 Pankaj Kumar Verma
Pankaj Kumar Verma Branden H. Watson
Branden H. Watson Venkateswara R. Sripathi
Venkateswara R. Sripathi Madhusudhana R. Janga
Madhusudhana R. Janga- 1Department of Plant and Soil Sciences, Institute of Genomics for Crop Abiotic Stress Tolerance, Texas Tech University, Lubbock, TX, United States
- 2Brownfield Seed & Delinting Co, Brownfield, TX, United States
- 3Center for Molecular Biology, Alabama A&M University, Normal, AL, United States
Background: Cotton leafroll dwarf virus (CLRDV), a member of the Polerovirus genus, is an emerging pathogen that threatens global cotton (Gossypium hirsutum) production. Since its first detection in Alabama in 2017, CLRDV has spread rapidly to several states of the United States of America, including Texas.
Methods: In 2024, symptomatic cotton plants were collected from Brownfield, Texas. Total RNA was extracted, and RT-PCR was performed to amplify the viral genome and sequenced. The complete sequence (5,838 bp) was obtained and compared with existing CLRDV genomes from the U.S.
Results: The Brownfield isolate displayed typical CLRDV genome features but also showed genetic differences compared to isolates from neighboring regions. Phylogenetic analysis indicated regional diversification, possibly due to environmental pressures or host cultivar variability.
Conclusion: This study highlights the presence and evolution of CLRDV in Texas and neighboring states. Ongoing surveillance and development of resistant cotton cultivars are essential to mitigate yield losses.
1 Introduction
Cotton (Gossypium hirsutum L.) stands as one of the world’s most valuable fiber crops, grown in over 80 countries and serving as a cornerstone of the global textile industry (1). During the 2023–2024 season, worldwide cotton production exceeded 24.67 million metric tons, equivalent to 113.29 million bales (1 bale = 480 pounds) (2). The United States plays a significant role in global production, ranking fourth with approximately 14.41 million bales, around 12% of the global total, and contributing nearly $6.62 billion to the national economy by cotton export (3). Within the U.S., Texas dominates national production with approximately 3.2 million hectares under cultivation, followed by Georgia, Arkansas, Mississippi, North Carolina, and Alabama. Cotton cultivation in the U.S. faces persistent threats from a range of biotic stresses, including pests and pathogenic organisms such as bacteria, fungi, nematodes, and viruses. In 2023, biotic stresses alone contributed to a 7.4% reduction in yield nationwide, resulting in an estimated loss of approximately 1.4 million bales (4). Although viral diseases currently contribute minimally to an overall yield loss of about 2,994 bales, the increasing spread of viruses such as cotton leafroll dwarf virus (CLRDV) raises concern for future outbreaks and economic consequences (4).
CLRDV, a member of the Polerovirus genus within the Solemoviridae family, was initially identified in Africa in 1949 and has since been reported in parts of Asia and South America (5). The virus was first detected in the U.S. in 2017 in Alabama (6) and has now been confirmed in at least 14 cotton-growing states, including Georgia, Mississippi, and Texas (7). Its prevalence is variable, with incidence rates ranging from below 1% to over 20%, depending on the region (5, 8–14). The first genomic insights of CLRDV in the U.S. emerged from partial sequences obtained in Alabama by Avelar, et al. (6), followed by a complete genome sequence from a Georgia isolate by Tabassum, et al. (13). Cotton leafroll dwarf virus (CLRDV) poses a growing challenge to cotton production across the U.S., yet critical gaps remain in our understanding of its genetic landscape. Although its incidence has increased in recent years, there is a need for more sequenced genome data to identify genetic variation and population structure of CLRDV strains affecting U.S. cotton fields. Moreover, uncovering new viral isolates and obtaining their full-length genome sequences are essential steps toward understanding how the virus evolves, adapts, and spreads. Such insights are not only vital for accurate diagnostics and targeted disease management but also for guiding resistance breeding efforts aimed at safeguarding cotton crops against emerging viral threats.
Building on this foundation, the present study aims to detect new CLRDV isolates and obtain their full-length sequence from the Brownfield, Texas samples. To achieve these objectives, symptomatic cotton leaf samples were systematically collected from commercial fields and subjected to amplification of the full-length sequence. Further, the comparative analyses with previously reported CLRDV sequences enabled the identification of nucleotide variations and phylogenetic relationships among isolates. These approaches collectively contribute to a more comprehensive understanding of CLRDV evolution and distribution in a major cotton-producing region of the southern United States. Our findings enhance the current understanding of CLRDV genetic diversity and provide valuable aid to support the development of effective monitoring strategies and the breeding of resistant cotton cultivars.
2 Materials and methods
2.1 Sample collection
During the 2024 growing season, leaf and petiole samples were collected from a commercial field located at Brownfield, Texas. The samples collected from cotton plants exhibiting symptoms indicative of CLRDV infection (5) which ranged from mild chlorosis to severe stunting and leaf curling (Figures 1A-F). To preserve RNA integrity, samples were flash-frozen in liquid nitrogen immediately after collection and stored at -80°C until further processing.

Figure 1. Identification and molecular confirmation of cotton leafroll dwarf virus (CLRDV) in symptomatic cotton plants from Brownfield, Texas. (A–F) Cotton plants showed characteristic symptoms of CLRDV infection: (A) leaf rolling and cupping, (B) reddening of stems and petioles, (C) interveinal chlorosis and vein yellowing, and (D–F) stunted growth with shortened internodes. (G) RT-PCR detection of CLRDV using specific primer sets: lane M, 1 kb Plus DNA ladder; lane 1, 312 bp amplicon with primers JL0067/JL0068; lane 3, 432 bp amplicon with primers JL0063/JL0068; lane 5, 2,173 bp amplicon with primers JL0100/JL0068. Lanes 2, 4, and 6 represent corresponding no-template controls. (H) An annotated full-length CLRDV genome map generated using SnapGene software (version 8.0.3).
