 Kamil Seyrek
Kamil Seyrek Johannes Espe
Johannes Espe Fabian Wohlfromm
Fabian Wohlfromm Inna N. Lavrik
Inna N. Lavrik- Translational Inflammation Research, Medical Faculty, Center of Dynamic Systems, Otto von Guericke University Magdeburg, Magdeburg, Germany
Apoptosis is a form of programmed cell death that eliminates excessive and damaged cells. It can be conducted by two ways: the extrinsic and the intrinsic (mitochondrial) pathways. The extrinsic death pathway is triggered by activation of the death receptors (DRs), while the intrinsic pathway is initiated by changes at the mitochondria. The induction of life and death signals via DRs requires an intricate regulation of signal transduction. In this regard, an optimal conformation of the extracellular domain of DR is required for the Death Ligand (DL) binding and initiation of DR signaling. Glycosylation, the enzymatic attachment of carbohydrates to proteins, can influence DR conformation and thereby receptor-ligand interaction. Due to the tremendous structural diversity of glycans attached to DRs, little is known about the role of specific glycosylation subtypes influencing functions of DRs. Deciphering the role of specific glycan signatures, so-called “glyco-code”, on DRs is important to understand how glycans are involved in signal transduction. Although apoptosis has been shown to be associated with altered glycosylation patterns of glycoproteins, our understanding how glycosylation modulates apoptosis is still limited. This review focuses on summarizing our current knowledge on the modulation of cell signaling via glycosylation of DRs.
1 Introduction
Apoptosis is a tightly regulated type of programmed cell death that can be induced via two pathways: the intrinsic and the extrinsic pathway. The intrinsic apoptosis pathway is activated via mitochondria upon various cell stress signals such as DNA damage, whereas the extrinsic pathway is mediated by activation of DRs, including cluster of differentiation 95 (CD95/Fas/APO-1, DR2), tumor necrosis factor related apoptosis inducing ligand receptor-1/2 (TRAIL-R1/2, DR4/5) and tumor necrosis factor receptor 1 (TNFR1, DR1) (Krammer et al., 2007). The activation of DRs via binding of cognate DLs leads to the assembly of apoptosis-initiating complexes. For instance, upon activation of CD95 or TRAIL-Rs, interactions between death domains (DDs) of DD-containing adaptor proteins such as Fas-associated protein with death domain (FADD) and DR are initiated (Krammer et al., 2007; Kischkel et al., 1995; Seyrek et al., 2020). DR-bound FADD then recruits Death Effector Domain (DED) proteins: procaspase-8a/b, −10a/d, or c-FLIPL/S/R, which leads to the assembly of the death-inducing signaling complex (DISC) (Seyrek et al., 2020; Scott et al., 2009; Ivanisenko et al., 2022). At the DISC, procaspase-8a/b assembles via its DEDs into a macromolecular platform, DED filaments, which is essential for activation of caspase-8 (Dickens et al., 2012; Schleich et al., 2012; Hughes et al., 2016; Yang et al., 2024). Besides DISC, the activation of caspase-8 upon DR stimulation might take place at a secondary signaling complex, termed complex II which contains procaspase-8a/b, c-FLIPL/S/R, FADD and RIPK1 (Lavrik et al., 2008; Geserick et al., 2009; Lavrik and Krammer, 2012). Activated caspase-8, in turn, cleaves procaspase-3 and -7 leading to their activation. This is followed by the cleavage of multiple cellular substrates leading to apoptosis and demolition of the cell (Schleich et al., 2012).
Glycosylation refers to the process of covalent modification by sugar moieties that are added to proteins or lipids through the catalytic action of glycosyltransferases. Glycosylation may involve inter- and intramolecular protein interactions that are critical for the optimal tertiary structure and function of proteins (Caramelo and Parodi, 2015). The addition of sugar residues to the proteins takes place in the endoplasmic reticulum (ER) and Golgi apparatus. Based on the linkage between the amino acid and the sugar, glycosylation is categorized mainly into two types, N-linked and O-linked (Figures 1A, B). The attachment of N-acetylglucosamine (GlcNAc) to an amide group of an asparagine side-chain via a glycosidic link is defined as N-linked glycosylation. Modification through a glycosidic bond between N-acetylgalactosamine (GalNAc) and a hydroxyl group of serine or threonine side chain is termed as O-linked glycosylation (Lehrman, 1991; Hart, 1997; Cheng et al., 2000; Ferreira et al., 2017). Attachment of N-glycans starts with the transfer of the common Glc3Man9GlcNAc2 precursor oligosaccharide onto nascent proteins, which are entering into the lumen of the ER. A fourteen monosaccharides long precursor oligosaccharide is pre-assembled on a dolichol phosphate membrane anchor before it is transferred to the nascent protein (Li et al., 2019). Further processing of high mannose-glycans by the action of several glycosidases and glycosyltransferases gives rise to complex- or hybrid-type N-glycans (Moriwaki et al., 2021). The different linkages generated by further modification via sialic acid and fucose lead to an immense structural diversity (Moremen et al., 2012). Contrary to the N-glycans, the initial GalNAc residue of O-glycans is transferred onto a folded protein in the Golgi followed by sequential attachment of other monosaccharides including β1,3-galactose or β1,3-GlcNAc by glycosyltransferases (Moriwaki et al., 2021). Another reversible form of protein glycosylation is O-GlcNAcylation (Zeidan and Hart, 2010). O-GlcNAcylation includes a single transfer of GlcNAc to the hydroxyl group of threonine or serine. Compared to O/N-Glycosylation this transfer only includes one single GlcNAc while in O/N-Glycosylation a complex structure via elongation is formed (Hart, 1997; Comer and Hart, 2000; Seyrek et al., 2024; Frenkel-Pinter et al., 2017). Even if it is only one single GlcNAc, it has a great impact on the signaling in apoptosis induction (Seyrek et al., 2024).
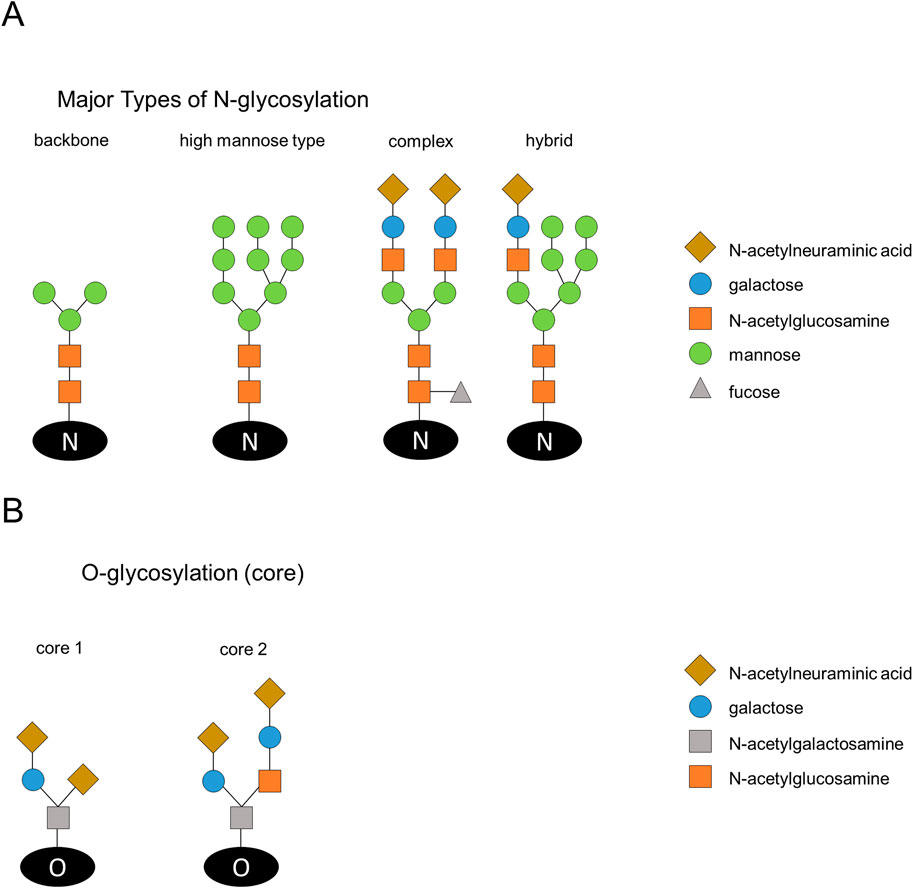
Figure 1. Schematic illustration of N-glycosylation structures and typical cores of O-glycosylation. (A) The main types of N-glycosylation are introduced. Addition of N-acetylglucosamine (GlcNAc) to an amide group of an asparagine results in N-linked glycosylation. The N-glycans have a backbone of N-acetylglucosamine and mannose (left). The high mannose type consists of N-acetylglucosamine and mannose. Modification of high-mannose glycans results in complex and hybrid-type N-glycans. Complex and hybrid glycans have the backbone of N-acetylglucosamine and mannose, but also have galactose and N-acetylneuraminic acid. In addition, fucose can be added to N-acetylglucosamine. (B) O-glycosylation cores are shown and divided into the two main types with N-acetylgalactosamine, galactose and N-acetylneuraminic acid (left side) or addition of N-acetylglucosamine (right side).
