 Samuel Narvey
Samuel Narvey Alex Vandenakker
Alex Vandenakker Megan Rempel
Megan Rempel Renée N. Douville
Renée N. Douville- 1Department of Biology, University of Winnipeg, Winnipeg, MB, Canada
- 2Division of Neurodegenerative Disorders, St. Boniface Hospital Albrechtsen Research Centre, Winnipeg, MB, Canada
Retroviral proteases are essential enzymes for viral replication and drive changes within the cellular proteome. While several studies have demonstrated that protease (PR) enzymes from exogenous retroviruses cleave cellular proteins and modulate cellular signaling, the impact of PRs encoded by endogenous retroviruses within the human genome has been largely overlooked. One human symbiont called Endogenous retrovirus-K (ERVK) is pathologically associated with both neurological disease and cancers. Using a computational biology approach, we sought to characterize the ERVK PR interactome. The ERVK PR protein sequence was analyzed using the Eukaryotic Linear Motif (ELM) database and results compared to ELMs of other betaretroviral PRs and similar endogenated viral PRs. A list of putative ERVK PR cellular protein interactors was curated from the ELM list and submitted for STRING analysis to generate an ERVK PR interactome. Reactome analysis was used to identify key pathways potentially influenced by ERVK PR. Network analysis postulated that ERVK PR interacts at the apex of several ubiquitination pathways, as well as has a role in the DNA damage response, gene regulation, and intracellular trafficking. Among retroviral PRs, a predicted interaction with proliferating cell nuclear antigen (PCNA) was unique to ERVK PR. The most prominent disease-associated pathways identified were viral carcinogenesis and neurodegeneration. This strengthens the role of ERVK PR in these pathologies by putatively driving alterations in cellular signaling cascades via select protein-protein interactions.
1. Introduction
Viral proteins frequently interact with host proteins to interfere with cellular signaling pathways. For exogenous viruses, these interferences modify pathways relating to host immunity, cell survival, virion production, and dissemination, resulting in greater replicative success (1, 2). The manner through which endogenous retroviruses (ERVs) interact with host proteomes is less understood.
ERVs are genomic symbionts that comprise approximately 8% of human DNA, with most being defective and non-infectious due to the accumulation of nonsense mutations and deletions since their integration (3, 4). In recent years, ERVs have been implicated in multiple pathological conditions, including cancer (5), autoimmune diseases (6), and neurological conditions (7). Endogenous retrovirus-K (ERVK/HERV-K) is of particular interest as proviral elements of the HML-2 clade are upregulated in a variety of disease conditions, including human immunodeficiency virus (HIV) infection (8), a variety of cancers (8, 9), rheumatoid arthritis (10), and amyotrophic lateral sclerosis (ALS) (8), potentially contributing to the pathology of these diseases (3, 7). ERVK polymorphisms have also been directly linked to genomic alterations and instabilities that are observed in these pathologies (11).
Retroviral proteases are essential enzymes for viral replication and drive changes within the cellular proteome (12, 13); thus, they are prime targets for antiviral therapy (14). The ERVK protease (PR) has been characterized (15–18), and a variety of ERVK transcripts are predicted to encode variants of this enzyme in human cells (19). The human genome comprises approximately 480 ERVK loci containing PR sequences, with the majority of them classified in the HML-3 clade and remainder classified as either HML-2 or other HML clades (19). While their capacity to complete the viral life cycle is not intact (20, 21), several ERVK loci can nonetheless generate transcripts that produce PR protein. Conformational analyses of ERVK PR have revealed a typical A2 aspartic acid protease dimer, consisting of two 106-residue monomers (19, 22). Each monomer consists of helical, flap, and β-sheet motifs. Active site motifs contain aspartic acid residues and are located within the interior pocket of the protease dimer (17, 19, 22). Despite diversity among ERVK clades, several ERVK loci encode a PR enzyme containing all the domain structures and motifs that are required for retroviral PR function (19).
The PR enzyme of exogenous retroviruses plays an essential role in facilitating viral protein maturation and replication. This maturation is accomplished by PR-mediated catalytic cleavage of precursor proteins (19). One of the primary functions of ERVK PR includes the proteolytic processing of Gag-Pol precursors into mature Gag proteins within the cytoplasm (23). Gag retroviral proteins are responsible for mediating intracellular transport to the cell membrane, coordinating viral particle assembly, and facilitating particle budding (14, 24, 25). ERVK PR inhibitors have effectively blocked the proteolytic processing of Gag-Pol precursors, demonstrating the role of ERVK PR in viral protein processing and maturation (17). In addition to viral protein processing, select retroviral PRs are also capable of processing host proteins, such as serine-threonine kinases and the precursor of NF-κB (23, 26). Host protein interactions such as these have been localized in both the nucleus and cytoplasm and are implicated in the disruption of immune signaling and the progression of human disease pathology (23).
Altered cell signaling, protein mislocalization and proteinopathy are recognized as hallmarks of HIV-associated neurocognitive disorder (HAND) (27) and ALS (28) neuropathology. One mechanism by which retroviral PRs can contribute to redistribution of cellular proteins is seen in HIV infection. Specifically, the HIV PR contributes to the sequestration of the innate immune sensor RIG-I, resulting in its relocalization to lysosomal structures and subsequent degradation (29). The consequence of this interference with innate immune signaling is that it prevents the cell from detecting HIV genomic RNA. At this time, it remains unclear if ERVK PR may also interact with select cellular proteins and contribute to their deregulation through interference with protein-protein interactions, sequestration or facilitating degradative processes, as seen in HIV infection (12, 13). HIV-1 PR has also been shown to cleave various host proteins, including actin, tropomyosins, and eIF3D (eukaryotic initiation factor 3D), thus altering cellular complexes and impacting cellular functions such as protein translation, RNA splicing, and apoptosis under appropriate conditions (12, 30, 31). Although ERVK PR has been shown to share substrates with HIV PR (16), it is unclear if it participates in the same processes, and its full interactome remains unknown.
We also seek to understand how ERVK PR interacts with cellular proteins and pathways, as has been done with other retroviral PRs (12, 30–33). Given the knowledge of how retroviral PRs impact cellular functions, we hypothesized that potential ERVK PR interaction partners could be identified using a computational biology approach, which would further provide clues to its effects on cellular pathways. A comparison with similar PRs from eukaryotic organisms and model species may also provide information required for the future development of in vivo models for ERVK PR-driven pathology.
2. Methods
2.1. Database curation
The National Centre for Biotechnology Information (NCBI) Protein-protein Basic Local Alignment Search Tool (BLASTp) (34) within the non-redundant (nr) database was used to identify PRs with sequences resembling ERVK PR (reference sequence HERV-K10, UniProtKB P10265.2). Default algorithm parameters were used, with cut-offs as follows: E value < 1.0X10-17 and identity of > 40%. Sequences were grouped based on phylogeny as informed by ICTV [International Committee on Taxonomy of Viruses; https://talk.ictvonline.org/ (accessed on 06 December 2021)] or OneZoom [OneZoom Tree of Life Explorer; version 3.4.1; Software for Technical Computation; United Kingdom, 2021, https://www.onezoom.org/ (accessed on 12 December 2021)] (35) and are listed in Tables S1-S6.
2.2. Protein alignments and eukaryotic linear motif annotation
Geneious Prime (version 2021.0.3) software (36) was used to align the ERVK PR protein sequence, as well as select representative PRs from exogenous Betaretroviruses (Figure 1) or endogenous retroviruses (Figure 2). A global alignment with free end gaps using BLOSUM62 matrix was performed. Longer sequences were truncated to overlap with the ERVK PR reference sequence. Figures depict sequence logo and protease active site DTGAD, flap VGVG, and GRDLL regions highlighted based on Conserved Domains Database (CDD) annotation (19, 37).
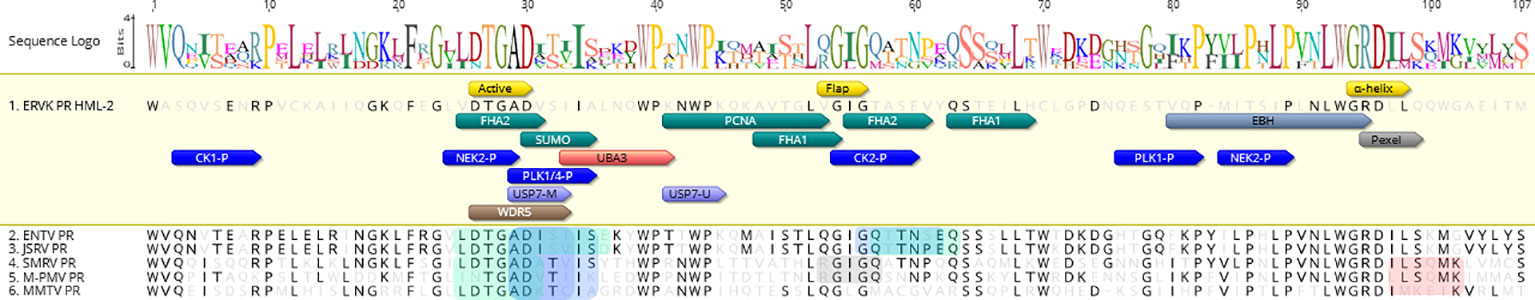
Figure 1 ERVK and exogenous betaretrovirus proteases share common eukaryotic linear motifs. The The ERVK10 protease (PR) sequence was aligned with PRs from Enzootic Nasal Tumor Virus (ENTV), Jaagsiekte sheep retrovirus (JSRV), Squirrel monkey virus (SMRV), Mason–Pfizer monkey virus (M-PMV), and Mouse mammary tumor virus (MMTV), showing the enzymatic active site conservation in yellow. ELMs associated with ERVK PR are positioned within the protein sequence, with colours respective of the functional clusters within the network in Figure 3. Sequence alignment and annotation performed using Geneious Prime software.
