 Muhammad W. Luqman1,2†‡
Muhammad W. Luqman1,2†‡ Piroon Jenjaroenpun3‡
Piroon Jenjaroenpun3‡ Jessica Spathos1Nikhil Shingte4Mitchell Cummins1,5Pattaraporn Nimsamer3Lars M. Ittner1Thidathip Wongsurawat3*
Jessica Spathos1Nikhil Shingte4Mitchell Cummins1,5Pattaraporn Nimsamer3Lars M. Ittner1Thidathip Wongsurawat3* Fabien Delerue1*†
Fabien Delerue1*†- 1Dementia Research Centre, Macquarie Medical School, Faculty of Medicine, Health and Human Sciences, Macquarie University, Sydney, NSW, Australia
- 2Khyber Medical University, Institute of Medical Sciences, Kohat, Pakistan
- 3Division of Medical Bioinformatics, Research Department, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand
- 4FOXG1 Research Foundation, New York, NY, United States
- 5School of Biotechnology and Biomolecular Sciences, The University of New South Wales, Sydney, NSW, Australia
Over the last decade CRISPR gene editing has been successfully used to streamline the generation of animal models for biomedical research purposes. However, one limitation to its use is the potential occurrence of on-target mutations that may be detrimental or otherwise unintended. These bystander mutations are often undetected using conventional genotyping methods. The use of Adeno-Associated Viruses (AAVs) to bring donor templates in zygotes is currently being deployed by transgenic cores around the world to generate knock-ins with large transgenes (i.e., 1–4 kb payloads). Thanks to a high level of efficiency and the relative ease to establish this technique, it recently became a method of choice for transgenic laboratories. However, a thorough analysis of the editing outcomes following this method is yet to be developed. To this end, we generated three different types of integration using AAVs in two different murine genes (i.e., Ace2 and Foxg1) and employed Oxford Nanopore Technologies long read sequencing to analyze the outcomes. Using a workflow that includes Cas9 enrichment and adaptive sampling, we showed that unintended on-target mutations, including duplication events and integration of viral sequences (sometimes reported using other workflows) can occur when using AAVs. This work highlights the importance of in-depth validation of the mutant lines generated and informs the uptake of this new method.
Introduction
Genetically modified (GM) animals, particularly mice, are powerful models to understand the mechanisms underlying physiological processes and human disorders. They are also invaluable tools to develop and test novel treatment strategies.
Transgenic laboratories around the world generate these models for biomedical research purposes, using either microinjection techniques (Delerue and Ittner, 2017), or more recently electroporation of fertilized zygotes (Qin et al., 2015; Kaneko and Mashimo, 2015). Electroporation is less challenging than microinjection and allows for high-throughput transformation of zygotes, whereas microinjection requires manipulation of zygotes one at a time. Moreover, survival and development rates are comparatively higher because electroporation is less invasive and damaging to the embryos than microinjection (Kaneko and Mashimo, 2015). As such, electroporation of zygotes is widely used to generate knockouts (KO) and small nucleotides changes, such as point mutations or base pair exchanges. We, and others, generated such mouse models using electroporation of one-cell embryos (Klugmann et al., 2022; Morey et al., 2022).
However, electroporation remains largely inefficient at driving the targeted integration of large transgenes to generate knock-in (KI) mouse lines, presumably because the zona pellucida (thick glycoprotein membrane protecting the embryos at the preimplantation stages) prevents the shuttling of these large transgenes inside the embryos. To the best of our knowledge, there has been no report to date of a successful transformation of double stranded DNA (e.g., plasmid or transgene) using electroporation of such zygotes, and the largest insertion reported so far by electroporation of single-stranded oligo is 1 kb in length (Miyasaka et al., 2018).
Therefore, a new method based on infection of zygotes with Adeno-Associated Viruses (AAVs) to bring the donor template inside the zygotes, coupled with electroporation of CRISPR ribonucleoprotein (RNP) complexes to induce Homology Directed Repair (HDR) has recently been developed (Yoon et al., 2018; Mizuno et al., 2018; Chen et al., 2019). Such method, coined “CRISPR-READI” shows high level of efficiency (up to 100% in our hands) and is relatively easy to implement in transgenic laboratories. Romeo et al. showed that AAVs could diffuse through the zona pellucida (Romeo et al., 2020), while the Rivera-Perez lab demonstrated that the transfection efficiency varies depending on the serotype used, serotype 6 having one of the highest levels of transduction in mouse zygotes (Yoon et al., 2018).
Over the last decade CRISPR gene editing has been extensively used to generate GM mice, however, one limitation to its use is the potential occurrence of on-target mutations that are detrimental or otherwise unintended. These bystander mutations are typically undetected using conventional genotyping (i.e., PCR) and routine (i.e., Sanger) sequencing (Simkin et al., 2022; Sailer et al., 2021). As such, an in-depth analysis of the editing outcomes is highly recommended (Lintott and Nutter, 2023) to ensure that GM animals are validated before extensive breeding.
Recently, Oxford Nanopore Technologies (ONT©) Long Read Sequencing (LRS) has been used in mice to identify the insertion site of randomly integrated transgenes (Bryant et al., 2023), targeted insertions (McCabe et al., 2024), and to confirm integration following recombinase-mediated cassette exchange (RMCE) (Low et al., 2022). However, to the best of our knowledge, LRS has not yet been used to perform quality control (QC) following AAV-driven gene editing in zygotes. To this end, we performed CRISPR-READI to target two murine genes (i.e., Ace2 and Foxg1) with transgenes of various sizes. We then analyzed these knock-ins using the Oxford Nanopore Technologies (ONT©) MinION Mk1c and/or GridION, following a Cas9-based enrichment method that we previously applied to cells (Wongsurawat et al., 2020). Using this method, we identified instances of concatemerization with partial AAV vector sequences integration (particularly Inverted Terminal Repeat sequences - ITR) in two out of five (40%) mouse lines generated.
Results
In our workflow, following PCR genotyping of the Founder mice, Long Read Sequencing was performed either at the F1 or the F2 generation (see Supplementary Figure S1 for the identification of each selected mouse in this study).
Generation of KI mouse lines
We performed AAV-driven gene editing on two different murine genes: the angiotensin I converting enzyme 2 (i.e., Ace2) and the forkhead box G1 (i.e., Foxg1) to insert three different transgenes.
First, we targeted the start codon of the murine Ace2 gene and inserted the human ACE2 (hACE2) coding sequence upstream of a polyadenylation signal (SV40 pA) (Figure 1a), using a combination of CRISPR/Cas9 and TALEN nucleases (no suitable sgRNA could be identified to target this genomic sequence, see Methods).