Cell surface glycans are not random polymers of monosaccharides but rather well-determined distinct glycans. Sugar moieties on proteins might be altered in several diseases including cancer (Holmes et al., 1987; Kunzke et al., 2017). Particularly, terminal monosaccharides substantially influence the function of proteins. Given that the glycosyltransferase profile could be different depending on the cell type, glycosylation pattern can also be dissimilar. DRs can also be modified with specific sugars, which can affect the signal transduction. Without the correct sugar modifications DRs cannot mediate the apoptotic signal properly (Pan et al., 2019; Lee et al., 2019; Newson et al., 2019). Even a small variation in subtypes of glycosylation may influence a death signal. It has to be highlighted that glycans on DRs can be characterized by strong diversity, which leads to challenges in defining specific functions of glycosylation in extrinsic apoptosis. Given that receptor-bound glycans are involved in the binding of the cognate DL and folding of the receptor to achieve an optimal tertiary structure, it is not surprising that glycosylation of DRs alters DR-mediated cell signaling (Wagner et al., 2007).
The majority of human cells were shown to express DRs, however not all the cells are susceptible to DR-mediated cell death, suggesting the existence of additional mechanisms diverting the death signal into alternative signaling pathways (Ivanisenko et al., 2022; Irmler et al., 1997). Indeed, the sensitivity of cells toward DR-mediated apoptotic cell death is regulated by multiple checkpoints in order to prevent the triggering of spontaneous apoptosis. Extensive work has revealed that glycosylation presents one of these checkpoints and plays a pivotal role in the modulation of DR signaling by allowing or sterically hindering the binding of their respective ligands. Moreover, depending on the cellular context or on the strength of the activation signal, stimulation of DRs may lead to apoptosis, regulated necrosis or even to cell survival (Lafont et al., 2017; Voigt et al., 2014). Glycosylation of DRs may play the key regulatory role in these decisions. In this context, this review focuses on the modulation of cell signaling via glycosylation of DRs.
2 Glycosylation of CD95 in signal transduction
The mature human CD95 has the molecular weight ranging from 45 to 54 kDa due to post-translational glycosylation (Shatnyeva et al., 2011; Seyrek et al., 2019; García-Fuster et al., 2004; Neumann et al., 2010; Bodmer et al., 2002; Seyrek et al., 2022). Activated by a binding of CD95L, CD95 undergoes conformational changes, which are essential for an optimal CD95 DISC and DED filament formation (Lavrik and Krammer, 2012) (Figure 2A). CD95 DED filament assembly leads to caspase-8 activation, which triggers the apoptotic cascade. Downstream of the DISC, activation of caspase cascade might lead to apoptosis via type I or type II signaling. In the latter case, i.e., for type II cells, apoptosis execution requires activation of the so-called mitochondrial amplification loop, which involves mitochondrial pore formation and release of cytochrome C from mitochondria leading to apoptosome formation with subsequent activation of caspase-3 (Figure 2A). Despite the fact that the DISC is the main site of action in the regulation of apoptosis, the molecular mechanisms that determine whether cells after activation of CD95 induce apoptotic, necroptotic or cell-survival signals are not yet fully understood. Extensive work on CD95 indicates that glycosylation can affect the localization as well as association of CD95 with other DISC components (Shatnyeva et al., 2011; Li et al., 2007). A series of elegant experiments showed that the extracellular domain of CD95 contains two N-glycosylation sites at N118 and N136 (Shatnyeva et al., 2011; Seyrek et al., 2019; Lichtenstein and Rabinovich, 2013) (Figure 2B). In addition, further potential glycosylation sites were identified by in silico predictions at N233 and T214, located in the intracellular DD of CD95 (Figure 2B) (Shatnyeva et al., 2011). The significant decrease in CD95-mediated apoptosis in K562 leukemia cells with CD95 N118A/N136A double mutant indicates that glycosylation at these sites is important for the induction of apoptosis (Wu et al., 2021). Further, N-glycosylation of CD95 was shown to be crucial for the CD95 ligand (CD95L) recognition and binding (Shatnyeva et al., 2011; Lichtenstein and Rabinovich, 2013).
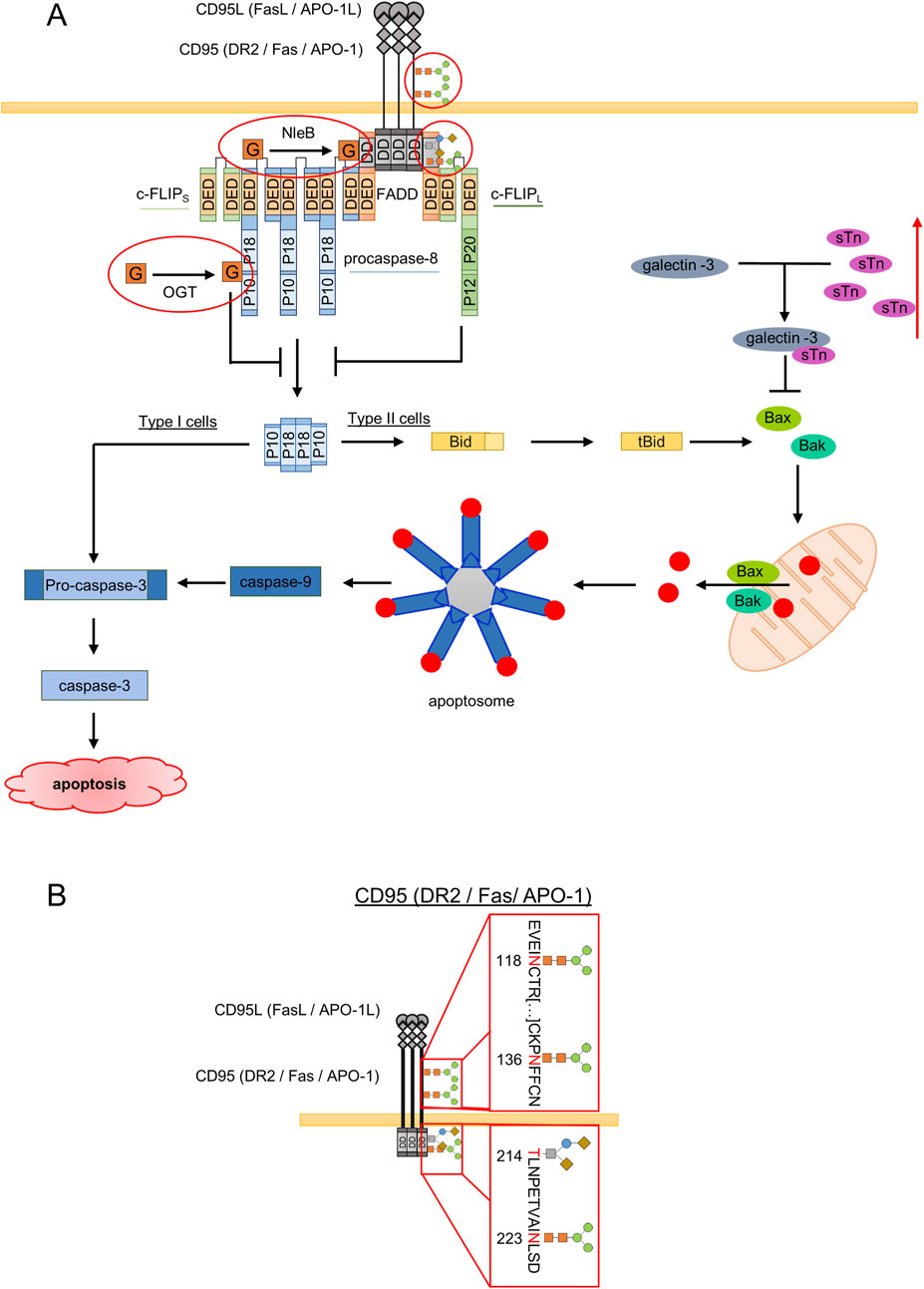
Figure 2. Major aspects of influence of glycosylation on the CD95L-induced signaling pathway. (A) CD95L binding leads to DISC (death-inducing signaling complex) formation. DISC comprises CD95, FADD, procaspase-8 and c-FLIPL/S/R. DISC assembly leads to autocatalytic activation of procaspase-8 at the DED filaments and subsequent cleavage to caspase-8 p182/p102 heterotetramers, which results in cleavage of procaspase-3 followed by its activation and apoptosis induction (Type I cells). In Type II cells, activated caspase-8 cleaves Bid to tBid, which supports mitochondrial pore formation by Bax and Bak. This leads to release of pro-apoptotic proteins from the mitochondria and apoptosome formation, which results in caspase-9 and caspase-3 activation. Sites of CD95 glycosylation are shown. An increase in sTn leads to accumulation of intracellular galectin-3 and its inhibitory effects on cell death pathways via inhibition of Bax oligomerization and subsequent Bax/Bak pore formation. Red outlines represent glycosylation of DR at N118 and N136 as well as the in silico predicted glycosylation at N223 and T214. Further red outlines are O-GlcNAc (O-linked β-N-acetylglucosamine) via NleB at FADD or at procaspase-8 via OGT (O-GlcNAc transferase). (B) Glycosylation sites on CD95. The receptor can be N-glycosylated at N118 and N136, which was shown experimentally The in silico predicted glycosylation sites: O-GlcNAcylation or O-glycosylation site at T214 and N-glycosylation site at N233 are shown. The N118 and N136 glycosylation sites are located in the extracellular part of the receptor. The in silico predicted N223 and T214 sites are located in the intracellular DD of the receptor.
