 Emma M. Bentley1*
Emma M. Bentley1* Samuel Richardson1
Samuel Richardson1 Eleanor Atkinson1
Eleanor Atkinson1 Peter Rigsby1Devy M. Emperador2Marian Killip3
Peter Rigsby1Devy M. Emperador2Marian Killip3 Mark Page1
Mark Page1 Giada Mattiuzzo1
Giada Mattiuzzo1- 1Medicines and Healthcare Products Regulatory Agency, South Mimms, United Kingdom
- 2Foundation for Innovative New Diagnostics, Geneva, Switzerland
- 3High Containment Microbiology, UK Health Security Agency, London, United Kingdom
Introduction: Recent public health emergencies of international concern (PHEIC) have highlighted the need to develop prophylactic treatments and effective diagnostics for pandemic preparedness. In particular, the 100 Days Mission, an initiative to respond to a PHEIC within the first 100 days after declaration, has put into focus the importance of the early availability of effective diagnostic tests. Reference materials are valuable to support the development, assessment and calibration of these tests. The use of World Health Organization (WHO) International Standards (IS), the highest order calibrant, allows for the harmonisation of data reporting which assists performance evaluation. WHO IS for emerging virus molecular testing are usually made by inactivating the pathogen of interest and they undergo extensive evaluation in multi-laboratory collaborative studies. However, the development of such reference material for Lassa virus (LASV) is challenging due to the requirement for high containment level facilities to handle LASV. Further, the development of molecular tests for the detection of LASV RNA is confounded by the high sequence diversity amongst circulating lineages and a need to evaluate performance against them.
Methods: To circumvent this, we have developed a low containment alternative using chimeric lentiviral particles packaging the RNA of five prototype LASV lineages. These were evaluated alongside the whole inactivated virus (Josiah strain) by 18 laboratories, across 25 methods, as part of an International multi-lab collaborative study.
Results: The data showed equivalent performance to the authentic virus in reducing interlaboratory variability; also, they highlighted the variability in detection across LASV lineages and supported the establishment of the chimeric particles as a WHO Reference Panel for Lassa virus RNA, alongside a WHO IS. This will greatly facilitate molecular test development and evaluation.
Discussion: Overall, the strategy has the flexibility to be applied to a range of high priority viral families and can rapidly be implemented to expand to new viral sequences of high consequence which may impact molecular test performance. This represents an important pandemic preparedness initiative to support the response to the next outbreak, including Disease X.
Introduction
Lassa fever is an acute viral haemorrhagic illness caused by Lassa virus (LASV) of the Arenaviridae family, which persists in Mastomys spp rats and is transmitted to humans primarily via exposure to their excrement (1, 2). Human-to-human transmission through contact with body fluids occurs in a proportion of cases, particularly in healthcare settings. Although it is reported approximately 80% of cases are asymptomatic, the remaining 20% result in severe disease (1). Lassa fever is widely reported in West Africa and is considered endemic in Benin, Ghana, Guinea, Liberia, Mali, Sierra Leone, and Nigeria. In recent years there has been a growing number of cases, and it is suspected that the annual Lassa fever incidence is up to, or in excess of 300,000 infections globally (3, 4) and that the population at risk is expanding either due to population increase or an expanding geographic range (5); also, imported Lassa fever cases in non-endemic regions are not a rare event (6–8). Lack of licensed medical countermeasures and the high outbreak potential has led the World Health Organization (WHO) to list Lassa virus as priority and prototype virus for the Arenaviridae family in their R&D Blueprint Pathogen Prioritization report (9). Clinical presentation of Lassa fever is variable and difficult to distinguish from other febrile illnesses circulating in endemic regions (1, 4). Rapid, early, diagnosis of a LASV infection is crucial to case management, including treatment and isolation measures. The availability of reliable molecular diagnostics, detecting LASV RNA by reverse-transcription (RT)-PCR, have proven valuable in the acute phase (10–13). These are frequently based on in-house assays (14, 15), however there is an increasing availability of commercial platforms (14, 16, 17).
LASV is a single stranded, ambisense RNA virus. The genome is split into two segments, the small segment (S) coding for the nucleoprotein and glycoprotein precursor and the large segment (L) coding for the enzymatic proteins. Historically, assays targeting the S-segment of the LASV genome have required updating because of false negative results due to newly discovered strains (12). Instead, targets within the L-segment are usually more conserved, yet dependent on primer design may detect other related Old World arenaviruses which reduces test specificity (11). The high genetic diversity of LASV, which has been phylogenetically grouped into 7 lineages (18–22), with lineages II, III, IV, V and VII being the most wide spread (23), presents a challenge to the development of nucleic acid amplification technique (NAT)-based assays, which should be capable of detecting multiple lineages. This is further complicated by the designation of LASV as a hazard group 4 pathogen and, in some countries, inclusion in Bioterrorism agent listings, which limits sample access and restricts the evaluation of new and existing NAT assays.
WHO International Standards (IS) are recognised as the highest order of reference materials for biological substances, with an assigned potency in International Units (IU) (24, 25). Calibration of assays to the WHO IS, and reporting of results in IU, allows for better compatibility between reported data and facilitates more robustly defined measures of assay analytical performance such as limits of detection and ultimately reliability of results. The availability of an IS for LASV RNA, which can be used in lower biosafety level laboratories, will allow for equitable comparability of available NAT-based assays and those under development. Given the high genetic diversity of LASV, which complicates the development of pan-lineage assays, the additional availability of a Reference Panel formed of representative isolates from other LASV lineages can assist both assay development and validation efforts, as well as laboratory proficiency and quality assessments. Such an approach has been applied to NAT-based assays for other diverse blood-borne pathogens, such as HIV-1 and Hepatitis B virus, with panels representing variant isolates being updated over time to help continually monitor assay performance against a changing clinical landscape (26, 27).
The candidate material to serve as a WHO IS should be as similar as possible to the clinical sample, to perform in a comparable manner in the assays and act as primary calibrant (28). The process for the development of WHO IS is also critical and it usually takes 2–3 years from endorsement to adoption (25). These timelines do not align with the rapid response needed during an outbreak scenario. Even during the COVID-19 pandemic, the development of the WHO IS for SARS-CoV-2 RNA took 10 months following the WHO Public Health Emergency of International Concern (PHEIC) declaration (29, 30). Alternative strategies for the rapid production of reference material in a pandemic situation are therefore needed. The use of synthetic armoured LASV RNA preparations, which controls the entire diagnostic process from extraction to amplification of the target viral sequences, offers flexibility, through speed of development and when access to live virus is restrictive. To this end, we have previously generated chimeric lentiviral particles (LVPs) containing Ebola virus sequences as a reference reagent for the molecular detection of Ebola virus (31) which were established as a WHO Reference Reagent following evaluation in a multi-laboratory collaborative study (32). A limit of this study was the lack of comparison with the authentic, inactivated, Ebola virus.
In this study, an inactivated LASV preparation was evaluated for the ability to harmonise results from assays currently in use in laboratories worldwide for the detection of LASV infection. Furthermore, LVPs containing LASV RNA are compared to the authentic virus to validate their use as a reference material which can be produced in a rapid manner without the constraint of high containment levels. Indeed, the LASV-LVPs enabled the generation of a panel of reference material for each of the LASV lineages which can be used to assess the suitability of diagnostic methods.
Materials and methods
Lassa virus amplification and inactivation
The Josiah strain of LASV was selected to represent Lineage IV (NCBI reference sequence: HQ688672 (S-segment); JN650518 (L-segment)). The viral stock was amplified and inactivated by the UK Health Security Agency (UKHSA) within a Containment Level 4 facility, following validated in-house methods. Briefly, the viral stock was amplified on Vero-E6 cells, in MEM + Glutamax (Sigma-Aldrich) supplemented with 5% foetal bovine serum (Sigma-Aldrich) and 1% antibiotic/antimycotic (Sigma-Aldrich), harvesting the supernatant 3 days post infection and clarifying by centrifugation at 3,000xg for 10 minutes. The titre of the amplified stock was measured as 5.5x105 plaque-forming units (PFU)/mL. Inactivation was performed by first incubating the viral stock with 3% (v/v) glacial acetic acid (Merck) at ambient temperature for 15 minutes, followed by pH neutralisation through the addition of a 1M sodium hydroxide solution (Life Technologies). Subsequently, the neutralised viral stock was heat treated at 60 °C for 60 minutes using a heat block and temperature control probes to ensure the required temperature was maintained during incubation. Inactivation was verified in vitro by the addition of 50 µL aliquots of the inactivated viral stock, testing 13% of the batch, onto Vero-E6 cells seeded within a 24-well plate. Testing was performed in parallel to a positive control of triplicates of a 10–7 dilution series of untreated viral stock. Following 4 days incubation with an Avicel overlay the plates were fixed with formaldehyde and stained with crystal violet to confirm the absence of plaques indicating infectious virus presence. Additionally, 11% of the inactivated viral stock was blind passaged by incubating 0.5 mL aliquots of inactivated stock with 2.5x106 Vero-E6 cells seeded within 12.5 cm2 flasks alongside positive and negative controls, incubating for 7 days. As well as visually monitoring for signs of cytopathic effect, samples were tested at the beginning and end of the passage by an in-house Lassa virus specific RT-PCR assay (33) to confirm no decrease in cycle threshold (Ct) values was detected.
