tight junction proteins were obviously down-regulated in the colon after DSS treatment, which further proved the gut barrier dysfunction in the colon. However, it is worth noting that CCl4 alone also caused the bacterial translocation and the elevated LPS level, but not the alterations in tight junction proteins in colon. Actually, this finding is not an exception and agrees with the results of other studies (Yao et al., 2006; Fouts et al., 2012). As Fouts et al. reported, occludin was decreased in the small intestine, but not the colon after injection of CCl4 as compared to control (Fouts et al., 2012), suggesting that the CCl4-induced toxic liver injury could induce the increase of intestinal tight junction permeability, and then promote bacterial translocation. Additionally, Yao et al. have revealed that CCl4-treated mice exhibited shortened and thinned microvilli, proposing that the damaged microvillus environment might be also responsible for the early onset of bacterial translocation (Yao et al., 2006). According to the newest evidence, we speculated that the reduction in the thickness of the colonic mucus layer may be another explanation for the bacterial translocation in CCl4-treated mice (Haderer et al., 2021). Furthermore, other viewpoints proposed that intestinal bacterial overgrowth and deficiencies in localhost immune defenses are the major mechanisms postulated to favor bacterial translocation in liver disease (Gómez-Hurtado et al., 2011). However, since we mainly focused on the effects of DSS-induced colitis on liver fibrosis, the mechanisms of CCl4-induced bacterial translocation were not investigated here. Even so, the present study confirmed that DSS-induced colitis could promote bacterial translocation by inducing the gut barrier dysfunction in colon in CCl4-induced liver fibrosis, which is in accord with our hypothesis.
Numerous studies unraveled critical functions for HSCs in the pathogenesis of liver diseases (Khomich et al., 2019). HSCs express neural markers (e.g., GFAP, synemin, and synaptophysin), mesenchymal/mesodermal markers (e.g., desmin vimentin, α-SMA, and Col) (Hyun et al., 2016; Tsuchida and Friedman, 2017). As previously reported, upon activation, HSCs down-regulate neural markers and up-regulate mesenchymal markers, such as vimentin, COl I, and α-SMA (Scholten et al., 2010), being consistent to our present results that GFAP was down-regulated while vimentin, COl I and α-SMA were up-regulated in the DSS + CCl4-treated mice. This evidence suggest that the colitis might facilitate the activation of HSCs. Additionally, TGF-β1 is reported to induce the HSC activation (Tsuchida and Friedman, 2017). Here, we observed that DSS induced the further up-regulation of TGF-β1 in the liver fibrosis mice, supporting the activation of HSCs. HSCs are the main cell type that responsible for collagen deposition and fibrogenesis in the liver (Carson et al., 2018). Thus, the increased activation of HSCs accelerated fibrogenesis. Liver fibrosis is the consequence of chronic liver injury and ECM accumulation, such as collagen proteins (Khurana et al., 2021). In the process of liver injury, HSCs are activated to fibroblasts expressing muscle α-SMA under the action of various cytokines and inflammatory mediators, and a large number of ECM components are synthesized during migration and proliferation, mainly Col I and Col III (Gómez-Hurtado et al., 2011; Khurana et al., 2021). The expression of collagen is affected by its related collagenases (MMP-2 and TIMP-2), which regulated collagen fibers (Laronha and Caldeira, 2020). With consistent this mechanism, our study revealed the up-regulation of Col I, Col III, and TIMP-2 and down-regulation of MMP-2 in DSS + CCl4-treated mice. Actually, the activation of HSCs is recognized to be mediated by the LPS-activated TLR4 signaling in fibrogenesis (Mann and Marra, 2010). In our study, accompanied by the elevated LPS levels, the up-regulation of TLR4, TRAF6, and NF-κB p65 in TLR4 signaling were also detected, implicating the activation of TLR4/NF-κB signaling. TLR4/NF-κB signaling plays an important role in regulating immune and inflammatory responses (Peng et al., 2014; Khan et al., 2020). Thus, more pro-inflammatory cytokines such as TNF-α, TGF-β, IFN-γ, and IL-17A were released in the liver of DSS + CCl4-treated mice. Overall, DSS-induced colitis promoted the activation of HSCs and TLR4 signaling through the gut-liver axis in CCl4-induced liver fibrosis mice. These findings were consistent with the data from experimental NASH (Gäbele et al., 2011) and the widely recognized mechanisms (Lee and Friedman, 2011). Thus, the fibrosis associated with different etiologies might share the integrated signaling networks that regulate the ECM deposition. Nevertheless, the investigation of activation of HSCs and signaling mechanism is preliminary, and still remain to be further studied in detail.