The oligosaccharide chains of glycoproteins are often terminated with sialic acids, a family of sugars with nine carbons mediating important biological roles (Harduin-Lepers et al., 2012). However, hypersialylation of CD95, which can account for up to 8 kDa increase of its molecular weight (Keppler et al., 1999), was shown to prevent its oligomerization and DISC formation (Charlier et al., 2010). In fact, N-Glycans on human CD95 were shown to be the substrates for β-galactoside α2,6-sialyltransferase I (ST6Gal1), which is located in the Golgi to catalyze the addition of α2,6-linked sialic acid onto subterminal galactose residues of N-glycans (Keppler et al., 1999; Swindall and Bellis, 2011; Peter et al., 1995). Importantly, increased ST6Gal1 activity was shown to suppress CD95-mediated apoptosis (Swindall and Bellis, 2011; Peter et al., 1995). In support with this, desialylation of CD95 glycans by neuraminidases in B-lymphocytic cells increased the sensitivity to CD95-mediated apoptosis (Keppler et al., 1999). In parallel, the expression of sialylated glycans is often augmented in cancer. It might be suggested that the negative charge of sialic acid would prevent receptor-ligand interaction and thereby reduce CD95-induced apoptosis, however the detailed molecular mechanisms of these events still have to be uncovered. Within this line, malignant cells are reported to use a distinct glycosylation machinery to decorate DRs with unique oligosaccharides to increase the resistance to apoptosis. In line with this, oncogenic Ras was shown to increase ST6Gal1 expression, which in turn downmodulates cell death (Bellis, 2004).
CD95 signaling network can be effected by glycosylation at the different regulatory stages. Specifically, it was shown that galectin-3 might play an important role in CD95 signaling. In particular, it was demonstrated that exogenously added galectin-3, a galactose-specific lectin, potentiates the chemotherapeutics-induced cytotoxicity, but not in cells overexpressing truncated sTn (Siaα2-6 GalNAcα1-Ser/Thr, sialyl-Tn) (Santos et al., 2016). Remarkably, the expression of sTn was shown to be associated with a reduction in galectin-3 in human gastric cancer cells (Santos et al., 2016). By contrast, knockdown of ST6GalNAc-I, a sialyltransferase responsible for sTn synthesis, restored the binding sites of galectin-3 on the cell surface and thereby increased the sensitivity of malignant cells to chemotherapeutic drugs (Santos et al., 2016). Strikingly, impairment of the binding of galectin-3 to glycoproteins through sTn overexpression leads to intracellular accumulation of galectin-3, which has a unique aspartate-tryptophane-glycine-arginine (NWGR) motif, which was shown to be essential for binding of key pro-apoptotic Bcl-2 family member Bax (Harazono et al., 2014). In this way, the interaction with galectin-3 inhibits Bax oligomerization via its NWGR motif, which prevents Bax-mediated pore formation at the mitochondria and subsequent apoptosis (Figure 2A). Subsequently, galectin-3 could strongly affect CD95-induced apoptosis induction in type II cells via targeting Bax as well as inhibit the intrinsic apoptosis pathway (Figure 2A). Accordingly, intracellular accumulation of galectin-3 is suggested to be responsible for the increased drug resistance in gastric carcinoma cells expressing aberrant sTn (Santos et al., 2016). From another side, as highlighted above, the extracellular galectin-3 has a pro-apoptotic role. Although numerous studies reported that extracellular galectin-3 promotes apoptosis in colon cancer cells (Zhuo et al., 2008), leukemia T cells (Xue et al., 2013) and in human B cell lymphoma (Suzuki and Abe, 2008), the detailed molecular mechanisms are still unknown. It is tempting to assume that extracellular galectin-3 might also directly influence sensitivity towards CD95-mediated apoptosis or the other DR-induced pathways via stabilization of receptor oligomers at the membrane. Taken together, the degree of sialylation of CD95 appears to be a checkpoint in the regulation of CD95-mediated apoptosis. Nevertheless, the specific regulation of CD95-induced apoptosis by sialylation and the involvement of galectin-3 remains to be fully investigated.
3 Glycosylation of DR4 and DR5 in signal transduction
TRAIL eliminates transformed cells by inducing apoptosis without significant toxicity towards normal cells in most tissues. Thus, TRAIL-induced cell signaling has attracted considerable attention in cancer therapy (Lemke et al., 2014). Although TRAIL is cytotoxic in various cancer cells, certain cancer cells are resistant to TRAIL-induced apoptosis (Deng and Shah, 2020). The reason why TRAIL triggers apoptosis in certain malignant cells while some other malignant or healthy cells are resistant to TRAIL-treatment is not fully understood. Further, certain cancer cells induce cell death via DR4, while some others undergo solely DR5-mediated apoptosis. ER stress has been reported to sensitise human cells to the TRAIL-induced apoptosis, suggesting that the ER stress might disturb checkpoints, which normally protect cells from cytotoxic effects of TRAIL (Martin-Perez et al., 2012). It is also suggested that glycosylation of DRs may be one of the checkpoints and distinct glycosylation patterns of DR4 versus DR5 may be the leading cause in the selectivity towards a particular TRAIL-R in different cells. Within this line, human DR4 was shown to be N-glycosylated at N156, whereas no consensus sequence for N-glycosylation on DR5 was revealed (Moriwaki et al., 2021; Yoshida et al., 2007; Dufour et al., 2017). Interestingly, modification at N156 by sugar moieties is reported to be not essential for DR4 cell surface distribution and ligand binding. However, when taken into consideration that the cytomegalovirus (CMV) UL141 protein restricts cell-surface expression of DR4 by promoting its intracellular retention and UL141 shows higher affinity for non-glycosylated DR4 (Smith et al., 2013), the role of N-glycans in DR4 translocation to the cell membrane, can not be excluded. DR4 glycosylation was shown to enhance TRAIL-induced apoptosis by promoting receptor clustering that leads to an increased DISC formation and caspase-8 activation (Dufour et al., 2017). Furthermore, mouse TRAIL-R was shown also to be N-glycosylated at N99, N122, and N150. Modification of mouse TRAIL-R promotes TRAIL-induced apoptosis in B16 melanoma cells (Dufour et al., 2017). On the other side, cells expressing the N-glycosylation-defective N156A mutant of DR4 and the N99A/N122A/N150A mutant mouse TRAIL-R were less sensitive to TRAIL-induced apoptosis than that of their wild-type counterparts (Dufour et al., 2017). By contrast, another study showed that mouse embryonic fibroblast cells with unglycosylated TRAIL-R mutant are more sensitive to TRAIL-induced apoptosis (Estornes et al., 2018). As the amount and profile of glycosyltransferases could be different depending on the cell type, the possibility of differences in linkage, monosaccharide composition, branching and terminal modifications of the glycan structures on TRAIL-Rs can not be excluded in these different experimental models. Hence, it is no surprise then that emergence of discrepancies between these studies results from using different cell lines.
Deficiency of a single monosaccharide may impair TRAIL-induced apoptosis and thereby facilitating cancer progression (Moriwaki et al., 2009). For example, fucosylation, a common oligosaccharide modification, was shown to influence the ligand-independent receptor association that leads to enhanced DISC formation (Zhang et al., 2019). By contrast, deficiency of fucose residues leads to acquisition of resistance to TRAIL-induced apoptosis and thereby cells escape from NK cell-mediated tumor surveillance (Moriwaki et al., 2009; Moriwaki et al., 2011). Further, colon cancer cell lines highly expressing α3/4-Fuc-transferease (FUT3) and α3-Fuc-transferase (FUT6) were shown to be TRAIL-sensitive, while knockdown of fucosyltransferases in several cancer lines reduced sensitivity of cells to TRAIL (Wagner et al., 2007; Zhang et al., 2019). Moreover, introduction of mutant GDP-mannose-4,6-dehydratase (GMD), an enzyme required for GDP-Fuc biosynthesis into HCT116 colon cancer cells, resulted in tumor progression and metastasis (Moriwaki et al., 2009). However, in GMD-rescued HCT116 cells fucosylation was restored and tumor growth decreased dramatically (Moriwaki et al., 2009). Fucosylation of DR4 in GMD-rescued cells disappeared upon PNGase F treatment, which removes all N-linked oligosaccharides from glycoproteins (Moriwaki et al., 2011), indicating that fucose residues are added to N-glycans, but not O-glycans. Accordingly, no fucosylation was observed on DR5, which was not reported to undergo N-glycosylation (Fukuoka et al., 2022). Although fucosylation of DR4 is revealed, the molecular architecture of fucosylation linkage and the exact glycan structure still remain unknown. Hence, this issue has to be addressed in future studies.