Chimeric lentiviral particles containing Lassa virus RNA
The preparation of glycoprotein-deficient lentiviral particles (LVPs) in which the Human Immunodeficiency virus (HIV-1) genes have been substituted with the S- or L-segment of LASV RNA was performed following a similar approach to that described previously (31). Briefly, the S- or L-segment of Lineage II, III, IV, V and VII strains (Table 1) were inserted between the long terminal repeats (LTRs) of the lentiviral packaging plasmid (OG269, Oxford Genetics) between the SalI and NheI restriction sites to remove the CMV promoter. Safety precautions were taken to prevent protein expression, including inserting single nucleotide mutations randomly within structural genes and the removal of the CMV promoter of the U3-defective lentiviral plasmid to dampen expression. Next generation sequencing (NGS) was performed on the plasmid to confirm insertion of the S- and L-segment sequences and removal of the CMV promoter, with the sequences deposited to GenBank (Table 1). The lentiviral plasmids containing the LASV genes were then individually transfected into HEK293T/17 cells together with a HIV-1 packaging plasmid p8.91, kindly donated by Prof. D. Trono (34). The LVPs harvested within the culture supernatant of the transfected cells were treated with 200U/mL of DNase I (Invitrogen) to remove residual plasmid DNA and purified by ultracentrifugation over a 20%-sucrose cushion in PBS. The quantity of S- and L-segment LVPs was evaluated via real-time RT-PCR targeting the HIV-U5 region common to each of the particles (31). An equimolar mix of the two chimeric LVPs containing the corresponding S- and L-segments of LASV RNA was formulated to a target of 1x109 genomes/mL for the Lineage IV LVPs and 2x108 genomes/mL for the remaining Lineages II, III, V and VII, within universal buffer (10 mM Tris-HCl (pH 7.4), 0.5% human serum albumin and 1% D-(+)- Trehalose dehydrate) containing a background of 1x105 copies/mL of human genomic DNA (Roche). The use of universal buffer allows for the dilution of the samples in the appropriate matrix for each method (35).
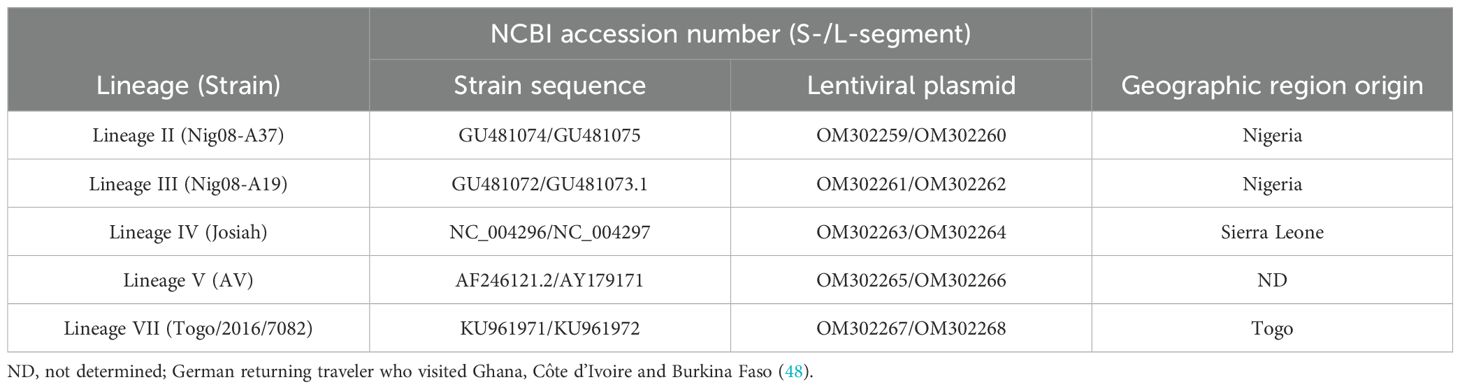
Table 1. Sequence information for the Lassa virus strains used to produce LVPs.
Lassa virus RNA extracts
Whole viral RNA extracts for matched strains of the Lassa virus lineages II, III, IV, V and VII used within this study (Table 1) were obtained from the Bernhard Nocht Institute for Tropical Medicine, provided via the European Virus Archive (EVAg). The RNA extracts were provided within AVE buffer.
Lassa virus genome detection
LASV genome was quantified by both an in-house real time RT-PCR and the commercial RealStar® Lassa Virus RT-PCR Kit 2.0 (Altona). The in-house assay used published primers and probe set (15) targeting the L-segment and followed a protocol optimised for Invitrogen SuperScript III Platinum One-Step qRT-PCR reagents (Invitrogen) (36). Viral RNA extraction was performed using the QIAamp viral RNA mini kit (Qiagen).
Formulation of the reference material
The candidate WHO IS was formulated by diluting the inactivated Lineage IV, Josiah strain, virus within universal buffer (10 mM Tris-HCl (pH 7.4), 0.5% human serum albumin and 1% D-(+)- Trehalose dehydrate) containing a background of 1x105 copies/mL of human genomic DNA (Sigma-Aldrich). The dilution was calculated to result in approximately 1.5x107 genomes/mL based on testing by RT-PCR assays detecting the Lassa virus genome, as described.
Both the inactivated Josiah LASV and the chimeric LVPs were then filled and freeze-dried at scale within an ISO9001 dedicated manufacturing facility. The samples were dispensed in 0.5mL volumes into 2.5mL glass DIN ampoules at 4°C on an AVF5090 filling line (Bausch & Stroebel, Ilshofen, Germany). The homogeneity of the fill was maintained by on-line check-weighing of a proportion of the filled ampoules. Filled ampoules were partially stoppered with halobutyl 13mm diameter igloo closures and lyophilised in a CS100 freeze dryer. Ampoules were loaded onto the shelves at 4°C and primary freezing was performed to -50°C over 1.5 hours. Primary drying was performed at -30°C for 40 hours at 30μb vacuum, then raising the shelf temperature to 25°C and holding vacuum in secondary drying for at least 15 hours, before releasing the vacuum and back-filling the vials with nitrogen. Ampoules were flame sealed on the same filling line. The sealed vials were stored at -20°C under continuous temperature monitoring.
Multi-Lab collaborative study samples
To test the performance of the formulated reference materials, they were included in a panel of samples that were de-identified by blind coding for testing purposes (Table 2). The panel included the formulated and freeze-dried inactivated LASV Lineage IV (WHO IS LASV-IV), and chimeric LVPs, as well as liquid frozen 1:10 and 1:50 diluted samples of the inactivated LASV and the corresponding chimeric LVP respectively. Both were formulated within universal buffer containing a background of 1x105 copies/mL of human genomic DNA, with the buffer also included as a negative sample.
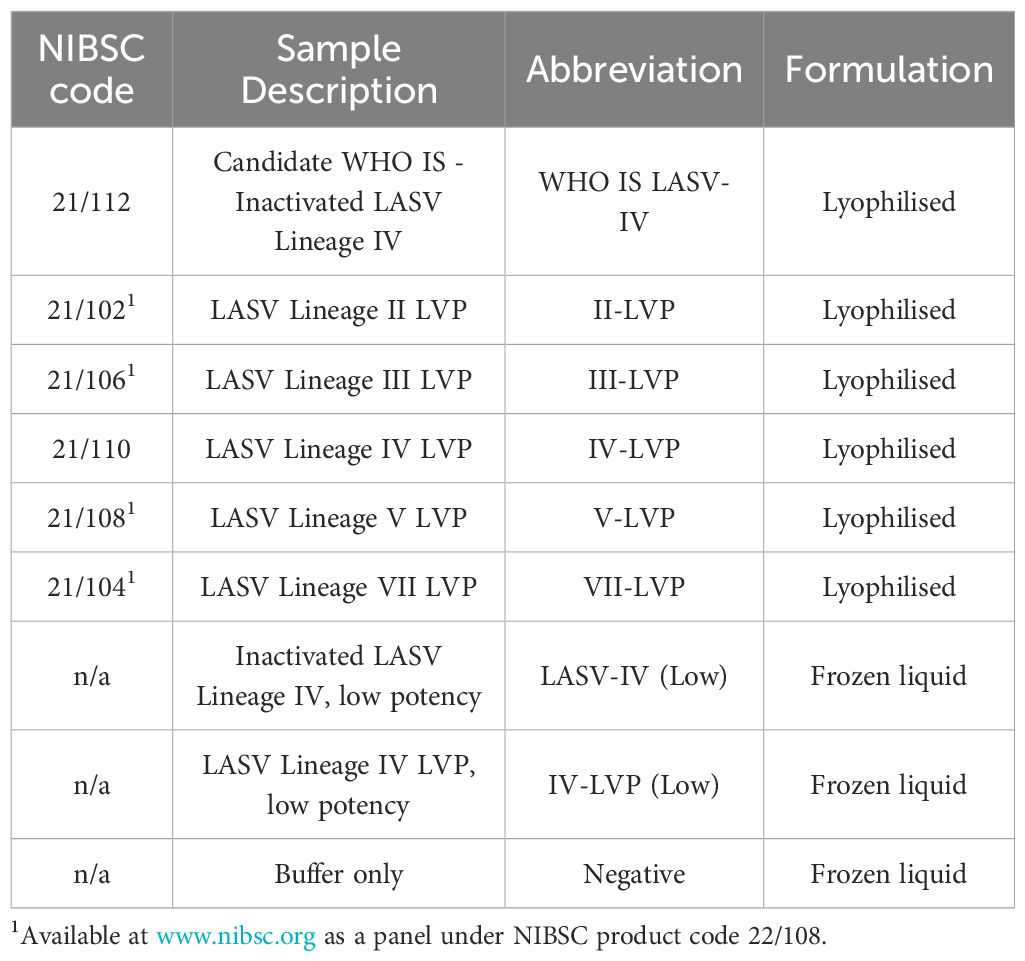
Table 2. Samples provided as part of multi-lab study.