Conclusion
The present study provided the first direct evidence for the pathogenesis of CCl4-induced liver fibrosis. We confirmed that intestinal inflammation aggravated liver damage, inflammation, and fibrosis in CCl4-induced liver fibrosis mice. More importantly, the results highlighted the role of gut-liver axis in its pathogenesis. Due to the altered intestinal permeability, intestinal inflammation caused by DSS led to the passage of bacterial products into liver, and in turn might promote the activation of TLR4 signaling and HSCs to induce the liver inflammation and fibrogenesis. These direct evidence increased our understanding of liver fibrosis pathogenesis and provided an additional perspective to the development of anti-fibrotic therapies.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by The Second Hospital of Hebei Medical University.
Author Contributions
Y-FL and X-LZ: conceptualization. Y-FL and G-CN: methodology and writing-original draft preparation. Y-FL, C-YL, and J-BG: material preparation. X-LZ: administrative support. G-CN, JS, and HL: formal analysis and data collection. Y-FL, G-CN, and HL: data analysis and interpretation. C-YL and J-BG: writing-review and editing. All authors read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Achiwa, K., Ishigami, M., Ishizu, Y., Kuzuya, T., Honda, T., Hayashi, K., et al. (2016). DSS colitis promotes tumorigenesis and fibrogenesis in a choline-deficient high-fat diet-induced NASH mouse model. Biochem. Biophys. Res. Commun. 470, 15–21. doi: 10.1016/j.bbrc.2015.12.012
Albanis, E., and Friedman, S. L. (2001). HEPATIC FIBROSIS: pathogenesis and Principles of Therapy. Clin. Liver Dis. 5, 315–334. doi: 10.1016/S1089-3261(05)70168-9
Alexopoulou, A., Agiasotelli, D., Vasilieva, L. E., and Dourakis, S. P. (2017). Bacterial translocation markers in liver cirrhosis. Ann. Gastroenterol. 30, 486–497. doi: 10.20524/aog.2017.0178
Aranda, R., Sydora, B. C., McAllister, P. L., Binder, S. W., Yang, H. Y., Targan, S. R., et al. (1997). Analysis of intestinal lymphocytes in mouse colitis mediated by transfer of CD4+, CD45RBhigh T cells to SCID recipients. J. Immunol. 158, 3464–3473.