It might be expected that DR4 alone would contribute to TRAIL-induced apoptosis, given that only DR4, but not DR5, is fucosylated. However, it is well established that both DR4 and DR5 can transduce apoptotic signal (von Karstedt et al., 2017). Importantly, considering the glycosylation-regulated activity of DR4 and DR5, GMD deficiency inhibits not only DR4-but also DR5-mediated apoptosis (Moriwaki et al., 2009). This suggests the existence of other molecular mechanisms, including glycosylation, that influence TRAIL signaling. Indeed, certain other fucosylated glycan structures, known as Lewis glycans (Lea, Leb, Lex, Ley and sialylated version sLex and sLea) attached to lacto/neolacto glycosphingolipids, but not to DR4 or DR5, were shown to increase TRAIL-induced apoptosis. In fact, sLea, Lex, and sLex on lacto/neolacto glycosphingolipids enhanced TRAIL-induced complex II formation without affecting the oligomerization of DR4 or DR5, and DISC formation (Fukuoka et al., 2022). Accordingly, the downmodulation of the level of Lewis glycans in DLD1 and SW480 colon cancer cell lines significantly decreases TRAIL-induced caspase-8 activation (Fukuoka et al., 2022). Interestingly, reintroduction of FUT3, which rescues the expression of sLea, Lex, and sLex, increased caspase-8 activation and apoptosis (Fukuoka et al., 2022). Similarly, introduction of FUT6, which rescues Lex and sLex, also enhanced TRAIL-induced apoptosis (Fukuoka et al., 2022). Interestingly, activation of intrinsic apoptosis by staurosporine or ABT-737 is unaffected in the absence of Lewis glycans. Overall, the increase in the amount of Lewis glycans on lacto/neolacto glycosphingolipids was shown to promote TRAIL-induced apoptotic cell death. Remarkably, Lewis glycans are shown to be increased during cancer progression and their expression level has shown a correlation with cancer malignancy. Therefore, sLea and sLex have been used as tumor markers for several cancers in the clinics (Fukuoka et al., 2022; Miyoshi et al., 2008). Furthermore, elevated glycosylation of ceramide, the building block of sphingolipids, was observed in multidrug resistant cancer cells (Morad et al., 2017). Hence, the dual function of Lewis glycans can be attributed to the distinct carriers or different binding sites of Lewis glycans within the same carrier. Furthermore, the further analysis of their influence on TRAIL-induced apoptosis is highly necessary.
Similar to the role of N-glycans in DR-mediated apoptosis, O-glycans on DR5 were shown to promote TRAIL-induced receptor clustering, DISC formation and caspase-8 activation (Wagner et al., 2007). In parallel, N-acetylgalactosaminyltransferase 14 (GALNT14), which is transferring an initial GalNAc (T-antigen) residue on serine or threonine residues, was reported to be critical for ligand-dependent clustering of DR5 (Wagner et al., 2007). The abnormal expression of GALNT14 was shown to be associated with tumorigenesis and progression of cancer (Huanna et al., 2015; Chu et al., 2022). Of note is that T-synthase requires a chaperone called Cosmc for its correct folding and optimal activation (Ju and Cummings, 2002). Mutations in Cosmc and methylation of its promoter are reported to play a role in the expression of Tn in pancreatic and cervical cancer (Ju et al., 2008; Radhakrishnan et al., 2014). In addition, the initial monosaccharide GalNAc may be modified by T-synthase, which transfers galactose residues from a donor (UDP-gal) to GalNAc to form Galβ1-3GalNAc-Ser/Thr (T antigen, core 1). T antigen structures can be further modified by addition of other monosaccharides including sialic acid (Ding et al., 2021). One to four Galβ1-3GalNAc structures with up to two sialic acid residues were detected on DR5 in Chinese hamster ovary cells (Wagner et al., 2007). Galβ1-3GalNAc chains on DR5 has been shown to induce the DR5 clustering and apoptosis (Gao et al., 2021; Jiang et al., 2020). By contrast, truncated O-glycans, Tn (GalNAcα1-Ser/Thr), and sTn, have been reported to impair homo-oligomerization and stability of DRs (Jiang et al., 2020). On the other hand, modulation of DR4-mediated apoptosis is not limited to the presence of N-glycans. In fact, fully N-glycosylated DR4 is also O-glycosylated, which contributes to the stability of DR4 (Jiang et al., 2020). In a similar manner, the presence of truncated O-glycans compromises DR4 stability (Jiang et al., 2020). In support with this, high expression of Tn and Sialyl-Tn is reported to be often expressed in human carcinoma cells (Radhakrishnan et al., 2014; Ju and Cummings, 2005; Sun et al., 2018). Further, they can also promote hetero-oligomerization between DR5 and decoy receptor 2 (DcR2), thereby reducing the death signal of DR5 (Jiang et al., 2020). Taken together, Tn/sTn antigens could confer a survival advantage to tumor cells.
Apart from classical O- and N-linked glycosylation, modification of DR4 by a single O-GlcNAcylation, a dynamic and reversible post-translational modification, was shown to be critical in DR4-mediated signal transduction (Lee et al., 2019). In fact, upon activation, DR4 is O-GlcNAcylated at S424 in its DD that leads to DR4 translocation into lipid rafts, which was reported to be required for more efficient DISC formation (Lee et al., 2019). Interestingly, TRAIL-resistant cancer cells were found to undergo apoptosis upon the modification of DR4 by O-linked GlcNAc (Lee et al., 2019). Moreover, in cells with non- O-GlcNAcylated DR4, probably due to impaired conformation of the receptor or lack of DR4 oligomerization, the assembly of the DISC was blocked, leading to the inhibitory effects on apoptosis induction (Lee et al., 2019). Interestingly, an elevated O-GlcNAcylation was also linked to resistance towards TRAIL treatment in pancreatic cancer cells. Specifically, DR5 was shown to be O-GlcNAcylated, which regulates its oligomerization, an essential step in the initiation of apoptosis (Pan et al., 2019; Yang et al., 2020; Lee et al., 2021). O-GlcNAcylated DR5 was shown to lack its oligomerization while the inhibition of O-GlcNAcylation of DR5 promoted oligomerization of DR5 (Yang et al., 2020). In this way, O-GlcNAcylation seems to differently act on DR4 versus DR5, which has to be investigated in the future studies. Moreover, O-GlcNAcylation also controls TRAIL-induced as well as CD95L-induced apoptosis downstream from DR. In particular, FADD has been reported to undergo O-GlcNAcylation. In the course of C. rodentium infection, FADD can be O-GlcNAcylated by the bacterial arginine glycosyltransferase effector NleB (Scott et al., 2017). NleB O-GlcNAcylates FADD in the DD at Arg117 (Figure 2B) (Scott et al., 2017; Pearson et al., 2013). This O-GlcNAcylation blocks the recruitment of caspase-8 to FADD which impairs the dimerization and subsequent activation of caspase-8 leading to a blockage of apoptosis (Scott et al., 2017). Besides FADD also other DD containing proteins like TRADD and RIPK1 are known to be O-GlcNAcylated leading to impaired apoptosis (Scott et al., 2017; Pearson et al., 2013; Li et al., 2013; Xue et al., 2020). Furthermore, caspases can also be O-GlcNAcylated. In particular caspase-8, -3 and -9 were reported to be O-GlcNAcylated which was shown by using a chemical reporter 6-Alkynyl-6-deoxy-GlcNAc (6AlkGlcNAc) (Chuh et al., 2017). Caspase-8 is O-GlcNAcylated near its cleavage sites D374 and D384, which was shown to inhibit its cleavage upon TNFα treatment. Similar mechanisms can be suggested to take place upon TRAIL or CD95L stimulation since caspase-8 activation plays a pivotal role in these pathways (Figure 2B). Thus, besides classical O- and N-linked glycosylation, modification of TRAIL signaling network by GlcNAc acts as an additional checkpoint in the regulation of TRAIL -mediated signal transduction.
4 Glycosylation of TNFR1 in signal transduction
The mechanisms through which TNFR1 signaling is regulated are not entirely defined, however it is well-established that depending on cellular context or downstream signaling events, stimulation of TNFR1 may lead to NF-κB activation, apoptosis or necroptosis. TNFR1 mediates the formation of two distinct signaling complexes, i.e., complex I and complex II. In detail, upon the acquisition of stimulatory signals from TNFα, the complex I is assembled at TNFR1. The complex I comprises the DD-containing proteins TRADD and RIPK1 (Chen and Goeddel, 2002). TNF receptor associated protein 2 (TRAF2) is subsequently bound to TRADD (Park et al., 2000; Hsu et al., 1996). TRAF2 recruitment is accompanied by cellular inhibitor of apoptosis protein 1 (cIAP1; also known as BIRC2) and cIAP2 (also known as BIRC3), translocation to the TNFR1 signaling core, which accomplishes the complex I formation, which is essential for the induction of anti-apoptotic signals (Chen and Goeddel, 2002; Wajant and Scheurich, 2011; Karin, 2006; Muppidi et al., 2004). On the other hand, depletion of cIAP1 and cIAP2 or removal of K63- and M1-linked ubiquitin chains from RIPK1 induces the formation of complex IIa consisting of RIPK1, TRADD, FADD, procaspase-8 and c-FLIPL (Micheau and Tschopp, 2003; Brenner et al., 2015). Complex II in the presence of RIPK3 and caspase inhibition can drive necroptosis induction and is named complex IIb or necrosome (Wang et al., 2008; Tenev et al., 2011). As the nucleation of complex IIa formation relies on RIPK1, complex IIa is also called the ripoptosome (Brenner et al., 2015; Tenev et al., 2011). Thus, complex I is associated with survival signals, whereas complex II promotes cell death.
