 Shengchen Shan1*
Shengchen Shan1* Michael T. Pisias2
Michael T. Pisias2 Evgeny V. Mavrodiev1
Evgeny V. Mavrodiev1 Jonathan P. Spoelhof1
Jonathan P. Spoelhof1 Bernard A. Hauser3
Bernard A. Hauser3 W. Brad Barbazuk3
W. Brad Barbazuk3 Pamela S. Soltis1,4,5
Pamela S. Soltis1,4,5 Douglas E. Soltis1,3,4,5
Douglas E. Soltis1,3,4,5 Bing Yang2,6
Bing Yang2,6- 1Florida Museum of Natural History, University of Florida, Gainesville, FL, United States
- 2Division of Plant Science and Technology, University of Missouri, Columbia, MO, United States
- 3Department of Biology, University of Florida, Gainesville, FL, United States
- 4Genetics Institute, University of Florida, Gainesville, FL, United States
- 5Biodiversity Institute, University of Florida, Gainesville, FL, United States
- 6Donald Danforth Plant Science Center, St. Louis, MO, United States
Polyploidy, or whole-genome duplication (WGD), is a significant evolutionary force. Following allopolyploidy, duplicate gene copies (homeologs) have divergent evolutionary trajectories: some genes are preferentially retained in duplicate, while others tend to revert to single-copy status. Examining the effect of homeolog loss (i.e., changes in gene dosage) on associated phenotypes is essential for unraveling the genetic mechanisms underlying polyploid genome evolution. However, homeolog-specific editing has been demonstrated in only a few crop species and remains unexplored beyond agricultural applications. Tragopogon (Asteraceae) includes an evolutionary model system for studying the immediate consequences of polyploidy in nature. In this study, we developed a CRISPR-mediated homeolog-specific editing platform in allotetraploid T. mirus. Using the MYB10 and DFR genes as examples, we successfully knocked out the targeted homeolog in T. mirus (4x) without editing the other homeolog (i.e., no off-target events). The editing efficiencies, defined as the percentage of plants with at least one allele of the targeted homeolog modified, were 35.7% and 45.5% for MYB10 and DFR, respectively. Biallelic modification of the targeted homeolog occurred in the T0 generation. These results demonstrate the robustness of homeolog-specific editing in polyploid Tragopogon, laying the foundation for future studies of genome evolution following WGD in nature.
1 Introduction
Polyploidy, also known as whole-genome duplication (WGD), is a major evolutionary force in plants (Soltis et al., 2015; Van de Peer et al., 2021; Morris et al., 2024). WGDs generate genetic, phenotypic, and metabolic diversity and are associated with increased evolvability and diversification (Wendel, 2015; Soltis and Soltis, 2016; Landis et al., 2018; Doyle and Coate, 2019; Fox et al., 2020; Van de Peer et al., 2021; Morris et al., 2024). All living angiosperms are of ancient polyploid ancestry (Jiao et al., 2011), and 35% of extant vascular plant species may have originated via polyploidy (Wood et al., 2009). In addition, most crops are polyploids (Renny-Byfield and Wendel, 2014), and polyploidy plays an important role in plant breeding (Udall and Wendel, 2006; Sattler et al., 2016). Therefore, a better understanding of polyploid genome function and evolution is essential for both comprehending plant diversity and facilitating crop improvement.
In newly formed allopolyploids (those that arose through hybridization of closely related species and associated genome doubling), there are duplicate gene copies (homeologs) with redundant or overlapping expression and function. Over time, these duplicates experience various fates, ranging from retention of both copies with original function to homeolog divergence to loss of one copy via fractionation (Papp et al., 2003; Freeling et al., 2012; Tang et al., 2012; De Smet et al., 2013; Wendel et al., 2018). Some genes are consistently conserved as singletons, implying their single-copy status is advantageous and favored by selection (Paterson et al., 2006; De Smet et al., 2013; Li et al., 2016). In addition, following WGD, genes from one parent may be more highly retained than those from the other (i.e., subgenomic dominance) (Chaudhary et al., 2009; Evangelisti and Conant, 2010; Wendel et al., 2012).
For many other genes, duplicate copies are preserved following WGD, a phenomenon that may be explained by the dosage balance hypothesis, which argues that genes encoding subunits of protein complexes tend to be retained following polyploidy; loss of one copy of these genes may be selected against because of the disrupted stoichiometry of members of multi-subunit protein complexes (Birchler and Veitia, 2007; 2012; 2021). Additionally, genes encoding transcription factors are usually dosage-sensitive and are more likely to be retained in duplicate following WGD: loss of one copy of transcription factor genes may have a pleiotropic effect on many downstream genes involved in the same pathway; this effect would not be observed when losing one copy of genes acting at the termini of genetic networks (Blanc and Wolfe, 2004; Freeling et al., 2012; Birchler and Veitia, 2021).
Despite these observed gene retention patterns, the mechanisms underlying these patterns remain elusive, and the phenotypic consequences of changes in gene dosage are still largely unknown. This is primarily due to: 1) the lack of a homeolog-specific editing system for manipulating gene dosage across polyploid plants, except in a few crop species, including hexaploid wheat (Triticum aestivum) (Zhang et al., 2016; Liang et al., 2017) and tetraploid cotton (Gossypium hirsutum) (Chen et al., 2025; Yu et al., 2025); and 2) the absence of functional studies in organisms that best exemplify the earliest phases of WGD, especially species from natural systems. A deeper insight into gene fate following recent WGD is critical for understanding the genetic basis of the success of polyploids.
The diploid-polyploid system in North American Tragopogon (Asteraceae) represents an evolutionary model for studying the immediate consequences of polyploidy (Ownbey, 1950; Soltis et al., 2004; 2012). The naturally occurring allotetraploids Tragopogon miscellus and T. mirus formed in the last 95–100 years. The diploid parents of T. miscellus are T. dubius and T. pratensis, and those of T. mirus are T. dubius and T. porrifolius. Previous studies have demonstrated that novel arrays of karyotypes, gene content and expression, and epigenetic regulation were generated in these newly formed Tragopogon polyploids (Tate et al., 2009; Buggs et al., 2011; Chester et al., 2012; Spoelhof et al., 2017; Shan et al., 2020; 2024; Yoo et al., 2024). In addition, an efficient CRISPR/Cas9-mediated gene editing system has been recently developed in T. porrifolius (2x) and T. mirus (4x), allowing simultaneous editing of both alleles in diploid Tragopogon and all four alleles in allotetraploid Tragopogon (Shan et al., 2018; Shan et al., 2025).
