1
University of California, Los Angeles, Los Angeles, CA, USA
2
Harvard School of Public Health, Boston, MA, USA
A review of the human developmental neuroimaging literature that investigates outcomes following exposure to psychosocial adversity is presented with a focus on two subcortical structures – the hippocampus and the amygdala. Throughout this review, we discuss how a consideration of developmental timing of adverse experiences and age at measurement might provide insight into the seemingly discrepant findings across studies. We use findings from animal studies to suggest some mechanisms through which timing of experiences may result in differences across time and studies. The literature suggests that early life may be a time of heightened susceptibility to environmental stressors, but that expression of these effects will vary by age at measurement.
Early adverse social environments such as abuse and neglect have been associated with a wide range of negative outcomes, including a dramatically increased risk for a variety of mental disorders (Breslau et al., 1999
; Brewin et al., 2000
). These often include, but are not limited to, anxiety, depression, ADHD, substance use disorders, and tobacco dependence. The link between childhood trauma and adult risk for mental health disorders has been described in a variety of ways but fundamentally, this link is biological in nature. These negative social environments become biologically embedded as changes in neural structure and function and, ultimately, the behaviors that lead to mental illness. Although initial susceptibilities for exposure to adversity may contribute to this association, pressures from the environment can alter neural development leading to negative outcomes. Describing the mechanisms by which adverse experiences during childhood lead to changes in neural development is an important step for understanding both brain development and ultimately for developing tools for clinical intervention.
In this review we will attempt to link the timing of negative childhood psychosocial stress exposure to differences in neural structure and function during childhood and adolescence. We restrict this review to empirical articles that address psychosocial trauma of abuse and neglect. A paper by Raizada & Kishiyama (this issue) reviews the literature on cognitive deprivation (e.g., low socio-economic status of the family), and therefore, this manuscript will not focus on early adversity of that type. We further limit our review to the neural development of two subcortical structures: the hippocampus and amygdala. We focus on these two regions because, based on a large adult human and non-human animal literature, we would expect significant environmental influence on these structures. The hippocampus and amygdala are important for socio-emotional functioning throughout development and are closely linked with the activity of the hypothalamic pituitary adrenocortical (HPA) axis, a significant neuroendocrine mediator of stressful events for humans. We purposefully narrow our focus to provide a richer discussion of the amygdala and hippocampus and how developmental timing interacts with environmental influences. Because the systemic output of the HPA axis, glucocorticoids (cortisol in humans), can pass through the blood-brain barrier, the HPA axis is one of the major pathways through which the effects of stress can shape brain development. The amygdala and hippocampus are rich with receptors for cortisol and are therefore major targets of the HPA axis. Thus, we see narrowing our review to the amygdala and hippocampus as one reasonable way to limit the scope of the neural effects of adversity that we examine here. We will describe some specific examples of when amygdala and hippocampal development are disrupted by negative psychosocial environments. By describing these associations, we hope to distill potential mechanisms by which exposure to adversity could become biologically embedded resulting in increased susceptibility to mental illness. Finally we describe potential future directions for research. Throughout the review, we will be making the argument that the effects of adversity will vary as a function of developmental timing, such that regionally defined effects will depend on the age at which exposures occurred and when neural outcomes were measured.
This focus on timing is consistent with the notion of sensitive periods identified in other developmental processes, such as vision or language, where developmental timing modifies the environment’s impact on neural development. A fundamental precept of developmental studies is that the timing of a particular exposure matters for the expression of a phenotype. Not all neural regions follow the same developmental trajectory (Giedd et al., 1996
; Bourgeois, 1997
; Huttenlocher and Dabholkar, 1997
). For example, primary sensory cortex such as V1 (visual cortex) appears to undergo important structural changes in the first year leading to life-long differences in visual perception, whereas other cortical regions (e.g., prefrontal cortex) continue to show structural development into adulthood. In the case of binocular vision or phoneme perception, timing of environmental exposure and timing of measurement is critical for observation of normal perceptual development (Hubel and Wiesel, 1970
; Kuhl, 2004
). These are powerful examples of the concept of a developmental sensitive period, which are periods of life when a system exhibits increased plasticity and therefore, susceptibility, to environmental influences. Although the effects of sensitive periods are observed in behavior, they are properties of neural circuits (see Knudsen, 2004
). Central to this concept is the notion that the process of development itself may increase the system’s likelihood of being shaped by the environment (Casey et al., 2000
). These periods often coincide with rapid development of a brain system, and therefore, individual neural systems will have their own sensitive periods (Lupien et al., 2009
). Once environmental exposure occurs, it modifies the architecture of the circuit in such a way that certain patterns of future activity are preferred (Knudsen, 2004
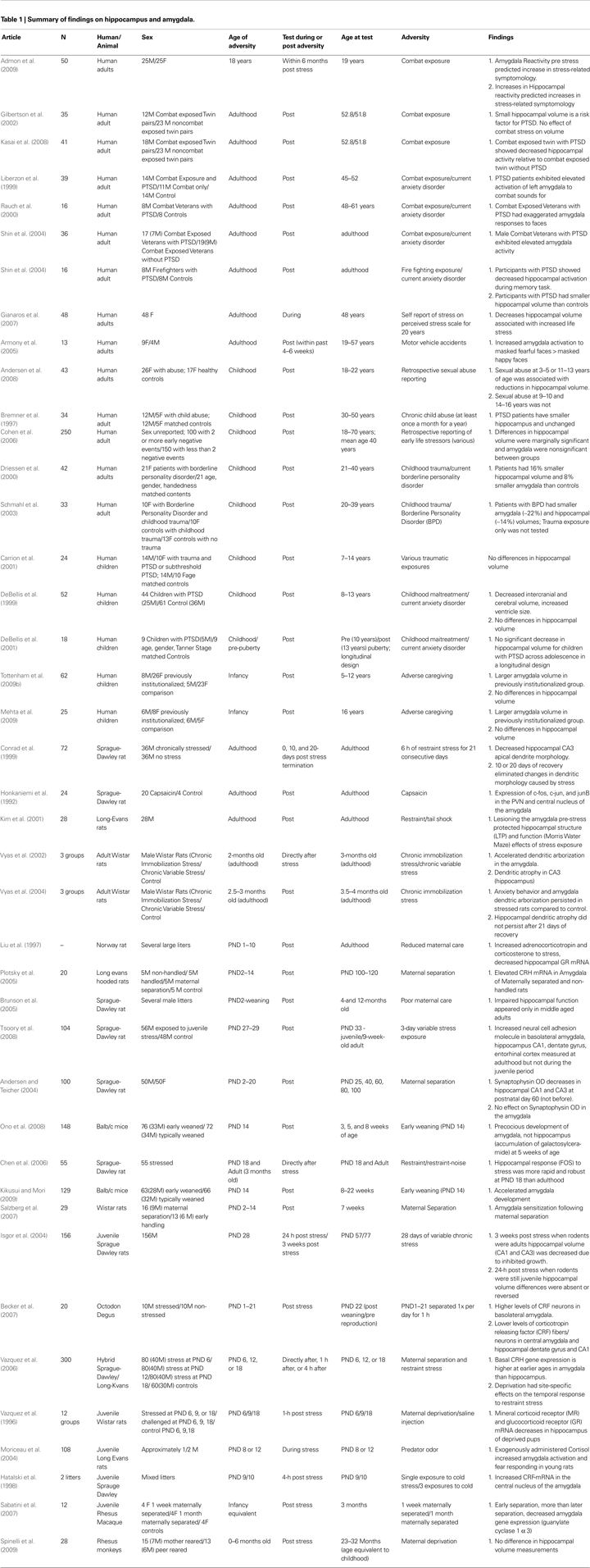
). Therefore, knowing the developmental timing of environmental exposures is critical when evaluating its effects. Beyond the timing of exposure, the timing of measurement can influence how we interpret the effects of environmental exposures, like adversity. Because compensatory neural mechanisms, which were not present during the stressor, may emerge once the adverse experience has terminated, timing should be well-characterized to disambiguate the effects of stress versus the effects of recovery. Moreover, as will be discussed below, the effects of an environmental exposure may not emerge for some time after the termination of the exposure. In the case of adversity we predict that the timing of exposure (i.e., age at which exposure occurred) and the timing of measurement (time since stressor) will matter for neural structure and function. Therefore, we will present human developmental neuroimaging studies to support this hypothesis and use findings from the animal literature to provide some description of mechanisms for developmental timing (literature summarized in Table 1
).