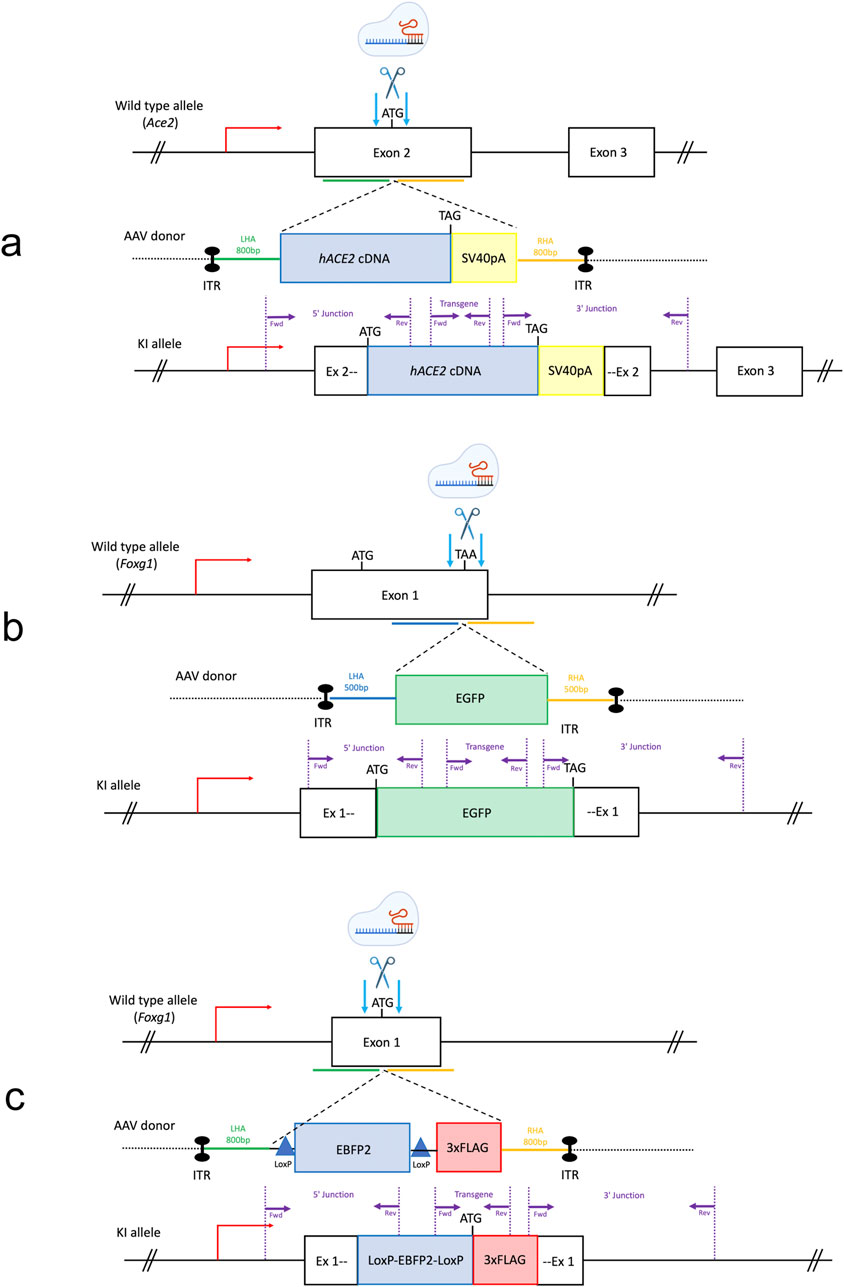
Figure 1. Generation of KI mouse lines by AAV-driven gene editing. The gene editing strategy for each knock-in (KI) line is illustrated. (a) The start codon of the murine Ace2 gene was targeted for Homology Directed Repair (HDR) with a donor template carrying the hACE2 coding DNA sequence upstream of a PolyA. (b) The stop codon of the mouse Foxg1 gene was targeted for HDR with an EGFP sequence at the c-terminus. (c) The start codon of the murine Foxg1 gene was targeted to insert a stop cassette and a triple FLAG tag. LHA = left homology arm, RHA = right homology arm, ITR = inverted terminal repeat sequence, hACE2 = human ACE2 coding sequence, SV40 pA = simian virus 40 polyadenylation signal, EGFP = Enhanced Green Fluorescence Protein sequence, EBFP2 = Enhanced Blue Fluorescent Protein sequence, 3xFLAG = triple FLAG tag, LSL = Lox-Stop-Lox. Purple arrows: location of the genotyping primers used in this study for the transgene-specific and the junction PCRs. Light blue arrows: endonucleases-induced double strand breaks.
We next targeted the stop codon of the endogenous Foxg1 gene to insert an Enhanced Green Fluorescent Protein (EGFP) sequence in frame (Figure 1b). Finally, we also targeted the start codon of the Foxg1 gene to generate a conditional KI by inserting a Lox-Stop-Lox (LSL) cassette (made of an EBFP2 coding sequence flanked by two LoxP sites) upstream of a triple FLAG sequence (Figure 1c).
The size of the transgenes ranged from ∼700 bp to 2.5 kb, and 5 h infection with high titers of recombinant AAV6 was performed for all KIs (a summary of the strategy used for the three KI approaches is detailed in Table 1).
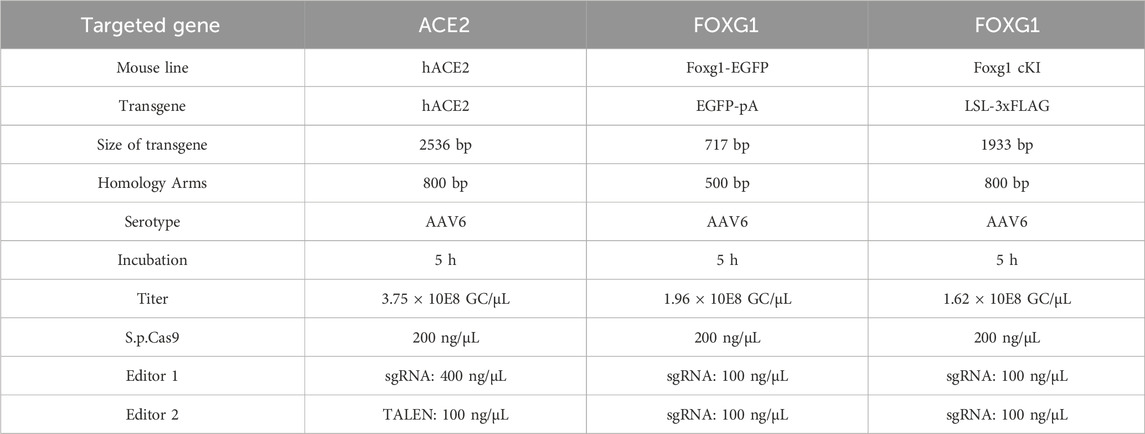
Table 1. CRISPR-READI strategy to generate the KI mouse lines. Details of the transgenes and editing specifications used to generate the mouse lines by AAV-driven gene editing. bp = base pairs, GC/μL = genome-copy per microliter, ng/μL = nanogram per microliter.
PCR genotyping of the KI mice does not identify any illegitimate mutation
hACE2 KI mice
To identify potential founders, we first ran a transgene-specific PCR. Out of eight pups, four (50%) carried the transgene (Figure 2a). To validate the insertion of the transgene at the endogenous Ace2 locus, we then performed 5′ and 3′ junction PCRs (Figures 2b,c). Out of four potential founders, three carried the transgene at the insertion site. The fourth one (#43215) may display random insertion or episomal presence of the transgene. Although not always completely specific (e.g., Figure 2a presents with extra bands), the PCR genotyping did not reveal any apparent illegitimate event at the targeted site. We next selected two founders (#43204 and 43205) to establish the colonies.

Figure 2. PCR Genotyping outcome for the KI mouse lines. Illustrative cropped gel electrophoresis for the three KI mouse lines generated by CRISPR-READI. (a) Transgene-specific genotyping of the hACE2 founders (G0) showed that 4 out of 8 pups generated (50%) carried the hACE2 transgene. Targeted integration of the hACE2 transgene was confirmed by 5’ (b) and 3’ (c) junction PCRs. Founder #43215 may carry the AAV template as an episome or be randomly integrated. (d) Transgene-specific genotyping of the selected Foxg1-EGFP line (G1 generation). Targeted integration for the Foxg1-EGFP line was confirmed by 5’ (e) and 3’ (f) junction PCRs. (g) Transgene-specific genotyping of the selected Foxg1 conditional KI (Foxg1 cKI) line (G1 generation). Targeted integration for the Foxg1 cKI line was confirmed by 5’ (h) and 3’ (i) junction PCRs. L1kb: Ladder (Bioline, HyperLadder 1 kb); L100: Ladder (Bioline, HyperLadder 100 bp).
Foxg1 KI mice
Similar to the hACE2 KI mice, PCRs identified several candidate Foxg1 KI Founders. Indeed, transgene-specific PCR identified sixteen potential founders out of 41 live pups (39%) for the Foxg1-EGFP; and three potential founders out of 18 live pups (17%) for the Foxg1 cKI, respectively.
We selected one Foxg1-EGFP founder and two Foxg1 cKI founders and bred them with wildtype C57BL/6J mice to establish the colonies. We performed transgene-specific and junction PCRs on F1 mice and observed a normal genotyping profile with expected band sizes for all Foxg1 KI mice (Figures 2d–i). The sequence of all primers used in this study can be found in Supplementary Table S1.
Cas9 enrichment generates adequate level of coverage
We performed genomic enrichment for five mice, two homozygous (#64005 from Founder #43205 and #65209 from Founder #43204) and three heterozygous (#80297, #88164 and #88312), and designed four sgRNAs for each locus (see Methods) to enrich the genomic regions of interest (ROI), following the nanopore Cas9-targeted sequencing (nCATS) method (Gilpatrick et al., 2020). The enrichment targeting a ∼3–5 kb ROI centered around the integration site (Figures 3a–c), yielded an on-target read depth ranging from 11 to 252x, representing 0.24%–2.28% of all reads (Figures 3a–c; Supplementary Table S2). Most samples yielded over 40x coverage, enough to reconstruct the KI consensus sequences using a de novo assembly approach, except for mouse #64005, which only achieved 11x coverage.
