 Jan Hofman1,2
Jan Hofman1,2 Petra Brenerova
Petra Brenerova Petra Borilova Linhartova
Petra Borilova Linhartova- 1RECETOX, Faculty of Science, Masaryk University, Brno, Czechia
- 2Department of Histology and Embryology, Faculty of Medicine, Masaryk University, Brno, Czechia
Although the understanding of the causes of infertility is the key to its successful treatment, recent studies have shown that as many as 50% of male-caused infertility cases are considered idiopathic. The microbial colonization of the male reproductive system was shown to be associated with reduced male reproductive fitness. Investigation of the seminal microbiome, however, remains challenging. This article aimed to improve this situation by creating the first comprehensive review of literature on the metagenomic methods (including the pre-analytical and analytical approaches) used in the research on human seminal bacteriome (total bacterial DNA in the matrix), published in 2018–2024. A total of 29 studies addressing the analysis of the human seminal bacteriome were identified. The analysis typically involved DNA extraction from the supernatant using commercial kits, amplification of the gene for 16S rRNA, and sequencing of amplicons. Where the separation of seminal plasma was performed, centrifugation was the dominant method used for this purpose. The significant heterogeneity in individual steps of methodological approaches in the analysis of the human seminal bacteriome complicates the comparison of results among studies and the establishment of standard procedures, hindering clinical advancements. For this reason, a protocol for the analysis of the human seminal plasma bacteriome is proposed here, which could lead to improved comparability of results among studies and make future research more efficient. This protocol is founded on rigorous quality control measures, compliance with the WHO laboratory manual for sample collection, extensive pretreatment involving mechanical and enzymatic lysis, DNA extraction using the QIAamp DNA Mini Kit (Qiagen), and short-read sequencing conducted on the MiSeq platform (Illumina).
1 Introduction
Since the early 1950s, a noticeable decline in fertility rates across Europe has been observed, irrespective of cultural or social contexts (1). A surge in research focused on reproductive health has followed, intending to shed light on this phenomenon. Recent meta-analyses have reinforced the hypothesis that the seminal microbiota plays a crucial role in reproductive health (2). As a part of this effort, the reproductive microbiome, particularly the bacteriome, i.e., the total bacterial genetic information in a matrix (in this case, seminal plasma) at a given time, has emerged as a possible culprit of the decline. Infections of the urogenital tract directly associated with its microbial colonization are responsible for approximately 15% of male infertility cases (3–6). However, this number should not be regarded as definitive, as research (in particular) on asymptomatic infections, remains limited, and such infections are currently regarded as having minimal clinical significance (7, 8). On the other hand, certain bacteria have been associated with various mechanisms that negatively impact reproductive capacity. These mechanisms include the direct adhesion of bacteria to spermatozoa (9–11), effects mediated by their products (12), or adverse outcomes resulting from the increased presence of leucocytes (13–16) despite the absence of clinical manifestations. An improved understanding of the interactions between the seminal fluid and spermatozoa could lead to better identification of infections and, in effect, the possibility of successful treatment. In general, symptoms tend to increase with increasing bacterial abundance (13, 17–19). Conversely, reduced diversity of seminal bacteriome is generally considered a sign of dysbiosis (20). The interest in this field has increased over the last decade, with the current experimental studies primarily focusing on the correlation between sperm quality and bacteriome diversity and composition (21–44).
With this increasing intensity of research on microbial colonization of the male reproductive tract, there is an urgent need to evaluate and standardize methodological approaches to enhance the accuracy of microbiome analyses. The advent of next-generation sequencing (NGS) pushed traditional microbiological examination methods using culture media and biochemical tests into the background for research purposes, although they still hold an important place in clinical practice. Nevertheless, Mardanov et al. reported that culture methods can detect only approximately 1% of all microbial taxa (45). This, in conjunction with the problematic culture of anaerobic bacteria (46) that are ever-present in seminal bacteriome (17, 22, 34), makes these methods unsuitable for the analysis of bacteriome in a matrix as complex as seminal fluid. Therefore, the authors of this review decided to focus on culture-independent methods used for bacteriome research. Presently, 16S rRNA amplicon sequencing is the predominant method employed in the study of the bacteriome due to a combination of analytical precision and cost-effectiveness. Such analysis of the human seminal bacteriome can be supplemented or substituted with targeted analyses of bacterial DNA, paving the way for clinical applications in diagnosing reproductive tract diseases and predicting male fertility.
This review article aims to (i) summarize the pre-analytical and analytical approaches used in the metagenomic analyses of the human seminal bacteriome, (ii) critically evaluate their pros and cons, and (iii) propose a workflow for the metagenomic analyses of seminal bacteriome reflecting the current state of the art, which could be used for future experimental studies. All of this should contribute to more accurate research on the seminal bacteriome in male reproductive health (while, of course, leaving space for individual researchers to adjust the workflow to their needs). This would increase the legitimacy of the studies performed in this field (47) as well as the comparability of results for future meta-analyses.
2 Literature research
The PubMed database was searched using keywords “(seminal OR sperm OR ejaculate) AND (microbiome OR microbiotic OR microbiotix OR microbiota OR microbiomes OR bacterium OR microbiology OR bacterial) OR (bacteriospermia)”, with additional criteria for article selection as follows: (i) only original articles, (ii) published from 2018 onwards to ensure the up-to-date metagenomic methods and knowledge in the dynamic field of molecular biology, and (iii) conducted on human subjects. Subsequently, titles and abstracts were screened to identify relevant articles and only relevant studies that describe the metagenomic analysis of the complex bacteriome (not just the analysis of individual clinically significant species) were used as information sources. This methodological algorithm is illustrated in Figure 1.
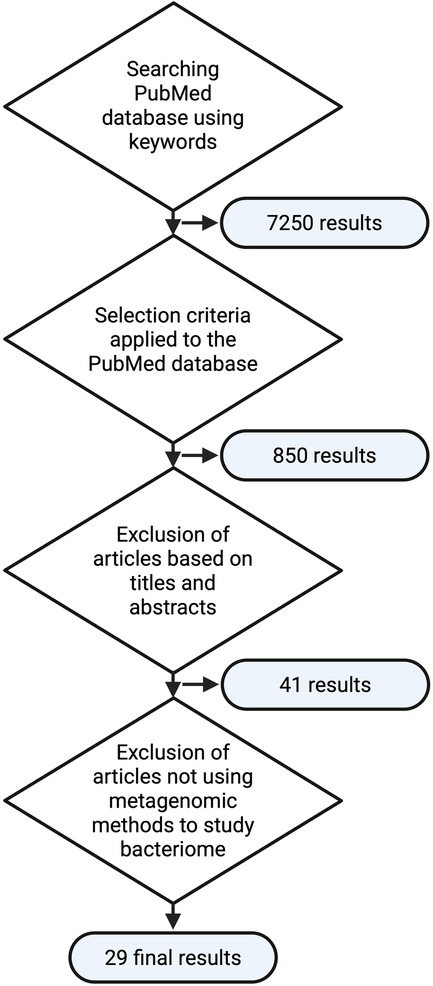
Figure 1. Flowchart of the methodological approach used in the literature review (created with bioRender.com).
3 Pre-analytical and analytical approaches used in the metagenomic analyses of the human seminal bacteriome
The initial literature search yielded 850 results. After scanning the titles and abstracts, 41 relevant original articles (13, 14, 19, 21–44, 46, 48–60) listed in the PubMed database and published since 2018 were identified. Twelve of these studies (13, 14, 19, 48–50, 52–57), however, did not analyze the microbiome but used microbiological and analytical methods to determine cultured bacteria; these studies with an alternative approach using inoculation of the sample on bacteriological media and analysis of the resulting colonies [mass spectrometry MALDI-TOF (48–50), VITEK II (13, 14, 19, 52, 54), or microscopic analysis (13, 14, 19, 48, 50, 53–57)] were excluded from further analysis, yielding a final set of 29 studies. The predominant focus of these studies was to explore the relationship between the seminal bacteriome and various health conditions, particularly infertility (21–25, 27–31, 33–39, 41–44, 46, 58, 59), prostatitis (40, 42), vasectomy (51), or cancerous diseases (26, 32). One study focused on the forensic evaluation of ejaculate (60). The sample sizes in these studies varied greatly, ranging from five to 285 volunteers/patients. The median size of the study sample was 56 participants.
3.1 Pre-analytical phase—collection of ejaculate sample, its processing, and storage
Out of the final set of 29 studies, the samples were collected through ejaculation into a sterile container in 27 studies. Depending on the design, this was in some studies supplemented by testicular sperm extraction (TESE) (21, 26, 27), percutaneous epididymal sperm aspiration (PESA) (21), or micro epididymal sperm aspiration (MESA) (42). These modified collection methods were used due to obstructive azoospermia or the need to collect a sample from a specific part of the male reproductive tract.
Sample collection through ejaculation into a sterile container was performed, with few exceptions, according to the principles recommended by the World Health Organization (WHO) (61), which entails (a) minimizing temperature fluctuation and time before and during analysis; (b) 2–7 days of sexual abstinence before the collection (maintaining a constant interindividual duration); (c) urination, washing the glans penis and hands with soap, rinsing everything well and drying before the actual collection; (d) collecting the entire ejaculate fraction into a sterile plastic or glass container tempered to 20–37°C; (e) processing the samples within three hours at room temperature; (f) placing the samples into an environment with a temperature of 37°C as soon as possible for precise evaluation of the liquefaction time; and (g) exclusive use of sterile laboratory material, and classification of all samples as biohazard.
Immediately after collection, the liquefaction of the ejaculate (i.e., the rate of proteolytic processes transforming a gel-like ejaculate into a watery one) at 37°C was usually evaluated. The evaluation of liquefaction must be completed within one hour of collection (61), and processing of the ejaculate sample itself immediately follows (within three hours at room temperature). Alternatively, the ejaculate samples were stored using cryopreservation methods—usually at −80°C (21, 27, 28, 38, 44), alternatively at −20°C (33, 46); in one study, samples were frozen at −20°C without any cryopreservative and then transferred to storage at −196°C (34).
