1
Department of Chemical Engineering, Stanford University, Stanford, CA, USA
2
Department of Materials Science and Engineering, Stanford University, Stanford, CA, USA
Neural prosthetic implants are currently being developed for the treatment and study of both peripheral and central nervous system disorders. Effective integration of these devices upon implantation is a critical hurdle to achieving function. As a result, much attention has been directed towards the development of biocompatible coatings that prolong their in vivo lifespan. In this work, we present a novel approach to fabricate such coatings, which specifically involves the use of surface-adsorbed, nanoscale-designed protein polymers to prepare reproducible, customized surfaces. A nanoscale modular design strategy was employed to synthesize six engineered, recombinant proteins intended to mimic aspects of the extracellular matrix proteins fibronectin, laminin, and elastin as well as the cell–cell adhesive protein neural cell adhesion molecule. Physical adsorption isotherms were experimentally determined for these engineered proteins, allowing for direct calculation of the available ligand density present on coated surfaces. As confirmation that ligand density in these engineered systems impacts neuronal cell behavior, we demonstrate that increasing the density of fibronectin-derived RGD ligands on coated surfaces while maintaining uniform protein surface coverage results in enhanced neurite extension of PC-12 cells. Therefore, this engineered protein adsorption approach allows for the facile preparation of tunable, quantifiable, and reproducible surfaces for in vitro studies of cell–ligand interactions and for potential application as coatings on neural implants.
Many recent experimental findings have emphasized the importance of surface properties for the optimization and control of cell adhesion, morphology, motility, proliferation, and differentiation in both in vitro and in vivo systems (Gordon et al., 2007
; Kasemo, 2002
; Stevens and George, 2005
). Of specific interest is the role that nanoscale cell–surface interactions play in the biocompatibility of neural implants. Neural prosthetic implants have shown promise in applications involving recording neural activity and the stimulation of function in both the central and peripheral nervous systems for the treatment of deafness and neurological diseases such as Parkinson’s and cerebral palsy (Branner et al., 2001
; Gordon et al., 2007
; He and Bellamkonda, 2005
; Kipke et al., 2003
; Olanow et al., 2000
). A major roadblock in the success of these implants has been the effective integration of the implant surface with the surrounding tissue (Anderson et al., 2008
).
Studies have shown that upon implantation of a foreign material, proteins rapidly adsorb to the implant bio-interface and that these proteins greatly affect the subsequent cellular and inflammatory responses (Anderson et al., 2008
; Wilson et al., 2005
). As a result, implants are now routinely modified to present thin nanostructures on their surfaces that aim to selectively bind beneficial molecules while minimizing non-specific adsorption (Ai et al., 2003
). Already established approaches for the fabrication of biocompatible implant surfaces include layer-by-layer electrostatic adsorption (Ai et al., 2003
; Ariga et al., 2007
), electrochemical polymerization (Cui et al., 2001
), covalent molecule attachment (Huber et al., 1998
; Massia et al., 2004
), self-assembling monolayers (Pakalns et al., 1999
), microcontact printing (James et al., 1998
), and surface adsorption of proteins and synthetic polymers (Elbert and Hubbell, 1996
; Giorgetti et al., 2008
; Wang, 2007
).
Here we present a new approach to create enhanced neural implant bio-interfaces that offers nanoscale precision and is reproducible, simple to apply, quantifiable, and customizable. This approach involves the preparation of thin films using surface-adsorbed, nanoscale-designed protein polymers (Heilshorn et al., 2005
). Our proteins are modular in design, consisting of alternating structural and biofunctional nanoscale domains that are intended to mimic aspects of extracellular matrix (ECM) proteins (Heilshorn et al., 2003
). The proteins are synthesized using recombinant, genetic engineering techniques, allowing for the creation of biocompatible polymers with nanoscale precision that impart highly specific protein functionalities. We make use of a highly flexible multi-step cloning strategy in order to allow for the rapid synthesis of new recombinant proteins that can introduce many different biofunctionalities. The engineered proteins discussed in this work contain identical nanoscale structural domains, similar to a repeated sequence found in the ECM protein elastin, and have been designed to present different biofunctional nanodomains taken from the ECM proteins fibronectin and laminin, and the cell–cell adhesion protein neural cell adhesion molecule (NCAM) (Meiners and Mercado, 2003
; Soroka et al., 2002
; Urry et al., 1991
).
Proteins are widely accepted to be highly surface active molecules, and their adsorption affinity to a given surface is determined by a combination of thermodynamic driving forces, such as electrostatic interactions and changes in system entropy due to structural alterations or protein/surface dehydration (Wilson et al., 2005
). This phenomenon provides for a relatively simple surface adsorption approach that addresses many current limitations of existing bioactive, thin-film preparation techniques. Most of these techniques focus on enhancing implant biocompatibility by either grafting short peptides or entire ECM proteins to surfaces. Methods that involve grafting peptides to surfaces typically focus on applying a uniform layer of a particular ligand rather than specified densities, are difficult to quantify, often involve a complex series of steps, and can result in decreased biofunctionality due to restricted conformational flexibility (Huber et al., 1998
; Massia et al., 2004
). In contrast, methods that rely on surface immobilization of entire ECM proteins suffer from difficulties in harvesting pure protein of a single splice variant and cannot be used to identify specific interactions integral for implant biocompatibility (El-Ghannam et al., 1998
; Gray, 2004
). Our surface modification approach offers several key improvements, including the inherent simplicity of the protein adsorption process, the relative ease of controlling and quantifying peptide ligand density, the design of conformational flexibility into the polymer, and the ability to prepare combinatorial mixtures of nanodomains for the identification of optimal peptide ligands and densities.