The emerging evidence has been obtained that glycosylation is also one of the crucial checkpoints in TNFR signaling. In this regard, the major role in regulation of TNFR networks belongs to O-GlcNAcylation. Specifically, TRADD was shown to be O-GlcNAcylated at Arg235 (Li et al., 2013; Xue et al., 2020). Arg235 is located at the position in the DD, which is well conserved in many DD proteins (Xue et al., 2020). O-GlcNAcylation by NleB completely alters the DD of TRADD and other DD-proteins, which blocks subsequent signal transduction (Li et al., 2013). TRADD can also be O-GlcNAcylated by SseK1 at Arg235 as well as Arg245 (Newson et al., 2019; Xue et al., 2020). These O-GlcNAcylation events inhibit apoptosis as well as NF-κB signaling (Xue et al., 2020). RIPK1 was also shown to be O-GlcNAcylated at Ser331, Ser440 and Ser669, which regulates its ubiquitination and binding to macromolecular complexes activating caspase-8 (Zeng et al., 2024). In this way, it was shown that O-GlcNAcylation of RIPK1 blocks apoptosis (Zeng et al., 2024). RIPK1 phosphorylation is a key step in induction of necroptosis (Laurien et al., 2020; Zhang et al., 2017). The O-GlcNAcylation of RIPK1 at Ser331 subsequently blocks its phosphorylation at Ser166 and necroptosis induction (Seo et al., 2023). Finally, SseK3 was reported to O-GlcNAcylate the DD of mTNFR1 and mTRAIL-R at Arg376 and Arg293, which corresponds to O-GlcNAcylation hTRAIL-R at Arg359 (Seyrek et al., 2024; Newson et al., 2019). These events also block DR-mediated signal transduction. Taken together, O-GlcNAcylation has an important regulatory role in TNFR-mediated pro- and anti-apoptotic branches of the pathway via acting on TNFR itself as well as on the core components of the network.
TNFR1 was shown to be N-glycosylated in the extracellular domain at N151 and N202 in mouse microglia cells (Han et al., 2015). The N-glycosylation of TNFR1 has been suggested to facilitate TNFα binding without affecting its surface localization. In line with this, the decrease in p65 translocation to the nucleus, which is a hallmark of NF-κB activation, was observed in cells with N151/N202Q mutant of TNFR1 (Han et al., 2015). Thus, N-glycosylation of TNFR1 is suggested to promote NF-κB signaling. Supportingly, upon TNFα stimulation, excessive microglia activation and increased central nervous system (CNS) inflammation was observed, whereas in cells with glycomutant of TNFR1 these signals were inhibited. N-glycosylation at N151 and N202 is suggested to enhance NF-κB signaling by promoting the inflammatory TNFα autocrine loop (Han et al., 2015). Importantly, no difference in cell surface expression between wild type and mutant TNFR1 was observed, which indicates that glycosylation at N151 and N202 is not a prerequisite for a proper intracellular trafficking of TNFR1 and the major role in the signaling outcome upon perturbation of these glycosylation sites belongs to interactions of TNFα with TNF-R1 (Han et al., 2015). Further studies have reported that human TNFR1 has three potential N-glycosylation sites at N54, N145 and N151 (Köhne et al., 1999). Although the functional significance of protein glycosylation in TNFR1 signaling is revealed, at least in part, further experiments are needed to investigate how glycosylation at these sites promotes TNFα binding and molecular mechanisms by which glycosylation modulates TNFR1 conformation.
In spite of the fact that N-glycosylation of TNFR1 at N151 and N202 promotes TNFα binding, excessive terminal modification of N-glycans by α2,6-bound sialic acid has been reported to protect cells from TNFα-induced apoptosis (Moriwaki et al., 2021; Liu et al., 2011; Holdbrooks et al., 2018; Alexander et al., 2020). Within these lines, removing of sialic acid residues by sialidase treatment enhanced sensitivity to TNFR1-induced apoptosis (Liu et al., 2011). It is important to highlight that increased expression of ST6Gal1 affects TNFR1-as well as CD95-mediated apoptosis, but not TRAIL-induced apoptosis (Swindall and Bellis, 2011). The α2,6-sialylation of TNFR1 reportedly does not influence ligand binding or subsequently initial receptor activation, and recruitment of complex I proteins. In contrary, excessive α2,6-sialylation prevents TNFR1 internalization, which is essential for TNFR1-induced apoptosis induction, but not for the activation of survival signaling (Micheau and Tschopp, 2003; Holdbrooks et al., 2018). Considering that extracellular galectin-3 promotes apoptosis and inactivates Akt (Leffler et al., 2002; Mazurek et al., 2007), this process might be impaired through increased α2,6-sialylation. This might be due to the inhibitory effects on the binding of galectin-3 to the cell surface oligosaccharides. Subsequently, it is not surprising that downregulation of TNFR1-mediated apoptosis in oversialylated cells may be associated to the decreased galectin-3 binding affinity to TNFR1, at least in part (Liu et al., 2011). Overall, upon α2,6-sialylation of TNFR1 NF-κB-signaling seems to be unchanged, while sialylation blocks the apoptotic TNFR1-signaling (Holdbrooks et al., 2018). Hence, α2,6-sialylation may contribute to the evasion of cancer cells from apoptosis. In line with this statement, ST6Gal1 expression was reported to be overexpressed in pancreatic, ovarian, and colon cancer, whereas its levels are low in healthy tissues of these organs (Swindall and Bellis, 2011; Schultz et al., 2016).
5 Future perspectives
The DR-mediated induction of life and death signals requires an intricate regulation of the macromolecular complexes assembled at the DRs. At the stage of initiation of the apoptotic signaling the proper tertiary structure of DRs is a prerequisite for optimal DR function and correct DISC formation. Without the correct sugar modifications, DRs can not trigger an efficient death signaling. Comparing the glycosylation network of CD95 with DR4/5 and TNFR1 similar features can be found as well as some differences such as different core proteins of the network undergo distinct types of glycosylation. All four receptors undergo glycosylation, which is proven to be one of the important regulatory checkpoints in DR signaling. In fact, glycans attached to the DRs can promote or restrain apoptotic signals and changes of their glycosylation status modulate the sensitivity of cells toward DR-mediated apoptosis. However, so far the role of the particular glycosylation site in the function of the particular receptor has revealed distinct mechanisms in the regulation of DR signaling by glycosylation, which reflects the certain diversity in the function of glycosylation sites. In the future studies, it is highly necessary to determine the precise glycan structures on DRs and characterize the influence of these structures on the binding of DL to the corresponding DR. Moreover, of a major importance is to understand their tissue- and cell type-specific variations, especially in the normal versus malignant cells. This would allow to get more specific insights into the DR-mediated network and develop new therapeutic approaches against diseases associated with deregulation of the DR network. Another important aspect in the future research belongs to unraveling more details in the intracellular glycosylation of DD that controls DR-mediated cell death and inflammation networks and thereby uncovering new targets for therapeutic intervention.
Author contributions
KS: Conceptualization, Writing – original draft. JE: Investigation, Project administration, Resources, Software, Writing – original draft. CK: Conceptualization, Funding acquisition, Resources, Validation, Writing – original draft. FW: Visualization, Writing – original draft. IL: Conceptualization, Supervision, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by European Regional Development Fund (project ALBB) and DFG (LA 2386).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alexander, K. L., Serrano, C. A., Chakraborty, A., Nearing, M., Council, L. N., Riquelme, A., et al. (2020). Modulation of glycosyltransferase ST6Gal-I in gastric cancer-derived organoids disrupts homeostatic epithelial cell turnover. J. Biol. Chem. 295 (41), 14153–14163. doi:10.1074/jbc.RA120.014887