Processing of the ejaculate includes separation of seminal plasma by centrifugation (22, 32, 38, 46), which partially removes spermatozoa, and, thus, human genetic information, from the sample. This step is vital in in vitro fertilization procedures; from this perspective, including this step is highly suitable if aiming to examine the microbiome directly associated with these procedures. The process takes place at room temperature. Unfortunately, centrifugation parameters were mentioned in three studies only and the protocol for separating spermatozoa from seminal plasma was not specified in any of them. Where centrifugation parameters were provided, the relative centrifugal forces were 800 g for 15 minutes with repeated centrifugation of the supernatant at 10,000 g for 10 minutes (32), and 7,000 g for 10 minutes (22). Gradient centrifugation was used in two studies (38, 46). Samples of seminal plasma for subsequent extraction of microbial DNA were stored at −80°C (22, 32) or were immediately used in the analytical phase.
Centrifugation was in two instances supplemented with the addition of dithiothreitol (DTT; 43 mM) as a reducing agent (23, 33). The purpose of DTT is to disrupt disulfide bridges in the protein structure, thus lysing the nuclear membrane of incompletely removed spermatozoa. This leads to the release of human DNA into the sample; it is, however, necessary to emphasize that unless whole-genome sequencing is used as the subsequent analysis, this poses no problem for bacterial identification using other methods (such as 16S rRNA PCR).
3.2 Analytical phase—extraction of microbial DNA from human seminal plasma
In all included bacteriome studies, the analysis was based on evaluating bacterial DNA (21–44, 46, 51, 58–60). In 22 cases, a commercial kit based on the spin column principle (21–28, 30, 31, 34, 35, 37–42, 46, 58–60) was used. Alternative solutions included the phenol-chloroform extraction (36, 43, 44), the Trizol LS reagent protocol (Invitrogen, USA) (32), a protocol utilizing cetrimonium bromide (CTAB) (29), and the NucliSENS easyMAG system (BioMèrieux, USA) (33) working on the principle of binding magnetic silica particles to the nucleic acid and their subsequent separation in the magnetic field.
The extraction procedures for obtaining microbial DNA from seminal plasma can be modified or combined. To achieve better cell lysis prior to the DNA extraction, pre-treatment using proteinase K with phosphate buffer and/or bead beating methods using glass, ceramic, or metal particles was utilized in eleven studies (21, 23, 25, 27–30, 34, 39, 41, 59).
The QIAamp DNA mini kit (Qiagen, USA/Germany/Switzerland) was the most commonly used one (22, 23, 35, 46). The manufacturer's instructions were typically adhered to, except for one study where (due to the low total amount of bacterial DNA in the sample), the amounts of reagents were changed while maintaining ratios (27). In another study, an additional step of washing during centrifugation (8,000 g; 1 min) prior to the use of the ZymoBIOMICS DNA Microprep kit (Zymo Research, USA) was added (59). The CTAB method was used in one study (29), the Trizol LS Reagent protocol (Invitrogen, USA) for the extraction of small RNA in another (32).
In several studies, Tris buffer and proteinase K were used for sample pre-treatment before phenol-chloroform extraction (36, 43, 44). In one study (32), DNA extraction was preceded by sample homogenization and cell lysis using the TissueLyser LT (Qiagen) homogenizer with the addition of 400 μl of Tris buffer.
Besides the aforementioned methods, in one case, the NucliSENS® easyMAG® (Biomerieux, USA) system was used according to the manufacturer's instructions (33).
3.3 Analytical phase—analysis of DNA extracted from human seminal plasma
The methods of bacteriome analysis used in the analyzed studies differed in their ability to determine taxonomic units. In four studies, PCR served for qualitative and/or quantitative determination of bacterial DNA. These included single-target qPCR (24, 25, 46) and multiplex qPCR (25, 31). Multiplex qPCR was used in studies targeting the analysis of specific difficult-to-culture bacteria, especially from the Mycoplasmataceae family, as well as Gardenella vaginalis, Sneathia sanguinegens, Bacteroides fragilis, and Megasphaera (25, 31). Amplification of the gene for 16S rRNA followed by NGS was the most common method (23 studies) (22, 23, 27–30, 32–41, 43, 44, 46, 51, 58–60). In some cases, it was supplemented with real-time (RT) qPCR (30, 33). In one study, these PCR products were analyzed using denaturing high-performance liquid chromatography (DHPLC) (42).
When amplifying the gene for 16S rRNA for subsequent sequencing (22, 23, 27–30, 32–41, 43, 44, 46, 51, 58–60), conditions were set depending on the chosen primers and master mix requirements. The choice of variable regions for PCR amplification itself presents a source of a certain variability among studies. Predominantly, PCRs focused on the V3–V4 (27, 28, 35, 37, 38, 40, 60) and V1–V9 regions (34, 59), although some researchers focused on the V3 (33), V4 (21, 30), V6 (58), V3–V6 (22), V3-V5 (26), V1-V3 (41), V1–V2 (23, 29, 39, 51), V2–V3 (46), or V4–V6 (44) regions. Nested PCR was used in two studies to reduce non-specific primer binding to DNA sequences outside the observed segments (26, 33). In both instances, the DNA was amplified to be sequenced [pyrosequencing and sequencing on the Ion Torrent PGM platform (Life Technologies, USA)]. One of these studies employed the V3–5 region for amplification (26), the other used just the V3 area (33).
Before the actual analysis, an additional step of amplicon purification was mentioned in 15 studies. AMPure XP beads (Beckman Coulter, Italy/USA) in one (23, 27, 32, 37–39, 44, 60) or two (26) cycles were the most commonly used of these methods. Other techniques included AxyPrep DNA Gel extraction kit (Axygen) (35), QIAquick column (Qiagen, USA) (51), NucleoMag NGS Clean-up (Macherey-Nagel, France) (41), and, in two cases, Zymo-Spin IC column (Zymo Research, USA) (34, 59).
Sequencing techniques play a dominant role in seminal microbiome analysis. Illumina products are the most commonly used for this purpose, including platforms such as the MiSeq system (fourteen studies) (23, 27, 28, 30, 34–36, 38–41, 44, 51, 60), HiSeq 2000 (two studies) (21, 58), HiSeq 2500 (one study) (29), HiSeq 4000 (one study) (32), and NovaSeq 6000 (Illumina Co., USA) (one study) (37). Besides Illumina products, sequencing using MinION (Oxford Nanopore Technologies) was employed as an alternative in one case (59). Pyrosequencing was applied in two studies, using the platforms Roche 454-GS Junior (Roche, USA) (26) and Roche 454 FLX (Roche, USA) (46). The Ion Torrent PGM (Thermo Fisher Scientific, USA) procedure was employed in two studies (22, 33).
Various library preparation kits were used to prepare libraries, including the NGSgo kit (GenDx, Netherlands) (34), CATS small RNA Quick-16S NGS (Diagenode, Belgium) (32), Quick-16S NGS library prep kit (Zymo Research, USA) (36), Ion Plus Fragment Ion PGM Hi-Q (Thermo Fisher Scientific, USA) (33), or Nextera XT (Illumina, USA) (27, 28, 41). Five studies used, in addition to the above, a control sequencing library, specifically PhiX (Illumina) (23, 32, 38, 41, 44). The final analysis of outputs also varied according to the respective bioinformatics pipelines.
Based on the literature review, the general workflow for analyses of seminal bacteriome is shown in Figure 2, and methods used in each study are highlighted in Supplementary Table 1 within Supplementary 1.
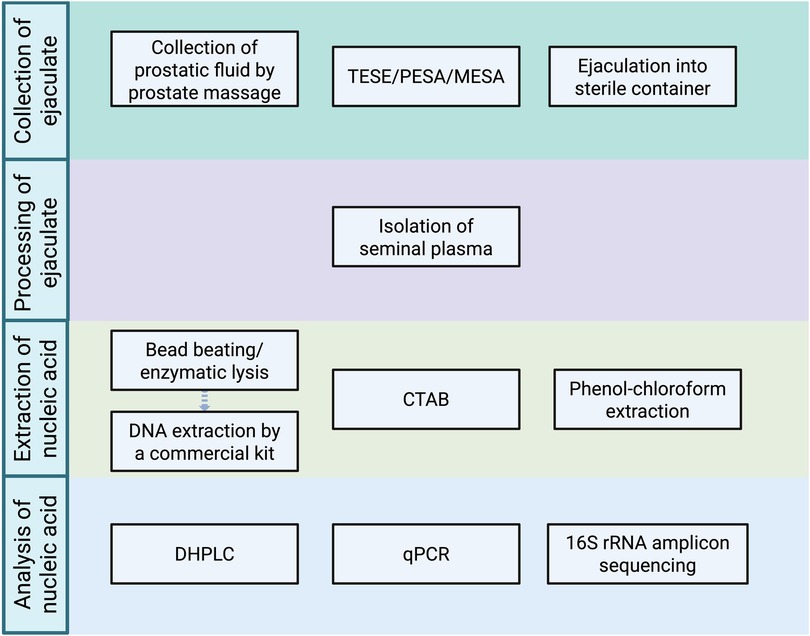
Figure 2. Methods previously used in individual steps of the analysis of human seminal bacteriome (created with bioRender.com). (TESE, testicular sperm extraction; PESA, percutaneous epididymal sperm aspiration; MESA, microsurgical epididymal sperm aspiration; CTAB, cetrimonium bromide method; qPCR, quantitative polymerase chain reaction; DHPLC, denaturing high-performance liquid chromatography.).
4 Critical evaluation of the approaches used in the metagenomic analyses of the human seminal bacteriome
4.1 The WHO manual on semen collection and its application in studying the seminal microbiome
The WHO laboratory manual for the examination and processing of human semen has become the gold standard in the field (61). However, it is necessary to keep in mind that the manual primarily focuses on sperm quality assessment and processing protocols, not on the analysis of seminal plasma. In the reviewed studies, sample collection was consistently executed through ejaculation into a sterile container, adhering to WHO guidelines within the environments of reproductive health clinics.
WHO guidelines recommend the start of the microbial analysis or freezing of the samples within 3 hours of collection (61). This seems reasonable as the collection of samples usually happens in external infrastructures such as in vitro fertilization (IVF) clinics and adhering to WHO recommendations generally seems to be a good way of ensuring standardization across studies. This, of course, does not preclude researchers from more rapid processing, which could be an even better option if feasible. However, studies examining bacteriome change in skin swabs and stool samples have not found any statistically significant change even after 24 hours (62, 63). The only study focusing on the temporal change in seminal samples focused on forensic analysis, considering a much longer time frame (weeks) (64). Still, as a precaution against the introduction of error, it is reasonable to keep the time from sampling to analysis/storage constant among samples within a single study. Similarly, collecting samples at the same time of day could also be recommended to account for any possible intrapersonal diurnal changes in the microbiome composition (although no studies have examined this in seminal samples so far). Lastly, repeated thawing and re-freezing of samples is not recommended (65).
