1
Department of Psychiatry, School of Medicine, Yale University, New Haven, CT, USA
2
Brain Mapping Unit, Department of Psychiatry, Behavioural and Clinical Neurosciences Institute, School of Clinical Medicine, University of Cambridge, Cambridge, UK
Delusions are bizarre and distressing beliefs that characterize certain mental illnesses. They arise without clear reasons and are remarkably persistent. Recent models of delusions, drawing on a neuroscientific understanding of learning, focus on how delusions might emerge from abnormal experience. We believe that these models can be extended to help us understand why delusions persist. We consider prediction error, the mismatch between expectancy and experience, to be central. Surprising events demand a change in our expectancies. This involves making what we have learned labile, updating and binding the memory anew: a process of memory reconsolidation. We argue that, under the influence of excessive prediction error, delusional beliefs are repeatedly reconsolidated, strengthening them so that they persist, apparently impervious to contradiction.
In a recent and highly influential paper (Kapur, 2003
), Kapur related the aberrant experiences characteristic of early psychosis to inappropriate dopamine signaling by appealing to the concept of motivational salience (Berridge and Robinson, 1998
), a quality bestowed on objects or events by virtue of their behavioral relevance. Salient stimuli grab attention, drive action and influence goal-directed behavior; processes that are usurped by drug related stimuli in addiction (Berridge and Robinson, 1998
). Due to dopamine dysregulation, psychotic individuals attribute salience inappropriately to stimuli, thoughts and percepts. The world of an individual with emerging psychosis is strange and sinister, pregnant with new meaning. These experiences can crystallize into bizarre causal attributions and beliefs: delusions. Under the influence of antipsychotic medication, aberrant salience is dampened, patients are less perturbed by their experiences and symptom resolution begins (Kapur, 2003
). That is, the treatment alters the experiences such that the delusion can extinguish.
Kapur’s ideas relate to a number of previous suggestions that key symptoms of schizophrenia emerge from abnormal attention and learning processes (Corlett et al., 2007a
). This re-consideration of symptoms in the context of a contemporary model for the role of dopamine has had a big impact on cognitive neuroscientists studying psychosis and supportive evidence has already emerged (Corlett et al., 2007b
, Murray et al., 2008
). We believe that it can be taken further and embedded more deeply in the principles of formal animal learning theory. In doing so, we provide an account for features of delusions that are not readily explicable in terms of motivational salience alone: their persistence, which is often as striking as their bizarreness.
The salience of an event can be parsed into several components (Horvitz, 2002
), some of which are represented by dissociable neural substrates (El-Amamy and Holland, 2007
). Salience can relate to attention and motivation (Redgrave and Gurney, 2006
). Moreover, it is apparent that prediction error, a fundamental parameter in associative learning models, is a driving force in salience attribution, an observation that is reflected in a growing literature within computational modeling (Smith et al., 2006
) and empirical investigation (Menon et al., 2007
). Prediction error represents the mismatch between what we expect in a given situation and what we actually experience. By working to reduce this mismatch, we improve our understanding of the causal structure of the world (Dickinson, 2001
). That is, a prediction error is a signal that our understanding of, or belief about, the world must be updated. Furthermore, those stimuli that engender prediction errors become more salient and this will be reflected in greater allocation of attention when they next occur (Schultz and Dickinson, 2000
).
Prediction error theories of delusion formation suggest that under the influence of inappropriate prediction error signal, possibly as a consequence of dopamine dysregulation, events that are insignificant and merely coincident seem to demand attention, feel important and relate to each other in meaningful ways. Delusions ultimately arise as a means of explaining these odd experiences (Kapur, 2003
; Maher, 1974
). The insight relief gained by arriving at an explanatory scheme leads to strong consolidation of the scheme in memory (Figure 1
).