Psychosocial Stress Exposure: HPA Axis Function
Psychosocial stress can adversely impact brain development, and the literature on stress suggests that these changes occur largely through the HPA axis (reviewed in Loman and Gunnar, 2010
). We will begin by briefly reviewing the structure of the HPA axis. A stressor sufficiently strong will elicit a full stress response (Kemeny, 2009
) which includes activation of both the sympathetic nervous system and activation of the HPA axis. The latter, which produces a longer-term response to a stressor than the former (hours rather than seconds to minutes), begins with signals from the amygdala (reviewed in Herman and Cullinan, 1997
), which lead to peripheral (systemic) glucocorticoid increases via hypothalamus, pituitary, and adrenal gland activity and increases in corticotropin-releasing hormone in the brain (CRH; including in the amygdala; Makino et al., 1994a
). Eventually peripheral glucocorticoids make their way to the brain. Glucocorticoids easily pass through the blood-brain barrier (Zarrow et al., 1970
), and because the amygdala and hippocampus have a high density of receptors for unbound glucocorticoids, they are regions that are highly susceptible to the products of the HPA axis. When the stressor is removed and high circulating glucocorticoids are no longer necessary, glucocorticoids suppress HPA axis activation by occupying receptors in the hippocampus eventually inhibiting activity of the HPA axis (van Haarst et al., 1997
). In contrast, glucocorticoid occupation of amygdala receptors can have a facilitating effect on the activity of the HPA axis, often increasing CRH production within the amygdala. Therefore, circulating glucocorticoids can have contrasting effects in the amygdala and hippocampus, and these two structures can play contrasting roles in the activity of the HPA axis.
As discussed in Gunnar and Quevedo (2007)
, stress is a psychological condition in which the individual experiences challenges to their well-being that overwhelm their resources for coping. Although this construct can be studied behaviorally and biologically (Dantzer, 1991
), behavioral distress does not always mirror physiological stress reactions. For example, infants’ physiological (cortisol) response to inoculation is not perfectly correlated with amount or intensity of crying (Gunnar et al., 1989
). However, elevated HPA axis activity can provide one biological index of stress, and importantly, one that can shape brain development.
There is much evidence that children who are exposed to early adverse experiences, such as poverty (Lupien et al., 2000
), abuse (Tarullo and Gunnar, 2006
), or orphanage rearing (Gunnar et al., 2001
; Dobrova-Krol et al., 2008
), have increased stress reactivity and corresponding increased GR exposure. Adverse caregiving is a type of stress also used in animal models. These models provide the opportunity, usually not available in humans, for examination of stress effects at the cellular level. A variety of stressors (Bonaz and Rivest, 1998
), as well as administration of high levels of glucocorticoids (Makino et al., 1994a
), result in increased mRNA for CRH receptors hypothalamus and amygdala. This upregulation can lead to an increased fear response and/or a lowered threshold for the fear response to occur. High levels of glucocorticoids will also result in CRH mRNA level increases in the amygdala (Makino et al., 1994a
), thereby potentiating the fear response. The amygdala has been understood to be functionally dormant in the rat neonatal period. However, significant stressors and/or GR administration can precociously activate the amygdala (Moriceau et al., 2004
), indicating that the amygdala is biologically prepared to be activated early in life under the right conditions. In addition, CRH receptors are maximally expressed in the amygdala and hippocampus early in development (reviewed in Baram and Hatalski, 1998
), a finding that may provide insight into why young animals are especially vulnerable to adversity. Moreover, high elevations of glucocorticoids can downregulate hippocampal receptors that normally aid in the negative feedback to the HPA axis (van Haarst et al., 1997
), thereby resulting in a dysregulated axis. The process of glucocorticoids increasing hippocampal receptors occurs throughout development, including early in life (Vazquez, 1998
). We will discuss in greater detail below how the products of the HPA axis specifically affects both the hippocampus and the amygdala.
The hippocampus has been implicated in learning and memory in adults and children. In adults, when the hippocampus is removed surgically, encoding of long term memories is disrupted resulting in anterograde amnesia: new memories cannot be formed (Markowitsch and Pritzel, 1985
). Initial findings such as these in neuropsychological research were the result of bilateral hippocampal resection as a treatment for epilepsy (including the famously described patient HM; Zola-Morgan et al., 1982
). When the hippocampus is lesioned in children, a similar specific deficit in episodic memory is observed (Vargha-Khadem et al., 1997
; Brizzolara et al., 2003
; Temple and Richardson, 2004
).
As described above, in the context of stress exposure, the hippocampus has another important role; it provides a negative feedback mechanism, which modifies the HPA axis response (as reviewed in Kim and Yoon, 1998
). This negative feedback mechanism is accomplished via activation of glucocorticoid (GR) and mineralocorticoid receptors (MR) by circulating levels of glucocorticoids. Within the brain, MR have a 90% higher affinity for glucocorticoids than GR. At basal (non-stress) levels, the majority of MR are occupied by circulating levels of glucocorticoids. During a typical HPA axis stress response, increased availability of glucocorticoids leads to GR occupation, activating a negative feedback loop and decreasing the HPA axis response. Early in life, the MR/GR ratio is lower than later in life, with MR mRNA increasing with age and GR mRNA decreasing with age (Vazquez et al., 1996
). The immature ratio of MR to GR may result in unique hippocampal vulnerability to stress early in life. As has been demonstrated in older animals, chronic occupation of GR may impair the hippocampally-mediated negative feedback process resulting in extended HPA axis activation following stressful events and dysregulation of the HPA axis. Down-regulation of hippocampal MR has been identified in very young rats as well (reviewed in Vazquez, 1998
), which may increase the likelihood of GR occupation for young animals.
Hippocampal Development
In developmental neuroimaging studies, there is some evidence for age-related change (increases) in recruitment of the hippocampus during long-term memory tasks across late childhood (Paz-Alonso et al., 2008
). However, other studies observed unchanging hippocampal recruitment across 8–24 years of age during scene memory encoding (Ofen et al., 2007
), supporting the idea that hippocampal function is intact at least by childhood. Structural studies using magnetic resonance imaging (MRI) have revealed developmental differences in the volume of the hippocampus from birth through young adulthood. From birth to year 2, the hippocampus shows relatively little growth (Knickmeyer et al., 2008
). More substantial structural changes tend to be observed later in development. In an initial cross-sectional study of children it was determined that the hippocampus showed protracted volumetric growth across childhood for girls but not boys (Giedd et al., 1996
). In subsequent studies, however, this gender difference was not replicated, and instead, a longitudinal analysis of hippocampal growth between the ages of 4–25 years has shown continued developmental change into adulthood, where that the anterior hippocampus decreases in volume across childhood while the posterior hippocampus increased in volume (Gogtay et al., 2006
).