The length of sexual abstinence before collection presents another aspect of data collection. WHO recommends 2–7 days of sexual abstinence prior to sampling (61). Shorter abstinence has been associated with decreasing bacterial abundance and diversity (48). Moreover, other factors play a role in changing the bacteriome composition with abstinence (48). Because of this, it is reasonable to keep the abstinence period preceding the sample collection as consistent as possible to ensure the same time for bacterial growth. This being said, the period should fall within the WHO-recommended time frame to support comparison among studies and to reflect the general clinical practice at IVF clinics. If choosing between shorter and longer abstinence within this timeframe, the longer time might be beneficial by leading to higher bacterial abundance, which could be advantageous considering that the ejaculate is a low-abundant matrix.
In the pre-analytical phase, the separation of the seminal plasma and microbial DNA from spermatozoa is an optional approach for minimizing the load of human DNA. However, the WHO guidelines lack a detailed methodological workflow for processing seminal plasma samples for microbiome analysis; to make things worse, most studies fail to specify their processing techniques. Various methods, such as magnet-activated cell sorting (MACS), discontinuous density gradient, or direct swim-up, are available for separating motile and morphologically normal spermatozoa (61). Nonetheless, these methods can significantly reduce sperm yield (66), rendering them unsuitable for simultaneous spermatozoa and microbiome analyses. Furthermore, any such manipulation of the sample may alter the bacterial composition. Reports of spermiogram analyses conducted according to WHO principles are not common in the included studies, despite the adherence to the general guideline. The analytical methods employed for the microbiome studies varied considerably compared to the more standardized sample collection procedures.
Samples obtained via TESE, PESA, and MESA methods possess unique characteristics that must be acknowledged during processing. For instance, the prostatic fluid´s role in ejaculate liquefaction is significant (67). If any ejaculate fraction is absent, the workflow needs to be adjusted to account for the physiological properties of the matrix. TESE, PESA, and MESA sampling methods are primarily suitable for microbiome analysis from specific regions of the male reproductive tract or in cases of obstructive azoospermia precluding traditional collection methods (42).
4.2 How to process an ejaculate sample and obtain seminal plasma for microbiome analysis?
Immediately after collection, the ejaculate sample must be liquefied to allow the extraction of the microbial genetic material. Liquefaction is a process facilitated by prostatic serine proteases capable of breaking down the (predominantly) fibrin matrix present in the ejaculate. The duration of liquefaction is a critical diagnostic criterion linked to spermiogram quality, with certain microbial taxa potentially influencing the speed and quality of this process (68). Maintaining the optimal conditions for liquefaction, particularly the temperature of 37°C immediately after collection, is essential. The liquefaction typically occurs within 15–30 minutes; failure of its completion within 60 minutes may indicate a pathological condition (61). Orbital movement (e.g., using an orbital mixer) can make the process more efficient.
To ensure representative sampling, the entire volume of the sample should be homogenized before further processing. This can be achieved through 15–30 s of swirling movement (manually or with an orbital mixer). Working with ejaculate can pose ethical complexities compared to working with seminal plasma alone, necessitating the removal of human DNA prior to nucleic acid extraction, although lysed spermatozoa do not interfere with the PCR amplification of the gene for 16S rRNA (21, 23–30, 33–44, 46, 51, 58–60). Centrifugation methods are commonly employed for this purpose, but there is considerable variability in parameters across studies, particularly regarding the relative centrifugal force (RCF). Temperature conditions are usually not described, with room temperature being the standard when mentioned.
Two primary centrifugation approaches are recognized: gradient centrifugation and simple centrifugation. However, only two reviewed studies reported the use of a density gradient (38, 46). Gradient centrifugation is more complex and costly; however, if using parameters according to the specific parameters outlined in the WHO manual (200–400 g for 15–20 minutes using 40% and 80% gradients) (61), a lower fraction of spermatozoa is separated from seminal plasma (note the different purpose of the WHO guideline). This, on the one hand, better prevents undesirable separation of the microbiota but, on the other, does not reliably remove spermatozoa and human DNA from the sample. However, as mentioned above, this does not necessarily pose a problem as from the perspective of 16S rRNA amplification, no separation of spermatozoa from the seminal plasma is necessary. Hence, if aiming to analyze the ejaculate itself, none of the centrifugation methods described in the literature can be recommended as the risk-to-benefit ratio is unreasonable. The risk of removing an unknown amount of bacteria with unknown composition is too high; moreover, the elimination of sperm cells is ineffective, leaving a significant amount of sperm cells as well as of free human DNA in the supernatant (69), which prevents meaningful metagenomic analysis anyway.
If the methodology (such as the utilization of a metagenomic approach) or external factors (such as ethical concerns) demand the removal of spermatozoa, it would be probably better to resort to methods that have been successfully used in other matrices, despite the fact that, with one exception, they have not yet been tested on ejaculate samples (which, in any case, calls for performing such methodological studies). Even there, however, it is necessary to be careful regarding the choice of the separation method. Filtration utilized, for example, for the depletion of epithelial cells from saliva (70) might be suboptimal as it cannot remove free DNA (69). Selective depletion of host DNA in seminal samples might be a more suitable approach. However, this approach has been sparsely tested on ejaculate samples. This depletion can be achieved by selective lysis of eukaryotic cell wall using reagents such as saponin, Triton X-100, or Tween 20 (71–73), followed by the degradation of free-floating DNA using either Dnase I (74, 75) or benzonase nuclease that has greater toleration to working conditions (69). The intricacy of this approach, however, lies in the composition of the sperm cell wall. Spermatozoa have a specific cell wall structure containing a higher percentage of polyunsaturated fatty acids (76), which makes the success of the lysis likely; so far, nevertheless, it has been verified in just a single study using the QIAmp DNA Microbiome kit (Qiagen, USA) (28). Moreover, it reportedly decreases bacterial abundance in low-abundant samples (69) and can be only applied on fresh samples (not frozen as freezing could cause the disruption of the bacterial cell wall as well).
Enzymatic depletion of eukaryotic DNA based on specific enzymes targeting methylated CpG regions after the DNA release from a cell is another (at least theoretically) possible option (77). The pitfall of this method, however, might lie in the specific way human DNA is stored within the spermatozoa. This DNA not only possesses characteristic methylome (78) but is also stored in a protamin-bound structure, which could complicate the process. However, no commercially available kit has been so far tested for this application, which, again, calls for methodological research in this area. Finally, the utilization of Oxford Nanopore MinION and Crisp-Cas9 technology represent promising concepts for the future; they need to be validated before use (69).
4.3 DNA extraction from human seminal plasma
The extraction of microbial nucleic acid from the sample is an essential step in the analysis of the human seminal microbiome. PCR-based amplification techniques surpass the culture-based techniques in allowing the detection of non-culturable microorganisms, thus providing a more comprehensive view of the microbial landscape within the seminal fluid.
The efficacy of microbial DNA extraction is influenced by several factors, including sample quality, microorganism lysis (often supplemented with bead beating), and the purification of genetic material from proteins and other substances.
The lysis of bacterial cells was suggested to be a more important step for maximizing the DNA yield than recovery steps (79–82). Utilizing enzymatic (83, 84) and/or mechanical lysis (81, 85, 86) was reported to yield higher amounts of bacterial DNA from the sample compared to no pretreatment. The combination of the aforementioned procedures is reported to slightly reduce the abundance of bacterial DNA; on the other hand, it improves the representativeness (and, therefore, the accuracy of subsequent determination) of microbiota in oral (83, 87) and gut (84) samples. The increase in bacterial DNA abundance after the use of enzymatic lysis is likely because of the ability of lysozymes to disrupt peptidoglycans of Gram-positive bacteria; on the other hand, the slight decrease in DNA abundance after the mechanical lysis is probably associated with a loss of a part of the sample during the procedure (83). A combination of several lysis enzymes (lysozyme, mutanolysin, staphilysin) seems to perform even better than the use of lysozyme alone (84). The use of the correct size of beads for mechanical lysis is also an important factor, as a size too big can reduce the amount of extracted DNA (84). It needs to be, however, mentioned that no methodological study directly comparing the use of multiple pretreatment techniques in ejaculate samples has been published so far.
The extraction of microbial DNA itself is typically performed using commercial kits, in some studies supplemented with a reducing agent (e.g., DTT) (23, 33) for better protein denaturation and removal. The selection of commercial DNA extraction kits is often guided by factors such as ease of use, reproducibility, and compatibility with downstream applications. Due to these advantages, commercial kits have become a preferred method for investigating the seminal bacteriome. However, the suitability of a specific kit depends on several factors, particularly the intended method of the extracted DNA evaluation. As discussed previously, kits that facilitate both the depletion of host (human) DNA and the extraction of high-molecular-weight DNA are advantageous for metagenomic applications. Gant et al. evaluated six commercial kits for their performance in host DNA depletion and suitability for long-read sequencing using the Oxford Nanopore MinION platform. The kits included the NucleoSpin Food Kit, Quick-DNA HMW MagBead Kit, ZymoBIOMICS DNA Miniprep Kit, QIAamp PowerFecal Pro Kit, Moss protocol, and DNAexpress Kit. Among these, the Quick-DNA HMW MagBead Kit was identified as the most suitable for this application (88). Notably, according to the manufacturer, this kit is compatible with a variety of biological fluids, further supporting its versatility for microbiome research. In another comparative study, Wright et al. evaluated the performance of several commercial DNA extraction kits—Qiagen DNeasy PowerSoil Pro Kit, HostZERO Microbial DNA Kit, PureLink Microbiome Kit, and Qiagen DNeasy Blood and Tissue Kit—using vaginal samples as the test matrix (89). Their findings revealed that all tested kits introduced some level of bias in microbial community profiles. However, the HostZERO Microbial DNA Kit was identified as the least suitable for vaginal samples due to its pronounced impact on microbial composition.
