 Nathaniel Felbinger1
Nathaniel Felbinger1 David Trudil1Lawrence Loomis1Richard Ascione1,2
David Trudil1Lawrence Loomis1Richard Ascione1,2 Gregory Siragusa1,3Seiji Haba1
Gregory Siragusa1,3Seiji Haba1 Shruti Rastogi1Aidan Mucci1Mark Claycomb1
Shruti Rastogi1Aidan Mucci1Mark Claycomb1 Sebastian Snowberger1Brian Luke4
Sebastian Snowberger1Brian Luke4 Stephen Francesconi5
Stephen Francesconi5 Shirley Tsang1*
Shirley Tsang1*- 1New Horizons Diagnostics, Inc., Arbutus, MD, United States
- 2Georgetown University School of Medicine, Washington, DC, United States
- 3Scout Microbiology LLC, Waukesha, WI, United States
- 4Brian Luke Consulting LLC, Frederick, MD, United States
- 5DTRA (Defense Threat Reduction Agency), Ft., Belvoir, VA, United States
Previous studies have attempted to characterize the antibody response of individuals to the SARS-CoV-2 virus on a linear peptide level by utilizing peptide microarrays. These studies have helped to identify epitopes that have potential to be used for diagnostic tests to identify infected individuals. The immunological responses of individuals who have received the two most popular vaccines available in the US, the Moderna mRNA-1273 or the Pfizer BNT162b2 mRNA vaccines, have not been characterized. We aimed to identify linear peptides of the SARS-CoV-2 spike protein that elicited high IgG or IgA binding activity and to compare the immunoreactivity of infected individuals to those who received both doses of either vaccine by utilizing peptide microarrays. Our results revealed peptide epitopes of significant IgG binding among recently infected individuals. Some of these peptides are located near variable regions of the receptor binding domains as well as the conserved region in the c-terminal of the spike protein implicated in the high infectivity of SARS-CoV-2. Vaccinated individuals lacked a response to these distinct markers despite the overall antibody binding activity being similar.
1 Introduction
We are identifying B cell epitopes that bind to anti-SARS-CoV-2 IgG and IgA in infected and vaccinated individuals by using a linear peptide microarray to study IgG and IgA binding in the surface spike glycoprotein. These epitopes are important because the sequences can be used to develop diagnostic assays and therapeutic agents to identify and treat SARS-CoV-2 or any future SARS viruses, including the new variants of SARS-CoV-2 which continue to emerge and rapidly spread (1). These variants contain mutations that play a role in viral infectivity and decrease the effectiveness of the current mRNA-based vaccines (2, 3). The surface spike glycoprotein mediates SARS-CoV-2 entry into the cell by binding the S1 subunits to the host receptor angiotensin-converting Enzyme 2 (ACE2) and subsequently promoting viral and cellular membrane fusion by its S2 subunit, leading to the release of the viral genome into the cytoplasm. The immunodominant neutralizing epitopes have been identified primarily in the S1 subunit containing the receptor-binding-domain (RBD) and N-terminal-domain (NTD). The S2 subunit, in contrast, is buried inside the S protein in a prefusion conformation unlikely to induce neutralizing antibodies (nAbs) against SARS-CoV-2 after infection or post-vaccinations with its full amino acid sequences. The S2 contains two important domains of heptad repeat 1 (HR1) and heptad repeat 2 (HR2) as well as the fusion peptide (FP), which bridge the viral infection to host cells. Studying epitopes at the peptide level of the spike protein helps to reveal which mutations in currently prevalent variants may increase infectivity, transmissibility, and vaccine evasion. In addition, epitopes that are found to have consistent antibody binding may serve as a useful diagnostic target to assess previous infection or the general protective status of individuals. Viral load detected using qPCR testing can fall to undetectable levels within one to two weeks after symptom onset (4) and antigen tests are sometimes plagued by low sensitivity (5). Gaining an understanding of the epitopes that elicit significant antibody binding may prove exceptionally useful for improving diagnostic methods and vaccine development.
Peptide microarrays have many immunological applications. We used this tool to screen antibody binding activity against viral peptide sequences of the entire SARS-CoV-2 viral proteome. By using linear peptide heat mapping, we identified strong B cell epitope responses. The linear peptide has limitations, notably missing binding activity upon the structure formation. ELISA can be used to verify the activity and recapitulate the epitopes of the linear peptide antigen used to ensure PTM detection or other tertiary structure effects. The advantages of using linear peptides are their simple structure, ease of synthesis, and ability to stimulate immune responses without requiring complex 3D conformation. As effect of tertiary cannot be tested with linear peptides, assay-based verification (e.g. ELISA) was used to confirm antibody binding activity.
Previous works utilizing peptide microarrays have focused on mapping the humoral responses to the SARS-CoV-2 proteome using samples from individuals currently or recently infected with SARS-CoV-2 (6–12). SARS-CoV-2 epitopes that elicit a significant antibody response in infected samples have been identified in the spike (S), nucleocapsid (N), membrane (M), ORF1ab, and ORF3a proteins. As some of these proteins are shared with or have close analogs in other common human coronaviruses (hCoVs), some cross-reactivity has been observed (5, 13). There are many epitopes that demonstrate highly specific reactivity with SARS-CoV-2 antibodies distributed across the SARS-CoV-2 proteome. Many epitopes have been identified in previous studies (8–10), both in commercially available and independently designed peptide microarrays. These findings support the use of the peptide microarray approach in locating highly specific and sensitive SARS-CoV-2 human B cell epitopes.
Microarray data can provide answers regarding the presence of potential epitopes and the characterization of the target pathogen’s mechanisms of infection. As entire targeted viral proteomes can be bound to microarrays, the process of screening for antibody binding is both fast and reproducible. These findings indicate that there may exist a linear epitope or group of linear epitopes that can serve as a marker of the protective status of individuals against SARS-CoV-2 infection. While this is an exciting prospect, previous microarray epitope mapping studies have been focused exclusively on the comparison of infected and naive individuals, and did not examine differences between the antibody response in infected and vaccinated individuals.
Our systematic approach to assessing individual peptides as potential epitopes using linear peptide microarrays, may prove especially important with the global relevance of the Delta variants and the emergence of the Omicron variants. The Omicron variants, which surfaced at the end of 2021, display lowered pathogenicity (14) while maintaining high transmissibility and infectivity, resulting in it becoming the dominant strain of SARS-CoV-2. Omicron is poised to continue as the main variant as its subvariants have a higher rate of mutations (15), which allow the subvariants to continue to increase in number. The majority of SARS-CoV-2 variants harbor a high number of mutations (15) in RBD of S1, and remain conserved in the S2 region at the C-terminal end of the surface glycoprotein. By analyzing the reactivity of neutralizing antibodies to peptides identified in parental variants, immune response to Omicron and future variants can be estimated by analyzing differences in the relevant regions of the proteome.
Our study explores IgG and IgA antibody reactivity of individuals vaccinated with the Moderna mRNA-1273 or the Pfizer BNT162b2 mRNA vaccines by testing serum samples against linear peptides of the whole proteome of SARS-CoV-2. We then compared the antibody binding of these vaccinated individuals with a set of infected and negative samples. To our knowledge, no study has utilized a peptide microarray approach to study individuals who received either the Moderna or Pfizer vaccine. As the Moderna and Pfizer vaccines contain the sequences of the SARS-CoV-2 spike protein, our study has focused mainly on the spike protein region. Data herein identify epitopes that could potentially serve as markers to assess protection against SARS-CoV-2, and generally characterize IgG and IgA antibody response against SARS-CoV-2 in vaccinated individuals. IgA, as a principal antibody class in the mucosal surface, acts as a first line of defense against respiratory infections such as SARS-CoV-2. IgA is also an important serum immunoglobulin which mediates a variety of protective functions through interaction with specific receptors and immune mediators. Using sera to perform IgA epitope profiling is difficult without additional purification, since IgG makes up the majority of the antibody content in serum whereas IgA makes up less than 10%. As some epitopes of the spike protein have been found to react with IgA (8), preliminary work was done using saliva samples to more easily test for IgA-specific epitopes. Optimization of these methods for use of saliva is ongoing, with goals of characterizing the initial immune response to respiratory infection and screening for protective mucosal IgA.