As will be discussed further below, the animal literature suggests that hippocampal development lags slightly behind amygdala development. For example, whereas learning to pair a cue with a shock (cued fear conditioning – an amygdala-dependent function) is present by postnatal day 18 in the rat, the same aged rats are unable to pair a context to a shock (contextual fear conditioning – a hippocampus dependent function; Rudy, 1993
). These findings have been interpreted as occurring because of the relative immaturity of the hippocampus (Cotman et al., 1973
; Wilson, 1984
; Rudy and Morledge, 1994
) Similarly, a recent non-human primate finding obtained with longitudinal structural MRI showed that, although both structures showed early development during an age-equivalent to early childhood, hippocampal development lagged behind the amygdala as indexed by age at which the slope of change leveled off (Payne et al., 2010
).
Despite the protracted development of the hippocampus, behavioral evidence suggests that some aspects of hippocampal function are present early in life. Across the first year of life, memory becomes increasingly context-independent, which is evidence of increased relational memory and involvement of the hippocampus (Robinson and Pascalis, 2004
). Extending putative hippocampal development into early childhood, Sluzenski and colleagues demonstrated that 4-year olds were not able to perform a relational memory task binding together pictures of animals and backgrounds whereas 6-year olds could and showed adult-like performance (Sluzenski et al., 2006
). However, when using more familiar objects (faces and scenes), 9-month-old infants showed evidence of intact relational memory (Richmond and Nelson, 2009
). Taken together, these findings are consistent with the notion that the basic relational function of the hippocampus is present early in life, although the hippocampus and its connections continue to show developmental change into adulthood.
Hippocampus and Stress
Evidence from adult rodent models shows that stress exposure alters hippocampal volume and function in adulthood (McEwen, 1999
, 2007
). At baseline levels, glucocorticoids appear to aid memory formation by enhancing hippocampal excitability (Diamond et al., 1992
; Pavlides et al., 1993
, 1994
). However, during stress-induced HPA axis activation, hippocampal function is disrupted (Diamond et al., 1992
; Pavlides et al., 1993
), and prolonged exposure to glucocorticoids from chronic stress is harmful to the hippocampus, resulting in reduced dendritic spines and eventually apoptosis of hippocampal neurons (Sapolsky, 1996
; Kim and Yoon, 1998
). Rodents exposed to early stress also demonstrate dendritic atrophy in hippocampal cells and decreased amplitude of long term potentiation in the CA3 area of the hippocampus, leading to deficits in memory formation (Brunson et al., 2005
).
Most animal studies of stress exposure examine pre- and early postnatal stress exposure or chronic stress exposure in the mature animal. To keep the parallels to human psychosocial trauma exposure as consistent as possible, we will not review the effects of prenatal stress here. Poor or absent maternal care has lasting effects on the hippocampus. Early stress exposure of this variety is associated with decreased hippocampal volume and function and dysregulated HPA function in adulthood (Liu et al., 1997
; for review, see Sanchez et al., 2001
). Few rodent studies have examined the effect of stress experienced or measured during the child/adolescent equivalent of the juvenile phase. In one study of juvenile rodents, early exposure to chronic stress did not result in differences in the hippocampus 24-h post stress but did 3-weeks post exposure when these rodents had reached adulthood (Isgor et al., 2004
), suggesting that hippocampal effects may not be readily apparent until animals are mature. In studies of mature rodents, chronic stress exposure is followed by hippocampal volume reductions 24-h post stress exposure. Adult rodent stress exposure confers risk for short-term differences in hippocampal structure, which within ten stress-free days, reverse (Conrad et al., 1999
). Intervention during the adolescent phase of development may modify the behavioral effects of juvenile stress exposures in rodents (Francis et al., 2002
), but does not alter the hippocampal pathology in adulthood associated with exposure to stress during the juvenile phase. Thus, it appears that stress-related reductions in hippocampal volume and changes in HPA axis activation incurred as a result of juvenile stress exposure are more permanent than those incurred during adulthood (Seckl and Meaney, 2004
). This may be the result of a greater number of stress hormone receptors early in life (reviewed in Baram and Hatalski, 1998
) and/or decreased MR (higher affinity)/GR(lower affinity) ratios early in life (Vazquez et al., 1996
).
In human adults, stress-related pathologies, such as major depressive disorder and post-traumatic stress disorder (PTSD), correlate with decreased hippocampal volume (Sheline et al., 2003
; Campbell et al., 2004
; Geuze et al., 2005
; Kitayama et al., 2005
; Smith, 2005
) and altered activity (Bremner, 2006
; often decreased activity Etkin and Wager, 2007
, but increased activity has also been identified – Shin et al., 2004
). Even in healthy middle-aged adults, self-reported stress over 12 years in a longitudinal study was associated with decreases in hippocampal grey matter volume acquired at year 13 in women aged 54 years (Gianaros et al., 2007
). In aging populations (60–90 years), high baseline and chronically increasing exposure to glucocorticoids measured yearly across a 6-year period predicted decreases in hippocampal volume relative to moderate baseline and decreasing glucocorticoids across the same time period (Lupien et al., 2005
). Patients taking high doses of corticosteroids for long periods demonstrate hippocampally-mediated memory deficits (Keenan et al., 1996
), and healthy volunteers given GR agonists perform more poorly on memory tasks within a few days (Wolkowitz et al., 1990
; Newcomer et al., 1994
). Finally, studies of adults who were exposed to abuse during childhood reveal decreases in hippocampal volume (Bremner et al., 1997
) and increased stress hormone production (Heim et al., 2002
).
Because toxic levels of stress cannot be experimentally manipulated in humans, these studies cannot causally link stress exposure to hippocampal volume. Recent data has called into question the direction or timing of the association between adult stress exposure and hippocampal volume; for instance small hippocampal volume may confer risk for acquiring PTSD or reporting greater stress instead of vice-versa. One study attempted to address this problem of directionality by showing that for monozygotic twins one of whom was in combat and one who was not, volume of the hippocampus in the non-combat exposed twin predicted PTSD symptomology in the individual exposed to combat (Gilbertson et al., 2002
; Kasai et al., 2008
). This finding points to the potential importance of early-shared environment and/or genetics in determining hippocampal volume separate from chronic exposure to toxic levels of stress in adulthood. Early environment in the form of childhood trauma exposure is a predictor of PTSD risk in combat exposed veterans (Brewin et al., 2000
; Dedert et al., 2009
). How early stress predisposes an individual to hippocampal alterations and increases stress reactivity is unclear, but a recent study showed that the association between hippocampus and emotional reactivity is predicted by pre-existing amygdala reactivity (Admon et al., 2009
). This prospective study suggests a specified temporal relationship between amygdala and hippocampus – one potential mechanism by which hippocampal volume indexes risk for PTSD.
