Laboratory for Neuronal Circuit Dynamics, RIKEN Brain Science Institute, Saitama, Japan
Genetically encoded fluorescent protein (FP) voltage sensors are promising tools for optical monitoring of the electrical activity of cells. Over the last decade, several designs of fusion proteins have been explored and some of them have proven to be sensitive enough to record membrane voltage transients from single mammalian cells. Most prominent are the families of voltage sensitive fluorescent proteins (VSFPs) that utilize the voltage sensor domain (VSD) of Ciona intestinalis voltage sensor-containing phosphatase (Ci-VSP). The voltage sensitivity of the fluorescence readout of these previously reported membrane potential indicators is achieved either via a change in the efficiency of fluorescence resonance energy transfer between two FP spectral variants or via modulation in the fluorescence intensity of a single FP. Here, we report our exploration on a third VSFP design principle based on circularly permuted fluorescent protein (cpFP) variants. Using circularly permuted EGFP derived from GCaMP2 and two newly generated circularly permuted variants of the far-red emitting protein named mKate, we generated and characterized a series of voltage-sensitive probes wherein the cpFPs were fused to the VSD of Ci-VSP. The most promising variants were based on circularly permuted mKate with new N- and C-termini given by residues 180 and 182. Even so their voltage sensitivity was relatively modest, they constitute a proof of principle for this novel protein design.
Several designs of genetically encoded optical probes for membrane potential have been explored during the last decade. The first genetically encoded membrane voltage sensor, denoted Flash, was based on molecular fusion of Aequorea victoria green fluorescent protein (GFP) to the C-terminus of Drosophila Shaker potassium channel in which voltage-driven rearrangements of the channel alter the brightness of GFP (Siegel and Isacoff, 1997
). The second voltage sensor, named voltage-sensitive fluorescent protein 1 (VSFP1), was developed in our laboratory and is composed of the voltage sensor domain (VSD) from the Kv2.1 potassium channel fused to a pair of cyan and yellow fluorescent proteins (Sakai et al., 2001
). The third voltage sensor, denoted SPARC, was based on the molecular fusion of GFP into a skeletal muscle sodium channel (Ataka and Pieribone, 2002
). These three voltage sensors were limited by their poor plasma membrane targeting in mammalian cells (Baker et al., 2006
).
Lately, a homolog to the VSD of voltage-gated potassium (Kv) channels was found in the non-ion channel protein Ciona intestinalis voltage sensor-containing phosphatase (Ci-VSP, Murata et al., 2005
). Replacing the Kv VSD in VSFP1 with the self-contained VSD of Ci-VSP resulted in the first fluorescent protein (FP) voltage sensor that showed excellent membrane targeting in PC12 cells or hippocampal neurons and that reliably reported membrane voltage changes in several types of mammalian cells (Dimitrov et al., 2007
). The most promising variant from the first series of these constructs was termed VSFP2.1. Optimizing the interdomain linkers in VSFP2.1 and using alternative FP variants resulted in functionally improved versions termed VSFP2.3 and VSFP2.4. Using a single FP instead of the fluorescence resonance energy transfer (FRET) pair yielded VSFP3.1. These latest versions of the second (VSFP2s) and third (VSFP3s) generation FP voltage sensors exhibit an improved targeting to the plasma membrane, dynamic range and kinetics of voltage-dependent fluorescent changes (Lundby et al., 2008
; Mutoh et al., 2009
).
In spite of this initial breakthrough, development of optimized sensors with superior signal-to-noise ratio and optimized spectral properties remains imperative for in depth analysis of neuronal circuit dynamics. One of the most straightforward approaches to develop genetically encoded sensors with superior dynamic range and sensitivity is based on circularly permuted fluorescent proteins (cpFP) (Baird et al., 1999
; Nagai et al., 2001
; Nakai et al., 2001
; Knöpfel et al., 2003
). Circular permutation of a protein consists of connecting the native amino (N-) and carboxyl (C-) termini covalently with a peptide linker and cleaving the peptide backbone at one specific site (Luger et al., 1989
). Pioneering work of Baird et al. (1999)
have demonstrated circular permutation sites within Aequorea victoria GFP variants that preserve the original folding and formation of fluorophore. In these fluorescent circularly permuted GFP variants, new termini are introduced into alpha helices and beta sheets. Circular permutation thus allows placing the fusion of a sensor domain in close proximity to the chromophore environment, which can lead to direct changes in the fluorescent signal upon structural rearrangements initiated by the sensor domain. Based on this concept, several Ca2+ sensors have been developed, the best of which show much greater fractional fluorescent changes than FRET-based Ca2+ probes (Nagai et al., 2001
; Nakai et al., 2001
; Tallini et al., 2006
; Souslova et al., 2007
). Expanded dynamic range and good brightness values of these reported calcium sensors inspired us to explore a voltage sensor design based on cpFP. Here, we report the development of cpFP-based voltage sensors by using a known circularly permuted version of EGFP (Baird et al., 1999
) and two new cpFPs generated from the far red mKate (Shcherbo et al., 2007
). We conclude that the red-shifted cpFP-based voltage sensors will be of particular interest for in vivo imaging due to the elimination of green/yellow autofluorescence and for deep tissue imaging using two-photon excitation microscopy.
Generation of EGFP-Based Circularly Permuted Fluorescent Protein Variants
The cDNA encoding the circularly permuted EGFP of GCaMP2 (cpEGFP, 149–144; Diez-Garcia et al., 2005
; Tallini et al., 2006
) was used as a template for PCR amplification using a sense primer containing a NotI site (5′-GTCTGCGGCCGCCTGAGCTCACTCGAGAACG TCTATATCATGG-3′) and a reverse primer comprising an EcoRI site (5′-CTTTGAATTCATCAGTACGCCCCAGGATGTTGCC-3′).
VSFP(A)cpEGFP, VSFP(B)cpEGFP, VSFP(C)cpEGFP and VSFP(D)cpEGFP were obtained by substituting the FRET pair in VSFP2A(R217Q) to VSFP2D(R217Q) (Dimitrov et al., 2007
) with cpEGFP using NotI and EcoRI restriction endonucleases.
