



- 1 Section on In Vivo Neural Function, Laboratory for Integrative Neuroscience, NIAAA, NIH, Bethesda, USA
- 2 Laboratory for Integrative Neuroscience, NIAAA, NIH, Bethesda, USA
Extended training can induce a shift in behavioral control from goal-directed actions, which are governed by action-outcome contingencies and sensitive to changes in the expected value of the outcome, to habits which are less dependent on action-outcome relations and insensitive to changes in outcome value. Previous studies in rats have shown that interval schedules of reinforcement favor habit formation while ratio schedules favor goal-directed behavior. However, the molecular mechanisms underlying habit formation are not well understood. Endocannabinoids, which can function as retrograde messengers acting through presynaptic CB1 receptors, are highly expressed in the dorsolateral striatum, a key region involved in habit formation. Using a reversible devaluation paradigm, we confirmed that in mice random interval schedules also favor habit formation compared with random ratio schedules. We also found that training with interval schedules resulted in a preference for exploration of a novel lever, whereas training with ratio schedules resulted in less generalization and more exploitation of the reinforced lever. Furthermore, mice carrying either a heterozygous or a homozygous null mutation of the cannabinoid receptor type I (CB1) showed reduced habit formation and enhanced exploitation. The impaired habit formation in CB1 mutant mice cannot be attributed to chronic developmental or behavioral abnormalities because pharmacological blockade of CB1 receptors specifically during training also impairs habit formation. Taken together our data suggest that endocannabinoid signaling is critical for habit formation.
Introduction
We can learn to perform particular actions to obtain specific outcomes in our environments through a process of trial and error. These actions are goal-directed, and their performance is highly sensitive to changes in the incentive value of the outcome, and also to changes in the contingency between the action and the outcome. With repetition, however, actions can become not only more efficient but also more automatic and habitual (Dickinson, 1985 ; Foerde et al., 2007 ; Miyachi et al., 1997 ). Previous studies in rats have shown that extensive training on an instrumental task where animals lever press for particular food reinforcements can lead to a shift from goal-directed responding, which is sensitive to changes in the value of the outcome, to habitual responding which is insensitive to outcome devaluation and can be elicited by antecedent stimuli (Adams, 1982 ; Adams and Dickinson, 1981b ). Interestingly, shifts from goal-directed to habitual responding can be produced not only by extended training, but also by different schedules of reinforcement, with random interval schedules favoring the formation of habits compared with random ratio schedules (Adams and Dickinson, 1981b ; Dickinson, 1985 ; Dickinson et al., 1983 ).
The neuroanatomical circuits that support the learning and the performance of goal-directed actions are different than those supporting the formation of habits (Balleine and Dickinson, 1998 ; Yin and Knowlton, 2006 ). The acquisition of goal-directed actions appears to rely on the associative cortico-basal ganglia circuit involving the dorsomedial or associative striatum (Yin et al., 2005a ,b ), the pre-limbic cortex (Balleine and Dickinson, 1998 ), and the mediodorsal thalamus (Corbit et al., 2003 ). On the other hand, the formation of habits depends upon the dorsolateral or sensorimotor striatum (Yin et al., 2004 ) and the infralimbic cortex (Killcross and Coutureau, 2003 ). The molecular mechanisms underlying the switch between goal-directed and habitual behavior have been less studied. Dopamine may have multiple roles in this process (Costa, 2007 ; Hitchcott et al., 2007 ; Wickens et al., 2007b ). Amphetamine sensitization has been shown to lead to increased predisposition for habit formation (Nelson and Killcross, 2006 ). Interestingly, amphetamine sensitization can increase spine density in medium spiny neurons in dorsolateral striatum, which is necessary for habit formation, and at the same time decrease spine density in dorsomedial striatum, which is critical for goal-directed instrumental behavior (Jedynak et al., 2007 ). There are several possible reasons for the dissociable effects of training and dopamine in dorsolateral and dorsomedial striatum. For example, the regulation of dopamine re-uptake seems to be different in different striatal regions. The dopamine transporter (DAT), which is one of the targets of amphetamine, is highly expressed in the dorsolateral striatum, and less expressed in more medial and ventral regions of the striatum and in the pre-frontal cortex, where Catechol-O-methyl transferase (COMT) is more prevalent (Matsumoto et al., 2003 ; Wickens et al., 2007a ). Interestingly, lesions of the nigrostriatal input to dorsolateral striatum impair habit formation (Faure et al., 2005 ), while infusion of dopamine into the ventral medial prefrontal cortex seems to favor goal-directed behavior (Hitchcott et al., 2007 ).
Endocannabinoid release in the striatum is modulated by dopamine signaling (Giuffrida et al., 1999 ; Kreitzer and Malenka, 2005 ; Yin and Lovinger, 2006 ) and necessary for the induction of long-term depression (LTD) (Gerdeman and Lovinger, 2001 ; Gerdeman et al., 2002 ). Endocannabinoid signaling through the cannabinoid receptors type 1 (CB1) has been implicated in reward and addiction (Caille et al., 2007 ; Cossu et al., 2001 ; De Vries et al., 2001 ; Di Marzo et al., 2001 ; Gerdeman et al., 2003 ; Hansson et al., 2007 ; Houchi et al., 2005 ; Sanchis-Segura et al., 2004 ; Wang et al., 2003 ). The expression of CB1 receptors in the brain displays an interesting gradient across the striatum, with a very high level of expression in the dorsolateral striatum (Gerdeman et al., 2003 ; Herkenham et al., 1991 ), at both excitatory and inhibitory terminals (Uchigashima et al., 2007 ). Interestingly, recent studies have shown that amphetamine sensitization depends upon endocannabinoid signaling in the dorsal striatum (Corbille et al., 2007 ).
We therefore decided to investigate if endocannabinoid signaling is involved in habit formation by using mice with genetically targeted mutations in the CB1 gene (Zimmer et al., 1999 ). We first showed, using a reversible devaluation paradigm, that in mice random interval schedules also promoted habit formation while random ratio schedules promoted the acquisition of goal-directed actions. In addition, interval schedules promoted the exploration of a novel lever while ratio schedules promoted the exploitation of the reinforced lever. In addition, CB1 mutant mice showed impaired habit formation and enhanced exploitation. Finally, blocking CB1 receptors specifically during training (Gatley et al., 1996 ) was sufficient to impede habit formation in animals trained under interval schedules. Our data suggest that endocannabinoid signaling is critical for habit formation and for the increased exploration observed in interval schedules of reinforcement.
Materials and Methods
Animals
All experiments were approved by the NIAAA ACUC. C57Bl6/J mice between 2 and 6 months old were used in the experiments. WT male mice purchased from the Jackson laboratory at 8 weeks of age were used in the experiments comparing ratio versus interval schedules and in the experiments investigating the effects of pharmacological blockade of CB1. Mice were allowed to acclimate for at least 1 week before experiments started. Forty mice were used in the experiments using different reinforcement schedules. Twenty-four (12 per group) were used to assess the effect of different schedules of reinforcement on habit formation and a different group of 16 (8 per group) were employed to investigate the effect of different schedules of reinforcement on the exploration/exploitation test. Fifty-nine mice were employed in the experiments with the CB1 receptor antagonist AM25: saline (n = 21), 3 mg/Kg of AM251 (n = 21); and with 6 mg/Kg of AM251 (n = 17). A subgroup (saline n = 6; 3 mg/Kg of AM251 n = 4; and 6 mg/Kg of AM251 n = 9) was tested on the exploration/exploitation paradigm. CB1 mutant mice were generated as previously described (Zimmer et al., 1999 ). CB1 animals were obtained as homozygous mutants backcrossed into C57Bl6/J background, and were bred with C57Bl6/J WT mice to obtain CB1+/− mice. CB1+/− mice were bred with each other to generate experimental animals: WT, CB1+/−, and CB1−/− littermates. This ensured that any potential genetic drift due to previous homozygous breeding was identical among the experimental animals of different genotypes, and also that the maternal care and environment were similar between the different experimental groups. Both males and females were used, since the general effects of interval schedule training on habit formation were observed in both sexes. WT (21), CB1+/− (21), and CB1−/− (16) were used in the devaluation test. WT (10), CB1+/− (14), and CB1−/− (8) were tested on the exploration/exploitation paradigm.
Behavioral procedures
Behavioral training and testing took place in operant chambers (21.6 cm L × 17.8 cm W × 12.7 cm H) housed within sound attenuating chambers (Med-Associates, St. Albans, VT). Each chamber was equipped with two retractable levers on either side of the food magazine and a house light (3 W, 24 V) mounted on the opposite side of the chamber. Reinforcers were delivered into the magazine through a pellet dispenser or a pump with a syringe that delivered sucrose solution (20–30 μl of 10% solution per reinforcer). Magazine entries were recorded using an infrared beam and licks using a contact lickometer. Before training started mice were placed on a food deprivation schedule, receiving 1.5–2 g of food per day allowing them to maintain a body weight above 85% of their baseline weight. Throughout training mice were fed daily after the training session. Water was removed for 4–6 hour before each daily session. Mice were trained with two reinforcers: either regular “chow” pellets (Bio-Serv formula F05684) or sucrose (10% solution or 20 mg pellets). One reinforcer was delivered in the operant chamber contingent upon lever pressing, and the other reinforcer was presented freely in their home cage and used as a control for the devaluation test. The reinforcer and lever used were counterbalanced across groups.