In this study, we developed a homeolog-specific editing system in allotetraploid Tragopogon using MYB10 and DFR (dihydroflavonol 4-reductase) genes as examples. Both genes are involved in the well-characterized anthocyanin biosynthesis pathway (Elomaa et al., 2023). This represents the first such system established in a non-model polyploid plant. Our work provides the foundation for future investigations into gene retention patterns following polyploidy, with broad implications for both agricultural applications and evolutionary biology.
2 Materials and methods
2.1 Tragopogon material and seed germination
The allotetraploid T. mirus individual used in this study was grown from seed collected from a natural population in Pullman, WA, United States (Soltis and Soltis collection ID: 3091-3). Seed sterilization and germination followed the protocols described in Shan et al. (2018).
2.2 Identification of candidate genes in Tragopogon
The DFR genes in diploid Tragopogon species were identified in Shan et al. (2025). The process of identifying MYB10, an R2R3 MYB transcription factor gene, is described below. All T. dubius R2R3 MYB genes were identified using the Gerbera hybrida (Asteraceae) R2R3 domain (accession no. CAD87010; Elomaa et al., 2023) as the query in a BLASTP search (e-value = 1e-10) against the T. dubius annotated protein sequences (Spoelhof et al., in prep.). Sequences from Tragopogon and 17 additional R2R3 MYB gene sequences from model species, including Solanum lycopersicum, Mimulus lewisii, G. hybrida, and Vitis vinifera (Lin-wang et al., 2010; Yuan et al., 2014; Naing and Kim, 2018), were aligned using MAFFT (v.7.520; Katoh and Standley, 2013). A maximum likelihood tree was constructed using IQ-TREE (v.2.2.2.7; Minh et al., 2020), and the resulting phylogeny included a clade containing the Gerbera MYB10 gene and its T. dubius orthologs. Six R2R3 MYB genes were identified in T. dubius. Using the same approach, four R2R3 MYB genes were found in T. porrifolius. Reciprocal best BLAST hits confirmed orthologous relationships between the MYB candidates from T. dubius and T. porrifolius. All scripts used in this step are available at: https://github.com/GatorShan/EDGE_Project/tree/main/MYB10.
Expression profiles of T. dubius and T. porrifolius R2R3 MYB genes were examined using leaf transcriptomes (BioProject accession: PRJNA210897; Yoo et al., 2024). The trimmed reads from T. dubius and T. porrifolius were mapped to their respective reference genomes using STAR (v.2.7.11b; Dobin et al., 2013). Mapped reads were quantified using the featureCounts program from the Subread package (v.2.0.6; Liao et al., 2014). Raw counts were then converted to normalized expression values in FPKM (fragments per kilobase of exon per million fragments mapped). In both T. dubius and T. porrifolius, one R2R3 MYB gene exhibited dominant expression in the leaf transcriptomes. This gene was designated as MYB10 in both diploid species. Additionally, the expression profiles of both MYB10 homeologs were examined in allotetraploid T. mirus using leaf transcriptome data from Yoo et al. (2024): both homeologs were present and expressed in T. mirus. All scripts used in this step are available at: https://github.com/GatorShan/EDGE_Project/tree/main/Trag_leaf_transcriptome.
2.3 Plasmid construction
Construction of plasmids followed protocols described in Shan et al. (2018). Oligonucleotide sequences used are listed in Supplementary Table S1. For each candidate gene (e.g., MYB10 and DFR), a plasmid was constructed to specifically target the T. porrifolius homeolog.
For the plasmid targeting the T. porrifolius MYB10 homeolog, the cloning process started with two constructs: pCAMBIA1300-Cas9-GFP (destination vector) and pENTR4-AtU6-26 (entry vector). Oligonucleotides gTpoMYB10-F1 and gTpoMYB10-R1 were annealed to generate the double-stranded oligo gTpoMYB10-1. A single SNP between the two homeologs was present at this CRISPR target site. The gTpoMYB10-1 oligo was then integrated into the entry vector, resulting in construct pENTR4-AtU6-26-gTpoMYB10-1.
The sgRNA cassette from the entry vector was then mobilized into the destination vector via a Gateway LR reaction (Thermo Fisher Scientific, Waltham, MA, United States). The final plasmid was named pCAMBIA1300-Cas9-GFP-AtU6-26-gTpoMYB10-1; its sequence was confirmed through whole plasmid sequencing (performed by Plasmidsaurus using Oxford Nanopore Technology). The final construct was introduced into Agrobacterium tumefaciens strain EHA105 by electroporation.
For the plasmid targeting the T. porrifolius DFR homeolog, the process of plasmid construction was the same except for the use of a different entry vector (pENTR4-AtU6-1-AtU6-29). Oligonucleotides gTpoDFR1-F and gTpoDFR1-R were annealed to form gTpoDFR1; similarly, gTpoDFR2-F and gTpoDFR2-R were annealed to generate gTpoDFR2. The CRISPR target site for gTpoDFR1 contained five SNPs that differed between the two homeologs, while the site for gTpoDFR2 had one SNP. gTpoDFR1 and gTpoDFR2 were then integrated into the entry vector following AtU6-1 and AtU6-29 promoters, respectively. The final entry vector was pENTR4-AtU6-1-gTpoDFR1-AtU6-29-gTpoDFR2. Following the Gateway LR reaction, the final plasmid was named pCAMBIA1300-Cas9-GFP-AtU6-1-gTpoDFR1-AtU6-29-gTpoDFR2.
2.4 Agrobacterium-mediated transformation and plant regeneration
We followed the methods described in Shan et al. (2025) for transformation and regeneration. Briefly, leaf explants from 4-8-week-old T. mirus seedlings were placed on callus induction medium for 2 days. After co-cultivation with Agrobacterium (OD600 = 0.15–0.20), explants were transferred to co-cultivation medium for 3 days, then moved to selective callus induction medium. After 3 weeks, developing calli were transferred to selective shooting medium to initiate shoot formation. After 2–3 weeks, calli with emerging shoots were moved to shoot elongation medium. Two weeks later, elongated shoots were gradually transferred to selective rooting medium.