Generation of mKate-Based Circularly Permuted Fluorescent Protein Variants
The cDNA coding for cpmKate(144) was generated by three separate PCRs. First, the cDNA of the 3′ portion of mKate comprising amino acids 144–231 (Shcherbo et al., 2007
) was amplified with a sense primer containing a NotI site (5′-ATAAGAATGCGGCCGCACCGAGATGCTGTACCCCG-3′) and an antisense primer encoding a GGSGGT peptide linker along with the last two amino acids of TagRFP (Merzlyak et al., 2007
) (5′-GGTACCGCCACTGCCGCCATTAAGTTTGTGCCCCAG-3′). Second, the cDNA of the 5′ portion of mKate including amino acids 1–143 was amplified with a forward primer encoding the GGSGGT peptide linker combined with a GFP-type N-terminal end (5′-GGCGGCAGTGGCGGTACCGTGTCTAAGGGCG AAGAGCTG-3′) and a reverse primer comprising both a HindIII site and a point mutation (alanine to threonine) at position 145 (5′-CCCAAGCTTTCAGGAGGTCTCCCAGCCGAGTG-3′) as reported for the Ca2+ indicator Case12 wherein the combination of Glu148/Thr145 resulted in a significant increase in the dynamic range of the sensor (Souslova et al., 2007
). Finally, the entire cDNA of cpmKate(144) was amplified with the NotI and HindIII site-containing primers by using a mixture of the first and second PCR fragments as a template. VSFP(A)cpmKate(144) and VSFP(D)cpmKate(144) constructs were obtained by substituting the FP pair in VSFP2A(R217Q) and VSFP2D(R217Q) (Dimitrov et al., 2007
) with cpmKate(144), respectively, using the NotI and HindIII restriction sites.
The coding sequence for cpmKate(180) was obtained using a strategy similar to that mentioned above. Briefly, the cDNA of the 3′ portion of mKate comprising amino acids 180–231 was amplified with a sense primer comprising a NotI site (5′-ATAAGAATGCGGCCGCTCCAAGAAACCCGCTAAGAAC-3′) and an antisense primer encoding a GGTGGS peptide linker and the last two amino acids of TagRFP (Merzlyak et al., 2007
) (5′-ACTACCGCCTGTTCCGCCATTAAGTTTGTGCCCCAGTTTGC-3′). The cDNA of the 5′ portion of mKate containing amino acids 1–182 was then amplified with a forward primer encoding the GGTGGS peptide linker along with a GFP-type N-terminal end (5′-GGCGGAACAGGCGGTAGTATGGTGTCTAAGGGCGAAGAG-3′) and a reverse primer comprising a HindIII site (5′-CCCAAGCTTTCATTTCTTGGATCTGTATGTGGTCTTCAAGTTGC-3′). Finally, the entire cDNA of cpmKate(180) was amplified with the NotI and HindIII site-containing primers by using a mixture of the first and second PCR fragments as a template. The primers were designed to include the duplication of three residues at both N- and C-terminal ends of the circular protein variant to favor a native-like conformation as described in Li et al. (2008)
. The amplified PCR fragment was then digested with NotI and HindIII restriction endonucleases and subcloned in either VSFP2A(R217Q), VSFP2B(R217Q), VSFP2C(R217Q) or VSFP2D(R217Q), resulting in VSFP(A)cp-mKate(180), VSFP(B)cpmKate(180), VSFP(C)cpmKate(180) and VSFP(D)cpmKate(180), respectively.
In order to generate the cytosolic cpmKate(180) construct, site-directed mutagenesis was first carried out to introduce a NotI site after the methionine start codon of the VSD of VSFP2D(R217Q) by using the forward primer (5′-CTAGCGCCACCATGGGCGGCCGCGACGGTTCAGATTTTAGTCC-3′) and reverse primer (5′-CTGAACCGTCGCGGCCGCCCATGGTGGCGCTAGCCAGCTTG-3′). The underlined nucleotides correspond to mutations introducing a NotI site. The cDNA corresponding to cpmKate(180) was then subcloned into the site-directed mutagenesis PCR product using NotI and HindIII restriction endonucleases, resulting in a cytosolic version of cpmKate(180), we termed pcDNA3.1(-)cpmKate(180).
Cell Culture and Transfection
PC12 cells were grown in high glucose Dulbecco’s modified Eagle’s medium supplemented with 5% fetal calf serum and 10% horse serum. Cells were grown on poly-D-lysine-coated coverslips and transfections were performed 24 h after plating using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions (Lipofectamine™ 2000, Invitrogen).
Electrophysiology and Optical Recordings
Coverslips with transfected PC12 cells were placed into a recording chamber mounted on the stage of an inverted microscope (Eclipse TE-2000 U, Nikon) and voltage-clamp recordings in whole-cell patch-clamp configuration were performed using an Axopatch 200B patch-clamp amplifier (Axon Instruments, Sunnyvale, CA, USA). Glass pipettes (3.5–5 MW) were pulled from borosilicate glass using a two-stage vertical puller (PP-830, Narishige, Japan). The pipette solution contained (in mM): CsCl 130, MgCl2 1, HEPES 20, EGTA 5, MgATP 3 at pH 7.2. External solution contained (in mM): NaCl 150, KCl 4, CaCl2 2, MgCl2 1, Glucose 5, and HEPES 5. Fluorescence transients were acquired using a photodiode-based photometry system (TILL Photonics). Clampex software (Axon Instruments) was used for data acquisition, voltage command pulses and fluorescence excitation. Excitation light was passed through a band pass filter and dichroic mirror (542/50 and DCLP 593 nm). Emission light was detected via a long pass filter (LP 610 nm). Clampfit (Axon Instruments) and Origin software (OriginLab, Northampton, MA, USA) were used to analyze the fluorescence and electrophysiological signals.