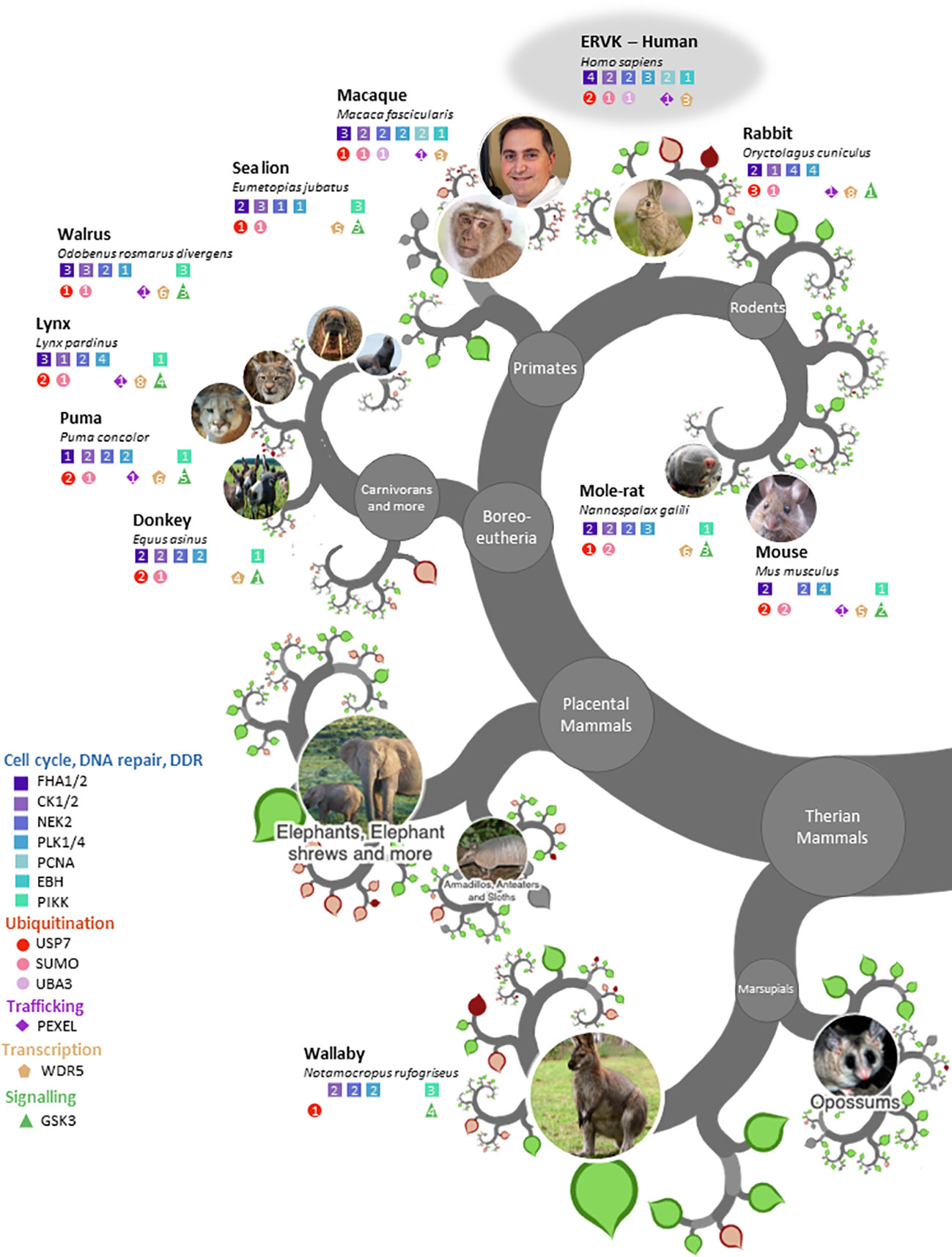
Figure 2 ERVK protease and similar endogenous proteases share eukaryotic linear motifs patterns. Modified OneZoom image illustrating the conservation of ELM motifs in proteases from eukaryotic organisms (Homo sapiens, Macaca fascicularis, Oryctolagus cuniculus, Mus musculus, Nannospalax galili, Puma concolor, Lynx pardinus, Equis asinus, Odobenus rosmarus divergens, Eumetopias jubatus, Notamocropus rufogriseus). Motifs are color-grouped according to function; Cell cycle/DNA repair/DDR (blue), ubiquitination (red), trafficking (purple), transcription (gold), and signaling (green). The number in each colored shape refers to the number of motifs with the respective protease enzyme. Note: Aquila chrysaetos (Table 2) was excluded to maintain figure clarity.
Aligned PR sequences were submitted to the Eukaryotic Linear Motif (ELM; http://elm.eu.org/, accessed March 2022) resource (38). A complete listing of ELMs identified in each PR is presented in Tables 1 and 2. ELMs unique to ERVK PR, as well as ELM sites exhibiting motif consensus above 70% with other PRs were annotated in Figures 1 and 2.

Table 1 ELM motifs in proteases from ERVK and exogenous betaretroviruses.
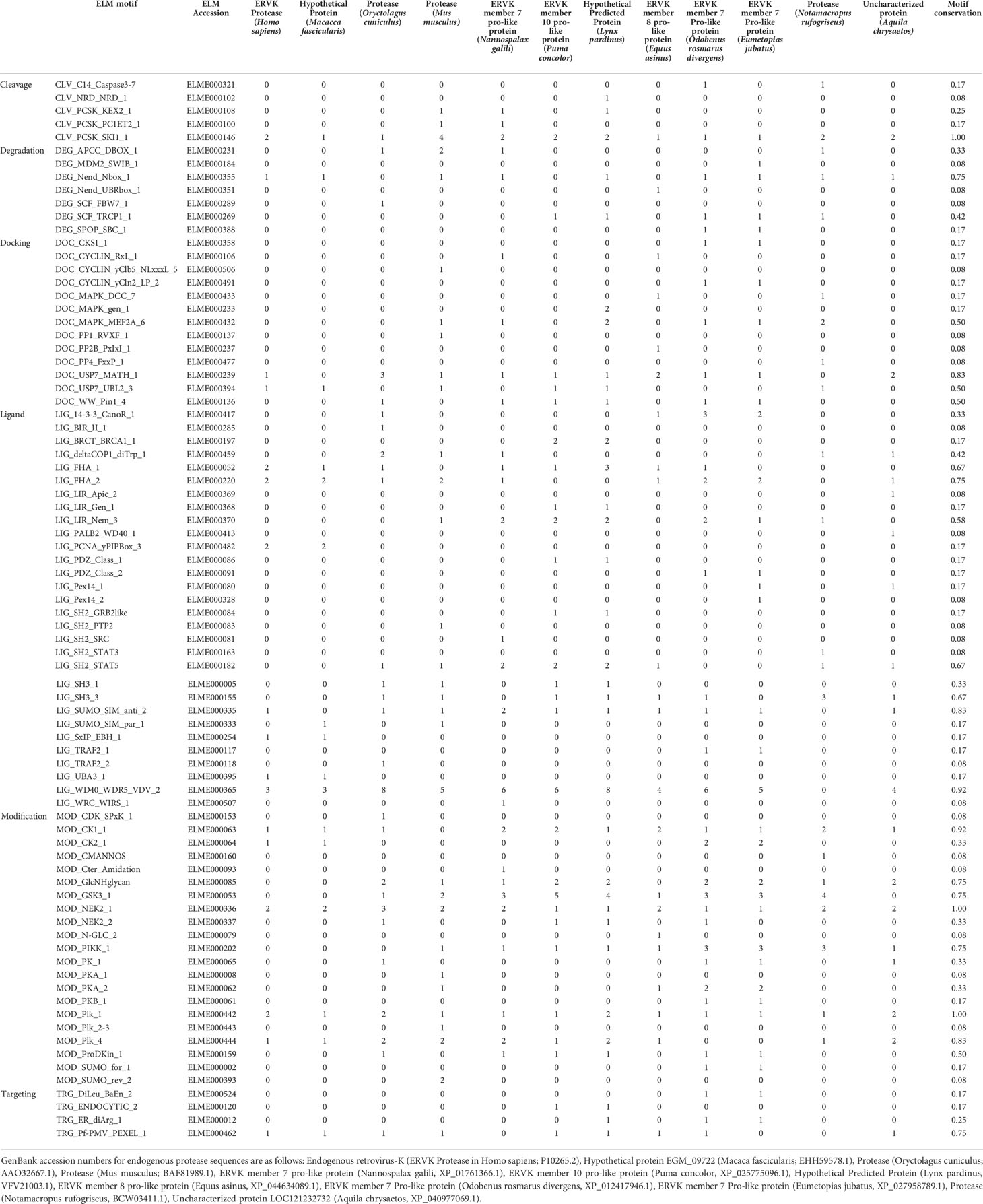
Table 2 ELM motifs in ERVK protease and similar endogenous proteases in eukaryotes.
2.3. STRING analysis and reactome pathways
The names of interacting proteins were curated from each ELM reference page to identify potential ERVK PR binding partners based on ELM motifs. When only a general interaction domain for a given ELM was listed, it was further linked to the InterPro database to curate a list of human proteins containing the interaction domain. Based on the 17 ELMs identified in ERVK PR, a total of 101 putative human protein interaction partners were identified. The list was submitted to STRING version 11.5 (https://string-db.org/) for network analysis (Table S7). Full network analysis was performed using Experiment and Databases as active interaction sources. Submitted query proteins are indicated by coloured nodes, with edges indicating confidence lines with a minimum interaction score of 0.4 (medium confidence). First shell interactors were limited to 10 and are indicated by uncoloured nodes. Query proteins unlinked to the network were omitted from analysis. A payload list was used to colour hub proteins based on cellular function. Reactome pathways associated with the network analysis were presented in a heatmap using GraphPad Prism (version 9.1.1) software and in a complementary list (Table S8). Additional network analysis based on KEGG pathways, UniProt keywords and Gene Ontology (GO) function are provided in supplementary tables (Tables S9-S11).
3. Results and discussion
Both cellular and retroviral aspartic proteases are known to impact many cellular signaling pathways, including those implicated in homeostasis (39, 40), immunity (41–43), DDR (44), autophagy (45), neurological disease (39, 46), and oncogenesis (39). Herein, we describe putative cellular interactors for the ERVK PR and how those interactions may impact cellular function and viral replication.
3.1. Characterization of eukaryotic linear motifs in ERVK protease and other exogenous betaretrovirus proteases
To determine which exogenous and endogenous retroviruses encode PR sequences that are most similar to ERVK PR, we performed BLASTp searches using the non-redundant (nr) NCBI database. As expected, exogenous Betaretroviruses were identified through BLASTp search, which included multiple hits for Enzootic Nasal Tumor Virus (ENTV), Mason–Pfizer monkey virus (M-PMV), Jaagsiekte sheep retrovirus (JSRV), Squirrel monkey virus (SMRV) and Mouse mammary tumor virus (MMTV) (Table 1).
ELM analysis of a representative sequence from each genus was compared with ERVK PR and revealed the conservation of select protein motifs (Table 1, Figure 1). Apart from a general conservation of the DTGAD active site motif (19), all betaretroviral PRs contained several interaction motifs related to DNA damage response (DDR), including interaction with Forkhead-associated (FHA) domain proteins and WD40 repeat domain WDR5 proteins (47, 48). Modulation of ubiquitination was also evident as a strategy of betaretroviral PRs. Putative docking with cell signaling associated ubiquitin-specific protease 7 [USP7/HAUSP (49, 50)] was also a conserved prediction for all betaretroviral PRs. Specifically, ERVK PR contains a conserved USP7 TRAF domain (MATH) P/A-xx-S binding motif (ADVS, aa. 29-32), as well as a USP7 UBL2 domain interaction site (aa. 41-45). Other frequently observed ELM motifs in betaretroviral PRs included a binding motif for ubiquitin-activating enzyme-3 (UBA3) adenylation domain found in several ubiquitin-like (UBL) E1 enzymes and the presence of a small ubiquitin-like modifier (SUMO)-interacting motif (SIM).