Aydın, M. M., and Akçalı, K. C. (2018). Liver fibrosis. Turk. J. Gastroenterol. 29, 14–21. doi: 10.5152/tjg.2018.17330
Barrett, R., Zhang, X., Koon, H. W., Vu, M., Chang, J. Y., Yeager, N., et al. (2012). Constitutive TL1A expression under colitogenic conditions modulates the severity and location of gut mucosal inflammation and induces fibrostenosis. Am. J. Pathol. 180, 636–649. doi: 10.1016/j.ajpath.2011.10.026
Berumen, J., Baglieri, J., Kisseleva, T., and Mekeel, K. (2021). Liver fibrosis: pathophysiology and clinical implications. Wiley Interdiscip. Rev. Syst. Biol. Med. 13:e1499. doi: 10.1002/wsbm.1499
Carson, J. P., Ramm, G. A., Robinson, M. W., McManus, D. P., and Gobert, G. N. (2018). Schistosome-Induced Fibrotic Disease: the Role of Hepatic Stellate Cells. Trends Parasitol. 34, 524–540. doi: 10.1016/j.pt.2018.02.005
Chassaing, B., Aitken, J. D., Malleshappa, M., and Vijay-Kumar, M. (2014). Dextran Sulfate Sodium (DSS)-Induced Colitis in Mice. Curr. Protoc. Immunol. 104, 15.25.11–15.25.14. doi: 10.1002/0471142735.im1525s104
Elpek, G. Ö (2014). Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: an update. World J. Gastroenterol. 20, 7260–7276. doi: 10.3748/wjg.v20.i23.7260
Fouts, D. E., Torralba, M., Nelson, K. E., Brenner, D. A., and Schnabl, B. (2012). Bacterial translocation and changes in the intestinal microbiome in mouse models of liver disease. J. Hepatol. 56, 1283–1292. doi: 10.1016/j.jhep.2012.01.019
Frasinariu, O. E., Ceccarelli, S., Alisi, A., Moraru, E., and Nobili, V. (2013). Gut-liver axis and fibrosis in nonalcoholic fatty liver disease: an input for novel therapies. Dig. Liver Dis. 45, 543–551. doi: 10.1016/j.dld.2012.11.010
Gäbele, E., Dostert, K., Hofmann, C., Wiest, R., Schölmerich, J., Hellerbrand, C., et al. (2011). DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J. Hepatol. 55, 1391–1399.
Gómez-Hurtado, I., Santacruz, A., Peiró, G., Zapater, P., Gutiérrez, A., Pérez-Mateo, M., et al. (2011). Gut microbiota dysbiosis is associated with inflammation and bacterial translocation in mice with CCl 4-induced fibrosis. PLoS One 6:e23037. doi: 10.1371/journal.pone.0023037
Haderer, M., Neubert, P., Rinner, E., Scholtis, A., Broncy, L., Gschwendtner, H., et al. (2021). Novel pathomechanism for spontaneous bacterial peritonitis: disruption of cell junctions by cellular and bacterial proteases. Gut 1–13. doi: 10.1136/gutjnl-2020-321663 [online ahead of print]
Hintermann, E., and Christen, U. (2019). The Many Roles of Cell Adhesion Molecules in Hepatic Fibrosis. Cells 8:1503. doi: 10.3390/cells8121503
Hu, H., Lin, A., Kong, M., Yao, X., Yin, M., Xia, H., et al. (2020). Intestinal microbiome and NAFLD: molecular insights and therapeutic perspectives. J. Gastroenterol. 55, 142–158. doi: 10.1007/s00535-019-01649-8
Hyun, J., Wang, S., Kim, J., Rao, K. M., Park, S. Y., Chung, I., et al. (2016). MicroRNA-378 limits activation of hepatic stellate cells and liver fibrosis by suppressing Gli3 expression. Nat. Commun. 7:10993. doi: 10.1038/ncomms10993
Ji, Y., Dai, Z., Sun, S., Ma, X., Yang, Y., Tso, P., et al. (2018). Hydroxyproline Attenuates Dextran Sulfate Sodium-Induced Colitis in Mice: involvment of the NF-κB Signaling and Oxidative Stress. Mol. Nutr. Food Res. 62:e1800494. doi: 10.1002/mnfr.201800494
Karlsson, A., Jagervall, A., Pettersson, M., Andersson, A. K., Gillberg, P. G., and Melgar, S. (2008). Dextran sulphate sodium induces acute colitis and alters hepatic function in hamsters. Int. Immunopharmacol. 8, 20–27. doi: 10.1016/j.intimp.2007.10.007
Kasarala, G., and Tillmann, H. L. (2016). Standard liver tests. Clin. Liver Dis. 8, 13–18. doi: 10.1002/cld.562
Khan, M. Z., Khan, A., Xiao, J., Ma, J., Ma, Y., Chen, T., et al. (2020). Overview of Research Development on the Role of NF-κB Signaling in Mastitis. Animals 10:1625.