Training started with a 30 minute magazine training session in which one reinforcer was delivered on a random time schedule on average every 60 second (30 reinforcers). The following day lever-pressing training started, in which each animal learned to press one lever to obtain a specific reinforcer. Each daily session began with the illumination of the house light and insertion of the lever, and ended with the retraction of the lever and the offset of the house light. Typically, lever-pressing training commenced with three sessions of continuous reinforcement (CRF) in the first 3 days. The first CRF sessions lasted 90 minute or until the mice received five reinforcers, the second CRF session lasted 90 minute or until the mice received 15 reinforcers, and the last CRF session lasted 90 minute or until the mice received 30 reinforcers. After CRF, animals were trained in either ratio or interval schedules, with all the sessions lasting 90 minute or until mice received 30 reinforcers. For random ratio training, after the last session of CRF mice were given one session of random ratio 10 (RR-10) and then switched to random ratio 20 (RR-20; on average one reinforcer every 20 lever presses). For interval training, after the last session of CRF, mice were then given one session of random interval 30 (RI-30) and then switched to random interval 60 (RI-60; on average one reinforcer delivered upon the first press after 60 second since the last reinforcer). In the experiments with CB1 mutant mice the CRF phase lasted longer than 3 days and animals were only switched to interval schedules after they responded consistently during the CRF sessions (some animals received training with FI-20 during the CRF phase before transitioning to RI-30 in Figure 4 ; also, see difference in breeding scheme and genetic background).
The devaluation test commenced 24 hour after the last training day, and lasted 2 days. On each day mice were given ad libitum exposure to one of the reinforcers for 1 hour in a separate cage. Mice were allowed to consume either the reinforcer earned by lever pressing (devalued condition), or the one they received for free in their home cage (valued condition), so devaluation was achieved by sensory-specific satiety. The amount of reinforcer consumed during the ad libitum session was recorded, and mice that did not consume a minimum of 0.4 g of each reinforcer were not included in the analyses. Immediately after the ad libitum feeding session, mice were given a 5 minute test in extinction with the training lever extended. No extra training was conducted on probe days. The order of the valued and devalued condition tests (day 1 or day 2) was counterbalanced across animals, and the number of presses on the training lever for each condition was recorded. The devaluation index was calculated as (presses valued condition − presses devalued condition)/(presses valued condition + presses devalued condition).
The exploration test was a 5 minute extinction test not preceded by feeding in which two levers were presented—the lever on which the animals were trained and a novel lever which was identical to the training lever but located in a different position inside the box. The number of presses on each lever was recorded. Lever presses during the devaluation or exploration tests were normalized to the number of lever presses during the last day of training previous to the extinction test. The exploration test measured generalization to a different lever that was identical (similar stimulus) and involved a similar response as the training lever. The rationale for the design of the exploration test was the following. If responding in ratio-trained animals is goal-directed and dependent on the contingency between the response and the outcome (Colwill and Rescorla, 1985 ), then ratio-trained animals would press mostly the lever that was reinforced during training. Conversely, if responding in interval-trained animals is more habitual and more dependent on the stimulus-response relation than on the expected value of the outcome (Adams and Dickinson, 1981a ), then interval-trained animals would generalize and press the novel lever that was never paired with the outcome.
Genotyping
Genotyping of the CB1 mutant mice was done using the following primers: Forward primer (CB1F): 5′ GTA CCA TCA CCA CAG ACC TCC TC 3′; Reverse primer KO (CNKO3): 5′ AAG AAC GAG ATC AGC AGC CTC TGT T 3′; Reverse primer WT (CB1wt): 5′ GGA TTC AGA ATC ATG AAG CAC TCC A 3′. Annealing 60°C for 1′.
Drugs
AM251 (A6226, Sigma) was suspended in saline with 1% DMSO at the concentrations of 0.3 mg/ml and 0.6 mg/ml. Control mice were injected with saline with 1% DMSO. Saline and AM251, either 3 mg/Kg or 6 mg/Kg, were injected intra-peritoneally (i.p.) 30 minute before training only during the RI-30 and RI-60 training days. The CRF training, and the devaluation and exploration tests were done without any previous injections.
Statistics
Statistical analyses were done using SPSS. Acquisition of lever presses, head entries, and reinforcement rate were analyzed using Repeated Measures Analyses of Variance ANOVA. As per the experimental design, during the devaluation test planned comparisons using a paired t-test were made between the devalued and valued conditions for each group with the null hypothesis being that there is no statistical difference between valued and devalued conditions, and the alternative hypothesis that the two conditions are different. Similarly, planned comparisons with a paired t-test were used for analyzing the responding on the two levers (same or different) for the exploration test. Correlation analyses were performed using Pearson's correlation coefficient test. α = 0.05 for all tests performed. Mean and standard error of the mean (SEM) are presented on each graph (although SEM are not indicative of the variability in paired tests).
Results
Effect of different schedules of reinforcement on habit formation
We first examined if in mice different schedules of reinforcement lead to differences in habit formation. We trained different groups of mice in an operant task where animals had to press one lever for a particular outcome under either ratio or interval schedules of reinforcement (Figure 1 ). Animals trained in a random ratio schedule had 3 days of CRF training, followed by 1 day of RR-10 and 3 days of RR-20. Animals trained in a random interval schedule underwent 3 days of CRF training, followed by 1 day of RI-30 and 3 days of RI-60. All groups increased lever pressing throughout training (F6,132 = 37.9, p < 0.001), and there was no significant interaction between training and schedule of reinforcement (F6,17 = 1.05, p = 0.43) (Figure 1 A). Although there was a tendency for random ratio-trained animals to press at higher rates during training, there was no main effect of training schedule (F1,22 = 2.00, p = 0.17). We examined the average rate of head entries into the magazine to determine if the two schedules would produce different patterns of magazine exploration. We found that the average rate of head entry changed with training (F6,132 = 9.06, p < 0.001), and there was no effect of schedule (F1,22 = 0.65, p = 0.43), or interaction between schedule and training (F6,17 = 2.43, p = 0.07) (Figure 1 B). We also investigated if the average rate of reinforcement was different for the different training schedules. The average rate of reinforcement changed significantly throughout training (F6,132 = 61.4, p < 0.001), though there was no significant effect of schedule (F1,22 = 1.01, p = 0.33), or interaction between training and schedule of reinforcement (F6,17 = 2.17, p = 0.10). Finally, we examined if the rate of reinforcements per lever press would differ between ratio and interval schedules (Figure 1 C). The rate of reinforcements per lever press changed with training (F6,132 = 3716.66, p < 0.001), and there was a significant difference between the ratio and interval groups (F1,22 = 10.7, p = 0.003; post hoc analyses show a difference between schedules in training days 4 and 5), although there was no interaction between training and schedule of reinforcement (F4,19 = 2.80, p = 0.06) (Figure 1 D).
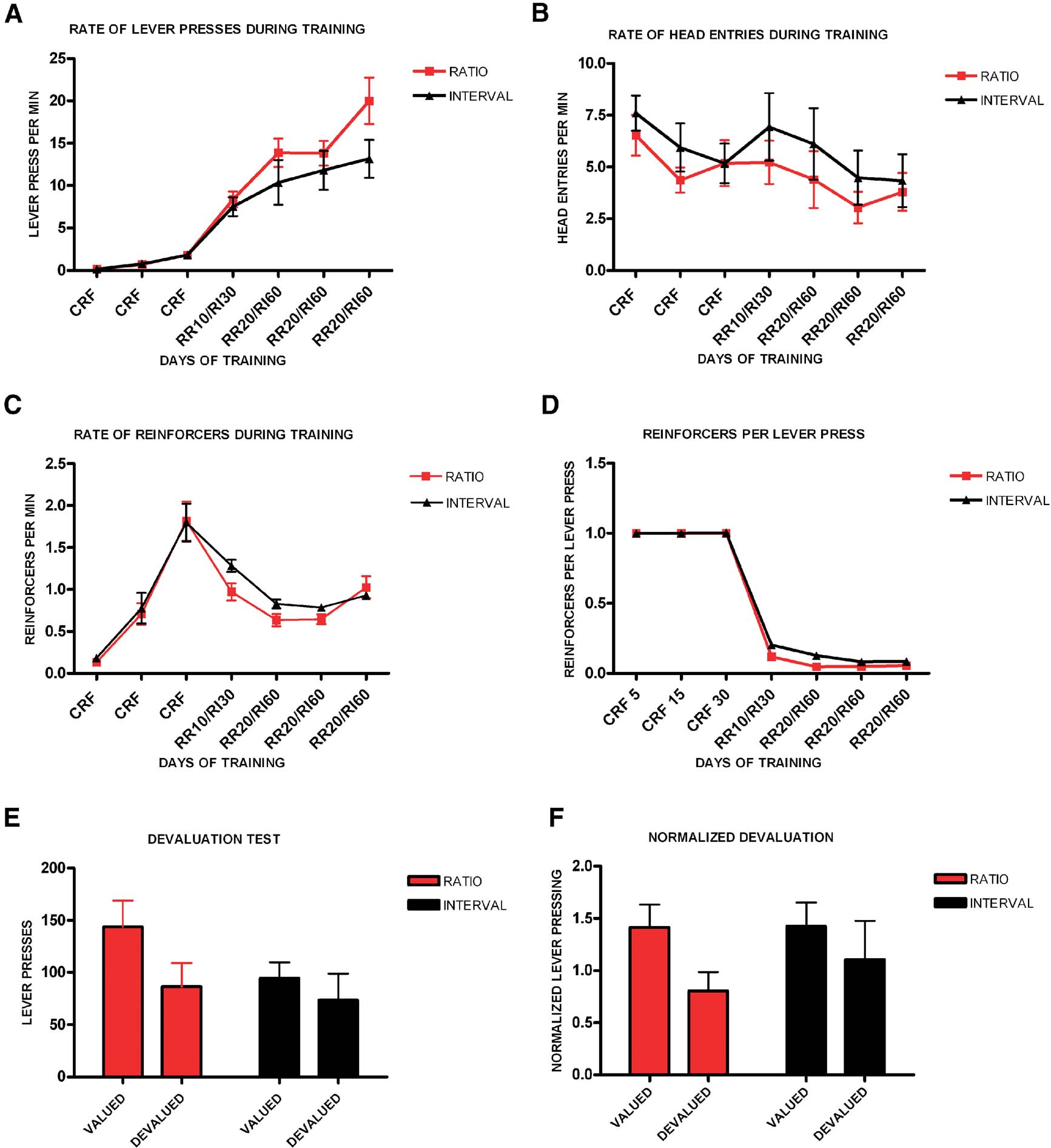
Figure 1. Different schedules of reinforcement produce different predisposition to habit formation in C57B16/J mice. (A) Acquisition of the lever-pressing task in animals trained on random ratio and random interval schedules. The rate of lever pressing (per minute) for each daily session is depicted. (B) Average rate of head entry throughout training for the random interval and random ratio groups. (C) Average rate of reinforcement throughout training for the random interval and random ratio groups. (D) Rate of reinforcement per lever press throughout training for the random interval and random ratio groups. (E) Absolute number of lever presses during the valued versus the devalued condition for the different training schedules. (F) Lever pressing during the valued versus the devalued condition normalized to the lever pressing of the last day of training.