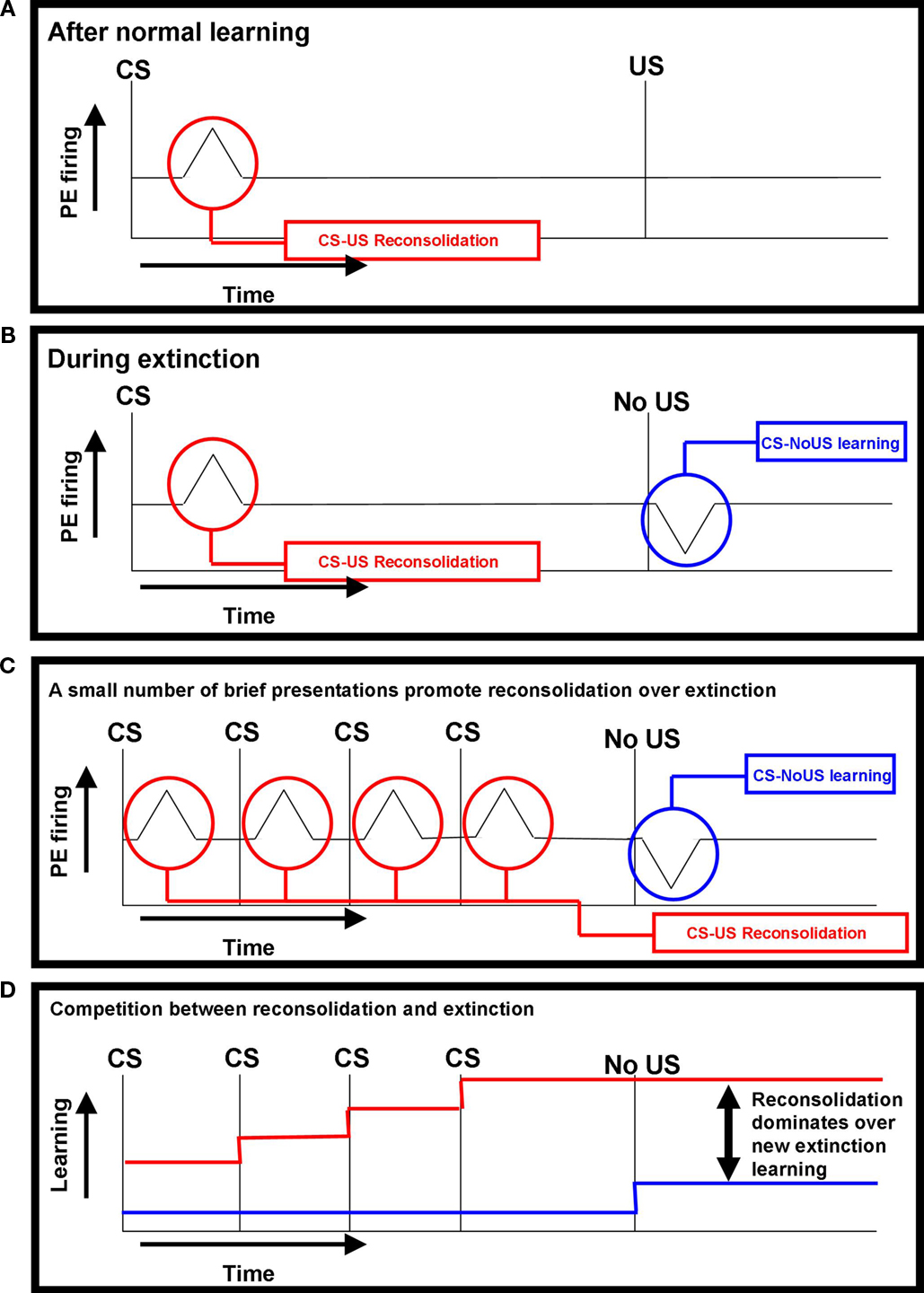
Figure 1. Prediction error responses during learning, extinction and reconsolidation. In these schematics, an upward deflection from the baseline reflects a positive prediction error and a downward deflection represents a negative prediction error (PE). (A) After normal learning – Dopamine neurons come to respond to the conditioned stimulus, the best predictor of the salient outcome (Schultz and Dickinson, 2000
). When the CS is presented, the organism is reminded of the salient event (represented in red). (B) During extinction – When the salient event does not occur, a negative prediction error promotes new extinction learning: the formation of a competing CS–NoUs association (represented in blue) (Bouton, 2000
; Eisenhardt and Menzel, 2007
). (C) A small number of brief presentations promote reconsolidation over extinction – According to the IRH, brief presentations of the conditioned stimulus serve as a reminder and promote reconsolidation of the CS–US association, which overrides any new extinction learning that occurs when the salient event does not occur (Eisenhardt and Menzel, 2007)
. (D) Competition between reconsolidation and extinction – This schematic shows the competition between representations of the CS–US association (strengthened by reconsolidation) and the CS–NoUS association (formed during extinction). With brief reminder presentations, reconsolidation dominates over extinction.
In support of this view, aberrant prediction error signals during learning in patients with first-episode psychosis have been confirmed experimentally (Corlett et al., 2007b
, Murray et al., 2008
). Furthermore, the magnitude of aberrant prediction error signal correlated with delusion severity across a group of patients with first-episode psychosis (Corlett et al., 2007b
). However, there are important characteristics of delusions that still demand explanation: notably their persistence. Normal associations can extinguish if they prove erroneous, normal beliefs can be challenged and modified. But delusions are noteworthy for the fact that they remain even in the absence of support and in the face of strong contradictory evidence. We believe that this striking clinical phenomenon can be explained within the same framework by considering key findings from the animal learning literature, a literature that has been formerly invoked to explain chronic relapse to drug abuse; extinction (Crombag et al., 2008
) and reconsolidation (Lee et al., 2005
).