Bellis, S. L. (2004). Variant glycosylation: an underappreciated regulatory mechanism for beta1 integrins. Biochimica biophysica acta 1663 (1-2), 52–60. doi:10.1016/j.bbamem.2004.03.012
Bodmer, J. L., Schneider, P., and Tschopp, J. (2002). The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 27 (1), 19–26. doi:10.1016/s0968-0004(01)01995-8
Brenner, D., Blaser, H., and Mak, T. W. (2015). Regulation of tumour necrosis factor signalling: live or let die. Nat. Rev. Immunol. 15 (6), 362–374. doi:10.1038/nri3834
Caramelo, J. J., and Parodi, A. J. (2015). A sweet code for glycoprotein folding. FEBS Lett. 589 (22), 3379–3387. doi:10.1016/j.febslet.2015.07.021
Charlier, E., Condé, C., Zhang, J., Deneubourg, L., Di Valentin, E., Rahmouni, S., et al. (2010). SHIP-1 inhibits CD95/APO-1/Fas-induced apoptosis in primary T lymphocytes and T leukemic cells by promoting CD95 glycosylation independently of its phosphatase activity. Leukemia 24 (4), 821–832. doi:10.1038/leu.2010.9
Chen, G., and Goeddel, D. V. (2002). TNF-R1 signaling: a beautiful pathway. Sci. (New York, NY) 296 (5573), 1634–1635. doi:10.1126/science.1071924
Cheng, X., Cole, R. N., Zaia, J., and Hart, G. W. (2000). Alternative O-glycosylation/O-phosphorylation of the murine estrogen receptor beta. Biochemistry 39 (38), 11609–11620. doi:10.1021/bi000755i
Chu, Y. D., Fan, T. C., Lai, M. W., and Yeh, C. T. (2022). GALNT14-mediated O-glycosylation on PHB2 serine-161 enhances cell growth, migration and drug resistance by activating IGF1R cascade in hepatoma cells. Cell Death Dis. 13 (11), 956. doi:10.1038/s41419-022-05419-y
Chuh, K. N., Batt, A. R., Zaro, B. W., Darabedian, N., Marotta, N. P., Brennan, C. K., et al. (2017). The new chemical reporter 6-Alkynyl-6-deoxy-GlcNAc reveals O-GlcNAc modification of the apoptotic caspases that can block the cleavage/activation of caspase-8. J. Am. Chem. Soc. 139 (23), 7872–7885. doi:10.1021/jacs.7b02213
Comer, F. I., and Hart, G. W. (2000). O-Glycosylation of nuclear and cytosolic proteins. Dynamic interplay between O-GlcNAc and O-phosphate. J. Biol. Chem. 275 (38), 29179–29182. doi:10.1074/jbc.R000010200
Deng, D., and Shah, K. (2020). TRAIL of hope meeting resistance in cancer. Trends Cancer 6 (12), 989–1001. doi:10.1016/j.trecan.2020.06.006
Dickens, L. S., Boyd, R. S., Jukes-Jones, R., Hughes, M. A., Robinson, G. L., Fairall, L., et al. (2012). A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol. Cell 47 (2), 291–305. doi:10.1016/j.molcel.2012.05.004
Ding, R., Hu, X., Hu, W., Du, Z., Huang, P., Wang, M., et al. (2021). Cosmc transfection decreases malignant behavior of Tn(+) cells and enhances sensitivity to apoptosis when induced by Apo2L/TRAIL via alteration of O-glycan structure. Aging 13 (19), 23393–23406. doi:10.18632/aging.203633
Dufour, F., Rattier, T., Shirley, S., Picarda, G., Constantinescu, A. A., Morle, A., et al. (2017). N-glycosylation of mouse TRAIL-R and human TRAIL-R1 enhances TRAIL-induced death. Cell Death Differ. 24 (3), 500–510. doi:10.1038/cdd.2016.150
Estornes, Y., Dondelinger, Y., Weber, K., Bruggeman, I., Peall, A., MacFarlane, M., et al. (2018). N-glycosylation of mouse TRAIL-R restrains TRAIL-induced apoptosis. Cell Death Dis. 9 (5), 494. doi:10.1038/s41419-018-0544-7
Ferreira, J. A., Magalhães, A., Gomes, J., Peixoto, A., Gaiteiro, C., Fernandes, E., et al. (2017). Protein glycosylation in gastric and colorectal cancers: toward cancer detection and targeted therapeutics. Cancer Lett. 387, 32–45. doi:10.1016/j.canlet.2016.01.044
Frenkel-Pinter, M., Shmueli, M. D., Raz, C., Yanku, M., Zilberzwige, S., Gazit, E., et al. (2017). Interplay between protein glycosylation pathways in Alzheimer's disease. Sci. Adv. 3 (9), e1601576. doi:10.1126/sciadv.1601576
Fukuoka, T., Moriwaki, K., Takamatsu, S., Kondo, J., Tanaka-Okamoto, M., Tomioka, A., et al. (2022). Lewis glycosphingolipids as critical determinants of TRAIL sensitivity in cancer cells. Oncogene 41 (38), 4385–4396. doi:10.1038/s41388-022-02434-3
Gao, Y., Luan, X., Melamed, J., and Brockhausen, I. (2021). Role of glycans on key cell surface receptors that regulate cell proliferation and cell death. Cells 10 (5), 1252. doi:10.3390/cells10051252
García-Fuster, M. J., Ferrer-Alcón, M., Miralles, A., and García-Sevilla, J. A. (2004). Deglycosylation of Fas receptor and chronic morphine treatment up-regulate high molecular mass Fas aggregates in the rat brain. Eur. J. Pharmacol. 496 (1-3), 63–69. doi:10.1016/j.ejphar.2004.06.018
Geserick, P., Hupe, M., Moulin, M., Wong, W. W., Feoktistova, M., Kellert, B., et al. (2009). Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J. Cell Biol. 187 (7), 1037–1054. doi:10.1083/jcb.200904158
Han, L., Zhang, D., Tao, T., Sun, X., Liu, X., Zhu, G., et al. (2015). The role of N-glycan modification of TNFR1 in inflammatory microglia activation. Glycoconj J. 32 (9), 685–693. doi:10.1007/s10719-015-9619-1
Harazono, Y., Nakajima, K., and Raz, A. (2014). Why anti-Bcl-2 clinical trials fail: a solution. Cancer Metastasis Rev. 33 (1), 285–294. doi:10.1007/s10555-013-9450-8
Harduin-Lepers, A., Krzewinski-Recchi, M. A., Colomb, F., Foulquier, F., Groux-Degroote, S., and Delannoy, P. (2012). Sialyltransferases functions in cancers. Front. Biosci. Elite Ed. 4 (1), 499–515. doi:10.2741/396
Hart, G. W. (1997). Dynamic O-linked glycosylation of nuclear and cytoskeletal proteins. Annu. Rev. Biochem. 66, 315–335. doi:10.1146/annurev.biochem.66.1.315
Holdbrooks, A. T., Britain, C. M., and Bellis, S. L. (2018). ST6Gal-I sialyltransferase promotes tumor necrosis factor (TNF)-mediated cancer cell survival via sialylation of the TNF receptor 1 (TNFR1) death receptor. J. Biol. Chem. 293 (5), 1610–1622. doi:10.1074/jbc.M117.801480
Holmes, E. H., Ostrander, G. K., Clausen, H., and Graem, N. (1987). Oncofetal expression of Lex carbohydrate antigens in human colonic adenocarcinomas. Regulation through type 2 core chain synthesis rather than fucosylation. J. Biol. Chem. 262 (23), 11331–11338. doi:10.1016/s0021-9258(18)60963-9
Hsu, H., Shu, H. B., Pan, M. G., and Goeddel, D. V. (1996). TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 84 (2), 299–308. doi:10.1016/s0092-8674(00)80984-8
Huanna, T., Tao, Z., Xiangfei, W., Longfei, A., Yuanyuan, X., Jianhua, W., et al. (2015). GALNT14 mediates tumor invasion and migration in breast cancer cell MCF-7. Mol. Carcinog. 54 (10), 1159–1171. doi:10.1002/mc.22186
Hughes, M. A., Powley, I. R., Jukes-Jones, R., Horn, S., Feoktistova, M., Fairall, L., et al. (2016). Co-Operative and hierarchical binding of c-FLIP and caspase-8: a unified model defines how c-FLIP isoforms differentially control cell fate. Mol. Cell 61 (6), 834–849. doi:10.1016/j.molcel.2016.02.023
Irmler, M., Thome, M., Hahne, M., Schneider, P., Hofmann, K., Steiner, V., et al. (1997). Inhibition of death receptor signals by cellular FLIP. Nature 388 (6638), 190–195. doi:10.1038/40657
Ivanisenko, N. V., Seyrek, K., Hillert-Richter, L. K., Konig, C., Espe, J., Bose, K., et al. (2022). Regulation of extrinsic apoptotic signaling by c-FLIP: towards targeting cancer networks. Trends Cancer 8 (3), 190–209. doi:10.1016/j.trecan.2021.12.002
Jiang, Y., Wen, T., Yan, R., Kim, S. R., Stowell, S. R., Wang, W., et al. (2020). O-glycans on death receptors in cells modulate their sensitivity to TRAIL-induced apoptosis through affecting on their stability and oligomerization. Faseb J. 34 (9), 11786–11801. doi:10.1096/fj.201900053RR
Ju, T., and Cummings, R. D. (2002). A unique molecular chaperone Cosmc required for activity of the mammalian core 1 beta 3-galactosyltransferase. Proc. Natl. Acad. Sci. U. S. A. 99 (26), 16613–16618. doi:10.1073/pnas.262438199
Ju, T., and Cummings, R. D. (2005). Protein glycosylation: chaperone mutation in Tn syndrome. Nature 437 (7063), 1252. doi:10.1038/4371252a
Ju, T., Lanneau, G. S., Gautam, T., Wang, Y., Xia, B., Stowell, S. R., et al. (2008). Human tumor antigens Tn and sialyl Tn arise from mutations in Cosmc. Cancer Res. 68 (6), 1636–1646. doi:10.1158/0008-5472.CAN-07-2345
Karin, M. (2006). Nuclear factor-kappaB in cancer development and progression. Nature 441 (7092), 431–436. doi:10.1038/nature04870