In order to investigate if lever pressing in the mice trained in different schedules was goal-directed or habitual we performed a devaluation test (Figure 1 E). During the devaluation test, random ratio-trained animals responded significantly less during the devalued condition, when the outcome they pressed for during training was devalued by sensory-specific satiety, than during the non-devalued condition (t11 = 4.15, p = 0.002) (see section “Materials and Methods ”). In contrast, mice trained in a random interval schedule of reinforcement failed to show sensitivity to changes in value during the test, and pressed equally during the valued and devalued conditions (t11 = 1.61, p = 0.14). Because the level of lever pressing after training was different between the ratio and interval groups, we normalized the rate of responding during the devaluation test to the rate of responding during the last day of training (Figure 1 F). The normalized data confirmed that the random ratio group showed significant devaluation while the random interval group did not (t11 = 4.16, p = 0.002; t11 = 1.65, p = 0.13).
To investigate further if ratio-trained animals devalue more because they have higher levels of lever pressing and interval-trained animals are less sensitive to devaluation because of a floor effect, we analyzed the correlation between the levels of lever pressing and the levels of devaluation for each of the training schedules (Figure 2 ). There was no significant correlation between the total number of lever presses during the last day of training and the amount of devaluation for both random ratio (r = −0.13, p = 0.69) (Figure 2 A) and random interval (r = −0.32, p = 0.31) (Figure 2 B) schedules. Furthermore, there was no correlation between the number of lever presses during the valued condition and the amount of devaluation for both the random schedule-trained mice (r = −0.25, p = 0.43) (Figure 2 C), and the random interval-trained mice (r = −0.54, p = 0.07) (Figure 2 D). Additionally, there was no significant correlation between the total number of lever presses during devaluation (valued + devalued condition) and the amount of devaluation in mice trained in the random ratio schedule (r = −0.47, p = 0.13) (Figure 2 E). For interval schedule-trained animals there was even a significant negative correlation between the total number of lever presses during devaluation and the amount of devaluation (r = −0.68, p = 0.01) (Figure 2 F). These data suggest that the different sensitivity to devaluation of animals trained in ratio and interval schedules cannot be explained by the overall amounts of lever pressing during training or testing.
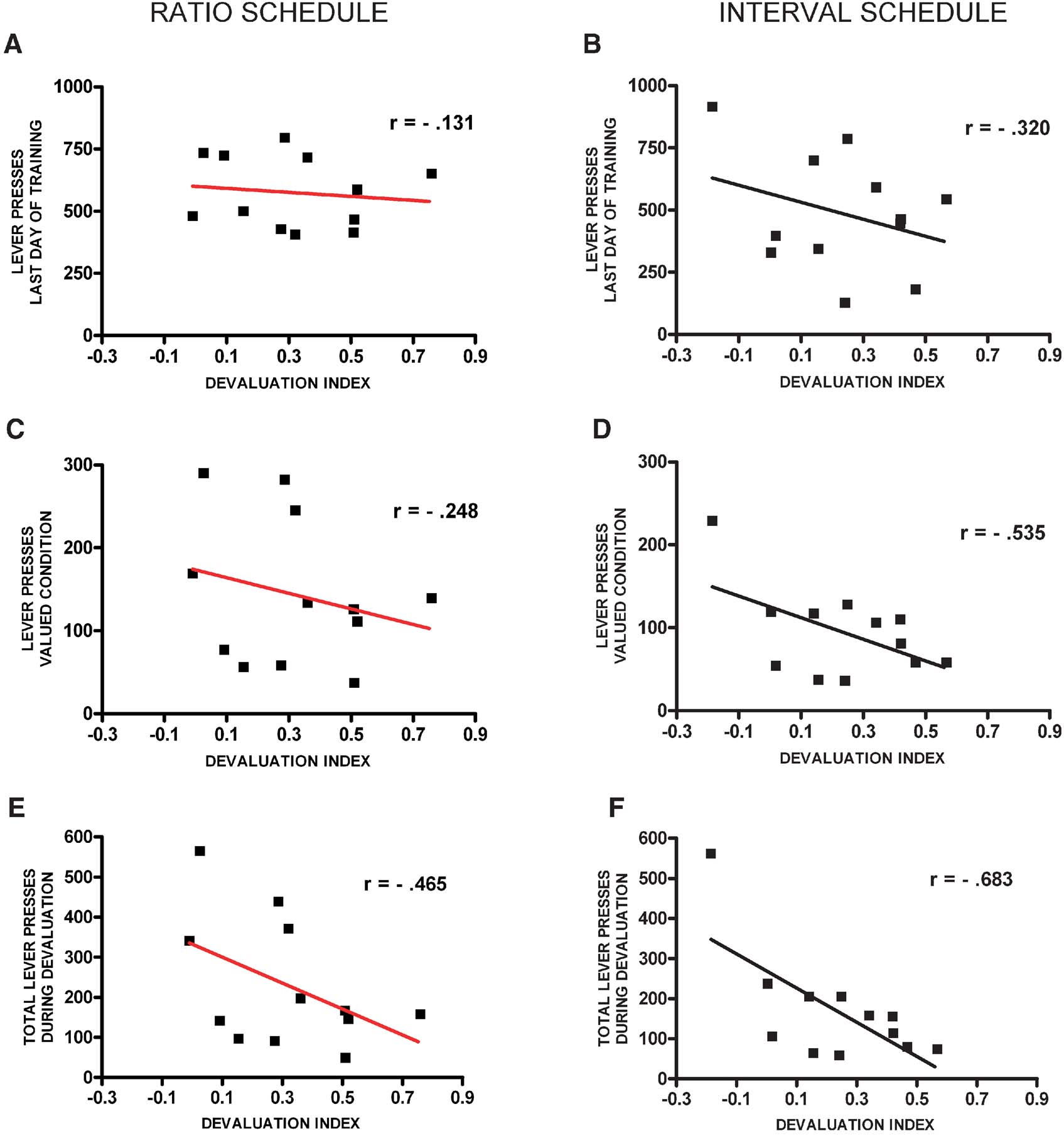
Figure 2. Correlation between the levels of lever pressing and devaluation for random ratio and random interval trained animals. (A) Correlation between the total number of lever presses during the last day of training and the devaluation index in animals trained on random ratio. (B) Correlation between the total number of lever presses during the last day of training and the devaluation index in animals trained on random interval. (C) Correlation between the total number of lever presses during the valued condition and the devaluation index in animals trained on random ratio. (D) Correlation between the total number of lever presses during the valued condition and the devaluation index in animals trained on random interval. (E) Correlation between the total number of lever presses during both days of the devaluation test and the devaluation index in animals trained on random ratio. (F) Correlation between the total number of lever presses during both days of the devaluation test and the devaluation index in animals trained on random interval.
Finally, and following a reviewer's suggestion, we analyzed the sensitivity to devaluation in a subset of animals matched for performance during random ratio and random interval training. Animals increased their rate of lever pressing during training (F6,48 = 22.75, p < 0.001), and there was no significant interaction between training and schedule of reinforcement (F6,3 = 1.40, p = 0.99), and no significant effect of schedule (F1,8 = 0.005, p = 0.95) (Figure 3 A). There was no significant difference in the rate of head entries during training between ratio and interval schedules (F1,8 = 0.03, p = 0.86), and no interaction between training and the type of schedule (F6,3 = 0.98, p = 0.55) (Figure 3 B). The average rate of reinforcement changed throughout training (F6,48 = 23.7, p = 0.01), but there was no interaction between training and schedule (F6,3 = 1.32, p = 0.44), and no main effect of schedule (F1,8 = 0.03, p = 0.86) (Figure 3 C). Furthermore, although the rate of reinforcements per lever press changed throughout training (F6,48 = 21 144, p < 0.001) there was no interaction between training and schedule of reinforcement (F4,5 = 0.77, p = 0.59), and there was no main effect of schedule of reinforcement (F1,8 = 2.27, p = 0.17) (Figure 3 D). Nonetheless, during the devaluation test, random ratio-trained mice showed significant devaluation (t4 = 3.30, p = 0.03) while random interval-trained mice did not (t4 = −0.022, p = 0.98) (Figure 3 E). The normalized devaluation showed the same effect (Figure 3 F).