If delusion formation may be explained in terms of associative learning then perhaps extinction may represent the process through which delusions are resolved (Miller, 1989
). Extinction involves a decline in responding to a stimulus that has previously been a consistent predictor of a salient outcome (Lovibond, 2004
).
Prediction error is also central to extinction (Pedreira et al., 2004
). A recent account (Redish et al., 2007
) suggests that negative prediction error (a reduction in baseline firing rate of prediction error coding neurons) leads the organism to categorize the extinction situation as different from the original, reinforced, situation and it now learns not expect the salient event in that situation. This learning focuses on contextual cues, allowing the animal to distinguish the newly non-reinforced context from the old, reinforced one. Extinction does not involve unlearning of the original association, but rather the formation a new association between the absence of reinforcement and the extinction situation (Pavlov, 1927
). Extinction experiences (the absence of expected reinforcement) invoke an inhibitory learning process which eventually overrides the original cue response in midbrain dopamine neurons (Pan et al., 2008
). Individuals with psychosis do not learn well from these absent but expected events (Waltz et al., 2007
), nor do they consolidate the learning that does occur (Holt et al., 2009
).
But there is more to delusion maintenance than persistence in the absence of supportive evidence: delusions persist even when there is evidence that directly contradicts them. When confronted with counterfactual evidence, deluded individuals do not simply disregard the information. Rather, they may make further erroneous extrapolations and even incorporate the contradictory information into their belief (Joseph, 1986
). So, while delusions are fixed, they are also elastic and may incorporate new information without shifting their fundamental perspective. We now go on to consider these characteristics with respect to the internal reinforcement hypothesis (IRH) of extinction learning and memory reconsolidation (Eisenhardt and Menzel, 2007
).
The traditional view of memory as a store that consolidates over time has been challenged by demonstrations that recall or reactivation of a memory briefly restores it into a labile state, rendering it susceptible to interference by amnestic agents. The process – through which memories are recalled, become labile, are combined with new information and finally consolidated once more – has been termed reconsolidation (Misanin et al., 1968
; Nader et al., 2000
).
This phenomenon suggests a more dynamic aspect to memory function, one perhaps geared toward dealing with incumbent information rather than merely retrospection. Furthermore, reconsolidation has been argued to result in the enhancement of salient memories (Tronson and Taylor, 2007
), a claim supported by empirical data. (Lee, 2008
; Tronson et al., 2006
). Since beliefs must overlap with memories (Eichenbaum and Bodkin, 2000
), what might we learn from the brain bases of memory formation and maintenance that will enhance our understanding of delusions?
The IRH contends that extinction and reconsolidation are two parallel processes and that an organism’s behavior is based upon the balance between the strength of their two memory representations following memory retrieval. This balance may be modulated by prediction error (Eisenhardt and Menzel, 2007
). During conditioning, an organism learns a relationship between a previously neutral stimulus (CS; e.g., a tone) and a reinforcer (food or electric shock, US). In a subsequent training session, an unreinforced exposure to the CS (presenting the tone in the absence of the food reward) might, besides inducing extinction learning, remind the animal about the reinforced situation (when food followed the tone). This kind of reminder might itself lead to new learning and new consolidation, i.e., reconsolidation (Eisenhardt and Menzel, 2007
). This reconsolidation process appears to be preferentially engaged in situations when updating occurs, that is, when additional information needs to be incorporated into memory (Morris et al., 2006
; Pedreira et al., 2004
).
The midbrain dopamine neuron response to conditioned stimuli (Schultz, 1998
) might drive reminder learning and reconsolidation of the CS–US relationship. In parallel, the surprising absence of a US engages extinction, through which a new CS–noUS association is formed and consolidated. Which of these representations controls behavior depends on the temporal characteristics of the learning situation (Pedreira et al., 2004
). If the CS is presented exactly as it was during training, but no reinforcer is presented, then the organisms’ expectancies are definitively disconfirmed and extinction dominates. However, if the CS is presented, say, more briefly than previously, the organism’s expectancies are neither definitively confirmed nor completely violated. In this case, reconsolidation of the CS–US association dominates, driven by the midbrain dopamine neuron response which reminds the organism of the reinforced situation (Eisenhardt and Menzel, 2007
).
We have conceptualized delusions as a network of associations formed by aberrant prediction error signals (Corlett et al., 2007a
). Given the features of the IRH, perhaps we might interpret the persistence of delusions, even in the absence of supportive evidence and the presence of contradictory evidence, by positing inappropriate activity in the midbrain reminder system. That is, aberrant prediction errors might re-evoke the representation of the delusion without definitively disconfirming it. This would drive preferential reconsolidation over and above any new extinction learning (Figure 2
). The net effect would be a strengthening of the delusion through reconsolidation rather than a weakening by extinction.