No studies comparing the performance of multiple kits for extracting DNA for microbiome profiling using short-read 16S rRNA amplicon sequencing in ejaculate samples have been published so far. However, cautious extrapolation from studies on vaginal and cervical microbiome may provide some information as sexual partners were shown to share approximately 56% of their genital microbiota (30), including many clinically relevant pathogens. In vaginal microbiome studies, the DNeasy Blood and Tissue Kit has been reported to yield a higher DNA quantity compared to the MoBio PowerSoil Kit, although the latter provided greater microbial diversity in the resulting data (90). This highlights a trade-off between DNA yield and community diversity that must be considered when selecting a kit for specific research objectives. Shibata et al. conducted a comparative analysis of several DNA isolation kits on cervical microbiota samples, including the ZymoBIOMICS DNA Miniprep Kit, QIAamp PowerFecal Pro DNA Kit, QIAamp DNA Mini Kit, and the IndiSpin Pathogen Kit (91). Their findings indicated that all tested kits produced comparable results in terms of microbiome composition, suggesting a degree of interchangeability for studies involving cervical samples. In the context of low-biomass samples from the urogenital tract, such as urine, multiple studies have identified the DNeasy Blood and Tissue Kit to be the most suitable option for DNA extraction. Its effectiveness in such matrices has been consistently demonstrated (92, 93), further supporting its potential application in studies of the seminal microbiome.
When considering DNA extraction kits for long-read sequencing of the seminal microbiome, products from Zymo Research appear to be particularly suitable. On a sample consisting of Gram-positive Bacillus subtilis and Gram-negative Escherichia coli, the Quick-DNA HMW MagBead Kit was reported to be the most effective among several compared in a recent benchmarking study (88). In ejaculate samples, the ZymoBIOMICS DNA Miniprep Kit has been recently successfully employed in the analysis of ejaculate samples (59).
To date, the QIAamp DNA Mini Kit has been the most commonly used extraction method for short-read 16S rRNA amplicon sequencing in seminal microbiome studies (22, 23, 35, 46). When comparing 16S rRNA amplicon sequencing in ejaculate and vaginal samples, it must be noted that ejaculate typically exhibits lower bacterial load but higher diversity than vaginal samples (30, 58, 94), which complicates a direct comparison of the results of different extraction methods. This makes it unclear whether higher-yielding kits, such as the DNeasy Blood and Tissue Kit, or a diversity-optimizing kit, such as the MoBio PowerSoil Kit, would be more appropriate for ejaculate samples. Moreover, the ejaculate microbiome shares significant bacterial overlap with the urinary microbiome (17), for which the DNeasy Blood and Tissue Kit has been repeatedly recommended, making this kit another promising candidate for seminal microbiome studies. At present, however, due to the limited number of comparative studies directly evaluating extraction methods in ejaculate samples, it is not yet possible to make definitive recommendations. However, based on current literature, the QIAamp DNA Mini Kit is the most commonly used kit and, therefore, can be considered a reasonable standard for ongoing research. Still, continued investigation is critically needed to determine the optimal extraction protocol for ejaculate samples, which would ultimately contribute to methodological standardization and improved reproducibility in the field.
4.4 Sequencing analyses of human seminal bacteriome
The conditions of PCR done on the microbial DNA preceding sequenation need to be chosen depending on the specific master mix and primers. The advent of long-read sequencing technologies has opened new possibilities for studying bacterial communities by allowing the amplification and analysis of the full-length gene for 16S rRNA, rather than focusing solely on its hypervariable regions. This approach enhances taxonomic resolution, particularly at the species level, while maintaining comparable accuracy at higher taxonomic levels such as genus and family (95–99). However, long-read sequencing remains technically demanding and requires the use of third-generation sequencing platforms such as PacBio (95) or Oxford Nanopore Technologies’ MinION (96, 100). To date, the number of studies investigating the use of sequencing platforms on seminal microbiome is severely limited. A comparison of the Illumina MiSeq and the ONT MinION platform reported no significant differences in the identification of abundant taxa between platforms (34, 59). However, differences in low-abundant taxa were observed, likely due to the disproportionate impact of sequencing errors on taxa with low relative abundance. Notably, both studies detected distinct bacterial community structures at the genus level, which may reflect sequencing biases intrinsic to each platform (101, 102). Still, looking forward, long-read sequencing appears to be a promising direction for seminal microbiome research.
Nevertheless, in short-read sequencing workflows, the choice of hypervariable region for PCR amplification remains an important consideration. Currently, no consensus exists on the optimal region, and different regions offer varying degrees of taxonomic resolution and bacterial coverage (103–107). Moreover, there are no comparative data specifically evaluating hypervariable regions in the context of ejaculate samples. When using short-read sequencing, it is well-established that no single hypervariable region can capture the full diversity of bacterial taxa (83). It was suggested that a correct combination of hypervariable regions could improve final results (108); however, multiple studies indicate that the impact of hypervariable region selection is generally less significant than that of DNA extraction methods (83, 109). Furthermore, the optimal region may vary depending on the biological sample (83, 104, 109–111).
As there are no studies directly addressing the performance of individual hypervariable regions in ejaculate samples, cautious extrapolation from vaginal microbiome research can help here as mentioned above. Even though the V1–V3 (112, 113) regions are the most commonly used in vaginal microbiome studies, the V3–V4 regions were shown to perform better than V1–V2 regions (112, 113).
Unlike in vaginal samples, the V3–V4 regions are currently the most frequently employed in ejaculate microbiome studies. Importantly, none of these studies reported issues regarding the resolution power of the V3–V4 region in practice. Given its consistent performance, widespread use, and supportive findings from related vaginal microbiome studies, it can be concluded that the V3–V4 region appears to be a suitable target for bacterial profiling of ejaculate samples. Nevertheless, long-read sequencing technologies offer superior taxonomic resolution and represent a promising advancement for future seminal microbiome research.
The analysis of microbial DNA is typically conducted following standardized recommendations by the manufacturers of the sequencing tools. The dominance of Illumina (used in more than 75% of studies performing sequencing analyses of seminal bacteriome) underscores the status of that company as the leader in (not only) seminal microbiome analysis.
Based on current evidence, the continued use of Illumina MiSeq for seminal microbiome studies can be recommended due to its established performance, broad adoption, and availability of validated protocols. Looking ahead, a gradual shift toward third-generation sequencing platforms can be anticipated, driven by improving error rates, enhanced protocols, and the superior resolution they offer. Nevertheless, it is true that platform selection is often determined not only by technical performance but also by availability, cost, and institutional bioinformatic support. Importantly, with the exception of two seminal microbiome studies, most comparative data cited here are derived from other body site microbiomes, highlighting the need for further research specifically targeting the seminal microbiome to optimize platform selection.
4.5 Quality control
Contamination remains a persistent concern in microbiome research, particularly in studies involving low-biomass samples, where the relative contribution of contaminants increases as the endogenous microbial signal decreases (114–116). Alarmingly, contaminants are frequently detected within laboratory reagents, especially DNA extraction kits, which can introduce exogenous bacterial DNA and compromise data integrity (115). Additional major sources of contamination include well-to-well cross-contamination during sample processing (117) and environmental contamination originating from laboratory surfaces and the surrounding air (115).
To mitigate contamination and improve laboratory reliability, several best practices have been established. These include regular environmental (surface wipe) testing, routine monitoring of sample positivity rates, incorporation of process controls, tracking of laboratory issues or complaints, and ensuring sufficient personnel training. The physical separation of workstations based on the workflow stage is another practice recommended to prevent cross-contamination (118). Additionally, randomization of sample placement on plates has been shown to reduce well-to-well contamination (117). Strict sterilization protocols and the use of laminar flow hoods or biosafety cabinets during sample handling should be considered essential components of contamination prevention, particularly in 16S rRNA amplicon sequencing workflows.
For each experiment, the use of negative and positive controls plays a crucial role in ensuring valid interpretation of results and prevention of erroneous conclusions (116, 119). Negative controls help identify environmental contamination, while positive controls or an internal standard ensure that a negative result is not due to procedural errors. These controls were widely utilized in the studies, both during microbial DNA extraction (23, 25–28, 34, 37–39, 59, 60) and during PCR amplification (25, 35, 38, 39, 41, 43, 44). In several papers, authors even resorted to using alternative analytical methods to validate their results and to rule out the inadequacy of their primary techniques (25, 59). The incorporation of MOCK microbial communities (MOCKs) as in situ positive standards is a strategy widely adopted for contamination control, assessment of protocol-associated biases, and validation of bioinformatic pipelines (116, 120, 121). MOCKs, composed of known microbial taxa, provide a benchmark for evaluating experimental and analytical steps, allowing researchers to detect unexpected taxa indicative of contamination and to gauge the reliability of sequencing and taxonomic assignment.
Post-sequencing computational tools also play a crucial role in identifying and removing contaminant sequences. Tools such as Decontam use statistical models to distinguish true biological signals from contaminants based on the frequency and prevalence across sample types (116, 122, 123), GRIMER (124) offers interactive tools for identifying potential sources of contamination and visualizing microbial profiles, while SourceTracker employs Bayesian methods to trace the origin of microbial communities, aiding in the attribution of contamination to environmental or reagent sources (116). The significance of these controls cannot be overstated, and the fact that some studies failed to report them does not necessarily imply non-use.
At this point, it is crucial to emphasize the importance of performing analyses that would provide a head-to-head comparison of the approaches currently employed in studying the human seminal bacteriome. Such a comprehensive evaluation would enhance analytical accuracy as well as the reliability and comparability of findings across studies. These efforts can pave the way for more robust conclusions and foster a deeper understanding of the human seminal microbiota, ultimately contributing to advancements in both basic and applied microbiological research.
5 Recommended workflow for the metagenomic analysis of human seminal bacteriome
Standardization of protocols is a central theme highlighted throughout this work. Based on the considerations discussed, it is recommended that sample collection follows WHO guidelines, particularly in maintaining a consistent abstinence period that falls within the range of 2–7 days as recommended by WHO; within a single study, however, an even narrower window should be aimed for to minimize interindividual variability. It could be assumed that the duration of abstinence towards the upper limit of the WHO range may be beneficial as it might increase the absolute amount of bacteria in the ejaculate samples. Figure 3 shows the workflow of the most commonly utilized methodical approach for analyzing the human seminal bacteriome.