Statistical methods
Potency estimates for each of the study samples were calculated from raw data returned by the study participants. A different approach was applied dependent on whether the data was quantitative or qualitative. For quantitative data, reported as copies, the geometric mean of within-assay replicates was taken, correcting for sample input, dilution factor and linearity of dilutions where appropriate. A potency estimate was reported as Log10 copies/mL calculated from the geometric mean across the independent assays. Qualitative data, reported as Ct values and their positive/negative interpretation, was evaluated to provide a potency estimate in Log10 NAT detectable units/mL (NDU/mL) which is corrected for the equivalent volume of sample amplified. This was calculated by pooling sample data across independent assays within a laboratory, with an acceptance criterion of at least 4 replicates at each dilution point, to provide a number positive out of number tested at each dilution step. A single endpoint for each sample dilution series was calculated using the method of maximum likelihood (11). The model assumes that the probability of a positive result at a given dilution follows a Poisson distribution and that a single ‘copy’ will provide a positive result. The estimated endpoint is equivalent to the dilution at which there is an average of a single copy per sample tested, or the dilution at which 63% of samples tested are positive. Calculations were performed using the R package ‘glm2’ (37).
Potencies relative to samples WHO IS LASV-IV or IV-LVP were calculated by parallel line analysis (PLA) using the raw data for sample dilutions provided either in copies or Ct values. All calculations were performed using Combistats (38). Linearity was assessed by visual assessment. Test samples with response ranges that did not overlap with the range for the sample selected as reference standard were also excluded from the calculation of relative potency. Non-parallelism was assessed by calculating the ratio of fitted slopes for the test and candidate reference sample under consideration. Samples were concluded to be non-parallel when the slope ratio was outside of the range 0.67 – 1.50 and the resulting relative potency estimate was considered not valid. All potencies are presented in Log10 units.
Overall analysis was based on the Log10 estimates of copies/mL, ‘Log10 NAT detectable units/mL’ or Log10 relative potency, as required. Overall mean estimates were calculated as the arithmetic mean across all laboratories, combined, as well as segregated based on assay target. Statistical analysis was performed on combined data to evaluate inter-laboratory variation. This was expressed as the standard deviation (s) of the Log10 estimates, the % Geometric Coefficient of Variation: and the Interquartile Range (UQ/LQ) of the untransformed estimates. Further analysis of variability was undertaken by calculating the proportion of laboratory mean potency estimates within 0.3 Log10 (2-fold) of the study median estimate for the sample.
Results
Collaborative study design
Eighteen participants joined the collaborative study using methods for the detection of LASV RNA in routine use in their laboratories (Supplementary Table S1). Participants were asked to perform 3 independent assay runs, and for each assay, to prepare at least two independent dilution series of the samples, within a matrix specific to their assay, which would optimally cover 5 to 6 dilution points, with at least one point beyond the endpoint dilution. Data was returned from 25 methods with 11 methods involving the detection of 2 target genes. Where laboratories performed multiple assay methods, or a method provided a readout against multiple targets, laboratory codes are appended by a letter indicating the different methods/targets e.g. 1a, 1b. The methods used include both in-house and commercial kits, with technologies based on block/gel-based RT-PCR, real-time RT-PCR and digital PCR. Four of the datasets provided quantitative results expressed in copies, with the remaining datasets providing qualitative results as either pos/neg or Ct values.
All the study samples were correctly identified by dual target LASV NAT methods
Initially we focused on the correct identification of positive and negative panel samples by the participants (Table 3). Eleven participants processed the samples with a dual target method (Table 3, highlighted in grey) and following the manufacturer’s instructions, a sample is scored positive if at least one of the target genes is detected. The majority of the participants used the same platform, RealStar® Lassa Virus RT-PCR Kit 2.0, and correctly identified samples as positive or negative across both targets; while an alternative commercial platform (Lab 15) did not detect the four LVP samples for lineages II, III, V and VII with the S-segment target, however the samples were still determined as positive due to detection via the second, L-segment, target.
Table 3. Scoring of study samples as positive or negative by the different LASV RNA Assay methods.
The inactivated Josiah LASV (WHO IS LASV-IV) and the IV-LVP were detected across all single target assays, with just Lab 8a recording a false negative result in one out of 3 replicates for the inactivated LASV-IV. The lower potency dilutions of the WHO IS LASV-IV and the IV-LVP were also detected efficiently by all but one (IV-VLP low) or two Labs (LASV-IV low), which likely reflects a lower sensitivity of these methods. The LASV Lineage V-LVP sample was detected by all but one assay (Lab 11d) which uses a Lineage IV-specific primer set and thus also failed to detect the samples for the other LASV Lineages. Overall, around half of the single target methods failed or could not efficiently detect Lineage III (8/14) and Lineage VII (7/14), while the Lineage II sample was detected more efficiently, with just 3/14 methods recording a false negative result. Only one single target method returned a false negative result for the Linage V sample. There was a higher detection failure rate for assays targeting the S-segment, as expected based on the known genetic diversity within this target across the divergent Lineages. There were no cases of false positive results reported for the negative control.
There is a high variability in LASV RNA quantification between laboratories
The majority of methods (21/25) within the study reported a qualitative result readout (threshold cycle, Ct) across end-point dilutions. Using this data, a quantitative raw potency estimate was calculated as Log10 NAT detectable units (NDU)/mL and the data was analysed alongside that of quantitative results reported in Log10 copies/mL (Supplementary Table S2). It is important to note that NDU/mL and copy numbers are not necessarily equivalent and therefore the two units may not be directly comparable. Overall, the reported potency estimates varied considerably for each of the samples with more than 1000-fold difference across the majority of samples, although the lowest potency sample (LASV-IV low) had just a 107-fold spread in estimated potencies (Figure 1; Supplementary Table S2). The data did not show a clear distinction between detection capabilities amongst the different lineages or potency estimates for assays based on whether they were targeting the S- or L-segment, however there was a general trend for higher estimates for assays targeting the S-segment. Although this was not the case for the Lineage III and VII-LVP samples, which had lower potency estimates for assays targeting the S-segment. The LVP samples for Lineage II, III, V and VII were formulated to be equivalent potencies based on common sequence in lentiviral vector, yet the average LASV-specific potency estimate is only equivalent for the Lineage II and V sample. The estimated potency of the WHO IS LASV-IV is 85-fold lower than the corresponding chimeric IV-LVP, which is close to the expected 65-fold difference based on the formulation of the samples (2x109 genome/mL versus 1.65x107 genomes/mL).