We ultimately intend to use this robust adsorption process to more rationally design optimized implant bio-interfaces that offer precise and customized nanoscale content and display either single or multiple peptide ligands. This manuscript presents the development of these surfaces by first introducing the modular design, synthesis, and purification of a set of recombinant, nanoscale-designed protein polymers from Escherichia coli bacteria. To demonstrate the flexibility and robustness of this strategy, we designed, cloned, expressed, and purified six new engineered proteins. These engineered proteins were used to prepare adsorbed surfaces on glass substrates that exhibited controlled peptide content and similar surface saturation levels when quantified with physical adsorption isotherms. The initial in vitro growth of neuronal-like PC-12 cells on the adsorbed protein coatings exhibited a high level of cell–surface biocompatibility, and neurite elongation was shown to be directly correlated to the surface density of specific biofunctional nanodomains. These results set the stage for future combinatorial analysis of surface ligand compositions with potential applications in the study of cell–surface interactions in vitro or as neural implant bio-interfaces.
Protein Synthesis and Purification
Expression plasmids were constructed in pET15b vectors (Novagen) using multi-step recombinant DNA cloning techniques and transformed into the E. coli expression strain BL21(DE3) (Straley and Heilshorn, 2009
). Protein expression was induced under the control of the T7-lac promoter at an OD600 of about 0.6 (37°C, LB medium) with 1 mM β-isopropyl thiogalactoside and allowed to express for 3-h post induction.
The RGD, RDG, NCAM, and NCAM Neg proteins were purified using an inverse temperature cycling process (Meyer and Chilkoti, 1999
). For this process, the wet cell pellet was resuspended in TEN Buffer [1 g/mL and 1-mM phenylmethylsulphonyl fluoride (PMSF)], sonicated, and agitated overnight at 4°C. The pH was adjusted to 9 with 4 N NaOH, incubated at 4°C for 1 h, and centrifuged at 4°C. NaCl was added to the supernatant at a final concentration of 1 M. This solution was agitated overnight at 4°C, incubated at 37°C shaking for 3 h, and centrifuged at 37°C. The pellet was resuspended in water (0.1 g/mL), agitated overnight at 4°C, adjusted to a pH of about 9 with 1 N NaOH, incubated at 4°C shaking for 1 h, and centrifuged at 4°C. Warm (37°C, 1 M NaCl) and cold (4°C, pH ∼ 9) purification cycles were each repeated twice. The final supernatant was desalted using size exclusion centrifugation (10,000 MWCO, Millipore Amicon) and lyophilized. Typical protein yields were around 25–50 mg/L.
The lower critical solution temperature (LCST) transitions of the YIGSR and GRYSI proteins necessitated purification using His-tag separation with nickel affinity columns in order to achieve similar protein yields. For this process, the wet cell pellet was resuspended in native lysis buffer at 4°C (50-mM NaH2PO4, 300-mM NaCl, 10-mM imidazole, pH = 8, 100-mL lysis buffer per 1-L original cell culture volume, 1-mM PMSF), sonicated, and centrifuged for 30 min at 4°C. The supernatant was added to Ni-NTA affinity resin (Qiagen, 0.25-mL resin/mL lysis buffer), incubated at 4°C shaking for 1 h, and gravity filtered in a protein column (4°C). The resin was washed twice with native wash buffer (50-mM NaH2PO4, 300-mM NaCl, 20-mM imidazole, pH = 8, 100-mL wash buffer per 1-L original cell culture volume, 4°C) and then the bound protein was eluted using native elution buffer (50-mM NaH2PO4, 300-mM NaCl, 250-mM imidazole, pH = 8, 50-mL elution buffer per 1-L original cell culture volume, 4°C). The final eluent was concentrated and then buffer exchanged for water using size exclusion centrifugation (10,000 MWCO, Millipore Amicon, 4°C).
Lower Critical Solution Temperature Measurement
Lyophilized protein was resuspended at a concentration of 10 mg/mL in phosphate buffered saline (PBS, pH = 7.2, 4°C). Turbidity readings at 300 nm were monitored as a function of temperature using a Molecular Devices SpectraMax Plus384 Spectrophotometer. The temperature was increased at a rate of 0.1°C/min with an equilibration time of 30 s at each step.
Substrate Preparation
All adsorbed substrates were prepared on glass surfaces by dissolving lyophilized protein in PBS (pH = 7.4), pipetting these solutions on top of the glass surfaces, and incubating the samples overnight at 4°C. Control surfaces were similarly prepared using either no protein or 50-μg/mL rat tail collagen I (Sigma). The coverslips were then washed thrice with PBS buffer at room temperature.
Bicinchoninic Acid Assay
Adsorbed protein substrates were prepared using the specified protein concentrations as described above on glass-bottom 24-well plates (Matek) and covered with 250 μL of PBS and 250 μL of bicinchoninic acid (BCA) assay working reagent (Sigma). Simultaneously, a set of standard solutions containing known concentrations of the protein of interest were prepared in PBS buffer (0, 0.5, 5, 10, 20, and 30 μg/mL) and mixed with an equal volume of BCA working reagent. All reactions were sealed with tape and incubated for 1.5 h, shaking, at 50°C. The solutions were then equilibrated at room temperature for ∼20 min and absorbance readings were taken at 562 nm.