2.2 RNA extraction and reverse transcription-PCR
Total RNA was isolated from symptomatic leaf and petiole tissue using the Spectrum™ Plant Total RNA Kit (Sigma-Aldrich, St. Louis, MO, USA), following the manufacturer’s instructions. To eliminate potential genomic DNA contamination, the RNA was treated with RNase-free DNase I (Qiagen, USA). Complementary DNA (cDNA) was synthesized using the GoScript™ Reverse Transcriptase cDNA Synthesis Kit (Promega, USA) and a gene-specific reverse primer (Table 1). The reverse transcription protocol involved the denaturation of RNA and gene-specific reverse primer at 70°C for 5 minutes, followed by the immediate addition of the RT master mix and incubation at 42°C for 90 minutes to ensure optimal primer annealing and cDNA synthesis. PCR amplification was conducted using primer pairs JL0067-JL0068 and JL0063-JL0068, targeting conserved regions of the viral movement and coat protein genes for diagnostic detection of CLRDV (13, 15). To amplify a longer genomic fragment spanning partial ORF1 and ORF3 regions (~2132 nt), the JL0100-JL0068 primer pair was employed. Amplicons were sequenced using Oxford Nanopore sequencing, which was carried out by Plasmidsaurus (https://plasmidsaurus.com/). The resulting sequences were aligned using NCBI’s BLASTn tool and BioEdit (16).
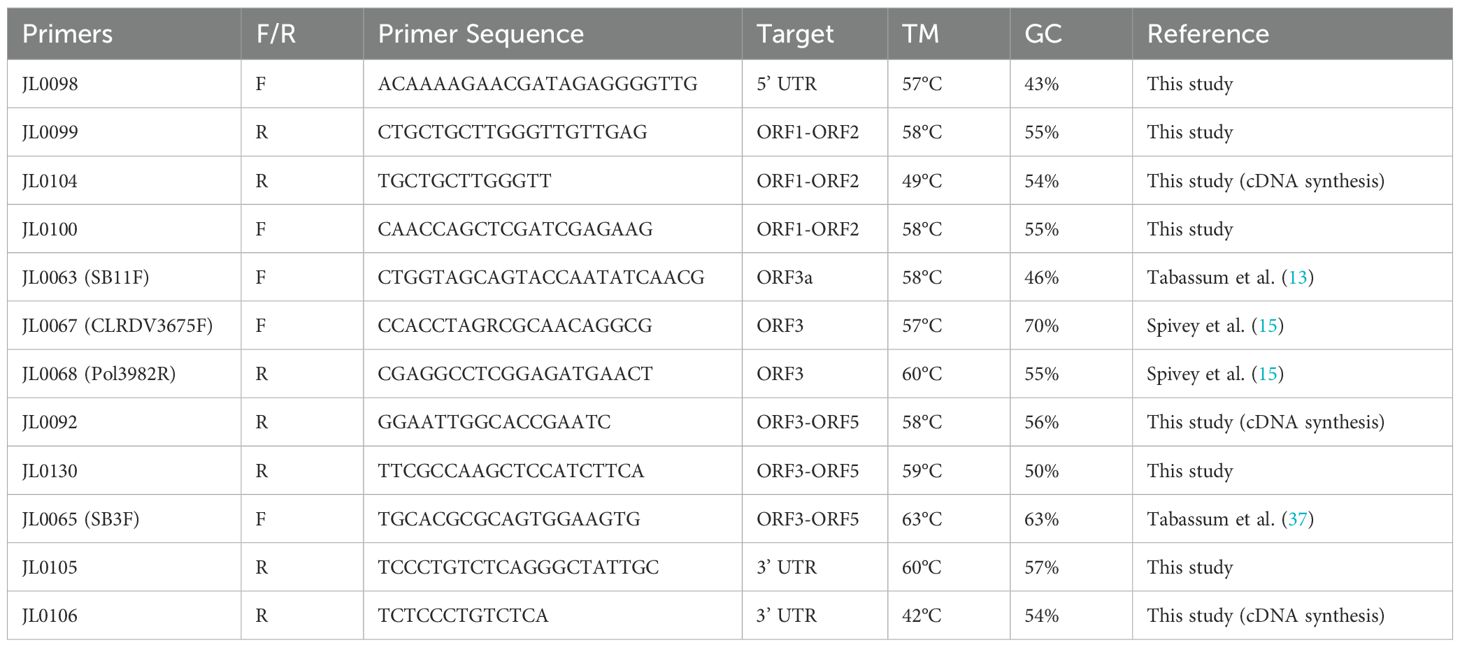
Table 1. List of primers used in this study.