2 Materials and methods
2.1 Infected, vaccinated, and control serum samples
Serum samples were purchased from RayBiotech (Atlanta, GA) and Reprocell (Beltsville, MD). Samples were divided into three groups:
- SARS-CoV-2 positive individuals (n=30)
- Individuals who received either the Moderna or Pfizer mRNA vaccines (n=17)
- A naive negative control group (n=10)
Among the 10 negative controls, five serum samples were collected prior to the pandemic before the end of 2019 (n=5). There are five paired vaccinated individuals whose serum samples were collected one to three days before receiving the first dose of either the Moderna (n=2) or Pfizer (n=3) vaccine. These five were reported as never having been infected before their vaccination. The post-vaccinated sera were collected between 6 - 44 days after receiving their second dose of either Moderna or Pfizer vaccine. Basic demographic information and the date of sample collection relative to symptom onset for infected patients can be found in Supplemental Table S1. Demographic information for vaccinated and negative subjects can be found in Supplemental Table S2. Available symptom information for all infected samples is available in Supplemental Table S3. All purchased infected sera were inactivated with a 4.0% Triton X-100 treatment prior to their arrival at our facility. All samples were immediately aliquoted and stored at -80° C upon arrival at our facility.
2.2 Peptide microarrays
For our systematic proteome analysis, PEPperCHIP® (Heidelberg, Germany) SARS-CoV-2 Proteome Microarray slides were used for all samples. These slides contain a series of linear peptide sequences that cover the entire SARS-CoV-2 proteome. Each of these peptides is printed in duplicate and consists of 15 amino acids in total, 13 of which overlap with neighboring SARS-CoV-2 peptide sequences. Additionally, the slides contain certain mutated peptide sequences found in SARS-CoV-2 variants and a set of influenza and polio peptides which were utilized as internal controls.
Prior to performing the procedure, solutions of PBST wash buffer (Phosphate-buffered saline with 0.05% Tween20, at 7.4 pH), and pH 7.4 1mM Tris dipping buffer were prepared. All solutions were filtered using a 0.44-micron vacuum filter kit and pH was adjusted to 7.4 with the addition of 3M HCl if necessary. Microarray slides were initially incubated in a solution of 0.05% PBST (7.4 pH) and 10% blocking buffer (Rockland Immunochemicals, Inc., Pottstown, PA) for 15 minutes at room temperature. Microarray slides were aspirated and then blocked with a blocking buffer for 30 minutes at room temperature. After blocking, slides were again aspirated and probed with a mixture of fluorescent secondary antibodies that would be used for peptide binding detection consisting of Rabbit Anti-Human IgG DyLight™ 800 (Rockland Immunochemicals), Alexa Conjugated Goat Anti-Human IgA 680 (Jackson ImmunoResearch Labs, West Grove, PA), and Mouse Anti-HA (influenza virus hemagglutinin) Dylight™ 680 (PEPperCHIP®). This prescreen of secondary antibody treatment was used to reveal any non-specific secondary antibody interactions with the slide.
After performing three PBST washes of one minute each, slides were treated with sera diluted 1:500 (16–18) in a 10% blocking buffer/PBST solution and incubated overnight at 4°C. Following this overnight incubation, slides were washed three times with PBST for one minute each and dipped into the previously prepared 1mM Tris Solution. Detection of IgG was performed via treatment with Rabbit Anti-Human IgG DyLight™ 800 for 45 minutes at room temperature. After performing three PBST washes of one minute each, slides were then treated a second time with samples diluted 1:200 in 10% blocking buffer/PBST and incubated overnight at 4°C. After the second overnight incubation, samples were washed three times with PBST for one minute each and treated with Alexa Conjugated Goat Anti-Human IgA 680 for 45 minutes at room temperature for detection of IgA. Before scanning, a final set of three one-minute washes with PBST was performed followed by dipping the slides three times in the 1mM Tris Solution.
Fluorescent signals were acquired using the InnoScan 710-IR microarray scanner from Innopsys (Chicago, IL). Slides were scanned at wavelengths of 670 nm for IgA detection and 785 nm for IgG detection and at a resolution of 30uM for both wavelengths. Fluorescent values were retrieved using Mapix Microarray Image Acquisition and Analysis Software (Innopsys).
2.3 Epitope validation
The tiff image files, acquired after probing slides with secondary antibody solution, were analyzed for any slide artifacts prior to data analysis (19). Artifact sequences from the prescreen of secondary antibody treatment were excluded from further analysis. Any microarray slides that did not display sufficient reactivity with polio and HA-positive control peptide spots were considered invalid.
For data analysis, we used the background-subtracted median intensity values acquired from the Mapix software. To identify potential epitopes, we first averaged these median fluorescent intensity values of each peptide found within the spike protein for each experimental group. We then selected peptides in which the average median intensity value of the infected group was at least 1.5-fold greater and had p-values of < 0.05 from an unpaired t-test comparison to the average log2-normalized median intensity value of the negative controls. Among these selected peptides, we compared the average log2-normalized median intensity value of each peptide to their neighboring peptides. Peptides that had significantly different fluorescent intensity values [α = 0.05] were screened out and the remaining peptides were recognized as potential epitopes (5, 20).
2.4 ELISA testing
In-house ELISA tests (21) were utilized to detect levels of IgG and IgA antibodies against SARS-CoV-2 RBD antigen. Serum samples were diluted at 1:10,000 in a 1% milk PBST solution. 100 µl of serum samples (in duplicate) were added to ELISA plates that were coated with 150 ng of SARS-CoV-2 RBD antigen per well (Cat#40592-Vo8H, Sino Biological, Wayne, PA). The plates were incubated overnight at 4oC and washed three times in a 0.1% PBST solution. Secondary detection antibody solutions of Mouse Anti-Human IgG Fc-HRP (SouthernBiotech, Birmingham, AL) diluted 1:10,000 in 1% milk PBST and Mouse Anti-Human IgA1-HRP/Mouse Anti-Human IgA2-HRP (SouthernBiotech) diluted 1:4,000 in 1% milk PBST solution were used to detect IgG and IgA, respectively. Secondary antibody solutions were incubated at room temperature for one hour on an orbital shaker set at 200 rpm. Plates were washed three times with a 1% PBST solution and developed for 10 minutes with a TMB substrate solution. 3M HCl was added and plates were scanned using the SpectraMax® iD3 Multi-Mode Microplate Reader (Molecular Devices, San Jose, CA) (Table S4).
3 Results
3.1 SARS-CoV-2 spike epitopes exhibiting significant IgG binding identified in infected and vaccinated samples
To identify reactive epitopes, we looked at peptides in which the average log2-normalized intensity value of either of the two infected sample groups had a fold-change value of at least 1.5 and yielded adjusted p-values of < 0.05
[α = 0.05] from an unpaired t-test when compared to the mean log2-normalized intensity value from the negative control group (12, 20). Using these criteria, nine peptides with significantly higher IgG antibody binding were identified in the spike protein region (Figure 1 and Table 1) among infected individuals.