Most betaretroviral PRs were also predicted to be phosphorylated by the cell cycle checkpoint kinases NIMA (Never In Mitosis Gene A)-Related Kinase 2 (NEK2) (51) and polo-like kinases (PLK-1 and PLK-4) (52) (Table 1). Additionally, ERVK, ENTV, and JRSV PRs were predicted to be phosphorylated by casein kinases (CK-1 and CK-2) via an S-X-X-S/T site (53). All betaretroviral PRs were predicted substrates for glycogen synthase kinase 3 (GSK3) (54) (Table 1), except for ERVK PR. Additionally, ELMs for MAPK docking motifs and SH3 ligands were not present in ERVK PR.
ERVK PR was unique in its propensity to putatively interact with proliferating cell nuclear antigen (PCNA) (55), as it contained two overlapping PCNA-interacting protein (PIP) motifs (aa. 41-55), also known as a PIP box, just prior to the PR flap domain (Figure 1, Table 1). In addition, only ERVK PR contained an N-terminal SxIP motif (Figure 1, aa. 80-94) that binds to EBH domains in end-binding proteins involved in microtubule transport (56), similar to what is predicted for ERVK integrase (57). This pattern of consistent SxIP motifs in ERVK proteins may point to a unique usage of microtubule networks through EBH domain interaction.
It is important to note that despite the similar complement of ELM motifs in betaretro viral PRs, some sites were positioned differently than in ERVK PR. Additional ELMs and their motif frequencies in individual betaretroviral PRs are listed in Table 1.
3.2. Characterization of eukaryotic linear motifs in ERVK protease and other endogenous proteases
ERVK-like endogenous PRs were evident in much of the tree of life, including eutherians, marsupials, aves, ecdysozoa, and eubacteria (Figure 2, Tables S2-S6). ELM conservation in these PRs for select protein interaction partners frequently included binding motifs for deubiquitinating enzyme USP7, FHA 1 and 2 proteins, SUMO motifs, WD40 repeat domain protein WDR5, as well as PR phosphorylation sites for cell cycle proteins CK1 and PLK4. All endogenous PRs exhibited phosphorylation sites for NEK2 and PLK1. ELMs identified in ERVK PR but uncommon in other endogenous PRs were PCNA interaction sites, UBA3 binding, and SxIP motifs for interaction with end-binding proteins.
ELMs that were highly conserved in most endogenous PRs but are absent in ERVK PR were also observed. Among these signatures was the frequent presence of phosphotyrosine ligands bound by SH2 domains, SH3 ligands, GSK3 phosphorylation sites, PIKK phosphorylation sites, and glycosaminoglycan attachment sites. Additional ELMs and their frequencies in endogenous PRs similar to ERVK can be found in Table 2.
3.3. Unlike similar enzymes, the ERVK protease contains distinct ELM signatures
Two motifs in ERVK PR stand out as distinct for this virus, while other signatures are differentially expressed in ERVK PR as compared with similar PRs.
3.3.1. Among betaretroviruses, ERVK protease has a unique predicted interaction with PCNA
PCNA is a cellular hub protein for DNA replication and repair, as well as epigenetic control of chromatin remodeling, with over 100 known cellular interactors (58, 59). Its many protein-protein interactions are facilitated by short linear motifs, including PIP boxes in cognate ligands. The canonical PIP-box motif is Qxx(L/M/I/V)xx(Y/F)(Y/F) (55, 59, 60); here, ERVK PR presents with a non-canonical PIP-box QKAVTGLV signature for binding the DNA clamp PCNA (Figure 1, aa. 45-53, yeast-like PIP box variant). The glutamine in position 1 fits the front face of the PCNA ring called the “Q pocket” (59, 60). The residues in positions 4-8 generally form a helix. The aliphatic hydrophobic valine in position 4 is typical of canonical PIP boxes, whereas the hydrophobic residues (LV) in position 7 and 8 are similar in composition to the non-canonical PIP box of the yeast anti-recombinogenic helicase Srs2, which places the terminal two residues into a large hydrophobic pocket on the front face of PCNA called the “three-forked plug” (58–60).
Among betaretroviruses, ERVK PR was exceptional in its predicted interaction with PCNA, which has several potential implications related to DDR and viral replication. Key ERVK PR hub protein UBE2N acts with HLTF and SHPRH to mediate Lys-63-linked poly-ubiquitination of PCNA for engagement of DDR during genotoxic stress (61). In the case of EBV infection, the EBV deubiquitinating enzyme BPLF1 targets ubiquitinated PCNA, which disrupts DDR (62). A similar mechanism is also employed by the Herpes simplex virus 1 (HSV-1) UL36USP protein (63). Deubiquitination of PCNA is typically performed by USP1; however, the deubiquitinating function of USP7 is important for suppressing PCNA activity during DNA repair and, unlike USP1, acts independently of cell cycle processes (64). Both EBV BPLF1 and HSV-1 UL36USP viral proteins can also perform this task and therefore can modulate the function of PCNA in the translesion synthesis (TLS) DNA damage tolerance pathway. K164 monoubiquitination of PCNA recruits polymerases η, ι, and κ to DNA replication forks and activates TLS, thus allowing DNA synthesis despite damaged DNA templates (65). Extension of K63-linked polyubiquitin on K164 activates the template switch pathway allowing for error-free DNA repair (65). Thus, deubiquitination of PCNA results in increased cell sensitivity to DNA-damage but can facilitate viral DNA replication (63).
The herpesvirus EBV BPLF1 and HSV-1 UL36USP proteins each contain a PIP box motif to facilitate their interaction with PCNA (62, 63), opening the possibility that the ERVK PR PIP box may also enable a similar contact of this viral protein with PCNA. Given that ERVK PR was also predicted to interact with USP7, a molecular bridge of USP7-ERVK PR-PCNA may be formed preventing PCNA ubiquitination, resulting in a similar inhibitory effect on TLS during ERVK reactivation. However, it is now appreciated that PIP boxes may bind a more diverse set of proteins than just PCNA (58, 59), thus potentially allowing ERVK PR to interact with additional cellular partners.
The ELM profile of ERVK PR has additional implications for PCNA interaction due to the presence of the SIM motif for SUMO recognition. PCNA can be both ubiquitinated and SUMOylated, which impacts its function in DDR pathways (60). Srs2 specifically recognizes SUMO-PCNA using both its non-canonical PIP box and SIM motif (60, 66). As ERVK PR also contains both a non-canonical PIP box and SIM motifs, it is likely that ERVK PR may also specifically interact with the SUMOylated form of PCNA. Unlike Ub-PCNA, which is involved in recruiting translesion DNA polymerases for TLS, the SUMOylation of PCNA is recognized by the anti-recombinogenic DNA helicase Srs2 in yeast. Srs2 limits homologous recombination and facilitates synthesis-dependent single-strand annealing (SDSA) for double-stranded DNA break repair (66). Notably, ERVK PR exhibits a tandem SIM motif and non-canonical PIP box arrangement, similar to Srs2 and potentially its human orthologues (66, 67). Experimental validation will be required to confirm ERVK PR and SUMO-PCNA interaction, as well as the cellular impact of a putative partnership. Considering that ERVK activity is frequently associated with cellular DNA damage and oncogenesis (9), future studies into the impact of ERVK PR on PCNA activity are warranted.
One reason that viruses may target PCNA is its differential impact on viral replication. In the case of DNA viruses, PCNA often acts as a cofactor for viral genome replication in the nucleus (63, 68–71). Although PCNA is largely considered a nuclear protein, it has also been shown to interfere with ssRNA virus replication by forming a complex with genomic viral RNA in the cytoplasm, which suppressed viral polymerase activity and blocked the replication of Potexviruses (72). It remains unclear whether PCNA impacts betaretroviral replication.
3.3.2. ERVK protease may preferentially use end-binding protein-based transport
Cellular transport of viral proteins is essential for viral replication and pathology (73). ERVK PR contains an N-terminal SxIP motif (Figure 1, aa. 80-94) for binding to EBH domains in end-binding proteins involved in microtubule transport (56). Of note, ERVK IN is also predicted to interact with end-binding proteins (57), and the same SxIP motif (ELME000254) is found in both ERVK enzymes. The cluster related to the plus-end tracking proteins (+TIP) consist of only microtubule associated protein RP/EB family members (MAPRE1, MAPRE2, and MAPRE3) and the microtubule motor protein kinesin family member 14 (KIF14), making it by far the smallest grouping within the ERVK PR interactome (Figure 3). This grouping is connected to the rest of the network through one connection with SUMO3, which is known to be an important factor in microtubule-mediated chromosome separation during cell division (74). Together, this suggests the use of the cellular centrosome for viral trafficking and viral aggresome formation (73). Deregulation of the centrosome during viral replication may underlie aberrant cell division and transformation (73); thus, this could be investigated as a potential pathway associated with ERVK-driven oncogenesis.