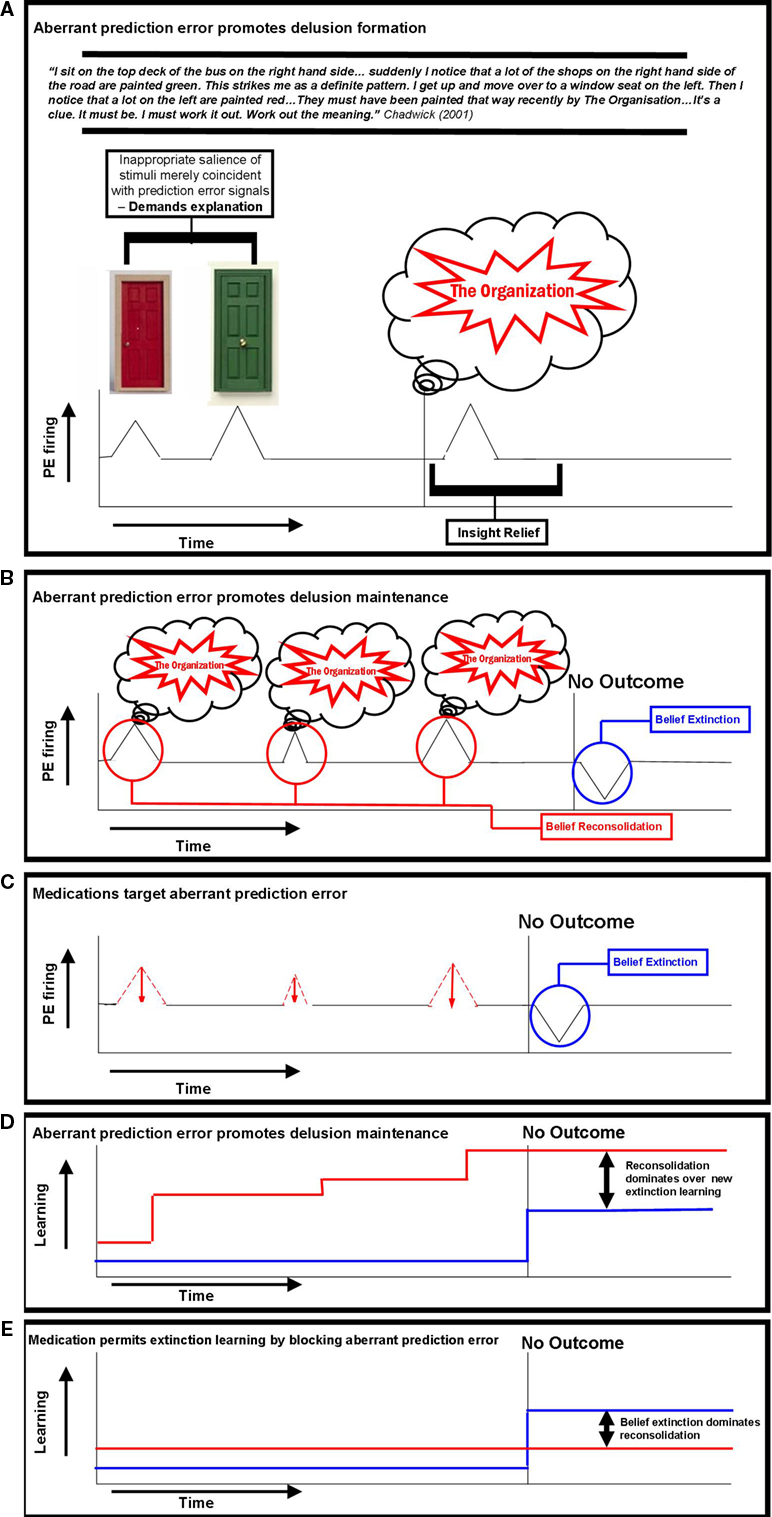
Figure 2. Prediction error firing during delusion formation, maintenance and resolution. (A) Aberrant prediction error promotes delusion formation – Aberrant, internally generated prediction error signals imbue mere coincidences (the colors of doors and the sides of the street) with significance which demands explanation (Chadwick, 2001)
. When an explanation is arrived at, the insight relief may engender dopamine firing which further stamps in the association between the odd experiences and the Organization (Miller, 1993)
. (B) Aberrant prediction error promotes delusion maintenance – Once the explanatory scheme has been developed, it is invoked any time an aberrant prediction error occurs. These reminders, serve to reconsolidate the belief that odd experiences are a result of the malevolent intervention of The Organization. This reconsolidation overcomes any extinction learning that may occur when no harm comes to the sufferer from The Organization or when others try to convince the sufferer that The Organization is not ministrating against them. (C) Medications target aberrant prediction error – Antipsychotic medications target aberrant prediction error signals, reminders do not occur and as such reconsolidation is blocked. New extinction learning is allowed to prevail and the individual recovers from their delusion. The mechanism for this targeting of prediction error induced reconsolidation may be at the level of neurotransmission in the synapse (e.g., Aripiprazole blocking phasic dopamine signaling (Hamamura et al., 2007
) or it may be intracellular, targeting the way in which the experience-explanation belief is stored epigenetically (Bredy and Barad, 2008
). (D) Aberrant prediction error promotes delusion maintenance – Prediction errors modulate the representations that govern behavior. Under aberrant prediction error, beliefs reconsolidate rather than extinguish. (E) Medication permits extinction learning by blocking aberrant prediction error – Under antipsychotic medication, aberrant prediction errors are blocked, allowing extinction learning to dominate and the resolution of the delusional belief.