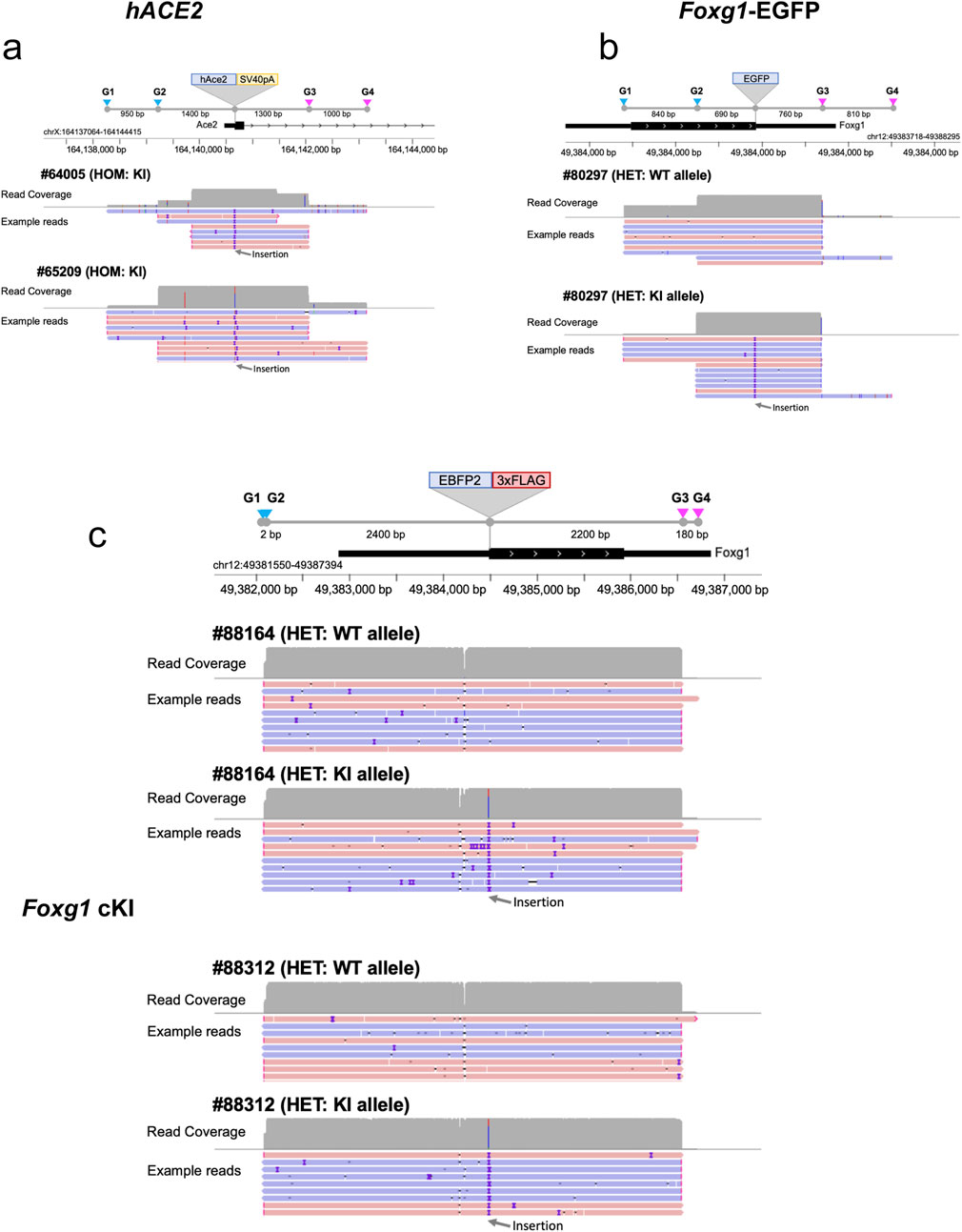
Figure 3. Cas9 enrichment strategy and resulting read depth for each KI. Illustration of the Cas9-based enrichment (nCATs method) of genomic DNA using four sgRNA and resulting read coverage. (a) Enrichment of the murine Ace2 targeted region using four sgRNA encompassing the insertion site of the hACE2 transgene. Example of reads (pink = 5’ to 3’ reads, blue = 3’ to 5’ reads, purple = mismatch) for each homozygous hACE2 mouse tested (#64005 and #65209). (b) Enrichment of the murine Foxg1 targeted region using four sgRNA encompassing the insertion site of the EGFP transgene. Example of reads for the heterozygous Foxg1-EGFP mouse tested (#80297). (c) Enrichment of the murine Foxg1 targeted region using four sgRNA encompassing the insertion site of the EBFP2-3xFLAG transgene. Example of reads for the two heterozygous Foxg1 cKI mouse tested (#88164 and #88312). G1-G4 = sgRNA1 to sgRNA4, WT = wildtype, KI = knock-in.
When looking at allele specificity (i.e., read counts for both wildtype and KI alleles for the three heterozygous mice (#80297, #88164, #88312), there was a slightly higher read count for the KI allele for mouse #80297, but this difference was not confirmed for the other two heterozygous mice (see Table 2), suggesting that there is no bias towards one or the other allele when performing LRS downstream of Cas9 enrichment. This may not be the case for LRS performed downstream of PCR amplification, as PCR may preferentially amplify the shorter allele.
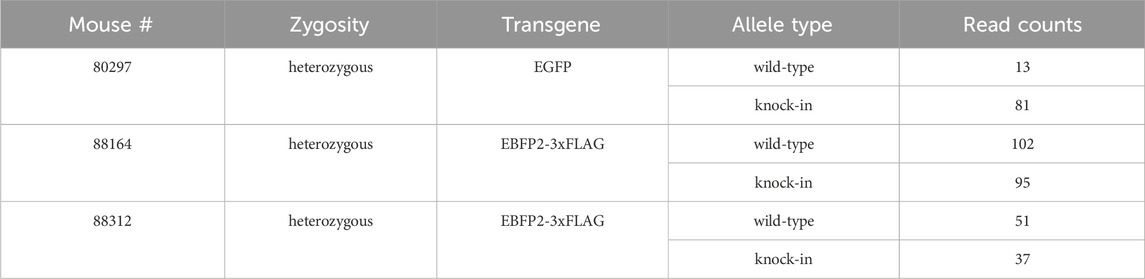
Table 2. Read counts per allele for each heterozygous mouse sequenced in this study. Allele-specific (wildtype and knock-in) read counts for the 3 heterozygous mice sequenced in this study following Cas9 enrichment.
Adaptive sampling (AS) is a bioinformatic feature that enhances targeted sequencing by selectively processing reads from predefined regions of interest. As DNA strands are read, the sequence is compared to the reference sequence in real time. Should the sequences not match, the nanopore closes and rejects the DNA strand, allowing another strand to be sequenced, aiming to increase on-target yield and coverage depth (Payne et al., 2020).
To evaluate whether adaptive sampling (AS) (Martin et al., 2022) could enhance coverage post Cas9-enrichment as previously assessed (Rubben et al., 2022), two mice (#64005 and #65209) were sequenced, both with and without AS (as referenced in Table 3).