Despite the numerous studies showing reduced hippocampal volume in adulthood following stress exposure in either childhood or adulthood, there is no evidence of hippocampal volume differences during childhood caused by stress. In fact, it has been concluded that adversity exposed individuals studied during childhood do not differ in hippocampal volume from their peers (De Bellis et al., 2001
; Woon and Hedges, 2008
), despite evidence that children who experience trauma have increased GR exposure (De Bellis et al., 1999a
). While it is possible that developmental timing of stress exposure is responsible for this lack of effect in the hippocampus, it is also possible that this discordance in findings is the result of timing of measurement. That is, hippocampal impacts could emerge late in life even when exposure to stressful life circumstances occurred early. Only one longitudinal study has attempted to directly test this hypothesis. De Bellis and colleagues imaged children before puberty (Tanner stage I or II) and 2 to 3 years later (Tanner stage III or IV) to determine if childhood stress-induced changes in hippocampal volume become evident during adolescence. This investigation did not support the hypothesis that early abuse exposure might ‘emerge’ as differences in hippocampal volume during adolescence (De Bellis et al., 2001
); however, in the final stage of this longitudinal study, these participants were not yet adults. A more recent study that extends the age at test has shown evidence of hippocampal changes that emerge in adulthood from childhood stress (Andersen et al., 2008
). Taken together, these data suggest that early-life stress results in hippocampal volume decreases and functional alterations when measured in adulthood, but these effects are difficult to observe or are not observable during childhood. The conclusion that childhood stress exposure leads to adult but not childhood differences in hippocampal volume leaves open certain possibilities. For instance, early stress exposure, while unrelated to childhood hippocampal volume, may result in other neurobiological changes (for example, in the amygdala) during childhood, which subsequently result in adulthood differences in hippocampal volume. We will discuss the impact of adversity on the amygdala during development in the subsequent section.
The amygdala has been implicated in learning about the emotional significance of stimuli (Davis and Whalen, 2001
). Having a mechanism to determine the relative safety or danger of situations is adaptive at any age. However, when less is known about the environment (e.g., early in life), the need to learn about the safety or danger of novel events will be greater (Tottenham et al. 2009a
), and this type of learning is heavily dependent on the amygdala (reviewed in Davis and Whalen, 2001
; LeDoux, 1993
). Neuroimaging studies have confirmed that, like in rodents and non-human primates, the human amygdala responds to negative as well as positively valenced stimuli (Breiter et al., 1996
; Somerville et al., 2004
; Hennenlotter et al., 2005
), suggesting it supports learning about the emotional significance of the environment in general.
Amygdala Development
The basic architecture of the human amygdala is present at birth (Humphrey, 1968
; Ulfig et al., 2003
). Nonetheless, the amygdala undergoes structural and functional change across an extended developmental period (reviewed in Tottenham et al., 2009a
). Structurally, the amygdala exhibits a protracted period of development, extending from year one through late childhood. In an initial study, the amygdala was observed to grow in volume across 4–18 years only for male subjects (Giedd et al., 1996
); structurally, the female amygdala reached adult-like volume by age 4-years old. This growth trajectory was confirmed in subsequent studies (Schumann et al., 2004
) and observed developmental change even when children 2 years of age were included (Mosconi et al., 2009
). Notably, many studies of amygdala volume in children have focused on autism, therefore limiting the control sample to male children, which may be problematic given the sexual dimorphism identified in structural studies (Giedd et al., 1996
). Amygdala volume development for female children is understudied, and sex-hormones influence amygdala development in human populations (Rose et al., 2004
), indicating that future research should carefully consider the role of sex in modifying differences in amygdala function and structure in response to stress. Despite these structural changes observed during childhood, a recent longitudinal non-human study showed that the most rapid rate of primate amygdala development occurs during the early postnatal period, leveling off soon after (Payne et al., 2010
). This rapid rate of change may heighten the vulnerability of the amygdala to environmental influence early in life.
Similar to structural development, the amygdala shows early functionality that is followed by developmental change across childhood and adolescence. Like adults (e.g., Breiter et al., 1996
), children and adolescents reliably recruit the amygdala when processing emotion from facial expressions. Greater amygdala activity for fearful faces relative to fixation is observed in both adolescents (Baird et al., 1999
; Killgore et al., 2001
) and children (Thomas et al., 2001a
). Adolescents also demonstrate greater activity in response to these faces than adults, activity that is not generated by differential visual scanning of the faces (Guyer et al., 2008
). However, the response pattern differs for younger subjects where children, unlike adults, exhibit greater amygdala recruitment for neutral faces than fearful (Thomas et al., 2001b
) and other facial expressions (Lobaugh et al., 2006
), suggesting that the amygdala (and most likely its connections with cortical regions) undergoes refinement over childhood and adolescence.
Fear conditioning experiments with adolescents (e.g., pairing a neutral cue with an air blast directed at the larynx) have shown that before adulthood, individuals can learn to associate a neutral stimulus with a negative one via increased amygdala activity to the conditioned stimulus (Monk et al., 2003a
). This type of amygdala-dependent cued fear conditioning is similar to the process identified in adults (LaBar et al., 1998
; Critchley et al., 2002
; Knight et al., 2005
; Delgado et al., 2006
). In adults, both positively valenced (e.g., reward, happy and/or attractive faces) and negatively valenced events recruit amygdala activity (Breiter et al., 1996
; LaBar et al., 1998
; Baxter and Murray, 2002
; Hamann et al., 2002
; Somerville et al., 2004
; Hennenlotter et al., 2005
; Belova et al., 2007
). However, many studies with adults find that negative events activate amygdala response above and beyond positive events (Morris et al., 1998
; Hamann et al., 2002
; Whalen et al., 2004
) – suggesting that the adult amygdala discriminates based not only on arousal properties but also on the valence of stimuli. It is not yet established whether early in life the amygdala discriminates stimuli of different valence as well as it does to arousal properties.
The amygdala appears to be more reactive earlier in life than in adulthood. This claim is supported by the repeated findings that amygdala activity continues to change from childhood to adulthood, peaking in activity during adolescence before it declines in adulthood (Monk et al., 2003b
; Killgore and Yurgelun-Todd, 2007
; Guyer et al., 2008
; Hare et al., 2008
) and this activity is tightly coupled to the modulatory connections of the ventromedial prefrontal cortex (vmPFC) (Hare et al., 2008
). This increased susceptibility to emotionally-relevant events would aid in learning about the environment, at a time in life when relatively little is known about the world. Moreover, the functional precocity of the amygdala may reflect the relative importance of establishing competence in recognizing and learning about emotionally-relevant stimuli prior to establishing competence in other developmental domains (e.g., cognitive development).
Consistent with the notion that the amygdala is highly active early in life, lesion studies that have isolated the timing of lesions to discrete periods of life suggest that lesions early in life often have a more dramatic effect on behavior than those that occur later. Naturally occurring lesion studies in humans show that early amygdala lesions (i.e., congenital) significantly impair processing of facial expressions, particularly fearful ones (Adolphs et al., 1994
). However, amygdala lesions occurring later in life (i.e., adulthood) appear to have less of an effect on processing these expressions (Hamann and Adolphs, 1999
). These developmental differences are most apparent when the task is non-verbal (e.g., judgments of perceived similarity between expressions). Presumably, the amygdala is important during developmental periods when learning about the meaning of relevant social stimuli, like facial expressions, is occurring, but may be less critical once these associations have been formed. More precise lesion studies in non-human primates when the effects of timing can be manipulated also lend support to the idea that the amygdala of young animals is uniquely important in learning about the environment. Amygdala lesions that occur both in neonatal or adult macaques result in the animals showing less fear of non-social items. However, these lesions produce distinct responses to social stimuli that vary as a function of the timing of the lesion. Amygdala lesions in adult animals result in an increase in affiliative social behaviors (e.g., less distance from peers, more affiliative vocalization coos, more walk bys; Emery and Amaral, 1999
), but when they occur in infancy, these lesions result in exaggerated fear responses during social interactions (e.g., decreased exploration, increased fear grimaces and screams; Bauman et al., 2004
). Prather et al. (2001)
suggest that the exaggerated social fear in monkeys with early amygdala lesions is the result of these monkeys never having the capacity to appropriately learn any social signal from conspecifics, and therefore, are left unable to recognize social cues that signal safety. To summarize, the literature suggests that the amygdala becomes functionally active early in life although it demonstrates continued refinement (largely through increased cortical connections) throughout childhood and adolescence. How this trajectory interacts with exposure to stress will be discussed in the next section.