2.3 Full-length sequence amplification and sequencing
Further, for the full-length CLRDV sequence amplification, a strategic primer design approach was implemented based on multiple sequence alignment (MSA). Coding sequences from closely related CLRDV strains were retrieved from the NCBI Virus database based on the alignment with an amplified 2.1 kb partial fragment. These sequences were aligned using ClustalW with default settings to identify conserved regions, particularly at the 5′ and 3′ termini of the target open reading frame (17). Regions with ≥90% conservation were selected as candidate primer binding sites. SnapGene version 8.0.3 was used to design 4 pairs of primer sets within these regions. These primers were used to amplify the full-length CLRDV sequence in 4 overlapping fragments using RT-PCR reaction using Phusion High-Fidelity DNA Polymerase (Thermo Scientific, USA), and PCR products were sequenced at Plasmidsaurus (https://plasmidsaurus.com/) using the Oxford nanopore technique. All four fragments were assembled after removing the overlapped region to get the full-length sequence. Viral coding regions were predicted using SnapGene’s import feature function (SnapGene Version 8.0.3) from the annotation GFF3 file of the closest aligned sequences.
2.4 Sequence alignment and phylogenetic analysis
Phylogenetic relationships of amplified sequence in this study (accession number: PV548928) and existing full-length CLRDV sequences retrieved from the NCBI Virus database (https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/virus?SeqType_s=Nucleotide&VirusLineage_ss=Cotton%20leafroll%20dwarf%20virus,%20taxid:312295) were analyzed using the phangorn R package (18) after the multiple sequence alignment with the msa package (19). Sequences were aligned using the ClustalW algorithm with default substitution parameters (20). The aligned sequences were converted into phyDat format for phylogenetic inference. Model selection was performed using the modelTest function in the phangorn R package, and the best-fit evolutionary model (TIM2e+G(4)+I) was selected based on the Akaike Information Criterion (AIC) and Bayesian Information Criterion (BIC) (18). A maximum likelihood (ML) tree was inferred using the pml_bb function under the selected model. Node support was assessed via 1000 bootstrap replicates using the bootstrap.pml function with nearest-neighbor interchange (NNI) optimization. The resulting phylogenetic tree was midpoint rooted and visualized using the plotBS function with ultrafast bootstrap values. Tip labels were color-coded based on the geographic origin of the state to highlight the spatial distribution of viral lineages. A consensus network was constructed using the consensusNet function with a 20% threshold to display topological variation among bootstrap replicates.
Pairwise nucleotide sequence comparisons were conducted between the CLRDV isolate identified in this study (accession number: PV548928) and previously reported CLRDV genome sequences retrieved from the NCBI Virus database (accession numbers: OK185946, OK185945, OQ107471, PP556773, PP556772, PP556774, OK185941, OQ107470, OK185944, OK185943, OK185942, MN872302, OM687235), which were identified from Texas and surrounding regions. The ORFs 0–5 sequence alignment was performed using ClustalW, and pairwise distance was calculated using the dist.DNA function of the Ape R package was plotted using the pheatmap R package (21).
2.5 Amino acid sequence analysis
A multiple sequence alignment (MSA) was carried out using MAFFT (22), followed by the computation of site-specific conservation scores via the bio3d package in R (23) to explore the evolutionary dynamics and functional conservation among the aligned protein sequences. A maximum likelihood tree was also constructed using amino acid alignment using IQTree-3.0.0 (24). To characterize the putative proteins encoded by the complete CLRDV genome, amino acid sequences were analyzed using MEME Suite with default parameters for the identification of conserved motifs (25).
3 Results
3.1 New CLRDV isolate from Brownfield, Texas, identified by RT-PCR and sequencing
In this study, we identified an unreported viral isolate from cotton samples collected in Brownfield, Texas. Initial detection was achieved using PCR amplification, which revealed diagnostic bands of expected sizes 312 bp and 432 bp (Figure 1G). The partial CLRDV sequence of a 2,173 bp amplicon was obtained using primer pair JL0100 and JL0068 (Figure 1G, Supplementary Table S1), which shares the highest similarity with known CLRDV isolates from Oklahoma, Kansas, and Texas (GenBank accessions: OM687235.2 and PP556774.1) (26). Further, the full-length CLRDV genome was obtained by amplifying and sequencing four overlapping fragments. Assembly of these fragments yielded a complete genome sequence of 5838 nt, which represents a typical CLRDV isolate (Figure 1H). Sequence analysis revealed a 90.58%-99.69% identity with previously reported CLRDV genomes (Supplementary Table S2). The assembled sequence was submitted to NCBI under accession number PV548928, which was used in all subsequent analysis.
3.2 Sequence analysis reveals genetic variation among CLRDV isolates
Since the first identification of CLRDV, 834 CLRDV-related sequences have been deposited in the NCBI Virus Database, including 60 complete or near-complete genomes of these; 50 full-length sequences originate from U.S. samples, including the isolate identified in this study (PV548928) (27). Supplementary Figure S1 illustrates the geographic spread of both complete and partial CLRDV sequences in the USA, specifically showing their detected states of origin as submitted to NCBI. In order to establish the relationship of the identified CLRDV isolate from Brownfield, Texas, with previously identified CLRDV isolates, a phylogenetic analysis was conducted using the Maximum Likelihood (ML) approach. The full-length genome sequence obtained (5838nt) was aligned with a set of CLRDV isolates having full-length sequences from various geographical regions retrieved from the NCBI virus database. Multiple sequence alignment was performed using ClustalW, showing the variation among the sequences from samples collected from Texas and neighboring states (Supplementary Figure S2). The phylogenetic tree was constructed in the best-fit evolutionary model (TIM2e+G(4)+I), which was determined to be the best-fitting substitution model based on the Akaike Information Criterion (AIC) and Bayesian Information Criterion (BIC) with 1,000 bootstrap replicates. The resulting ML tree revealed that the Brownfield isolates clustered within a clade comprising isolates from the southern Great Plains region of the United States (Figure 2A). Within this clade, the Brownfield isolate showed the closest evolutionary relationship to the CLRDV isolate EC4 (GenBank: OM687235.2), which was previously identified in cotton samples collected from Oklahoma. To find the variation among the regional population, we reconstructed the phylogenetic tree with isolates identified in Texas and neighboring states (Figure 2B).