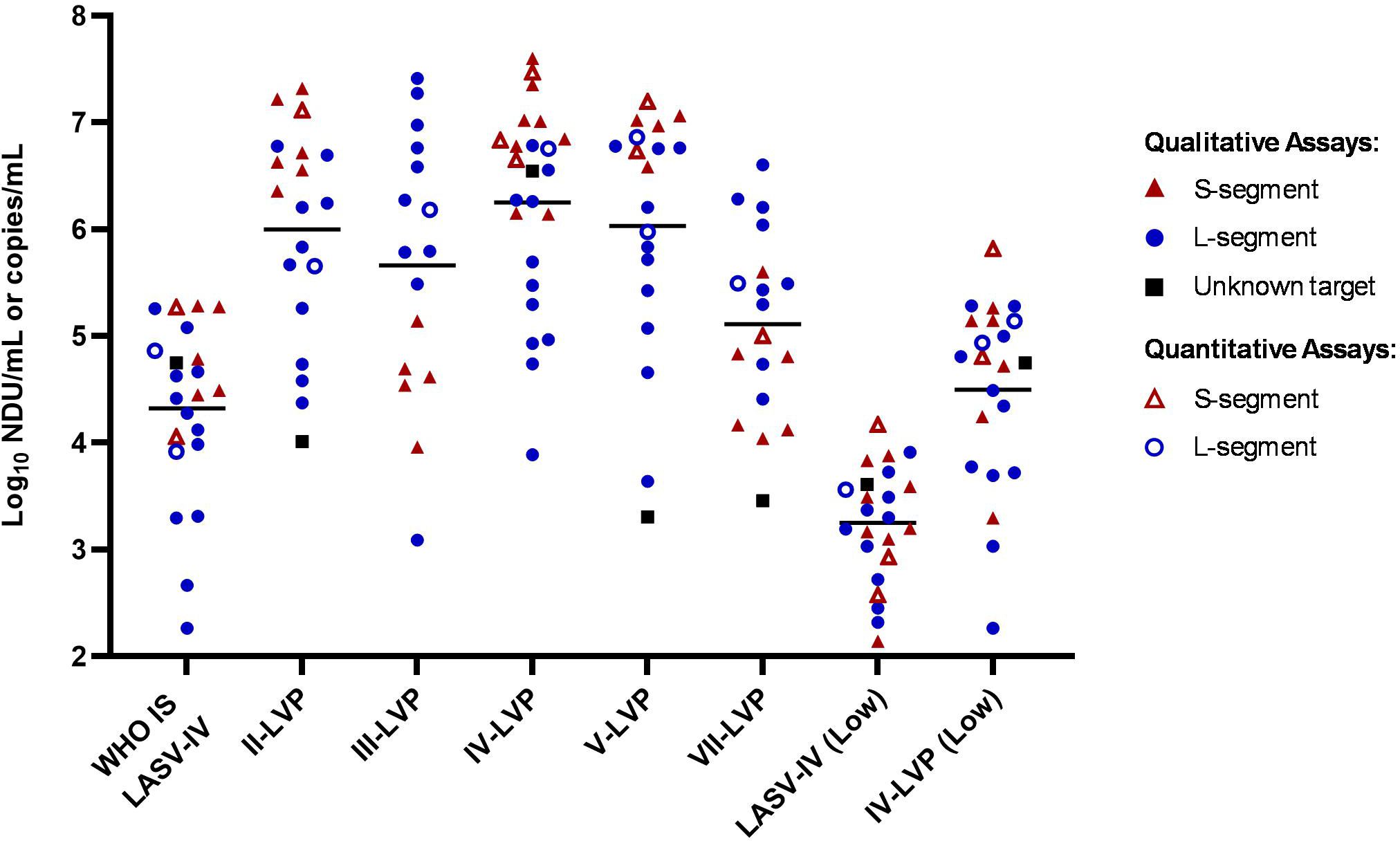
Figure 1. Laboratory reported potency estimates for the study samples. Data is reported as Log10 NDU/mL for labs reporting qualitative data (full symbols) with endpoint Ct values and Log10 copies/mL for labs reporting quantitative data (open symbols). Black lines denote the average potency estimate.
Inter-laboratory variation is reduced by reporting sample potencies relative to a WHO IS
The intended use of an WHO IS is to increase comparability of results from different assays targeting the same analyte by providing an independent reference, which allows data to be harmonised by expressing the values in the same International Unitage (IU) calculated relative to the WHO IS. To assess the suitability of the inactivated LASV-IV to serve as a WHO IS, the results from the participants were calculated relative to the WHO IS LASV-IV by parallel line analysis for datasets associated with numerical values (Ct value or copies) (Figure 2; Supplementary Table S3). Except for sample VII-LVP and III-LVP, all other samples showed a reduction in the spread of data for the combined S- and L-segment targets compared to the raw potency estimates. This is reduced from more than 1000-fold (Figure 1) to within 70-fold for the Lineage IV samples (Figure 2). The spread of data for the Lineage II-LVP is reduced from 2000-fold to within 180-fold. The spread of data for Lineage V-LVP sample is affected by an outlying data point, without which the potencies fall within 18-fold. With the data expressed relative to the WHO IS, the disagreement in sample potency estimates between the S- and L-segment target assays for samples III-LVP and VII-LVP becomes distinct. This is due to the lower detection efficiency of assays targeting the S-segment for Lineages III and VII, while for the other Lineage II, IV and V samples the expected potency is detected, and it is similar (within 3-fold) to the that obtained by targeting the L-segment.
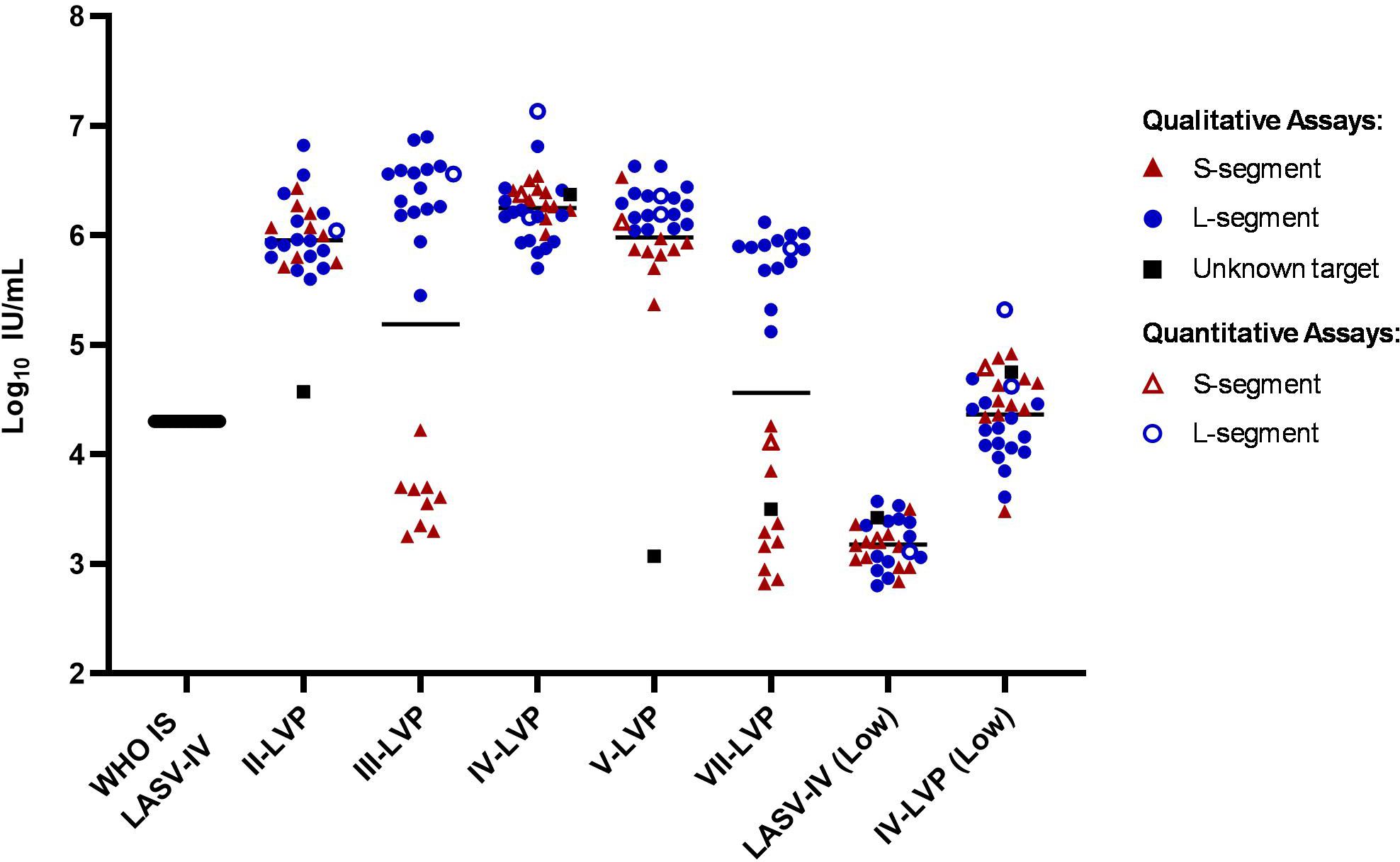
Figure 2. Potency estimates of the study samples reported relative to the WHO IS LASV-IV. Potency estimates were determined by parallel line analysis based on Ct values (qualitative) or copies (quantitative), with an assigned value of 4.3 Log10 IU/mL for the WHO IS. Black lines denote the average potency estimate.
Chimeric LVP achieves similar levels of data harmonisation to an inactivated virus based WHO IS
To evaluate whether the chimeric LASV IV-LVP can be used as a suitable alternative to inactivated virus for reducing inter-laboratory variation, the LASV RNA quantification results from the participants were also expressed relative to the IV-LVP sample (Figure 3; Supplementary Table S4). The level of data harmonisation achieved is similar to that observed with the WHO IS LASV-IV, with potency estimates between methods also falling into a 70-fold range for the Lineage IV samples, a lower 112-fold range for the Lineage II-LVP and a comparable 23-fold for the Lineage V-LVP samples once the outlying datapoint is removed. Similar to the inactivated virus WHO IS, the data harmonised to the IV-LVP shows a clearer distinction in the detection of the S- and L-segment, with the results clustering based upon assay target and demonstrating the lower detection efficiency for the S-segment.