PC-12 Cell Culture
Adsorbed protein substrates were prepared as described above on sterile glass coverslips using protein solutions with 1-mg/mL total protein (1:0, 1:1, and 0:1 RDG:RGD protein), blocked with bovine serum albumin (BSA, fraction V, 0.2% in PBS, heat inactivated at 85°C for 10 min) to prevent non-specific binding, and rinsed three times with PBS. PC-12 cells (ATCC) were prepared in F12 Kaighn’s complete media (10% horse serum, 5% fetal bovine serum, and 1% penicillin-streptomycin) at 37°C, 5% CO2. Cells were added to the coverslips at a density of 15,000 cells/cm2 and differentiated in F12 Kaighn’s differentiation media [1% penicillin-streptomycin, 50 ng/mL recombinant human β-NGF (R&D Systems)] for 6 days, at 37°C, 5% CO2. Media was changed every 2 days.
Immunostaining
After 6 days of differentiation in F12 Kaighn’s differentiation media, samples were rinsed twice with PBS, fixed with 4% paraformaldehyde in PBS for 15 min at room temperature, and rinsed twice more with PBS. Cells were permeabilized with 0.1% Triton X-100 in PBS buffer (PBST, 15 min, room temperature), blocked with 10% normal goat serum (PBST, 1 h, room temperature), and incubated with rabbit monoclonal neuronal class III β-tubulin antibody (dilution factor of 1:500, Covance, PBST containing 5% normal goat serum, room temperature, 2 h). Samples were then rinsed with PBST, incubated with goat anti-rabbit IgG Alexa Fluor 488 (Invitrogen, PBST containing 5% goat serum, 1 h), and rinsed with PBST. The samples were then incubated with 1 μg/mL of 4′,6-diamidino-2-phenylindole (DAPI, Roche) in methanol for 15 min at 37°C, washed with methanol, and mounted on microscope slides using Prolong Gold Antifade Reagent (Invitrogen). The fluorescently labeled cells were visualized with an inverted Zeiss Axiovert 200 microscope (40× objective) and digital images were captured with a CCD camera.
Quantification of Neurite Extension
Fluorescent images were obtained as described above. A total of 20 images (total area = 7.22 mm2) were analyzed in triplicate for the 1:0, 1:1, and 0:1 RDG:RGD protein samples. For each image, cells with neurites greater than one cell diameter and cells with neurites greater than two cell diameters were counted. Statistical significance between data sets was determined using a paired, two-tailed Student’s t-test.
Material Design and Synthesis
A schematic depicting the modular genetic design and exact amino acid content of our engineered proteins is shown in Figure 1
; a total of six novel protein polymers were synthesized for subsequent study. Each recombinant protein contains two peptide tag sequences for identification, an enterokinase cleavage site for tag removal, and four repeats of a sequence containing a nanoscale biofunctional domain followed by a nanoscale structural domain. The multi-step cloning strategy used to synthesize these proteins (Figure 2
) first required the design and commercial synthesis of oligonucleotide strands corresponding to each protein module. Single-stranded oligonucleotide sequences were designed to encode a specific domain sequence and to contain flanking regions which introduced key enzyme restriction sites for the remainder of the cloning process (see Figure 1 in Supplementary Material for a detailed plasmid map). Each of these sequences was optimized for dominant codon usage in E. coli in an effort to increase final protein expression yields.
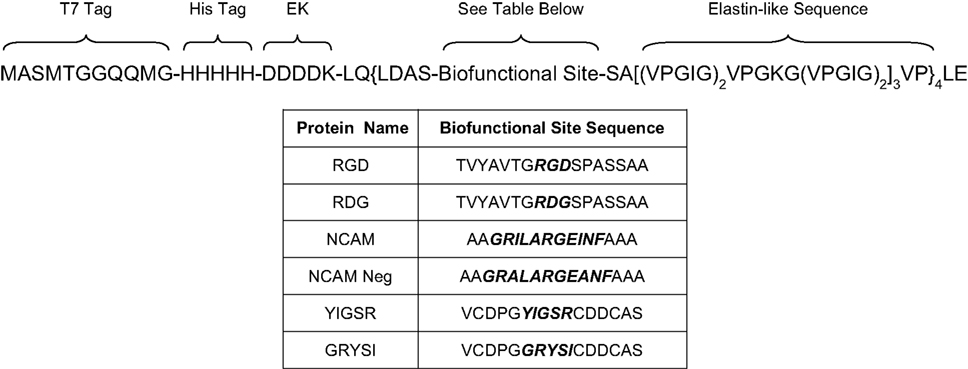
Figure 1. Amino acid sequences of the nanoscale-designed proteins (EK refers to an enterokinase cleavage site).