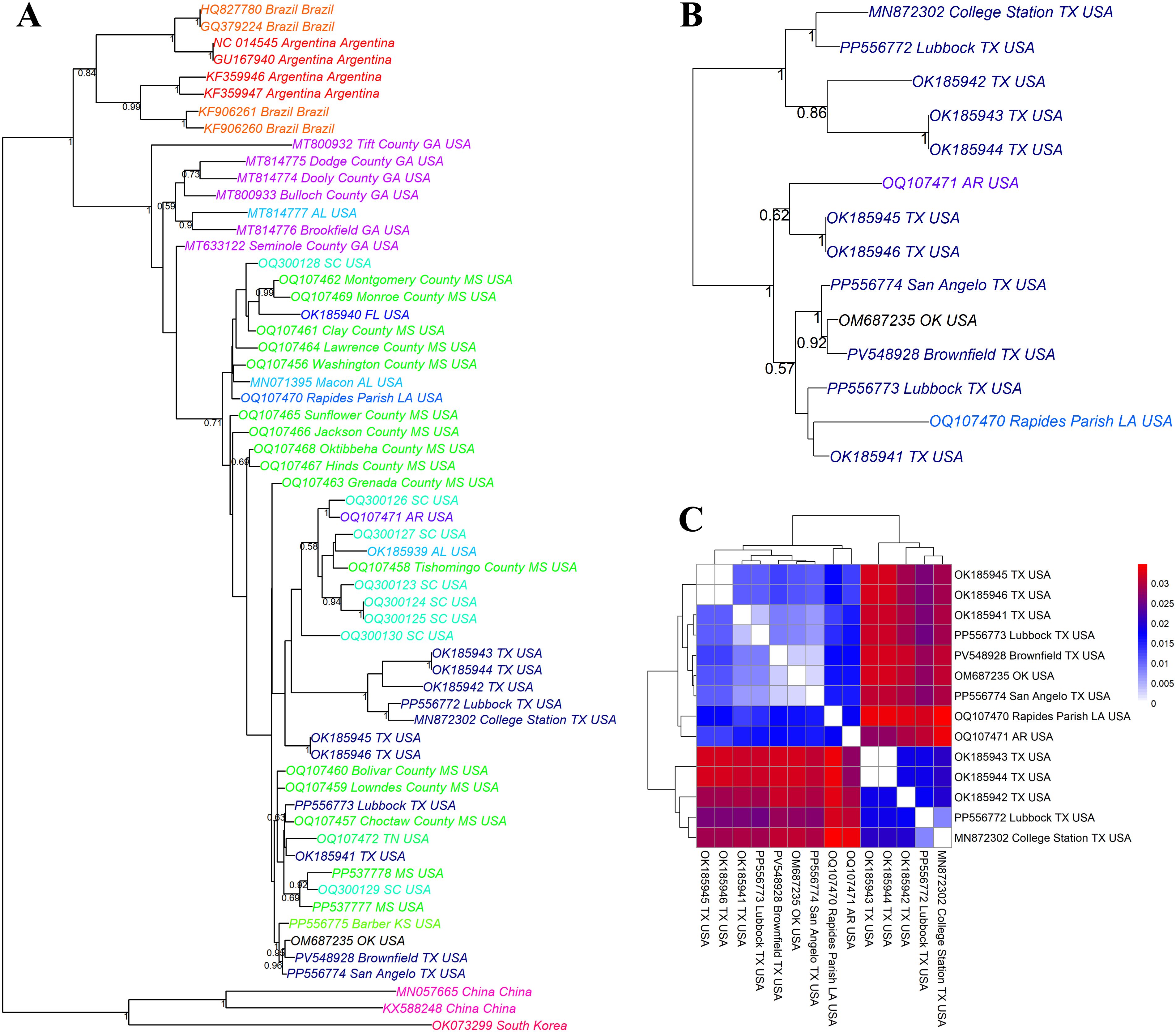
Figure 2. Phylogenetic analysis of cotton leafroll dwarf virus (CLRDV) isolates from Texas and neighboring regions. Maximum likelihood phylogenetic tree based on ClustalW alignment of (A) all available full-length nucleotide sequences of CLRDV isolates retrieved from the NCBI Virus database, (B) Full-length CLRDV sequences of the samples collected from a specific region of Texas and surrounding states. Tree nodes are annotated with GenBank accession numbers and geographic origins, with color-coding used to represent different geographic regions at the state level. (C) Pairwise nucleotide sequence dissimilarity analysis among CLRDV isolates from Texas and adjacent states indicates regional sequence variation. The color scale represents nucleotide divergence, with blue indicating high similarity (low genetic distance) and red representing greater divergence.
To assess the genetic relationships among CLRDV isolates, a pairwise distance matrix was generated using full-length genome sequences. The resulting heatmap (Figure 2C) illustrates the genetic divergence among 14 CLRDV isolates from Texas and neighboring states (Oklahoma, Arkansas, and Louisiana). The color scale represents nucleotide divergence, with blue indicating high similarity (low genetic distance) and red representing greater divergence. The Brownfield isolate (PV548928) clustered closely with isolates from Oklahoma (OM687235), Lubbock (PP556773), and San Angelo (PP556774), showing minimal genetic divergence (dark blue). This suggests a high degree of sequence conservation and possible regional movement or common ancestry among these isolates. In contrast, isolates from College Station (MN872302), Rapides Parish, LA (OQ107470), and Arkansas (OQ107471) displayed greater divergence (red shades), indicating broader genetic variation across geographical regions. These findings support the existence of region-specific CLRDV lineages and highlight the close genetic similarity of the Brownfield isolate with other West Texas variants (Figure 2C, Supplementary Table S3).