This hypothesis derives support from the phenomenology of delusional beliefs; with medication, delusions resolve gradually (Stanton and David, 2000
), and, during that resolution patients describe an intermediate stage, a duality of belief and disbelief [“a part of me wants to dispel the delusions whilst a part of me is frightened and resists” (Ruocchio, 1991
)].
During this phase, patients may describe how much less occupied they are by their delusions. They become less salient (Kapur, 2004
). This transition would be aided by medications which prevent aberrant prediction error firing. The absence of aberrant error signals would gradually shift the balance in favor of the extinction of delusion-related material (Figure 2
). The duality of patients’ beliefs is consistent with our suggestion that multiple representations (in the simplest sense endorsing the belief and not endorsing it) compete for expression in behavior. With medication, the extinction trace (not endorsing the delusion) comes to win that competition. Some more chronically ill patients appear to show another kind of double-awareness, known as double-bookkeeping (Sass, 2004
). Here, patients appear to regain a modicum of insight: delusions persist but the patient does not consistently act upon them. This may be because chronically ill patients experience severe negative symptoms including avolition and emotional blunting, with the consequence that delusional beliefs cannot maintain their previously high degree of salience. Thus, they no longer feel the compulsion to act or ruminate upon the beliefs, though the memory of how salient the belief used to be is sufficient to maintain it (Sass, 2004
). We argue, after Alberini (2007
), that this salience is sufficient to ensure that the belief is not extinguished.
With medication, the memory trace mediating belief in the delusion is not erased. It is overshadowed by extinction learning. Hence, if medications are ceased, the delusion returns (Chadwick, 2001
). Moreover, this model could explain why the contents of patients’ delusions persist across consecutive psychotic episodes (Sinha and Chaturvedi, 1989
): since the association persists, albeit in attenuated form, recurrence of the neurochemical disturbance that provoked it in the first place will now allow it to re-emerge. State dependency may provide further explanatory insight into the re-emergence of delusional beliefs when antipsychotic drugs are stopped. That is, when the neurochemical sequelae in which a memory was formed are reinstated, the probability and richness with which that memory is recalled are enhanced (Goodwin et al., 1969
). While most experimental investigations of state-dependency have focused on administration of compounds that alter cognitive and mood states, the endogenous altered states of consciousness that attend mental illness have been suggested to engender state-dependent recall and therefore alter prognosis (Reus et al., 1979
). Finally, extinguished behaviors and delusions spontaneously recover. Even after extinction, the passage of time and the presence of interoceptive or exteroceptive contextual cues can cause the old behavior or belief to re-occur (Bouton, 2000
; see Box 1
).
Box 1. Reconsolidation based therapies for delusions
Classical demonstrations of reconsolidation were based on observations of amnesia for associations that animals had been reminded about just prior to electroconvulsive shock (ECS, Misanin et al., 1968
). ECS has been administered therapeutically in schizophrenia (electroconvulsive therapy or ECT). Whilst effective in first-episode patients, it is less so in the more chronically ill (Fink and Sackeim, 1996
), perhaps because the intervention is applied after a delusion has become strongly consolidated and is less susceptible to disruption.
ECT is usually administered under general anesthesia making its targeted application to a pathogenic memory problematic. However, ECT has been applied, without anesthesia, with a reminder treatment, to patients with fixed intrusive beliefs (Rubin, 1976
). Patients had their attention directed toward their most disturbing beliefs, and were instantly given ECT. All patients improved dramatically for periods of up to 10 years at the time of publication.
Of course, this approach would no longer be considered ethically acceptable. However, the findings lend some support to the notion that the reconsolidation/extinction balance is important for delusion maintenance. A less invasive intervention such as transcranial magnetic stimulation might be employed to modulate reactivated memories, an approach that has also been indicated for motor memory rehabilitation following stroke (Bernad and Doyon, 2008
).