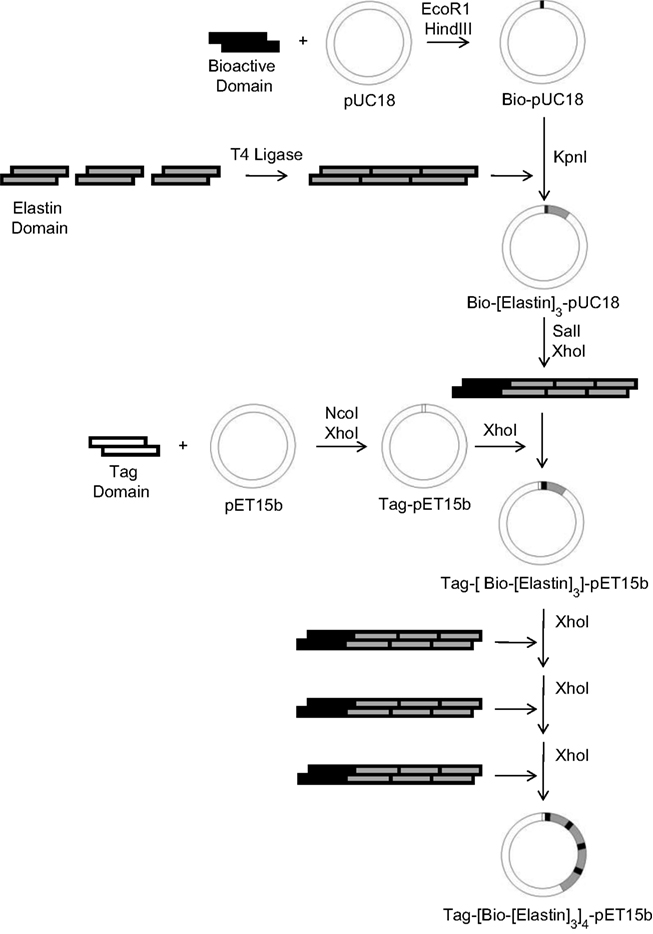
Figure 2. Multi-step cloning procedure for construction of protein expression plasmids (Tag: tag domain, Bio: biofunctional nanodomain, Elastin: structural nanodomain).
As shown in Figure 2
, the synthesis of the first protein expression plasmid required a total of 11 steps (including annealing the synthetic oligonucleotide strands), but for each additional recombinant protein only six steps were required since the structural and tag sequences are identical. The multi-step cloning process makes use of two commercially available vectors, the high-copy number pUC18 cloning vector and the pET15b expression vector, which places a T7lac promoter upstream of the target protein sequence providing a mechanism for inducible expression in bacterial culture. In order to allow for multimerization of the biofunctional and structural domains within the final expression vector, SalI and XhoI restriction sites were positioned on either side of each repeated unit. This design takes advantage of the recognition properties of the SalI and XhoI restriction enzymes, which cut different DNA sequences but create identical sticky ends, thus allowing for repeated insertion of the domain sequences by destroying the previous XhoI sites. Directly following tetramerization of the biofunctional and structural domains for each of the six proteins, the DNA sequence of the resultant expression plasmid was confirmed with sequencing analysis.
The inherent flexibility of this cloning strategy allowed for the custom preparation of six proteins containing different nanoscale biofunctional peptide domains. In specific, three domains, previously shown to affect neuronal cell behavior, were selected for synthesis: the RGD domain of fibronectin, the YIGSR domain in the β1 chain of laminin, and the P2 domain of NCAM. The RGD domain of fibronectin, a protein found in the developing central nervous system at sites of axonal extension, has been previously shown to promote neurite attachment and elongation due to interaction with a variety of integrins displayed on the surfaces of neurites (Meiners and Mercado, 2003
). The YIGSR domain of laminin, a protein that serves as a major component of the neuronal basement membrane and that has been found to be present during development and upon injury, has been linked to increased neuronal attachment through interaction with an hypothesized neuronal receptor homologous to one identified in epithelial cells (Meiners and Mercado, 2003
). The P2 peptide found in NCAM, a glycoprotein displayed on the surfaces of neuronal cells involved in cell–cell and cell–matrix interactions, has been shown to inhibit cellular aggregation and to increase neurite outgrowth (Soroka et al., 2002
). Amino acid sequences beyond the minimal active peptide sequence were used to construct the nanoscale biofunctional domains in order to promote bioactivity within the protein polymer by facilitating conformational flexibility and potentially preserving the secondary structure of the peptide (Heilshorn et al., 2003
). In addition, sequences corresponding to nanoscale negative controls of the RGD, YIGSR, and NCAM domains were incorporated into three additional proteins by either scrambling the primary amino acid sequence of the domain or including previously identified negative sequences (Liu et al., 2004
; Soroka et al., 2002
).
All six proteins contain identical nanoscale structural domain content, which mimics the repeated VPGVG sequence found in the ECM protein elastin, a fibrous protein naturally found in connective tissue that possesses known properties of elasticity and resilience (Figure 1
) (Nicol et al., 1992
). The elastin-like sequence is not known to affect neuronal cell behavior and is, therefore, meant to serve as a relatively bio-inert backbone for the presentation of nanoscale biofunctional peptide domains and to facilitate reproducible physical adsorption of the nanoscale-designed proteins. Previous characterization of elastin-like proteins have demonstrated biocompatibility, high expression levels in bacterial fermentations, and cost-effective purification (Heilshorn et al., 2005
; Meyer and Chilkoti, 1999
).