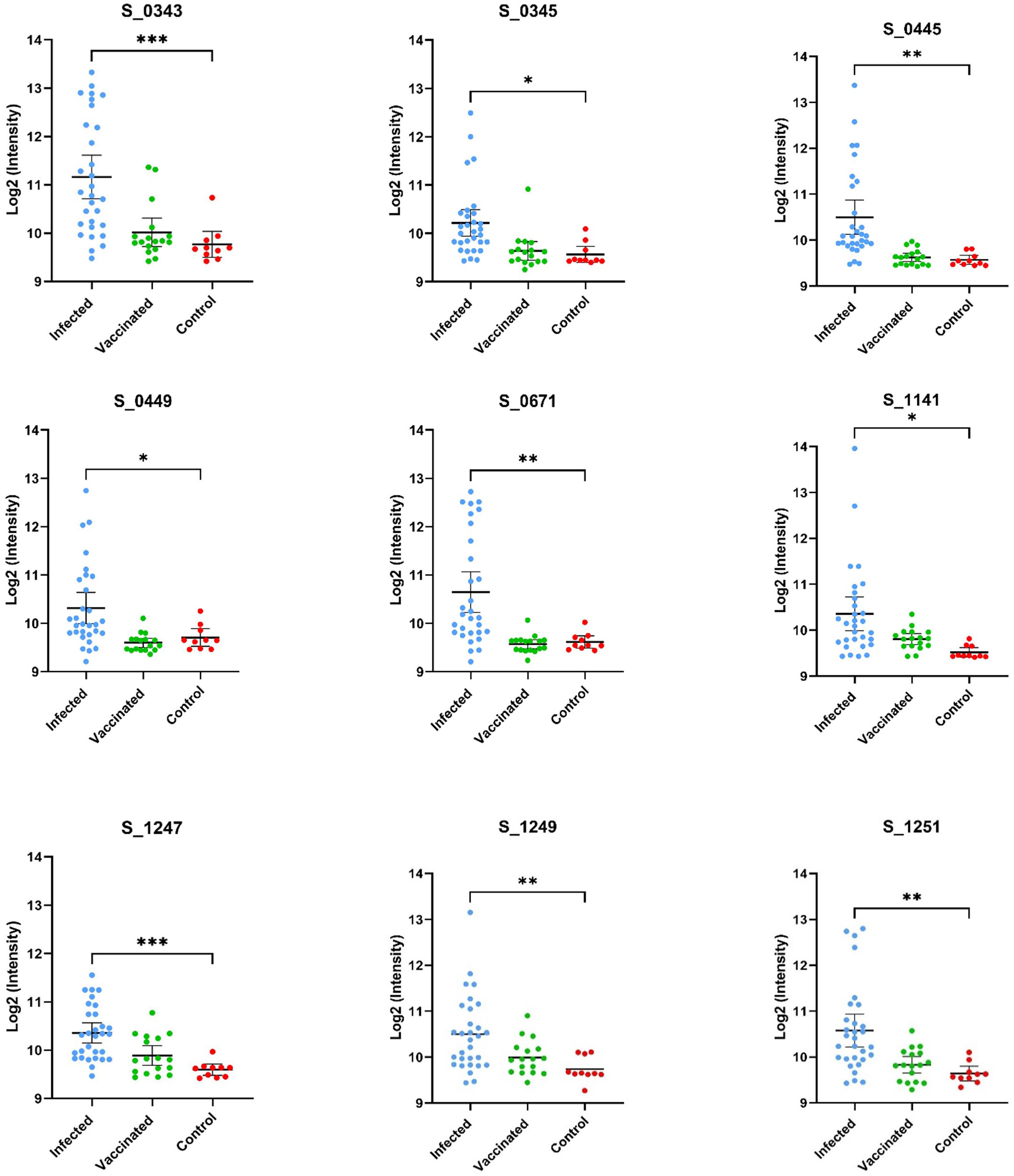
Figure 1 Displayed are the log2 normalized individual fluorescent intensity values of infected (n=30), vaccinated (n=17), and control (n=10) sample groups. The peptide ID along with the results of the unpaired t-test comparing the infected and control groups can be found above the data. The results of the t-test comparing the fluorescent intensities of the infected and negative control groups values are shown (* <.05, ** <.01, *** <.001, **** <.0001).
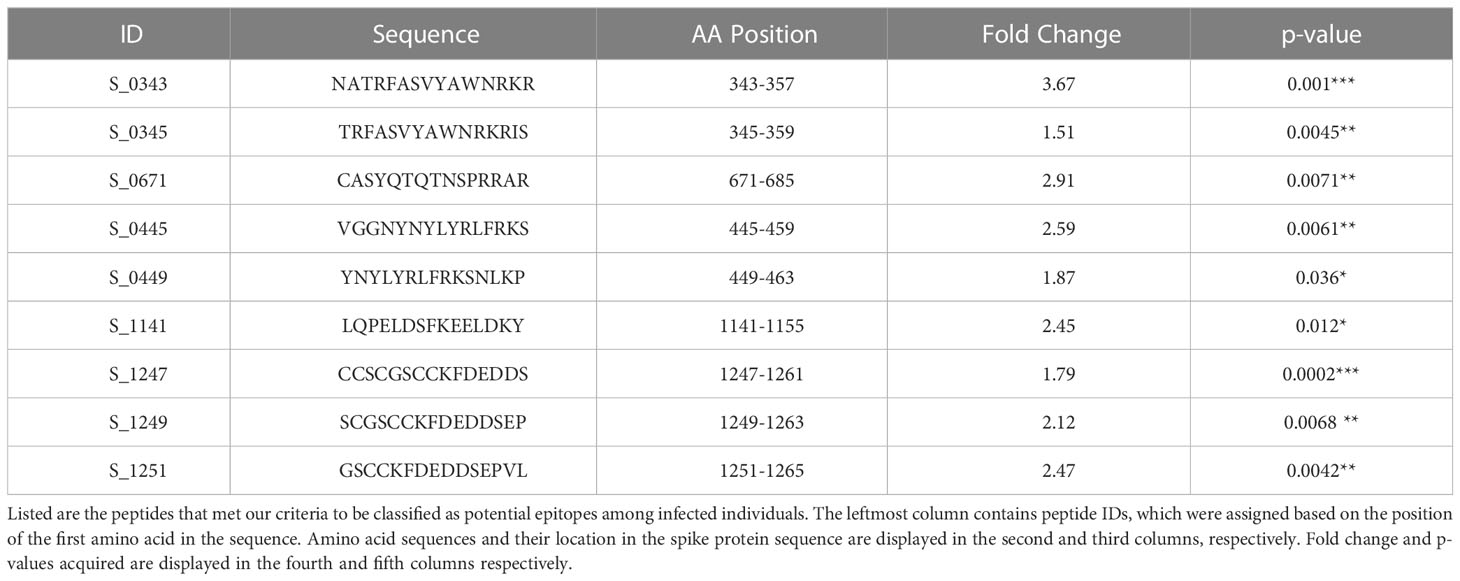
Table 1 Highly responsive peptide sequences of spike protein.
Using the same approach of epitope identification with our vaccinated groups, no peptides were found to meet the criteria for significant IgG binding activity. Two peptides were found to have p-values less than 0.05 [α = 0.05], but these peptides did not meet our fold change criteria as described in our methods (Figure 2). Of the nine peptides identified as significant in the infected sample group, five were also found to have significantly greater IgG binding than the vaccine group. Peptides with no significant differences in binding activity between the recently infected group and vaccinated group were peptides S_1141, S_1247, S_1249, and S_1251. The SARS-CoV-2 RBD IgG titers of the vaccinated individuals were comparable to the IgG titers of infected samples according to data supplied by the sample provider, RayBiotech, and our own in-house RBD ELISA testing.