All selection and regeneration steps were carried out in a tissue culture incubator under controlled conditions (14 h light/10 h dark, 25 °C; model CU36L4C8, Percival Scientific, Perry, IA, United States). Rooted shoots were then transferred to soil and grown in a growth chamber under the same conditions (model AR-1110, Percival Scientific).
2.5 Genotyping regenerated T. mirus
To detect the presence of the transgene, a fragment of the transfer DNA (T-DNA) was amplified using genomic DNA extracted from regenerated T. mirus plants. For the experiment targeting the MYB10 gene, primers gTpoMYB10-F1 and GmUbi-R2 were used to amplify a 616-bp DNA fragment of the T-DNA (Supplementary Figure S1A). Each PCR reaction (25 μL) contained 1 μL of genomic DNA (25–125 ng), 1 × Green GoTaq Reaction Buffer (Promega, Madison, WI, United States), 2.5 mM MgCl2, 200 μM dNTPs, 0.5 μM of each primer, and 1.25 U Apex Taq DNA polymerase (Genesee Scientific, El Cajon, CA, United States). PCR conditions were as follows: initial denaturation at 95 °C for 3 min; 32 cycles at 95 °C for 30 s, 53 °C annealing for 30 s, and 72 °C extension for 1 min; final extension at 72 °C for 5 min; and hold at 4 °C. Plants were considered transgenic if a band of the expected size (616 bp) was detected by gel electrophoresis. For the experiment targeting the DFR gene, primers AtU6-F2 and AtU6-R2 were used to amplify a 476-bp T-DNA fragment (Supplementary Figure S1B). The PCR conditions were the same as above, except the annealing temperature was set at 68.1 °C.
To evaluate editing results for the two candidate genes in allopolyploid T. mirus, we genotyped each homeolog separately (Supplementary Figures S2, S3). For the MYB10 gene, primers TduMYB10-F1 and TduMYB10-R1 were used to amplify the T. dubius homeolog (amplicon size: 895 bp) (Supplementary Figure S2A), and primers TpoMYB10-F1 and TpoMYB10-R1 were used to amplify the T. porrifolius homeolog (amplicon size: 936 bp) (Supplementary Figure S2B). Each 20-μL PCR reaction contained 1 μL of genomic DNA (20–100 ng), 1 × Phusion HF Buffer (New England Biolabs, Ipswich, MA, United States), 200 μM dNTPs, 0.5 μM of each primer, and 0.4 U Phusion DNA polymerase (New England Biolabs). PCR conditions for both primer sets were as follows: initial denaturation at 98 °C for 30 s; 32 cycles at 98 °C for 10 s, 61 °C annealing for 30 s, and 72 °C extension for 45 s; final extension at 72 °C for 10 min; and hold at 4 °C.
For genotyping the two homeologs of the DFR gene in T. mirus, primers Tdu-sub_DFR_F1 and Tdu-sub_DFR_R1 were used to amplify the T. dubius homeolog (amplicon size: 992 bp) (Supplementary Figure S3A), and Tpo-sub_DFR_F1 and Tpo-sub_DFR_R3 were used for the T. porrifolius homeolog (amplicon size: 1,053 bp) (Supplementary Figure S3B). The PCR setting was identical to that used for MYB10 genotyping, except the annealing temperature was adjusted to 61.5 °C and the extension time was increased to 1 min.
PCR products were sent for Sanger sequencing at Eurofins Genomics, Louisville, KY, United States. Genotypes were inferred by manually inspecting the chromatograms, following the approach described in Shan et al. (2018), with the assistance of the Synthego ICE Analysis tool (v.3; https://ice.editco.bio/#/).
3 Results
3.1 Homeolog divergence for MYB10 and DFR genes in allotetraploid Tragopogon mirus
For MYB10, we identified one CRISPR target site in exon 1 (Figure 1A). There is one SNP at this site between the two homeologs in T. mirus: A in the T. dubius homeolog and G in the T. porrifolius homeolog. This SNP is three nucleotides (nt) upstream of the protospacer adjacent motif (PAM) sequence (i.e., TGG), which is present in both homeologs. Utilizing this SNP, we designed single-guide RNA (sgRNA) gTpoMYB10-1 to specifically target the T. porrifolius homeolog. Although the CRISPR/Cas9 complex can bind both homeologs (as each contains a PAM site), only the targeted T. porrifolius homeolog–where the genomic sequence perfectly matches the spacer sequence of the sgRNA–is expected to be edited.
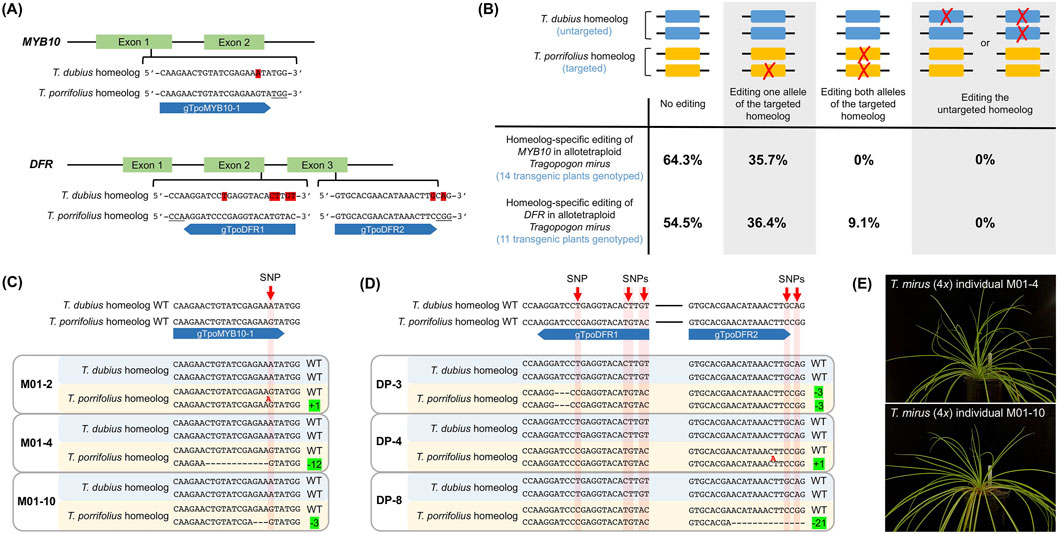
Figure 1. Homeolog-specific editing in allotetraploid Tragopogon mirus. (A) One CRISPR/Cas9 target site was located within exon 1 of MYB10, and the sgRNA gTpoMYB10-1 was designed to specifically target the T. porrifolius homeolog in T. mirus (4x). A single SNP distinguishing the two homeologs at the target site is highlighted in red. For DFR, two CRISPR/Cas9 target sites were selected–one in exon 2 and the other in exon 3. Two sgRNAs, gTpoDFR1 and gTpoDFR2, were designed to specifically target the T. porrifolius homeolog. SNPs distinguishing the T. dubius and T. porrifolius homeologs at these target sites are also highlighted in red. PAM sequences are underlined; sgRNAs are represented by blue bars, with the flat end indicating the 5′ end and the arrowhead indicating the 3′ end. (B) Genotyping results confirmed homeolog-specific editing of the MYB10 and DFR genes in T0 transgenic T. mirus (4x) plants. Both alleles from the T. dubius homeolog are shown as blue rectangles, and those from the T. porrifolius homeolog