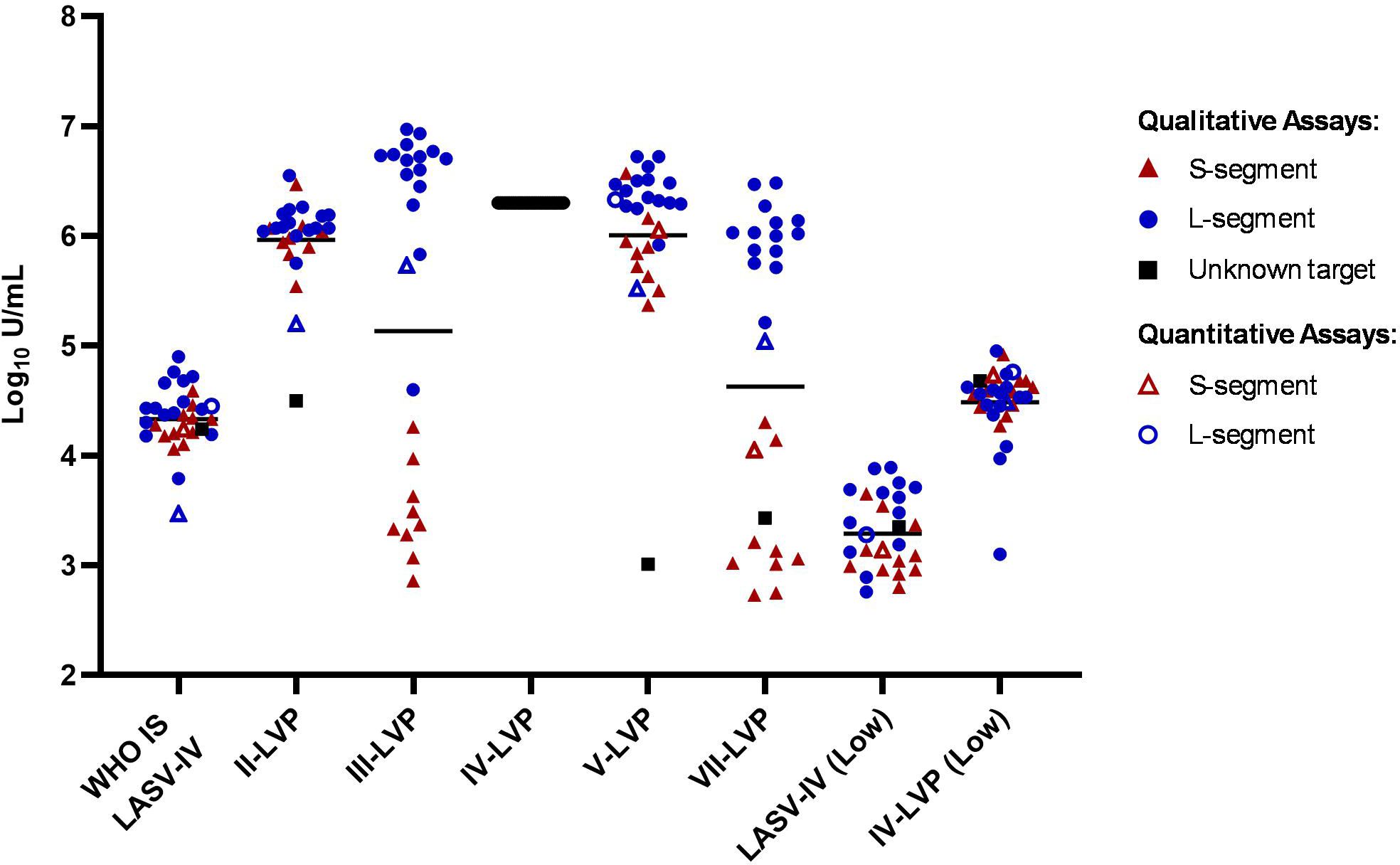
Figure 3. Potency estimates of the study samples reported relative to the chimeric lineage IV-LVP. Potency estimates are determined by parallel line analysis based on Ct values (qualitative) or copies (quantitative), with an assigned arbitrary value of 6.3 Log10 units/mL for the chimeric Lineage IV-LVP. Black lines denote the average potency estimate.
The level of harmonisation achieved by WHO IS LASV-IV and IV-VLP was compared using statistical measures of interlaboratory variation which included the Geometric Coefficient of Variation (%GCV), the proportion of potencies within 0.3 Log10 (2-fold) of the median (GM: Med<2) and the interquartile range (UQ/LQ) (Table 4). Overall, a comparable performance between the inactivated virus WHO IS and the chimeric IV-LVP is observed, demonstrating suitability of the chimeric LVP to act as a reference material. Whilst harmonization is not observed for the Lineage III- and VII-LVP when considering the combined S- and L-segment target datasets, when the L-segment data is evaluated in isolation there is a similar improvement in the statistical measures of data harmonization (Supplementary Table S5). Altogether, this highlights that when LASV NAT assays are calibrated to the WHO IS LASV-IV data reporting when testing other divergent Lineages is harmonized.
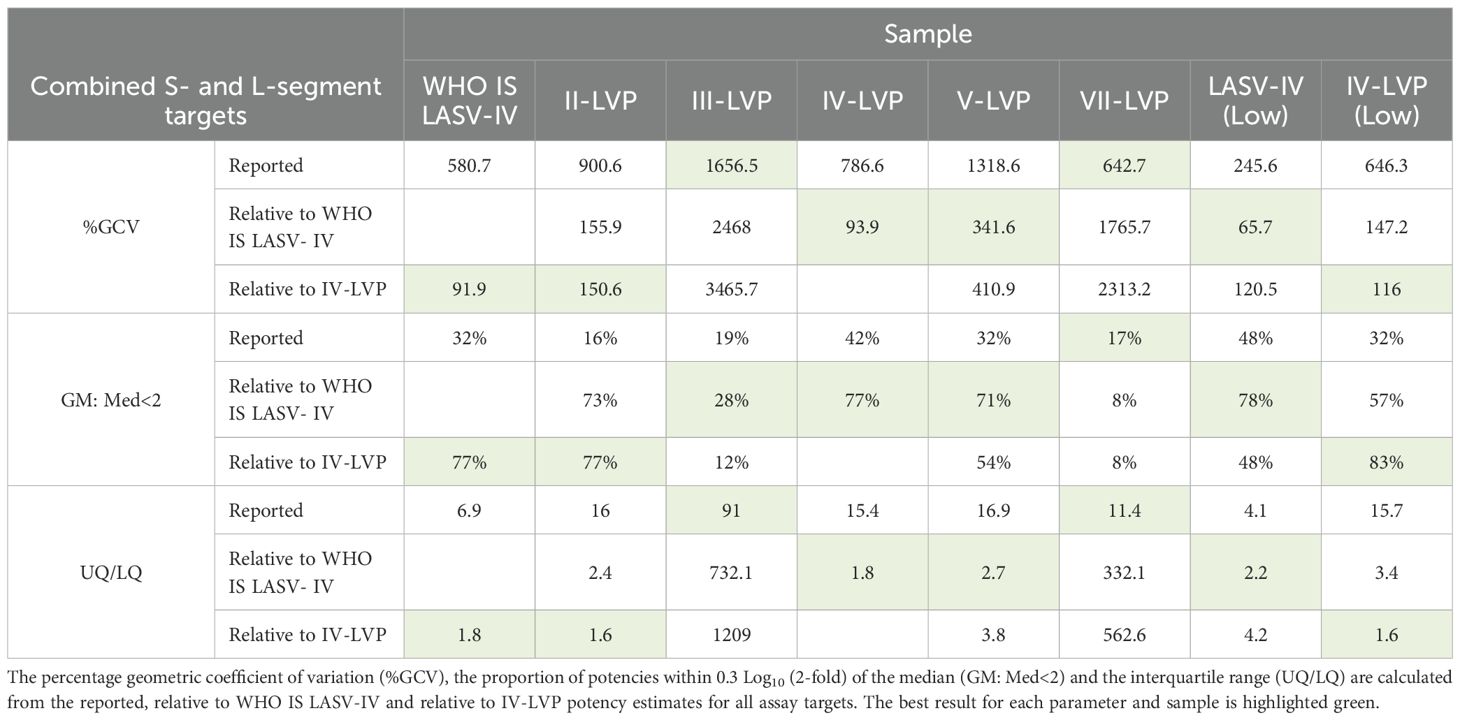
Table 4. Interlaboratory variation of the study sample potency estimates.