Amygdala and Stress
The amygdala and the extended amygdala (the bed nucleus of the stria terminalis; Davis, 1994
), appear to be critical in activating the HPA axis in response to cognitive-emotional challenge and threat (reviewed in Dedovic et al., 2009
). Stress and glucocorticoids have been found to increase CRH levels and upregulate CRH receptors in the amygdala of both mature (Makino et al., 1994b
) and developing rodents (Hatalski et al., 1998
) lowering the threshold for the fear response to occur. Chronic GR administration acts to augment the amygdala-mediated startle response in rats (Lee et al., 1994
) and CRH antagonist administration (Swerdlow et al., 1989
) suppresses the fear response in primates. Lesions of the amygdala prevent elevations in glucocorticoids in response to psychological stressors, such as restraint in rats, but do not prevent elevations in response to physiological stressors, such as illness or injury (Feldman et al., 1994
; Herman and Cullinan, 1997
). There are a large number of CRH producing neurons and CRH receptors in the amygdala, and infusion of CRH to the amygdala (Rosen and Schulkin, 1998
), amygdala stimulation (Mason, 1959
), and stress (Baram and Hatalski, 1998
; Bonaz and Rivest, 1998
) produce large increases in glucocorticoids and corresponding increases in behaviors indicative of fear and anxiety. Developmental human neuroimaging studies have also shown that naturally occurring elevations in glucocorticoids are associated with amygdala hyperactivity (Maheu et al., 2008
). Taken together, these findings strongly support the notion that the HPA axis and amygdala CRH are involved in potentiated fear responses following stress. Adverse experiences produce long-term changes in the amygdala structurally and functionally via high levels of circulating glucocorticoids and endogenously produced CRH, decreasing an individual’s threshold for reacting to emotional events. Animal models have provided evidence for this type of stress-induced kindling of the amygdala (where continual stimulation produces greater future excitability; Adamec and Shallow, 2000
).
Numerous neuroimaging studies have demonstrated that the amygdala is altered structurally and functionally by psychosocial stress in humans as well. Many of these studies have been with adults who have experienced extremely traumatic events (e.g., combat, physical assault) and show that in adults the amygdala is both smaller (Driessen et al., 2000
; Schmahl et al., 2003
) and more reactive to emotional stimuli (Liberzon et al., 1999
; Rauch et al., 2000
; Shin et al., 2004
; Armony et al., 2005
; reviewed in Shin et al., 2006
). The amygdala is influenced by trauma of less intensity as well. In one study, first and second year undergraduates showed a correlation between their parent’s social status, linked in previous studies to stress reactivity (Lupien et al., 2000
; Evans and Kim, 2007
) and their amygdala activation while viewing angry faces (Gianaros et al., 2008
). Although it is difficult to draw conclusions about causality from most human studies, directionality between adversity and amygdala structure and function has been confirmed with animal models of stress, which have routinely identified changes in the amygdala following stress administration (reviewed in Roozendaal et al., 2009
).
Developmentally, there are some similarities and some differences in the associations between amygdala and stress. Studies that have manipulated the timing of stress, typically in non-human animals, have found that the amygdala is particularly sensitive to stress early in life. Poor caregiving in rodents results in increased anxiety- and aggressive-behaviors in adulthood, which is associated with acceleration of amygdala development (Kikusui and Mori, 2009
), including early myelination (Ono et al., 2008
), increases in CRH-containing neurons (Becker et al., 2007
), and sensitization of the amygdala in adulthood (Salzberg et al., 2007
). Although the amygdala is functionally dormant in the rat neonatal period, significantly stressful events and/or GR administration can precociously activate the amygdala (Moriceau et al., 2004
), perhaps because of the early presence of CRH mRNA [present in day 2 (Avishai-Eliner et al., 1996
; Fenoglio et al., 2004
; Vazquez et al., 2006
)], and such amygdala effects can last until adulthood (elevated CRH mRNA; Plotsky et al., 2005
; reviewed in Caldji et al., 2000
). Non-human primate work shows that maternal deprivation stress also alters amygdala development. These effects can be more devastating the earlier they in life they occur, and they include deficits in socio-emotional behaviors (e.g., decreased social behavior and increased self-comforting), which are mediated by stress-induced changes in amygdala gene expression (Sabatini et al., 2007
). These stress-related timing effects coincide with the early development of the amygdala (Payne et al., 2010
), and suggests that amygdala vulnerability to stressors may be at a peak during the early postnatal period primates. This position is supported as well by rat models of early stress, which find that the amount of CRH required to produce amygdala-originating seizures in developing animals is 200 times smaller than required for adult animals (reviewded in Baram and Hatalski, 1998
) – another suggestion that the juvenile amygdala has heightened susceptibility to environmental pressure.
While there are a growing number of studies that have examined the role of stress on amygdala structure and function during development, most in humans have not been able to isolate the timing of stress to a discrete period in development (the way that animal models can). This difficulty is common in human studies since adversity that occurs at one point in development is rarely isolated and typically is accompanied by a lifetime of adverse rearing environment, making temporally-defined periods of stress difficult to identify. Studies in adults have often found decreased amygdala volumes in individuals with a history of stressful childhoods (Driessen et al., 2000
; Schmahl et al., 2003
– note: these individuals also had borderline personality disorder). This type of finding seems to contradict animal studies, which have identified larger amygdala cell size in addition to exaggerated amygdala activity (Vyas et al., 2002
, 2004
). In one human study that examined the effects of adverse caregiving restricted to the postnatal period (i.e., children reared in and then removed from orphanages) also found amygdala volume to be larger (once controlling for total brain volume), and did not find hippocampal differences (Tottenham et al., 2009b
). A second independent study replicated these findings of amygdala volume increases and a lack of hippocampal differences in adolescents who experienced institutional rearing in infancy (Mehta et al., 2009
). Importantly, these populations of previously institutionalized children were studied years after the adversity ended, and children were adopted into families of very high socio-economic status. These data suggest that the amygdala can change as a function of early adverse experiences, but once it does it is resistant to ameliorative environmental influences – an interpretation supported by animal work showing that cellular growth in the amygdala following adult stress, unlike the hippocampus, fails to reverse during a recovery period (Vyas et al., 2004
; Yang et al., 2007
). Perhaps this heightened and biased sensitivity to emotionally-relevant events is one way the organism ensures that it is prepared for future adversity, in an environment that has already proved to be threatening. Longitudinal studies that vary the timing of stress in humans to include the neonatal period, childhood, adolescence, and adulthood are required to fully understand sensitive periods for environmental stress.