are shown in yellow. Red crosses indicate sequence modifications. Off-target edits (i.e., modification of the untargeted T. dubius homeolog) were not detected in either experiment; all editing events occurred in the T. porrifolius homeolog. For MYB10, 64.3% of transgenic plants showed no edits in either homeolog, while 35.7% had a monoallelic edit in the T. porrifolius homeolog. For DFR, 54.4% of plants were unedited, 36.4% had monoallelic in the T. porrifolius homeolog, and 9.1% exhibited biallelic edits in the T. porrifolius homeolog. (C) Genotypes of representative T. mirus individuals with edits in the T. porrifolius MYB10 homeolog. The SNP distinguishing wildtype T. dubius and T. porrifolius homeologs is indicated by the red arrow. Nucleotide insertion is shown in red. (D) Genotyping results of representative T. mirus individuals with edits in the T. porrifolius DFR homeolog. For individual DP-8, not all deleted nucleotides (nt) in the “-21” allele are shown. “-x” denotes an x-nt deletion (e.g., 3-, 12-, or 21-nt), and “+y” indicates a y-nt insertion (e.g., 1-nt). WT: wildtype. (E) Photographs of two T. mirus (4x) individuals (M01-4 and M01-10) with the T. porrifolius homeolog of MYB10 successfully edited.
In terms of the DFR gene, two CRISPR target sites from the T. porrifolius homeolog were chosen: one is in exon 2, and the other in exon 3 (Figure 1A). We designed a sgRNA, named gTpoDFR1, to specifically target the T. porrifolius homeolog in exon 2. At this site, the PAM sequence (TGG) was present on both homeologs. There were five SNPs in the 21-nt protospacer region upstream of the PAM between the two homeologs (Figure 1A). For the second target site in exon 3, CGG PAM was only present in the targeted T. porrifolius homeolog. Another sgRNA, named gTpoDFR2, was designed to match the sequence upstream of PAM in the T. porrifolius homeolog. One SNP was found between the T. porrifolius homeolog and the corresponding region in the T. dubius homeolog (Figure 1A).
3.2 Homeolog-specific editing of MYB10 in allotetraploid T. mirus
Of the 14 regenerated plants (with both shoot and root) of T. mirus on selective media, all were transgenic (Supplementary Figure S1A). Among the 14 plants, five (35.7%) showed edits in the targeted T. porrifolius homeolog, while none had edits in the T. dubius homeolog (the untargeted copy) (Figures 1B,C). All five genome-edited plants carried a monoallelic mutation, with one of the two alleles from the T. porrifolius homeolog modified. The most common mutation was a 12-nt deletion (i.e., −12), observed in three T. mirus individuals: M01-4, M01-6, and M01-12 (Figures 1C,E); all three shared the exact same 12-nt deletion. In addition, one mutant T. mirus individual (M01-2) had 1-nt insertion (i.e., +1) located three nucleotides upstream of PAM; individual M01-10 had a 3-nt deletion (Figures 1C,E).
3.3 Homeolog-specific editing of DFR in allotetraploid T. mirus
Of the 12 plants of T. mirus that we genotyped, 11 were transgenic (Supplementary Figure S1B). Among the 11 plants, five (45.4%) had edits in the targeted T. porrifolius homeolog, while none showed modifications in the untargeted T. dubius homeolog (Figure 1B). One T. mirus individual (DP-3) contained biallelic mutation of the T. porrifolius homeolog: 3-nt deletions were found in both alleles at the site targeted by sgRNA gTpoDFR1 (Figure 1D). In addition, four plants had monoallelic mutations at the site targeted by gTpoDFR2: three individuals (DP-4, DP-5, and DP-7) shared the same 1-nt insertion, and one plant (DP-8) had a 21-nt deletion (Figure 1D).
4 Discussion
Although CRISPR/Cas-mediated gene editing has been applied in many allopolyploid plants (e.g., Braatz et al., 2017; Sánchez-Gómez et al., 2023; Xu et al., 2024; Zhang et al., 2025), these studies knocked out all copies of the target gene without differentiating between individual homeologs. Homeolog-specific editing has only been reported in a few polyploid crop species, including hexaploid wheat (T. aestivum) and allotetraploid cotton (G. hirsutum), to study function of genes associated with agronomic traits (Liang et al., 2017; Chen et al., 2025; Yu et al., 2025). GW2 controls grain weight in wheat. Liang et al. (2017) designed a sgRNA that perfectly matched the GW2-B1 and GW2-D1 homeologs but had a single nucleotide mismatch at the target site in the GW2-A1 homeolog. Following biolistic delivery of the CRISPR/Cas9 ribonucleoprotein complexes, 0%, 2.2%, and 4.4% of the T0 wheat plants showed editing events at the A, B, and D homeologs, respectively (Liang et al., 2017). By leveraging SNPs between the two GhMML3 homeologs, sgRNAs were designed to specifically knock out either the A-subgenome copy (GhMML3_A12) or the D-subgenome copy (GhMML3_D12) in G. hirsutum (4x) (Chen et al., 2025). This study demonstrated that GhMML3_A12 and GhMML3_D12 regulate fiber development in a dosage-dependent manner. Yu et al. (2025) used CRISPR to edit either the GhHD1A or the GhHD1D homeolog in G. hirsutum and found functional divergence between the two homeologs in regulating trichome and fiber initiation.