The properties of these proteins allow for purification using two different strategies, either a simple inverse temperature cycling technique or standard affinity column purification (Meyer and Chilkoti, 1999
). The hydrophobic elastin-like structural sequence in the proteins causes the polymers to exhibit LCST behavior (Urry et al., 1992
). At temperatures below the LCST, the proteins readily dissolve in water and at temperatures above the LCST, the proteins aggregate into a protein-rich coacervate, allowing for purification by centrifugation. The LCST behavior of the RGD protein is depicted in Figure 3
, showing an LCST value of approximately 33.5°C. Based on previous work relating elastin-like sequences to LCST behavior, the elastin-like sequence VPGIG (Figure 1
) was chosen to potentially allow for temperature cycling between 4°C (solubilization) and 37°C (precipitation), convenient temperatures commonly found in a molecular biology laboratory (Urry et al., 1992
). The inverse temperature cycling purification procedure worked effectively for the RGD, RDG, NCAM, and NCAM Neg proteins, resulting in un-optimized yields of approximately 25–50 mg/mL. The properties of the YIGSR and GRYSI protein required His-tag affinity column purification to achieve similar yields. Purity of all nanoscale-designed recombinant proteins produced in E. coli bacteria was confirmed using protein electrophoresis gels, Figure 4
. The tag domains were left attached to all proteins in this work but could be removed in future applications through cleavage with enterokinase.
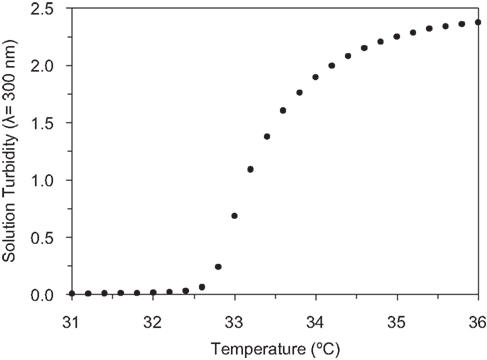
Figure 3. Measurement of the LCST of the RGD protein by monitoring changes in solution turbidity (λ = 300 nm) as a function of temperature.
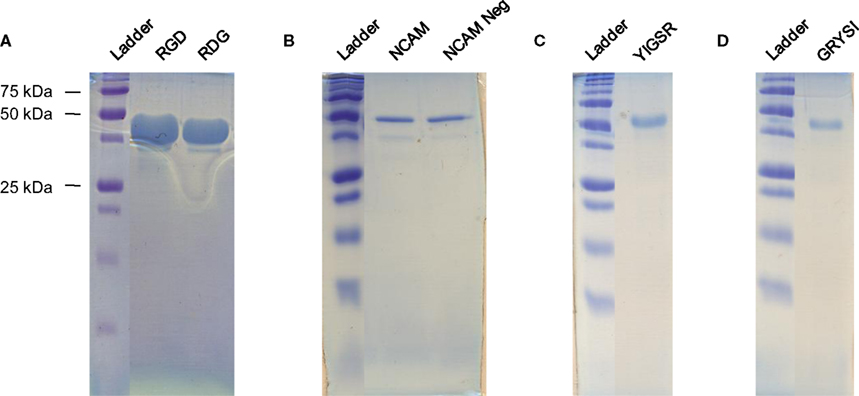
Figure 4. Protein electrophoresis gels of purified proteins. (A) RGD and RDG, (B) NCAM and NCAM Neg, (C) YIGSR, and (D) GRYSI.
Quantification of Adsorbed Protein Surfaces
The hydrophobic nature of the nanoscale elastin-mimetic domain was hypothesized to enable formation of reproducible thin films of engineered proteins through simple physical adsorption. To probe this technique, thin films of adsorbed, engineered proteins were prepared on glass substrates by dissolving purified protein in PBS buffer (pH = 7.4) at known concentrations and incubating these solutions on surfaces overnight at 4°C, a temperature at which the proteins are known to be soluble. Following adsorption, the surfaces were thoroughly washed to remove any unbound protein polymers. Adsorbed protein content was quantified by performing the BCA protein concentration assay (Heilshorn et al., 2005
; Ishii-Watabe et al., 2007
; Puleo et al., 2002
; Shoichet et al., 1994
). In this assay, polypeptides (more than three peptide bonds) reduce a copper reagent (Cu2+ to Cu1+) within an alkaline medium, and the reduced copper state is detected by a sensitive colorimetric detection reagent. The spectrophotometric absorbance (λ = 562 nm) of the reacted solution is proportional to the amount of protein present on the surface; the absolute concentration is determined using soluble calibration standards of the protein of interest (Heilshorn et al., 2005
; Ishii-Watabe et al., 2007
; Puleo et al., 2002
; Shoichet et al., 1994
; Stich, 1990
).
Physical adsorption isotherms were prepared using this assay for the RGD, YIGSR, and NCAM proteins by adsorbing proteins on glass surfaces using solutions containing 0–6 mg/mL protein, Figure 5
. Despite differences in their bioactive domain content, the engineered proteins adsorbed to the glass substrate at similar saturation levels, 30–44 ng/mm2, suggesting that the hydrophobic nature of the nanoscale elastin-mimetic structural domains and/or the overall protein molecular weight may be the primary driving forces for physical adsorption.