Khomich, O., Ivanov, A. V., and Bartosch, B. (2019). Metabolic Hallmarks of Hepatic Stellate Cells in Liver Fibrosis. Cells 9:24. doi: 10.3390/cells9010024
Khurana, A., Sayed, N., Allawadhi, P., and Weiskirchen, R. (2021). It’s all about the spaces between cells: role of extracellular matrix in liver fibrosis. Ann. Transl. Med. 9, 728–728. doi: 10.21037/atm-20-2948
Kisseleva, T., and Brenner, D. (2021). Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 18, 151–166. doi: 10.1038/s41575-020-00372-7
Koyama, Y., Xu, J., Liu, X., and Brenner, D. A. (2016). New Developments on the Treatment of Liver Fibrosis. Dig. Dis. 34, 589–596. doi: 10.1159/000445269
Kummen, M., Schrumpf, E., and Boberg, K. M. (2013). Liver abnormalities in bowel diseases. Best Pract. Res. Clin. Gastroenterol. 27, 531–542. doi: 10.1016/j.bpg.2013.06.013
Laronha, H., and Caldeira, J. (2020). Structure and Function of Human Matrix Metalloproteinases. Cells 9:1076. doi: 10.3390/cells9051076
Lee, U. E., and Friedman, S. L. (2011). Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 25, 195–206. doi: 10.1016/j.bpg.2011.02.005
Ma, J.-Q., Ding, J., Zhang, L., and Liu, C.-M. (2014). Hepatoprotective properties of sesamin against CCl4 induced oxidative stress-mediated apoptosis in mice via JNK pathway. Food Chem. Toxicol. 64, 41–48.
Mann, D. A., and Marra, F. (2010). Fibrogenic signalling in hepatic stellate cells. J. Hepatol. 52, 949–950. doi: 10.1016/j.jhep.2010.02.005
Marotto, D., Atzeni, F., Ardizzone, S., Monteleone, G., Giorgi, V., and Sarzi-Puttini, P. (2020). Extra-intestinal manifestations of inflammatory bowel diseases. Pharmacol. Res. 161:105206. doi: 10.1016/j.phrs.2020.105206
Matsuda, M., and Seki, E. (2020). Hepatic Stellate Cell-Macrophage Crosstalk in Liver Fibrosis and Carcinogenesis. Semin. Liver Dis. 40, 307–320. doi: 10.1055/s-0040-1708876
Nii, T., Bungo, T., Isobe, N., and Yoshimura, Y. (2020). Intestinal inflammation induced by dextran sodium sulphate causes liver inflammation and lipid metabolism disfunction in laying hens. Poult. Sci. 99, 1663–1677. doi: 10.1016/j.psj.2019.11.028
Okasha, H., Elgohary, A., Abd El Moety, A., and Elbadawy, A. (2000). Diagnostic and prognostic value of serum presepsin in cirrhotic patients with spontaneous bacterial peritonitis. GROUP 2, 1000.1000.
Pellicoro, A., Ramachandran, P., Iredale, J. P., and Fallowfield, J. A. (2014). Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 14, 181–194. doi: 10.1038/nri3623
Peng, Q., Liu, H., Shi, S., and Li, M. (2014). Lycium ruthenicum polysaccharide attenuates inflammation through inhibiting TLR4/NF-κB signaling pathway. Int. J. Biol. Macromol. 67, 330–335.
Poeta, M., Pierri, L., and Vajro, P. (2017). Gut–Liver Axis Derangement in Non-Alcoholic Fatty Liver Disease. Children 4:66.