A reference panel for LASV lineages supports evaluation of NAT-based assay performance
As the testing of the chimeric LVPs for LASV Lineages III and VII revealed a high variability in detection, with a separation of potency estimates for the S- or L-segment, additional characterization experiments were performed to investigate whether the preparation of the LVP samples caused the poor performance of the S-target assay. Each of the chimeric LVPs was tested alongside matched whole viral RNA extracts obtained though EVAg using the RealStar® Lassa Virus RT-PCR kit, which targets both gene segments. The results are reported relative to the WHO IS LASV-IV and expressed in IU/mL, to facilitate comparison between the data. It was found that both the chimeric LVP and whole virus RNA performed similarly, with the same lower potency detection for the S-segment of the Lineage III and VII samples (Figure 4). This data demonstrates a different efficiency in the detection of the LASV RNA in the dual-target assay for Lineage III and VII and highlights the value in the availability of a well characterized Reference Panel comprising the other Lineage samples (II, III, V and VII) to support evaluation of assay performance.
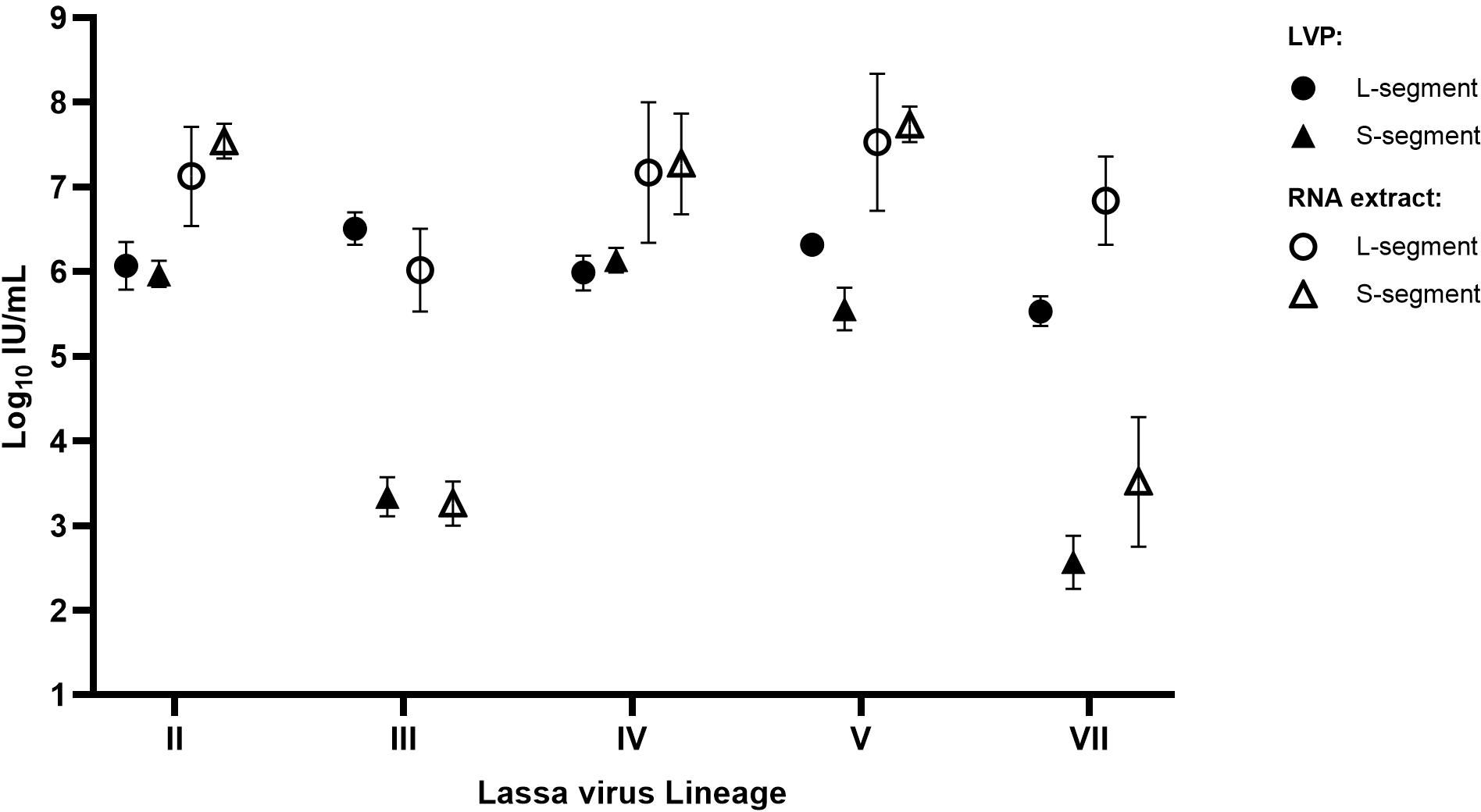
Figure 4. Potency of the LASV chimeric LVP samples compared to matched whole viral RNA extracts. Each sample was quantified in parallel, alongside the WHO IS, with potencies reported relative to the Standard in IU/mL based on parallel line analysis of sample Ct values. Error bars represent the 95% upper and lower confidence limits (n=2).
Discussion
Reference Materials are required to facilitate comparison of results from different methods and/or different laboratories. Harmonised calibration of assays is an essential step to optimising clinical management of patients, ensuring consistency in treatment and also effective diagnosis, which is particularly critical during outbreak scenarios which may occur for priority pathogens. WHO IS, established by the Expert Committee on Biological Standardization (ECBS), are the highest order of reference material and they define the unitage, International Unit (IU) to express the biological potency of a sample. The complex nature of clinical samples means nucleic acid amplification cannot be precisely determined by physiochemical means (25); indeed, the multi-step process from clinical sample collection to result, and the presence of potential contaminants in the samples means that there is greater inherent variability. Therefore, expressing results relative to a Standard calibrated in IU/mL prevents assay-bias and reduces inter-laboratory variation (24). However, it is important that the reference material is included in every step of the procedure to control the whole process, including the nucleic acid extraction. Authentic virus is therefore the ideal candidate to serve as WHO IS for viral NAT-based methods and this has been demonstrated for multiple infectious disease diagnostic NAT-based assays, particularly in the blood virology field (24, 25). As an example, the detection of human hepatitis C virus (HCV) by molecular methods has demonstrated a 30-fold improvement in comparability of data between different kits since a WHO IS was first made available in 1997 (39). In this study we have shown that a preparation of acid/heat inactivated LASV lineage IV (Josiah strain) is able to reduce the difference in the quantification of the LASV RNA between the 18 participants by more than 10-fold. The results from this collaborative study were discussed during the WHO ECBS meeting in April 2022 and the committee established the sample WHO IS LASV-IV, NIBSC product code 21/112, as the First WHO International Standard for Lassa virus RNA (40, 41).
Given the high genetic diversity of LASV and the challenges to develop pan-lineage assays, a further factor that was evaluated during this study was the utility of a WHO IS based on a Lineage IV, Josiah strain, to harmonise the reporting of data across other Lineages. The results showed that the variability of reported data when testing prototype strains for Lineages II, III, V and VII could be reduced if reported relative to the WHO IS LASV-IV (Figure 1 and 2). The majority of assay methods within the study rely on qualitative results determination with the dual-target assays being able to correctly detect positive/negative samples based on at least one of the targets being positive. The qualitative nature of these assays also overcomes probable difficulties in concordant quantification between targets for more divergent strains, highlighted by the different efficiency in the detection between targets in the S- and L-segment for the Lineage III and VII samples (Figures 3, 4). This could be due to primers mismatch, however, as the primer sequences are proprietary it was not possible to confirm. The issue of ensuring consistency in the detection and performance of assays for viruses which have a high level of genetic variability has previously been overcome with the availability of Reference Panels comprising representative variants, with examples including HIV-1 and Hepatitis B virus (HBV) (42–44). The panels can be updated as new variants arise and help facilitate improvements in assay development. The results of this study supported the establishment of a WHO International Reference Panel for Lassa virus RNA, NIBSC product code 22/108, which comprises Lineage II, III, V and VII samples (45) and can be used for the development and evaluation of both quantitative and qualitative assays.
The Reference Panel members were easily produced using the chimeric LVP strategy (31) based on published sequences (Table 1). Chimeric LVP can be produced within a few weeks from when a viral sequence is available and have a favourable safety profile. Further, they act as a whole process control due to being based on a lentivirus and thus require nucleic acid extraction. As a response to an outbreak, in the context of the 100 Days Mission, it is critical to ensure early availability to accurate and standardized diagnostics (46). This was highlighted during the response to the SARS-CoV-2 pandemic, where NAT-based assays flooded the market and an evaluation of their fitness for purpose was required (47). In this case, a chimeric LVP for SARS-CoV-2 was rapidly made available as a Working Standard within 8 weeks of the WHO declaring a PHEIC (NIBSC product 19/304), although technology is available to reduce the timeframe further. A single batch of approximately 1500 vials was provided free of charge and lasted more than one year to bridge until the WHO IS was available. Following establishment of the WHO IS based on inactivated virus, users of the Working Standard were able to back-calibrate assays to provide results in International Units (30). Similar to the conclusions from the SARS-CoV-2 RNA study, in this collaborative study we have demonstrated that a chimeric Lineage IV-LVP could harmonise data reporting in a comparable manner to the equivalent inactivated virus and act as a suitable reference material. A limitation of this study is the restrictions associated with handling of un-inactivated Lassa virus, which is a hazard group 4 pathogen, meaning it was not possible to fully assess other factors of performance such as the effect of the inactivation procedure or a comparison with clinical samples within the collaborative study.