Fluorescence Analysis
Confocal images were obtained with a Nikon confocal laser scanning microscope (C1si/FN1, Nikon, Japan) using a 40× water immersion objective. A 444-nm wavelength diode laser was used to excite the fusion proteins involving cpEGFP. Linear deconvolution of spectrally resolved images was performed using EZ-C1 3.50 imaging software (Nikon, Japan). A 543-nm wavelength He–Ne laser was used to excite the cpmKate variants. The fluorescence intensity was measured relative to the background signal using ImagePro software.
Design and Characterization of VSFP Variants Based on cpEGFP
We took the advantage of cDNA encoding the FRET-based or single FP-based voltage sensors available in our laboratory to replace their fluorescent reporter domain with a cpFP. We first explored the use of cpEGFP149–144 derived from GCaMP2, wherein the numbers indicate the amino acid residues positioned at the N- and C-terminal ends of cpEGFP, respectively (Nakai et al., 2001
). In this cpEGFP variant, the original N- and C-termini were connected by a six amino acid linker (GGTGGS). The N-terminal portion of this cpEGFP was then connected to the C-terminal end of the VSD of Ci-VSP. We generated four different fusion protein versions, denoted VSFP(A)cpEGFP, VSFP(B)cpEGFP, VSFP(C)cpEGFP and VSFP(D)cpEGFP. The letters A–D refer to the increasing length of the amino acid linker connecting the S4 helix of the VSD to the fluorescent reporter protein as previously described in Dimitrov et al. (2007
). Principal designs for FRET-, single FP- and cpFP-based VSFPs are illustrated in Figure 1
.
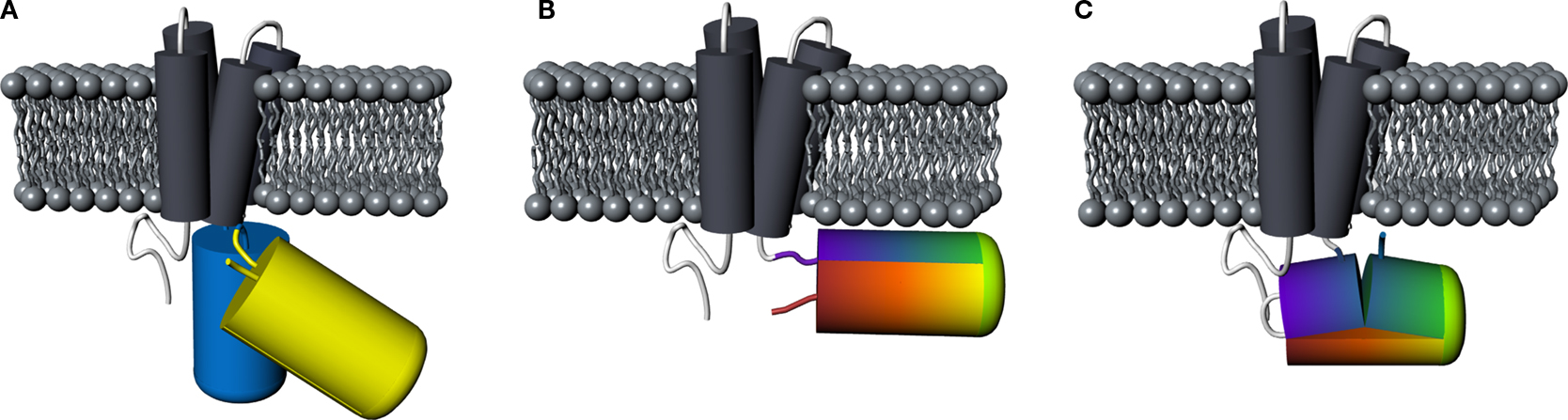
Figure 1. Comparison of principal designs for voltage-sensitive fluorescent proteins. (A) FRET-based voltage probe. Cyan- and yellow-emitting GFP variants are connected by a short linker. (B) Single FP-based voltage probe. (C) Circularly permuted FP-based voltage probe. The N- and C-terminal portions of GFP are swapped, resulting in new N- and C-terminal ends. The color gradient (from purple to red) shows the progression from the N- to the C-terminus of the original protein. Note the direct attachment of the GFP reporter to the S4 domain.
In order to investigate the fluorescence and membrane targeting of these proteins, we expressed VSFP(A)cpEGFP, VSFP(B)cpEGFP, VSFP(C)cpEGFP and VSFP(D)cpEGFP in PC12 cells and performed confocal imaging 3 days after transfection. Analysis of the confocal images revealed that all four fusion proteins were either non or only weakly fluorescent (Figure 2
). To minimize the influence of autofluorescence, linear spectral deconvolution was carried out as shown in the bottom row of Figure 2
. EGFP fluorescence (as identified by its spectral properties) was observed for VSFP(C)cpEGFP and VSFP(D)cpEGFP as shown in Figures 2
C,D, respectively. The fluorescence brightness of VSFP(C)cpEGFP and VSFP(D)cpEGFP was not noticeably different. In contrast, VSFP(A)cpEGFP was not fluorescent and VSFP(B)cpEGFP was only very weakly fluorescent. Due to the absence of a significant signal, the fluorescence intensity of VSFP(A)cpEGFP and VSFP(B)cpEGFP was enhanced by 10-fold. However, we found only green cellular autofluorescence with VSFP(A)cpEGFP and apparent (albeit dim) fluorescence with VSFP(B)cpEGFP in these conditions (shown in the white box of the first two images in the bottom row). While we could clearly observe VSFP(C)cpEGFP- and VSFP(D)cpEGFP-specific fluorescence, their membrane targeting was not efficient as revealed by comparing fluorescence and corresponding differential interference contrast images.