Keppler, O. T., Peter, M. E., Hinderlich, S., Moldenhauer, G., Stehling, P., Schmitz, I., et al. (1999). Differential sialylation of cell surface glycoconjugates in a human B lymphoma cell line regulates susceptibility for CD95 (APO-1/Fas)-mediated apoptosis and for infection by a lymphotropic virus. Glycobiology. 9 (6), 557–569. doi:10.1093/glycob/9.6.557
Kischkel, F. C., Hellbardt, S., Behrmann, I., Germer, M., Pawlita, M., Krammer, P. H., et al. (1995). Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 14 (22), 5579–5588. doi:10.1002/j.1460-2075.1995.tb00245.x
Köhne, C., Johnson, A., Tom, S., Peers, D. H., Gehant, R. L., Hotaling, T. A., et al. (1999). Secretion of glycosylation site mutants can be rescued by the signal/pro sequence of tissue plasminogen activator. J. Cell. Biochem. 75 (3), 446–461. doi:10.1002/(sici)1097-4644(19991201)75:3<446::aid-jcb10>3.0.co;2-i
Krammer, P. H., Arnold, R., and Lavrik, I. N. (2007). Life and death in peripheral T cells. Nat. Rev. Immunol. 7 (7), 532–542. doi:10.1038/nri2115
Kunzke, T., Balluff, B., Feuchtinger, A., Buck, A., Langer, R., Luber, B., et al. (2017). Native glycan fragments detected by MALDI-FT-ICR mass spectrometry imaging impact gastric cancer biology and patient outcome. Oncotarget 8 (40), 68012–68025. doi:10.18632/oncotarget.19137
Lafont, E., Kantari-Mimoun, C., Draber, P., De Miguel, D., Hartwig, T., Reichert, M., et al. (2017). The linear ubiquitin chain assembly complex regulates TRAIL-induced gene activation and cell death. EMBO J. 36 (9), 1147–1166. doi:10.15252/embj.201695699
Laurien, L., Nagata, M., Schunke, H., Delanghe, T., Wiederstein, J. L., Kumari, S., et al. (2020). Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation. Nat. Commun. 11 (1), 1747. doi:10.1038/s41467-020-15466-8
Lavrik, I. N., and Krammer, P. H. (2012). Regulation of CD95/Fas signaling at the DISC. Cell Death Differ. 19 (1), 36–41. doi:10.1038/cdd.2011.155
Lavrik, I. N., Mock, T., Golks, A., Hoffmann, J. C., Baumann, S., and Krammer, P. H. (2008). CD95 stimulation results in the formation of a novel death effector domain protein-containing complex. J. Biol. Chem. 283 (39), 26401–26408. doi:10.1074/jbc.M800823200
Lee, H., Oh, Y., Jeon, Y. J., Lee, S. Y., Kim, H., Lee, H. J., et al. (2019). DR4-Ser424 O-GlcNAcylation promotes sensitization of TRAIL-tolerant persisters and TRAIL-resistant cancer cells to death. Cancer Res. 79 (11), 2839–2852. doi:10.1158/0008-5472.CAN-18-1991
Lee, S. J., Lee, D. E., Choi, S. Y., and Kwon, O. S. (2021). OSMI-1 enhances TRAIL-induced apoptosis through ER stress and NF-κB signaling in colon cancer cells. Int. J. Mol. Sci. 22 (20), 11073. doi:10.3390/ijms222011073
Leffler, H., Carlsson, S., Hedlund, M., Qian, Y., and Poirier, F. (2002). Introduction to galectins. Glycoconj J. 19 (7-9), 433–440. doi:10.1023/B:GLYC.0000014072.34840.04
Lehrman, M. A. (1991). Biosynthesis of N-acetylglucosamine-P-P-dolichol, the committed step of asparagine-linked oligosaccharide assembly. Glycobiology. 1 (6), 553–562. doi:10.1093/glycob/1.6.553
Lemke, J., von Karstedt, S., Zinngrebe, J., and Walczak, H. (2014). Getting TRAIL back on track for cancer therapy. Cell Death Differ. 21 (9), 1350–1364. doi:10.1038/cdd.2014.81
Li, S., Zhang, L., Yao, Q., Li, L., Dong, N., Rong, J., et al. (2013). Pathogen blocks host death receptor signalling by arginine GlcNAcylation of death domains. Nature 501 (7466), 242–246. doi:10.1038/nature12436
Li, S. T., Lu, T. T., Xu, X. X., Ding, Y., Li, Z., Kitajima, T., et al. (2019). Reconstitution of the lipid-linked oligosaccharide pathway for assembly of high-mannose N-glycans. Nat. Commun. 10 (1), 1813. doi:10.1038/s41467-019-09752-3
Li, Y., Yang, X., Nguyen, A. H., and Brockhausen, I. (2007). Requirement of N-glycosylation for the secretion of recombinant extracellular domain of human Fas in HeLa cells. Int. J. Biochem. and cell Biol. 39 (9), 1625–1636. doi:10.1016/j.biocel.2007.04.002
Lichtenstein, R. G., and Rabinovich, G. A. (2013). Glycobiology of cell death: when glycans and lectins govern cell fate. Cell death Differ. 20 (8), 976–986. doi:10.1038/cdd.2013.50
Liu, Z., Swindall, A. F., Kesterson, R. A., Schoeb, T. R., Bullard, D. C., and Bellis, S. L. (2011). ST6Gal-I regulates macrophage apoptosis via α2-6 sialylation of the TNFR1 death receptor. J. Biol. Chem. 286 (45), 39654–39662. doi:10.1074/jbc.M111.276063
Martin-Perez, R., Niwa, M., and Lopez-Rivas, A. (2012). ER stress sensitizes cells to TRAIL through down-regulation of FLIP and Mcl-1 and PERK-dependent up-regulation of TRAIL-R2. Apoptosis Int. J. Program. cell death. 17 (4), 349–363. doi:10.1007/s10495-011-0673-2
Mazurek, N., Sun, Y. J., Liu, K. F., Gilcrease, M. Z., Schober, W., Nangia-Makker, P., et al. (2007). Phosphorylated galectin-3 mediates tumor necrosis factor-related apoptosis-inducing ligand signaling by regulating phosphatase and tensin homologue deleted on chromosome 10 in human breast carcinoma cells. J. Biol. Chem. 282 (29), 21337–21348. doi:10.1074/jbc.M608810200
Micheau, O., and Tschopp, J. (2003). Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114 (2), 181–190. doi:10.1016/s0092-8674(03)00521-x
Miyoshi, E., Moriwaki, K., and Nakagawa, T. (2008). Biological function of fucosylation in cancer biology. J. Biochem. 143 (6), 725–729. doi:10.1093/jb/mvn011
Morad, S. A. F., Davis, T. S., MacDougall, M. R., Tan, S. F., Feith, D. J., Desai, D. H., et al. (2017). Role of P-glycoprotein inhibitors in ceramide-based therapeutics for treatment of cancer. Biochem. Pharmacol. 130, 21–33. doi:10.1016/j.bcp.2017.02.002
Moremen, K. W., Tiemeyer, M., and Nairn, A. V. (2012). Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol. cell Biol. 13 (7), 448–462. doi:10.1038/nrm3383
Moriwaki, K., Chan, F. K. M., and Miyoshi, E. (2021). Sweet modification and regulation of death receptor signalling pathway. J. Biochem. 169 (6), 643–652. doi:10.1093/jb/mvab034
Moriwaki, K., Noda, K., Furukawa, Y., Ohshima, K., Uchiyama, A., Nakagawa, T., et al. (2009). Deficiency of GMDS leads to escape from NK cell-mediated tumor surveillance through modulation of TRAIL signaling. Gastroenterology 137 (1), 188–198. 98.e1-2. doi:10.1053/j.gastro.2009.04.002
Moriwaki, K., Shinzaki, S., and Miyoshi, E. (2011). GDP-mannose-4,6-dehydratase (GMDS) deficiency renders colon cancer cells resistant to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor- and CD95-mediated apoptosis by inhibiting complex II formation. J. Biol. Chem. 286 (50), 43123–43133. doi:10.1074/jbc.M111.262741
Muppidi, J. R., Tschopp, J., and Siegel, R. M. (2004). Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity 21 (4), 461–465. doi:10.1016/j.immuni.2004.10.001
Neumann, L., Pforr, C., Beaudouin, J., Pappa, A., Fricker, N., Krammer, P. H., et al. (2010). Dynamics within the CD95 death-inducing signaling complex decide life and death of cells. Mol. Syst. Biol. 6, 352. doi:10.1038/msb.2010.6
Newson, J. P. M., Scott, N. E., Yeuk Wah Chung, I., Wong Fok Lung, T., Giogha, C., Gan, J., et al. (2019). Salmonella effectors SseK1 and SseK3 target death domain proteins in the TNF and TRAIL signaling pathways. Mol. Cell Proteomics 18 (6), 1138–1156. doi:10.1074/mcp.RA118.001093
Pan, L., Fu, T. M., Zhao, W., Zhao, L., Chen, W., Qiu, C., et al. (2019). Higher-order clustering of the transmembrane anchor of DR5 drives signaling. Cell 176 (6), 1477–1489. doi:10.1016/j.cell.2019.02.001
Park, Y. C., Ye, H., Hsia, C., Segal, D., Rich, R. L., Liou, H. C., et al. (2000). A novel mechanism of TRAF signaling revealed by structural and functional analyses of the TRADD-TRAF2 interaction. Cell 101 (7), 777–787. doi:10.1016/s0092-8674(00)80889-2
Pearson, J. S., Giogha, C., Ong, S. Y., Kennedy, C. L., Kelly, M., Robinson, K. S., et al. (2013). A type III effector antagonizes death receptor signalling during bacterial gut infection. Nature 501 (7466), 247–251. doi:10.1038/nature12524
Peter, M. E., Hellbardt, S., Schwartz-Albiez, R., Westendorp, M. O., Walczak, H., Moldenhauer, G., et al. (1995). Cell surface sialylation plays a role in modulating sensitivity towards APO-1-mediated apoptotic cell death. Cell death Differ. 2 (3), 163–171.