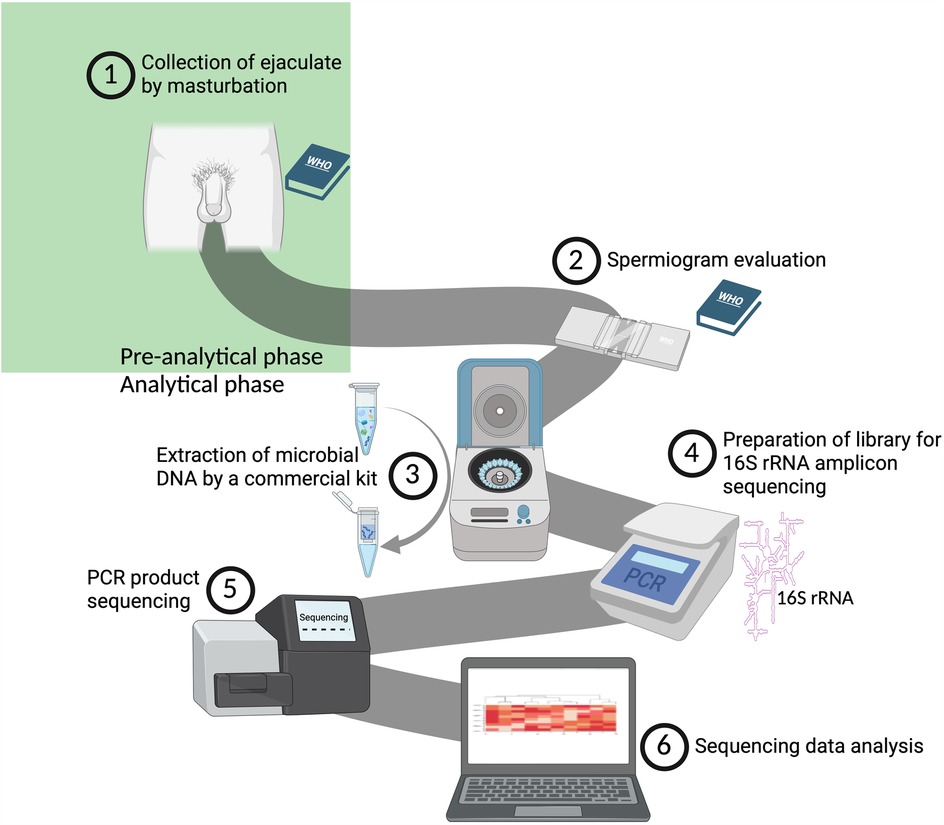
Figure 3. An illustration of our proposed workflow for analyzing the human seminal bacteriome (created with bioRender.com). (1) Collection of ejaculate following the guidelines outlined in the WHO Laboratory Manual for the Examination and Processing of Human Semen (61). (2) Assessment of ejaculate quality in adherence to the WHO Laboratory Manual (61). (3) Extraction of bacterial DNA using the QIAamp DNA Mini Kit (Qiagen). (4) Amplification of the gene for 16S rRNA, focusing on the V3-V4 variable region (27, 28, 35, 37, 38, 40, 60). (5) Sequencing of the amplified products on the MiSeq platform (Illumina, USA). (6) Bioinformatic and statistical analysis and interpretation of the raw data obtained.
Although third-generation sequencing platforms offer superior resolution and greater potential compared to second-generation technologies, their limited accessibility and adoption currently hinder their widespread use in standard workflows. The third-generation long-read sequencing should be, therefore, adopted in future studies as the technology matures and becomes more broadly available. For now, however, the authors of this review propose a standardized protocol based on short-read sequencing, which is more commonly used and supported by established workflows:
Samples should be processed within three hours of collection or stored at −80 °C to ensure sample integrity. Unless metagenomic analysis specifically requires otherwise, avoiding the separation of spermatozoa may be beneficial for microbiome analysis (especially when 16S rRNA is concerned, for which human DNA poses no interference) as partial removal of microbiota may occur during such separation. For sample pretreatment, a combined approach using bead-beating and enzymatic lysis is recommended, ideally utilizing a cocktail of lytic enzymes. DNA should then be extracted using the QIAamp DNA Mini Kit (Qiagen), which offers reliable performance in similar low-biomass samples.
For short-read sequencing, primers targeting the V3–V4 hypervariable regions of the gene for 16S rRNA appear to be the most suitable for seminal samples. Following library preparation, sequencing should be performed using the MiSeq platform (Illumina), as supported by current evidence.
To ensure data quality and monitor contamination, both positive and negative controls should be incorporated into the workflow, alongside the inclusion of a MOCK microbial community. When combined with strict adherence to good laboratory practices and, where possible, the application of appropriate bioinformatics tools, this approach should enable adequate contamination control and reproducibility in seminal microbiome studies.
6 Conclusion
The analysis of the human seminal bacteriome is an emerging field that has significant implications for understanding male fertility. Our review underscores the importance of methodological rigor in studying the seminal bacteriome, particularly through the amplification of the gene for 16S rRNA followed by sequencing, which has emerged as the standard approach.
However, a notable heterogeneity in the metagenomic approaches employed across studies calls for the standardization of protocols. This can facilitate performing meta-analyses combining individual studies, ultimately advancing our understanding of the role of the human seminal microbiota in fertility disorders and male reproductive health. To be able to create such standardized workflows, methodological studies comparing the performance of various protocols need to be performed. Such efforts will not only improve our understanding of the seminal microbiota but also pave the way for potential therapeutic interventions addressing male infertility.
As a standardized protocol for metagenomic analysis of human seminal bacteriome, we recommend that sample collection be performed in accordance with the WHO laboratory manual, followed by pretreatment combining mechanical disruption and enzymatic lysis. For DNA extraction, the QIAamp DNA Mini Kit (Qiagen) is advised. Library preparation should target the V3–V4 hypervariable regions of the gene for 16S rRNA, which, in our assessment, are best analyzed using the MiSeq sequencing platform (Illumina).
Author contributions
JH: Conceptualization, Visualization, Writing – original draft. PBr: Writing – review & editing. PBL: Supervision, Conceptualization, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
This publication was supported from the European Union’s Horizon 2020 Research and Innovation Programme under grant agreement No 857560. This publication reflects only the author's view, and the European Commission is not responsible for any use that may be made of the information it contains. Authors also thank the RECETOX Research Infrastructure (ID LM2023069) financed by the Ministry of Education, Youth and Sports for its supportive background. BioRender.com was utilized for the creation of all figures in this article. Authors thank Dr. Jaroslav Janosek for his valuable comments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. We acknowledge the assistance of OpenAI's ChatGPT-4 Turbo (solely for preliminary translations of the text, which were subsequently verified and refined).
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frph.2025.1557912/full#supplementary-material
Abbreviations
NGS, next-generation sequencing; TESE, testicular sperm extraction; PESA, percutaneous epididymal sperm aspiration; MESA, micro epididymal sperm aspiration; WHO, World Health Organization; CTAB, cetrimonium bromide DNA extraction method; MALDI-TOF, matrix-assisted laser desorption/ionization-time of flight mass spectrometry; WGS, Whole genome sequencing); DTT, dithiothreitol; RT qPCR, real-time quantitative polymerase chain reaction; DHPLC, denaturing high-performance liquid chromatography; MACS, magnet-activated cell sorting; RCF, relative centrifugal force.
References
1. ESHRE Capri Workshop Group. Fertility and ageing. Hum Reprod Update. (2005) 11(3):261–76. doi: 10.1093/humupd/dmi006
2. Farahani L, Tharakan T, Yap T, Ramsay JW, Jayasena CN, Minhas S. The semen microbiome and its impact on sperm function and male fertility: a systematic review and meta-analysis. Andrology. (2021) 9(1):115–44. doi: 10.1111/andr.12886
3. Pellati D, Mylonakis I, Bertoloni G, Fiore C, Andrisani A, Ambrosini G, et al. Genital tract infections and infertility. Eur J Obstet Gynecol Reprod Biol. (2008) 140(1):3–11. doi: 10.1016/j.ejogrb.2008.03.009
4. Solomon M, Henkel R. Semen culture and the assessment of genitourinary tract infections. Indian J Urol. (2017) 33(3):188–93. doi: 10.4103/iju.IJU_407_16
5. Ochsendorf FR. Sexually transmitted infections: impact on male fertility. Andrologia. (2008) 40(2):72–5. doi: 10.1111/j.1439-0272.2007.00825.x
6. Diemer T, Huwe P, Ludwig M, Hauck EW, Weidner W. Urogenital infection and sperm motility. Andrologia. (2003) 35(5):283–7. doi: 10.1111/j.1439-0272.2003.tb00858.x
7. Bar-Chama N, Fisch H. Infection and pyospermia in male infertility. World J Urol. (1993) 11(2):76–81. doi: 10.1007/BF00182033
8. Krissi H, Orvieto R, Ashkenazi J, Gilboa YC, Shalev J, Moscovitch I, et al. Effect of contaminated preprocessed semen on fertilization rate and embryo quality in assisted reproductive techniques. Gynecol Endocrinol. (2004) 18(2):63–7. doi: 10.1080/09513590310001651821
9. Ma X, Gao X. The effect of ureaplasma urealyticum on the level of P34H expression, the activity of hyaluronidase, and DNA fragmentation in human spermatozoa. Am J Reprod Immunol. (2017) 77(1):e12600. doi: 10.1111/aji.12600
10. Schulz M, Sánchez R, Soto L, Risopatrón J, Villegas J. Effect of Escherichia coli and its soluble factors on mitochondrial membrane potential, phosphatidylserine translocation, viability, and motility of human spermatozoa. Fertil Steril. (2010) 94(2):619–23. doi: 10.1016/j.fertnstert.2009.01.140
11. Fraczek M, Piasecka M, Gaczarzewicz D, Szumala-Kakol A, Kazienko A, Lenart S, et al. Membrane stability and mitochondrial activity of human-ejaculated spermatozoa during in vitro experimental infection with Escherichia coli, Staphylococcus haemolyticus and Bacteroides ureolyticus. Andrologia. (2012) 44(5):315–29. doi: 10.1111/j.1439-0272.2012.01283.x