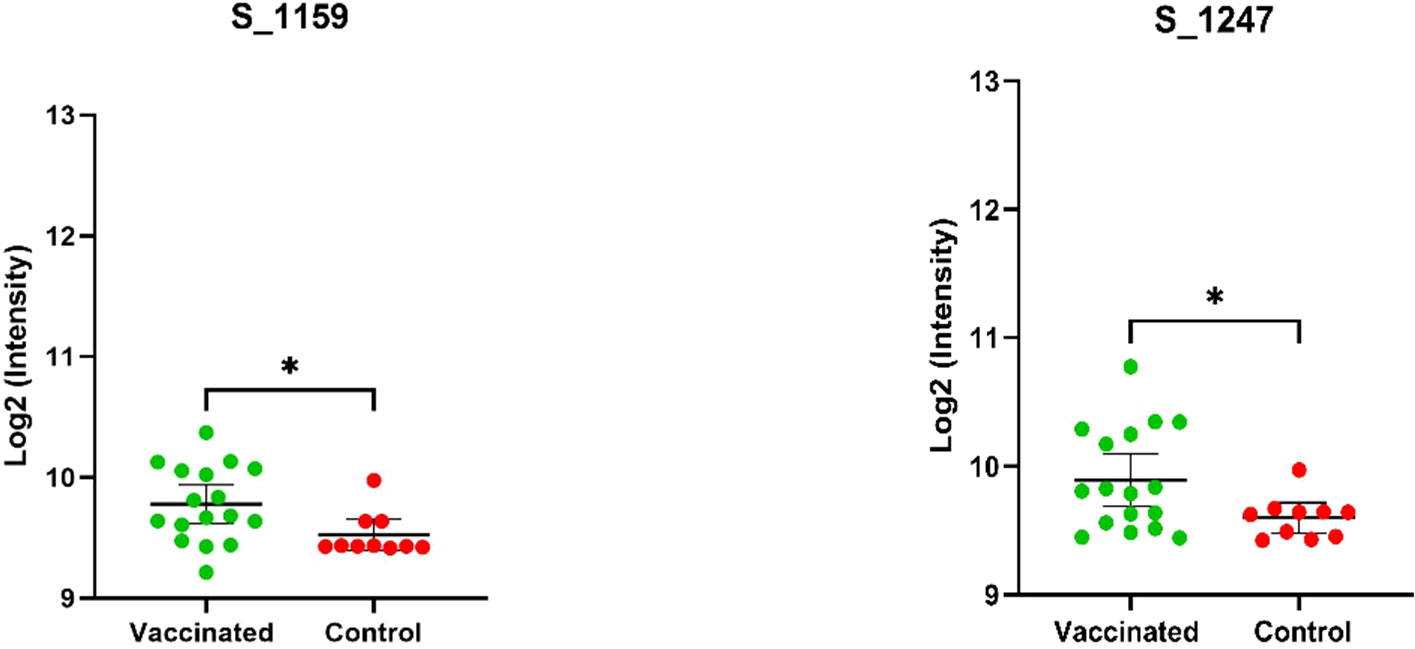
Figure 2 Depicted above are mean log2 normalized fluorescent intensities of the vaccinated and control groups that were found to be significantly different (p-value <.05) based on the results of an unpaired t-test. The ID and results of the t-test (* <.05) are displayed above the plots. The vaccinated group was not found to have a fold change greater than 1.5 and, therefore, these peptides were not recognized as epitopes of high interest based on our criteria.
Within the infected samples, peptides identified as having significant IgG binding activity appear to be distributed throughout the spike protein (Figure 1). Identified reactive epitopes that meet our criteria include two consecutive peptides in the RBD (S_0343 and S_0345), one located in the putative fusion peptide domain (S_0671), one found in proximity to the identified Heptad Repeat 2 sequence (S_1141), and three located within a cysteine-rich sequence (S_1247, S_1249, and S_1251) neighboring what is thought to be a transmembrane domain (22) located in the C terminal of the S2 subunit. Among these peptides, those that were found to have relatively higher intensity among the vaccinated group (S_1159 and S_1247) were both found in the S2 region.
3.2 No significant difference in IgG epitope profiles of individuals who received the moderna or pfizer vVaccine
No individual linear peptide exhibited significant IgG binding in our vaccinated sample group as per our criteria. Overall IgG binding activity between infected and vaccinated groups was comparable. However, no individual peptide with consistent binding was found in the vaccinated group with the level of statistical significance that was found with the infection-specific peptides. To ascertain differences in peptide epitope binding activity, we compared the IgG binding of individuals who received the Moderna (n=8) and the Pfizer vaccines (n=9). Comparison of the Moderna and Pfizer vaccinated sample groups revealed little in terms of significant differences in IgG epitope binding activity in samples between the two vaccines. Both vaccinated groups appeared to have more IgG binding activity to epitopes in the S2 region of the spike protein (Figure 3), although peptide S_0343 located in the RBD region appeared to have significant binding in certain individuals of both groups. Though no peptides qualified as potential epitopes according to the defined cutoff, the peptides that yielded the highest antibody titer in each group were compared to see if the binding patterns of the two vaccine groups were similar. To explore the identity of these peptides and potential differences in their distribution, highly fluorescent peptides were identified. Eleven peptides were found to have intensities above the cutoff in the Moderna sample group and two peptides were identified as being above this cutoff in the Pfizer sample group (Table 2). Both peptides above 3.0 standard deviations in the Pfizer vaccinated group also found among the peptides identified in the Moderna vaccinated group. Of the eleven peptides identified in the Moderna vaccinated sample group, six were either identified as having significant IgG binding activity or overlapped with a peptide found to have significant IgG binding activity within the infected group.
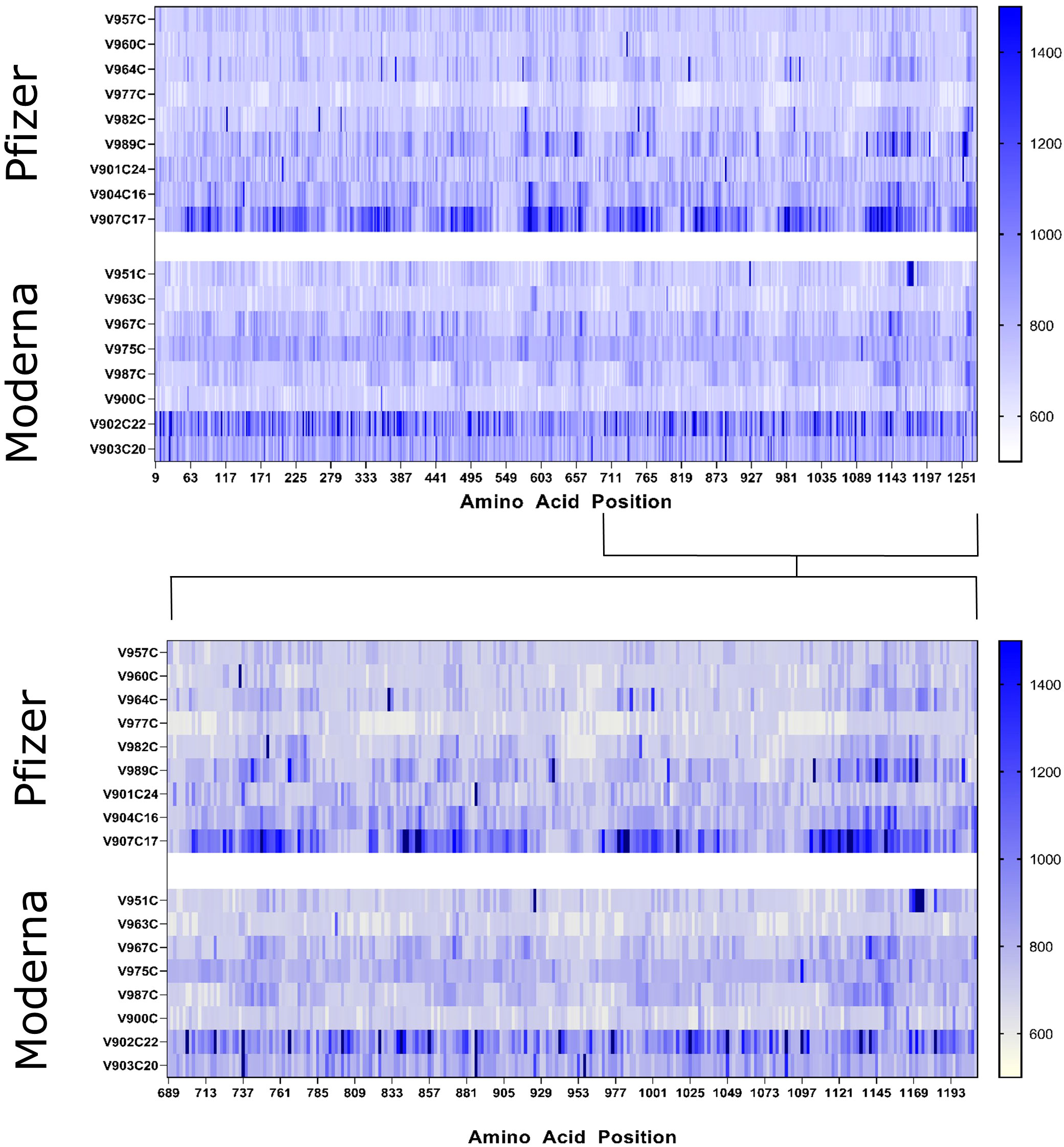
Figure 3 Heat maps of the whole SARS-CoV-2 spike protein (top) and S2 region (bottom) displaying the raw fluorescent intensity values of vaccinated samples were created to compare epitope binding of Moderna and Pfizer vaccinated individuals. The fluorescent intensity values were read at the 785 nm wavelength, which corresponds to IgG binding activity. Sample IDs are displayed to the left of their respective rows. Individuals who received the Pfizer vaccine are displayed in the upper panels while those who received the Moderna vaccine are found in the lower panels. The amino acid sequence of the entire spike protein is displayed in a linear manner with amino acid positions denoted below.