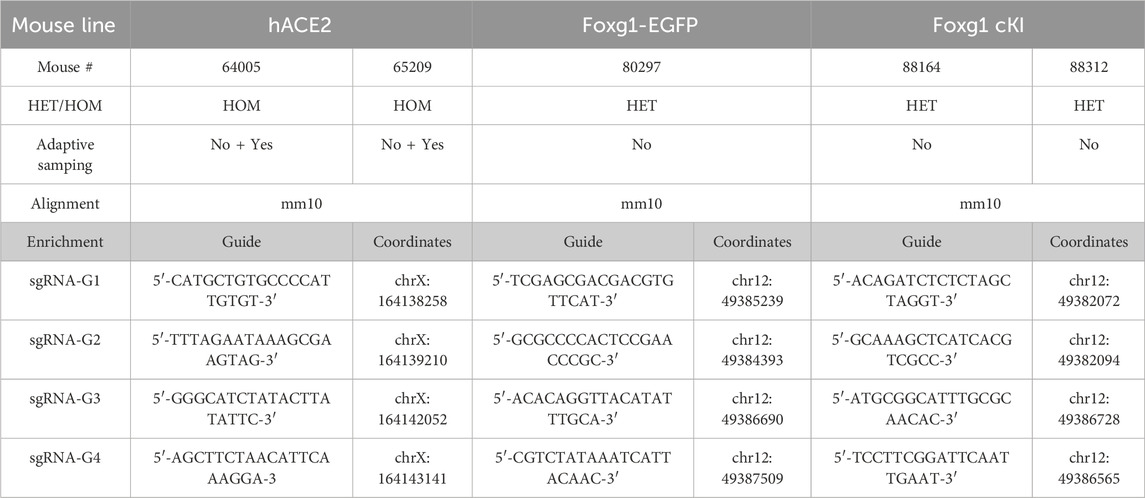
Table 3. Cas9 enrichment strategy for nanopore long read sequencing. The nanopore Cas9-targeted sequencing (nCATS) method requires enrichment of the genomic sequence of interest. This table details the sequence of the guides used to enrich specific regions centered around the transgene integration site. HOM = homozygous, HET = heterozygous, chr = chromosome.
AS was found to increase the number of on-target reads in both cases, allowing the production of a valid consensus sequence for mouse #64005. However, the degree of improvement following AS was only moderate, with an increase in depth from 1.3 to 4.7 times (Table 4). Nonetheless, Cas9 enrichment alone yielded a sufficient number of reads (Supplementary Table S2) across most experiments for de novo assembly, presenting a viable option to generate consensus sequences in scenarios with limited computational resources (adaptive sampling requires a workstation equipped with a high-performance GPU). Given that standard nCATS enrichment frequently provided sufficient coverage (∼40x) for successful de novo assembly and reliable detection of complex events like concatemers, the added complexity and computational requirements of AS is a trade-off to consider. While we recommend considering AS if computational resources are readily available, especially for challenging targets, our results suggest that standard nCATS sequencing remains a robust and viable approach for this type of analysis when AS implementation is not feasible.
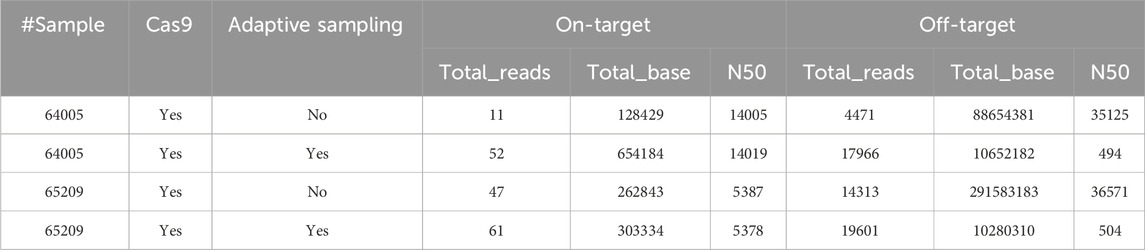
Table 4. Effect of adaptive sampling on sequencing outputs following Cas9-enriched nanopore sequencing. Details of the on-target (i.e., containing the region of interest) and off-target (i.e., not containing the region of interest) reads produced on the MinION for the same mice with and without adaptive sampling (AS). N50 = sequence length of the shortest contig at 50% of the total assembly length.
Long read sequencing identifies occurrences of concatemerization
All nCATS libraries were sequenced on either a MinION Mk1c or a GridION. The analysis of the consensus sequences was performed by aligning them to the mm10 reference assembly. Dot plots visualization (Figure 4) revealed the presence of concatemers at the insertion site.
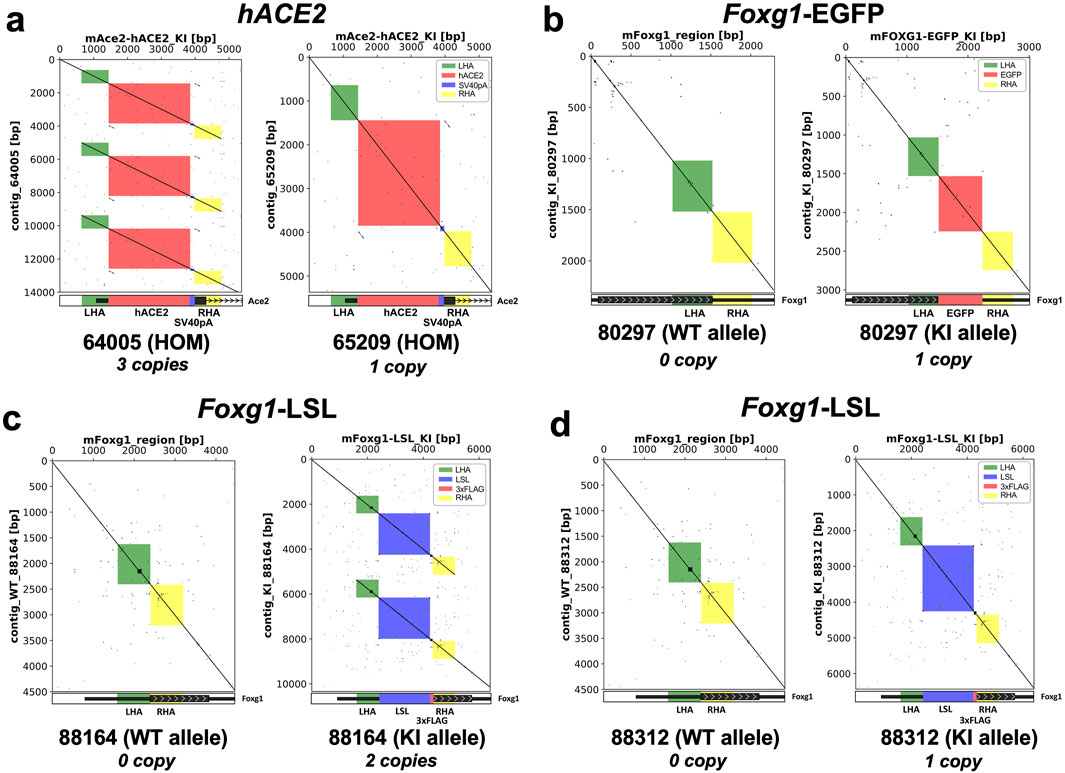
Figure 4. Visualization of the editing outcomes at the targeted loci following AAV-driven gene editing. Dot plots of the targeted loci show that 2 lines out of 5 (40%) carry an array of multicopies (i.e., concatemers) at the insertion site. x-axis = wildtype sequence of the targeted gene (left panels); theoretical KI sequences (right panels), y-axis = consensus sequences obtained by nanopore sequencing. A continuous line indicates full alignment, a broken line indicates a mismatch. (a) Dot plots analysis for the hACE2 homozygous mice reveals that mouse #65209 carries one copy whereas mouse #64005 carries three copies of the transgene. (b) Dot plots analysis for the Foxg1-EGFP heterozygous mouse reveals that mouse #80297 carries one copy of the transgene. Dot plots analysis for the Foxg1 cKI heterozygous mice reveals that mouse #88164 carries two copies (c) whereas mouse #88132 carries one copy of the transgene. (d) LHA = left homology arm, RHA = right homology arm, ITR = inverted terminal repeat sequence, hACE2 = human ACE2 coding sequence, SV40 pA = simian virus 40 polyadenylation signal, EGFP = Enhanced Green Fluorescence Protein sequence, 3xFLAG = triple FLAG tag, LSL = Lox-Stop-Lox.
Indeed, mouse #64005 (homozygous hACE2) carried three copies of the transgene (Figure 4a, left panel), while mouse #88164 (Heterozygous Foxg1 cKI) had two copies integrated at the cut site (Figure 4c, right panel). Overall, of the five Founder lines bred forward across all gene targets, we detected concatemer integration events in two of the established lines (Table 5). Moreover, when the consensus sequences were aligned against the theoretical KI sequences (Figure 4, right panels), we did not find any mismatch, suggesting precise integration of the donor, and sequence fidelity (when aligned to the respective predicted knock-in sequence).