12. Prabha V, Gupta T, Kaur S, Kaur N, Kala S, Singh A. Isolation of a spermatozoal immobilization factor from Staphylococcus aureus filtrates. Can J Microbiol. (2009) 55(7):874–8. doi: 10.1139/W09-032
13. Eini F, Kutenaei MA, Zareei F, Dastjerdi ZS, Shirzeyli MH, Salehi E. Effect of bacterial infection on sperm quality and DNA fragmentation in subfertile men with leukocytospermia. BMC Mol Cell Biol. (2021) 22(1):42. doi: 10.1186/s12860-021-00380-8
14. Zeyad A, Hamad M, Amor H, Hammadeh ME. Relationships between bacteriospermia, DNA integrity, nuclear protamine alteration, sperm quality and ICSI outcome. Reprod Biol. (2018) 18(1):115–21. doi: 10.1016/j.repbio.2018.01.010
15. Punab M, Lõivukene K, Kermes K, Mändar R. The limit of leucocytospermia from the microbiological viewpoint. Andrologia. (2003) 35(5):271–8. doi: 10.1111/j.1439-0272.2003.tb00856.x
16. Lackner JE, Herwig R, Schmidbauer J, Schatzl G, Kratzik C, Marberger M. Correlation of leukocytospermia with clinical infection and the positive effect of antiinflammatory treatment on semen quality. Fertil Steril. (2006) 86(3):601–5. doi: 10.1016/j.fertnstert.2006.01.032
17. Hou D, Zhou X, Zhong X, Settles ML, Herring J, Wang L, et al. Microbiota of the seminal fluid from healthy and infertile men. Fertil Steril. (2013) 100(5):1261–1269.e3. doi: 10.1016/j.fertnstert.2013.07.1991
18. Fraczek M, Hryhorowicz M, Gill K, Zarzycka M, Gaczarzewicz D, Jedrzejczak P, et al. The effect of bacteriospermia and leukocytospermia on conventional and nonconventional semen parameters in healthy young normozoospermic males. J Reprod Immunol. (2016) 118:18–27. doi: 10.1016/j.jri.2016.08.006
19. Zeyad A, Hamad MF, Hammadeh ME. The effects of bacterial infection on human sperm nuclear protamine P1/P2 ratio and DNA integrity. Andrologia. (2018) 50(2):e12841. doi: 10.1111/and.12841
20. D’Argenio V. Human microbiome acquisition and bioinformatic challenges in metagenomic studies. Int J Mol Sci. (2018) 19(2):383. doi: 10.3390/ijms19020383
21. Chen H, Luo T, Chen T, Wang G. Seminal bacterial composition in patients with obstructive and non-obstructive azoospermia. Exp Ther Med. (2018) 15(3):2884–90. doi: 10.3892/etm.2018.5778
22. Monteiro C, Marques PI, Cavadas B, Damião I, Almeida V, Barros N, et al. Characterization of microbiota in male infertility cases uncovers differences in seminal hyperviscosity and oligoasthenoteratozoospermia possibly correlated with increased prevalence of infectious bacteria. Am J Reprod Immunol. (2018) 79(6):e12838. doi: 10.1111/aji.12838
23. Baud D, Pattaroni C, Vulliemoz N, Castella V, Marsland BJ, Stojanov M. Sperm microbiota and its impact on semen parameters. Front Microbiol. (2019) 10:234. doi: 10.3389/fmicb.2019.00234
24. Ricci S, De Giorgi S, Lazzeri E, Luddi A, Rossi S, Piomboni P, et al. Impact of asymptomatic genital tract infections on in vitro fertilization (IVF) outcome. PLoS One. (2018) 13(11):e0207684. doi: 10.1371/journal.pone.0207684
25. Damke E, Kurscheidt FA, Irie MMT, Gimenes F, Consolaro MEL. Male partners of infertile couples with seminal positivity for markers of bacterial vaginosis have impaired fertility. Am J Mens Health. (2018) 12(6):2104–15. doi: 10.1177/1557988318794522
26. Alfano M, Ferrarese R, Locatelli I, Ventimiglia E, Ippolito S, Gallina P, et al. Testicular microbiome in azoospermic men—first evidence of the impact of an altered microenvironment. Hum Reprod. (2018) 33(7):1212–7. doi: 10.1093/humrep/dey116
27. Molina NM, Plaza-Díaz J, Vilchez-Vargas R, Sola-Leyva A, Vargas E, Mendoza-Tesarik R, et al. Assessing the testicular sperm microbiome: a low-biomass site with abundant contamination. Reprod Biomed Online. (2021) 43(3):523–31. doi: 10.1016/j.rbmo.2021.06.021
28. Lundy SD, Sangwan N, Parekh NV, Selvam MKP, Gupta S, McCaffrey P, et al. Functional and taxonomic dysbiosis of the gut, urine, and semen microbiomes in male infertility. Eur Urol. (2021) 79(6):826–36. doi: 10.1016/j.eururo.2021.01.014
29. Yang H, Zhang J, Xue Z, Zhao C, Lei L, Wen Y, et al. Potential pathogenic bacteria in seminal microbiota of patients with different types of dysspermatism. Sci Rep. (2020) 10(1):6876. doi: 10.1038/s41598-020-63787-x
30. Okwelogu SI, Ikechebelu JI, Agbakoba NR, Anukam KC. Microbiome compositions from infertile couples seeking in vitro fertilization, using 16S rRNA gene sequencing methods: any correlation to clinical outcomes? Front Cell Infect Microbiol. (2021) 11:709372. doi: 10.3389/fcimb.2021.709372
31. Pagliuca C, Cariati F, Bagnulo F, Scaglione E, Carotenuto C, Farina F, et al. Microbiological evaluation and sperm DNA fragmentation in semen samples of patients undergoing fertility investigation. Genes (Basel). (2021) 12(5):654. doi: 10.3390/genes12050654
32. Mørup N, Main AM, Jørgensen N, Daugaard G, Juul A, Almstrup K. The seminal plasma microbiome of men with testicular germ cell tumours described by small RNA sequencing. Andrology. (2023) 11(4):756–69. doi: 10.1111/andr.13305
33. Campisciano G, Iebba V, Zito G, Luppi S, Martinelli M, Fischer L, et al. Lactobacillus iners and gasseri, Prevotella bivia and HPV belong to the microbiological signature negatively affecting human reproduction. Microorganisms. (2020) 9(1):39. doi: 10.3390/microorganisms9010039
34. Garcia-Segura S, Del Rey J, Closa L, Garcia-Martínez I, Hobeich C, Castel AB, et al. Seminal microbiota of idiopathic infertile patients and its relationship with sperm DNA integrity. Front Cell Dev Biol. (2022) 10:937157. doi: 10.3389/fcell.2022.937157
35. Yao Y, Qiu XJ, Wang DS, Luo JK, Tang T, Li YH, et al. Semen microbiota in normal and leukocytospermic males. Asian J Androl. (2022) 24(4):398–405. doi: 10.4103/aja202172
36. Bukharin OV, Perunova NB, Ivanova EV, Chaynikova IN, Bekpergenova AV, Bondarenko TA, et al. Semen microbiota and cytokines of healthy and infertile men. Asian J Androl. (2022) 24(4):353–8. doi: 10.4103/aja202169
37. Chen P, Li Y, Zhu X, Ma M, Chen H, He J, et al. Interaction between host and microbes in the semen of patients with idiopathic nonobstructive azoospermia. Microbiol Spectr. (2023) 11(1):e04365–22. doi: 10.1128/spectrum.04365-22
38. Amato V, Papaleo E, Pasciuta R, Viganò P, Ferrarese R, Clementi N, et al. Differential composition of vaginal microbiome, but not of seminal microbiome, is associated with successful intrauterine insemination in couples with idiopathic infertility: a prospective observational study. Open Forum Infect Dis. (2020) 7(1):ofz525. doi: 10.1093/ofid/ofz525
39. Osadchiy V, Belarmino A, Kianian R, Sigalos JT, Ancira JS, Kanie T, et al. Semen microbiota are dramatically altered in men with abnormal sperm parameters. Sci Rep. (2024) 14(1):1068. doi: 10.1038/s41598-024-51686-4
40. Puerta Suárez J, Cardona Maya WD. Microbiota, prostatitis, and fertility: bacterial diversity as a possible health ally. Adv Urol. (2021) 2021:1–8. doi: 10.1155/2021/1007366
41. Gachet C, Prat M, Burucoa C, Grivard P, Pichon M. Spermatic microbiome characteristics in infertile patients: impact on sperm count, mobility, and morphology. J Clin Med. (2022) 11(6):1505. doi: 10.3390/jcm11061505
42. Pilatz A, Kilb J, Kaplan H, Fietz D, Hossain H, Schüttler CG, et al. High prevalence of urogenital infection/inflammation in patients with azoospermia does not impede surgical sperm retrieval. Andrologia. (2019) 51(10):e13401. doi: 10.1111/and.13401
43. Rivera VV, Cardona Maya WD, Puerta-Suárez J. The relationship between sexually transmitted microorganisms and seminal quality in asymptomatic men. Asian J Urol. (2022) 9(4):473–9. doi: 10.1016/j.ajur.2021.09.004
44. Veneruso I, Cariati F, Alviggi C, Pastore L, Tomaiuolo R, D’Argenio V. Metagenomics reveals specific microbial features in males with semen alterations. Genes (Basel). (2023) 14(6):1228. doi: 10.3390/genes14061228
45. Mardanov AV, Kadnikov VV, Ravin NV. Metagenomics: A Paradigm Shift in Microbiology. in: Metagenomics. London: Elsevier (2018). p. 1–13. Available online at: https://linkinghub.elsevier.com/retrieve/pii/B978008102268900001X (Accessed April 08, 2025).