3.3 Conservation and motif analysis of aligned protein-coding sequences
To further characterize the identified CLRDV isolate from Brownfield, Texas, we performed a detailed amino acid sequence analysis of the predicted viral proteins encoded by the sequenced genome. We translated the coding regions into their corresponding amino acid sequences using the nucleotide sequence data. The translation was carried out using the standard genetic code, and the individual open reading frames (ORFs) were identified based on the known genome organization of CLRDV. A multiple sequence alignment (MSA) was constructed using MAFFT. Further, the maximum likelihood tree constructed from amino acid alignments suggested the variation between sequences of CLRDV identified from a small geographical region, i.e. Texas and surrounding areas (Figure 3A). The computation of site-specific conservation scores, ranging from 0 (completely variable) to 1 (fully conserved), were assigned to each residue position bsed on the degree of sequence similarity at that site (Figure 3B). Conversely, multiple dips with scores below 0.4 reflect variable or evolutionarily flexible segments, potentially associated with surface loops, linker regions, or domains undergoing adaptive evolution. Further, we performed de novo motif discovery using the MEME Suite to pinpoint specific sequence elements that are recurrently conserved. This analysis uncovered ten highly significant motifs (E-values ranging from 7.08e-242 to 8.49e-242) shared among the analyzed sequences (Figure 3C). All motifs were reproducibly detected across the 14 viral isolates examined, including newly identified isolates and others, underscoring their likely evolutionary retention and biological importance. The consensus sequences of the motifs spanned 20 to 50 nucleotides, and several displayed notable features consistent with regulatory or structural roles. For example, Motif 1 (AGATAACTCAGTAGCTTGTTATAGCAGGAGCTCTTAA) and Motif 4 (AACAATTTTGGAACAGTTTTTCTCGAGATCTGAAATCA) were among the most broadly distributed and positionally conserved, suggesting their potential function as core regulatory elements or essential protein-binding sites. Repetitive motifs such as Motif 5 and Motif 7 exhibited features resembling G-rich tracts or tandem repeats, often implicated in RNA secondary structure formation or transcriptional control mechanisms. Overall, the combination of high conservation scores and consistently recurring motifs provides compelling evidence for the presence of functionally constrained domains and regulatory elements within these viral protein-coding regions. The strong statistical support (E-values < 1e-241) further emphasizes that these motifs likely contribute to conserved molecular functions such as replication, transcription regulation, or host interaction. At the same time, the variation in sequences suggests the evolution of the CLRDV genome under environmental pressure.
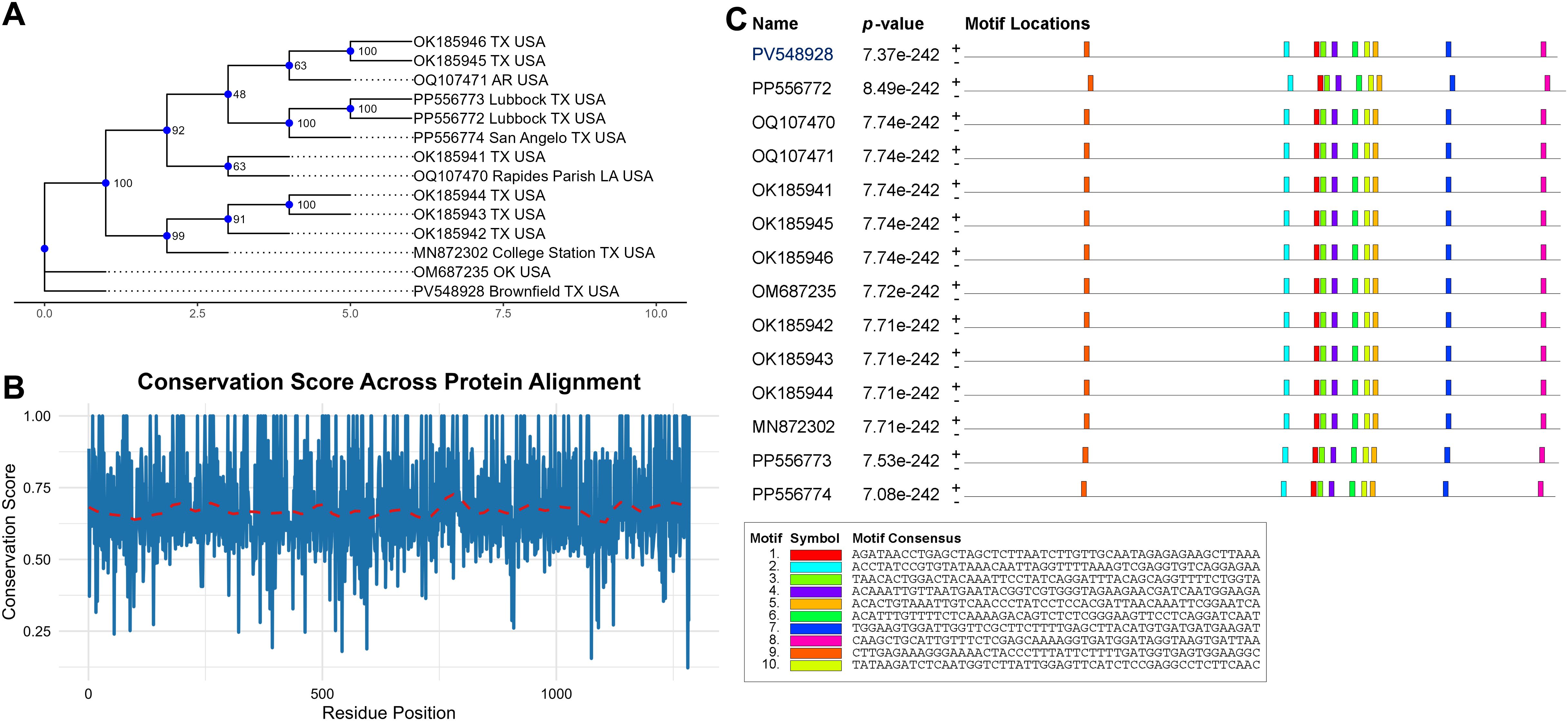
Figure 3. Conservation analysis of amino acid sequences. (A) maximum likelihood tree of amino acid sequences (B) Conservation score plot across the aligned protein sequences. The blue line represents residue-wise conservation scores, and the red dashed line denotes a loess-smoothed trend highlighting the general conservation pattern. (C) Motif distribution plot of conserved elements detected in protein-coding sequences. Colored boxes represent distinct motifs identified by MEME. The presence of these motifs across all input sequences, combined with extremely low E-values, indicates high statistical significance.