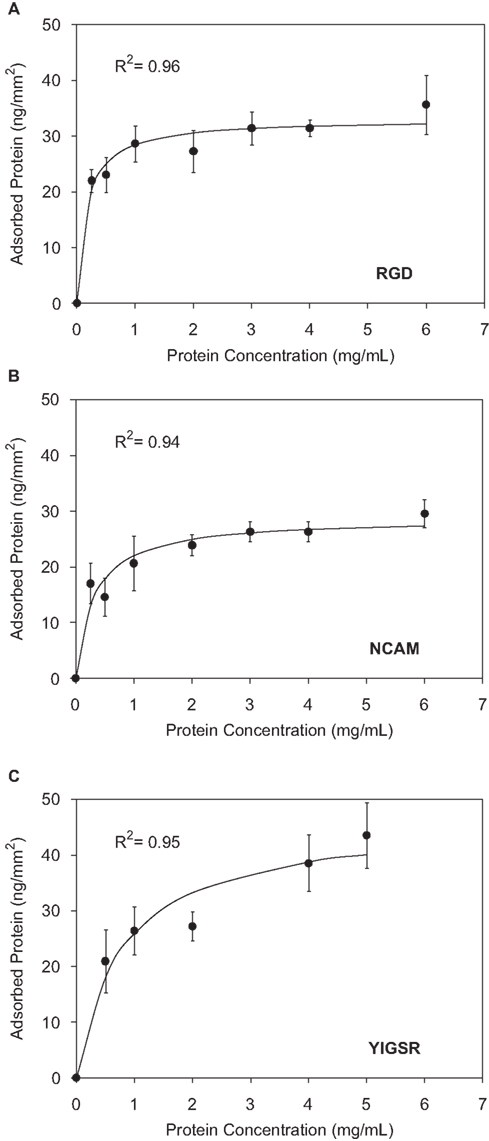
Figure 5. Physical adsorption isotherms as determined using the BCA assay for (A) RGD, (B) NCAM, and (C) YIGSR nanoscale-designed proteins.
Since recombinant protein production of rationally-designed proteins offers precise, nanoscale control over polymer content, the surface concentration of adsorbed, engineered protein is directly related to the surface concentration of available ligand; one protein polymer presents four nanoscale biofunctional domains. This high level of control over protein synthesis and the availability of a simple quantification reaction allows for the facile preparation of coated surfaces with reproducible and known biofunctional ligand concentrations. The amount of ligand present on a surface can be further tuned by preparing combinatorial surfaces containing mixtures of proteins with different nanoscale biofunctional domains, including the negative control domain sequences. This approach to fabricating bioactive surface coatings avoids techniques involving grafting synthetic peptides, which are difficult to control and quantify and often have limited biofunctionality, or immobilizing harvested ECM proteins, which present a complicated array of cell–surface interactions and may be present as multiple splice variants, confounding experimental results and complicating data analysis and reproducibility.
PC-12 Cell Culture on Adsorbed Protein Surfaces
The effective use of these surface-adsorbed proteins as neural implant coatings requires biocompatibility with neuronal cells and biofunctionality of the nanoscale domains intended to bind with specific cell–surface receptors. As an initial demonstration of in vitro cell–surface biocompatibility, a clonal line of rat pheochromocytoma cells known as PC-12 cells were grown on adsorbed RGD and collagen surfaces; adsorbed collagen is commonly used to promote adhesion of neuronal-like PC-12 cells (Banker and Goslin, 1998
) and has been considered for use in coating neural implants to enhance biocompatibility (Zhong et al., 2001
). While PC-12 cells are known to sometimes behave differently from primary neurons, they are often utilized for the preliminary evaluation of biomaterials (Arien-Zakay et al., 2009
; Cheng et al., 2003
; Gunn et al., 2005
).
Adsorbed protein films were prepared by dissolving RGD protein (1 mg/mL) and collagen in PBS buffer and incubating these solutions on glass coverslips overnight, at 4°C. These surfaces were then thoroughly washed and blocked with BSA to prevent non-specific cell adhesion. PC-12 cells were differentiated on the coverslips in the presence of 50 ng/mL nerve growth factor (NGF). Following 6 days of culture, the cells were analyzed for morphological differences between the engineered surfaces and the collagen positive-control surfaces using immunostaining (Figure 6
). Positive staining for neuron-specific β-tubulin on both surfaces indicated that the engineered surfaces supported cell proliferation, neuronal differentiation, and neurite outgrowth, and no evidence of cytotoxicity was observed.
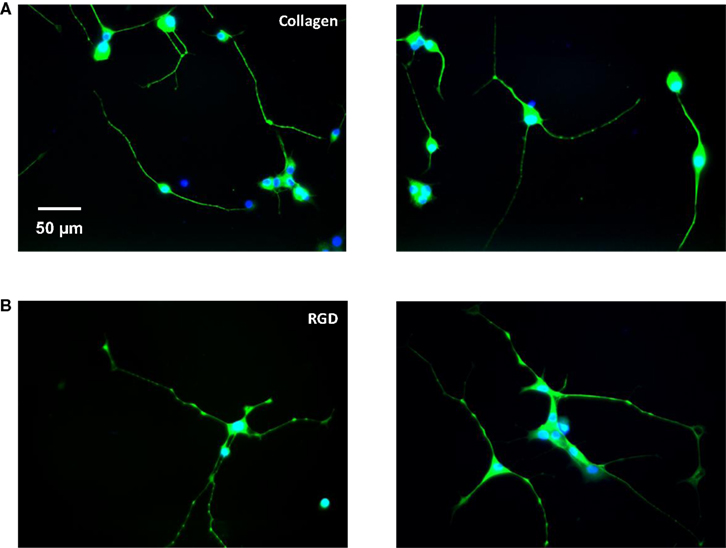
Figure 6. Representative fluorescent images of PC-12 cells differentiated for 6 days on (A) adsorbed collagen and (B) adsorbed RGD protein; nuclei stained with DAPI in blue, cytoplasm stained with neuron-specific β-tubulin in green.