46. Štšepetova J, Baranova J, Simm J, Parm Ü, Rööp T, Sokmann S, et al. The complex microbiome from native semen to embryo culture environment in human in vitro fertilization procedure. Reprod Biol Endocrinol. (2020) 18(1):3. doi: 10.1186/s12958-019-0562-z
47. Holmes C, McDonald F, Jones M, Ozdemir V, Graham JE. Standardization and omics science: technical and social dimensions are inseparable and demand symmetrical study. Omics J Integr Biol. (2010) 14(3):327–32. doi: 10.1089/omi.2010.0022
48. Tvrdá E, Ďuračka M, Benko F, Kováčik A, Lovíšek D, Gálová E, et al. Ejaculatory abstinence affects the sperm quality in normozoospermic men—how does the seminal bacteriome respond? Int J Mol Sci. (2023) 24(4):3503. doi: 10.3390/ijms24043503
49. Tvrdá E, Lovíšek D, Gálová E, Schwarzová M, Kováčiková E, Kunová S, et al. Possible implications of bacteriospermia on the sperm quality, oxidative characteristics, and seminal cytokine network in normozoospermic men. Int J Mol Sci. (2022) 23(15):8678. doi: 10.3390/ijms23158678
50. Ho CLT, Vaughan-Constable DR, Ramsay J, Jayasena C, Tharakan T, Yap T, et al. The relationship between genitourinary microorganisms and oxidative stress, sperm DNA fragmentation and semen parameters in infertile men. Andrologia. (2022) 54(2):e14322. doi: 10.1111/and.14322
51. Suarez Arbelaez MC, Israeli JM, Tipton CD, Loloi J, Deebel N, Leong JY, et al. Pilot study: next-generation sequencing of the semen microbiome in vasectomized versus nonvasectomized men. Eur Urol Focus. (2023) 9(1):75–82. doi: 10.1016/j.euf.2022.11.010
52. Iovene MR, Martora F, Bombace F, Montella F, Del Vecchio C, De Rosa M, et al. A new enrichment diagnostic platform for semen culture. J Microbiol Methods. (2018) 144:168–72. doi: 10.1016/j.mimet.2017.11.018
53. Berjis K, Ghiasi M, Sangy S. Study of seminal infection among an infertile male population in qom, Iran, and its effect on sperm quality. Iran J Microbiol. (2018) 10(2):111–6.29997751
54. Trinchieri A, Abdelrahman KM, Bhatti KH, Bello JO, Das K, Gatsev O, et al. Spectrum of causative pathogens and resistance rates to antibacterial agents in bacterial prostatitis. Diagnostics. (2021) 11(8):1333. doi: 10.3390/diagnostics11081333
55. Machen GL, Bird ET, Brown ML, Ingalsbe DA, East MM, Reyes M, et al. Time trends for bacterial species and resistance patterns in semen in patients undergoing evaluation for male infertility. Bayl Univ Med Cent Proc. (2018) 31(2):165–7. doi: 10.1080/08998280.2018.1444298
56. Shash RYM, Mohamed GAA, Shebl SE, Shokr M, Soliman SA. The impact of bacteriospermia on semen parameters among infertile Egyptian men: a case–control study. Am J Mens Health. (2023) 17(3):15579883231181861. doi: 10.1177/15579883231181861
57. Karthikeyan M, N.S K, Singh R. Association of semen bacteriological profile with infertility: – A cross-sectional study in a tertiary care center. J Hum Reprod Sci. (2021) 14(3):260–6. doi: 10.4103/jhrs.jhrs_49_21
58. Koort K, Sõsa K, Türk S, Lapp E, Talving E, Karits P, et al. Lactobacillus crispatus -dominated vaginal microbiome and Acinetobacter -dominated seminal microbiome support beneficial ART outcome. Acta Obstet Gynecol Scand. (2023) 102(7):921–34. doi: 10.1111/aogs.14598
59. Garcia-Segura S, Del Rey J, Closa L, Garcia-Martínez I, Hobeich C, Castel AB, et al. Characterization of seminal microbiome of infertile idiopathic patients using third-generation sequencing platform. Int J Mol Sci. (2023) 24(9):7867. doi: 10.3390/ijms24097867
60. Yao T, Han X, Guan T, Wang Z, Zhang S, Liu C, et al. Effect of indoor environmental exposure on seminal microbiota and its application in body fluid identification. Forensic Sci Int. (2020) 314:110417. doi: 10.1016/j.forsciint.2020.110417
61. WHO. Laboratory Manual for the Examination and Processing of Human Semen. 6th ed. Geneva: World Health Organization (2021). p. 1.
62. Lauber CL, Zhou N, Gordon JI, Knight R, Fierer N. Effect of storage conditions on the assessment of bacterial community structure in soil and human-associated samples: influence of short-term storage conditions on microbiota. FEMS Microbiol Lett. (2010) 307(1):80–6. doi: 10.1111/j.1574-6968.2010.01965.x
63. Kim D, Hofstaedter CE, Zhao C, Mattei L, Tanes C, Clarke E, et al. Optimizing methods and dodging pitfalls in microbiome research. Microbiome. (2017) 5(1):52. doi: 10.1186/s40168-017-0267-5
64. Gürsoy N, Karadayı S, Akmayan İ, Karadayı B, Özbek T. Time-dependent change in the microbiota structure of seminal stains exposed to indoor environmental. Int J Legal Med. (2024) 138(2):591–602. doi: 10.1007/s00414-023-03108-9
65. Song SJ, Amir A, Metcalf JL, Amato KR, Xu ZZ, Humphrey G, et al. Preservation methods differ in fecal microbiome stability, affecting suitability for field studies. mSystems. (2016) 1(3):e00021–16. doi: 10.1128/mSystems.00021-16
66. Hammadeh ME, Kuhnen A, Amer AS, Rosenbaum P, Schmidt W. Comparison of sperm preparation methods: effect on chromatin and morphology recovery rates and their consequences on the clinical outcome after in vitro fertilization embryo transfer. Int J Androl. (2001) 24(6):360–8. doi: 10.1046/j.1365-2605.2001.0317a.x
67. Anamthathmakula P, Winuthayanon W. Mechanism of semen liquefaction and its potential for a novel non-hormonal contraception†. Biol Reprod. (2020) 103(2):411–26. doi: 10.1093/biolre/ioaa075
68. Liu H, Song X, Huang M, Zhan H, Wang S, Zhu S, et al. Ureaplasma urealyticum induces polymorphonuclear elastase to change semen properties and reduce sperm motility: a prospective observational study. J Int Med Res. (2022) 50(6):03000605221106410. doi: 10.1177/03000605221106410
69. Shi Y, Wang G, Lau HCH, Yu J. Metagenomic sequencing for microbial DNA in human samples: emerging technological advances. Int J Mol Sci. (2022) 23(4):2181. doi: 10.3390/ijms23042181
70. Marotz CA, Sanders JG, Zuniga C, Zaramela LS, Knight R, Zengler K. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome. (2018) 6(1):42. doi: 10.1186/s40168-018-0426-3
71. Hasan MR, Rawat A, Tang P, Jithesh PV, Thomas E, Tan R, et al. Depletion of human DNA in spiked clinical specimens for improvement of sensitivity of pathogen detection by next-generation sequencing. J Clin Microbiol. (2016) 54(4):919–27. doi: 10.1128/JCM.03050-15
72. Fong W, Rockett R, Timms V, Sintchenko V. Optimization of sample preparation for culture-independent sequencing of Bordetella pertussis. Microb Genomics. (2020) 6(3):e000332. doi: 10.1099/mgen.0.000332
73. Yeoh YK. Removing host-derived DNA sequences from microbial metagenomes via mapping to reference genomes. In: Carvalhais LC, Dennis PG, editors. The Plant Microbiome. New York, NY: Springer US (2021). p. 147–53. Available at: http://link.springer.com/10.1007/978-1-0716-1040-4_13 (Accessed April 08, 2025).
74. Shi X, Shao C, Luo C, Chu Y, Wang J, Meng Q, et al. Microfluidics-Based enrichment and whole-genome amplification enable strain-level resolution for airway metagenomics. mSystems. (2019) 4(4):e00198–19. doi: 10.1128/mSystems.00198-19
75. Bruggeling CE, Garza DR, Achouiti S, Mes W, Dutilh BE, Boleij A. Optimized bacterial DNA isolation method for microbiome analysis of human tissues. MicrobiologyOpen. (2021) 10(3):e1191. doi: 10.1002/mbo3.1191
76. Martínez P, Proverbio F, Camejo MI. Sperm lipid peroxidation and pro-inflammatory cytokines. Asian J Androl. (2007) 9(1):102–7. doi: 10.1111/j.1745-7262.2007.00238.x
77. Willbanks A, Leary M, Greenshields M, Tyminski C, Heerboth S, Lapinska K, et al. The evolution of epigenetics: from prokaryotes to humans and its biological consequences. Genet Epigenetics. (2016) 8:GEG.S31863. doi: 10.4137/GEG.S31863
78. Åsenius F, Danson AF, Marzi SJ. DNA methylation in human sperm: a systematic review. Hum Reprod Update. (2020) 26(6):841–73. doi: 10.1093/humupd/dmaa025
79. Salonen A, Nikkilä J, Jalanka-Tuovinen J, Immonen O, Rajilić-Stojanović M, Kekkonen RA, et al. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J Microbiol Methods. (2010) 81(2):127–34. doi: 10.1016/j.mimet.2010.02.007
80. Scupham AJ, Jones JA, Wesley IV. Comparison of DNA extraction methods for analysis of Turkey cecal microbiota. J Appl Microbiol. (2007) 102(2):401–9. doi: 10.1111/j.1365-2672.2006.03094.x
81. Li F, Hullar MAJ, Lampe JW. Optimization of terminal restriction fragment polymorphism (TRFLP) analysis of human gut microbiota. J Microbiol Methods. (2007) 68(2):303–11. doi: 10.1016/j.mimet.2006.09.006
82. Anderson KL, Lebepe-Mazur S. Comparison of rapid methods for the extraction of bacterial DNA from colonic and caecal lumen contents of the pig: COMPARISON OF DNA EXTRACTION METHODS. J Appl Microbiol. (2003) 94(6):988–93. doi: 10.1046/j.1365-2672.2003.01917.x
83. Teng F, Darveekaran Nair SS, Zhu P, Li S, Huang S, Li X, et al. Impact of DNA extraction method and targeted 16S-rRNA hypervariable region on oral microbiota profiling. Sci Rep. (2018) 8(1):16321. doi: 10.1038/s41598-018-34294-x
84. Yuan S, Cohen DB, Ravel J, Abdo Z, Forney LJ. Evaluation of methods for the extraction and purification of DNA from the human microbiome. PLoS One. (2012) 7(3):e33865. doi: 10.1371/journal.pone.0033865
85. Nylund L, Heilig HGHJ, Salminen S, De Vos WM, Satokari R. Semi-automated extraction of microbial DNA from feces for qPCR and phylogenetic microarray analysis. J Microbiol Methods. (2010) 83(2):231–5. doi: 10.1016/j.mimet.2010.09.003
86. Ariefdjohan MW, Savaiano DA, Nakatsu CH. Comparison of DNA extraction kits for PCR-DGGE analysis of human intestinal microbial communities from fecal specimens. Nutr J. (2010) 9(1):23. doi: 10.1186/1475-2891-9-23