4 Discussion
CLRDV, the probable causative agent of Cotton Blue Disease, is becoming an increasing threat to cotton production in various regions, including the southeastern United States, parts of South America, and Africa (5). The virus is primarily transmitted by the cotton aphid (Aphis gossypii). Early-stage infections of CLRDV often go unnoticed due to their latent and asymptomatic nature, making early detection and intervention particularly challenging. Management strategies for CLRDV are hindered by several factors, including the absence of resistant cotton varieties and the aphid’s high reproductive rate, adaptability, and increasing resistance to chemical insecticides (28, 29). In addition, the virus’s ability to survive in alternate hosts, including weeds and volunteer cotton plants, enables it to overwinter and reemerge during the growing season, further complicating control efforts (30). Moreover, specific mutations in viral genes, such as P0, which plays a role in viral pathogenicity and suppression of host RNA silencing, have been identified as key targets for diagnostic advancements (31). Advances in genomic sequencing have provided new insights into the virus’s spread and evolution, offering opportunities to better understand and address CLRDV (32). Genome sequencing data are crucial for assessing genetic diversity among CLRDV strains in different regions, enabling the tracking of viral movement, detection of emerging virulent variants, and the development of more precise molecular diagnostic tools. Thus, combining genome-based surveillance with traditional agricultural practices and breeding initiatives offers a promising approach to more effectively manage CLRDV.
The high bootstrap value (>90%) associated with the phylogenetic grouping of certain isolates suggests a strong genetic similarity and a recent common ancestry between these strains. This close relationship implies that the EC4-like variant may have spread quickly, or that the isolates detected in Brownfield underwent mutations, possibly through aphid-mediated transmission or human activities, such as the movement of infected plant material. Detection of this isolate in Brownfield (TX) aligns with statewide reports of CLRDV circulation across commercial cotton fields and raises concerns that the virus may be more widespread than currently known. Moreover, genomic data from retrospective studies indicate CLRDV has been cryptically circulating in the U.S. since at least 2006, underscoring the potential genetic diversity yet to be uncovered (32). Although the sequence variation among isolates remains limited, this highlights the need for ongoing genomic surveillance to detect novel variants before they attain wider distribution. Further, the amino acid changes observed in the viral sequences provide important insights into potential viral adaptations (13, 33). Phylogenetic reconstruction using Maximum Likelihood methods demonstrated that CLRDV isolates form geographically distinct clades, indicating regional adaptation (34–36). The fact that isolates from the Brownfield cluster together suggests that local environmental pressures or varying resistance traits among cotton cultivars may be selecting for specific viral variants. Moreover, coordinated studies from sentinel plots across the U.S. cotton belt highlight cultivar-specific variation in disease incidence and severity, reinforcing the hypothesis that host resistance shapes viral evolution (35, 36). Collectively, these findings support the idea that regional environmental factors and cultivar composition are driving local adaptation of CLRDV, enhancing the virus’s ability to persist and spread. Ongoing monitoring of viral evolution and transmission dynamics is essential for understanding the virus’s adaptation to local conditions and for developing effective, region-specific management strategies and diagnostic tools.
5 Conclusion
Despite its economic importance, knowledge regarding the genetic diversity of CLRDV and the evolutionary forces shaping its variability remains limited. This study underscores the nucleotide diversity in relatively conserved sequences among closely related CLRDV isolates, primarily driven by mutations and recombination events in a small area. The amino acid sequence analysis of the new CLRDV isolate from Brownfield, Texas, highlighted both conserved and variable regions across the viral genome. The identified amino acid substitutions in the movement protein suggest potential implications for the virus’s ability to spread within cotton plants, warranting further investigation into their functional significance. Ongoing surveillance and functional studies of these amino acid changes are crucial for understanding CLRDV spread dynamics and developing more effective management strategies. These insights will aid in developing molecular diagnostic tools, strengthening resistance breeding, and improving epidemiological monitoring of CLRDV in the U.S. and South America. Further research should focus on virus transmission, genome variation, recombination, host interactions, and the role of aphid vectors in viral evolution.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: NCBI GenBank, accession PV548928.