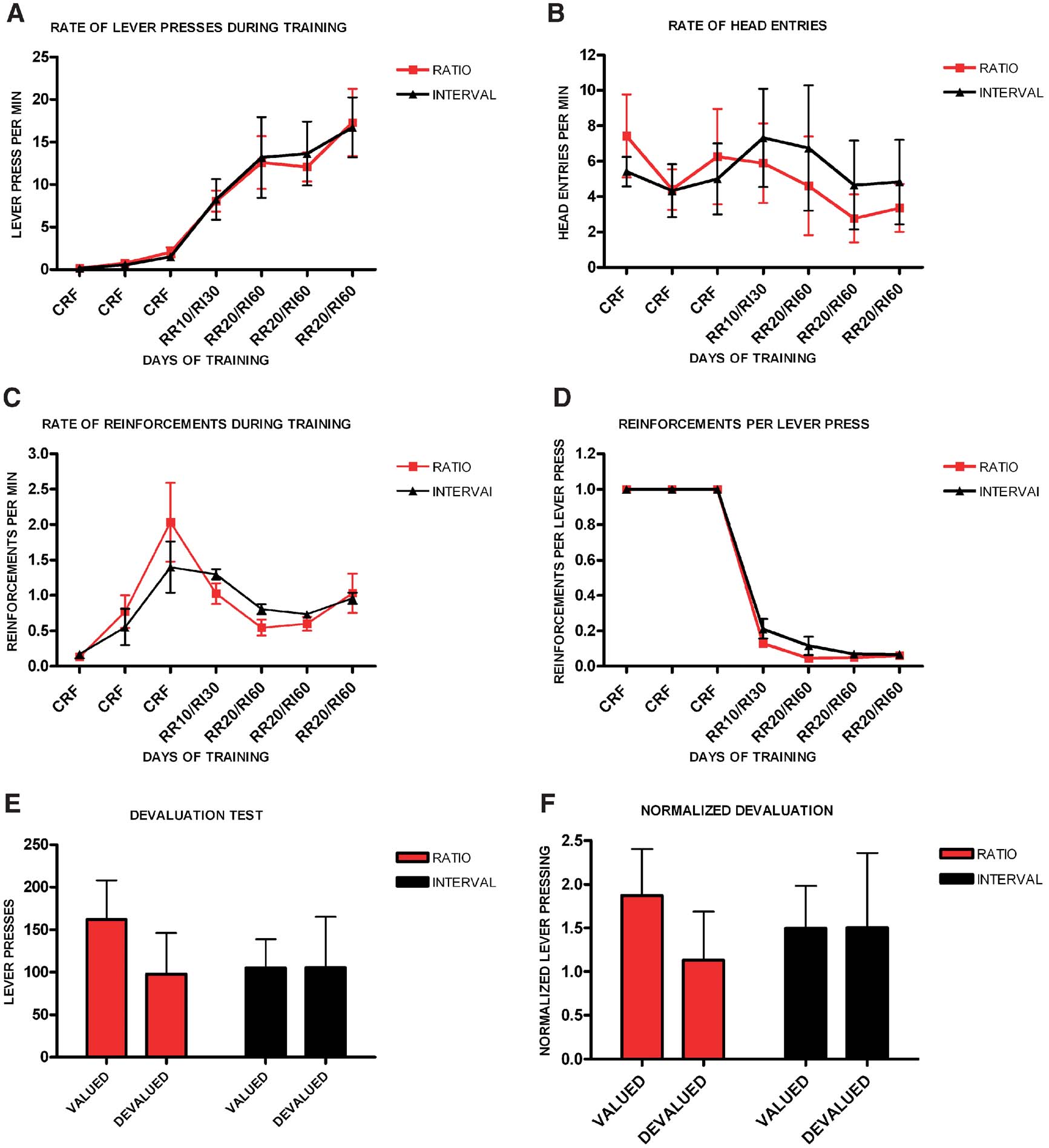
Figure 3. Different schedules of reinforcement induce different sensitivity to devaluation in a subgroup of C57B16/J mice matched for performance during training. (A) Acquisition of the lever-pressing task in animals trained on random ratio and random interval schedules. The rate of lever pressing per minute for each daily session is depicted. (B) Average rate of head entry throughout training for the random interval and random ratio groups. (C) Average rate of reinforcement throughout training for the random interval and random ratio groups. (D) Rate of reinforcement per lever press throughout training for the random interval and random ratio groups. (E) Absolute number of lever pressed during the valued versus the devalued condition for the different training schedules. (F) Lever pressing during the valued versus the devalued condition normalized to the lever pressing of the last day of training.
Taken together, these data suggest that random ratio-trained mice acquired goal-directed actions while random interval-trained animals became habitual.
Effect of different schedules of reinforcement on the exploration of a novel lever
It has been hypothesized that the shift from goal-directed responding to habitual responding corresponds to a shift from outcome driven actions to actions that are elicited by antecedent stimuli. We therefore examined to what extent animals trained in different schedules of reinforcement would press a novel lever identical to their training lever (Figure 4 ). We trained two different groups of mice in random ratio and random interval schedules and tested their propensity to exploit the training lever versus explore a novel lever. Consistently with the previous experiment, all animals acquired the task (F6,84 = 20.5, p < 0.001), with no significant interaction between acquisition and schedule of reinforcement (F6,9 = 2.89, p = 0.07). In this experiment mice trained on a random ratio schedule did press at higher rates than mice trained on a random ratio interval schedule (F1,14 = 12.8, p < 0.001), (Figure 4 A). Random ratio-trained animals pressed significantly more on the lever that was reinforced during training than on the novel lever (t7 = 4.35, p < 0.001, Figure 4 B). However, random interval-trained animals pressed the novel lever as much as the training lever (t7 = 1.23, p = 0.26). The normalized data confirmed that the random ratio group pressed mostly on the training lever (t7 = 3.88, p < 0.01), while the random interval group explored the novel lever as much as the training lever (t7 = 0.62, p = 0.56), (Figure 4 C).
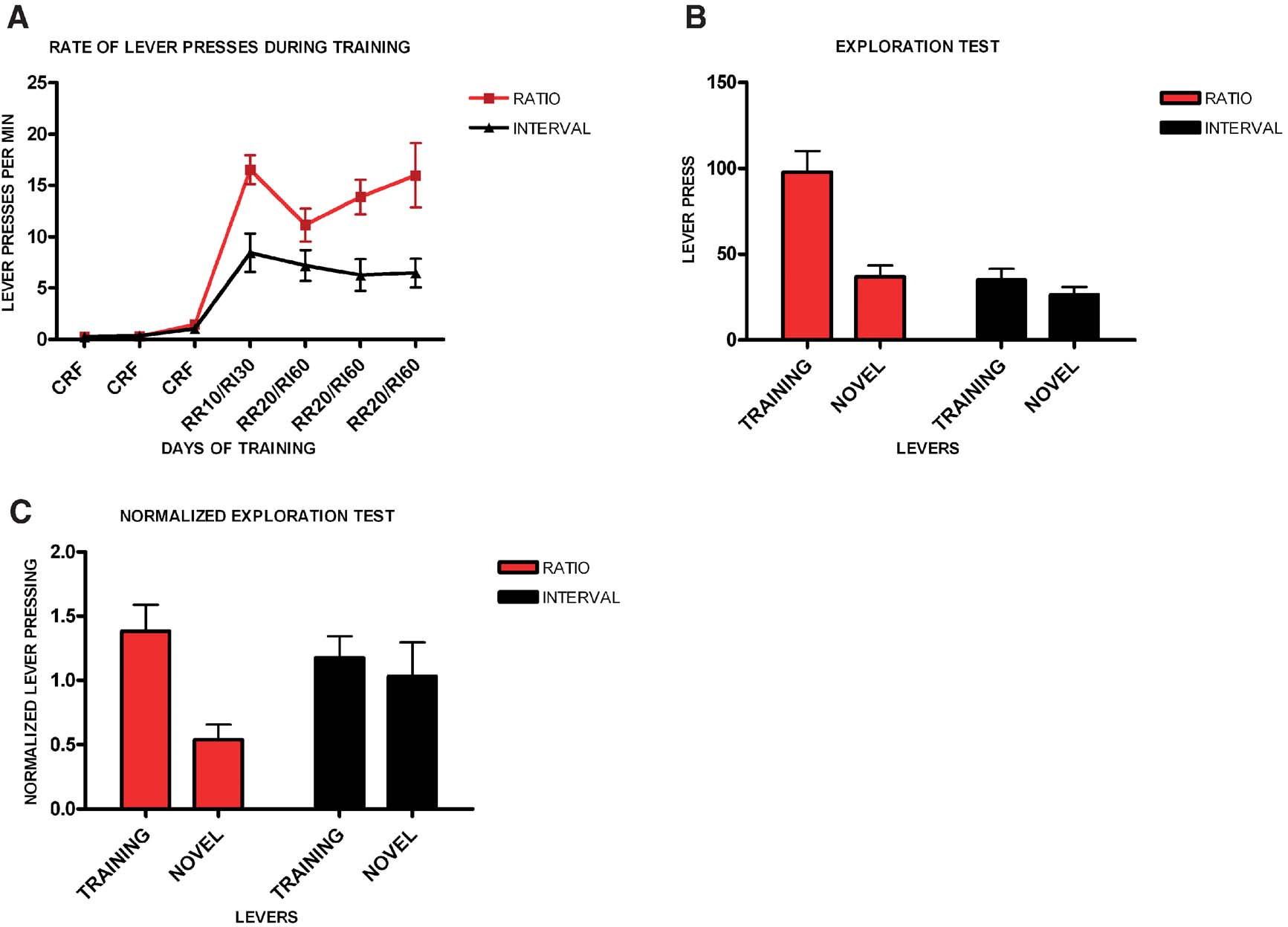
Figure 4. Different schedules of reinforcement produce different predisposition for exploration versus exploitation in C57B16/J mice. (A) Acquisition of the lever-pressing task in animals trained on random ratio and random interval schedules. The rate of lever pressing (per minute) for each daily session is depicted. (B) Absolute number of lever presses on the training lever versus a novel lever for the different training schedules. (C) Lever pressing on the training lever versus a novel lever normalized to the lever pressing of the last day of training.