87. Abusleme L, Hong BY, Dupuy AK, Strausbaugh LD, Diaz PI. Influence of DNA extraction on oral microbial profiles obtained via 16S rRNA gene sequencing. J Oral Microbiol. (2014) 6(1):23990. doi: 10.3402/jom.v6.23990
88. Gand M, Bloemen B, Vanneste K, Roosens NHC, De Keersmaecker SCJ. Comparison of 6 DNA extraction methods for isolation of high yield of high molecular weight DNA suitable for shotgun metagenomics nanopore sequencing to detect bacteria. BMC Genomics. (2023) 24(1):438. doi: 10.1186/s12864-023-09537-5
89. Wright ML, Podnar J, Longoria KD, Nguyen TC, Lim S, Garcia S, et al. Comparison of commercial DNA extraction kits for whole metagenome sequencing of human oral, vaginal, and rectal microbiome samples. bioRxiv [Preprint]. (2023). doi: 10.1101/2023.02.01.526597
90. Mattei V, Murugesan S, Al Hashmi M, Mathew R, James N, Singh P, et al. Evaluation of methods for the extraction of microbial DNA from vaginal swabs used for microbiome studies. Front Cell Infect Microbiol. (2019) 9:197. doi: 10.3389/fcimb.2019.00197
91. Shibata T, Nakagawa M, Coleman HN, Owens SM, Greenfield WW, Sasagawa T, et al. Evaluation of DNA extraction protocols from liquid-based cytology specimens for studying cervical microbiota. PLoS One. (2021) 16(8):e0237556. doi: 10.1371/journal.pone.0237556
92. Vendrell JA, Henry S, Cabello-Aguilar S, Heckendorn E, Godreuil S, Solassol J. Determination of the optimal bacterial DNA extraction method to explore the urinary Microbiota. Int J Mol Sci. (2022) 23(3):1336. doi: 10.3390/ijms23031336
93. Karstens L, Siddiqui NY, Zaza T, Barstad A, Amundsen CL, Sysoeva TA. Benchmarking DNA isolation kits used in analyses of the urinary microbiome. Sci Rep. (2021) 11(1):6186. doi: 10.1038/s41598-021-85482-1
94. Mändar R, Punab M, Borovkova N, Lapp E, Kiiker R, Korrovits P, et al. Complementary seminovaginal microbiome in couples. Res Microbiol. (2015) 166(5):440–7. doi: 10.1016/j.resmic.2015.03.009
95. Buetas E, Jordán-López M, López-Roldán A, D’Auria G, Martínez-Priego L, De Marco G, et al. Full-length 16S rRNA gene sequencing by PacBio improves taxonomic resolution in human microbiome samples. BMC Genomics. (2024) 25(1):310. doi: 10.1186/s12864-024-10213-5
96. Matsuo Y, Komiya S, Yasumizu Y, Yasuoka Y, Mizushima K, Takagi T, et al. Full-length 16S rRNA gene amplicon analysis of human gut microbiota using MinIONTM nanopore sequencing confers species-level resolution. BMC Microbiol. (2021) 21(1):35. doi: 10.1186/s12866-021-02094-5
97. Nygaard AB, Tunsjø HS, Meisal R, Charnock C. A preliminary study on the potential of nanopore MinION and illumina MiSeq 16S rRNA gene sequencing to characterize building-dust microbiomes. Sci Rep. (2020) 10(1):3209. doi: 10.1038/s41598-020-59771-0
98. Shin J, Lee S, Go MJ, Lee SY, Kim SC, Lee CH, et al. Analysis of the mouse gut microbiome using full-length 16S rRNA amplicon sequencing. Sci Rep. (2016) 6(1):29681. doi: 10.1038/srep29681
99. Wongsurawat T, Nakagawa M, Atiq O, Coleman HN, Jenjaroenpun P, Allred JI, et al. An assessment of Oxford nanopore sequencing for human gut metagenome profiling: a pilot study of head and neck cancer patients. J Microbiol Methods. (2019) 166:105739. doi: 10.1016/j.mimet.2019.105739
100. Miura H, Takeda M, Yamaguchi M, Ohtani Y, Endo G, Masuda Y, et al. Application of MinION amplicon sequencing to buccal swab samples for improving resolution and throughput of rumen microbiota analysis. Front Microbiol. (2022) 13:783058. doi: 10.3389/fmicb.2022.783058
101. Pfeiffer F, Gröber C, Blank M, Händler K, Beyer M, Schultze JL, et al. Systematic evaluation of error rates and causes in short samples in next-generation sequencing. Sci Rep. (2018) 8(1):10950. doi: 10.1038/s41598-018-29325-6
102. Delahaye C, Nicolas J. Sequencing DNA with nanopores: troubles and biases. PLoS One. (2021) 16(10):e0257521. doi: 10.1371/journal.pone.0257521
103. Yang B, Wang Y, Qian PY. Sensitivity and correlation of hypervariable regions in 16S rRNA genes in phylogenetic analysis. BMC Bioinformatics. (2016) 17(1):135. doi: 10.1186/s12859-016-0992-y
104. Claesson MJ, Wang Q, O’Sullivan O, Greene-Diniz R, Cole JR, Ross RP, et al. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. (2010) 38(22):e200–e200. doi: 10.1093/nar/gkq873
105. Cai L, Ye L, Tong AHY, Lok S, Zhang T. Biased diversity metrics revealed by bacterial 16S pyrotags derived from different primer sets. PLoS One. (2013) 8(1):e53649. doi: 10.1371/journal.pone.0053649
106. Tremblay J, Singh K, Fern A, Kirton ES, He S, Woyke T, et al. Primer and platform effects on 16S rRNA tag sequencing. Front Microbiol. (2015) 6:771. doi: 10.3389/fmicb.2015.00771
107. Barb JJ, Oler AJ, Kim HS, Chalmers N, Wallen GR, Cashion A, et al. Development of an analysis pipeline characterizing multiple hypervariable regions of 16S rRNA using mock samples. PLoS One. (2016) 11(2):e0148047. doi: 10.1371/journal.pone.0148047
108. Jones CB, White JR, Ernst SE, Sfanos KS, Peiffer LB. Incorporation of data from multiple hypervariable regions when analyzing bacterial 16S rRNA gene sequencing data. Front Genet. (2022) 13:799615. doi: 10.3389/fgene.2022.799615
109. Castelino M, Eyre S, Moat J, Fox G, Martin P, Ho P, et al. Optimisation of methods for bacterial skin microbiome investigation: primer selection and comparison of the 454 versus MiSeq platform. BMC Microbiol. (2017) 17(1):23. doi: 10.1186/s12866-017-0927-4
110. López-Aladid R, Fernández-Barat L, Alcaraz-Serrano V, Bueno-Freire L, Vázquez N, Pastor-Ibáñez R, et al. Determining the most accurate 16S rRNA hypervariable region for taxonomic identification from respiratory samples. Sci Rep. (2023) 13(1):3974. doi: 10.1038/s41598-023-30764-z
111. Fouhy F, Clooney AG, Stanton C, Claesson MJ, Cotter PD. 16S rRNA gene sequencing of mock microbial populations- impact of DNA extraction method, primer choice and sequencing platform. BMC Microbiol. (2016) 16(1):123. doi: 10.1186/s12866-016-0738-z
112. Hugerth LW, Pereira M, Zha Y, Seifert M, Kaldhusdal V, Boulund F, et al. Optimal protocols for sequence-based characterization of the human vaginal microbiome. bioRxiv [Preprint]. (2020). doi: 10.1101/2020.05.05.079996
113. Graspeuntner S, Loeper N, Künzel S, Baines JF, Rupp J. Selection of validated hypervariable regions is crucial in 16S-based microbiota studies of the female genital tract. Sci Rep. (2018) 8(1):9678. doi: 10.1038/s41598-018-27757-8
114. Stinson LF, Keelan JA, Payne MS. Identification and removal of contaminating microbial DNA from PCR reagents: impact on low-biomass microbiome analyses. Lett Appl Microbiol. (2019) 68(1):2–8. doi: 10.1111/lam.13091
115. Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, et al. Reagent contamination can critically impact sequence-based microbiome analyses. BMC Biol. (2014) 12:87. doi: 10.1186/s12915-014-0087-z
116. Karstens L, Asquith M, Davin S, Fair D, Gregory WT, Wolfe AJ, et al. Controlling for contaminants in low biomass 16S rRNA gene sequencing experiments. mSystems. (2018) 4(4):e00290–19. doi: 10.1128/mSystems.00290-19
117. Minich JJ, Sanders JG, Amir A, Humphrey G, Gilbert J, Knight R. Quantifying and understanding well-to-well contamination in microbiome research. mSystems. (2019) 4(4):e00186–19. doi: 10.1128/mSystems.00186-19
118. Starolis MW. The contamination monitoring toolbox: best practices for molecular microbiology testing. Clin Microbiol Newsl. (2024) 47:21–7. doi: 10.1016/j.clinmicnews.2024.07.001
119. Dyrhovden R, Rippin M, Øvrebø KK, Nygaard RM, Ulvestad E, Kommedal Ø. Managing contamination and diverse bacterial loads in 16S rRNA deep sequencing of clinical samples: implications of the law of small numbers. mBio. (2021) 12(3):e00598–21. doi: 10.1128/mBio.00598-21
120. Galla G, Praeg N, Colla F, Rzehak T, Illmer P, Seeber J, et al. Mock community as an in situ positive control for amplicon sequencing of microbiotas from the same ecosystem. Sci Rep. (2023) 13(1):4056. doi: 10.1038/s41598-023-30916-1
121. Tourlousse DM, Narita K, Miura T, Ohashi A, Matsuda M, Ohyama Y, et al. Characterization and demonstration of mock communities as control reagents for accurate human microbiome community measurements. Microbiol Spectr. (2022) 10(2):e0191521. doi: 10.1128/spectrum.01915-21
122. Jervis-Bardy J, Leong LEX, Marri S, Smith RJ, Choo JM, Smith-Vaughan HC, et al. Deriving accurate microbiota profiles from human samples with low bacterial content through post-sequencing processing of illumina MiSeq data. Microbiome. (2015) 3(1):19. doi: 10.1186/s40168-015-0083-8
123. Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. (2018) 6(1):226. doi: 10.1186/s40168-018-0605-2
Keywords: methodology, bacteriome, ejaculate, sperm, semen, bacteriospermia, spermiogram, fertility
Citation: Hofman J, Brenerova P and Borilova Linhartova P (2025) State-of-the-art approaches in the investigation of human seminal bacteriome using metagenomic methods. Front. Reprod. Health 7:1557912. doi: 10.3389/frph.2025.1557912
Received: 9 January 2025; Accepted: 16 May 2025;
Published: 5 June 2025.
Edited by:
Ariane Zamoner, Federal University of Santa Catarina, BrazilReviewed by:
Jorge Hallak, University of São Paulo, BrazilBerlin Pandapotan Pardede, National Research and Innovation Agency (BRIN), Indonesia
Copyright: © 2025 Hofman, Brenerova and Borilova Linhartova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Petra Borilova Linhartova, cGV0cmEubGluaGFydG92YUByZWNldG94Lm11bmkuY3o=