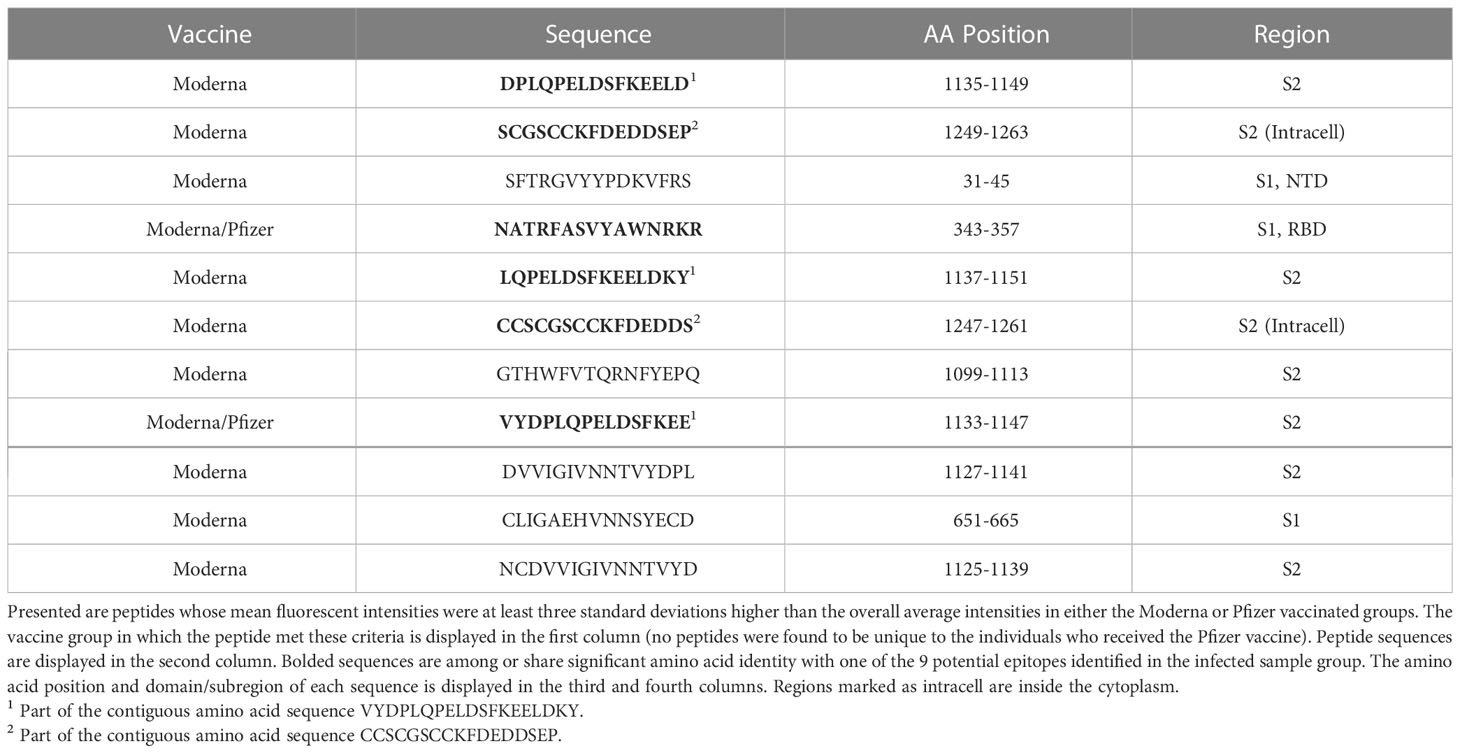
Table 2 Highly responsive peptides found in the vaccinated individual.
Interestingly, there are four frequent mutations in common variants found and identified within at least one peptide sequence (Table 3) of S_0671, which is right in the cleavage site for the S1 and S2 region (RRAR) of the spike protein.
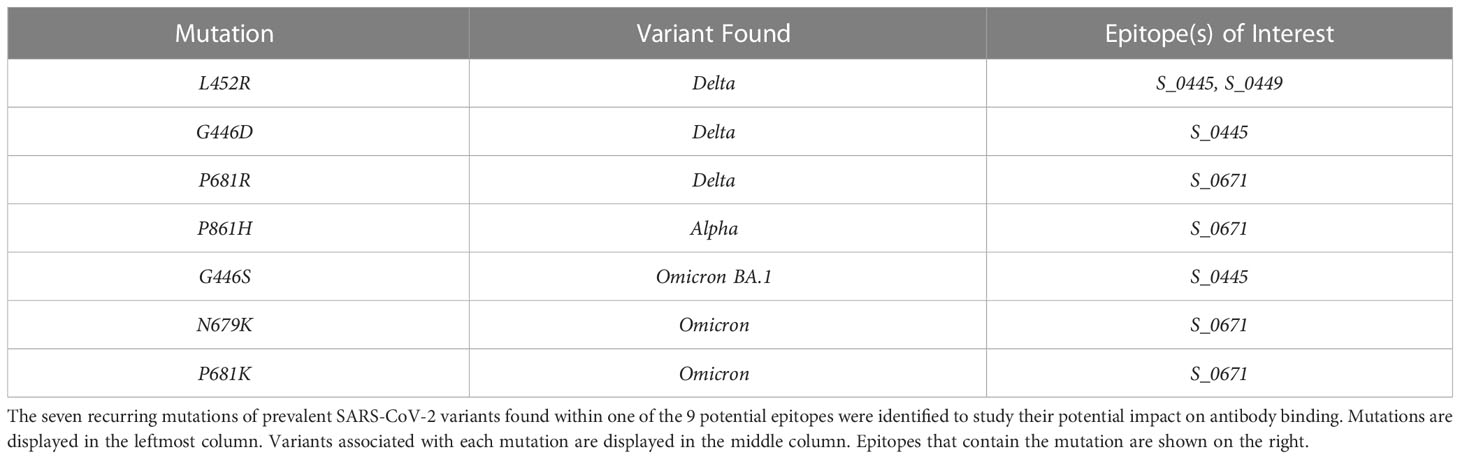
Table 3 Recurring mutations in S1.
3.3 No significant IgA binding activity to SARS-CoV-2 spike protein epitopes identified in infected or vaccinated individuals
The same approach used to identify IgG epitopes was utilized to identify IgA epitopes. However, no significant IgA epitopes were identified in either the infected group or the vaccinated group. The IgA titer information of the samples provided by RayBiotech was limited, but in-house ELISA testing of the RBD antigen revealed low levels of IgA binding (verified by additionally testing dilutions of 1:1000 to 1:10,000 of serum) throughout both infected and vaccinated groups (Table S4). The PEPperCHIP® microarrays contained the entire proteome of SARS-CoV-2, allowing for the identification of significant IgA binding to peptides outside of the spike protein. We found significant IgA binding in the ORF, and X region of the virus (data not shown and disclosed here). However, this report is solely focused on the spike protein, so these significant occurrences of IgA binding outside of the spike protein have been excluded.
4 Discussion
4.1 Alignment analysis of identified potential peptide epitopes
To assess the potential of incorporating the discovered peptide sequences into viable therapeutics or diagnostics, we investigated the conservation of sequences among other closely related coronaviruses and the proximity of the sequences to mutations found in current dominant variants. Previous studies have established that antibodies that bind to certain linear epitopes found in the SARS-CoV-2 proteome are found to be reactive to conserved peptide stretches in other hCoVs (14). Aligning the epitopes to closely related CoVs helped to assess the potential for nonspecific antibody binding by antibodies present due to exposure to other coronaviruses. With the current global prevalence of variant strains such as Delta and Omicron (Figure 4), it is important to investigate if the identified peptide sequences will be affected by common mutations of the spike protein. A 15mer linear peptide microarray was used to “walk along” the entire viral proteome and determine highly reactive linear epitopes. At the time of the study, only linear peptide slides were available for SARS-CoV-2. Areas of high activity were found, suggesting further study of tertiary structure using cyclic peptide arrays may be warranted in the future.