CB1 mutant mice have decreased predisposition for habit formation
We next investigated if endocannabinnoid signaling would be involved in habit formation. We trained WT, CB1+/−, and CB1−/− littermates on a random interval schedule (Figure 5 ). Animals from the different genotypes increased lever pressing across days (F7,385 = 6.8, p < 0.001), (Figure 5 A), and there was no effect of genotype on lever pressing (F1,55 = 0.75, p = 0.48), or interaction between training and genotype (F14,100 = 1.65, p = 0.08). The average rate of head entries into the magazine also increased throughout training (F7,385 = 9.18, p < 0.001), but there was no significant difference between the groups (F1,55 = 0.23, p = 0.79), or interaction between training and genotype (F14,100 = 0.65, p = 0.82), (Figure 5 B). As training progressed the average rate of reinforcement increased significantly for all genotypes (F7,385 = 15.3, p < 0.001), but there was no significant difference between genotypes (F1,55 = 0.25, p = 0.78), or interaction between training and genotype (F14,100 = 0.60, p = 0.86), (Figure 5 C). The average reinforcement per lever press decreased across training (F7.385 = 9.18, p < 0.001), but there was no interaction between training and genotype (F14,100 = 0.95, p = 0.5) or difference between genotypes (F1,55 = 0.19, p = 0.83), (Figure 5 D). As expected, during the devaluation test WT mice failed to show an effect of devaluation (t20 = 0.32, p = 0.76), indicating that their responding was habitual. However, both CB1+/− (t20 = 2.4, p = 0.03) and CB1−/− (t15 = 3.25, p = 0.01) mutants showed sensitivity to sensory-specific satiety, suggesting that their actions were goal-directed (Figure 5 E).
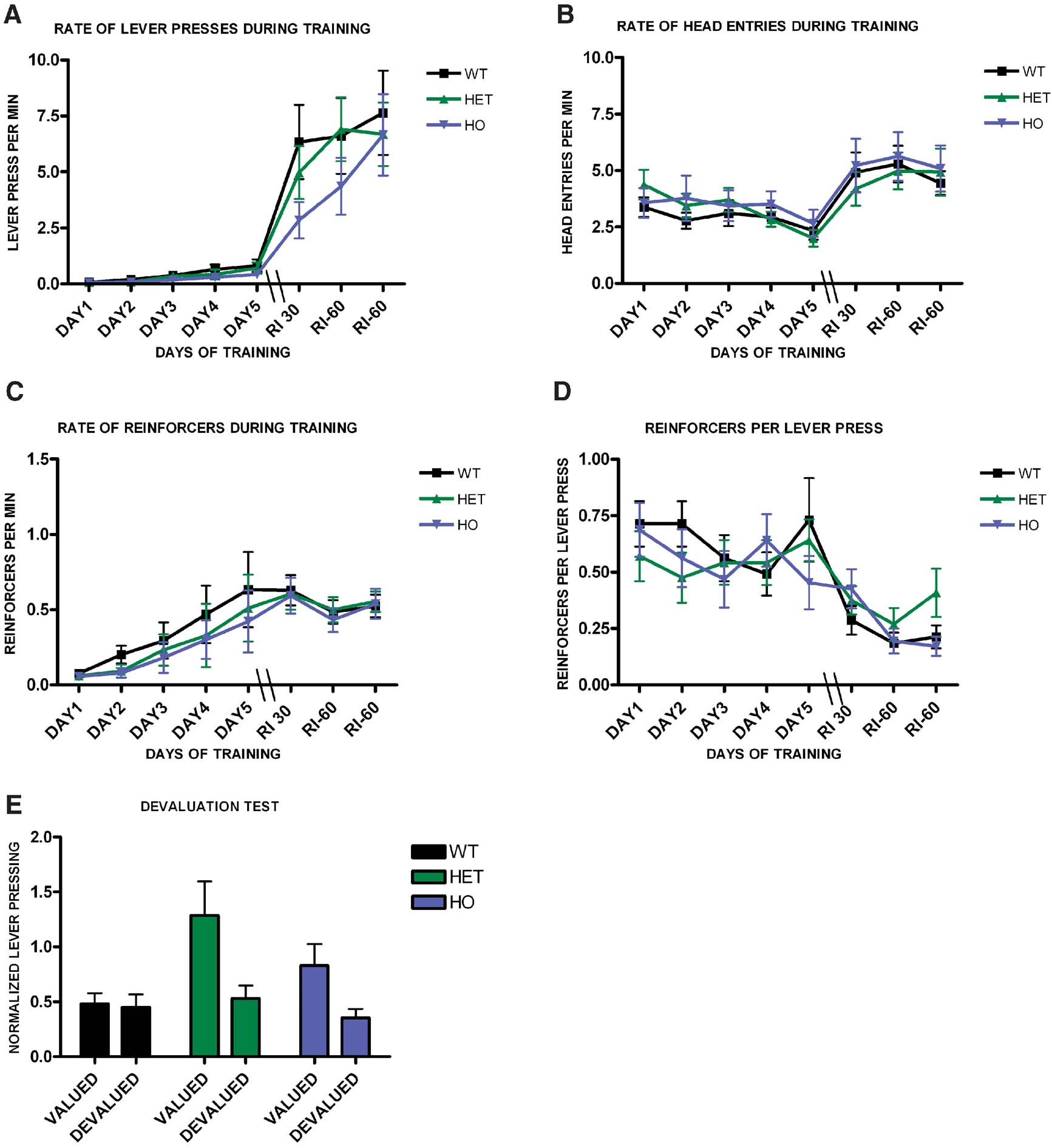
Figure 5. Decreased predisposition for habit formation in CB1 mutant mice. (A) Acquisition of the lever pressing task in WT, CB1+/−, and CB1−/− mice trained in a random interval schedule. The rate of lever pressing (per minute) for each daily session is depicted. (B) Average rate of head entry throughout training for the WT, CB1+/−, and CB1−/− mice. (C) Average rate of reinforcement throughout training for WT, CB1+/−, and CB−/− mice. (D) Rate of reinforcement per lever press throughout training for WT, CB+/−, and CB1−/− mice. (E) Normalized lever pressing during the valued versus the devalued condition for WT, CB+/−, and CB1−/− mice.
We trained a group of WT, CB1+/−, and CB1−/− mice in random interval schedules, and tested their propensity to explore a novel lever compared to the training lever. As before, we observed no difference in the acquisition of the task across genotypes (F2,29 = 0.94, p = 0.40) (Figure 6 A), and no significant interaction of training and genotype (F18,44 = 1.08, p = 0.40), while all groups learned the task (F9,261 = 13.8, p < 0.001). During the exploration test (Figure 6 B), WT mice showed substantial exploration of the novel lever and they pressed equally both levers (t9 = 1.92, p = 0.09). CB1+/− also pressed equally in both levers (t13 = 1.36, p = 0.19), which is different than what was observed in the devaluation test, and could reflect the fact that in this experiment animals were trained longer than in the devaluation experiment, or differences in the sensitivity of the exploration and the devaluation tests. In contrast, CB1−/− mice did press significantly more on the training lever than on the novel lever (t7 = 4.11, p = 0.005). Together, these data indicate that CB1 mutant mice show reduced habit formation.
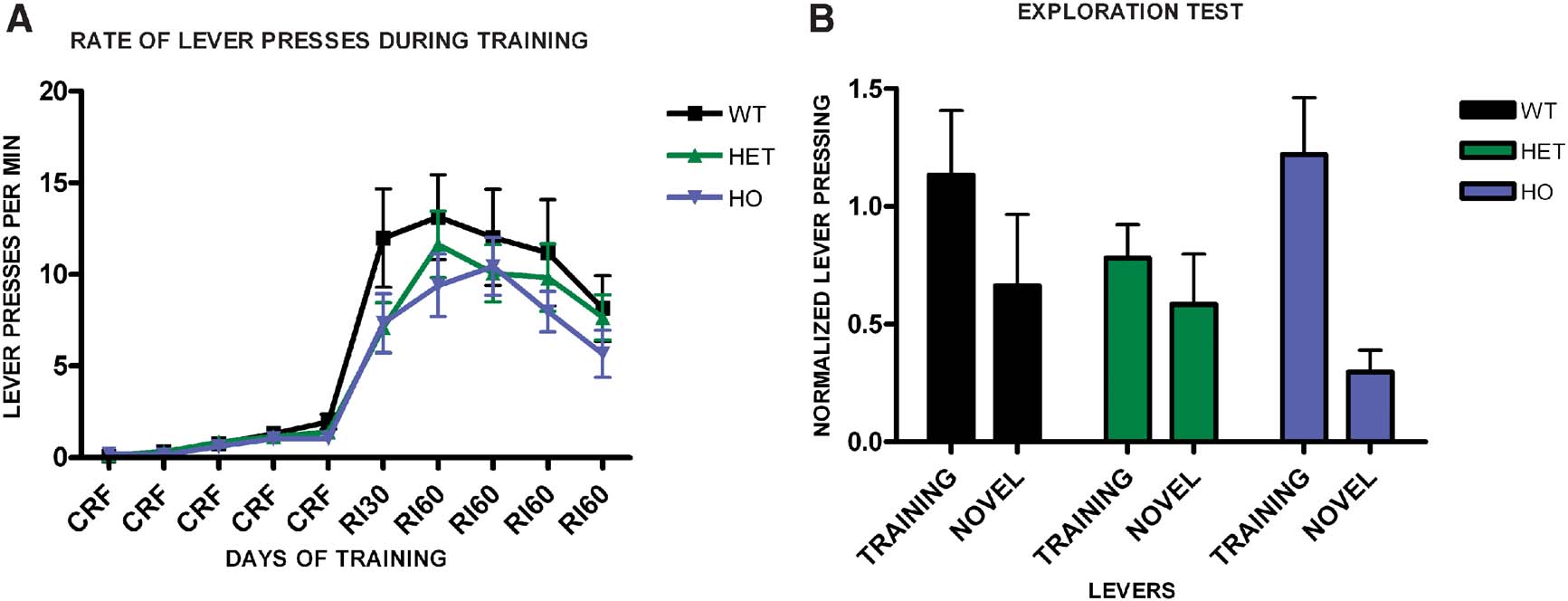
Figure 6. Exploration versus exploitation in WT, CB1+/−, and CB1−/− mice. (A) Acquisition of the lever pressing task in WT, CB1+/−, and CB1−/− mice animals trained on a random interval schedule. The rate of lever pressing per minute for each daily session is depicted. (B) Lever pressing (normalized) on the training lever versus a novel lever in WT, CB1+/−, and CB1−/− mice.