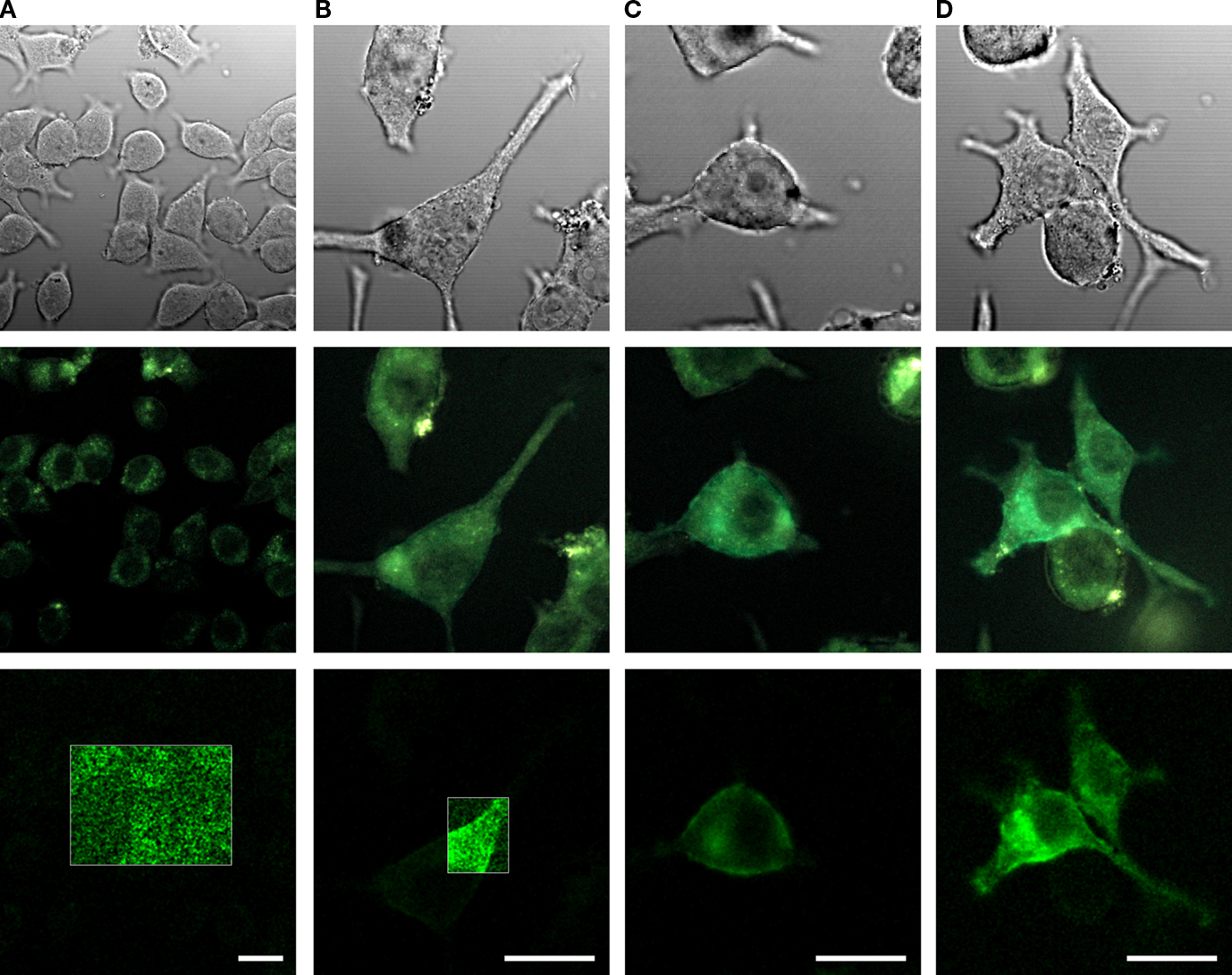
Figure 2. Comparison of the expression levels of cpEGFP-based VSFP variants in PC12 cells. Images of PC12 cells transfected with either VSFP(A)cpEGFP (A), VSFP(B)cpEGFP (B), VSFP(C)cpEGFP (C) or VSFP(D)cpEGFP (D). The first row illustrates the differential interference contrast images of PC12 cells. Second row, identical field of view, shows the real color fluorescence images of PC12 cells. The third row illustrates the GFP component of the fluorescence shown in the second row after linear spectral deconvolution of the real color images. The fluorescence intensity within the white box is represented with 10-fold larger scale. The cells were excited with a 444-nm wavelength diode laser. Scale bars = 20 μm.
Because of their very low fluorescence intensity, we did not test these constructs in further detail for voltage-dependent modulation of fluorescence.
Design and Characterization of VSFP Variants Based on cpmKate
The presence of significant background noise due to green tissue autofluorescence motivated us to develop a voltage sensor based on a red-shifted cpFP. To this aim, we took advantage of the newly developed far-red emitting FP named mKate (Shcherbo et al., 2007
) to explore its circularly permuted versions. The positions of the selected circular permutation sites for the generation of cpmKate variants were based on the three-dimensional structure of mKate (Pletnev et al., 2008
) and are illustrated in Figure 3
A.