Figure 4 To determine the conservation of the potential epitopes, the Wuhan SARS-CoV-2 sequence was aligned with the Delta and Omicron variant sequences, closely related Pangolin and bat-infecting (RaTG13) coronaviruses, and human SARS-CoV, MERS-CoV, HCOV-HKU1, -OC43, -NL63, and -2293. Peptide IDs are displayed above corresponding alignments, and the epitopes are highlighted for SARS-CoV-2 with corresponding peptides in bold for all sequences, with notable deviations between similar sequences underlined.
The spike glycoproteins of several other CoVs and hCoVs were compared to that of SARS-CoV-2. Four of these coronaviruses, the alphacoronaviruses HCoV-229E and HCoV-NL63 and betacoronaviruses HCoV-OC43 (lineage A) and HCoV-HKU1 (lineage A) are highly virulent and associated with causing upper respiratory illness in human adults (23). The other two hCoVs are SARS-CoV (lineage B) and MERS-CoV (lineage C), which are less virulent and more pathogenic (24). To predict potential antibodies that may exhibit cross-reactive binding with peptides of other CoVs, we created alignments of the spike protein sequences found in human as well as the bat-infecting CoV RaTG13 (lineage B) and Pangolin-CoV (lineage B) (Figure 4). The alignments revealed that the identified peptides were poorly conserved outside of the lineage B hCoVs. Of the nine peptides identified among the acutely infected group, the peptides S_1141 and S_1247-S_1251 were found to be the most conserved among the hCoVs. Peptide S_1141, located in proximity to the HR2 (heptad repeat domain 2) sequence, was entirely conserved among all lineage B CoVs analyzed. The sequence in proximity to S_1141 has also been identified as being potentially cross-reactive in previous microarray studies of SARS-CoV-2 (9). Peptides S_1247-S_1251 are located near the C-terminus of the spike protein just outside the transmembrane domain. These contiguous peptides contain highly conserved cysteine residues which are thought to be imperative for membrane fusion (25). Very recently, Pang et al. (26) suggested the conserved HR1 and HR2 domains present in the conformation of the fusion intermediate in the S2 subunit may serve as potential targets for vaccine development, which supports our findings at the C-terminal of the surface glycoprotein. These results warrant additional study on more individuals with complex immune status, such as vaccinated without infection or with infection or infected with different variants to identify epitopes specific to long-term B-cell response that could be used to accurately predict long-term immune status.
4.2 Peptide mutations of SARS-CoV-2 variants found in epitopes of interest
With the current prevalence of certain SARS-CoV-2 variants (27), it is important to assess whether their mutations are found in proximity to the identified peptides and whether these mutations have a role in increasing viral transmissibility. We discovered seven such variant mutation sites present in proximity to at least one of our peptides (S_0671) (Table 3) that are present in current prominent variants: Delta (B.1.617.2), Alpha (B.1.1.7), and Omicron (B 1.1.529).
The L452R and G446D mutations present in the Delta variant (28) were found within the region of the two overlapping identified epitopes S_0445 and S_0449. Although these mutations are not believed to directly affect the ACE2 binding activity of the virus, they are located in a peptide stretch that has been identified as an epitope for neutralizing antibodies and are thought to help inhibit neutralizing activity (29, 30). The G446S mutation found in the Omicron variant at the same position as the G446D mutation of the Delta variant is also connected with an escape from certain classes of neutralizing antibodies (31). Due to the limitation of the specimen, we are not able to perform further identification by generating mutagenesis to study the effect of these mutants on their binding.
Another set of mutations, P681R, P681H, P681K, and N679K are all located within the peptide of interest S_0671. These mutations are commonly found in either Delta (P681R), Alpha (P681H), or Omicron (P681K and N679K) variants, within or in proximity to the polybasic insertion sequence RRAR. This insertion sequence has been identified as the S1/S2 furin cleavage site that appears to be unique to SARS-CoV-2 among closely related hCoVs (32, 33). Cleavage of this site gives RBD flexibility to change between an open and closed conformation before ACE2 Binding and is thought to be essential in conformational changes prior to viral host cell insertion (32, 33). The P681R mutation found in the Delta variant is thought to enhance furin cleavage function (34) and likely contributes to increased infectivity. It has been recently reported that the band of cleaved Omicron and Alpha were obviously weak despite the P681H mutation, which implies other mutations found in proximity to the furin cleavage site (T716I and N679K) could aid in the regulation of the proteolytic cleavage of S protein (35). Due to the significance of these mutations in the Delta and Omicron variants, the S_0671 peptide may be an epitope of particular interest for future studies. Assessing whether antibody binding to the mutated linear epitope is observed could help assess whether the mutation may also have a more direct role in the enhanced viral escape of the Delta and Omicron variants. The peptides found to be significant epitopes in our infected patient group, S_1141 and S_1247-S_1251, are located in the S2 subregion which is thought to be a highly conserved region among hCoVs (36). The peptides do not appear to contain any common mutations observed in the currently relevant variants (37).
Notably, none of the nine epitope sequences with significant IgG binding identified in the infected group were present in the N-terminal domain region of the spike protein. Although the NTD region is thought to be a particularly important target of neutralizing antibodies (29, 30) and contains numerous mutations thought to help increase the infectivity of current variants (29, 31), these findings are corroborated by previous epitope mapping studies utilizing a proteome-wide microarray that have identified few to no potential epitopes in this region (6, 8).
4.3 Antibody binding profile comparison of vaccinated and infected individuals
Our analysis of antibody immunoreactivity to the SARS-CoV-2 spike protein revealed differences in antibody binding activity between the infected and vaccinated individuals. However, both groups showed overall higher IgG binding activity to epitopes throughout the spike protein relative to the naive samples. Despite similar patterns of IgG binding activity between the two groups, only samples from the infected group were found to bind to the distinct epitopes to an extent that met the statistical criteria of the study.
Previous studies have shown that vaccinated individuals produce a robust and easily detectable immune response that is comparable to natural infection (38, 39). We believe multiple factors contributed to the differences observed via linear peptide microarray between the infected and vaccinated groups. Many of the acutely infected samples were collected two to three weeks after infection, when infection-induced antibody production is thought to peak (40). Some of the vaccinated group had been previously infected, and it has been shown that previously infected individuals who receive a full vaccine treatment have extremely high antibody titers (41). This may have increased variability in response across the vaccinated group. Corresponding symptom information which may affect immune response, such as severity of symptoms, was only partially available for the infected group. It has been found that severe symptoms correspond to higher antibody production (42) and more distinct antibody binding patterns (7, 8). These variables limit comparisons between the infected and vaccinated groups in this study.
The peptides of S_1159 and S_1247-S_1251 (Figure 2), located in the C-terminal S2 protein, were identified in the vaccinated group and could potentially be used to distinguish between vaccinated and naive individuals. These peptides were not recognized as epitopes of high interest, but met the minimum statistical criteria (p-value <.05) with limited sample size, and were found to bind to antibodies in other assays such as in-house peptide ELISA and lateral flow immunoassays (data not shown). However, these peptides are well conserved among the hCoVs, so further analysis is warranted to determine if they are specific to the vaccinated group. To determine the full significance of this effect between populations and to potentially identify further distinct epitopes, an additional study with a larger sample pool is required.
The lack of significant IgA response within the spike region was somewhat surprising as previous studies utilizing longitudinal peptide microarrays had successfully identified IgA-reactive epitopes within the spike protein (8). The Schwarz et al. study notably revealed that IgA epitopes were more distinct with intense symptoms and peaked at three weeks post-infection and were mostly absent by ten weeks. Additional studies have found significant decreases in IgA levels as early as four weeks after infection (43). Additionally, IgA levels may have been low due to some of the infected group being asymptomatic or mildly symptomatic at the time of serum collection. Since the quantity of overall IgA in serum is typically found to be significantly lower than that of IgG, it is possible that levels of IgA were not high enough to discriminate from the naive samples. This is supported by ELISA data, which revealed consistently low IgA titers among the sera samples as opposed to simultaneously collected saliva samples (Table S4). These findings are supported by other studies, where it has been observed that the amount of IgA antibodies reactive to the SARS-CoV-2 spike protein can vary dramatically among infected individuals (41).