Beyond plant systems, CRISPR-mediated allele-specific editing in diploid species (Yoshimi et al., 2014; Smith et al., 2015; Hashizume et al., 2025) and homeolog-specific editing in polyploid species (Gan et al., 2021; 2023) have been reported in several animal models. In an allele-specific editing experiment in the diploid laboratory rat (Rattus norvegicus), sgRNAs were designed to target either the TyrC or Tyrc allele by exploiting one SNP at the CRISPR/Cas9 target site. Editing was found only in the targeted allele, with editing efficiencies of 30.4% and 28.6% for the TyrC and Tyrc alleles, respectively (Yoshimi et al., 2014). Smith et al. (2015) showed that CRISPR/Cas9 can distinguish between two alleles in human cells based on a single nucleotide difference. In polyploid carp (Carassius gibelio), sgRNAs were designed to target either the CgRunx2b-A or CgRunx2b-B homeolog to study their respective roles in intermuscular bone development (Gan et al., 2023).
Currently, homeolog-specific editing studies in polyploid species employ several approaches to differentiate between homeologs. First, the non-PAM approach takes advantage of presence/absence variation in the PAM sequence–only the homeolog containing the PAM site will be edited. In the current study, we used this approach to differentiate the two DFR homeologs at the target site in exon 3 (Figure 1A). This method has also been applied in the study of cotton by Chen et al. (2025). The second approach aims to maximize mismatches within the 10 nucleotides immediately upstream of PAM (also known as the seed sequence) in the sgRNA, as targeting specificity is largely determined by this region. Studies in tetraploid cotton (Chen et al., 2025) and hexaploid wheat (Zhang et al., 2016) have employed this strategy. In T. mirus, a single SNP within the seed sequence successfully differentiated the T. dubius and T. porrifolius MYB10 homeologs (Figures 1A,B). Finally, the use of premixed CRISPR ribonucleoprotein (RNP) has been shown to improve homeolog editing specificity compared to expression of the CRISPR DNA construct. In wheat, Liang et al. (2017) reported that expression of the CRISPR/Cas9 DNA construct led to a 3.8% editing rate in the untargeted homeolog, whereas the RNP approach showed no off-target edits.
Despite the novelty of applying homeolog-specific gene editing in allotetraploid cotton (G. hirsutum), neither Chen et al. (2025) nor Yu et al. (2025) reported homeolog editing efficiency or whether any off-target events (i.e., modification of the untargeted homeolog) were detected. In both cases, homozygous mutant lines were often obtained in subsequent generations, implying monoallelic mutation (i.e., editing one of two alleles of the target homeolog) may have been present in the T0 generation (Chen et al., 2025; Yu et al., 2025). In model crop species such as cotton, substantial resource investment has enabled the development of streamlined and highly efficient gene editing platforms, where reporting metrics such as editing efficiency and off-target rate may be considered less critical. However, for researchers working on non-model polyploid plants, including those of evolutionary significance, such information is essential for guiding future genome editing efforts.
In the present study, we demonstrated the feasibility of performing homeolog-specific editing in a non-model plant polyploid system. In allotetraploid Tragopogon mirus, the homeolog-specific editing efficiencies were 35.7% and 45.5% for MYB10 and DFR genes, respectively (Figure 1). Biallelic mutation of the target homeolog was detected in the T0 generation, and importantly, no transgenic plant showed editing of the untargeted homeolog. This detailed report of homeolog-specific editing in T. mirus (4x) provides a valuable reference for researchers interested in applying similar technologies to their own species of interest.
One limitation of the current work is the relatively low percentage of biallelic mutants in the T0 generation: 0% and 9.1% for MYB10 and DFR, respectively (Figure 1B). In diploid T. porrifolius (2x), the efficiency of creating DFR null mutants–where both alleles are edited–was 100% (Shan et al., 2025). Moreover, when using sgRNAs targeting conserved sequences shared by the T. dubius and T. porrifolius homeologs, 71.4% of the transgenic T. mirus showed edits in all four alleles (from both homeologs) (Shan et al., 2025). Therefore, the relatively low efficiency of homeolog-specific editing was unexpected.
Homeolog-specific editing efficiency in polyploid Tragopogon can be improved through several strategies. First, incorporating additional sgRNAs may enhance editing efficiency. Our study showed that, compared to using one sgRNA (targeting MYB10), using two sgRNAs (targeting DFR) increased the editing efficiency from 35.7% to 45.5% (Figure 1B). Future constructs could include three or four sgRNAs to further increase the frequency of biallelic mutants. Second, while only CRISPR/Cas9 was used here, combining CRISPR/Cas9 and CRISPR/Cas12a systems could broaden targeting options: Cas12a recognizes a TTTN PAM sequence, in contrast to the NGG PAM required by Cas9 (Zhang et al., 2023). Additionally, Cas12a crRNA arrays are more compact than Cas9 sgRNA arrays, allowing the incorporation of more guide RNAs in the construct (Port et al., 2020). Third, monoallelic mutants could serve as explants for a second round of transformation, increasing the likelihood of obtaining biallelic mutants. Finally, additional biallelic mutants are expected in the T1 generation, as 25% of progeny from a self-fertilized monoallelic T0 plant should carry biallelic mutations.
The availability of homeolog-specific editing in the evolutionary model T. mirus (4x) now enables us to address key questions in polyploid genome evolution and fractionation. Although the current work does not include a systematic phenotypic analysis of T. mirus mutants with modified DFR or MYB10 homeologs, such analyses will be performed in our future work. Several important questions will be addressed, including those listed here. For genes in which retention of both homeologs is predicted to be favored by selection (such as genes encoding subunits of protein complexes or those that are dosage-sensitive), what are the effects of losing one versus the other homeolog on co-expression network and phenotype? Does the parental copy matter in terms of resulting network and phenotypic response? Additionally, for genes for which reversion to singleton status is predicted to be favored by selection, both homeologs may still be present and expressed in the early stages of polyploidy, including 95–100-year-old T. mirus. How do the phenotypes and transcriptomes differ between plants retaining both homeologs and genome-edited individuals with only one homeolog? Finally, in cases where one parental homeolog is preferentially retained or expressed in polyploids, what are the consequences of retaining the non-preferred copy? In summary, homeolog-specific editing opens new avenues for investigating polyploid genome evolution, with significant implications for both basic and applied plant science.