Alternatively, it may be possible to target specific a reactivated memory pharmacologically, reactivating a memory and administering a drug to impair its reconsolidation whilst its representation is still labile. The impact of pharmacological manipulations on memory reconsolidation depends on the nature of what is recalled and how the memory is re-evoked (Lee et al., 2006
): The effects of a glutamatergic agonist and an antagonist on reconsolidation were modulated by the way in which memories were reactivated. Following a long reactivation session [favoring extinction], blocking NMDA receptors blocked extinction and enhancing their function promoted extinction. However, following a brief reactivation session [favoring reconsolidation], the opposite was true: NMDA blockade enhanced and an NMDA agonist impaired extinction.
D-cycloserine (DCS) an NMDA receptor partial agonist has been trialed as an adjunct to traditional dopaminergic antipsychotics, but only modest improvements were observed, and in some patients, DCS aggravated positive symptoms (van Berckel et al., 1999
). This inconsistency may have implications for the therapeutic targeting of delusions through reconsolidation blockade. When attempting to block reconsolidation, a brief re-exposure session should be used to render the belief labile. If the reminder session is too extensive, the amnestic agent will impair new extinction learning that is invoked and will lead to maintenance of the maladaptive memory underpinning the belief.
An alternative therapeutic approach might involve enhancing extinction of the delusional memory. However, this would have a limited effect as extinction learning is context dependent (Bouton, 2000
). Following the addiction example, an addict will extinguish drug seeking behaviors in a rehabilitation treatment center but they may relapse outside this context (Taylor et al., 2009
). Training in multiple different extinction contexts does not seem to improve this (Bouton et al., 2006
). The ideal therapeutic approach would involve a combined approach which impaired reconsolidation but also encouraged new extinction learning.
Despite the observation that directly challenging a delusion may lead to a strengthening of the belief (Milton et al., 1978
; Simpson and Done, 2002
) cognitive therapies for delusions have been applied with reported success (Chadwick and Lowe, 1990
, 1994
; Rector and Beck, 2001
; Turkington and Dudley, 2004
). It is telling, with respect to the current model, that certain principles have emerged, (Chadwick and Lowe, 1994
) notably:
1. Modification should begin with the least strongly held beliefs.
2. Patients are encouraged to consider the alternative to the delusional belief rather than to try to accept this alternative immediately.
3. Evidence for the belief, rather than the belief itself, should be challenged.
4. The patient should be encouraged to voice the arguments against his or her beliefs.
In the context of our model, principles 1 and 2 might attenuate the excessive reactivation and reconsolidation of the delusion while principles 3 and 4 would tip the balance in favor of its extinction.
Given our preference for a combined approach that enhances extinction and impairs reconsolidation, we note with interest the recent demonstration of significant weakening of a fear memory in rats by inducing extinction following reactivation (Monfils et al., 2009
). These data could perhaps be mapped onto the suggested parameters for CBT targeting delusions. That is, the delusional memory could be re-engaged and, whilst it is labile, the data that are considered supportive could be challenged resulting in its extinction.
Applying the IRH to delusions suggests that reminding patients of their delusions in a therapeutic setting, by trying to disconfirm their beliefs might, paradoxically, lead to an increase in the conviction of the belief – if that reminder treatment occurred whilst patients were experiencing inappropriate prediction error firing (see Figure 2
). There is some evidence that this may indeed be the case: confronting deluded patients with contradictory evidence does indeed strengthen delusional beliefs (Milton et al., 1978
). Indeed, as we outline above, they may even incorporate challenging information into their belief (Simpson and Done, 2002
). Aberrant prediction error driven reconsolidation might account for the voracity with which patients incorporate ostensibly unrelated information into their delusional scheme – since aberrant prediction error signals remind the patient of their delusion and render its representation highly associable, the subsequent reconsolidation process incorporates the new information, maintaining and strengthening the delusion rather than extinguishing it (Figure 2
).
Memory reconsolidation may underpin the transition from a salient episodic experience to a habitual belief about the world (Alberini, 2007
). The neurobiological systems responsible for coding the salience of an event are intimately related to those associated with learning and memory, such that modulations of those systems with pharmacological intervention can engender inappropriate salience attribution (Corlett et al., 2006
). Such interventions can be substituted for training trials in experimental animals, and can strengthen the memory trace to the same extent as it is strengthened by actual repeated experience (Alberini, 2007
). That is, pharmacological interventions which enhance the salience of certain associations will reinforce or strengthen those associations as if they had been repeatedly experienced. The effect of pharmacological interventions on the processing of reactivated memories depends on how the memory is reactivated (Lee et al., 2006
; see Box 1
). As such, novel therapeutic approaches may well involve bypassing the receptor level pharmacology of synaptic plasticity and instead targeting alternative mechanisms of memory maintenance (see Box 2
). It is possible that reactivation of a memory trace for a salient event increases the stabilization of that trace. The most salient memories would be reactivated most frequently and would therefore engender the most reconsolidation based stabilization, increasing their fixity. The net effect would be that a salient experience more rapidly and profoundly updates knowledge and perhaps is more likely to become enshrined in a belief.