The value of this study is not limited to Lassa virus diagnostics, but is applicable to priority pathogens which require high containment level or are not easily isolated and propagated in vitro or difficult to share globally. This work offers a strategy to develop essential tools to support the critical phases of a pandemic/epidemic where accurate and timely diagnosis is fundamental to identifying, monitoring and controlling pathogen transmission.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
EB: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Visualization, Writing – original draft, Writing – review & editing. SR: Investigation, Writing – review & editing. EA: Formal analysis, Writing – review & editing. PR: Formal analysis, Writing – review & editing. DE: Conceptualization, Funding acquisition, Writing – review & editing. MK: Investigation, Resources, Writing – review & editing. MP: Conceptualization, Writing – review & editing. GM: Conceptualization, Writing – original draft, Writing – review & editing, Funding acquisition.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. Support for this Project was provided through funding from The Foundation for Innovative New Diagnostics (FIND). The views expressed by the authors do not necessarily reflect the views of The Foundation for Innovative New Diagnostics. Production of inactivated Lassa virus was supported by the UK Health Security Agency.
Acknowledgments
We gratefully acknowledge the contributions of the collaborative study participants for generating data and meeting the tight timeframes of this study: Iain MacLeod (Aldatu Biosciences, USA), Alke Heitmann, Julian Tudrzierz (Altona Diagnostics GmbH, Germany), Masayuki Shimojima, Takeshi Kurosu, Tomoki Yoshikawa, Yuki Takamatsu (Department of Virology I, National Institute of Infectious Diseases, Japan), Zsofia Igloi, Richard Molenkamp, Freek van Loenen (Viroscience, Erasmus MC, The Netherlands), Marian Killip, Ulrike Arnold, David Jackson (High Containment Microbiology, UK Health Security Agency, United Kingdom), Jean-Claude Manuguerra, Valerie Caro, Jessica Vanhomwegen (Institut Pasteur, France), Misa Korva, Tatjana Avsic Zupanc (Institute of Microbiology and Immunology, University of Ljubljana, Slovenia), Rita Cordeiro, Ana Pelerito (Instituto Nacional de Saúde Doutor Ricardo Jorge, Portugal), Il-Hwan Kim, Seil Kim, Hee Min Yoo, Changwoo Park (Korea Research Institute of Standards and Science, South Korea), Fabrizio Carletti (National Institute for Infectious Diseases ‘Lazzaro Spallanzani’, Italy), John Dogbah, Josiah S. George (National Public Health Institute of Liberia, Liberia), Adeleye Semiu Adesola, Musa Abdulmajid Suleiman, Ifeanyichukwu Ameachi Paul, Madubuike Kingsley, Saadatu Aliyu Abubakar, Akabuike Ogechukwu M., Thomas Andrew, Munzali Shamsu, Atanda Zethar, Ahmad Adama, Catherine Okoi, Ahumibe Anthony, Akinpelu M. Afolabi, Olajumoke Babatunde, Nwando G. Mba, Ifedayo Adetifa (National Reference Laboratory, Nigeria Centre for Disease Control, Nigeria), Daniel Bailey (Rare and Imported Pathogens Laboratory, UK Health Security Agency, United Kingdom), Walter Zhang (Shanghai ZJ Bio-Tech Co., Ltd. Liferiver, China), Olivier Engler, Roland Züst, Denise Siegrist (Spiez Laboratory, Switzerland), Rosie Watts, Alfred Flomo, Amara S. Fofanah,Emmanuel Kerkula, Stanley Kerkula (UNC-Phebe Molecular Laboratory, Liberia), John Klena (Viral Special Pathogens Branch - US CDC, USA). This project was a collaborative effort between the MHRA and FIND.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2025.1668042/full#supplementary-material
References
1. WHO. Lassa fever (2024). Available online at: https://www.who.int/news-room/fact-sheets/detail/lassa-fever (Accessed June 11, 2025).
2. Garry RF. Lassa fever - the road ahead. Nat Rev Microbiol. (2023) 21:87–96. doi: 10.1038/s41579-022-00789-8
3. NCDC. An update of Lassa fever outbreak in Nigeria . Available online at: https://ncdc.gov.ng/diseases/sitreps/?cat=5&name=AnupdateofLassafeveroutbreakinNigeria (Accessed June 11, 2025).
4. Aloke C, Obasi NA, Aja PM, Emelike CU, Egwu CO, Jeje O, et al. Combating lassa fever in west african sub-region: progress, challenges, and future perspectives. Viruses. (2023) 15. doi: 10.3390/v15010146
5. Klitting R, Kafetzopoulou LE, Thiery W, Dudas G, Gryseels S, Kotamarthi A, et al. Predicting the evolution of the Lassa virus endemic area and population at risk over the next decades. Nat Commun. (2022) 13:5596. doi: 10.1038/s41467-022-33112-3
6. Bell-Kareem AR and Smither AR. Epidemiology of lassa fever BT - lassa fever: epidemiology, immunology, diagnostics, and therapeutics. In: Garry R, editor. Springer International Publishing, Cham (2023). volume 440 p. 87–109. doi: 10.1007/82_2021_234
7. Li C and Luo F. The first imported case of Lassa fever in China: a case description. Quant Imaging Med Surg. (2025) 15:1066–72. doi: 10.21037/qims-24-1784
8. ECDC. Communicable Disease Threats Report- week 18 (2024). Available online at: https://www.ecdc.europa.eu/sites/default/files/documents/communicable-disease-threats-report-week-18-2024.pdf (Accessed June 11, 2025).
9. WHO. Pathogens prioritization: a scientific framework for epidemic and pandemic research preparedness. Geneva (2024). Available online at: https://cdn.who.int/media/docs/default-source/consultation-rdb/prioritization-pathogens-v6final.pdf?sfvrsn=c98effa7_7&download=true (Accessed June 11, 2025).
10. Takah NF, Brangel P, Shrestha P, and Peeling R. Sensitivity and specificity of diagnostic tests for Lassa fever: a systematic review. BMC Infect Dis. (2019) 19:647. doi: 10.1186/s12879-019-4242-6
11. Vieth S, Drosten C, Lenz O, Vincent M, Omilabu S, Hass M, et al. RT-PCR assay for detection of Lassa virus and related Old World arenaviruses targeting the L gene. Trans R Soc Trop Med Hyg. (2007) 101:1253–64. doi: 10.1016/j.trstmh.2005.03.018
12. Stephan Ö, Michaela L, Petra E, Marcus P, Christian D, Meike H, et al. Improved detection of lassa virus by reverse transcription-PCR targeting the 5′ Region of S RNA. J Clin Microbiol. (2010) 48:2009–13. doi: 10.1128/jcm.02351-09
13. Asogun DA, Adomeh DI, Ehimuan J, Odia I, Hass M, Gabriel M, et al. Molecular diagnostics for lassa fever at Irrua specialist teaching hospital, Nigeria: lessons learnt from two years of laboratory operation. PloS Negl Trop Dis. (2012) 6:e1839. doi: 10.1371/journal.pntd.0001839
14. Mazzola LT and Kelly-Cirino C. Diagnostics for Lassa fever virus: a genetically diverse pathogen found in low-resource settings. BMJ Glob Heal. (2019) 4:e001116. doi: 10.1136/bmjgh-2018-001116
15. Nikisins S, Rieger T, Patel P, Müller R, Günther S, and Niedrig M. International external quality assessment study for molecular detection of Lassa virus. PloS Negl Trop Dis. (2015) 9:e0003793. doi: 10.1371/journal.pntd.0003793
16. Emperador D. LASSA FEVER DIAGNOSTICS- Performance evaluation and access (2022). Available online at: https://cdn.who.int/media/docs/default-source/blue-print/day2_session6_3_devy-emperador_lassa-vaccine-meeting_nigeria.pdf?sfvrsn=e8186329_3 (Accessed July 1, 2025).