Data availability statement
mRNA sequences of MYB10 from T. dubius (TduMYB10) and T. porrifolius (TpoMYB10) are deposited in GenBank (accession numbers PV761044 and PV761045, respectively); sequencing results of MYB10 and DFR from T. mirus mutants are deposited in Dryad (https://doi.org/10.5061/dryad.0vt4b8h9t).
Author contributions
SS: Visualization, Writing – original draft, Formal Analysis, Writing – review and editing, Data curation, Investigation, Software, Conceptualization, Methodology. MP: Writing – review and editing, Formal Analysis, Methodology. EM: Methodology, Writing – review and editing. JS: Formal Analysis, Methodology, Writing – review and editing. BH: Methodology, Resources, Writing – review and editing. WB: Funding acquisition, Writing – review and editing, Resources, Conceptualization. PS: Resources, Funding acquisition, Writing – review and editing, Conceptualization, Writing – original draft, Supervision. DS: Writing – review and editing, Conceptualization, Supervision, Resources, Funding acquisition, Writing – original draft. BY: Supervision, Funding acquisition, Writing – original draft, Formal Analysis, Resources, Conceptualization, Writing – review and editing, Methodology.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by National Science Foundation grants IOS-1923234 to DS, PS, BY, and WB, DEB-2043478 to DS and PS, and DBI-2320251 to PS, DS, and WB.
Acknowledgments
The authors thank Andre Chanderbali for helpful discussion.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author BY declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgeed.2025.1645542/full#supplementary-material
References
Birchler, J. A., and Veitia, R. A. (2007). The gene balance hypothesis: from classical genetics to modern genomics. Plant Cell. 19, 395–402. doi:10.1105/tpc.106.049338
Birchler, J. A., and Veitia, R. A. (2012). Gene balance hypothesis: connecting issues of dosage sensitivity across biological disciplines. Proc. Natl. Acad. Sci. U. S. A. 109, 14746–14753. doi:10.1073/pnas.1207726109
Birchler, J. A., and Veitia, R. A. (2021). One hundred years of gene balance: how stoichiometric issues affect gene expression, genome evolution, and quantitative traits. Cytogenet. Genome Res. 161, 529–550. doi:10.1159/000519592
Blanc, G., and Wolfe, K. H. (2004). Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell. 16, 1679–1691. doi:10.1105/tpc.021410
Braatz, J., Harloff, H. J., Mascher, M., Stein, N., Himmelbach, A., and Jung, C. (2017). CRISPR-Cas9 targeted mutagenesis leads to simultaneous modification of different homoeologous gene copies in polyploid oilseed rape (Brassica napus). Plant Physiol. 174, 935–942. doi:10.1104/pp.17.00426
Buggs, R. J., Zhang, L., Miles, N., Tate, J. A., Gao, L., Wei, W., et al. (2011). Transcriptomic shock generates evolutionary novelty in a newly formed, natural allopolyploid plant. Curr. Biol. 21, 551–556. doi:10.1016/j.cub.2011.02.016
Chaudhary, B., Flagel, L., Stupar, R. M., Udall, J. A., Verma, N., Springer, N. M., et al. (2009). Reciprocal silencing, transcriptional bias and functional divergence of homeologs in polyploid cotton (Gossypium). Genetics 182, 503–517. doi:10.1534/genetics.109.102608
Chen, R., Zhang, J., Li, J., Chen, J., Dai, F., Tian, Y., et al. (2025). Two duplicated GhMML3 genes coordinately control development of lint and fuzz fibers in cotton. Plant Commun. 6, 101281. doi:10.1016/j.xplc.2025.101281
Chester, M., Gallagher, J. P., Symonds, V. V., Cruz da Silva, A. V., Mavrodiev, E. V., Leitch, A. R., et al. (2012). Extensive chromosomal variation in a recently formed natural allopolyploid species, Tragopogon miscellus (Asteraceae). Proc. Natl. Acad. Sci. U. S. A. 109, 1176–1181. doi:10.1073/pnas.1112041109
De Smet, R., Adams, K. L., Vandepoele, K., Van Montagu, M. C., Maere, S., and Van de Peer, Y. (2013). Convergent gene loss following gene and genome duplications creates single-copy families in flowering plants. Proc. Natl. Acad. Sci. U. S. A. 110, 2898–2903. doi:10.1073/pnas.1300127110
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi:10.1093/bioinformatics/bts635
Doyle, J. J., and Coate, J. E. (2019). Polyploidy, the nucleotype, and novelty: the impact of genome doubling on the biology of the cell. Int. J. Plant Sci. 180, 1–52. doi:10.1086/700636
Elomaa, P., Uimari, A., Mehto, M., Albert, V. A., Laitinen, R. A., and Teeri, T. H. (2023). Activation of anthocyanin biosynthesis in Gerbera hybrida (Asteraceae) suggests conserved protein-protein and protein-promoter interactions between the anciently diverged monocots and eudicots. Plant Physiol. 133, 1831–1842. doi:10.1104/pp.103.026039
Evangelisti, A. M., and Conant, G. C. (2010). Nonrandom survival of gene conversions among yeast ribosomal proteins duplicated through genome doubling. Genome Biol. Evol. 2, 826–834. doi:10.1093/gbe/evq067
Fox, D., Soltis, D. E., Soltis, P. S., Ashman, T. L., and Van de Peer, Y. (2020). Polyploidy: a biological force from cells to ecosystems. Trends Cell. Biol. 30, 688–694. doi:10.1016/j.tcb.2020.06.006
Freeling, M., Woodhouse, M. R., Subramaniam, S., Turco, G., Lisch, D., and Schnable, J. C. (2012). Fractionation mutagenesis and similar consequences of mechanisms removing dispensable or less-expressed DNA in plants. Curr. Opin. Plant Biol. 15, 131–139. doi:10.1016/j.pbi.2012.01.015
Gan, R. H., Wang, Y., Li, Z., Yu, Z. X., Li, X. Y., Tong, J. F., et al. (2021). Functional divergence of multiple duplicated Foxl2 homeologs and alleles in a recurrent polyploid fish. Mol. Biol. Evol. 38, 1995–2013. doi:10.1093/molbev/msab002