CB1 blockade impairs habit formation
Since the CB1 null mutants that we used carry the mutation constitutively, we tested if blockade of CB1 receptors specifically during training was sufficient to impair habit formation. We trained three different groups of mice on interval schedules of reinforcement, and after CRF training we injected them with either saline, 3 mg/kg of the CB1 receptor antagonist AM251, or with 6 mg/kg of AM251. All treatment groups increased lever pressing across days (F6,336 = 42.2, p < 0.001) (Figure 7 A), and there was no effect of treatment on lever pressing (F2,56 = 0.82, p = 0.99), or interaction between training and treatment (F12,104 = 0.93, p = 0.53). There was a significant change in the rate of head entry during training (F6.336 = 7.4, p = 0.001), but there was no difference among treatments (F2,56 = 0.44, p = 0.64) and no interaction between training and treatment (F12,104 = 1.1, p = 0.37), (Figure 7 B). As training progressed the average rate of reinforcement changed significantly (F6,336 = 58.6, p < 0.001), but there was no significant effect of treatment (F2,56 = 0.97, p = 0.38), or interaction between training and treatment (F12,104 = 1.28, p = 0.00), (Figure 7 C). Similarly, the average rate of reinforcements per lever press changed during training (F6,336 = 3915, p < 0.001), but there was no significant difference between treatment groups (F2,56 = 0.10, p = 0.90), and no interaction between training and treatment (F8,108 = 0.82, p = 0.59), (Figure 7 D).
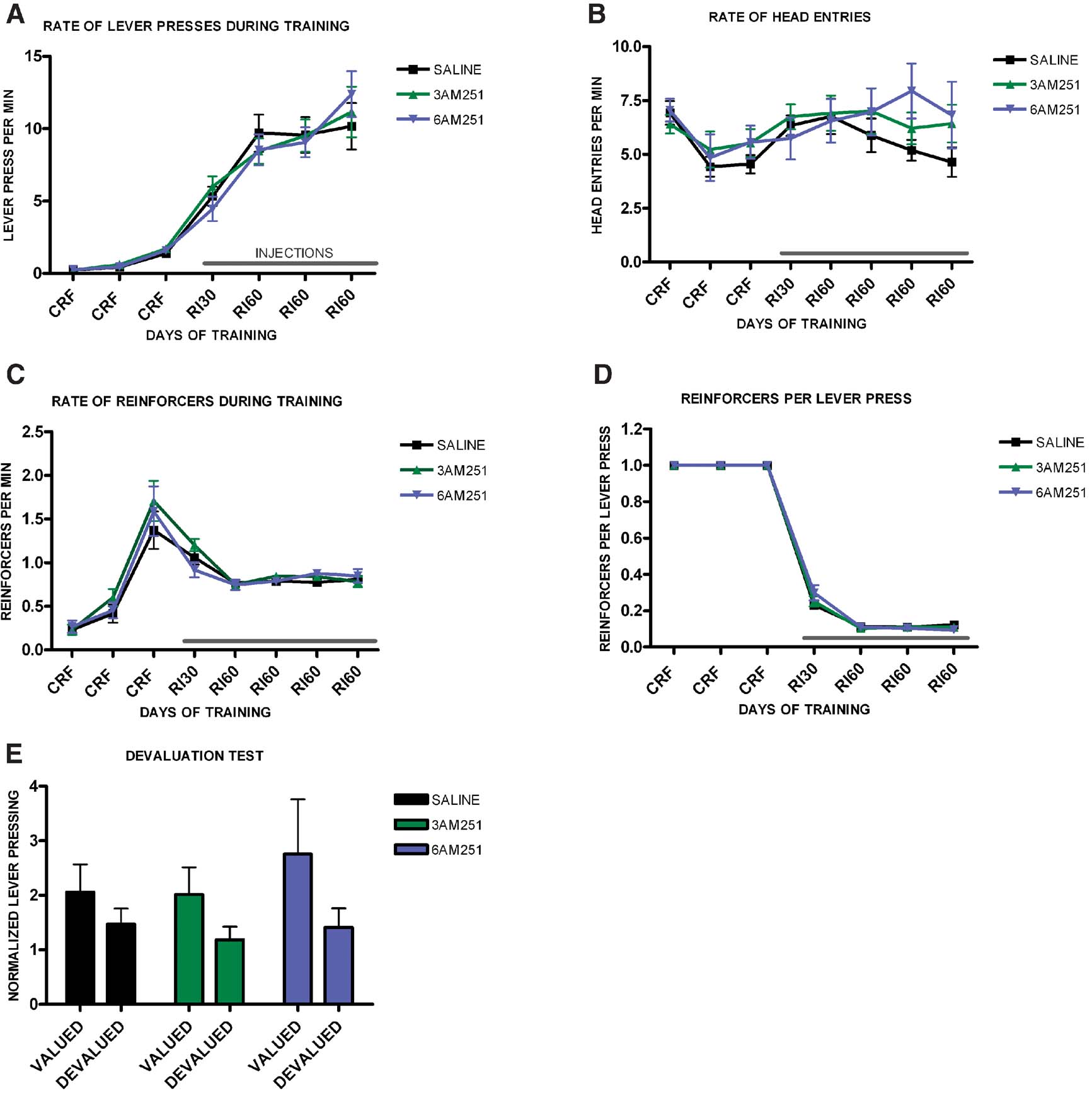
Figure 7. Decreased predisposition for habit formation in C57B16/J mice injected with CB1 antagonists during random interval training. (A) Acquisition of the lever-pressing task for animals injected with saline, 3 mg/kg AM251 or 6 mg/kg AM251. The rate of lever pressing (per minute) for each daily session is depicted. Note that animals were only injected during RI-30 and RI-60 training. (B) Average rate of head entry throughout training for mice injected with saline, 3 mg/kg AM251 or 6 mg/kg AM251. (C) Average rate of reinforcement throughout training for mice injected with saline, 3 mg/kg AM251 or 6 mg/kg AM251. (D) Rate of reinforcement per lever press throughout training for mice injected with saline, 3 mg/kg AM251 or 6 mg/kg AM251. (E) Normalized lever pressing during the valued versus the devalued condition for mice injected with saline, 3 mg/kg AM251 or 6 mg/kg AM251. The devaluation test was performed off drug.
In order to assess if the animals' behavior was goal-directed or habitual we performed a devaluation test off drug, (Figure 7 E). Mice injected with saline during interval training became habitual and did not show an effect of devaluation (t20 = 1.46, p = 0.16). Mice injected with 3 mg/kg of AM251 also did not show a devaluation effect (t20 = 1.78, p = 0.09) indicating that their responding was habitual. Mice injected with 6 mg/kg of AM251 did show significant devaluation, indicating that their lever pressing was goal-directed (t16 = 2.11, p = 0.04).
Using the same treatment procedure, we trained a different group of mice to test their tendency to explore a novel lever compared to the training lever. Again, the groups learned the task (F7,112 = 8.41, p < 0.001) and we observed no significant interaction of training and genotype (F14,22 = 0.91, p = 0.56) and no effect of treatment on the acquisition of the task (F2,16 = 0.24, p = 0.79), (Figure 8 A). During the exploration test, mice injected with saline pressed equally both levers (t5 = 0.33, p = 0.75), (Figure 8 B). However, both mice injected with 3 mg/kg of AM251 (t3 = 5.73, p = 0.01) and 6 mg/kg AM251 (t8 = 2.65, p = 0.03) pressed significantly more the training lever than the novel lever.
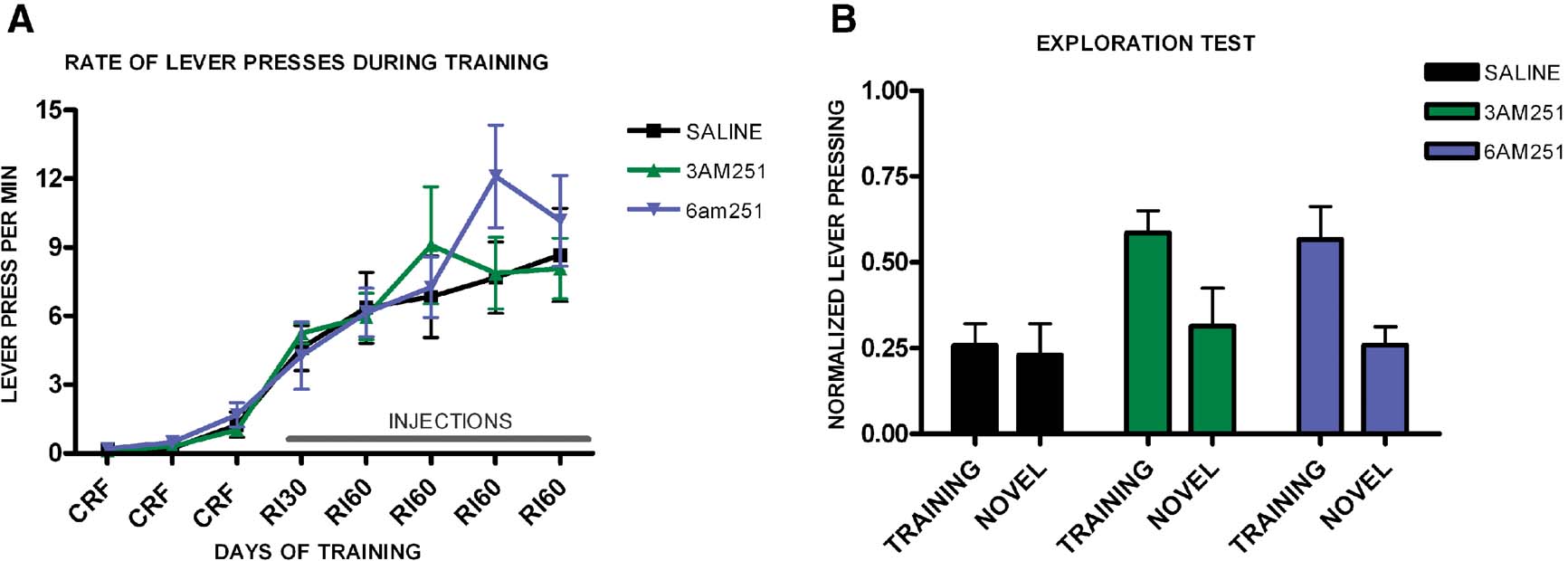
Figure 8. Exploration versus exploitation in C57B16/J mice injected with saline, 3 mg/kg AM251 or 6 mg/kg AM251. (A) Acquisition of the lever-pressing task for mice injected with saline, 3 mg/kg AM251 or 6 mg/kg AM251 trained on a random interval schedule. The rate of lever pressing (per minute) for each daily session is depicted. Note that animals were only injected during RI-30 and RI-60 training. (B) Lever pressing (normalized) on the training lever versus a novel lever in mice injected with saline, 3 mg/kg AM251 or 6 mg/kg AM251.