17. Luo X-L, Zhang X-D, Li B-J, Qin T, Cao Z-J, Fan Q-J, et al. Comparative evaluation of standard RT-PCR assays and commercial real-time RT-PCR kits for detection of lassa virus. Microbiol Spectr. (2023) 11:e0501122. doi: 10.1128/spectrum.05011-22
18. Ehichioya DU, Dellicour S, Pahlmann M, Rieger T, Oestereich L, Becker-Ziaja B, et al. Phylogeography of lassa virus in Nigeria. J Virol. (2019) 93. doi: 10.1128/JVI.00929-19
19. Whitmer SLM, Strecker T, Cadar D, Dienes H-P, Faber K, Patel K, et al. New lineage of lassa virus, Togo, 2016. Emerging Infect diseases. United States;. (2018) 24:599–602. doi: 10.3201/eid2403.171905
20. Wiley MR, Fakoli L, Letizia AG, Welch SR, Ladner JT, Prieto K, et al. Lassa virus circulating in Liberia: a retrospective genomic characterisation. Lancet Infect Dis. (2019) 19:1371–8. doi: 10.1016/S1473-3099(19)30486-4
21. Manning JT, Forrester N, and Paessler S. Lassa virus isolates from Mali and the Ivory Coast represent an emerging fifth lineage. Front Microbiol. (2015) 6:1037. doi: 10.3389/fmicb.2015.01037
22. Yadouleton A, Agolinou A, Kourouma F, Saizonou R, Pahlmann M, Bedié SK, et al. Lassa virus in pygmy mice, Benin, 2016-2017. Emerg Infect Dis. (2019) 25:1977–9. doi: 10.3201/eid2510.180523
23. Wang X, Ye X, Li R, Zai X, Hu M, Wang S, et al. Spatio-temporal spread and evolution of Lassa virus in West Africa. BMC Infect Dis. (2024) 24:314. doi: 10.1186/s12879-024-09200-8
24. WHO. WHO manual for the preparation of secondary reference materials for in vitro diagnostic assays designed for infectious disease nucleic acid or antigen detection: calibration to WHO International Standards (2017). Available online at: https://www.who.int/publications/m/item/annex-6-trs-no-1004 (Accessed June 11, 2025).
25. WHO. Recommendations for the preparation, characterization and establishment of international and other biological reference standards (revised 2004) (2004). Available online at: https://www.who.int/publications/m/item/annex2-trs932 (Accessed June 11, 2025).
26. Morris C, Govind S, Fryer J, and Almond N. SoGAT—25 years of improving the measurement of nucleic acids in infectious disease diagnostics (a review). Metrologia. (2019) 56:044007. doi: 10.1088/1681-7575/ab2aa3
27. Baylis SA, Wallace P, Mcculloch E, Niesters HGM, Nübling CM, Colleen E, et al. Standardization of nucleic acid tests: the approach of the world health organization (2019). Available online at: https://journals.asm.org/journal/jcm (Accessed June 11, 2025).
28. WHO. Consultation commutability of WHO biological reference preparation for in vitro detection of infectious markers. In: Technical Report Series, no. 987 (2014) WHO Press, Geneva, Switzerland, pg 26–27. Available online at: https://iris.who.int/bitstream/handle/10665/129494/TRS_987_eng.pdf?sequence=1 (Accessed June 11, 2025).
29. WHO. COVID 19 Public Health Emergency of International Concern (PHEIC) Global research and innovation forum: towards a research roadmap (2020). Available online at: https://www.who.int/publications/m/item/covid-19-public-health-emergency-of-international-concern-%28pheic%29-global-research-and-innovation-forum (Accessed June 11, 2025).
30. Bentley EM, Mee ET, Routley S, Mate R, Fritzsche M, Hurley M, et al. Collaborative study for the establishment of a WHO international standard for SARS-coV-2 RNA (2020). Available online at: https://www.who.int/publications/m/item/WHO-BS-2020.2402 (Accessed June 11, 2025).
31. Mattiuzzo G, Ashall J, Doris KS, MacLellan-Gibson K, Nicolson C, Wilkinson DE, et al. Development of lentivirus-based reference materials for ebola virus nucleic acid amplification technology-based assays. PloS One. (2015) 10:e0142751. doi: 10.1371/journal.pone.0142751
32. Wilkinson DE, Page M, Almond N, Anderson R, Ashall J, Berry N, et al. WHO collaborative study to assess the suitability of interim standards for Ebola virus NAT assays (2015). Available online at: https://iris.who.int/bitstream/handle/10665/197763/WHO_BS_2015.2279_eng.pdf?sequence=1 (Accessed June 11, 2025).
33. Trombley AR, Wachter L, Garrison J, Buckley-Beason VA, Jahrling J, Hensley LE, et al. Comprehensive panel of real-time TaqMan polymerase chain reaction assays for detection and absolute quantification of filoviruses, arenaviruses, and New World hantaviruses. Am J Trop Med Hyg. (2010) 82:954–60. doi: 10.4269/ajtmh.2010.09-0636
34. Zufferey R, Nagy D, Mandel RJ, Naldini L, and Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol. (1997) 15:871–5. doi: 10.1038/nbt0997-871
35. Fryer J, Heath AB, Anderson R, and Minor PD. Collaborative study to evaluate the proposed 1st WHO international standard for human cytomegalovirus (HCMV) for nucleic acid amplification (NAT)-based assays. (2010) WHO Press, Geneva, Switzerland. Available online at: https://iris.who.int/bitstream/handle/10665/70521/WHO_BS_10.2138_eng.pdf?sequence=1 (Accessed June 11, 2025).
36. Boisen ML, Uyigue E, Aiyepada J, Siddle KJ, Oestereich L, Nelson DKS, et al. Field evaluation of a Pan-Lassa rapid diagnostic test during the 2018 Nigerian Lassa fever outbreak. Sci Rep. (2020) 10:8724. doi: 10.1038/s41598-020-65736-0
37. The R project for statistical computing . Available online at: https://www.r-project.org/ (Accessed June 11, 2025).
38. EDQM. CombiStats. Available online at: https://www.edqm.eu/en/lp-combistats (Accessed June 11, 2025).
39. Saldanha J, Lelie N, and Heath A. Establishment of the first international standard for nucleic acid amplification technology (NAT) assays for HCV RNA. WHO Collab Study Group Vox Sang. (1999) 76:149–58. doi: 10.1046/j.1423-0410.1999.7630149.x
40. Bentley EM, Richardson S, Atkinson E, Rigsby P, Emperador D, Killip M, et al. Collaborative Study for the Establishment of a WHO International Standard and Reference Panel for Lassa virus RNA (2022). Available online at: https://www.who.int/publications/m/item/who-bs-2022.2419 (Accessed June 11, 2025).
41. WHO. Expert Committee on Biological Standardization, Seventy-fifth report (2022). Available online at: https://www.who.int/publications/i/item/9789240057081 (Accessed June 11, 2025).
42. Chudy M, Hanschmann K-M, Kress J, Nick S, Campos R, Wend U, et al. First WHO International Reference Panel containing hepatitis B virus genotypes A-G for assays of the viral DNA. J Clin Virol Off Publ Pan Am Soc Clin Virol. (2012) 55:303–9. doi: 10.1016/j.jcv.2012.08.013
43. Morris CL, Wigglesworth E, and Heath AB. Report on an international collaborative study to establish the 2nd WHO international subtype reference panel for HIV-1 NAT assays. WHO/BS/2012.2209. Geneva, Switzerland (2012). Available online at: https://iris.who.int/bitstream/handle/10665/78050/WHO_BS_2012.2209_eng.pdf?sequence=1 (Accessed June 11, 2025).
44. Morris CL, Wigglesworth E, and Heath AB. Report on an international collaborative study to establish the 1st WHO international reference panel for HIV-1 circulating recombinant forms for NAT assays. WHO/BS/2013.2226. Geneva, Switzerland (2013). Available online at: https://iris.who.int/bitstream/handle/10665/96352/WHO_BS_2013.2226_eng.pdf?sequence=1 (Accessed June 11, 2025).
45. Bentley EM. ADDENDUM to WHO/BS/2022.2419: Collaborative Study for the Establishment of a WHO International Standard and Reference Panel for Lassa virus RNA WHO/BS/2024.2468. Geneva, Switzerland (2024). Available online at: https://cdn.who.int/media/docs/default-source/biologicals/call-for-comments/who_bs_2024.2468_lassa_rna_addendum.pdf?sfvrsn=8fe9ac11_1 (Accessed June 11, 2025).
46. International pandemic preparedness secretariat- 100 days mission . Available online at: https://ippsecretariat.org/ (Accessed September 10, 2025).
47. Mercer T, Almond N, Crone MA, Chain PSG, Deshpande A, Eveleigh D, et al. The Coronavirus Standards Working Group’s roadmap for improved population testing. Nat Biotechnol. (2022) 40:1563–8. United States. doi: 10.1038/s41587-022-01538-1
Keywords: Lassa virus, diagnostics, international standards, reference materials, emerging virus
Citation: Bentley EM, Richardson S, Atkinson E, Rigsby P, Emperador DM, Killip M, Page M and Mattiuzzo G (2025) The development and evaluation of reference materials for Lassa virus molecular diagnostics. Front. Virol. 5:1668042. doi: 10.3389/fviro.2025.1668042
Received: 17 July 2025; Accepted: 24 September 2025;
Published: 03 November 2025.
Edited by:
Alessandra Ruggiero, University of Verona, ItalyReviewed by:
Antonino Di Caro, Saint Camillus International University of Health and Medical Sciences, ItalyMohammad Rizki Fadhil Pratama, Universitas Muhammadiyah Palangkaraya, Indonesia
Copyright © 2025 Bentley, Richardson, Atkinson, Rigsby, Emperador, Killip, Page and Mattiuzzo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emma M. Bentley, RW1tYS5CZW50bGV5QG1ocmEuZ292LnVr