The amygdala is part of an extended neural network. In particular, it has rich connections with the vmPFC (Amaral et al., 1992
; Milad and Quirk, 2002
; Ghashghaei et al., 2007
) and hippocampus (Ikegaya et al., 1996a
,b
). The vmPFC can modulate the activity of the amygdala through descending projections, perhaps via afferents to the intercalated cells of the amygdala that inhibit its own activity. Secondly, the amygdala and hippocampus co-modulate each other such that the amygdala can influence hippocampally-mediated memory formation and the hippocampus influences amygdala response when emotional stimuli are encountered (Phelps, 2004
). The three structures (amygdala, hippocampus, and vmPFC) coordinate during emotional learning. In new environmental contexts, the hippocampus inhibits the vmPFC, which releases the amygdala from vmPFC inhibition (reviewed in Kim and Richardson, 2010
). Like the amygdala and hippocampus, stress alters vmPFC activity in adults, where significantly stressful events leads to impaired vmPFC-dependent fear extinction learning (Milad et al., 2009
). It has been demonstrated that chronic stress impairs extinction learning (Miracle et al., 2006
) possibly via blockage of long-term potentiation (Maroun and Richter-Levin, 2003
) and dendritic reorganization (Wellman, 2001
; Izquierdo et al., 2006
), including shortening of dendritic length caused by stress-induced reduction of the growth factor brain-derived neurotrophic factor (reviewed in Czeh et al., 2008
). Also, chronic stress weakens vmPFC regulation over amygdala and results in enhanced amygdala sensitivity to footshock (Correll et al., 2005
). Additionally, the bidirectional and direct connections between amygdala and vmPFC suggest that stress-induced changes in the amygdala may, over the course of development, impair amygdala-vmPFC connectivity. Much less is known about the development of the vmPFC following early adversity, although a small number of rodent studies shows that early-life adversity alters vmPFC development both structurally (higher synaptic densities compared to controls – Ovtscharoff and Braun, 2001
) and functionally (reduced tyrosine hydroxylase-positive fiber innervation – Braun et al., 1999
). Because of its rich interconnectedness, early-life stress may act on this system through multiple pathways and future investigations will benefit from a systems-level approach. At the current time, the literature has primarily focused on the effects of stress on each area.
Taken together, the developmental data and the human adult data suggest that early in life, the amygdala undergoes expansive growth following a stressor. This growth is accompanied by hyperactivity, which after a prolonged period results in cellular atrophy and/or death (and smaller volumes as measured by MRI). Such a trajectory would explain why many studies in adults who experienced years of adversity show decreased amygdala volume. This hypothesized trajectory has been identified in studies with depressed patients, who after the initial depressive episode show enlarged amygdala volume, but after living with depression for an extended period of time, show amygdala volume decreases (Frodl et al., 2002
; Lange and Irle, 2004
). These findings point to the hypothesis that alterations of amygdala following neonatal stress are apparent early in life because of the amygdala’s early functional development, its early exposure to stress hormones (those associated with the HPA stress response), and its maximal expression of stress hormone (CRH) receptors early in life (reviewed in Baram and Hatalski, 1998
).
Stressful events do not impact the whole brain in a uniform fashion, but instead the effects are region specific, exhibiting some of the largest effects in the amygdala and hippocampus. The amygdala and the hippocampus exhibit differential effects of stress that occurs in adulthood, and often contrasting, such that stress decreases size, complexity, and activity of the hippocampus (reviewed in Lupien et al., 2007
; Bremner et al., 2008
) and shows the opposite effects in the amygdala (larger, more reactive amygdala) (Liberzon et al., 1999
; Rauch et al., 2000
; Armony et al., 2005
). The differences between the two structures seem to be partially related to the time course of the molecular events that occur between the two structures that follows stress. This cascade begins in the amygdala, which exerts molecular and chemical downstream effects on the hippocampus. Cells in the amygdala participate in the earliest reaction to environmental stressors, often initiating the HPA cascade. They are quickly activated by stress and express immediate-early genes (Honkaniemi et al., 1992
), whereas the hippocampus begins to play its role later in the cascade, negatively feeding back on the HPA axis to inhibit its activity (Herman and Cullinan, 1997
). CRH-induced seizures produce the earliest discharges in the amygdala (Baram et al., 1992
), which propagate later to the hippocampus (Haas et al., 1990
). Therefore, stress-induced changes in the amygdala may have downstream effects on the HPA axis that over time can change the structure and function of later stages in the axis (Brunson et al., 2001b
), like the hippocampus. These timing differences may contribute to the reasons why hippocampal alterations tend to developmentally follow amygdala alterations from stress (Admon et al., 2009
).
Observation of these effects may depend on when they are measured. The effects of early-life adversity have been observed in the amygdala during development (Sabatini et al., 2007
; Mehta et al., 2009
; Tottenham et al., 2009b
). Although there is evidence for molecular-level stress-induced changes in the immature hippocampus (e.g., Hatalski et al., 2000
; Chen et al., 2006
), stress-induced changes in the hippocampus are often difficult to observe during the juvenile period, but may be apparent later in life. Maternal deprivation stress in non-human primates failed to show hippocampal differences as measured by structural MRI in the age-equivalent of childhood (Spinelli et al., 2009
). Rodent studies that have manipulated timing of stress and time of measurement have found that hippocampal alterations may be developmentally delayed and appear later in life; neonatal maternal deprivation did not result in a drop in hippocampal synaptophysin (a synapse related protein) until postnatal day 60 (adulthood; Andersen and Teicher 2004
). Behaviors that depend on the hippocampus follow a similar trajectory; early-life insults often result in “acquired” cognitive decline, only appearing in older animals, not younger (Brunson et al., 2005
). During early-adulthood, the hippocampally-mediated cognitive decline associated with maternal deprivation was unobservable and were not apparent until animals were middle-aged. These behavioral impairments are accompanied by decreased hippocampal LTP, dendritic atrophy, and mossy fiber expansion – hippocampal phenotypes typically associated with stress that occurs in adulthood (McEwen, 1999
). It is noteworthy that, unlike with the mature hippocampus, which shows recovery after the termination of a stressor, stress administered early in life has long-term effects on the hippocampus.
Contributing to the differences between the two regions during development may be the differential emergence of HPA-related peptides following stress. Stress hormones and CRH mRNA production in amygdala have a developmentally early onset, appearing as early as postnatal day 2 in the rat (Avishai-Eliner et al., 1996
; Vazquez et al., 2006
), an earlier onset than in the hippocampus (Fenoglio et al., 2004
). This discrepant time course between the amygdala and hippocampus maybe related to the modulatory role that the amygdala has over the hippocampus (Packard and Teather, 1998
). These temporal discrepancies appear early in life, where stressful events produce elevations in CRH that occur first in the amygdala and are observed only afterwards in the hippocampus (reviewed in Baram and Hatalski, 1998
). Environmental manipulations that result in changes in stress-related gene expression (CRH/GR receptors) are apparent in the amygdala at an age when they are not present in the hippocampus (during the early postnatal period). Neonatal separation stress resulted in an increase in CRF containing neurons in the amygdala, but not the hippocampus in juvenile rodents (Becker et al., 2007
). Taken together these data illustrate the interaction between developmental timing and timing of stress exposure (Fenoglio et al., 2004
) and suggest that the amygdala response to stressors is a temporal prerequisite for hippocampal changes following stress (Kim et al., 2001
). In support of this postulate, a rare prospective study with new recruits to the Israeli Defense Forces showed that while amygdala and hippocampal activity closely correlated with subjective reported stress, hippocampal vulnerability to stress depended on how hyperactive the initial amygdala response was (Admon et al., 2009
).