Box 2. The biology of memory maintenance
Changes in synaptic plasticity are widely believed to underpin learning and memory. However, the role of synaptic changes in memory maintenance was ascertained from preparations assuming Hebbian rules; put simply, neurons that fire together wire together. Simple Hebbian rules deal well with learning about contiguous events but they cannot explain the role of prediction error in learning (McLaren and Dickinson, 1990
), nor can they explain the memory reconsolidation phenomena critical to the current hypothesis (Arshavsky, 2006
). Furthermore, changes in synaptic plasticity may not be the mechanism for memory (or belief) maintenance across the lifespan – the components of a synapse that confer its strength and excitability are frequently completely recycled (Arshavsky, 2006
). A more permanent memory storage mechanism is necessary to ensure the accurate restoration of synaptic proteins following such recycling events (Arshavsky, 2006
). One relatively stable and information rich candidate is DNA. Changes in how the genome can be expressed could mediate long-term retention of information by an organism (Crick, 1984
; Holliday, 1999
). Such epigenetic changes represent a mechanism through which an organism can adapt to its environment and maintain that adaptation throughout its lifetime, processes that are likely disturbed in a number of mental illnesses (Bredy, 2007
).
Much of the genome may be silenced in a particular cell, whilst other portions will be highly expressed; a process mediated in part through changes in proteins called histones which combine with DNA to form chromatin. Histone proteins can be modified in a variety of ways which impact upon how readily the DNA with which they are associated can be expressed as protein. Histone modification makes an important contribution to learning related gene expression. One particular histone modification, changes in acetylation, is mediated by an enzyme called histone deacetylase (HDAC). Inhibition of HDAC enhances long-term memory (Levenson et al., 2004
).
Barad and colleagues recently demonstrated that the HDAC inhibitor valproic acid modulate extinction and reconsolidation of conditioned fear. They hypothesize that HDAC inhibitors: “overcome the reconsolidation-like incubation of fear by spaced CS-presentation to allow effective extinction to take place” (Bredy and Barad, 2008
). This is exactly the desired effect for an antipsychotic drug within the context of the IRH. Whilst valproate, has not been trialed as a stand-alone antipsychotic, it does benefit patients with schizophrenia as an adjunct to antipsychotics (Basan and Leucht, 2004
), future research should explore the role of HDAC inhibition as an antipsychotic mechanism, particularly its potential application in combination with reminder treatments as a therapeutic approach to delusions.
A belief, once formed, can be conceived as an habitual way of looking at the world and of interpreting incoming sensory information (see Box 3
). It is, in other terms, a schema. Perhaps reconsolidation is a crucial component of building such schemata. The transition from salient experience to belief habit in psychotic states occurs rapidly due to excessive prediction error firing (Corlett et al., 2007a
) inducing repeated recall of delusion-related material and reconsolidation of that material, updated with new information, forming a “schema-like” representation through which incumbent information is then interpreted. To quote Jaspers; “the trail is blazed and the preparedness for the significant experience then permeates almost all perceived contents. The now dominant delusion motivates the apperceptive schema for all future percepts.”(Jaspers, 1963
) – see Box 1
.
Box 3. Perception, learning and belief
We have recently speculated on the interaction between prior beliefs and current sensory experiences (Corlett, ,2009
Fletcher and Frith, 2009
) and how delusions may arise from an imbalance in these interactions. We argue that, in the earliest stages of psychosis, prior to delusion formation, noise in the nervous system leads to excessive free energy or prediction error (Friston, 2005
). Subjectively, this experience is of a mutated world, one which feels strange, perhaps sinister and beset by new significance. Priors are updated in order to minimize free energy and this must necessarily lead to the formation of new beliefs and expectancies. The new model of the world is built to accommodate or explain away these strange experiences. With continued aberrant prediction error those new priors are strengthened further. In the terms of the present model we suggest that memory reconsolidation provides a potential mechanism for this updating process. With sufficient reconsolidation, the priors become so strong as to be resistant to contradictory evidence.