These data indicate that blockade of CB1 receptors specifically during training is sufficient to impair habit formation.
Discussion
In this study, using genetic and pharmacological tools in mice we showed that endocannabinoid signaling through CB1 receptors is critical for habit formation. We first showed that in mice, as in rats, training with different reinforcement schedules leads to distinct types of behavioral control and to different susceptibility to habit formation. While training on a random ratio schedule lead to the acquisition of goal-directed actions that are sensitive to the expected value of the outcome, training on a random interval schedule lead to less sensitivity to devaluation. Furthermore, we showed for the first time that random interval training also favored the exploration of a novel lever during an extinction test, while random ratio training promoted exploitation of the reinforced lever. These results suggest that in ratio-trained animals the behavior is governed by the action-outcome contingency because the animals decrease pressing specifically when the outcome they press for is devalued, and when given the choice between the training lever and a novel but identical lever, they tend to choose the lever that was previously associated with the outcome. On the other hand, the behavior of random interval-trained animals seems to be governed more by stimulus-response than action-outcome relations because responding in trained animals become less sensitive to devaluation, and they do generalize to an identical lever that never lead to delivery of the outcome (Dickinson, 1985 ; Dickinson and Balleine, 1995 , 2002 ). The differences observed in the type of learning favored by each schedule could not be attributed to trivial factors like different reinforcement rates because these were not different between random interval and random ratio schedules, which is consistent with previous studies (Dawson and Dickinson, 1990 ; Dickinson et al., 1983 ) Also, we did not observe significant differences in the rate of head entry although this “checking” behavior seemed to be more frequent in random interval-trained groups, which could reflect the uncertainty about the consequences of the action associated with the schedule (Dickinson et al., 1983 ). We did observe that random ratio-trained animals tended to press at higher rates than random interval-trained animals, which is consistent with previous studies comparing these schedules (Dawson and Dickinson, 1990 ; Dickinson et al., 1983 ). Consistent with the higher lever-pressing rates, animals trained on a ratio schedule tended to earn on average fewer reinforcers per lever press. However, the differences in lever-pressing rates were not observed in every group of animals (Figure 1 , Figure 3 ), while the differences in sensitivity to devaluation were. Therefore, it does not seem that higher lever-pressing rates could explain the difference in sensitivity to devaluation observed in the different schedules. Rather, variations in the correlation between the rate of responding and the rate of reinforcement during training under the different schedules may be more critical (Dickinson et al., 1983 ). We also showed that the exploration test can be used as a test to differentiate the behavior of ratio and interval-trained animals. This test may complement the devaluation test, and be of importance when examining mutant animals with different sensitivities to food reward. Interestingly, recent studies in humans showed that activity in the caudate nucleus of the dorsal striatum (roughly the dorsomedial striatum in rodents) is correlated with the value-based exploitation, in an exploitation-exploration task (Daw et al., 2006 ). However, it is important to note that although random ratio and random interval training bias the behavior of mice on both the devaluation and the exploration tests, these tests may measure slightly different processes. At any rate, our data suggests that these reinforcement schedules combined with different post-training probe tests are useful to study the molecular, cellular, and circuit mechanism underlying goal-directed actions and habit formation in mice (Wiltgen et al., 2007 ; Yin et al., 2006 ).
Using these assays to examine habit formation in mice, we showed that CB1 mutant mice have impaired habit formation. Although endocannabinoid signaling through CB1 has been implicated in eating and the rewarding aspects of food (Di Marzo et al., 2001 ; Osei-Hyiaman et al., 2005 ; Sanchis-Segura et al., 2004 ), the results could not be easily explained by different sensitivity of the CB1 mice during the devaluation test because the test was conducted in extinction, and more importantly because they also showed enhanced exploitation of the reinforced lever during the exploration test, in relation to WT littermates. Also, these results do not seem to be caused by developmental or behavioral abnormalities that may occur chronically in CB1 mutant mice due to the fact that their mutation is constitutive. Rather, it seems that endocannabinoid signaling through CB1 is necessary at the time of training, as injections of the CB1 antagonist AM251 specifically during random interval training blocked habit formation in normal mice. Finally, these effects cannot be due to blockade of CB1 receptors during the tests because both the devaluation and exploration tests were done off drug.
Because CB1 receptors are expressed almost ubiquitously in the brain, it remains unclear precisely where endocannabinoids act to promote habit formation. Previous work suggests that the requisite endocannabinoid signaling takes place in the dorsolateral striatum (Gerdeman et al., 2007 ). This striatal region is shown by lesion studies to be critical for habit formation: local depletion of dopamine as well as excitotoxic lesions render behavior goal-directed even with training schedules that lead to habitual behavior in control animals (Faure et al., 2005 ; Yin et al., 2004 ). Moreover, retrograde endocannabinoid signaling has been shown to be necessary for LTD at the corticostriatal synapse in this region (Gerdeman et al., 2002 ). It would be interesting to investigate if the effects observed in this study are caused by lack of CB1 receptors at terminals originating from specific cortical areas. Nevertheless, a number of other possibilities remain. For example, CB1 receptors are highly expressed by the GABAergic terminals of striatal medium spiny projection neurons, which send projections to the globus pallidus and substania nigra pars reticulata (Herkenham et al., 1991 ). These axons also have collaterals that synapse on neighboring spiny neurons. Endocannabinoid signaling at these synapses could also be involved in habit formation. One admittedly speculative possibility is that reduced GABA release at the collaterals caused by CB1 activation can also reduce lateral inhibition in the striatum and thus reduce the selectivity of actions, as shown by more action generalization and exploration of the novel lever in our study. CB1 receptors are also expressed in high levels in terminals from parvalbumin-positive interneurons (Uchigashima et al., 2007 ). Interestingly, as we observed, a heterozygous mutation in the CB1 receptor affected habit formation, suggesting that tight regulation of endocannabinoid signaling at one or several synapse types is important for behavioral control. As a new generation of genetic tools to investigate circuit function becomes available in mice, it will be important to investigate the brain region and the cell types where CB1 signaling is required for habit formation.
In summary, our data shows that endocannabinoid signaling through CB1 receptors is critical for habit formation, and that instrumental tasks in mouse models can be an important tool for investigating the molecular, cellular, and circuit mechanisms of habit formation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgements
We thank David M. Lovinger for comments on the manuscript, Guoxiang Luo for technical help, and S. Baktai and G. Kunos for providing the CB1 mutant mice. This research was supported by the NIAAA DICBR.
References
Adams, C. (1982). Variations in the sensitivity of instrumental responding to reinforcer devaluation. Q. J. Exp. Psychol. Comp. Physiol. Psychol. 34B, 77–98.
Adams, C., and Dickinson, A. (1981a). Actions and habits: variations in associative representations during instrumental learning. In Information Processing in Animals: Memory Mechanisms, N. Spear, and R. Miller, eds. pp. 143–166.
Adams, C. D., and Dickinson, A. (1981b). Instrumental responding following reinforcer devaluation. Q. J. Exp. Psychol. 33, 109–122.
Balleine, B. W., and Dickinson, A. (1998). Goal-directed instrumental action: contingency and incentive learning and their cortical substrates. Neuropharmacology 37, 407–419.
Caille, S., Alvarez-Jaimes, L., Polis, I., Stouffer, D. G., and Parsons, L. H. (2007). Specific alterations of extracellular endocannabinoid levels in the nucleus accumbens by ethanol, heroin, and cocaine self-administration. J. Neurosci. 27, 3695–3702.
Colwill, R. M., and Rescorla, R. A. (1985). Postconditioning devaluation of a reinforcer affects instrumental responding. J. Exp. Psychol. Anim. Behav. Process. 11, 120–132.
Corbille, A. G., Valjent, E., Marsicano, G., Ledent, C., Lutz, B., Herve, D., and Girault, J. A. (2007). Role of cannabinoid type 1 receptors in locomotor activity and striatal signaling in response to psychostimulants. J. Neurosci. 27, 6937–6947.
Corbit, L. H., Muir, J. L., and Balleine, B. W. (2003). Lesions of mediodorsal thalamus and anterior thalamic nuclei produce dissociable effects on instrumental conditioning in rats. Eur. J. Neurosci. 18, 1286–1294.
Cossu, G., Ledent, C., Fattore, L., Imperato, A., Bohme, G. A., Parmentier, M., and Fratta, W. (2001). Cannabinoid CB1 receptor knockout mice fail to self-administer morphine but not other drugs of abuse. Behav. Brain Res. 118, 61–65.
Costa, R. M. (2007). Plastic corticostriatal circuits for action learning: what's dopamine got to do with it? Ann. N. Y. Acad. Sci. 1104, 172–191.