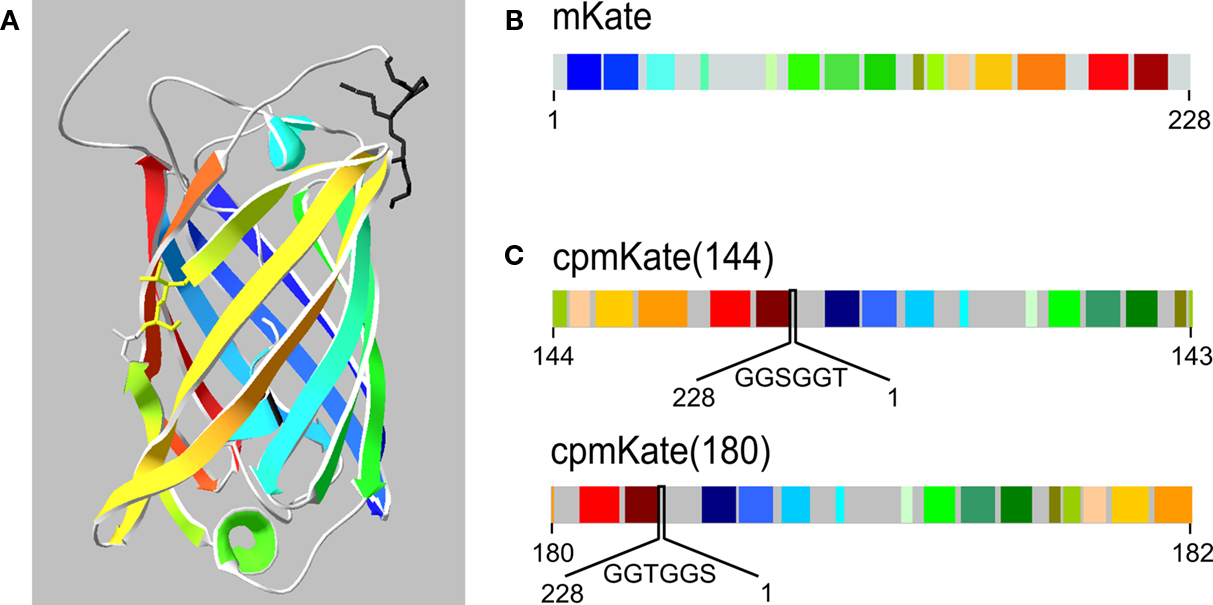
Figure 3. Schematic representation of mKate and cpmKate variants. (A) Ribbon representation of the crystal structure of mKate (Pletnev et al., 2008
). Note the color code for each beta sheet and alpha helix. The circular permutation sites at amino acid residues 144 and 180 are highlighted as a stick representation. (B) Schematic primary structure of mKate based on (A). (C) Design of cpmKate variants circularly permuted at amino acid residues corresponding to position 144 (upper line) and 180 (lower line). The linkers are shown in between the original N- and C-termini.
We first investigated circular permutation of mKate (Figure 3
B) at site 144–143 (Figure 3
C, upper line), wherein the numbers correspond to the amino acid residues located at the N- and C-termini of cpmKate(144). The original N- and C-terminal ends were connected by a six amino acid linker (GGSGGT). Fusion of cpmKate(144) into VSFP2A and VSFP2D scaffolds yielded VSFP(A)cpmKate(144) and VSFP(D)cpmKate(144), respectively. Nonetheless, both constructs did not display any fluorescence upon transfection in PC12 cells (data not shown).
Next, we circularly permutated mKate at amino acid residue 180 so that amino acid 180 and 182 became the new N- and C-terminus, respectively (Figure 3
C, lower line). The original N- and C-terminal ends were connected by a six amino acid linker (GGTGGS).
We first investigated the fluorescence properties of cpmKate(180) without fusion to the VSD of Ci-VSP. As expected, unfused cpmKate(180) was a soluble FP as indicated by its cytosolic expression pattern (Figure 4
A). The emission spectrum of cpmKate(180) was identical to that of the original mKate as shown in Figure 4
B. For both proteins, emission peaked around 625 nm. We then compared the fluorescence brightness of mKate and cpmKate(180) in transfected PC12 cells and found that cpmKate(180) was 24.6 ± 1.7 % less bright than the parent mKate (Figures 4
A,C).
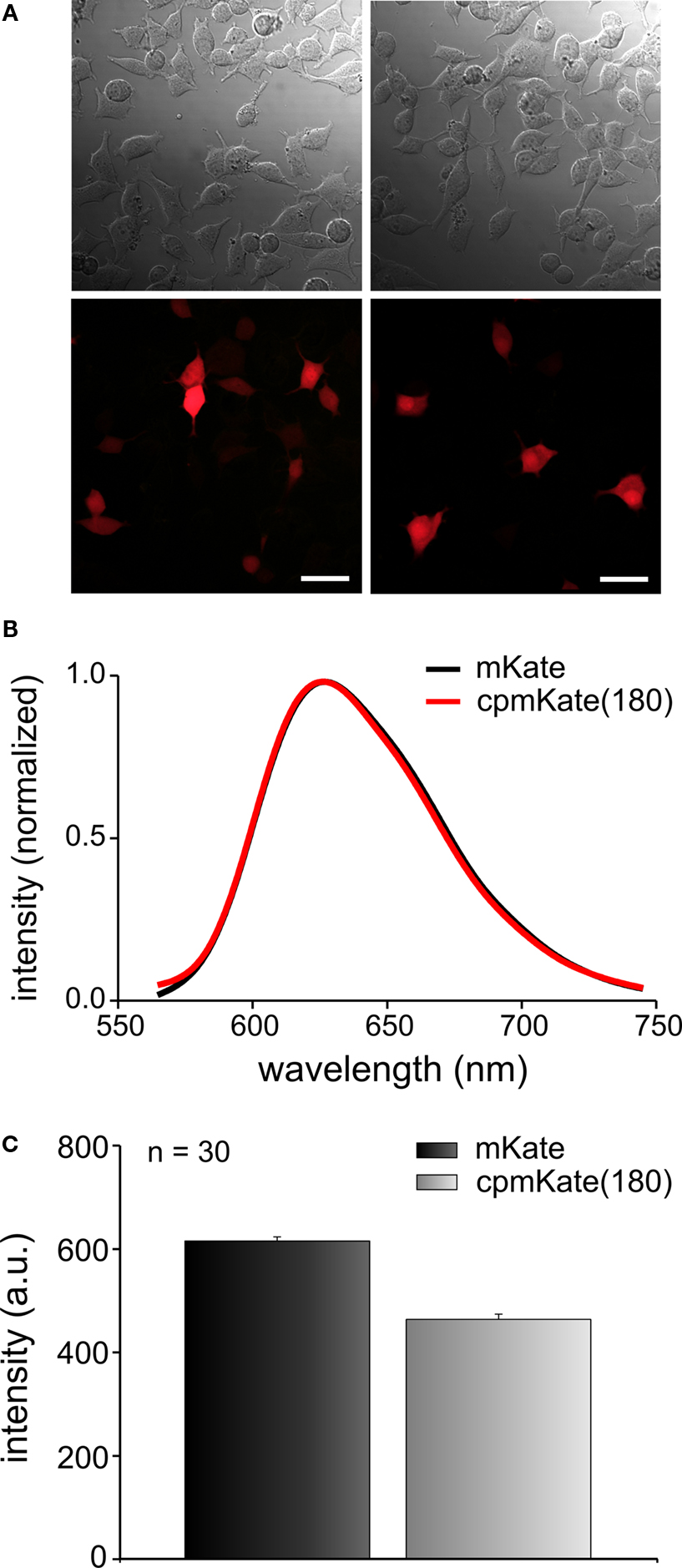
Figure 4. Comparison of fluorescence brightness and spectral properties of mKate and cpmKate(180) in transfected PC12 cells. (A) Differential interference contrast images (first row) and real color fluorescence images (second row) of PC12 cells transfected with pcDNA3.1(-)mKate (left column) and pcDNA3.1(-)cpmKate(180) (right column). (B) Normalized fluorescence emission spectra of mKate (black) and cpmKate(180) (red). (C) Comparison of mKate and cpmKate(180) fluorescence intensity. The error bars are standard error of mean (n = 30, number of cells). Scale bars = 30 μm.