If future studies continue to show that the effects of stress occur in the amygdala developmentally earlier than the hippocampus, it might shed some light on the timing effects often observed in human neuroimaging studies. Evidence from the human literature supporting this temporal hypothesis is mounting. The effects of adversity on these two regions vary by when the stressor was administrated and by age at which testing occurred. For example, larger amygdala volumes are observed during childhood following neonatal early adverse caregiving, although no hippocampal differences were observed (Tottenham et al., 2009b
). Age at test is critical since developmental studies that include adolescent subjects often do not find amygdala differences following trauma exposure (Carrion et al., 2001
; De Bellis et al., 2001
, 1999b
). These amygdala effects are typically unobservable, or have not been measured, in adults who experienced childhood stress (Bremner et al., 1997
; Cohen et al., 2006
), where it has even been reported that childhood stress may actually result in smaller amygdala volumes in adulthood (Driessen et al., 2000
). This pattern suggests that initial stress-induced hypertrophy and hyperactivity of amygdala neurons eventually leads to neuronal atrophy or cell death by adulthood (Teicher et al., 2003
), a pattern identified in other populations (e.g., autism spectrum disorders) who demonstrate amygdala hypertrophy in childhood followed by smaller volumes in adulthood (Schumann et al., 2004
). Effects of trauma do not appear in amygdala volumetric measures in adulthood, while such effects are observable in the hippocampus (reviewed in Lupien et al., 2007
; Bremner et al., 2008
), leaving open the possibility that hippocampal effects may not emerge until adulthood, possibly as a result of stress-induced changes in the amygdala.
It is also possible that the hippocampus (perhaps due unique developmental timelines) is less vulnerable to stress effects early in life than the amygdala (at least as measured by volume). This may be related to the developmental timing of the structural development of these two structures. As noted earlier, there are data consistent with the hypothesis that amygdala development slightly precedes hippocampal development, and this developmental differential may have consequences for each structure’s vulnerability. Alternatively, hippocampal effects may be masked during development. Perhaps measuring changes in volume caused by stress are difficult in the context of developmental changes (Giedd et al., 1996
) as measured by MRI. The assertion that the developing hippocampus is entirely invulnerable to environmental stressors is weakened by molecular studies in rodents that show that juvenile stress is followed by altered expression of molecules involved in neural development and synaptic plasticity (i.e., neural cell adhesion molecules), effects that extend into adulthood (Tsoory et al., 2008
) (Isgor et al., 2004
). Perhaps volumetric studies may not be sensitive enough to show stress-induced hippocampal changes in childhood, although the effects in the amygdala are robust enough to be observed by this measure.
Technological advances are improving our ability to better address these questions in humans. Using diffusion tensor imaging, DTI, studies have replicated and extended findings from volumetric studies of adversity. In one study young adults exposed to verbal abuse as children had significant differences in the white matter tracts associating medial temporal lobe structures to the prefrontal cortex (cingulum bundle) and the fornix fibers of the hippocampus (Choi et al., 2009
). DTI reveals the structural connectivity of neural structures and may aid researchers in better describing how associations between structures, such as the ones hypothesized here between the amygdala and hippocampus support developmental patterns in the effects of stress exposure. Other methods for exploring associations between structures such as resting state fMRI, or resting state functional connectivity have not yet been employed in the service of understanding the effect of adversity on neural development. But these techniques have been used to great effect in understanding development generally (Fair and Dosenbach, 2007
; Fair and Cohen, 2008
) and in defining functionally connected regions of interest in individual subjects (Cohen et al., 2008
) a technique which would be well suited to the investigation of individual differences associated with stress. In addition, resting state connectivity and DTI could be used in a range of ages down to children as young as 2 years who have been successfully studied while asleep (Redcay and Courchesne, 2008
). Thus, through a combination of connectivity and volumetric studies, it would be possible to, in children as young as 2 and extending through adulthood, examine the structural and functional networks that underlie the embedding of adversity, and directly test the hypothesis, proposed here, that amygdala changes lead and produce hippocampal effects of adversity.
The techniques proposed here have in common with volumetric studies that they can be performed in the same way with children, adolescents, and adults. Notably absent are functional neuroimaging techniques which require the use of a task. In examining the developmental effects of adversity the use of functional neuroimaging presents potential challenges. Examining functional differences across studies is difficult because subtle differences in experimental design disallow certain findings. In addition, across ages the same task may be more salient and/or more difficult. For example a young child may not understand a task which includes a great deal of peer feedback about behavior where as an adolescent will both understand this task and peer feed back will be a very salient emotional stimulus. Including information like this in the interpretation of results can be difficult. These caveats aside, neural activation is most closely linked to behavior and obtaining functional neuroimaging data as a function of early adversity would have the potential to expose important differences and similarities structure-function relationships.
Transitions between developmental stages have not been studied extensively with regard to early adversity, and longitudinal work that includes a close examination of transitions into childhood and adolescence is necessary to delineate the effects of early adversity. Indeed, there is some behavioral evidence that the effects of early adverse caregiving may exacerbate once children age into adolescence. In a longitudinal study of children adopted from Romanian orphanages, Rutter and colleagues have shown that unlike other domains of development (e.g., cognitive development, height, head circumference) which show massive catch-up following removal from orphanage care, there was a significant increase in emotional difficulties caused by institutional care once children transitioned into adolescence (Colvert et al. 2008
). These findings are consistent with the notion that the transition into adolescence potentially marks another period when the environment can exert large effects on the brain (Sisk and Foster, 2004
; Schulz et al., 2009
). Findings from the rodent suggest that pubertal onset may be an important agent in amplifying or making obvious environmental influences on brain development. For example, stress-induced sex difference in hippocampally-dependent trace learning only emerge after adolescence (Hodes and Shors, 2005
), perhaps as a result of the organizing effects of increased pubertal testosterone on the hippocampus (reviewed in Sisk and Zehr, 2005
.
Thus far we have considered the timing of stress exposure as a variable that may moderate the effect this variable has on the hippocampus and amygdala. There are various other aspects of stress that may modify its impact on neural structures. Chief among these are the kind and duration of stress. Because stress is chiefly a psychologically defined construct (Gunnar and Quevedo, 2007
), one that is subjective in nature, determining which stressors (e.g., abuse vs. neglect) are “the worst” is difficult since there are multiple stressors that can present challenges to the individual’s homeostasis. However, certain aspects of stress, for example, controllability and predictability may be particularly effective at influencing HPA activity (Davis and Levine, 1982
). Presumably the stressors described throughout this manuscript are unpredictable and uncontrollable from the standpoint of the infant or child experiencing them, thus increasing the potential impact of early stressors. Stress duration should additionally be considered as a factor, since chronic stressors may affect neural development differently, often detrimentally, than single instance traumatic events (reviewed in Brunson et al., 2003
). Again, the literature reviewed throughout this manuscript provides examples of chronically stressful environments. Nonetheless, dose-response relationships between stress duration and brain development have been reported (Brunson et al., 2001a
; Mehta et al., 2009
; Tottenham et al., 2009b
). A final variable that should be considered when examining the effect of adversity on neural development is the temporal context in which neural development is tested. For example we argue here that amygdala development and its modification by stress precedes that of the hippocampus. However many developmental studies of traumatic stressors (for example, studies of child abuse) may occur when children are still under a great deal of stress. Duration since stressor needs to be considered, since the amygdala and hippocampus may have different recovery rates (Vyas et al., 2004
; Yang et al., 2007
). For example, when measured almost a decade after removal from orphanages, post-institutionalized children show amygdala hypertrophy and hyperactivity, but do not show differences in hippocampal structure (Mehta et al. 2009
; Tottenham et al., 2009b
). However, differences in duration since stressor cannot account for all of the data since many studies (in older individuals) show decreased hippocampus years after stress termination (Andersen et al., 2008
), suggesting again that developmental timing of exposure will influence the outcome measure. Nonetheless, more control over duration since stressor is warranted in order to better understand the effects of adversity. To summarize, we suggest that (1) early life is a period of increased vulnerability although the effects of stress may be difficult to detect for years (as seems to be the case with the hippocampus) (2) stress-induced changes in amygdala (initial increases in activity and growth) are apparent earlier in life and more robustly than the hippocampus (decreases in growth), and (3) later in life, when hippocampal changes are finally apparent, the initial amygdala volume increases may ultimately change to volumetric decreases (although it may remain hyperactive).