Radhakrishnan, P., Dabelsteen, S., Madsen, F. B., Francavilla, C., Kopp, K. L., Steentoft, C., et al. (2014). Immature truncated O-glycophenotype of cancer directly induces oncogenic features. Proc. Natl. Acad. Sci. U. S. A. 111 (39), E4066–E4075. doi:10.1073/pnas.1406619111
Santos, S. N., Junqueira, M. S., Francisco, G., Vilanova, M., Magalhães, A., Dias Baruffi, M., et al. (2016). O-glycan sialylation alters galectin-3 subcellular localization and decreases chemotherapy sensitivity in gastric cancer. Oncotarget 7 (50), 83570–83587. doi:10.18632/oncotarget.13192
Schleich, K., Warnken, U., Fricker, N., Ozturk, S., Richter, P., Kammerer, K., et al. (2012). Stoichiometry of the CD95 death-inducing signaling complex: experimental and modeling evidence for a death effector domain chain model. Mol. Cell 47 (2), 306–319. doi:10.1016/j.molcel.2012.05.006
Schultz, M. J., Holdbrooks, A. T., Chakraborty, A., Grizzle, W. E., Landen, C. N., Buchsbaum, D. J., et al. (2016). The tumor-associated glycosyltransferase ST6Gal-I regulates stem cell transcription factors and confers a cancer stem cell phenotype. Cancer Res. 76 (13), 3978–3988. doi:10.1158/0008-5472.CAN-15-2834
Scott, F. L., Stec, B., Pop, C., Dobaczewska, M. K., Lee, J. J., Monosov, E., et al. (2009). The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature 457 (7232), 1019–1022. doi:10.1038/nature07606
Scott, N. E., Giogha, C., Pollock, G. L., Kennedy, C. L., Webb, A. I., Williamson, N. A., et al. (2017). The bacterial arginine glycosyltransferase effector NleB preferentially modifies Fas-associated death domain protein (FADD). J. Biol. Chem. 292 (42), 17337–17350. doi:10.1074/jbc.M117.805036
Seo, J., Kim, Y., Ji, S., Kim, H. B., Jung, H., Yi, E. C., et al. (2023). O-GlcNAcylation of RIPK1 rescues red blood cells from necroptosis. Front. Immunol. 14, 1160490. doi:10.3389/fimmu.2023.1160490
Seyrek, K., Ivanisenko, N. V., Konig, C., and Lavrik, I. N. (2024). Modulation of extrinsic apoptotic pathway by intracellular glycosylation. Trends cell Biol. 34 (9), 728–741. doi:10.1016/j.tcb.2024.01.003
Seyrek, K., Ivanisenko, N. V., Richter, M., Hillert, L. K., Konig, C., and Lavrik, I. N. (2020). Controlling cell death through post-translational modifications of DED proteins. Trends Cell Biol. 30 (5), 354–369. doi:10.1016/j.tcb.2020.02.006
Seyrek, K., Ivanisenko, N. V., Wohlfromm, F., Espe, J., and Lavrik, I. N. (2022). Impact of human CD95 mutations on cell death and autoimmunity: a model. Trends Immunol. 43 (1), 22–40. doi:10.1016/j.it.2021.11.006
Seyrek, K., Richter, M., and Lavrik, I. N. (2019). Decoding the sweet regulation of apoptosis: the role of glycosylation and galectins in apoptotic signaling pathways. Cell Death Differ. 26 (6), 981–993. doi:10.1038/s41418-019-0317-6
Shatnyeva, O. M., Kubarenko, A. V., Weber, C. E., Pappa, A., Schwartz-Albiez, R., Weber, A. N., et al. (2011). Modulation of the CD95-induced apoptosis: the role of CD95 N-glycosylation. PLoS One 6 (5), e19927. doi:10.1371/journal.pone.0019927
Smith, W., Tomasec, P., Aicheler, R., Loewendorf, A., Nemčovičová, I., Wang, E. C., et al. (2013). Human cytomegalovirus glycoprotein UL141 targets the TRAIL death receptors to thwart host innate antiviral defenses. Cell host and microbe 13 (3), 324–335. doi:10.1016/j.chom.2013.02.003
Sun, X., Ju, T., and Cummings, R. D. (2018). Differential expression of Cosmc, T-synthase and mucins in Tn-positive colorectal cancers. BMC cancer 18 (1), 827. doi:10.1186/s12885-018-4708-8
Suzuki, O., and Abe, M. (2008). Cell surface N-glycosylation and sialylation regulate galectin-3-induced apoptosis in human diffuse large B cell lymphoma. Oncol. Rep. 19 (3), 743–748. doi:10.3892/or.19.3.743
Swindall, A. F., and Bellis, S. L. (2011). Sialylation of the Fas death receptor by ST6Gal-I provides protection against Fas-mediated apoptosis in colon carcinoma cells. J. Biol. Chem. 286 (26), 22982–22990. doi:10.1074/jbc.M110.211375
Tenev, T., Bianchi, K., Darding, M., Broemer, M., Langlais, C., Wallberg, F., et al. (2011). The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 43 (3), 432–448. doi:10.1016/j.molcel.2011.06.006
Voigt, S., Philipp, S., Davarnia, P., Winoto-Morbach, S., Röder, C., Arenz, C., et al. (2014). TRAIL-induced programmed necrosis as a novel approach to eliminate tumor cells. BMC cancer 14, 74. doi:10.1186/1471-2407-14-74
von Karstedt, S., Montinaro, A., and Walczak, H. (2017). Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer 17 (6), 352–366. doi:10.1038/nrc.2017.28
Wagner, K. W., Punnoose, E. A., Januario, T., Lawrence, D. A., Pitti, R. M., Lancaster, K., et al. (2007). Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat. Med. 13 (9), 1070–1077. doi:10.1038/nm1627
Wajant, H., and Scheurich, P. (2011). TNFR1-induced activation of the classical NF-κB pathway. FEBS J. 278 (6), 862–876. doi:10.1111/j.1742-4658.2011.08015.x
Wang, L., Du, F., and Wang, X. (2008). TNF-alpha induces two distinct caspase-8 activation pathways. Cell 133 (4), 693–703. doi:10.1016/j.cell.2008.03.036
Wu, Y. X., Lu, H. F., Lin, Y. H., Chuang, H. Y., Su, S. C., Liao, Y. J., et al. (2021). Branched I antigen regulated cell susceptibility against natural killer cytotoxicity through its N-linked glycosylation and overall expression. Glycobiology 31 (5), 624–635. doi:10.1093/glycob/cwaa117
Xue, J., Gao, X., Fu, C., Cong, Z., Jiang, H., Wang, W., et al. (2013). Regulation of galectin-3-induced apoptosis of Jurkat cells by both O-glycans and N-glycans on CD45. FEBS Lett. 587 (24), 3986–3994. doi:10.1016/j.febslet.2013.10.034
Xue, J., Hu, S., Huang, Y., Zhang, Q., Yi, X., Pan, X., et al. (2020). Arg-GlcNAcylation on TRADD by NleB and SseK1 is crucial for bacterial pathogenesis. Front. cell Dev. Biol. 8, 641. doi:10.3389/fcell.2020.00641
Yang, C. Y., Tseng, Y. C., Tu, Y. F., Kuo, B. J., Hsu, L. C., Lien, C. I., et al. (2024). Reverse hierarchical DED assembly in the cFLIP-procaspase-8 and cFLIP-procaspase-8-FADD complexes. Nat. Commun. 15 (1), 8974. doi:10.1038/s41467-024-53306-1
Yang, S. Z., Xu, F., Yuan, K., Sun, Y., Zhou, T., Zhao, X., et al. (2020). Regulation of pancreatic cancer TRAIL resistance by protein O-GlcNAcylation. Laboratory investigation; a J. Tech. methods pathology 100 (5), 777–785. doi:10.1038/s41374-019-0365-z
Yoshida, T., Shiraishi, T., Horinaka, M., Wakada, M., and Sakai, T. (2007). Glycosylation modulates TRAIL-R1/death receptor 4 protein: different regulations of two pro-apoptotic receptors for TRAIL by tunicamycin. Oncol. Rep. 18 (5), 1239–1242. doi:10.3892/or.18.5.1239
Zeidan, Q., and Hart, G. W. (2010). The intersections between O-GlcNAcylation and phosphorylation: implications for multiple signaling pathways. J. Cell Sci. 123 (Pt 1), 13–22. doi:10.1242/jcs.053678
Zeng, X., Chen, Z., Zhu, Y., Liu, L., Zhang, Z., Xiao, Y., et al. (2024). O-GlcNAcylation regulation of RIPK1-dependent apoptosis dictates sensitivity to sunitinib in renal cell carcinoma. Drug Resist. Updat. Rev. Comment. Antimicrob. anticancer Chemother. 77, 101150. doi:10.1016/j.drup.2024.101150
Zhang, B., van Roosmalen, I. A. M., Reis, C. R., Setroikromo, R., and Quax, W. J. (2019). Death receptor 5 is activated by fucosylation in colon cancer cells. FEBS J. 286 (3), 555–571. doi:10.1111/febs.14742
Zhang, Y., Su, S. S., Zhao, S., Yang, Z., Zhong, C. Q., Chen, X., et al. (2017). RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 8, 14329. doi:10.1038/ncomms14329
Keywords: death receptor, post-translational modifications, glycosylation, CD95, DISC, TRAIL, TNFR1
Citation: Seyrek K, Espe J, König C, Wohlfromm F and Lavrik IN (2025) Glycans on death receptors as sweet markers and signaling checkpoints. Front. Cell Death 4:1585989. doi: 10.3389/fceld.2025.1585989
Received: 01 March 2025; Accepted: 26 March 2025;
Published: 08 April 2025.
Edited by:
Nicolas Bidere, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Olivier Micheau, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceTiphaine Douanne, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2025 Seyrek, Espe, König, Wohlfromm and Lavrik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Inna N. Lavrik, aW5uYS5sYXZyaWtAbWVkLm92Z3UuZGU=