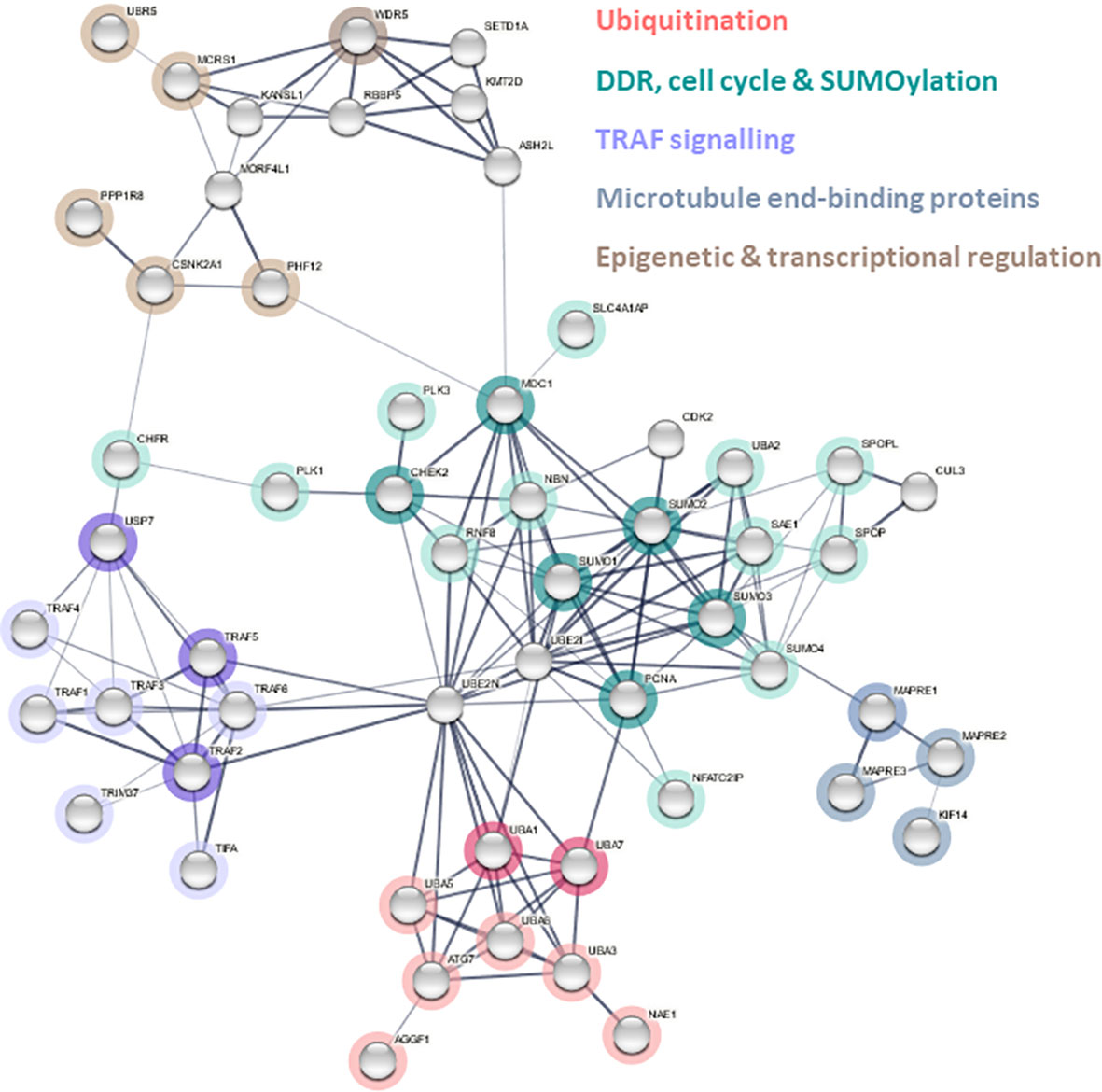
Figure 3 Predicted ERVK protease interactome. Cellular proteins containing complementary interaction motifs for ELMs identified in ERVK PR were used to identify potential protein interactors used as query proteins for STRING network analysis version 11.5. Edges indicate both functional and physical protein associations. A payload list was generated to colour nodes and hubs related to dominant pathways: ubiquitination (red), DNA damage response (DDR)/cell cycle/SUMOylation (teal), TRAF signaling (purple), microtubule end-binding protein (grey), epigenetic and transcriptional regulation (brown). Pale grey nodes indicate putative second shell interactors.
Of note, ERVK employs a different strategy for end-binding protein interaction than HIV. While ERVK viral enzymes contain an SxIP motif that binds EB proteins, conversely HIV capsid has EB-like motifs that interact with SxIP motifs in +TIP (75).
3.4. ERVK interactome reveals association with a diversity of cellular pathways
A list of potential interacting proteins was compiled based on the ELMs identified in ERVK PR, and an ERVK PR interactome network was constructed using STRING software (Figure 3). The ERVK PR network contained 102 nodes and 154 edges (expected number of edges 47), resulting in a significant PPI enrichment (p<1.0e-16). All direct ERVK PR interactor proteins (Table S7, Figure 3 coloured nodes) are shown with links to a maximum of 10 second shell interactions (uncoloured nodes).
Central hub proteins within the curated STRING network were identified based on a node degree ≥10 and included ubiquitin-conjugating enzyme E2 N (UBE2N), ubiquitin-conjugating enzyme E2 I (UBE2I), Small Ubiquitin Like Modifiers (SUMO-1, SUMO-2, and SUMO-3), mediator of DNA damage checkpoint 1 (MDC1), PCNA, and TNF receptor associated factor 6 (TRAF6).
Figure 4 depicts a heat map of the most significant Reactome pathways associated with the ERVK PR network. Major pathways emerge under groupings of cell cycle control, ubiquitination, DDR, transcriptional regulation, nervous system, immunity, and metabolism (see Figure 4 and Table S8 for full Reactome listing). Similarly, enriched KEGG pathways consisted of ubiquitin-mediated proteolysis (p=4.92 x 10-9), small cell lung cancer (p=1.4 x 10-4), NF-κB signaling pathway (p=1.7 x 10-4) and viral carcinogenesis (p=0.0049) (Table S9). Further analysis of the network included UniProt keywords and GO function analysis (Tables S10 and S11, respectively), which corroborated many of the pathways identified above.
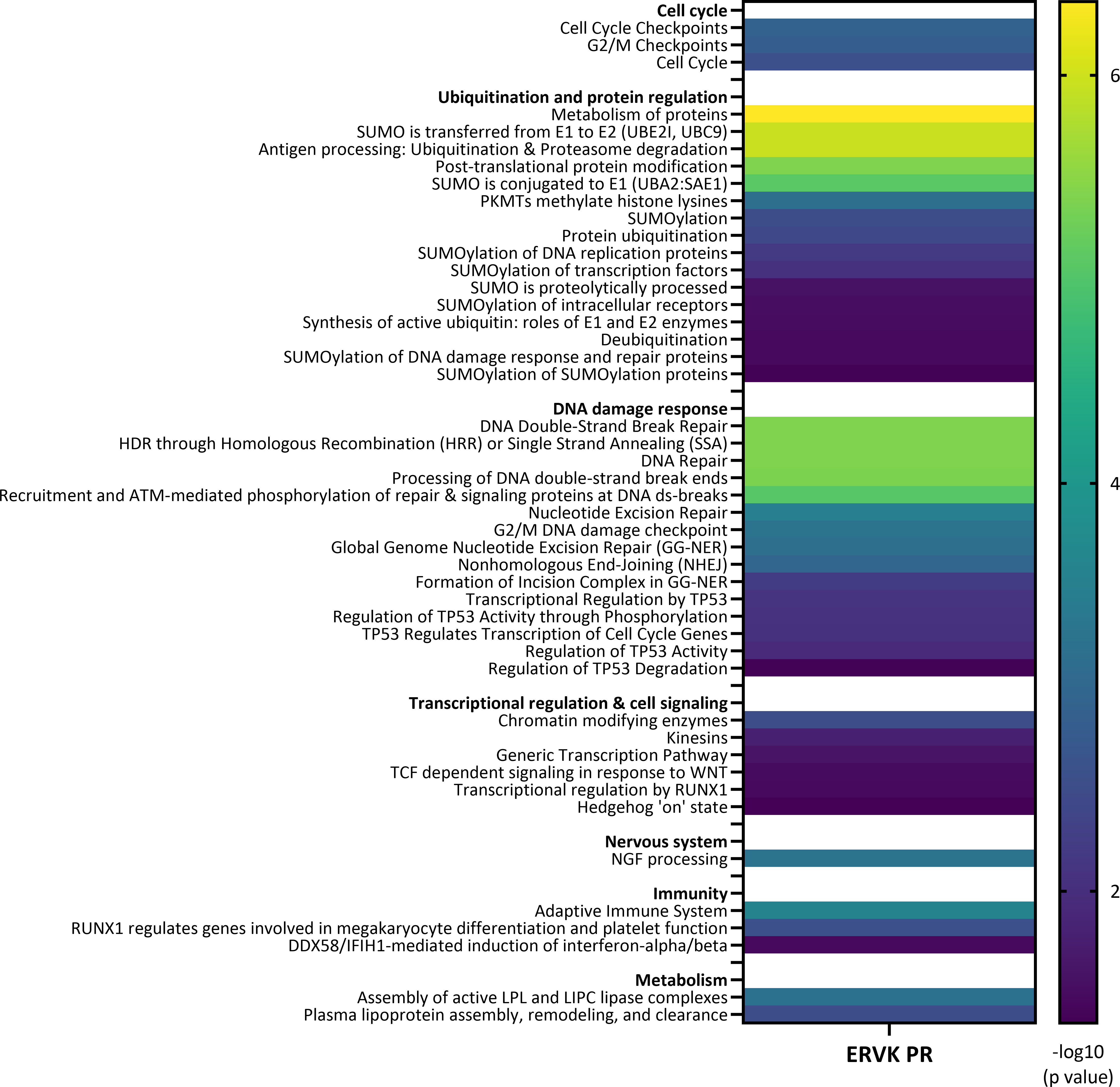
Figure 4 Pathways associated with ERVK protease interactome. Heat map of enriched Reactome pathways associated with the ERVK PR interactome based on STRING network analysis. Enriched terms are reported along with significance scores (-log10 p value). ERVK PR is predicted to interact with cellular pathways involved in cell cycle, ubiquitination and protein regulation, DNA damage response, transcriptional regulation and cell signaling, nervous system, immunity, and metabolism.
3.4.1 ERVK protease is associated with the apex of ubiquitin-like pathways
Ubiquitination is implicated in many cellular processes, including the cell cycle, DDR, and cell signaling; thus, its repeated implication in the ERVK PR interactome points to the importance of ubiquitin control in retroviral replication (76–78). The ERVK PR network highlights putative interaction partners at the apex of protein modification pathways, namely E1 ubiquitin-activating enzymes (UBA1, UBA3, UBA7, ATG7) and SUMO proteins (Figure 3). While UBA1 is implicated in ubiquitination processes related to protein homeostasis, UBA3 is a key component of the NEDDylation pathway, UBA7 activates interferon stimulated gene 15 (ISG15) to induce an antiviral response, and ATG7 activates autophagy related protein 8 (ATG8) to initiate autophagy (79). In contrast, SUMO modification often alters protein-protein interactions and protein affinity for a target (79). Together, this suggests that ERVK PR may modulate several ubiquitin-like protein (UBL) pathways by engaging with initiator E1 proteins.
Ubiquitin-like modifier-activating enzyme 1 (UBA1) was determined to be a significant node within the ERVK PR interactome. UBA1 is known to function as an essential component of ubiquitination, contributing to both ubiquitin-proteasome degradation and selective autophagy systems (80, 81). In addition to its function in protein degradation, UBA1-mediated ubiquitylation is also an important mechanism involved in cell cycle progression, DDR, and apoptosis (80). Furthermore, impaired UBA1 activity within the nervous system has proven to be detrimental to neuronal function and health (82). Murine, zebrafish, and fruit fly models of UBA1 impairment in motor neurons have demonstrated severe defects in locomotion and evidence of neurodegeneration (82, 83). We suspect that ERVK PR interactions with UBA1 directly interfere with its ability to facilitate ubiquitination in proteasomal degradation pathways, ultimately contributing to the neurodegeneration of motor neurons in Amyotrophic Lateral Sclerosis (ALS). When UBA1 activity within the nucleus is diminished, the repair of DNA double-stranded breaks is severely impaired (84) and associated with defects in apoptosis and enhanced tissue growth (85), suggesting an additional role in oncogenesis.