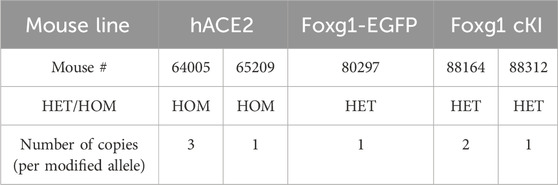
Table 5. Long read sequencing outcome for the selected KI mice. Details of the number of copies integrated at the insertion site for the five selected mice. Note that two out of five (40%) mice carry multicopies (i.e., concatemers).
Partial AAV vector sequences integration at the insertion site
Viral sequences were not detected in the three lines that did not have concatemer events. Conversely, both mice carrying concatemers also carried AAV vector sequences, essentially the Inverted Terminal Repeat (ITR) sequences (Figure 5). Mouse #64005 contained both ITRs (Figures 5a,b), whereas only the 3′ ITR was detected in the genome of mouse #88164 (Figure 5c). Importantly, these partial integrations were not found at the outermost extremities, where Homology Directed Repair occurred. As such, it is unlikely that the ITRs could be detected by junction PCRs, because only the biggest amplicons contain the ITR sequences, yet these amplicons are typically too big to be generated by junction PCRs (see Figure 6).

Figure 5. Alignment of the consensus sequences with the respective AAV vectors. The alignment identifies partial insertion of AAV vector sequences in concatemer carriers. The alignment between the hACE2 concatemer carrier consensus sequence (mouse #64005) and the AAV vector sequences reveals that both the 5’ ITR (a) and the 3’ ITR (b) sequences align partially, illustrating the integration of these ITR sequences together with a small part of the vector containing the cloning sites (i.e., XhoI and AgeI). The alignment between the Foxg1 cKI concatemer carrier consensus sequence (mouse #88164) and the sequence of the AAV vector used to generate this KI reveals that the 3’ ITR sequence (c) aligns partially, illustrating the integration of this ITR sequence together with a small part of the vector containing the cloning site (i.e., SphI). Note that the alignment of mice carrying single copies and wildtype alleles did not show any integration of the AAV vector sequences. LHA = left homology arm, RHA = right homology arm, ITR = inverted terminal repeat sequence.

Figure 6. The presence of ITR sequences is only found in between copies of concatemer carriers. (a) Consensus sequence for the homozygous hACE2 mouse #64005 showing the location of the ITR sequences. (b) Schematic representation of the concatemer (3 copies) in mouse #64005 and associated 5’ junction PCR design. Note that the ITR sequences are only found at the junction of each copy, but not at the at the outermost extremities, where HDR occurred. As such, junction PCRs may not identify the presence of these ITRs, assuming that amplicons 2 and 3 may be too big to be generated.
Discussion
Gene editing in mice to generate models of human disorders is critical to biomedical research. For instance, the insertion of the human ACE2 cDNA into its murine counterpart allows for the “humanization” of the Ace2 gene, contributing to the development of new therapeutics against Sars-CoV-2 infections (Sun et al., 2020). Similarly, rare genetic disorders such as Foxg1 syndrome require the development of mouse models to study the pathomechanisms underlying the disorders (Younger et al., 2022).
Although the disruption of genomic sequences (knock-out) is relatively straightforward in zygotes, the targeted insertion of large transgenes following Homology Directed Repair (HDR) remains challenging. Recently, AAV-driven electroporation of CRISPR RNP complexes in mouse zygotes proved a reliable and seamless method to generate KI mice, and transgenic cores around the world routinely use this method to generate mouse models (Duddy et al., 2024).
However, quality control (QC) for these models is typically done by transgene-specific and junction PCRs, followed by Sanger sequencing. Yet this method has been shown to be inadequate to detect potential on-target mutations (Simkin et al., 2022), and leading transgenic laboratories developed guidelines for thorough validation of the alleles following gene editing (Bunton-Stasyshyn et al., 2022; Codner et al., 2018). Although off-target mutations are typically rare (Peterson et al., 2023) and no more frequent than genetic drift in mice (Peterson et al., 2023; Iyer et al., 2015; Nakajima et al., 2016), on-target mutations may also occur. It is important to note that we limited our analysis to targeted LRS, rather than whole genome sequencing, and therefore cannot rule out any potential off-target effects.
To this end, we performed Long Read Sequencing (LRS) of the targeted loci following CRISPR-READI and found occurrences of on-target illegitimate mutations. Out of five mice analyzed by LRS, two carried multicopy integration (i.e., concatemers). Furthermore, the alignment of the consensus sequences with that of the respective AAV vectors revealed partial integration of the AAV vector sequences, essentially the Inverted Terminal Repeat (ITR) sequences. Such illegitimate on-target mutations were typically not detected when we used the traditional PCR genotyping strategy (Mizuno et al., 2018), because these mutations did not occur at the outermost boundaries of the integration site. Instead, these sequences were typically found in between copies of the transgene, suggesting that the concatemerization occurred before integration. Although LRS can be performed in a multiplexed manner downstream of PCR amplification (McCabe et al., 2024), LRS downstream of Cas9 enrichment presents with the advantage of not being limited by the performance of the DNA polymerase. Indeed, long range PCRs can be challenging, and a 3 kb KI carrying 3 copies (i.e., amplicon over 10 kb, including homology arms) would hardly be amplified by PCR, even using highly performant and processive DNA polymerases. Likewise, highly repetitive or GC reach regions are often not amplifiable by PCR (see Figure 6), whereas Cas9 enrichment can be performed to sequence DNA fragments of any size and any composition.
Although concatemers may affect the expression of the transgenes at the phenotypic level, this analysis is beyond the scope of this study and has not been carried out on the KI mice.
When comparing the efficacy of Cas9 enrichment alone versus its combination with adaptive sampling, our data suggested that adaptive sampling could enhance coverage. Therefore, we advocate for the use of adaptive sampling when resources permit. Nonetheless, Cas9 enrichment alone yielded a sufficient number of reads across most experiments, presenting a viable option in scenarios with limited computational resources.
Limitations
Although LRS downstream of Cas9 enrichment to analyze the outcome of editing events is an unbiased method that does not require PCR amplification (and as such avoids potential amplification mistakes or biases), its main limitation is its inability to be multiplexed (Scholz et al., 2024), compared to PCR-based methods (McCabe et al., 2024). Hence, because non-multiplexed analysis of the KI is substantially expensive (one mouse per flow cell), it is not possible to carry out statistical analysis on the limited number of mice (5) we sequenced. The development of a multiplexed method to run LRS downstream of Cas9 enrichment will allow for robust statistical comparison of outcome inter and intra experiments. Likewise, the limited number of mice sequenced in this study does not allow for a mechanistic analysis on the formation of concatemers and/or partial integrations. These intermolecular recombinations may originate from events such as ITR-mediated recombination (Duan et al., 1998), and/or from uncomplete quality controls during the manufacturing process of the AAV (Bai et al., 2024). Variables such as homology arm length, AAV titer, or other editing conditions may influence the frequency of concatemerization or vector integration. Further studies are required to identify the main molecular drivers of concatemerization, yet we recommend ensuring that a thorough quality control be carried out during the manufacture of AAV.
Finally, it is important to note that the High Molecular Weight (HMW) DNA used in this study was collected from the mouse kidney. A suitable HMW DNA extraction method using tail biopsies or ear notches would allow for the LRS analysis to be performed on Founder mice and avoid breeding unnecessary mice, which is a major ethical consideration. To center the analysis on the Founder mice, alternative methods such as digital droplet PCR (ddPCR) and quantitative real time PCR (qPCR) may be useful to identify copy number, and breed solely the KI mice that do not carry concatemers. Moreover, a thorough analysis of the frequency of the potential non-targeted events (both episomal presence and random integration) would inform transgenic laboratories as to the minimal number of Founders to analyze, to ensure at least one properly targeted line is generated.