As the saliva samples showed significantly higher IgA levels than the sera used in the main study, saliva samples were collected from a subset of the vaccinated group to potentially identify IgA-specific epitopes that may have been missed initially. Wisnewski et al.’s results supported our preliminary data from testing saliva (not shown) on the trends of IgG and IgA response in SARS-CoV-2 infected and vaccinated individuals. Specifically, salivary IgA response was found to significantly increase within 2-3 weeks of vaccination (44). Further development of a microarray method utilizing saliva samples may help to study IgA binding activity against SARS-CoV-2 epitopes. Our ongoing study of identifying salivary IgG and IgA binding from previously infected individuals has demonstrated antibody binding to some epitopes that were identified in this report.
5 Conclusion
Our study of the SARS-CoV-2 spike protein epitope profiles of vaccinated and infected individuals has led us to identify epitopes that differentiate antibody binding patterns resulting from naturally occurring infection and vaccination. These peptides could potentially be implemented in diagnostic tools that could test for the presence of protective antibodies or active infection.
The mRNA vaccines are primarily directed against the S1 RBD of the virus, a hypervariable region most prone to viral mutation which results in variants that are less affected by the host’s immune response. On the contrary, antibody response to linear peptides derived from conserved areas of the SARS virus in the S2 C-terminal region is significantly increased in infected individuals when compared to vaccinated individuals. These responses are often cross-reactive between SARS-CoV-1 and SARS-CoV-2, which could be attributed to long term B cell activity. We speculate that this is due to T cell and long-term B cell antibody responses by the infected individual which moderate the toxicity of the host response. Peptides with the potential to identify vaccine-only individuals have been found, but a further assessment of the use of said peptides to discriminate between vaccinated, infected, and recently exposed individuals is necessary. The peptides may also serve to study the effect that variant mutations have on antibody binding involved in the viral-escape mechanisms. This information could be crucial as new variants continue to develop in the pandemic landscape. Developing additional assays to screen saliva samples may be helpful with efforts to perform IgA epitope profiling of the SARS-CoV-2 proteome and will aid in the development of saliva-based diagnostics and immunity determinations. This could also include detecting the immune response from the presence of the virus in the upper respiratory system within asymptomatic and pre-symptomatic individuals, possibly prior to detection by other methods (45).
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
NF, ST, and DT: Created study framework and experimental design. SF, GS, and DT: Funding, resource acquisition and management. NF: Acquired data and performed statistical analysis. Wrote first draft of manuscript. Responsible for graphics visualization. All authors contributed to the article and approved the submitted version.
Funding
This study is supported by a contract from the U. S. Defense Threat Reduction Agency MCDC2007-003.
Conflict of interest
NF, DT, LL, RA, GS, SH, SR, AM, MC, SS and ST were employed by New Horizons Diagnostics, Inc. GS were employed by Scout Microbiology LLC. BL were employed by Brian Luke Consulting LLC.
The remaining author declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2023.988109/full#supplementary-material
References
1. Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM, et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol (2021) 19(7):409–24. doi: 10.1038/s41579-021-00573-0
2. Bian L, Gao F, Zhang J, He Q, Mao Q, Xu M, et al. Effects of SARS-CoV-2 variants on vaccine efficacy and response strategies. Expert Rev Vaccines (2021) 20(4):365–73. doi: 10.1080/14760584.2021.1903879
3. Planas D, Veyer D, Baidaliuk A, Staropoli I, Guivel-Benhassine F, Rajah MM, et al. Reduced sensitivity of SARS-CoV-2 variant delta to antibody neutralization. Nature (2021) 596(7871):276–80. doi: 10.1038/s41586-021-03777-9
4. Fröberg J, Gillard J, Philipsen R, Lanke K, Rust J, van Tuijl D, et al. SARS-CoV-2 mucosal antibody development and persistence and their relation to viral load and COVID-19 symptoms. Nat Commun (2021) 12(1):5621. doi: 10.1038/s41467-021-25949-x
5. Fernandez-Montero A, Argemi J, Rodríguez JA, Ariño AH, Moreno-Galarraga L. Validation of a rapid antigen test as a screening tool for SARS-CoV-2 infection in asymptomatic populations. In: Sensitivity, specificity and predictive values. EclinicalMedicine (The Lancet) (2021) 37:100954. Available at: https://www.thelancet.com/journals/eclinm/article/PIIS2589-5370(21)00234-0/fulltext.
6. Heffron AS, McIlwain SJ, Amjadi MF, Baker DA, Khullar S, Armbrust T, et al. The landscape of antibody binding in SARS-CoV-2 infection. PloS Biol (2021) 19(6):e3001265. doi: 10.1371/journal.pbio.3001265
7. Holenya P, Lange PJ, Reimer U, Woltersdorf W, Panterodt T, Glas M, et al. Peptide microarray-based analysis of antibody responses to SARS-CoV-2 identifies unique epitopes with potential for diagnostic test development. Eur J Immunol (2021) 51(7):1839–49. doi: 10.1002/eji.202049101
8. Schwarz T, Heiss K, Mahendran Y, Casilag F, Kurth F, Sander LE, et al. SARS-CoV-2 proteome-wide analysis revealed significant epitope signatures in COVID-19 patients. Front Immunol (2021) 12:765. doi: 10.3389/fimmu.2021.629185
9. Musicò A, Frigerio R, Mussida A, Barzon L, Sinigaglia A, Riccetti S, et al. SARS-CoV-2 epitope mapping on microarrays highlights strong immune-response to n protein region. Vaccines (2021) 9(1):35. doi: 10.3390/vaccines9010035
10. Li Y, Xu Z, Lei Q, Lai Dy, Hou H, Jiang Hw, et al. Antibody landscape against SARS-CoV-2 proteome revealed significant differences between non-structural/ accessory proteins and structural proteins. medRxiv (2020) 08:20246314. doi: 10.1101/2020.12.08.20246314
11. Mishra N, Huang X, Joshi S, Guo C, Ng J, Thakkar R, et al. Immunoreactive peptide maps of SARS-CoV-2. Commun Biol (2021) 4:225. doi: 10.1038/s42003-021-01743-9
12. Poh CM, Carissimo G, Wang B, Amrun SN, Lee CYP, Chee RSL, et al. Two linear epitopes on the SARS-CoV-2 spike protein that elicit neutralising antibodies in COVID-19 patients. Nat Commun (2020) 11(1):2806. doi: 10.1038/s41467-020-16638-2
13. Mohandas S, Yadav PD, Shete A, Nyayanit D, Jain R, Sapkal G, et al. Protective immunity of the primary SARS-CoV-2 infection reduces disease severity post re-infection with delta variants in Syrian hamsters. Viruses (2022) 14(3):596. doi: 10.3390/v14030596
14. Iuliano AD, Brunkard JM, Boehmer TK, Peterson E, Adjei S, Binder AM, et al. Trends in disease severity and health care utilization during the early omicron variant period compared with previous SARS-CoV-2 high transmission periods - united states, December 2020-January 2022. MMWR Morb Mortal Wkly Rep (2022) 71(4):146–52. doi: 10.15585/mmwr.mm7104e4
15. Desingu PA, Nagarajan K, Dhama K. Emergence of omicron third lineage BA.3 and its importance. J Med Virol (2022) 94(5):1808–10. doi: 10.1002/jmv.27601
16. Gnjatic S, Ritter E, Büchler MW, Giese NA, Brors B, Frei C, et al. Seromic profiling of ovarian and pancreatic cancer. Proc Natl Acad Sci (2010) 107(11):5088–93. doi: 10.1073/pnas.0914213107
17. Negm OH, Hamed M, Monaghan TM. A protein microarray assay for serological determination of antigen-specific antibody responses following clostridium difficile infection. J Vis Exp (2018) 136):57399. doi: 10.3791/57399
18. Sharon D, Snyder M. Serum profiling using protein microarrays to identify disease related antigens. Methods Mol Biol Clifton NJ. (2014) 1176:169–78. doi: 10.1007/978-1-4939-0992-6_14
19. Yu H, Nguyen K, Royce T, Qian J, Nelson K, Snyder M, et al. Positional artifacts in microarrays: experimental verification and construction of COP, an automated detection tool. Nucleic Acids Res (2007) 35(2):e8. doi: 10.1093/nar/gkl871
20. Liang T, Cheng M, Teng F, Wang H, Deng Y, Zhang J, et al. Proteome-wide epitope mapping identifies a resource of antibodies for SARS-CoV-2 detection and neutralization. Signal Transduct Target Ther (2021) 6(1):1–3. doi: 10.1038/s41392-021-00573-9
21. Crowther JR. The ELISA guidebook. New York: Humana Press. (2009) 566. Available at: https://isbnsearch.org/isbn/9781617378843.