Tuning Cellular Responses Through Combinatorial Presentation of Engineered Proteins
Once initial biocompatibility was established, the surface-adsorbed engineered proteins were further evaluated for their ability to control cell behavior by tuning ligand surface composition. In specific, adsorbed protein surfaces were prepared that contained variable nanoscale RGD biofunctional domain density. This was accomplished by preparing protein adsorption solutions containing a constant amount of total protein (1 mg/mL) but different ratios of RGD and RDG protein (1:0, 1:1, 0:1 RDG:RGD). Since the RGD and RDG proteins have the exact same amino acid composition, equivalent glass adsorption amounts can be accurately assumed for both polymers. This experimental design allows systematic tuning of the RGD biofunctional domain density while maintaining a uniform coverage of protein surface concentration. Therefore, effects of specific cellular interactions with the RGD biofunctional domain can be independently isolated from non-specific, physical cell–protein interactions.
Adsorbed surfaces were prepared on coverslips as described above and were again blocked with BSA; collagen positive-control surfaces and BSA negative-control surfaces were also prepared. PC-12 cells were differentiated on these films with 50 ng/mL NGF, and after 6 days, phase contrast images were taken of each surface sample; representative images are shown in Figure 7
. The available RGD ligand density was calculated for each substrate assuming a total protein surface concentration of 28.6-ng protein per mm2, the value measured for an adsorbed surface prepared from a 1-mg/mL solution of RGD protein (Figure 5
A). The quantified RGD ligand densities were determined to be 0, 0.91, and 1.82 RGD peptides/nm2 for the substrates prepared from 1:0, 1:1, 0:1 RDG:RGD solutions, respectively.
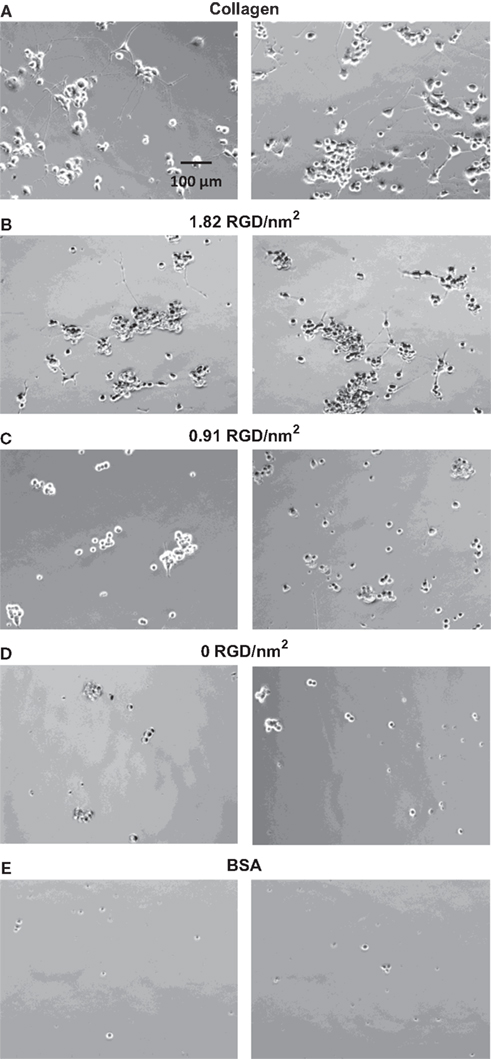
Figure 7. Phase contrast images of PC-12 cells differentiated for 6 days on control substrates and adsorbed protein films with varying RGD biofunctional nanodomain density and uniform overall protein surface coverage. (A) Collagen, (B) 1.82 RGD peptides/nm2, (C) 0.91 RGD peptides/nm2, (D) 0 RGD peptides/nm2, and (E) BSA.
PC-12 cell adhesion on the thin film containing a surface concentration of 1.82 RGD biofunctional nanodomains per nm2 showed enhanced cell adhesion relative to the negative BSA control but exhibited lower levels of cell adhesion and increased aggregation levels relative to the positive collagen control. This is an expected result, since the RGD films display only a single biofunctional nanodomain, while the collagen surfaces are known to display multiple beneficial nanodomains. Also readily apparent in the phase contrast images (Figure 7
) is the considerable difference in the number of neurites extended on the engineered surfaces as a function of RGD domain density. Image analysis and quantification from 20 fields of view, n = 3, (Figure 8
) demonstrated that PC-12 cells exhibited a statistically significant increase in total neurite extension (neurite length > 1 cell body diameter) and neurite length (neurite length > 2 cell body diameters) with increasing RGD surface concentration and that the highest level of neurite extension was achieved on the positive collagen control.