Generalization
Yet, the entire process, from DNA extraction to data analysis, was completed within 4 days. This work highlights the potential of this method to provide a rapid and efficient means of assessing transgene insertion in a timely manner. Moreover, this approach has the potential to be widely applied as a tool for identifying and characterizing structural rearrangements and repetitive regions following gene editing in mice. Although the present analysis is restricted to a limited number of mice, it has been previously demonstrated that genomic double-stranded breaks tend to capture foreign DNA (Sargent et al., 1997). As such, concatemers and/or illegitimate integrations have been observed with high frequency in different settings, including human cells edited with ZFN (Olsen et al., 2010; Radecke et al., 2010), murine embryonic stem cells edited with CRISPR (Erbs et al., 2023), C. Elegans (Dickinson et al., 2013) and Zebrafish (Gutierrez-Triana et al., 2018) edited with CRISPR, and cattle edited with TALEN (Norris et al., 2020). Even a typical workflow of microinjection of plasmids with CRISPR RNPs can generate up to 60% of the lines carrying multicopy integrations in mice (Skryabin et al., 2020). This could only be mitigated by biotinylation of the donor template (Medert et al., 2023). Importantly, high level of AAV vector integration (up to 47%) was also found when performing AAV-driven electroporation of CRISPR RNP complexes in cultured murine cells (Hanlon et al., 2019) and human hepatocytes (Ginn et al., 2020), in line with our findings in zygotes.
Conclusion
Therefore, we recommend using long read sequencing as a stringent QC for KI lines generated using CRISPR-READI, and potentially other methods. This work highlights the importance of in-depth validation of the mutant lines generated by transgenic cores, which is critical to ensure reproducibility of animal research, and as such, helps prevent animal wastage. Besides, this work also yields important information with regards to the uptake of this new method, given that CRISPR-Cas9/rAAV6 therapeutic strategies are currently implemented for monogenic diseases such as severe combined immunodeficiency (SCID) (Iancu et al., 2023) while recent studies found frequent concatemeric insertions in stem cells (Suchy et al., 2024) and partial AAV vector integration after gene therapy (Simpson et al., 2023).
Methods
Generation of KI mouse lines
The generation of KI mice was performed following the CRISPR-READI method (Chen et al., 2019). Briefly, C57BL/6J fertilized zygotes (obtained by superovulating 3–4-week-old females with 5IU of Pregnant Mare Serum Gonadotropin and Human Chorionic Gonadotropin 46 h apart) were infected for 5 h with AAV6 carrying the respective transgene of interest in modified culture medium (Embryotech Laboratories, ETECH-EL) before thorough washing and ex-vivo electroporation of CRISPR reagents, as previously reported (Morey et al., 2022). As a general rule for the design of the editing strategies, we tried to select CRISPR guides (and TALENs) overlapping the insertion site, ensuring that the induced double strand break is close enough to allow for HDR to occur on the released strand, while preventing any recut following HDR. Moreover, the highest concentrations possible for each reagent were selected, while always keeping a 10 μL final volume. Recombinant AAV6 was commercially sourced (Vector Builder, pilot scale packaging >2 × 1011 GC/mL). The zygotes were then reimplanted into pseudopregnant outbred mice (ARC(s), Ozgene) following conventional protocols (Delerue and Ittner, 2017).
hACE2 KI mice
The second exon of the Ace2 gene (ENSEMBL ENSMUSG00000015405) was targeted using a commercially synthesised (Integrated DNA Technologies, Inc.) guide (sequence in Supplementary Table S1). This single-guide RNA (sgRNA) was rationally designed using a computational tool (Oliveros et al., 2016) to minimize off-targets (https://bioinfogp.cnb.csic.es/tools/breakingcas/) and incubated for 10 min at room temperature with Alt-R™ S.p. Cas9 Nuclease V3 (IDT # 1081058) to form ribonucleoprotein (RNP) complexes. TALEN mRNA (ThermoFisher Scientific) targeting the same locus was also added to the editing mix, because no suitable sgRNA could be identified to target this genomic sequence.
Following AAV infection, this editing mix was electroporated (NEPA21, Nepagene) into fertilised C57BL/6 zygotes with the respective concentrations: 200 ng/μL Cas9, 400 ng/μL sgRNA, 100 ng/μL TALEN (Table 1). For all experiments, the same electroporation parameters were used. Poring pulses: 4 pulses, 25 V, 1.5msec, 50msec intervals, 10% decay, Polarity +; Transfer pulses: 4 pulses, 3V, 50msec, 50msec intervals, 40% decay, Polarity +/−.
Foxg1-EGFP KI mice
The first exon of the Foxg1 gene (ENSEMBL ENSMUSG00000020950) was targeted at the stop codon using two commercially synthesised (Integrated DNA Technologies, Inc.) guides (sequences in Supplementary Table S1). These single-guide RNAs (sgRNAs) were rationally designed using a computational tool (Oliveros et al., 2016) to minimize off-targets (https://bioinfogp.cnb.csic.es/tools/breakingcas/) and incubated for 10 min at room temperature with Alt-R™ S.p. Cas9 Nuclease V3 (IDT # 1081058) to form ribonucleoprotein (RNP) complexes.
Following AAV infection, this RNP mix was electroporated (NEPA21, Nepagene) into fertilised C57BL/6 zygotes with the respective concentrations: 200 ng/μL Cas9, 100 ng/μL each guide (Table 1).
Foxg1 cKI mice
These mice were generated using the exact same procedure (and concentrations) as the one used for the Foxg1-EGFP mice, however the sgRNAs targeted the start codon of the first exon of the Foxg1 gene.
For all KI lines, live pups were produced and bred to establish colonies. PCR genotyping on isopropanol-precipitated DNA from tail biopsies was performed to identify potential founders. Genotyping screen consisted in transgene-specific PCR, followed by 5′ and 3′ junctions PCRs (primer sequences in Supplementary Table S1). Imaging of the electrophoresis gels was done using a BioRad Gel Doc EZ imager with default parameters. Original gels are presented in Supplementary Figure S2.
Genomic DNA extraction
High Molecular Weight (HMW) DNA was obtained using the Monarch® HMW DNA extraction kit for Tissue (T3060L, New England Biolabs), as previously described (Low et al., 2022). Briefly, fresh kidney tissues from heterozygous (HET) or Homozygous (HOM) mice were harvested, kept on ice and 20 mg of tissue was immediately processed.
Library preparation
Enrichment of the genomic region of interest (ROI) was performed using the Cas9 Sequencing Kit (SQK-CS9109, ONT©) following the nCATS method (Gilpatrick et al., 2020). Briefly, following dephosphorylation, Cas9-guided adapter ligation and dA tailing were conducted on 5 μg of HMW genomic DNA for each mouse, using four rationally designed sgRNAs (Labun et al., 2016) (two upstream and two downstream of the ROI, approximately 0.7–2.4 kilobases from the insertion site (Figures 3a–c). Next, AMPure XP bead purification was performed, and final concentrations were measured using Qubit fluorometric quantification (Koetsier and Cantor, 2021) (Q33238, ThermoFisher Scientific) before loading each nCATS library on an R9.4.1 flow cell (FLO-MIN106D, ONT©).
Long read sequencing
Where applicable, adaptive sampling was employed to selectively capture reads belonging to the target regions of interest (ROI) defined in the corresponding BED files (see Supplementary Material) specifying 15 kilobases upstream and downstream from the ROI, using the Mus musculus C57BL/6J (mm10) as genomic reference. Nanopore sequencing was performed with or without adaptive sampling for 72 h on a MinION Mk1b or GridION without reloading. Fast5 files and FASTQ files were collected for analysis.
Data analysis and visualization
Data acquisition was performed using MinKNOW (v5.2.13). Base calling was executed using GUPPY (v6.4.6) with the High-accuracy Model (HAC). The resulting FASTQ files were then aligned to the mouse reference genome (mm10) using minimap2 (v2.24). The depth of reads targeting specific regions was assessed using BEDTools genomecov (v2.30). The reads that mapped to the target region encompassing sgRNAs cleavage sites were extracted using the intersect function of BEDTools (v2.30). These reads were then used for de novo assembly with Canu (v2.2) followed by subsequent polishing with nanopore FASTQ using two rounds of Flye-polishing (v2.9.2-b1786) and one round of Medaka-polishing (v1.8.0). Visualization of on-target reads and assembly outcomes was performed using the Integrative Genomics Viewer (IGV v2.16.2). Finally, dot plot graphs were generated using FlexiDot (v1.06) to analyse the editing outcomes at the insertion sites, while consensus sequences were aligned to the AAV vector sequences using Snapgene v7.1.1. (Figure 5).
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and the datasets can be found in the Supplementary Material.
Ethics statement
All procedures were approved by Macquarie University Animal Ethics Committees and were conducted in accordance with the Australian Code of Pracfor the Care and Use of Animals for Scientific Purposes. Furthermore, this study is reported in accordance with ARRIVE guidelines (https://arriveguidelines.org).