22. Izvorski A. Predicted 3D models of the SARS-CoV-2 spike protein membrane proximal external region and transmembrane domain. In: ChemRxiv (2020). Available at: https://chemrxiv.org/engage/chemrxiv/article-details/60c74f99842e651900db387b.
23. Liu DX, Liang JQ, Fung TS. Human coronavirus-229E, -OC43, -NL63, and -HKU1 (Coronaviridae). Encycl Virol (2021) 428–40. doi: 10.1016/B978-0-12-809633-8.21501-X
24. Thomas SR, Elkinton JS. Pathogenicity and virulence. J Invertebr Pathol (2004) 85(3):146–51. doi: 10.1016/j.jip.2004.01.006
25. Xia X. Domains and functions of spike protein in SARS-Cov-2 in the context of vaccine design. Viruses (2021) 13(1):109. doi: 10.3390/v13010109
26. Pang W, Lu Y, Zhao YB, Shen F, Fan CF, Wang Q, et al. A variant-proof SARS-CoV-2 vaccine targeting HR1 domain in S2 subunit of spike protein. Cell Res (2022) 1–18. doi: 10.1038/s41422-022-00746-3
27. Lambrou AS, Shirk P, Steele MK, Paul P, Paden CR, Cadwell B, et al. Genomic surveillance for SARS-CoV-2 variants: Predominance of the delta (B.1.617.2) and omicron (B.1.1.529) variants - united states, June 2021-January 2022. MMWR Morb Mortal Wkly Rep (2022) 71(6):206–11. doi: 10.15585/mmwr.mm7106a4
28. Cherian S, Potdar V, Jadhav S, Yadav P, Gupta N, Das M, et al. SARS-CoV-2 spike mutations, L452R, T478K, E484Q and P681R, in the second wave of COVID-19 in maharashtra, India. Microorganisms (2021) 9(7):1542. doi: 10.3390/microorganisms9071542
29. Greaney AJ, Starr TN, Barnes CO, Weisblum Y, Schmidt F, Caskey M, et al. Mapping mutations to the SARS-CoV-2 RBD that escape binding by different classes of antibodies. Nat Commun (2021) 12(1):4196. doi: 10.1038/s41467-021-24435-8
30. McCallum M, Bassi J, De Marco A, Chen A, Walls AC, Di Iulio J, et al. SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science (2021) 373(6555):648–54. doi: 10.1126/science.abi7994
31. Cao Y, Wang J, Jian F, Xiao T, Song W, Yisimayi A, et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature (2022) 602(7898):657–63. doi: 10.1038/s41586-021-04385-3
32. Xia S, Lan Q, Su S, Wang X, Xu W, Liu Z, et al. The role of furin cleavage site in SARS-CoV-2 spike protein-mediated membrane fusion in the presence or absence of trypsin. Signal Transduct Target Ther (2020) 5(1):1–3. doi: 10.1038/s41392-020-0184-0
33. Peacock TP, Goldhill DH, Zhou J, Baillon L, Frise R, Swann OC, et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat Microbiol (2021) 6(7):899–909. doi: 10.1038/s41564-021-00908-w
34. Saville JW, Mannar D, Zhu X, Srivastava SS, Berezuk AM, Demers JP, et al. Structural and biochemical rationale for enhanced spike protein fitness in delta and kappa SARS-CoV-2 variants. Nat Commun (2022) 13(1):742. doi: 10.1038/s41467-022-28324-6
35. Du X, Tang H, Gao L, Wu Z, Meng F, Yan R, et al. Omicron adopts a different strategy from delta and other variants to adapt to host. Signal Transduct Target Ther (2022) 7(1):1–3. doi: 10.1038/s41392-022-00903-5
36. Lei KC, Zhang XD. Conservation analysis of SARS-CoV-2 spike suggests complicated viral adaptation history from bat to human. Evol Med Public Health (2020) 2020(1):290–303. doi: 10.1093/emph/eoaa041
37. Centers for Disease Control and Prevention. SARS-CoV-2 variant classifications and definitions. (2021) 1–12. Available at: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html.
38. Favresse J, Gillot C, Di Chiaro L, Eucher C, Elsen M, Van Eeckhoudt S, et al. Neutralizing antibodies in COVID-19 patients and vaccine recipients after two doses of BNT162b2. Viruses (2021) 13(7):1364. doi: 10.3390/v13071364
39. Greaney AJ, Loes AN, Gentles LE, Crawford KHD, Starr TN, Malone KD, et al. Antibodies elicited by mRNA-1273 vaccination bind more broadly to the receptor binding domain than do those from SARS-CoV-2 infection. Sci Transl Med (2021) 13(600):eabi9915. doi: 10.1126/scitranslmed.abi9915
40. Wu J, Liang B, Chen C, Wang H, Fang Y, Shen S, et al. SARS-CoV-2 infection induces sustained humoral immune responses in convalescent patients following symptomatic COVID-19. Nat Commun (2021) 12(1):1813. doi: 10.1038/s41467-021-22034-1
41. Kent SJ, Juno JA. Vaccination after prior COVID-19 infection: Implications for dose sparing and booster shots. EBioMedicine (2021) 72:103586. doi: 10.1016/j.ebiom.2021.103586
42. Guthmiller JJ, Stovicek O, Wang J, Changrob S, Li L, Halfmann P, et al. SARS-CoV-2 infection severity is linked to superior humoral immunity against the spike. mBio (2021) 12(1):e02940–20. doi: 10.1128/mBio.02940-20
43. Quinti I, Mortari EP, Fernandez Salinas A, Milito C, Carsetti R. IgA antibodies and IgA deficiency in SARS-CoV-2 infection. Front Cell Infect Microbiol (2021) 11:655896. doi: 10.3389/fcimb.2021.655896
44. Wisnewski AV, Luna JC, Redlich CA. Human IgG and IgA responses to COVID-19 mRNA vaccines. PloS One (2021) 16(6):e0249499. doi: 10.1371/journal.pone.0249499
Keywords: B-cell epitope, variant mutations, peptide microarray, IgG, SARS-CoV-2, spike protein, hCoV
Citation: Felbinger N, Trudil D, Loomis L, Ascione R, Siragusa G, Haba S, Rastogi S, Mucci A, Claycomb M, Snowberger S, Luke B, Francesconi S and Tsang S (2023) Epitope mapping of SARS-CoV-2 spike protein differentiates the antibody binding activity in vaccinated and infected individuals. Front. Virol. 3:988109. doi: 10.3389/fviro.2023.988109
Received: 06 July 2022; Accepted: 09 February 2023;
Published: 03 March 2023.
Edited by:
Andrea Lombardi, University of Milan, ItalyReviewed by:
George William Carnell, University of Cambridge, United KingdomStephen Crooke, Centers for Disease Control and Prevention (CDC), United States
Copyright © 2023 Felbinger, Trudil, Loomis, Ascione, Siragusa, Haba, Rastogi, Mucci, Claycomb, Snowberger, Luke, Francesconi and Tsang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shirley Tsang, U2hpcmxleVRAbmhkaWFnLmNvbQ==