USP7 is a deubiquitinating enzyme that plays a significant role in cell signaling through its regulation of substrate stability and degradation (86). Predicted to interact with all betaretroviral PRs, USP7 is a ubiquitin-specific protease which plays a role in DDR, immune response, epigenetic control, and cancer (49, 50). USP7 is targeted in viral evasion of innate immunity, with specific strategies employed by Herpes simplex virus 1 (HSV-1) and Ebola virus (49, 87, 88). USP7 TRAF domain-mediated interactions are mutually exclusive as interaction partners all bind the identical interface (89). This can be seen in USP7 partner switches depending on the post-translational modification status of Mdm2 and p53 (89). Thus, when a viral protein interacts with USP7 via its TRAF domain, it may displace cellular proteins or covet the site thus preventing cellular interactors from engaging USP7. Indeed, the vIRF4 protein from Kaposi’s sarcoma-associated herpesvirus (KSHV) binds the USP7 TRAF domain and competitively blocks substrate binding of MDM2 and p53 (90). Similarly, the Epstein-Barr nuclear antigen 1 (EBNA1) protein of EBV also preferentially binds the TRAF domain of USP7, resulting in destabilizing p53 and reducing p53 levels (91, 92). Considering that ERVK PR contains a conserved USP7 TRAF domain binding motif, as well as a USP7 UBL2 domain binding site (similar to that used by HSV-1 ICP0 protein (93)), we predict that ERVK PR engages USP7 and may alter signaling pathways related to innate immunity and cancer. ERVK PR’s putative dual interactions with USP7 has implications for p53 regulation (Figure 4, Table S8), and thus oncogenic processes (86). Specifically, network analysis revealed putative p53 regulation occurring at the level of p53 transcription, phosphorylation, activity, degradation, as well as p53 involvement in control of cell cycle genes (Table S8).
Another pathway implicated in the ERVK PR interactome is NEDDylation, a form of post-translational modification where ubiquitin-like protein NEDD8 is conjugated to its target proteins. NEDD8-activating enzyme (NAE) is formed by a heterodimer of ubiquitin-like modifier activating enzyme 3 (UBA3, a putative betaretroviral PR interactor) and amyloid precursor protein binding protein-1 (APPBP1) (79). The PR-UBA3 interaction has notable implications for impacting antiviral immunity, based on strong evidence for a necessity of NEDDylation-associated cullin-RING E3 ubiquitin ligases in retroviral infections, as a means to overcome cellular viral restriction factors APOBEC and SAMHD1 (94). However, given the broad antiviral effect of NEDDylation inhibitor MLN4924 (95), and the role of NEDDylation in activating NF-κB, IRFs, and interferon production (96, 97), it remains unclear how PR involvement in NEDDylation may impact ERVK activity.
ERVK PR was also associated with ISGylation. Putative ERVK PR interaction partner UBA7 activates interferon−stimulated gene 15 (ISG15) conjugation to a variety of target proteins related to interferon response (98). Both cellular and viral proteins can be targeted for ISGylation, with impacts on innate immune signaling, as well as viral replication (98, 99). Select proteins associated with the ERVK PR interactome (PCNA, p53, NBN, UBA1, ATG7, SUMO2, TRAF2) are known to be modified by ISG15 (99), yet the downstream effects are still largely unknown. It is unclear whether ERVK viral proteins are ISGylated or what the influence may be on viral proteins in host cells.
An affinity for SUMO proteins was also evident given the SIM motifs within the ERVK PR, in conjunction with putative interaction with several SUMOylated proteins including PCNA and MDC1. Many viruses target UBC9/UBE2I as it is the sole E2 SUMO-conjugating enzyme in the SUMOylation pathway (100); therefore, its identification as a hub protein in the ERVK PR interactome deserves further investigation. SUMOylation is essential for HIV integrase-mediated proviral insertion into the host genome and virion infectivity, yet SUMO modification of cellular protein IκBα sequesters NF-κB in the cytoplasm and limits HIV-1 transcription (101). This indicates that control over SUMOylation processes is an important component of HIV replication, specifically as it pertains to regulation of DDR and innate immune signaling (102). Similarly, all betatretroviral PRs were predicted to encode SIM motifs, indicating a conservation for recognition of SUMOylated proteins among this group of PRs.
3.4.2 TRAF Signaling
Tumor necrosis factor receptor (TNF-R)-associated factors (TRAFs) are signaling adaptors that modulate the inflammatory response, often through their regulation of canonical and non-canonical NF-κB signaling (103). Moreover, USP7, TRAF2 and TRAF5 contain a RING domain, granting them E3 ubiquitin ligase activity (103, 104). Ubiquitin E3 ligases TRAF2, TRAF5, and TRAF6 appear to be key proteins that potentially link ERVK PR activity to NF-κB, MAPK, and RIP1 signaling pathways (103, 105). ERVK PR binding of the TRAF MATH domain may have agonistic or inhibitory effects on TRAF’s ability to modulate intracellular signaling. Both HIV-1 Nef protein and hepatitis C virus Core protein activate TRAF signaling, leading to a subsequent increase in NF-κB activation (106). It is possible that ERVK PR participates in a similar mechanism that recruits TRAFs to enhance NF-κB activation, which ultimately facilitates ERVK viral replication through transcription via NF-κB -binding sites on ERVK LTRs (107). Thus, USP7 and TRAF protein involvement in the ERVK PR interactome points to a potential impact on NF-κB signaling (Figure 3, Table S9), as identified by KEGG pathway analysis (hsa04064, p<0.00017).
3.4.3 DDR, cell cycle, and processes regulated by SUMOylation
DNA repair pathways were a strong component of the ERVK PR network (Figure 4, Table S8). Identified mechanisms included DNA double-strand break processing and repair, recruitment and ATM-mediated phosphorylation of repair and signaling proteins at DNA double- strand breaks. Homology directed repair (HDR) through homologous recombination (HR), or single strand annealing (SSA), were also identified as repair strategies for double-strand DNA lesions. Additionally, global genome nucleotide excision repair (GG-NER), nonhomologous end-joining (NHEJ), and cell cycle-related DNA damage checkpoints were identified as activities within the ERVK PR network. Phospho-Ser/Thr binding domain proteins are key hub proteins in cell cycle regulation and DDR (47); specifically, FHA domain protein interaction sites (4), WD40 repeat interaction sites (3), PIP box (1), and PLK phosphorylation sites (3) were identified in ERVK PR (Tables 1 and 2, Figure 1). In contrast to exogenous and most endogenous PRs, fewer WD40 repeat domain WDR5 interaction sites were found in ERVK PR (3 vs. 4-9 sites each). This suggests that ERVK PR has potentially shifted away from WD40 domain interaction in favour PCNA interaction as a means to connect with DDR pathways (47, 108). Involvement in these processes may be related to requirements for optimal ERVK integrase activity and ERVK integrase-mediated DNA damage caused during viral DNA integration in the host genome (109–111).
The SUMOylation cluster is the most widely connected throughout the ERVK PR interactome (Figure 3), displaying the greatest number of interactions with the other network clusters. The Reactome analysis identified SUMOylation of DNA replication proteins and DDR proteins as processes within the ERVK PR network (Figure 4, Table S8). MDC1 was identified as a hub protein of the ERVK PR interactome and is involved in ATM-mediated DNA repair mechanisms and cell cycle checkpoint control (112). SUMOylation of MDC1 is essential for effective DDR. MDC1 can only be SUMOylated and degraded at sites of DNA damage, and disruption of this process is associated with impaired HR repair (113). Neighbouring primary nodes to MDC1 included RNF8 and NBN, both involved in MDC1-mediated HR repair (114) and NHEJ (115), respectively (Figure 3). Deficiency or allelic variation in MDC1, RNF8, and NBN results in susceptibility to cancer (115–118).
3.4.4 Histone modification & transcriptional regulation
The high conservation of WDR5 among multicellular organisms may be foundational to its consistent targeting by retroviral PRs (Tables 1 and 2). WDR5 has a canonical role in histone methylation; however, it also plays a role as a histone tail reader and transcriptional regulator of protein synthesis genes (119, 120). While ERVK PR contains fewer WDR5 binding linear motifs than other PRs, its position in the interactome strongly suggests ties with both TRAF signaling and DDR (Figure 3).
3.5. Disease pathways implicated in the ERVK protease interactome
Disease-associated processes that were significantly enriched in the network included small cell lung cancer (KEGG, p=0.00014), host-virus interactions (UniProt, p=0.0029), viral carcinogenesis (KEGG, p=0.0049), Epstein-Barr virus (EBV) infection (KEGG, p=0.0355), and neurodegeneration (UniProt, p=0.0453). This points to ERVK PR having a role in virus-mediated oncogenesis and neurodegenerative disease, conditions previously associated with the activity of ERVK (121–127). Engagement of PCNA (55, 128), USP7 (89, 129) and TRAF signaling cascades (103) by ERVK PR may feed into NF-κB driven and pro-oncogenic pathways. Additionally, hub proteins PCNA (55), UBA1 (80), and UBA5 (130) were identified in the ERVK PR interactome and are implicated in neurodegeneration.
4. Conclusion
Improving our understanding of how ERVK PR modulates cellular pathways is a first step toward developing effective antiretroviral therapy for ERVK-associated diseases. Despite their efficiency in combating HIV infection (131), protease inhibitor drugs are significantly less effective at inhibiting ERVK PR (16, 132). Moreover, protease inhibitors are also candidate drugs for treating cancer (133). Our work highlights that ERVK PR likely modulates ubiquitin-like pathways and DDR, thus strengthening the association of ERVK with neurodegenerative disease and cancer. As such, ERVK PR may be useful as a target for future drug development in combating the progression of ALS and ERVK-driven oncogenesis. However, it is possible that the cellular impact of ERVK PR is not solely based on its enzymatic activity, and thus additional investigation into the molecular pathways perturbed by this viral protein is warranted.
Dedication
This study is dedicated to patients with ALS—we are working on it!
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
Data curation, methodology, validation, investigation, and formal analysis by SN, MR, AV, and RD. Conceptualization, supervision, project administration, resources, visualization, and funding acquisition by RD. Writing original draft preparation by RD, review and editing by SN, MR, AV, and RD. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by a Discovery grant from the Natural Sciences and Engineering Research Council of Canada (NSERC, RGPIN-2016-05761).
Acknowledgments
We apologize to all colleagues whose work was not cited due to space limitations or any oversights on our part. We thank Signy Brownell for initial investigation into the ERVK protease interactome. The authors appreciate input from Ilena Benoit and Dr. Domenico Di Curzio as part of their peer review of the manuscript. We also acknowledge Dr. Di Curzio for donating his likeness to represent the face of humanity for Figure 2.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2022.972156/full#supplementary-material
Supplementary Table 1 | Exogenous retroviruses with similarity to ERVK protease based on BLASTp search.
Supplementary Table 2 | Eutherian genomes with similarity to ERVK protease based on BLASTp searches.
Supplementary Table 3 | Marsupial genomes with similarity to ERVK protease based on BLASTp searches.
Supplementary Table 4 | Aves genomes with similarity to ERVK protease based on BLASTp searches.
Supplementary Table 5 | Ecdysozoa genomes with similarity to ERVK protease based on BLASTp searches.
Supplementary Table 6 | Eubacteria genomes with similarity to ERVK protease based on BLASTp searches.
Supplementary Table 7 | List of query proteins for STRING analysis based on ELM interaction motifs in ERVK protease.
Supplementary Table 8 | Full list of reactome pathways identified in the STRING analysis of the ERVK protease interactome.