Pavlov believed that one could equate his conditioned reflexes with Helmholtz’ learned perceptual expectancies (Barlow, 1990
; Helmholtz, 1871/1971
), an assertion supported by empirical evidence (Davies et al., 1982
). Recently, computational neuroscientists have also come to appreciate the overlap between learning and perception, emphasizing the consilience between formal theories of conditioning, Bayesian accounts of learning and signal detection theories of perceptual decision making (Dayan and Daw, 2008
). According to such combined models, organisms must learn the rules of the game they are playing and, in the context of those rules, they must discern meaningful events and contingencies from meaningless noise. Organisms achieve this by maintaining a robust but flexible set of expectancies about the world that tune their sensitivity and responses biases.
Estes drew similar parallels between learning, memory and signal detection theory in his stimulus sampling theory of memory (Estes, 1997
). Here, after a memory trace is encoded, the occurrence of the same or similar events reactivates the trace, necessitating some re-encoding or reconsolidation process, arguing that this process is subject to random error, due to the physiological noise that permeates the nervous system and provides the analytical basis for signal detection theory (Green and Swets, 1966
). Estes hypothesizes that random error (or physiological noise) at re-encoding may account for the loss of detailed information, the incorporation of spurious details and the shift toward familiar information that occur with successive reproductions of information from memory (Bartlett, 1932
). Unlike Estes’ model, in our account prediction error signal (both appropriate and inappropriate) drives memory reactivation and also impacts on how that memory is re-encoded.
We argue that delusions occur in the context of a noisy nervous system that is attempting to form and maintain a robust set of priors. Such excessive noise would engender more cycles of reactivation and subsequent reconsolidation, leading to a bizarre and maladaptive set of expectancies about the world, expectancies strong enough to vitiate normal sensory and cognitive experience. These learned expectancies have much in common with Jaspers’ apperceptive schema’ – beliefs so strong that they are impervious to sensation – which he believed were the basis for the maintenance of delusional beliefs (Jaspers, 1963
).
We have argued that aberrant prediction error signals may be important not only for delusion formation (Corlett et al., 2007a
) but also for delusion maintenance since they drive the retrieval and reconsolidation based strengthening (Lee, 2008
) of delusional beliefs, even in situations when extinction learning ought to dominate (Eisenhardt and Menzel, 2007
; Pedreira et al., 2004
). Given the proposed function of reconsolidation, in driving automaticity of behavior (Stickgold and Walker, 2007
) we argue that in an aberrant prediction error system, delusional beliefs rapidly become inflexible habits. Taking this translational approach will enhance our understanding of psychotic symptoms and may move us closer to the consilience between the biology and phenomenology of delusions that Kapur sought in his article (Kapur, 2003
).
The authors have no competing financial interests.
The authors would like to thank Trevor Robbins, Jonathan Lee, Amy Milton, David Shanks and Graham Murray who gave insightful criticism of the hypothesis. Philip Corlett is the University of Cambridge Parke Davis Exchange Fellow in Biomedical Sciences and a NARSAD Young Investigator. Jane Taylor is supported by NARSAD and NIDA. John Krystal is supported by NIMH, NIAAA and NARSAD, he reports the following: Consulting: AstraZeneca Pharmaceuticals, LP, Cypress Bioscience, Inc., HoustonPharma, Schering-Plough Research Institute, Shire Pharmaceuticals, and Pfizer Pharmaceuticals; Advisory Boards: Bristol-Myers Squibb, Eli Lilly and Co., Forest Laboratories, GlaxoSmithKline, Lohocla Research Corporation, Merz Pharmaceuticals, Takeda Industries, and Transcept Pharmaceuticals, Inc.; Exercisable Warrant Options: Tetragenex Pharmaceuticals Inc.; Research Support: Janssen Research Foundation; Pending Patents: glutamatergic agents for psychiatric disorders (depression, OCD), antidepressant effects of oral ketamine, and oral ketamine for depression. All other authors report no biomedical financial interests or potential conflicts of interest. Paul Fletcher is supported by the Bernard Wolfe Health Neuroscience Fund and the Wellcome Trust.
Corlett, P. R., Honey, G. D., Aitken, M. R., Dickinson, A., Shanks, D. R., Absalom, A. R., Lee, M., Pomarol-Clotet, E., Murray, G. K., McKenna, P. J., Robbins, T. W., Bullmore, E. T., and Fletcher, P. C. (2006). Frontal responses during learning predict vulnerability to the psychotogenic effects of ketamine: linking cognition, brain activity, and psychosis. Arch. Gen. Psychiatry 63, 611–621.
van Berckel, B. N., Evenblij, C. N., van Loon, B. J., Maas, M. F., van der Geld, M. A., Wynne, H. J., van Ree, J. M., and Kahn, R. S. (1999). D-cycloserine increases positive symptoms in chronic schizophrenic patients when administered in addition to antipsychotics: a double-blind, parallel, placebo-controlled study. Neuropsychopharmacology 21, 203–210.