Author contributions
ML: Investigation, Project administration, Writing – review and editing. PJ: Data curation, Formal Analysis, Methodology, Software, Validation, Writing – review and editing. JS: Methodology, Writing – review and editing. NS: Data curation, Methodology, Software, Writing – review and editing. MC: Data curation, Formal Analysis, Software, Writing – review and editing. PN: Data curation, Methodology, Software, Writing – review and editing. LI: Funding acquisition, Resources, Supervision, Writing – review and editing. TW: Data curation, Formal Analysis, Funding acquisition, Investigation, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review and editing. FD: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by a joint research seeding grant from Macquarie University and Mahidol University (round 2022). Further research support was from the Australian Research Council (DP210101957). Pattaraporn Nimsamer received financial support from the NSRF via the Program Management Unit for Human Resources and Institutional Development, Research Innovation (Grant No. B13F660073). The computing facilities were supported by Mahidol University and the Office of the Ministry of Higher Education, Science, Research and Innovation under the Reinventing University project: the Center of Excellence in AI-Based Medical Diagnosis (AI-MD) sub-project, NSTDA Supercomputer center (ThaiSC), and the National e-Science Infrastructure Consortium.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgeed.2025.1582097/full#supplementary-material
References
Bai, X., Hong, J. F., Yu, S., Hu, D. Y., Chen, A. Y., Rich, C. A., et al. (2024). Prevalence of errors in lab-made plasmids across the globe. bioRxiv 17, 596931. doi:10.1101/2024.06.17.596931
Bryant, W. B., Yang, A., Griffin, S. H., Zhang, W., Rafiq, A. M., Han, W., et al. (2023). CRISPR-Cas9 long-read sequencing for mapping transgenes in the mouse genome. CRISPR J. 6, 163–175. doi:10.1089/crispr.2022.0099
Bunton-Stasyshyn, R. K., Codner, G. F., and Teboul, L. (2022). Screening and validation of genome-edited animals. Lab. Anim. 56, 69–82. doi:10.1177/00236772211016922
Chen, S., Sun, S., Moonen, D., Lee, C., Lee, A. Y. F., Schaffer, D. V., et al. (2019). CRISPR-READI: efficient generation of knockin mice by CRISPR RNP electroporation and AAV donor infection. Cell Rep. 27, 3780–3789. doi:10.1016/j.celrep.2019.05.103
Codner, G. F., Mianné, J., Caulder, A., Loeffler, J., Fell, R., King, R., et al. (2018). Application of long single-stranded DNA donors in genome editing: generation and validation of mouse mutants. BMC Biol. 16, 70. doi:10.1186/s12915-018-0530-7
Delerue, F., and Ittner, L. M. (2017). Generation of genetically modified mice through the microinjection of oocytes. J. Vis. Exp., 55765. doi:10.3791/55765
Dickinson, D. J., Ward, J. D., Reiner, D. J., and Goldstein, B. (2013). Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat. Methods 10, 1028–1034. doi:10.1038/nmeth.2641
Duan, D., Sharma, P., Yang, J., Yue, Y., Dudus, L., Zhang, Y., et al. (1998). Circular intermediates of recombinant adeno-associated virus have defined structural characteristics responsible for long-term episomal persistence in muscle tissue. J. Virol. 72, 8568–8577. doi:10.1128/JVI.72.11.8568-8577.1998
Duddy, G., Courtis, K., Horwood, J., Olsen, J., Horsler, H., Hodgson, T., et al. (2024). Donor template delivery by recombinant adeno-associated virus for the production of knock-in mice. BMC Biol. 22 (1 22), 26–12. doi:10.1186/s12915-024-01834-z
Erbs, V., Lorentz, R., Eisenman, B., Schaeffer, L., Luppi, L., Lindner, L., et al. (2023). Increased on-target rate and risk of concatemerization after CRISPR-enhanced targeting in ES cells. Genes (Basel) 14, 401. doi:10.3390/genes14020401
Gilpatrick, T., Lee, I., Graham, J. E., Raimondeau, E., Bowen, R., Heron, A., et al. (2020). Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat. Biotechnol. 38, 433–438. doi:10.1038/s41587-020-0407-5
Ginn, S. L., Amaya, A. K., Liao, S. H. Y., Zhu, E., Cunningham, S. C., Lee, M., et al. (2020). Efficient in vivo editing of OTC-deficient patient-derived primary human hepatocytes. JHEP Rep. 2, 100065. doi:10.1016/j.jhepr.2019.100065
Gutierrez-Triana, J. A., Tavhelidse, T., Thumberger, T., Thomas, I., Wittbrodt, B., Kellner, T., et al. (2018). Efficient single-copy HDR by 5’ modified long dsDNA donors. Elife 7, e39468. doi:10.7554/eLife.39468
Hanlon, K. S., Kleinstiver, B. P., Garcia, S. P., Zaborowski, M. P., Volak, A., Spirig, S. E., et al. (2019). High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 10, 4439. doi:10.1038/s41467-019-12449-2
Iancu, O., Allen, D., Knop, O., Zehavi, Y., Breier, D., Arbiv, A., et al. (2023). Multiplex HDR for disease and correction modeling of SCID by CRISPR genome editing in human HSPCs. Mol. Ther. Nucleic Acids 31, 105–121. doi:10.1016/j.omtn.2022.12.006
Iyer, V., Shen, B., Zhang, W., Hodgkins, A., Keane, T., Huang, X., et al. (2015). Off-target mutations are rare in Cas9-modified mice. Nat. Methods 12, 479. doi:10.1038/nmeth.3408
Kaneko, T., and Mashimo, T. (2015). Simple genome editing of rodent intact embryos by electroporation. PLoS One 10, e0142755. doi:10.1371/journal.pone.0142755
Klugmann, M., Kalotay, E., Delerue, F., Ittner, L. M., Bongers, A., Yu, J., et al. (2022). Developmental delay and late onset HBSL pathology in hypomorphic Dars1 M256L mice. Neurochem. Res. 47, 1972–1984. doi:10.1007/s11064-022-03582-4
Koetsier, P. A., and Cantor, E. J. (2021). A simple approach for effective shearing and reliable concentration measurement of ultra-high-molecular-weight DNA. Biotechniques 71, 439–444. doi:10.2144/btn-2021-0051
Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B., and Valen, E. (2016). CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 44, W272–W276. doi:10.1093/nar/gkw398
Lintott, L. G., and Nutter, L. M. J. (2023). Genetic and molecular quality control of genetically engineered mice. Methods Mol. Biol., 2631 53, 101. doi:10.1007/978-1-0716-2990-1_3
Low, B. E., Hosur, V., Lesbirel, S., and Wiles, M. V. (2022). Efficient targeted transgenesis of large donor DNA into multiple mouse genetic backgrounds using bacteriophage Bxb1 integrase. Sci. Rep. 12, 5424. doi:10.1038/s41598-022-09445-w
Martin, S., Heavens, D., Lan, Y., Horsfield, S., Clark, M. D., and Leggett, R. M. (2022). Nanopore adaptive sampling: a tool for enrichment of low abundance species in metagenomic samples. Genome Biol. 23, 11. doi:10.1186/s13059-021-02582-x
McCabe, C. V., Price, P. D., Codner, G. F., Allan, A. J., Caulder, A., Christou, S., et al. (2024). Long-read sequencing for fast and robust identification of correct genome-edited alleles: PCR-based and Cas9 capture methods. PLoS Genet. 20, e1011187. doi:10.1371/journal.pgen.1011187
Medert, R., Thumberger, T., Tavhelidse-Suck, T., Hub, T., Kellner, T., Oguchi, Y., et al. (2023). Efficient single copy integration via homology-directed repair (scHDR) by 5′modification of large DNA donor fragments in mice. Nucleic Acids Res. 51, e14. doi:10.1093/nar/gkac1150