Supplementary Table 9 | Full list of KEGG pathways identified in the STRING analysis of the ERVK protease interactome.
Supplementary Table 10 | Full list of UniProt keywords identified in the STRING analysis of the ERVK protease interactome.
Supplementary Table 11 | Full list of gene ontology function terms identified in the STRING analysis of the ERVK protease interactome.
References
1. Christiaansen A, Varga SM, Spencer JV. Viral manipulation of the host immune response. Curr Opin Immunol (2015) 36:54–60. doi: 10.1016/j.coi.2015.06.012
2. McLean JE, Ruck A, Shirazian A, Pooyaei-Mehr F, Zakeri ZF. Viral manipulation of cell death. Curr Pharm Des (2008) 14:198–220. doi: 10.2174/138161208783413329
3. Subramanian RP, Wildschutte JH, Russo C, Coffin JM. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology (2011) 8:90. doi: 10.1186/1742-4690-8-90
4. Belshaw R, Dawson AL, Woolven-Allen J, Redding J, Burt A, Tristem M. Genomewide screening reveals high levels of insertional polymorphism in the human endogenous retrovirus family HERV-K(HML2): implications for present-day activity. J Virol (2005) 79:12507–14. doi: 10.1128/JVI.79.19.12507-12514.2005
5. Ruprecht K, Mayer J, Sauter M, Roemer K, Mueller-Lantzsch N. Endogenous retroviruses and cancer. Cell Mol Life Sci (2008) 65:3366–82. doi: 10.1007/s00018-008-8496-1
6. Balada E, Ordi-Ros J, Vilardell-Tarres M. Molecular mechanisms mediated by human endogenous retroviruses (HERVs) in autoimmunity. Rev Med Virol (2009) 19:273–86. doi: 10.1002/rmv.622
7. Romer C. Viruses and endogenous retroviruses as roots for neuroinflammation and neurodegenerative diseases. Front Neurosci (2021) 15:648629. doi: 10.3389/fnins.2021.648629
8. Garcia-Montojo M, Doucet-O'Hare T, Henderson L, Nath A. Human endogenous retrovirus-K (HML-2): a comprehensive review. Crit Rev Microbiol (2018) 44(6):1–24. doi: 10.1080/1040841X.2018.1501345
9. Downey RF, Sullivan FJ, Wang-Johanning F, Ambs S, Giles FJ, Glynn SA. Human endogenous retrovirus K and cancer: Innocent bystander or tumorigenic accomplice? Int J Cancer (2015) 137:1249–57. doi: 10.1002/ijc.29003
10. Freimanis G, Hooley P, Ejtehadi HD, Ali HA, Veitch A, Rylance PB, et al. A role for human endogenous retrovirus-K (HML-2) in rheumatoid arthritis: investigating mechanisms of pathogenesis. Clin Exp Immunol (2010) 160:340–7. doi: 10.1111/j.1365-2249.2010.04110.x
11. Mahmoudabadi G, Phillips R. A comprehensive and quantitative exploration of thousands of viral genomes. eLife (2018) 7:e31955. doi: 10.7554/eLife.31955
12. Jager S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, et al. Global landscape of HIV-human protein complexes. Nature (2011) 481:365–70. doi: 10.1038/nature10719
13. Li G, De Clercq E. HIV Genome-wide protein associations: a review of 30 years of research. Microbiol Mol Biol Rev (2016) 80:679–731. doi: 10.1128/MMBR.00065-15
14. Weber IT, Wang YF, Harrison RW. HIV Protease: Historical perspective and current research. Viruses (2021) 13(5):839. doi: 10.3390/v13050839
15. Mueller-Lantzsch N, Sauter M, Weiskircher A, Kramer K, Best B, Buck M, et al. Human endogenous retroviral element K10 (HERV-K10) encodes a full-length gag homologous 73-kDa protein and a functional protease. AIDS Res Hum Retroviruses (1993) 9:343–50. doi: 10.1089/aid.1993.9.343
16. Towler EM, Gulnik SV, Bhat TN, Xie D, Gustschina E, Sumpter TR, et al. Functional characterization of the protease of human endogenous retrovirus, K10: can it complement HIV-1 protease? Biochemistry (1998) 37:17137–44. doi: 10.1021/bi9818927
17. Kuhelj R, Rizzo CJ, Chang CH, Jadhav PK, Towler EM, Korant BD. Inhibition of human endogenous retrovirus-K10 protease in cell-free and cell-based assays. J Biol Chem (2001) 276:16674–82. doi: 10.1074/jbc.M008763200
18. Schommer S, Sauter M, Krausslich HG, Best B, Mueller-Lantzsch N. Characterization of the human endogenous retrovirus K proteinase. J Gen Virol (1996) 77(Pt 2):375–9. doi: 10.1099/0022-1317-77-2-375
19. Turnbull MG, Douville RN. Related endogenous retrovirus-K elements harbor distinct protease active site motifs. Front Microbiol (2018) 9:1577. doi: 10.3389/fmicb.2018.01577
20. Dewannieux M, Harper F, Richaud A, Letzelter C, Ribet D, Pierron G, et al. Identification of an infectious progenitor for the multiple-copy HERV-K human endogenous retroelements. Genome Res (2006) 16:1548–56. doi: 10.1101/gr.5565706
21. Lee YN, Bieniasz PD. Reconstitution of an infectious human endogenous retrovirus. PLoS Pathog (2007) 3:e10. doi: 10.1371/journal.ppat.0030010
22. Dunn BM, Goodenow MM, Gustchina A, Wlodawer A. Retroviral proteases. Genome Biol (2002) 3:REVIEWS3006. doi: 10.1186/gb-2002-3-4-reviews3006.
23. Rigogliuso G, Biniossek ML, Goodier JL, Mayer B, Pereira GC, Schilling O, et al. A human endogenous retrovirus encoded protease potentially cleaves numerous cellular proteins. Mob DNA (2019) 10:36. doi: 10.1186/s13100-019-0178-z
24. Kraus B, Boller K, Reuter A, Schnierle BS. Characterization of the human endogenous retrovirus K gag protein: identification of protease cleavage sites. Retrovirology (2011) 8:21. doi: 10.1186/1742-4690-8-21
25. Krupovic M, Blomberg J, Coffin JM, Dasgupta I, Fan H, Geering AD, et al. Ortervirales: A new viral order unifying five families of reverse-transcribing viruses. J Virol (2018); 92(12):e00515–18. doi: 10.1128/JVI.00515-18
26. Riviere Y, Blank V, Kourilsky P, Israel A. Processing of the precursor of NF-kappa b by the HIV-1 protease during acute infection. Nature (1991) 350:625–6. doi: 10.1038/350625a0
27. Mackiewicz MM, Overk C, Achim CL, Masliah E. Pathogenesis of age-related HIV neurodegeneration. J Neurovirol (2019) 25:622–33. doi: 10.1007/s13365-019-00728-z
28. Kwong LK, Neumann M, Sampathu DM, Lee VM, Trojanowski JQ. TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol (2007) 114:63–70. doi: 10.1007/s00401-007-0226-5
29. Solis M, Nakhaei P, Jalalirad M, Lacoste J, Douville R, Arguello M, et al. RIG-i-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J Virol (2011) 85:1224–36. doi: 10.1128/JVI.01635-10
30. Impens F, Timmerman E, Staes A, Moens K, Arien KK, Verhasselt B, et al. A catalogue of putative HIV-1 protease host cell substrates. Biol Chem (2012) 393:915–31. doi: 10.1515/hsz-2012-0168
31. Shoeman RL, Kesselmier C, Mothes E, Honer B, Traub P. Non-viral cellular substrates for human immunodeficiency virus type 1 protease. FEBS Lett (1991) 278:199–203. doi: 10.1016/0014-5793(91)80116-K
32. Tozser J, Weber IT. The protease of human T-cell leukemia virus type-1 is a potential therapeutic target. Curr Pharm Des (2007) 13:1285–94. doi: 10.2174/138161207780618849
33. Wlodawer A, Gustchina A. Structural and biochemical studies of retroviral proteases. Biochim Biophys Acta (2000) 1477:16–34. doi: 10.1016/S0167-4838(99)00267-8
34. BLASTp. Bethesda (MD: National Library of Medicine (US), National Center for Biotechnology Information (2004). 2021/12/01.
35. Rosindell J, Harmon LJ. OneZoom: a fractal explorer for the tree of life. PLoS Biol (2012) 10:e1001406. doi: 10.1371/journal.pbio.1001406
36. Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics (2012) 28:1647–9. doi: 10.1093/bioinformatics/bts199
37. Lu S, Wang J, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, et al. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res (2020) 48:D265–8. doi: 10.1093/nar/gkz991
38. Kumar M, Gouw M, Michael S, Sámano-Sánchez H, Pancsa R, Glavina J, et al. ELM-the eukaryotic linear motif resource in 2020. Nucleic Acids Res (2020) 48:D296–d306. doi: 10.1093/nar/gkz1030.