Daw, N. D., O'Doherty, J. P., Dayan, P., Seymour, B., and Dolan, R. J. (2006). Cortical substrates for exploratory decisions in humans. Nature 441, 876–879.
Dawson, G. R., and Dickinson, A. (1990). Performance on ratio and interval schedules with matched reinforcement rates. Q. J. Exp. Psychol. B 42, 225–239.
De Vries, T. J., Shaham, Y., Homberg, J. R., Crombag, H., Schuurman, K., Dieben, J., Vanderschuren, L. J., and Schoffelmeer, A. N. (2001). A cannabinoid mechanism in relapse to cocaine seeking. Nat. Med. 7, 1151–1154.
Di Marzo, V., Goparaju, S. K., Wang, L., Liu, J., Batkai, S., Jarai, Z., Fezza, F., Miura, G. I., Palmiter, R. D., Sugiura, T., and Kunos, G. (2001). Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature 410, 822–825.
Dickinson, A. (1985). Actions and habits: the development of behavioural autonomy. Philos. Trans. R. Soc. Lond. B 308, 67–78.
Dickinson, A., and Balleine, B. W. (1995). Motivational control of instrumental action. Curr. Dir. Psychol. Sci. 4, 162–167.
Dickinson, A., and Balleine, B. (2002). The role of learning in the operation of motivational systems. In Steven's handbook of experimental psychology. In Learning, Motivation, and Emotion, H. Pashler, and R. Gallistel, eds. (New York, Wiley), pp. 497–533.
Dickinson, A., Nicholas, D. J., and Adams, C. D. (1983). The effect of the instrumental training contingency on susceptibility to reinforcer devaluation. Q. J. Exp. Psychol. 35B, 35–35 I.
Faure, A., Haberland, U., Conde, F., and El Massioui, N. (2005). Lesion to the nigrostriatal dopamine system disrupts stimulus-response habit formation. J. Neurosci. 25, 2771–2780.
Foerde, K. E., Knowlton, B. J., and Poldrack, R. A. (2007). Overtraining reduces neural responses to feedback in probabilistic classification learning. Paper presented at: Society for Neuroscience 37th Annual Meeting (San Diego, CA, 2007 Neuroscience Meeting Planner).
Gatley, S. J., Gifford, A. N., Volkow, N. D., Lan, R., and Makriyannis, A. (1996). 123I-labeled AM251: a radioiodinated ligand which binds in vivo to mouse brain cannabinoid CB1 receptors. Eur. J. Pharmacol. 307, 331–338.
Gerdeman, G., and Lovinger, D. M. (2001). CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J. Neurophysiol. 85, 468–471.
Gerdeman, G. L., Ronesi, J., and Lovinger, D. M. (2002). Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat. Neurosci. 5, 446–451.
Gerdeman, G. L., Partridge, J. G., Lupica, C. R., and Lovinger, D. M. (2003). It could be habit forming: drugs of abuse and striatal synaptic plasticity. Trends Neurosci. 26, 184–192.
Gerdeman, G. L., Schechter, J. B., and and French, E. D. (2007). Endocannabinoid signaling at striatal CB1 receptors is critical for the consolidation of stimulus-response memories. Paper presented at: 17th Annual Symposium on the cannabinoids (St-Sauveur,. (Québec) Canada, The International Cannabinoid Research Society).
Giuffrida, A., Parsons, L. H., Kerr, T. M., Rodriguez de Fonseca, F., Navarro, M., and Piomelli, D. (1999). Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat. Neurosci. 2, 358–363.
Hansson, A. C., Bermudez-Silva, F. J., Malinen, H., Hyytia, P., Sanchez-Vera, I., Rimondini, R., Rodriguez de Fonseca, F., Kunos, G., Sommer, W. H., and Heilig, M. (2007). Genetic impairment of frontocortical endocannabinoid degradation and high alcohol preference. Neuropsychopharmacology 32, 117–126.
Herkenham, M., Lynn, A. B., Johnson, M. R., Melvin, L. S., de Costa, B. R., and Rice, K. C. (1991). Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J. Neurosci. 11, 563–583.
Hitchcott, P. K., Quinn, J. J., and Taylor, J. R. (2007). Bidirectional Modulation of Goal-Directed Actions by Prefrontal Cortical Dopamine. Cereb. Cortex.
Houchi, H., Babovic, D., Pierrefiche, O., Ledent, C., Daoust, M., and Naassila, M. (2005). CB1 receptor knockout mice display reduced ethanol-induced conditioned place preference and increased striatal dopamine D2 receptors. Neuropsychopharmacology 30, 339–349.
Jedynak, J. P., Uslaner, J. M., Esteban, J. A., and Robinson, T. E. (2007). Methamphetamine-induced structural plasticity in the dorsal striatum. Eur. J. Neurosci. 25, 847–853.
Killcross, S., and Coutureau, E. (2003). Coordination of actions and habits in the medial prefrontal cortex of rats. Cereb. Cortex 13, 400–408.
Kreitzer, A. C., and Malenka, R. C. (2005). Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J. Neurosci. 25, 10537–10545.
Matsumoto, M., Weickert, C. S., Akil, M., Lipska, B. K., Hyde, T. M., Herman, M. M., Kleinman, J. E., and Weinberger, D. R. (2003). Catechol O-methyltransferase mRNA expression in human and rat brain: evidence for a role in cortical neuronal function. Neuroscience 116, 127–137.
Miyachi, S., Hikosaka, O., Miyashita, K., Karadi, Z., and Rand, M. K. (1997). Differential roles of monkey striatum in learning of sequential hand movement. Exp. Brain Res. 115, 1–5.
Nelson, A., and Killcross, S. (2006). Amphetamine exposure enhances habit formation. J. Neurosci. 26, 3805–3812.
Osei-Hyiaman, D., DePetrillo, M., Pacher, P., Liu, J., Radaeva, S., Batkai, S., Harvey-White, J., Mackie, K., Offertaler, L., Wang, L., and Kunos, G. (2005). Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J. Clin. Invest. 115, 1298–1305.
Sanchis-Segura, C., Cline, B. H., Marsicano, G., Lutz, B., and Spanagel, R. (2004). Reduced sensitivity to reward in CB1 knockout mice. Psychopharmacology (Berl) 176, 223–232.
Uchigashima, M., Narushima, M., Fukaya, M., Katona, I., Kano, M., and Watanabe, M. (2007). Subcellular arrangement of molecules for 2-arachidonoyl-glycerol-mediated retrograde signaling and its physiological contribution to synaptic modulation in the striatum. J. Neurosci. 27, 3663–3676.
Wang, L., Liu, J., Harvey-White, J., Zimmer, A., and Kunos, G. (2003). Endocannabinoid signaling via cannabinoid receptor 1 is involved in ethanol preference and its age-dependent decline in mice. Proc. Natl. Acad. Sci. USA 100, 1393–1398.
Wickens, J. R., Budd, C. S., Hyland, B. I., and Arbuthnott, G. W. (2007a). Striatal contributions to reward and decision making: making sense of regional variations in a reiterated processing matrix. Ann. N. Y. Acad. Sci. 1104, 192–212.
Wickens, J. R., Horvitz, J. C., Costa, R. M., and Killcross, S. (2007b). Dopaminergic mechanisms in actions and habits. J. Neurosci. 27, 8181–8183.
Wiltgen, B. J., Law, M., Ostlund, S., Mayford, M., and Balleine, B. W. (2007). The influence of Pavlovian cues on instrumental performance is mediated by CaMKII activity in the striatum. Eur. J. Neurosci. 25, 2491–2497.
Yin, H. H., and Knowlton, B. J. (2006). The role of the basal ganglia in habit formation. Nat. Rev. Neurosci. 7, 464–476.
Yin, H. H., and Lovinger, D. M. (2006). Frequency-specific and D2 receptor-mediated inhibition of glutamate release by retrograde endocannabinoid signaling. Proc. Natl. Acad. Sci. USA 103, 8251–8256.
Yin, H. H., Knowlton, B. J., and Balleine, B. W. (2004). Lesions of dorsolateral striatum preserve outcome expectancy but disrupt habit formation in instrumental learning. Eur. J. Neurosci. 19, 181–189.
Yin, H. H., Knowlton, B. J., and Balleine, B. W. (2005a). Blockade of NMDA receptors in the dorsomedial striatum prevents action-outcome learning in instrumental conditioning. Eur. J. Neurosci. 22, 505–512.
Yin, H. H., Ostlund, S. B., Knowlton, B. J., and Balleine, B. W. (2005b). The role of the dorsomedial striatum in instrumental conditioning. Eur. J. Neurosci. 22, 513–523.
Yin, H. H., Zhuang, X., and Balleine, B. W. (2006). Instrumental learning in hyperdopaminergic mice. Neurobiol. Learn. Mem. 85, 283–288.
Keywords: habit, reward, decision-making, plasticity, action, dopamine, exploration
Citation: Monica R.F. Hilário, Emily Clouse, Henry H. Yin and Rui M. Costa (2007). Endocannabinoid signaling is critical for habit formation. Front. Integr. Neurosci. 1:6. doi: 10.3389/neuro.07/006.2007
Received: 31 August 2007;
Paper pending published: 7 September 2007;
Accepted: 2 October 2007;
Published online: 2 November 2007
Edited by:
Sidney A. Simon, Duke University Medical Center, Durham, USAReviewed by:
Geoffrey Schoenbaum, University of Maryland School of Medicine, Baltimore, USAAlbino J. Oliveira-Maia, Universidade do Porto, Porto, Portugal Duke University Medical Center, Durham, USA
Copyright: © 2007 Hilário, Clouse, Yin, Costa. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Rui M. Costa, Section on In Vivo Neural Function, Laboratory for Integrative Neuroscience, NIAAA, NIH, 5625 Fishers Lane, MSC 9411 Bethesda, MD 20852-9411. e-mail:Y29zdGFydWlAbWFpbC5uaWguZ292