After confirming that cytosolic cpmKate(180) is fluorescent, we fused it to the VSD of Ci-VSP, generating VSFP(A)cpmKate(180), VSFP(B)cpmKate(180), VSFP(C)cpmKate(180) and VSFP(D)cpmKate(180). The letters A–D denote different interdomain linkers as described above. PC12 cells were transfected with these constructs and confocal imaging was carried out 48 h after transfection. As shown in Figure 5
B, VSFP(A)cpmKate(180) did not show any fluorescence while VSFP(B)cpmKate(180) was found to be very weakly fluorescent. In contrast, both VSFP(C)cpmKate(180) and VSFP(D)cpmKate(180) displayed bright fluorescence and clear targeting to the plasma membrane (Figures 5
C,D). The fluorescence intensity of cpmKate(180)-based voltage sensor variants is illustrated in Figure 5
E. We could clearly observe a raise in fluorescence intensity as the length of the linker connecting the VSD to cpmKate(180) is increased.
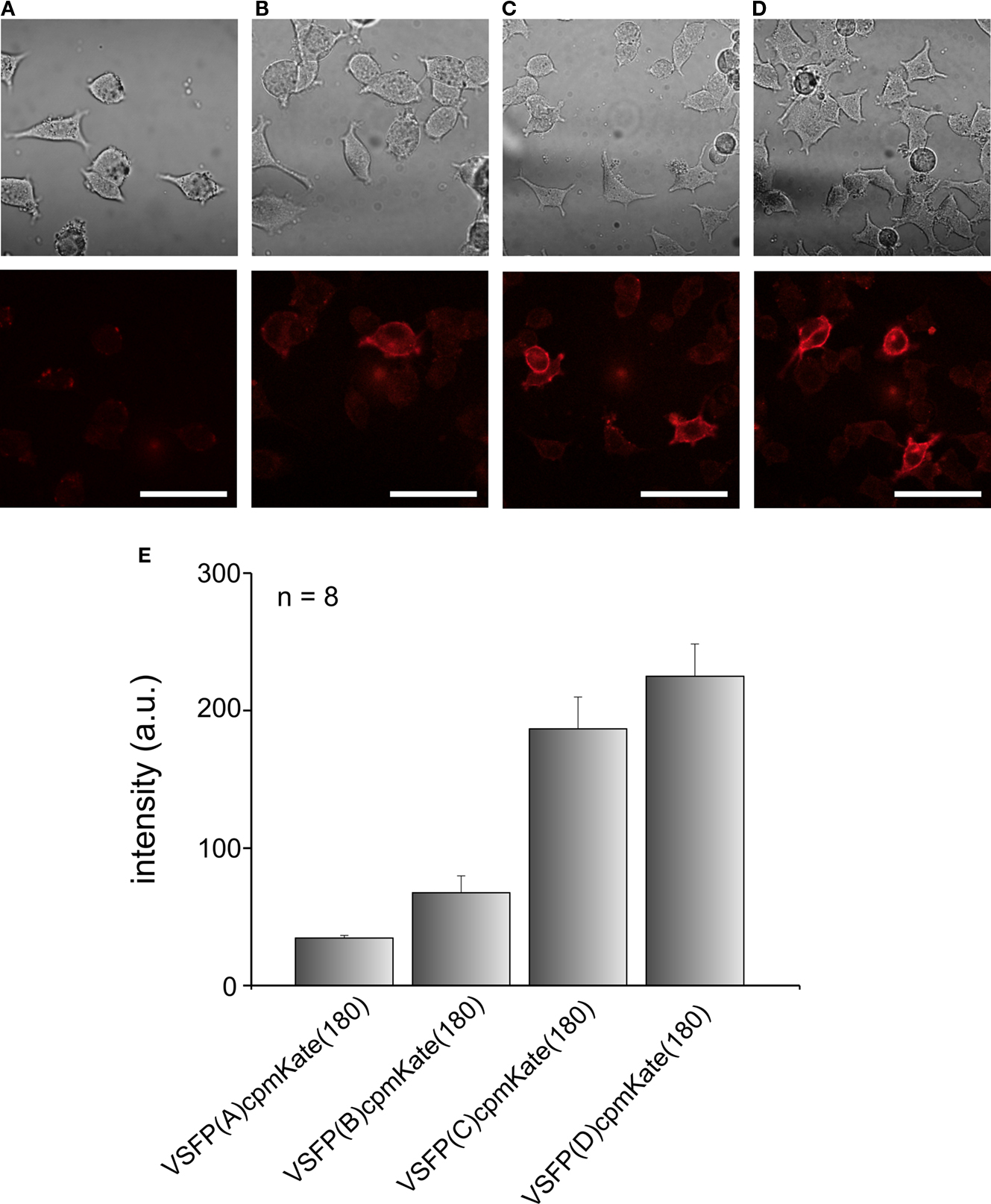
Figure 5. Comparison of the expression levels of VSFP variants derived from cpmKate(180) in PC12 cells. Differential interference contrast (upper row) and real color confocal fluorescence (lower row) images of PC12 cells expressing either VSFP(A)cpmKate(180) (A), VSFP(B)cpmKate(180) (B), VSFP(C)cpmKate(180) (C) or VSFP(D)cpmKate(180) (D). Note the targeting to the plasma membrane. (E) Comparison of the fluorescence intensity of cpmKate(180)-based VSFP variants. n = 8, number of cells used for each variant. Imaging was carried out using a 543-nm wavelength He–Ne laser. Scale bars = 50 μm.