Ethics statement
Ethical approval was not required for the studies on pants in accordance with the local legislation and institutional requirements.
Author contributions
PV: Formal analysis, Validation, Visualization, Data curation, Methodology, Writing – review & editing, Writing – original draft, Conceptualization, Investigation. BW: Writing – review & editing, Methodology, Formal analysis, Investigation. VS: Writing – review & editing. MJ: Supervision, Writing – review & editing, Funding acquisition, Resources, Formal analysis, Methodology, Data curation, Conceptualization, Investigation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Salary support by the Faculty Texas University Funds (TUF) Start Up under the supervision of the Operations Division of TTU.
Conflict of interest
The authors declare that the research was conducted without any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2025.1619281/full#supplementary-material
Supplementary Figure 1 | State-wise distribution of CLRDV genomic sequences submitted to the NCBI Virus database. This figure illustrates the number of Cotton leafroll dwarf virus (CLRDV) sequences reported from different U.S. states, as available in the NCBI Virus database. Each state’s contribution is represented based on the number of sequences linked to its geolocation, highlighting regional variations in sample collection and virus reporting. The data reflects current efforts in monitoring and documenting the spread of CLRDV across the country.
Supplementary Figure 2 | Multiple sequence alignment using ClustalW and visualized by NCBI multiple sequence alignment viewer. The grey dot shows the conserved sequence while red letters represent the variation in sequence.
References
1. Degefu DT and Gebregiorgis ZD. Cotton biotechnology. In: Cotton Sector Development in Ethiopia: Challenges and Opportunities. Springer Nature Singapore Pte Ltd. (2024). p. 65–88.
2. 2024/2025 cotton production(2025). Available online at: https://www.fas.usda.gov/data/production/commodity/2631000 (Accessed April 20, 2025).
3. U.S. Cotton exports in 2024(2025). Available online at: https://www.fas.usda.gov/data/commodities/cotton (Accessed April 20, 2025).
4. 2024 national disease losses, the crop protection network (Cpn)(2025). Available online at: https://cropprotectionnetwork.org/publications/cotton-disease-loss-estimates-from-the-united-states-2024 (Accessed April 20, 2025).
5. Edula SR, Bag S, Milner H, Kumar M, Suassuna ND, Chee PW, et al. Cotton leafroll dwarf disease: an enigmatic viral disease in cotton. Mol Plant Pathol. (2023) 24:513–26. doi: 10.1111/mpp.13335
6. Avelar S, Schrimsher D, Lawrence K, and Brown J. First report of cotton leafroll dwarf virus associated with cotton blue disease symptoms in Alabama. Plant Dis. (2019) 103:592–. doi: 10.1094/PDIS-09-18-1550-PDN
7. Koebernick J, Hagan A, Zaccaron M, Escalante C, Jacobson A, Bowen K, et al. Monitoring the distribution, incidence, and symptom expression associated with cotton leafroll dwarf virus in the southern United States using a sentinel plot system. PhytoFrontiers™. (2024) 4:671–81. doi: 10.1094/PHYTOFR-02-24-0008-R
8. Aboughanem-Sabanadzovic N, Allen T, Wilkerson T, Conner K, Sikora E, Nichols R, et al. First report of cotton leafroll dwarf virus in upland cotton (Gossypium hirsutum) in Mississippi. Plant Dis. (2019) 103:1798. doi: 10.1094/PDIS-01-19-0017-PDN
9. Alabi OJ, Isakeit T, Vaughn R, Stelly D, Conner KN, Gaytán BC, et al. First report of cotton leafroll dwarf virus infecting upland cotton (Gossypium hirsutum) in Texas. Plant Dis. (2020) 104:998. doi: 10.1094/PDIS-09-19-2008-PDN
10. Ali A and Mokhtari S. First report of cotton leafroll dwarf virus infecting cotton (Gossypium hirsutum) in Kansas. Plant Dis. (2020) 104:1880. doi: 10.1094/PDIS-12-19-2589-PDN
11. Ali A, Mokhtari S, and Ferguson C. First report of cotton leafroll dwarf virus from cotton (Gossypium hirsutum) in Oklahoma. Plant Dis. (2020) 104:2531. doi: 10.1094/PDIS-03-20-0479-PDN
12. Ferguson C and Ali A. Genetic diversity of cotton leafroll dwarf virus from the Southwestern United States and its implications for the multi-introduction event hypothesis and future evolution. Plant Dis. (2024) 108:3484–95. doi: 10.1094/PDIS-05-24-0952-SR
13. Tabassum A, Bag S, Suassuna ND, Conner KN, Chee P, Kemerait RC, et al. Genome analysis of cotton leafroll dwarf virus reveals variability in the silencing suppressor protein, genotypes and genomic recombinants in the USA. PloS One. (2021) 16:e0252523. doi: 10.1371/journal.pone.0252523
14. Wang H, Greene J, Mueller J, Conner K, and Jacobson A. First report of cotton leafroll dwarf virus in cotton fields of South Carolina. Plant Dis. (2020) 104:2532. doi: 10.1094/PDIS-03-20-0635-PDN
15. Spivey WW, Williamson Z, Seiter J, Abrahamian P, Wang H, Greene J, et al. Analysis of cotton leafroll dwarf virus P0 gene sequences from South Carolina reveals low variability among isolates. Plant Dis. (2023) 107:2613–9. doi: 10.1094/PDIS-10-22-2514-SR