Gan, R. H., Li, Z., Wang, Z. W., Li, X. Y., Wang, Y., Zhang, X. J., et al. (2023). Creation of intermuscular bone-free mutants in amphitriploid gibel carp by editing two duplicated runx2b homeologs. Aquaculture 567, 739300. doi:10.1016/j.aquaculture.2023.739300
Hashizume, R., Wakita, S., Sawada, H., Takebayashi, S. I., Kitabatake, Y., Miyagawa, Y., et al. (2025). Trisomic rescue via allele-specific multiple chromosome cleavage using CRISPR-Cas9 in trisomy 21 cells. PNAS Nexus 4, pgaf022. doi:10.1093/pnasnexus/pgaf022
Jiao, Y., Wickett, N. J., Ayyampalayam, S., Chanderbali, A. S., Landherr, L., Ralph, P. E., et al. (2011). Ancestral polyploidy in seed plants and angiosperms. Nature 473, 97–100. doi:10.1038/nature09916
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi:10.1093/molbev/mst010
Landis, J. B., Soltis, D. E., Li, Z., Marx, H. E., Barker, M. S., Tank, D. C., et al. (2018). Impact of whole-genome duplication events on diversification rates in angiosperms. Am. J. Bot. 105, 348–363. doi:10.1002/ajb2.1060
Li, Z., Defoort, J., Tasdighian, S., Maere, S., Van de Peer, Y., and De, S. R. (2016). Gene duplicability of core genes is highly consistent across all angiosperms. Plant Cell. 28, 326–344. doi:10.1105/tpc.15.00877
Liang, Z., Chen, K., Li, T., Zhang, Y., Wang, Y., Zhao, Q., et al. (2017). Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nat. Commun. 8, 14261. doi:10.1038/ncomms14261
Liao, Y., Smyth, G. K., and Shi, W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. doi:10.1093/bioinformatics/btt656
Lin-Wang, K., Bolitho, K., Grafton, K., Kortstee, A., Karunairetnam, S., McGhie, T. K., et al. (2010). An R2R3 MYB transcription factor associated with regulation of the anthocyanin biosynthetic pathway in Rosaceae. BMC Plant Biol. 10, 50. doi:10.1186/1471-2229-10-50
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., Von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi:10.1093/molbev/msaa015
Morris, J. P., Baslan, T., Soltis, D. E., Soltis, P. S., and Fox, D. T. (2024). Integrating the study of polyploidy across organisms, tissues, and disease. Annu. Rev. Genet. 58, 297–318. doi:10.1146/annurev-genet-111523-102124
Naing, A. H., and Kim, C. K. (2018). Roles of R2R3-MYB transcription factors in transcriptional regulation of anthocyanin biosynthesis in horticultural plants. Plant Mol. Biol. 98, 1–18. doi:10.1007/s11103-018-0771-4
Ownbey, M. (1950). Natural hybridization and amphiploidy in the genus Tragopogon. Am. J. Bot. 37, 487–499. doi:10.1002/j.1537-2197.1950.tb11033.x
Papp, B., Pál, C., and Hurst, L. D. (2003). Dosage sensitivity and the evolution of gene families in yeast. Nature 424, 194–197. doi:10.1038/nature01771
Paterson, A. H., Chapman, B. A., Kissinger, J. C., Bowers, J. E., Feltus, F. A., and Estill, J. C. (2006). Many gene and domain families have convergent fates following independent whole-genome duplication events in Arabidopsis, Oryza, Saccharomyces and Tetraodon. Trends Genet. 22, 597–602. doi:10.1016/j.tig.2006.09.003
Port, F., Starostecka, M., and Boutros, M. (2020). Multiplexed conditional genome editing with Cas12a in Drosophila. Proc. Natl. Acad. Sci. U. S. A. 117, 22890–22899. doi:10.1073/pnas.2004655117
Renny-Byfield, S., and Wendel, J. F. (2014). Doubling down on genomes: polyploidy and crop plants. Am. J. Bot. 101, 1711–1725. doi:10.3732/ajb.1400119
Sánchez-Gómez, C., Pose, D., and Martín-Pizarro, C. (2023). “Genome editing by CRISPR/Cas9 in polyploids,” in Polyploidy: methods and protocols. Editor Y. Yan de Peer (New York, NY: Humana), 459–473.
Sattler, M. C., Carvalho, C. R., and Clarindo, W. R. (2016). The polyploidy and its key role in plant breeding. Planta 243, 281–296. doi:10.1007/s00425-015-2450-x
Shan, S., Mavrodiev, E. V., Li, R., Zhang, Z., Hauser, B. A., Soltis, P. S., et al. (2018). Application of CRISPR/Cas9 to Tragopogon (Asteraceae), an evolutionary model for the study of polyploidy. Mol. Ecol. Resour. 18, 1427–1443. doi:10.1111/1755-0998.12935
Shan, S., Boatwright, J. L., Liu, X., Chanderbali, A. S., Fu, C., Soltis, P. S., et al. (2020). Transcriptome dynamics of the inflorescence in reciprocally formed allopolyploid Tragopogon miscellus (Asteraceae). Front. Genet. 11, 888. doi:10.3389/fgene.2020.00888
Shan, S., Gitzendanner, M. A., Boatwright, J. L., Spoelhof, J. P., Ethridge, C. L., Ji, L., et al. (2024). Genome-wide DNA methylation dynamics following recent polyploidy in the allotetraploid Tragopogon miscellus (Asteraceae). New Phytol. 242, 1363–1376. doi:10.1111/nph.19655
Shan, S., Pisias, M. T., Zhang, Z., Mavrodiev, E. V., Gitzendanner, M. A., Hauser, B. A., et al. (2025). Development of an efficient CRISPR-mediated genome editing platform in the diploid-polyploid model system Tragopogon (Asteraceae). J. Exp. Bot.
Smith, C., Abalde-Atristain, L., He, C., Brodsky, B. R., Braunstein, E. M., Chaudhari, P., et al. (2015). Efficient and allele-specific genome editing of disease loci in human iPSCs. Mol. Ther. 23, 570–577. doi:10.1038/mt.2014.226
Soltis, P. S., and Soltis, D. E. (2016). Ancient WGD events as drivers of key innovations in angiosperms. Curr. Opin. Plant Biol. 30, 159–165. doi:10.1016/j.pbi.2016.03.015
Soltis, D. E., Soltis, P. S., Pires, J. C., Kovarik, A., Tate, J. A., and Mavrodiev, E. (2004). Recent and recurrent polyploidy in Tragopogon (Asteraceae): cytogenetic, genomic and genetic comparisons. Biol. J. Linn. Soc. 82, 485–501. doi:10.1111/j.1095-8312.2004.00335.x
Soltis, D. E., Buggs, R. J., Barbazuk, W. B., Chamala, S., Chester, M., Gallagher, J. P., et al. (2012). “The early stages of polyploidy: rapid and repeated evolution in Tragopogon,” in Polyploidy and genome evolution. Editors P. S. Soltis, and D. E. Soltis (Berlin, Heidelberg: Springer), 271–292.