We then investigated the relationship between cpmKate(180) fluorescence and membrane potential in voltage-clamped PC12 cells transfected with the two brightest VSFP variants derived from cpmKate(180) [VSFP(C)cpmKate(180) and VSFP(D)cpmKate(180)]. As illustrated in Figure 6
, both VSFP(C)cpmKate(180) and VSFP(D)cpmKate(180) exhibited a significant change in fluorescence when tested with depolarizing voltage pulses from −140 to 60 mV (1.9 and 1.2%, respectively). Time constants (Figures 6
B,E) and amplitudes (Figures 6
C,F) associated with the ON and OFF responses were plotted as a function of step voltage for both voltage sensors. Our analysis revealed half maximal responses (V1/2) were slightly shifted towards negative potentials as compared to previously reported VSFP-based variants (Table 1
; Dimitrov et al., 2007
; Lundby et al., 2008
; Mutoh et al., 2009
) while activation [τon = 57.1 and 64.0 ms for VSFP(C)cpmKate(180) and VSFP(D)cpmKate(180), respectively] and deactivation [τoff = 125.0 and 174.4 ms for VSFP(C)cpmKate(180) and VSFP(D)cpmKate(180), respectively] kinetics were slower (Figures 6
B,E) than those of the best VSFP2 and VSFP3 variants.
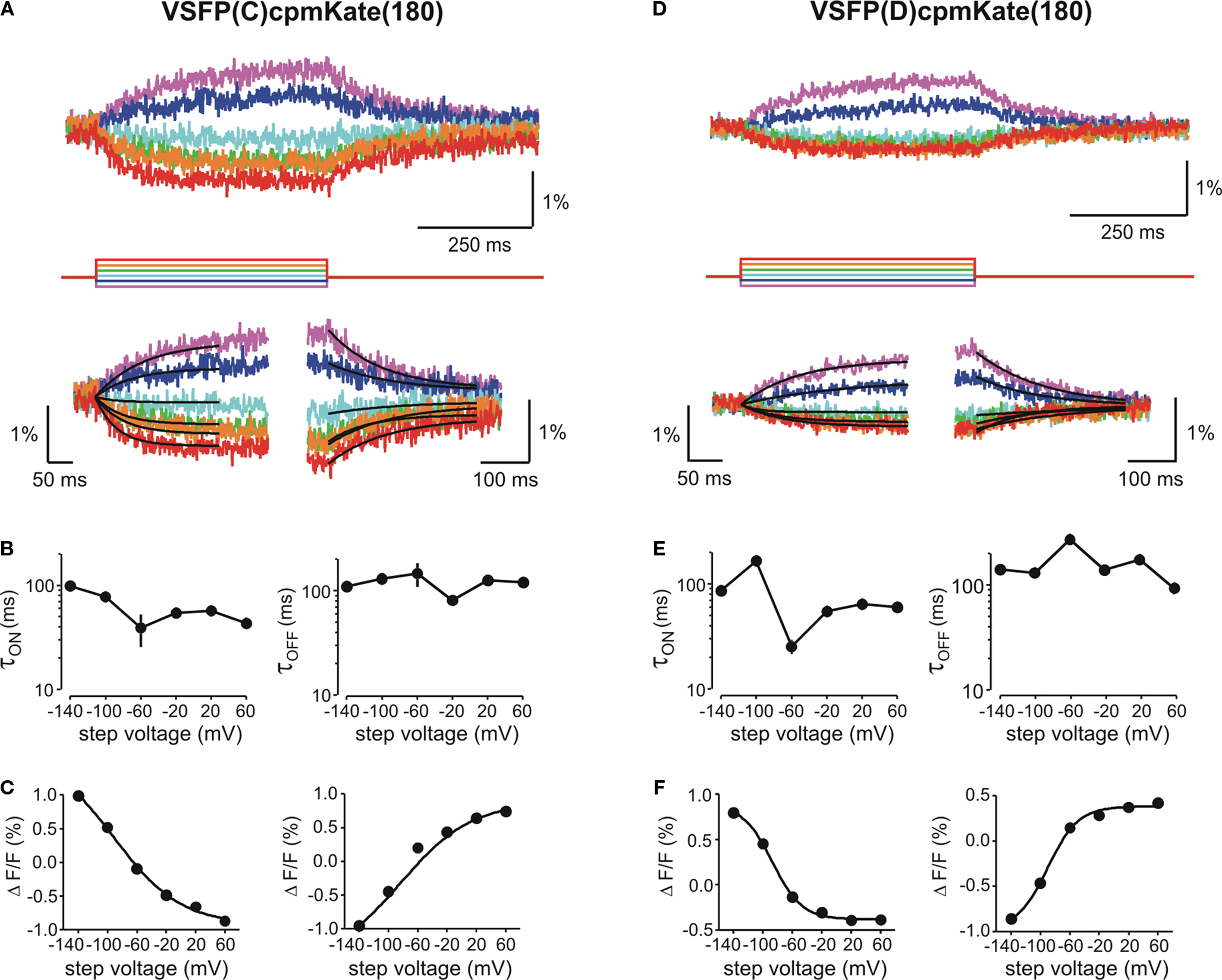
Figure 6. Functional characterization of cpmKate(180)-based VSFP variants in voltage-clamped PC12 cells. Changes in red fluorescence (upper traces) evoked by a family of voltage steps from a holding potential of −70 mV to test potentials ranging from −140 to 60 mV in 40 mV increments recorded from PC12 cells expressing VSFP(C)cpmKate(180) (A) or VSFP(D)cpmKate(180) (D). Lower panels show the single exponential fits of the ON (left) and OFF (right) time components of the fluorescence signal in response to each command potential. (B,E) Voltage dependence for the time constants of the ON (left) and OFF (right) responses. Note the slightly faster response of VSFP(C)cpmKate(180) as compared to VSFP(D)cpmKate(180). (C,F) Normalized fluorescence change versus step voltage during ON (left) and OFF (right) time responses. Lines are Boltzmann fits. Data are mean values obtained from five and six cells and 10–20 repetitions per cell for VSFP(C)cpmKate(180) and VSFP(D)cpmKate(180), respectively.