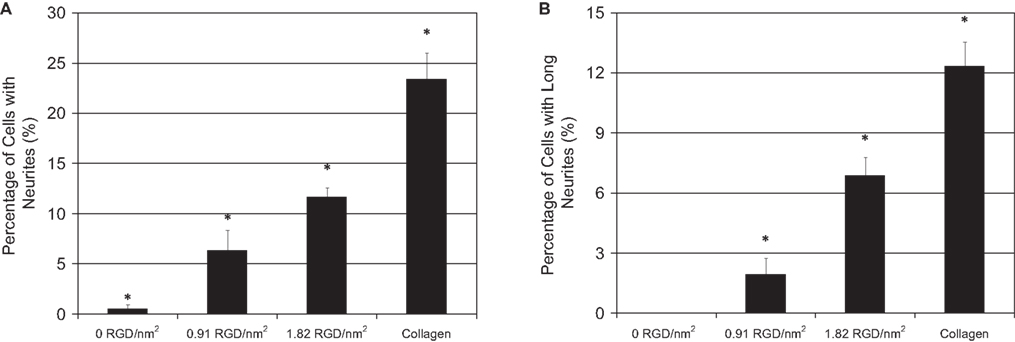
Figure 8. Quantification of PC-12 neurite extension after 6 days of differentiation on collagen positive-control substrates and adsorbed protein films with varying RGD biofunctional nanodomain density and uniform overall protein surface coverage. (A) Percentage of cells with neurites > 1 cell body diameter. (B) Percentage of cells with long neurites > 2 cell body diameters. Substrates are statistically different, p < 0.04.
The ultimate goal of this research is to create more reproducible, customizable, and quantifiable surface coatings, which can further enhance the biocompatibility of neural implants by studying specific cell–surface interactions. In this work, we have presented the design and synthesis of six recombinant, nanoscale-designed proteins that display various biofunctional ligands within an identical and relatively bio-inert elastin-mimic backbone. The specific biofunctional nanodomains chosen for integration within the rationally-designed proteins were derived from the naturally occurring proteins fibronectin, laminin, and NCAM and have been previously shown to both enhance neuronal cell adhesion and increase neurite extension. These engineered proteins were synthesized using a carefully designed sequence of flexible but robust cloning steps, making them highly amenable to sequence customization for the presentation of other biofunctional ligand domains already identified in the literature (Meiners and Mercado, 2003
). Using this framework, additional proteins can be easily engineered to display ligands that may play important roles in controlling the inflammatory response upon implantation of a neural device.
Due to a combination of thermodynamic interactions, most proteins show high surface affinity and are, therefore, easily adsorbed to surfaces via physical interactions (Hlady and Buijs, 1996
; Nakanishi et al., 2001
; Wahlgren and Arnebrant, 1991
; Wilson et al., 2005
). The adsorption process is very robust and as a result can be uniformly applied to implant surfaces of various shapes. In particular, the use of adsorbed engineered proteins allows for the preparation of surfaces with highly reproducible and quantifiable nanoscale ligand content. Such precision is made possible by the inherent, molecular-level control over protein composition offered by recombinant bacterial expression. Unlike ECM proteins harvested from natural mammalian sources, these nanoscale-designed proteins contain a known number of specific biofunctional ligands, and this relationship can be used to experimentally determine available ligand density on adsorbed surfaces using a straightforward and potentially high-throughput protein concentration assay. The rational-protein-design strategy also enables the incorporation of novel nanoscale ligands, such as the P2 domain of NCAM included here, that are not normally presented within the natural ECM. Physical adsorption isotherms were prepared for the RGD, YIGSR, and NCAM proteins and revealed similar levels of protein surface saturation independent of biofunctional domain content. This strategy will enable the preparation of surface coatings with controlled surface concentrations of multiple nanoscale ligands for specific cell–material interactions. The ability to accurately characterize and control surface content across a wide variety of potential ligands is a significant improvement over current techniques that involve grafting peptides or entire ECM proteins.
Initial studies of these nanoscale-designed protein surfaces in two-dimensional, in vitro PC-12 cell culture experiments demonstrated a high level of cell–surface biocompatibility and similar cellular morphology to that seen on collagen positive-control surfaces. Also important for eventual in vivo application is ligand biofunctionality within the engineered protein coating so that the surfaces can be used to stimulate cell behavior. It was shown that by simply increasing the density of nanoscale RGD domains within a surface coating of uniform protein coverage, PC-12 neurite extension was significantly increased. Future studies involving known mixtures of multiple ligands and varied ligand densities with more therapeutically relevant neuronal cell lines will be conducted to help elucidate the optimal surface composition for a given neural implant application. In order to fully characterize these combinatorial ligand displays, techniques must first be developed to accurately quantify the presence of multiple ligands on a single surface. The additional NCAM and YIGSR proteins introduced in this work represent initial efforts toward this goal and serve to demonstrate the versatility of our protein-design strategy for inclusion and presentation of a variety of biofunctional ligands within a standardized biocompatible polymer.
Taken together, these results provide much motivation for the future study and characterization of surface-adsorbed, nanoscale-designed proteins. Thin film coatings prepared by physical adsorption of engineered proteins possess highly desirable properties such as sequence tunability, reproducible surface content, multiple ligand display, and quantifiable ligand density. These coatings have demonstrated their use as a reproducible, quantifiable, and tunable strategy to directly affect cellular behavior and show potential for many future applications including in vitro cell screening, fundamental studies of cell–ligand interactions, and eventual use as biocompatible in vivo coatings for neural implants.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors acknowledge funding support from the National Academies Keck Futures Initiative, the John and Ulla deLarios Scholar Fund, and the Hellman Faculty Scholar Fund.
The Supplementary Material for this article can be found online at http://www.frontiersin.org/neuroengineering/paper/10.3389/neuro.16/009.2009
.
Soroka, V., Kiryushko, D., Novitskaya, V., Ronn, L. C. B., Poulsen, F. M., Holm, A., Bock, E., and Berezin, V. (2002). Induction of neuronal differentiation by a peptide corresponding to the homophilic binding site of the second Ig module of the neural cell adhesion molecule. J. Biol. Chem. 277, 24676–24683.