16. Hall TA ed. Bioedit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/Nt. Nucleic Acids Symp Ser. (1999) 41:95–8.
17. Thompson JD, Gibson TJ, and Higgins DG. Multiple sequence alignment using clustalw and clustalx. Curr Protoc Bioinf. (2003) 1):2.3. 1–2.3. 22. doi: 10.1002/0471250953.bi0203s00
18. Schliep KP. Phangorn: phylogenetic analysis in R. Bioinformatics. (2010) 27:592–3. doi: 10.1093/bioinformatics/btq706
19. Bodenhofer U, Bonatesta E, Horejš-Kainrath C, and Hochreiter S. Msa: an R package for multiple sequence alignment. Bioinformatics. (2015) 31:3997–9. doi: 10.1093/bioinformatics/btv494
20. Thompson JD, Higgins DG, and Gibson TJ. Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. (1994) 22:4673–80. doi: 10.1093/nar/22.22.4673
21. Kolde R. pheatmap: Pretty Heatmaps. R package version 1.0.13. (2025). Available online at: https://github.com/raivokolde/pheatmap.
22. Katoh K and Standley DM. Mafft multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. (2013) 30:772–80. doi: 10.1093/molbev/mst010
23. Grant BJ, Rodrigues AP, ElSawy KM, McCammon JA, and Caves LS. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. (2006) 22:2695–6. doi: 10.1093/bioinformatics/btl461
24. Nguyen L-T, Schmidt HA, von Haeseler A, and Minh BQ. Iq-tree: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. (2014) 32:268–74. doi: 10.1093/molbev/msu300
25. Bailey TL, Johnson J, Grant CE, and Noble WS. The meme suite. Nucleic Acids Res. (2015) 43:W39–49. doi: 10.1093/nar/gkv416
26. Ferguson C and Ali A. Global genetic diversity of tobacco ringspot virus including newly reported isolates from cotton (Gossypium hirsutum) in Oklahoma. Plant Dis. (2024) 108:635–46. doi: 10.1094/PDIS-07-23-1251-RE
27. Brister JR, Ako-Adjei D, Bao Y, and Blinkova O. Ncbi viral genomes resource. Nucleic Acids Res. (2015) 43:D571–D7. doi: 10.1093/nar/gku1207
28. Mahas JW. Management of Aphis Gossypii Populations and the Spread of Cotton Leafroll Dwarf Virus in Southeastern Cotton Production Systems. Auburn, Alabama: Auburn University (2020).
29. Heilsnis BJ. Transmission and Host Plant Resistance of Cotton Leafroll Dwarf Virus (Clrdv) in the Southeastern United States. Auburn, Alabama: Auburn University (2020).
30. Pandey S, Bag S, Roberts P, Conner K, Balkcom KS, Price AJ, et al. Prospective alternate hosts of an emerging polerovirus in cotton landscapes in the Southeastern United States. Viruses. (2022) 14:2249. doi: 10.3390/v14102249
31. Akinyuwa MF and Kang S-H. Functional characterization of Rna silencing suppressor encoded by cotton leafroll dwarf virus. Agriculture. (2024) 14:194. doi: 10.3390/agriculture14020194
32. Olmedo-Velarde A, Shakhzadyan H, Rethwisch M, West-Ortiz M, Waisen P, and Heck M. Data mining redefines the timeline and geographic spread of cotton leafroll dwarf virus. Plant Dis. (2025) 109:992–7. doi: 10.1094/pdis-06-24-1265-sc
33. Ramos-Sobrinho R, Adegbola RO, Lawrence K, Schrimsher DW, Isakeit T, Alabi OJ, et al. Cotton leafroll dwarf virus us genomes comprise divergent subpopulations and harbor extensive variability. Viruses. (2021) 13(11):2230. doi: 10.3390/v13112230
34. Akinyuwa MF, Price BK, Martin KM, and Kang S-H. A newly isolated cotton-infecting polerovirus with cryptic pathogenicity encodes a weak suppressor of Rna silencing. Front Agron. (2023) 5:1235168. doi: 10.3389/fagro.2023.1235168
35. Adegbola RO, Kitchen NT, Lawrence KS, Conner K, Mulvaney M, Small I, et al. (2022). Surveillance of cotton leafroll dwarf virus (Clrdv) in the U.S. By molecular diagnostics and phylogeny of Orf0 and Orf3 fragments, In: Proceedings of 2022 Beltwide Cotton Conferences, San Antonio, Texas, USA, January 4-6, 2022.
36. Adegbola RO, Ponvert ND, and Brown JK. Genetic variability among U.S.-sentinel cotton plot cotton leafroll dwarf virus and globally available reference isolates based on Orf0 diversity. Plant Dis. (2024) 108:1799–811. doi: 10.1094/pdis-02-23-0243-re
Keywords: cotton leafroll dwarf virus (CLRDV), cotton (Gossypium hirsutum), RNA virus in cotton, genetic diversity, virus detection
Citation: Verma PK, Watson BH, Sripathi VR and Janga MR (2025) Identification and genomic profiling of a cotton leafroll dwarf virus isolate from Brownfield, TX. Front. Virol. 5:1619281. doi: 10.3389/fviro.2025.1619281
Received: 28 April 2025; Accepted: 07 July 2025;
Published: 23 July 2025.
Edited by:
Cecilia Bender, ABC-bioresearch, ItalyReviewed by:
Amit Kumar Kesharwani, Washington State University, United StatesHarvinder Bennypaul, Canadian Food Inspection Agency (CFIA), Canada
Copyright © 2025 Verma, Watson, Sripathi and Janga. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Madhusudhana R. Janga, bWphbmdhQHR0dS5lZHU=