Soltis, P. S., Marchant, D. B., Van de Peer, Y., and Soltis, D. E. (2015). Polyploidy and genome evolution in plants. Curr. Opin. Genet. and Dev. 35, 119–125. doi:10.1016/j.gde.2015.11.003
Spoelhof, J. P., Chester, M., Rodriguez, R., Geraci, B., Heo, K., Mavrodiev, E., et al. (2017). Karyotypic variation and pollen stainability in resynthesized allopolyploids Tragopogon miscellus and T. Mirus. Am. J. Bot. 104, 1484–1492. doi:10.3732/ajb.1700180
Tang, H., Woodhouse, M. R., Cheng, F., Schnable, J. C., Pedersen, B. S., Conant, G., et al. (2012). Altered patterns of fractionation and exon deletions in Brassica rapa support a two-step model of paleohexaploidy. Genetics 190, 1563–1574. doi:10.1534/genetics.111.137349
Tate, J. A., Joshi, P., Soltis, K. A., Soltis, P. S., and Soltis, D. E. (2009). On the road to diploidization? Homoeolog loss in independently formed populations of the allopolyploid Tragopogon miscellus (Asteraceae). BMC Plant Biol. 9, 80. doi:10.1186/1471-2229-9-80
Udall, J. A., and Wendel, J. F. (2006). Polyploidy and crop improvement. Crop Sci. 46, S3–S14. doi:10.2135/cropsci2006.07.0489tpg
Van de Peer, Y., Ashman, T. L., Soltis, P. S., and Soltis, D. E. (2021). Polyploidy: an evolutionary and ecological force in stressful times. Plant Cell. 33, 11–26. doi:10.1093/plcell/koaa015
Wendel, J. F. (2015). The wondrous cycles of polyploidy in plants. Am. J. Bot. 102, 1753–1756. doi:10.3732/ajb.1500320
Wendel, J. F., Flagel, L. E., and Adams, K. L. (2012). “Jeans, genes, and genomes: cotton as a model for studying polyploidy,” in Polyploidy and genome evolution. Editors P. S. Soltis, and D. E. Soltis (Berlin, Heidelberg: Springer), 181–207.
Wendel, J. F., Lisch, D., Hu, G., and Mason, A. S. (2018). The long and short of doubling down: polyploidy, epigenetics, and the temporal dynamics of genome fractionation. Curr. Opin. Genet. and Dev. 49, 1–7. doi:10.1016/j.gde.2018.01.004
Wood, T. E., Takebayashi, N., Barker, M. S., Mayrose, I., Greenspoon, P. B., and Rieseberg, L. H. (2009). The frequency of polyploid speciation in vascular plants. Proc. Natl. Acad. Sci. U. S. A. 106, 13875–13879. doi:10.1073/pnas.0811575106
Xu, S., Li, F., Zhou, F., Li, J., Cai, S., Yang, S., et al. (2024). Efficient targeted mutagenesis in tetraploid Pogostemon cablin by the CRISPR/Cas9-mediated genomic editing system. Hortic. Res. 11, uhae021. doi:10.1093/hr/uhae021
Yoo, M. J., Koh, J., Boatwright, J. L., Soltis, D. E., Soltis, P. S., Barbazuk, W. B., et al. (2024). Investigation of regulatory divergence between homoeologs in the recently formed allopolyploids, Tragopogon Mirus and T. Miscellus (Asteraceae). Plant J. 117, 1191–1205. doi:10.1111/tpj.16553
Yoshimi, K., Kaneko, T., Voigt, B., and Mashimo, T. (2014). Allele-specific genome editing and correction of disease-associated phenotypes in rats using the CRISPR-Cas platform. Nat. Commun. 5, 4240. doi:10.1038/ncomms5240
Yu, L., Zhao, X., Hua, L., Yuan, R., Wei, Y., Luo, K., et al. (2025). Functionally differentiated GL2-interacting-repressor 1 homoeologs regulate epidermal hair development of Gossypium hirsutum. Plant Physiol. 198, kiaf184. doi:10.1093/plphys/kiaf184
Yuan, Y. W., Sagawa, J. M., Frost, L., Vela, J. P., and Bradshaw, Jr H. D. (2014). Transcriptional control of floral anthocyanin pigmentation in monkeyflowers (Mimulus). New Phytol. 204, 1013–1027. doi:10.1111/nph.12968
Zhang, Y., Liang, Z., Zong, Y., Wang, Y., Liu, J., Chen, K., et al. (2016). Efficient and transgene-free genome editing in wheat through transient expression of CRISPR/Cas9 DNA or RNA. Nat. Commun. 7, 12617. doi:10.1038/ncomms12617
Zhang, L., Li, G., Zhang, Y., Cheng, Y., Roberts, N., Glenn, S. E., et al. (2023). Boosting genome editing efficiency in human cells and plants with novel LbCas12a variants. Genome Biol. 24, 102. doi:10.1186/s13059-023-02929-6
Keywords: CRISPR, evolutionary model, genome evolution, homeolog-specific editing, polyploidy, Tragopogon
Citation: Shan S, Pisias MT, Mavrodiev EV, Spoelhof JP, Hauser BA, Barbazuk WB, Soltis PS, Soltis DE and Yang B (2025) Development of a homeolog-specific gene editing system in an evolutionary model for the study of polyploidy in nature. Front. Genome Ed. 7:1645542. doi: 10.3389/fgeed.2025.1645542
Received: 11 June 2025; Accepted: 04 August 2025;
Published: 29 August 2025.
Edited by:
Jaindra Nath Tripathi, International Institute of Tropical Agriculture (IITA), KenyaReviewed by:
Rui Zhang, Chinese Academy of Sciences (CAS), ChinaSamwel Muiruri, Kenyatta University, Kenya
Copyright © 2025 Shan, Pisias, Mavrodiev, Spoelhof, Hauser, Barbazuk, Soltis, Soltis and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shengchen Shan, c2hhbjE1ODUzOEB1ZmwuZWR1