39. Eder J, Hommel U, Cumin F, Martoglio B, Gerhartz B. Aspartic proteases in drug discovery. Curr Pharm Des (2007) 13:271–85. doi: 10.2174/138161207779313560
40. Yang H, Nkeze J, Zhao RY. Effects of HIV-1 protease on cellular functions and their potential applications in antiretroviral therapy. Cell biosci (2012) 2:32. doi: 10.1186/2045-3701-2-32
41. Colbert JD, Matthews SP, Miller G, Watts C. Diverse regulatory roles for lysosomal proteases in the immune response. Eur J Immunol (2009) 39:2955–65. doi: 10.1002/eji.200939650
42. Wang Q, Gao H, Clark KM, Mugisha CS, Davis K, Tang JP, et al. CARD8 is an inflammasome sensor for HIV-1 protease activity. Science (2021) 371(6535):eabe1707. doi: 10.1126/science.abe1707
43. Jeremiah SS, Miyakawa K, Matsunaga S, Nishi M, Kudoh A, Takaoka A, et al. Cleavage of TANK-binding kinase 1 by HIV-1 protease triggers viral innate immune evasion. Front Microbiol (2021) 12:643407. doi: 10.3389/fmicb.2021.643407
44. Serbyn N, Noireterre A, Bagdiul I, Plank M, Michel AH, Loewith R, et al. The aspartic protease Ddi1 contributes to DNA-protein crosslink repair in yeast. Mol Cell (2020) 77:1066–1079.e9. doi: 10.1016/j.molcel.2019.12.007
45. Kaminskyy V, Zhivotovsky B. Proteases in autophagy. Biochim Biophys Acta (2012) 1824:44–50. doi: 10.1016/j.bbapap.2011.05.013
46. Dominguez DI, Hartmann D, De Strooper B. BACE1 and presenilin: two unusual aspartyl proteases involved in alzheimer's disease. Neurodegener Dis (2004) 1:168–74. doi: 10.1159/000080982
47. Reinhardt HC, Yaffe MB. Phospho-Ser/Thr-binding domains: navigating the cell cycle and DNA damage response. Nat Rev Mol Cell Biol (2013) 14:563–80. doi: 10.1038/nrm3640
48. Mohammad DH, Yaffe MB. 14-3-3 proteins, FHA domains and BRCT domains in the DNA damage response. DNA Repair (Amst) (2009) 8:1009–17. doi: 10.1016/j.dnarep.2009.04.004
49. Bhattacharya S, Chakraborty D, Basu M, Ghosh MK. Emerging insights into HAUSP (USP7) in physiology, cancer and other diseases. Signal Transduct Target Ther (2018) 3:17. doi: 10.1038/s41392-018-0012-y
50. Zhou L, Ouyang T, Li M, Hong T, Mhs A, Meng W, et al. Ubiquitin-specific peptidase 7: A novel deubiquitinase that regulates protein homeostasis and cancers. Front Oncol (2021) 11:784672. doi: 10.3389/fonc.2021.784672
51. Fry AM, O'Regan L, Sabir SR, Bayliss R. Cell cycle regulation by the NEK family of protein kinases. J Cell Sci (2012) 125:4423–33. doi: 10.1242/jcs.111195
52. Kumar S, Sharma G, Chakraborty C, Sharma AR, Kim J. Regulatory functional territory of PLK-1 and their substrates beyond mitosis. Oncotarget (2017) 8:37942–62. doi: 10.18632/oncotarget.16290
53. Knippschild U, Kruger M, Richter J, Xu P, Garcia-Reyes B, Peifer C, et al. The CK1 family: Contribution to cellular stress response and its role in carcinogenesis. Front Oncol (2014) 4:96. doi: 10.3389/fonc.2014.00096
54. Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther (2015) 148:114–31. doi: 10.1016/j.pharmthera.2014.11.016
55. Choe KN, Moldovan GL. Forging ahead through darkness: PCNA, still the principal conductor at the replication fork. Mol Cell (2017) 65:380–92. doi: 10.1016/j.molcel.2016.12.020
56. Kumar P, Wittmann T. +TIPs: SxIPping along microtubule ends. Trends Cell Biol (2012) 22:418–28. doi: 10.1016/j.tcb.2012.05.005
57. Benoit I, Brownell S, Douville RN. Predicted cellular interactors of the endogenous retrovirus-K integrase enzyme. Microorganisms (2021) 9(7):1509. doi: 10.3390/microorganisms9071509
58. Boehm EM, Washington MT. R.I.P. to the PIP: PCNA-binding motif no longer considered specific: PIP motifs and other related sequences are not distinct entities and can bind multiple proteins involved in genome maintenance. Bioessays (2016) 38:1117–22. doi: 10.1002/bies.201600116
59. Prestel A, Wichmann N, Martins JM, Marabini R, Kassem N, Broendum SS, et al. The PCNA interaction motifs revisited: thinking outside the PIP-box. Cell Mol Life Sci (2019) 76:4923–43. doi: 10.1007/s00018-019-03150-0
60. Armstrong AA, Mohideen F, Lima CD. Recognition of SUMO-modified PCNA requires tandem receptor motifs in Srs2. Nature (2012) 483:59–63. doi: 10.1038/nature10883
61. Motegi A, Liaw HJ, Lee KY, Roest HP, Maas A, Wu X, et al. Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc Natl Acad Sci U S A (2008) 105:12411–6. doi: 10.1073/pnas.0805685105.
62. Whitehurst CB, Vaziri C, Shackelford J, Pagano JS. Epstein-Barr Virus BPLF1 deubiquitinates PCNA and attenuates polymerase eta recruitment to DNA damage sites. J Virol (2012) 86:8097–106. doi: 10.1128/JVI.00588-12
63. Dong X, Guan J, Zheng C, Zheng X. The herpes simplex virus 1 UL36USP deubiquitinase suppresses DNA repair in host cells via deubiquitination of proliferating cell nuclear antigen. J Biol Chem (2017) 292:8472–83. doi: 10.1074/jbc.M117.778076
64. Kashiwaba S, Kanao R, Masuda Y, Kusumoto-Matsuo R, Hanaoka F, Masutani C. USP7 is a suppressor of PCNA ubiquitination and oxidative-Stress-Induced mutagenesis in human cells. Cell Rep (2015) 13:2072–80. doi: 10.1016/j.celrep.2015.11.014
65. Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature (2002) 419:135–41. doi: 10.1038/nature00991
66. Marini V, Krejci L. Srs2: the "Odd-job man" in DNA repair. DNA Repair (Amst) (2010) 9:268–75. doi: 10.1016/j.dnarep.2010.01.007
67. Bacquin A, Pouvelle C, Siaud N, Perderiset M, Salome-Desnoulez S, Tellier-Lebegue C, et al. The helicase FBH1 is tightly regulated by PCNA via CRL4(Cdt2)-mediated proteolysis in human cells. Nucleic Acids Res (2013) 41:6501–13. doi: 10.1093/nar/gkt397
68. Postigo A, Ramsden AE, Howell M, Way M. Cytoplasmic ATR activation promotes vaccinia virus genome replication. Cell Rep (2017) 19:1022–32. doi: 10.1016/j.celrep.2017.04.025
69. Adeyemi RO, Fuller MS, Pintel DJ. Efficient parvovirus replication requires CRL4Cdt2-targeted depletion of p21 to prevent its inhibitory interaction with PCNA. PloS Pathog (2014) 10:e1004055. doi: 10.1371/journal.ppat.1004055
70. Daikoku T, Kudoh A, Sugaya Y, Iwahori S, Shirata N, Isomura H, et al. Postreplicative mismatch repair factors are recruited to Epstein-Barr virus replication compartments. J Biol Chem (2006) 281:11422–30. doi: 10.1074/jbc.M510314200
71. Bagewadi B, Chen S, Lal SK, Choudhury NR, Mukherjee SK. PCNA interacts with Indian mung bean yellow mosaic virus rep and downregulates rep activity. J Virol (2004) 78:11890–903. doi: 10.1128/JVI.78.21.11890-11903.2004
72. Lee CC, Wang JW, Leu WM, Huang YT, Huang YW, Hsu YH, et al. Proliferating cell nuclear antigen suppresses RNA replication of bamboo mosaic virus through an interaction with the viral genome. J Virol (2019) 93(22):e00961–19. doi: 10.1128/JVI.00961-19
73. Naghavi MH, Walsh D. Microtubule regulation and function during virus infection. J Virol (2017) 91(16):e00538-17. doi: 10.1128/JVI.00538-17
74. Wan J, Subramonian D, Zhang XD. SUMOylation in control of accurate chromosome segregation during mitosis. Curr Protein Pept Sci (2012) 13:467–81. doi: 10.2174/138920312802430563
75. Santos da Silva E, Shanmugapriya S, Malikov V, Gu F, Delaney MK, Naghavi MH. HIV-1 capsids mimic a microtubule regulator to coordinate early stages of infection. EMBO J (2020) 39:e104870. doi: 10.15252/embj.2020104870.
76. Patnaik A, Chau V, Wills JW. Ubiquitin is part of the retrovirus budding machinery. Proc Natl Acad Sci U S A (2000) 97:13069–74. doi: 10.1073/pnas.97.24.13069.
77. Vogt VM. Ubiquitin in retrovirus assembly: actor or bystander? Proc Natl Acad Sci U S A (2000) 97:12945–7. doi: 10.1073/pnas.97.24.12945.
78. Colomer-Lluch M, Castro-Gonzalez S, Serra-Moreno R. Ubiquitination and SUMOylation in HIV infection: Friends and foes. Curr Issues Mol Biol (2020) 35:159–94. doi: 10.21775/cimb.035.159
79. Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat Rev Mol Cell Biol (2009) 10:319–31. doi: 10.1038/nrm2673
80. Lambert-Smith IA, Saunders DN, Yerbury JJ. The pivotal role of ubiquitin-activating enzyme E1 (UBA1) in neuronal health and neurodegeneration. Int J Biochem Cell Biol (2020) 123:105746. doi: 10.1016/j.biocel.2020.105746
81. Groen EJN, Gillingwater TH. UBA1: At the crossroads of ubiquitin homeostasis and neurodegeneration. Trends Mol Med (2015) 21:622–32. doi: 10.1016/j.molmed.2015.08.003
82. Liu HY, Pfleger CM. Mutation in E1, the ubiquitin activating enzyme, reduces drosophila lifespan and results in motor impairment. PloS One (2013) 8:e32835. doi: 10.1371/journal.pone.0032835
83. Wishart TM, Mutsaers CA, Riessland M, Reimer MM, Hunter G, Hannam ML, et al. Dysregulation of ubiquitin homeostasis and beta-catenin signaling promote spinal muscular atrophy. J Clin Invest (2014) 124:1821–34. doi: 10.1172/JCI71318
84. Moudry P, Lukas C, Macurek L, Hanzlikova H, Hodny Z, Lukas J, et al. Ubiquitin-activating enzyme UBA1 is required for cellular response to DNA damage. Cell Cycle (2012) 11:1573–82. doi: 10.4161/cc.19978
85. Pfleger CM, Harvey KF, Yan H, Hariharan IK. Mutation of the gene encoding the ubiquitin activating enzyme ubal causes tissue overgrowth in drosophila. Fly (Austin) (2007) 1:95–105. doi: 10.4161/fly.4285
86. Wang Z, Kang W, You Y, Pang J, Ren H, Suo Z, et al. USP7: Novel drug target in cancer therapy. Front Pharmacol (2019) 10:427. doi: 10.3389/fphar.2019.00427
87. Daubeuf S, Singh D, Tan Y, Liu H, Federoff HJ, Bowers WJ, et al. HSV ICP0 recruits USP7 to modulate TLR-mediated innate response. Blood (2009) 113:3264–75. doi: 10.1182/blood-2008-07-168203
88. Vidal S, El Motiam A, Seoane R, Preitakaite V, Bouzaher YH, Gomez-Medina S, et al. Regulation of the Ebola virus VP24 protein by SUMO. J Virol (2019) 94(1):e01687-19. doi: 10.1128/JVI.01687-19