Miyasaka, Y., Uno, Y., Yoshimi, K., Kunihiro, Y., Yoshimura, T., Tanaka, T., et al. (2018). CLICK: one-step generation of conditional knockout mice. BMC Genomics 19, 318. doi:10.1186/s12864-018-4713-y
Mizuno, N., Mizutani, E., Sato, H., Kasai, M., Ogawa, A., Suchy, F., et al. (2018). Intra-embryo gene cassette knockin by CRISPR/Cas9-Mediated genome editing with adeno-associated viral vector. iScience 9, 286–297. doi:10.1016/j.isci.2018.10.030
Morey, N., Przybyla, M., van der Hoven, J., Ke, Y. D., Delerue, F., van Eersel, J., et al. (2022). Treatment of epilepsy using a targeted p38γ kinase gene therapy. Sci. Adv. 8, eadd2577. doi:10.1126/sciadv.add2577
Nakajima, K., Kazuno, A. A., Kelsoe, J., Nakanishi, M., Takumi, T., and Kato, T. (2016). Exome sequencing in the knockin mice generated using the CRISPR/Cas system. Sci. Rep. 6, 34703. doi:10.1038/srep34703
Norris, A. L., Lee, S. S., Greenlees, K. J., Tadesse, D. A., Miller, M. F., and Lombardi, H. A. (2020). Template plasmid integration in germline genome-edited cattle. Nat. Biotechnol. 38, 163–164. doi:10.1038/s41587-019-0394-6
Oliveros, J. C., Franch, M., Tabas-Madrid, D., San-León, D., Montoliu, L., Cubas, P., et al. (2016). Breaking-Cas-interactive design of guide RNAs for CRISPR-Cas experiments for ENSEMBL genomes. Nucleic Acids Res. 44, W267–W271. doi:10.1093/nar/gkw407
Olsen, P. A., Gelazauskaite, M., Randøl, M., and Krauss, S. (2010). Analysis of illegitimate genomic integration mediated by zinc-finger nucleases: implications for specificity of targeted gene correction. BMC Mol. Biol. 11, 35. doi:10.1186/1471-2199-11-35
Payne, A., Holmes, N., Clarke, T., Munro, R., Debebe, B. J., and Loose, M. (2020). Readfish enables targeted nanopore sequencing of gigabase-sized genomes. Nat. Biotechnol. 39 (4 39), 442–450. doi:10.1038/s41587-020-00746-x
Peterson, K. A., Khalouei, S., Hanafi, N., Wood, J. A., Lanza, D. G., Lintott, L. G., et al. (2023). Whole genome analysis for 163 gRNAs in Cas9-edited mice reveals minimal off-target activity. Commun. Biol. 6, 626. doi:10.1038/s42003-023-04974-0
Qin, W., Dion, S. L., Kutny, P. M., Zhang, Y., Cheng, A. W., Jillette, N. L., et al. (2015). Efficient CRISPR/cas9-mediated genome editing in mice by zygote electroporation of nuclease. Genetics 200, 423–430. doi:10.1534/genetics.115.176594
Radecke, S., Radecke, F., Cathomen, T., and Schwarz, K. (2010). Zinc-finger nuclease-induced gene repair with oligodeoxynucleotides: wanted and unwanted target locus modifications. Mol. Ther. 18, 743–753. doi:10.1038/mt.2009.304
Romeo, C., Chen, S. H., Goulding, E., Van Gorder, L., Schwartz, M., Walker, M., et al. (2020). AAV diffuses across zona pellucida for effortless gene delivery to fertilized eggs: AAV diffuses across mouse ZP. Biochem. Biophys. Res. Commun. 526, 85–90. doi:10.1016/j.bbrc.2020.03.026
Rubben, K., Tilleman, L., Deserranno, K., Tytgat, O., Deforce, D., and Van Nieuwerburgh, F. (2022). Cas9 targeted nanopore sequencing with enhanced variant calling improves CYP2D6-CYP2D7 hybrid allele genotyping. PLoS Genet. 18, e1010176. doi:10.1371/journal.pgen.1010176
Sailer, S., Coassin, S., Lackner, K., Fischer, C., McNeill, E., Streiter, G., et al. (2021). When the genome bluffs: a tandem duplication event during generation of a novel Agmo knockout mouse model fools routine genotyping. Cell Biosci. 11, 54. doi:10.1186/s13578-021-00566-9
Sargent, R. G., Brenneman, M. A., and Wilson, J. H. (1997). Repair of site-specific double-strand breaks in a mammalian chromosome by homologous and illegitimate recombination. Mol. Cell Biol. 17, 267–277. doi:10.1128/mcb.17.1.267
Scholz, V., Schönrock, V., Erdmann, H., Prokosch, V., Schoedel, M., Almus, M., et al. (2024). Parallel in-depth analysis of repeat expansions: an updated Clin-CATS workflow for nanopore R10 flow cells. bioRxiv 11 (05), 622099. doi:10.1101/2024.11.05.622099
Simkin, D., Papakis, V., Bustos, B. I., Ambrosi, C. M., Ryan, S. J., Baru, V., et al. (2022). Homozygous might be hemizygous: CRISPR/Cas9 editing in iPSCs results in detrimental on-target defects that escape standard quality controls. Stem Cell Rep. 17, 993–1008. doi:10.1016/j.stemcr.2022.02.008
Simpson, B. P., Yrigollen, C. M., Izda, A., and Davidson, B. L. (2023). Targeted long-read sequencing captures CRISPR editing and AAV integration outcomes in brain. Mol. Ther. 31, 760–773. doi:10.1016/j.ymthe.2023.01.004
Skryabin, B. V., Kummerfeld, D. M., Gubar, L., Seeger, B., Kaiser, H., Stegemann, A., et al. (2020). Pervasive head-to-tail insertions of DNA templates mask desired CRISPR-Cas9-mediated genome editing events. Sci. Adv. 6, eaax2941. doi:10.1126/sciadv.aax2941
Suchy, F. P., Karigane, D., Nakauchi, Y., Higuchi, M., Zhang, J., Pekrun, K., et al. (2024). Genome engineering with Cas9 and AAV repair templates generates frequent concatemeric insertions of viral vectors. Nat. Biotechnol. 43, 204–213. doi:10.1038/s41587-024-02171-w
Sun, S. H., Chen, Q., Gu, H. J., Yang, G., Wang, Y. X., Huang, X. Y., et al. (2020). A mouse model of SARS-CoV-2 infection and pathogenesis. Cell Host Microbe 28, 124–133. doi:10.1016/j.chom.2020.05.020
Wongsurawat, T., Jenjaroenpun, P., De Loose, A., Alkam, D., Ussery, D. W., Nookaew, I., et al. (2020). A novel Cas9-targeted long-read assay for simultaneous detection of IDH1/2 mutations and clinically relevant MGMT methylation in fresh biopsies of diffuse glioma. Acta Neuropathol. Commun. 8, 87. doi:10.1186/s40478-020-00963-0
Yoon, Y., Wang, D., Tai, P. W. L., Riley, J., Gao, G., and Rivera-Pérez, J. A. (2018). Streamlined ex vivo and in vivo genome editing in mouse embryos using recombinant adenoassociated viruses. Nat. Commun. 9, 412. doi:10.1038/s41467-017-02706-7
Keywords: long read sequencing (LRS), CRISPR, adeno-associated-virus (AAV), mice, zygotes, concatemers
Citation: Luqman MW, Jenjaroenpun P, Spathos J, Shingte N, Cummins M, Nimsamer P, Ittner LM, Wongsurawat T and Delerue F (2025) Long read sequencing reveals transgene concatemerization and vector sequences integration following AAV-driven electroporation of CRISPR RNP complexes in mouse zygotes. Front. Genome Ed. 7:1582097. doi: 10.3389/fgeed.2025.1582097
Received: 23 February 2025; Accepted: 06 May 2025;
Published: 04 June 2025.
Edited by:
Ahmed Elaswad, Sultan Qaboos University, OmanReviewed by:
Jun Song, Northeast Agricultural University, ChinaZacharias Kontarakis, ETH Zurich, Switzerland
Antony Adamson, The University of Manchester, United Kingdom
Copyright © 2025 Luqman, Jenjaroenpun, Spathos, Shingte, Cummins, Nimsamer, Ittner, Wongsurawat and Delerue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabien Delerue, ZmRlbGVydWVAbWRhbmRlcnNvbi5vcmc=; Thidathip Wongsurawat, dGhpZGF0aGlwLndvbkBtYWhpZG9sLmVkdQ==
†Present addresses: Muhammad W. Luqman, The University of Texas M.D. Anderson Cancer Center, Houston, United States Fabien Delerue, The University of Texas M.D. Anderson Cancer Center, Houston, United States
‡These authors have contributed equally to this work and share first authorship