Adverse experiences shape brain development, and these changes are often global in nature. However in this manuscript, we have focused on the effects on amygdala and hippocampal development, two regions that are particularly vulnerable to stress in adult populations. We argue that a developmental approach is necessary in understanding how environmental conditions such as trauma can impact outcome in these structures. While this review is limited in its scope, the restriction was purposeful in that it allowed for greater discussion of how timing of exposure and age at measurement can differentially influence these neural phenotypes. The hypothesis driving the manuscript was that developmental timing of adverse experiences and the age at testing would influence structure and function of the amygdala and hippocampus. The literature discussed in this review suggest that the amygdala and hippocampus are highly vulnerable to the effects of adverse early environments, although these effects may be more evident in the amygdala early in life, whereas the hippocampus effects may be more subtle early in life and may be more readily observable in humans studies later in life.
We included data from animal models of stress to address issues of timing and biological mechanism that are difficult to address with most human studies. Two of these issues in the human literature include methodological differences between studies and the methods of identifying the timing of stressful experiences. First, studies differ on how/whether they control for total brain volume when examining the effects of stress on regional morphometry. This difference will have a significant impact on volume findings since children with a history of trauma are more likely to have a smaller total brain volume relative to control samples (Casey et al., 2009
; De Bellis et al., 1999b
). In addition, recognizing changes in the volume of subcortical structures over time may be difficult because cortical volume decreases rapidly across development. Controlling for total cortical volume across development while measuring changes in structure volume may inadvertently increase observed structure development.
Secondly, the overwhelming majority of studies on stress rely on retrospective data. This approach, while usually a necessary nuisance in human studies of stress, is problematic because researchers must rely on subject reporting, which may be inaccurate (Maughan and Rutter, 1997
), particularly with respect to timing of events. Moreover, even if reporting is accurate, the varying times and durations of the stressful events in most studies make it unclear how timing played a role in the observed effects. Human developmental studies are critical in order to understand how stressful experiences specific to infants and children impact neural development. Drawing firm conclusions about the impact of stressful and traumatic events will rely on methodological advances. Longitudinal studies, where observation occurs at multiple time points allows for direct observation of change over time. Ideally, these studies would be prospective in nature, a design aspect that further complicates data collection. These types of studies are difficult and costly, but provide valuable information about developmental trajectories not often provided by cross-sectional design (Sowell et al., 2004
; Durston et al., 2006
; Gogtay et al., 2006
; Giedd et al., 2008
).
The main purpose of this manuscript is to highlight the importance of considering developmental timing, and the investigation into the short-term and long-term effects of adversity on neural development is only at its earliest stages. The hypotheses posited here will continue to be shaped by future results, and approaches like those described in this paper will help constrain interpretations of those data. This approach will be informative regarding the neural mechanisms that underlie mental health problems following early adversity and provide insight into the sensitive periods that define the windows of opportunity and vulnerability to environmental events, both adverse as well as ameliorative.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Bremner, J. D., Randall, P., Vermetten, E., Staib, L., Bronen, R. A., Mazure, C., Capelli, S., McCarthy, G., Innis, R. B., and Charney, D. S. (1997). Magnetic resonance imaging-based measurement of hippocampal volume in posttraumatic stress disorder related to childhood physical and sexual abuse–a preliminary report. Biol. Psychiatry 41, 23–32.
Brunson, K. L., Eghbal-Ahmadi, M., Bender, R., Chen, Y., and Baram, T. Z. (2001b). Long-term, progressive hippocampal cell loss and dysfunction induced by early-life administration of corticotropin-releasing hormone reproduce the effects of early-life stress. Proc. Natl. Acad. Sci. U.S.A. 98, 8856–8861.
Casey, B. J., Glatt, C. E., Tottenham, N., Soliman, F., Bath, K., Amso, D., Altemus, M., Pattwell, S., Jones, R., Levita, L., McEwen, B., Magarinos, A. M., Gunnar, M., Thomas, K. M., Mezey, J., Clark, A. G., Hempstead, B. L., and Lee, F. S. (2009). Brain-derived neurotrophic factor as a model system for examining gene by environment interactions across development. Neuroscience 164, 108–120.
Cohen, R. A., Grieve, S., Hoth, K. F., Paul, R. H., Sweet, L., Tate, D., Gunstad, J., Stroud, L., McCaffery, J., Hitsman, B., Niaura, R., Clark, C. R., McFarlane, A., Bryant, R., Gordon, E., and Williams, L. M. (2006). Early life stress and morphometry of the adult anterior cingulate cortex and caudate nuclei. Biol. Psychiatry 59, 975–982.
Colvert, E., Rutter, M., Beckett, C., Castle, J., Groothues, C., Hawkins, A., Kreppner, J., O’Connor T, G., Stevens, S., and Sonuga-Barke, E. J. (2008). Emotional difficulties in early adolescence following severe early deprivation: findings from the English and Romanian adoptees study. Dev. Psychopathol. 20, 547–567.
Dedert, E. A., Green, K. T., Calhoun, P. S., Yoash-Gantz, R., Taber, K. H., Mumford, M. M., Tupler, L. A., Morey, R. A., Marx, C. E., Weiner, R. D., and Beckham, J. C. (2009). Association of trauma exposure with psychiatric morbidity in military veterans who have served since September 11, 2001. J. Psychiatr. Res. 43, 830–836.
Mehta, M. A., Golembo, N. I., Nosarti, C., Colvert, E., Mota, A., Williams, S. C., Rutter, M., and Sonuga-Barke, E. J. (2009). Amygdala, hippocampal and corpus callosum size following severe early institutional deprivation: the English and Romanian Adoptees study pilot. J. Child. Psychol. Psychiatry 50, 943–951.
Shin, L. M., Orr, S. P., Carson, M. A., Rauch, S. L., Macklin, M. L., Lasko, N. B., Peters, P. M., Metzger, L. J., Dougherty, D. D., Cannistraro, P. A., Alpert, N. M., Fischman, A. J., and Pitman, R. K. (2004). Regional cerebral blood flow in the amygdala and medial prefrontal cortex during traumatic imagery in male and female Vietnam veterans with PTSD. Arch. Gen. Psychiatry 61, 168–176.
Shin, L. M., Shin, P. S., Heckers, S., Krangel, T. S., Macklin, M. L., Orr, S. P., Lasko, N., Segal, E., Makris, N., Richert, K., Levering, J., Schacter, D. L., Alpert, N. M., Fischman, A. J., Pitman, R. K., and Rauch, S. L. (2004). Hippocampal function in posttraumatic stress disorder. Hippocampus 14, 292–300.
Tottenham, N., Hare, T. A., Quinn, B. T., McCarry, T., Nurse, M., Gilhooly, T., Millner, A., Galvan, A., Davidson, M. C., Eigsti, I. M., Thomas, K. M., Gunnar, M. R., Altemus, M., Aronson, J., and Casey, B. J. (2009b). Prolonged institutional rearing is associated with atypically large amygdala volume and emotion regulation difficulties. Dev. Sci. 13, 46–61.