89. Valles GJ, Bezsonova I, Woodgate R, Ashton NW. USP7 is a master regulator of genome stability. Front Cell Dev Biol (2020) 8:717. doi: 10.3389/fcell.2020.00717
90. Lee HR, Choi WC, Lee S, Hwang J, Hwang E, Guchhait K, et al. Bilateral inhibition of HAUSP deubiquitinase by a viral interferon regulatory factor protein. Nat Struct Mol Biol (2011) 18:1336–44. doi: 10.1038/nsmb.2142
91. Saridakis V, Sheng Y, Sarkari F, Holowaty MN, Shire K, Nguyen T, et al. Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. Mol Cell (2005) 18:25–36. doi: 10.1016/j.molcel.2005.02.029
92. Holowaty MN, Zeghouf M, Wu H, Tellam J, Athanasopoulos V, Greenblatt J, et al. Protein profiling with Epstein-Barr nuclear antigen-1 reveals an interaction with the herpesvirus-associated ubiquitin-specific protease HAUSP/USP7. J Biol Chem (2003) 278:29987–94. doi: 10.1074/jbc.M303977200
93. Pfoh R, Lacdao IK, Georges AA, Capar A, Zheng H, Frappier L, et al. Crystal structure of USP7 ubiquitin-like domains with an ICP0 peptide reveals a novel mechanism used by viral and cellular proteins to target USP7. PLoS Pathog (2015) 11:e1004950. doi: 10.1371/journal.ppat.1004950
94. Nekorchuk MD, Sharifi HJ, Furuya AK, Jellinger R, de Noronha CM. HIV Relies on neddylation for ubiquitin ligase-mediated functions. Retrovirology (2013) 10:138. doi: 10.1186/1742-4690-10-138
95. Le-Trilling VT, Megger DA, Katschinski B, Landsberg CD, Ruckborn MU, Tao S, et al. Broad and potent antiviral activity of the NAE inhibitor MLN4924. Sci Rep (2016) 6:19977. doi: 10.1038/srep19977
96. Han K, Zhang J. Roles of neddylation against viral infections. Cell Mol Immunol (2018) 15:292–4. doi: 10.1038/cmi.2017.100
97. Zhao M, Zhang Y, Yang X, Jin J, Shen Z, Feng X, et al. Myeloid neddylation targets IRF7 and promotes host innate immunity against RNA viruses. PLoS Pathog (2021) 17:e1009901. doi: 10.1371/journal.ppat.1009901
98. Perng YC, Lenschow DJ. ISG15 in antiviral immunity and beyond. Nat Rev Microbiol (2018) 16:423–39. doi: 10.1038/s41579-018-0020-5
99. Thery F, Eggermont D, Impens F. Proteomics mapping of the ISGylation landscape in innate immunity. Front Immunol (2021) 12:720765. doi: 10.3389/fimmu.2021.720765
100. Imbert F, Langford D, Viruses SUMO. And immunity: the interplay between viruses and the host SUMOylation system. J Neurovirol (2021) 27:531–41. doi: 10.1007/s13365-021-00995-9
101. Proulx J, Borgmann K, Park IW. Post-translational modifications inducing proteasomal degradation to counter HIV-1 infection. Virus Res (2020) 289:198142. doi: 10.1016/j.virusres.2020.198142
102. Garvin AJ, Morris JR. SUMO, a small, but powerful, regulator of double-strand break repair. Philos Trans R Soc Lond B Biol Sci (2017) 372(1731):20160281. doi: 10.1098/rstb.2016.0281
103. Dhillon B, Aleithan F, Abdul-Sater Z, Abdul-Sater AA. The evolving role of TRAFs in mediating inflammatory responses. Front Immunol (2019) 10:104. doi: 10.3389/fimmu.2019.00104
104. Park HH. Structure of TRAF family: Current understanding of receptor recognition. Front Immunol (2018) 9:1999. doi: 10.3389/fimmu.2018.01999
105. Shi JH, Sun SC. Tumor necrosis factor receptor-associated factor regulation of nuclear factor kappaB and mitogen-activated protein kinase pathways. Front Immunol (2018) 9:1849. doi: 10.3389/fimmu.2018.01849
106. Khan KA, Abbas W, Varin A, Kumar A, Di Martino V, Dichamp I, et al. HIV-1 nef interacts with HCV core, recruits TRAF2, TRAF5 and TRAF6, and stimulates HIV-1 replication in macrophages. J Innate Immun (2013) 5:639–56. doi: 10.1159/000350517
107. Manghera M, Ferguson-Parry J, Lin R, Douville RN. NF-kappaB and IRF1 induce endogenous retrovirus K expression via interferon-stimulated response elements in its 5' long terminal repeat. J Virol (2016) 90:9338–49. doi: 10.1128/JVI.01503-16
108. Hayashi A, Giakoumakis NN, Heidebrecht T, Ishii T, Panagopoulos A, Caillat C, et al. Direct binding of Cdt2 to PCNA is important for targeting the CRL4(Cdt2) E3 ligase activity to Cdt1. Life Sci Alliance (2018) 1:e201800238. doi: 10.26508/lsa.201800238.
109. Lesbats P, Engelman AN, Cherepanov P. Retroviral DNA integration. Chem Rev (2016); 116(20):12730–12757. doi: 10.1021/acs.chemrev.6b00125
110. Yoder KE, Espeseth A, Wang XH, Fang Q, Russo MT, Lloyd RS, et al. The base excision repair pathway is required for efficient lentivirus integration. PloS One (2011) 6:e17862. doi: 10.1371/journal.pone.0017862
111. Knyazhanskaya E, Anisenko A, Shadrina O, Kalinina A, Zatsepin T, Zalevsky A, et al. NHEJ pathway is involved in post-integrational DNA repair due to Ku70 binding to HIV-1 integrase. Retrovirology (2019) 16:30. doi: 10.1186/s12977-019-0492-z
112. Coster G, Goldberg M. The cellular response to DNA damage: a focus on MDC1 and its interacting proteins. Nucleus (2010) 1:166–78. doi: 10.4161/nucl.11176
113. Luo K, Zhang H, Wang L, Yuan J, Lou Z. Sumoylation of MDC1 is important for proper DNA damage response. EMBO J (2012) 31:3008–19. doi: 10.1038/emboj.2012.158
114. Zhang F, Bick G, Park JY, Andreassen PR. MDC1 and RNF8 function in a pathway that directs BRCA1-dependent localization of PALB2 required for homologous recombination. J Cell Sci (2012) 125:6049–57. doi: 10.1242/jcs.111872
115. Uzunoglu H, Korak T, Ergul E, Uren N, Sazci A, Utkan NZ, et al. Association of the nibrin gene (NBN) variants with breast cancer. Biomed Rep (2016) 4:369–73. doi: 10.3892/br.2016.579
116. Ruff SE, Logan SK, Garabedian MJ, Huang TT. Roles for MDC1 in cancer development and treatment. DNA Repair (Amst) (2020) 95:102948. doi: 10.1016/j.dnarep.2020.102948
117. Wang B, Zhang L, Qiu F, Fang W, Deng J, Zhou Y, et al. A newfound association between MDC1 functional polymorphism and lung cancer risk in Chinese. PLoS One (2014) 9:e106794. doi: 10.1371/journal.pone.0106794
118. Zhou T, Yi F, Wang Z, Guo Q, Liu J, Bai N, et al. The functions of DNA damage factor RNF8 in the pathogenesis and progression of cancer. Int J Biol Sci (2019) 15:909–18. doi: 10.7150/ijbs.31972
119. Aho ER, Weissmiller AM, Fesik SW, Tansey WP. Targeting WDR5: A WINning anti-cancer strategy? Epigenet Insights (2019) 12:2516865719865282. doi: 10.1177/2516865719865282
120. Guarnaccia AD, Tansey WP. Moonlighting with WDR5: A cellular multitasker. J Clin Med 7 (2018); 7(2):21. doi: 10.3390/jcm7020021
121. Wang-Johanning F, Frost AR, Johanning GL, Khazaeli MB, LoBuglio AF, Shaw DR, et al. Expression of human endogenous retrovirus k envelope transcripts in human breast cancer. Clin Cancer Res (2001) 7:1553–60.
122. Buscher K, Trefzer U, Hofmann M, Sterry W, Kurth R, Denner J. Expression of human endogenous retrovirus K in melanomas and melanoma cell lines. Cancer Res (2005) 65:4172–80. doi: 10.1158/0008-5472.CAN-04-2983
123. Tai AK, O'Reilly EJ, Alroy KA, Simon KC, Munger KL, Huber BT, et al. Human endogenous retrovirus-K18 env as a risk factor in multiple sclerosis. Mult Scler (2008) 14:1175–80. doi: 10.1177/1352458508094641
124. Douville R, Liu J, Rothstein J, Nath A. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann Neurol (2011) 69:141–51. doi: 10.1002/ana.22149
125. Agoni L, Guha C, Lenz J. Detection of human endogenous retrovirus K (HERV-K) transcripts in human prostate cancer cell lines. Front Oncol (2013) 3:180. doi: 10.3389/fonc.2013.00180
126. Phan K, He Y, Fu Y, Dzamko N, Bhatia S, Gold J, et al. Pathological manifestation of human endogenous retrovirus K in frontotemporal dementia. Commun Med (Lond) (2021) 1:60. doi: 10.1038/s43856-021-00060-w
127. Contreras-Galindo R, Kaplan MH, Leissner P, Verjat T, Ferlenghi I, Bagnoli F, et al. Human endogenous retrovirus K (HML-2) elements in the plasma of people with lymphoma and breast cancer. J Virol (2008) 82:9329–36. doi: 10.1128/JVI.00646-08
128. Cardano M, Tribioli C, Prosperi E. Targeting proliferating cell nuclear antigen (PCNA) as an effective strategy to inhibit tumor cell proliferation. Curr Cancer Drug Targets (2020) 20:240–52. doi: 10.2174/1568009620666200115162814
129. Lu J, Zhao H, Yu C, Kang Y, Yang X. Targeting ubiquitin-specific protease 7 (USP7) in cancer: A new insight to overcome drug resistance. Front Pharmacol (2021) 12:648491. doi: 10.3389/fphar.2021.648491
130. Ebstein F, Kury S, Papendorf JJ, Kruger E. Neurodevelopmental disorders (NDD) caused by genomic alterations of the ubiquitin-proteasome system (UPS): the possible contribution of immune dysregulation to disease pathogenesis. Front Mol Neurosci (2021) 14:733012. doi: 10.3389/fnmol.2021.733012
131. Agbowuro AA, Huston WM, Gamble AB, Tyndall JDA. Proteases and protease inhibitors in infectious diseases. Med Res Rev (2018) 38:1295–331. doi: 10.1002/med.21475
132. Tyagi R, Li W, Parades D, Bianchet MA, Nath A. Inhibition of human endogenous retrovirus-K by antiretroviral drugs. Retrovirology (2017) 14:21. doi: 10.1186/s12977-017-0347-4
Keywords: endogenous retrovirus-K (ERVK), protease, interactome, eukaryotic linear motif, amyotrophic lateral sclerosis (ALS), ubiquitination, DNA damage response (DDR), cancer
Citation: Narvey S, Vandenakker A, Rempel M and Douville RN (2022) Predicted cellular interactors of the endogenous retrovirus-K protease enzyme. Front. Virol. 2:972156. doi: 10.3389/fviro.2022.972156
Received: 17 June 2022; Accepted: 05 September 2022;
Published: 23 September 2022.
Edited by:
Diego Forni, Eugenio Medea (IRCCS), ItalyReviewed by:
Lorenzo Agoni, Fondazione Poliambulanza Istituto Ospedaliero, ItalyJoseph Mathew Antony, University of Toronto, Canada
Copyright © 2022 Narvey, Vandenakker, Rempel and Douville. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renée N. Douville, ci5kb3V2aWxsZUB1d2lubmlwZWcuY2E=