Fluorescence response properties of cpmKate(180)-based voltage sensors are summarized in Table 1
.

Based on the knowledge and advantages derived from previously reported cpFP-based Ca2+ sensors, we used cpFPs to investigate their suitability as a voltage reporter in VSFP2/3-like constructs. Although cpEGFP149–144 was previously reported to produce significant responses to Ca2+ fluctuations as a FP reporter in GCaMP (Nakai et al., 2001
) and GCaMP2 (Diez-Garcia et al., 2005
; Tallini et al., 2006
), cpEGFP-based VSFP variants did not lead to sufficiently bright fusion proteins (Figure 2
). The reason of the low fluorescence intensity likely lies in the fact that folding and formation of the cpFP fluorophore is very sensitively affected by the nature of the fusion partners (Baird et al., 1999
; Akemann et al., 2001
; Nagai et al., 2001
; Nakai et al., 2001
). Accordingly, the fluorescence intensity of the brightest VSFP-cpEGFP variants did not exceed green cellular autofluorescence, which therefore considerably limits their application in animal tissues.
To overcome this issue, we developed a new series of cpFP-based voltage sensors derived from the far-red shifted FP named mKate whose emission maxima reaches the 600-nm barrier (Shcherbo et al., 2007
). The 600–1100-nm wavelength range, in particular, constitutes an optimal window for visualization in living tissues since the absorption of melanin and hemoglobin is considerably reduced (König, 2000
; Shcherbo et al., 2007
). In addition, long-wavelength light is usually associated with improved tissue penetration due to an increase in the mean free path before scattering (Helmchen and Denk, 2005
). In this study, we generated two circularly permuted variants of mKate namely cpmKate(144) and cpmKate(180) based on the crystal structure of mKate (Pletnev et al., 2008
). The first version, cpmKate(144) did not show any fluorescence, suggesting that cpFP variants with termini located within the β-barrel (Figure 5
A) are likely to loose their ability to form a chromophore, which is consistent with the notion that residues within β-strands are critical for protein folding to a functional three-dimensional structure (Topell et al., 1999
). Amino acid residue 180 lies in the loop region between two β-strands (Figure 5
A) and according to Topell et al. (1999
), new termini in loops are less critical for protein folding. Indeed, circular permutation of mKate at amino acid residue 180 [pcDNA3.1(-)cpmKate(180)] was found to be significantly fluorescent. However, cpmKate(180) was dimmer than its parent mKate, which is consistent with observations on cpFP variants of GFP and mCherry reported by Baird et al. (1999) and Li et al. (2008)
. Based on this result, we hypothesized that cpmKate(180) may be a good candidate for a red cpFP-based voltage sensor and fused cpmKate(180) to the VSD of Ci-VSP, resulting in VSFP(A)cpmKate(180), VSFP(B)cpmKate(180), VSFP(C)cpmKate(180) and VSFP(D)cpmKate(180). The two variants with the longest interdomain linkers [VSFP(C)cpmKate(180) and VSFP(D)cpmKate(180)] were fluorescent and exhibited proper trafficking to the plasma membrane. Moreover, VSFP(C)cpmKate(180) and VSFP(D)cpmKate(180) displayed a change in fluorescence upon a change in membrane potential. However, the voltage-dependent fluorescence signal was small and the response kinetics were slower than that of VSFP2 and VSFP3 voltage probes which were previously described by this laboratory. The response of VSFP(C)cpmKate(180) was slightly faster than that of VSFP(D)cpmKate(180), which is consistent with other VSFP-based constructs (Dimitrov et al., 2007
; Lundby et al., 2008
).
Based on these results, we believe that cpFPs hold great promise for the development of voltage-sensitive probes with enhanced optical properties. We are currently using site-directed and random mutagenesis approaches to improve the brightness and dynamic range of the VSFP variants derived from cpmKate(180). In addition, exploration of other permissive sites within mKate might be advantageous.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank all members of the Knöpfel laboratory for discussions and support. The work of this laboratory is supported by grants from RIKEN BSI (TK), the RIKEN BSI directors fund (Thomas Knöpfel), NIH grant NS057631 (under a sub award granted by Yale University to Thomas Knöpfel), the JSPS (Japanese Society for the Promotion of Science)-CIHR (Canadian Institutes of Health Research) postdoctoral fellowship program (Amelie Perron), a grant-in-aid for JSPS fellows (Amelie Perron, Thomas Knöpfel) and a grant-in-aid for young scientists from MEXT (Ministry of Education, Culture, Sports and Technology) (Hiroki Mutoh, Thomas Knöpfel).
Tallini, Y. N., Ohkura, M., Choi, B.R., Ji, G., Imoto, K., Doran, R., Lee, J., Plan, P., Wilson, J., Xin, H. B., Sanbe, A., Gulick, J., Mathai, J., Robbins, J., Salama, G., Nakai, J., and Kotlikoff, M. I. (2006). Imaging cellular signals in the heart in vivo: cardiac expression of the high-signal Ca2+ indicator GCaMP2. Proc. Natl. Acad. Sci. U.S.A. 103, 4753–4758.