Porras, M., Martín, M. T., Yang, P.-C., Jury, J., Perdue, M. H., and Vergara, P. (2006). Correlation between cyclical epithelial barrier dysfunction and bacterial translocation in the relapses of intestinal inflammation. Inflamm. Bowel Dis. 12, 843–852. doi: 10.1097/01.mib.0000231571.88806.62
Povero, D., Busletta, C., Novo, E., di Bonzo, L. V., Cannito, S., Paternostro, C., et al. (2010). Liver fibrosis: a dynamic and potentially reversible process. Histol. Histopathol. 25, 1075–1091. doi: 10.14670/HH-25.1075
Scholten, D., Osterreicher, C. H., Scholten, A., Iwaisako, K., Gu, G., Brenner, D. A., et al. (2010). Genetic labeling does not detect epithelial-to-mesenchymal transition of cholangiocytes in liver fibrosis in mice. Gastroenterology 139, 987–998. doi: 10.1053/j.gastro.2010.05.005
Seki, E., and Schnabl, B. (2012). Role of innate immunity and the microbiota in liver fibrosis: crosstalk between the liver and gut. J. Physiol. 590, 447–458. doi: 10.1113/jphysiol.2011.219691
Shiha, G., Ibrahim, A., Helmy, A., Sarin, S. K., Omata, M., Kumar, A., et al. (2017). Asian-Pacific Association for the Study of the Liver (APASL) consensus guidelines on invasive and non-invasive assessment of hepatic fibrosis: a 2016 update. Hepatol. Int. 11, 1–30.
Stevens, B. R., Goel, R., Seungbum, K., Richards, E. M., Holbert, R. C., Pepine, C. J., et al. (2018). Increased human intestinal barrier permeability plasma biomarkers zonulin and FABP2 correlated with plasma LPS and altered gut microbiome in anxiety or depression. Gut 67, 1555–1557.
Szabo, G. (2015). Gut–Liver Axis in Alcoholic Liver Disease. Gastroenterology 148, 30–36. doi: 10.1053/j.gastro.2014.10.042
Trivedi, P. P., and Jena, G. B. (2013). Ulcerative colitis-induced hepatic damage in mice: studies on inflammation, fibrosis, oxidative DNA damage and GST-P expression. Chem. Biol. Interact. 201, 19–30. doi: 10.1016/j.cbi.2012.12.004
Tsuchida, T., and Friedman, S. L. (2017). Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 14, 397–411. doi: 10.1038/nrgastro.2017.38
Vandenbroucke, R. E., Dejonckheere, E., Van Hauwermeiren, F., Lodens, S., De Rycke, R., Van Wonterghem, E., et al. (2013). Matrix metalloproteinase 13 modulates intestinal epithelial barrier integrity in inflammatory diseases by activating TNF. EMBO Mol. Med. 5, 1000–1016. doi: 10.1002/emmm.201202100
Wiest, R., Albillos, A., Trauner, M., Bajaj, J. S., and Jalan, R. (2017). Targeting the gut-liver axis in liver disease. J. Hepatol. 67, 1084–1103. doi: 10.1016/j.jhep.2017.05.007
Yao, G.-X., Shen, Z.-Y., Xue, X.-B., and Yang, Z. (2006). Intestinal permeability in rats with CCl4-induced portal hypertension. World J. Gastroenterol. 12, 479–481. doi: 10.3748/wjg.v12.i3.479
Keywords: liver fibrosis, ulcerative colitis, intestinal homeostasis, hepatic stellate cells, intestinal tight junction, TLR4 signaling, gut-liver axis
Citation: Liu Y-F, Niu G-C, Li C-Y, Guo J-B, Song J, Li H and Zhang X-L (2021) Mechanism of Ulcerative Colitis-Aggravated Liver Fibrosis: The Activation of Hepatic Stellate Cells and TLR4 Signaling Through Gut-Liver Axis. Front. Physiol. 12:695019. doi: 10.3389/fphys.2021.695019
Received: 14 April 2021; Accepted: 25 August 2021;
Published: 17 September 2021.
Edited by:
Kusum K. Kharbanda, University of Nebraska Medical Center, United StatesReviewed by:
JinGui Li, Yangzhou University, ChinaKarsten Gülow, University Medical Center Regensburg, Germany
Copyright © 2021 Liu, Niu, Li, Guo, Song, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-Lan Zhang, eGlhb2xhbnpoQDEyNi5jb20=
†These authors have contributed equally to this work and share first authorship