 Linda Resar1,2,3*
Linda Resar1,2,3* Tania Jain2
Tania Jain2- 1Department of Medicine (Hematology), The Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 2Department of Oncology, Sidney Kimmel Comprehensive Cancer Center, The Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 3Department of Pathology, The Johns Hopkins University School of Medicine, Baltimore, MD, United States
Myeloproliferative neoplasms (MPNs) are chronic, clonal blood disorders characterized by overproduction of mature blood cells. MPNs present with protean clinical manifestations, including vascular complications, with both venous and arterial thromboses, bleeding diathesis, which occur in the setting of extreme thrombocytosis and acquired von Willebrand disease, and an increased risk of transformation to acute myeloid leukemia (AML). While previously diagnosed primarily in middle-aged and older adults, MPNs are increasingly being recognized in children and young adults. Indeed, studies of clonal evolution trace the development of MPN driver mutations to in utero in some cases. However, prior diagnostic criteria and treatment guidelines have evolved primarily from studies of older MPN patients. Thus, research focused on MPN in younger patients is warranted. Here, we review prior studies of clinical manifestations, outcomes, and therapy for younger patients with MPN.
Introduction
The myeloproliferative neoplasms (MPNs) are clonal blood disorders characterized by acquired mutations in hematopoietic stem cells (HSCs) that lead to hyperactive signaling through Janus kinase-2 and signal activator of transcription (JAK/STAT), resulting in overproduction of myeloid lineages (1–10). The classical MPN phenotypes include: 1) essential thrombocytosis (ET), characterized by thrombocytosis, 2) polycythemia vera (PV), defined by erythrocytosis and frequently associated with thrombocytosis and leukocytosis, and, 3) primary myelofibrosis (PMF), marked by bone marrow scarring (myelofibrosis) and abnormal blood counts, which can include leukocytosis and thrombocytosis with progression to anemia and other cytopenias as bone marrow fibrosis increases (1–10). About 20% of adult patients with chronic MPN will evolve to secondary MF and 5% will progress to secondary AML, although the risk of leukemic transformation varies depending on presenting clinical phenotypes and bone marrow morphology (3, 5–16). For example, leukemic transformation risk is greatest in PMF, estimated to be 10-20% over 10 years in prior studies (3, 6–16). By contrast, the risk is much lower in PV, estimated to be 2-4% over 10-15 years, and even lower for ET, estimated to be 1-2% at 15 years in a prior study (3, 6–16). In contrast to de novo AML, secondary AML that arises in the setting of MPN is unresponsive to induction chemotherapy and therefore almost universally fatal (11).
MPNs are most frequently caused by mutations in JAK2 (namely JAK2V617F) (2–9). In older adults with MPN, JAK2V617F is detected in ~95% of all PV cases, whereas alternative JAK2 mutations occur in <5% of PV patients (2–9). JAK2V617F is also detected in ~50% of ET and MF in adults (2–9). Unexpectedly, rigorous population studies also found JAK2V617F mutations in the general population at rates far above those previously detected (17, 18) and a subset of these individuals do not develop MPN. Similar to MPN incidence, however, JAK2V617F mutations in the general population are more common in older individuals and occur with greater allele frequencies with increasing age. Why only a subset of patients with JAK2V617F mutations develop MPN remains unclear.
In addition to JAK2V617F, mutations in CALR or MPL also cause ET and PMF. A subset of adult MPN patients harbors no demonstrable mutations in JAK2, CALR, or MPL, and are therefore considered “triple-negative” (TN). In older patients, TN disease occurs with PMF and is associated with poor outcomes. By contrast, in adolescents and young adults (AYAs), TN disease is common in ET, where it is associated with favorable outcomes. Additional features related to the mutational status in children and young adults with MPN are distinct from those of adults and will be discussed here (3, 12–25).
Despite their young age, children and AYAs with MPN often present with symptomatic disease and serious complications (16, 19–32). As physicians and providers caring for patients with MPN, our treatment goals focus on minimizing complications and prolonging healthy lifespans. Thus, appropriate diagnosis and optimal management are particularly important in our youngest patients who may have more than eight decades of life ahead of them. Paradoxically, MPN in younger patients is complicated by both thromboses and hemorrhage. Bleeding diatheses occur when patients develop acquired von Willebrand disease with extreme thrombocytosis. Because school-aged children and adolescents are often expected to participate in physical education classes and may also be active in organized sports, bleeding complications are particularly relevant to those engaging in sports at risk for trauma.
In both younger and older MPN patients, clinical features are protean, and highly malleable over time, ranging from a complete absence of symptoms to those that significantly impact quality of life, including headaches, fatigue, pruritis, and abdominal pain (1–16, 19–32). Similar to older adult patients, MPN in younger patients exists on a continuum. While studies in younger patients are limited, it is clear that transformation occurs, albeit predominantly after pediatric and young adult patients reach middle age or later adulthood. Accordingly, treatment strategies that mitigate the risk of transformation are of paramount importance for our youngest patients. Here, we outline clinical and molecular features of MPN in younger patients, long-term outcomes, and treatment options based on available research from our group and many others (Figure 1).
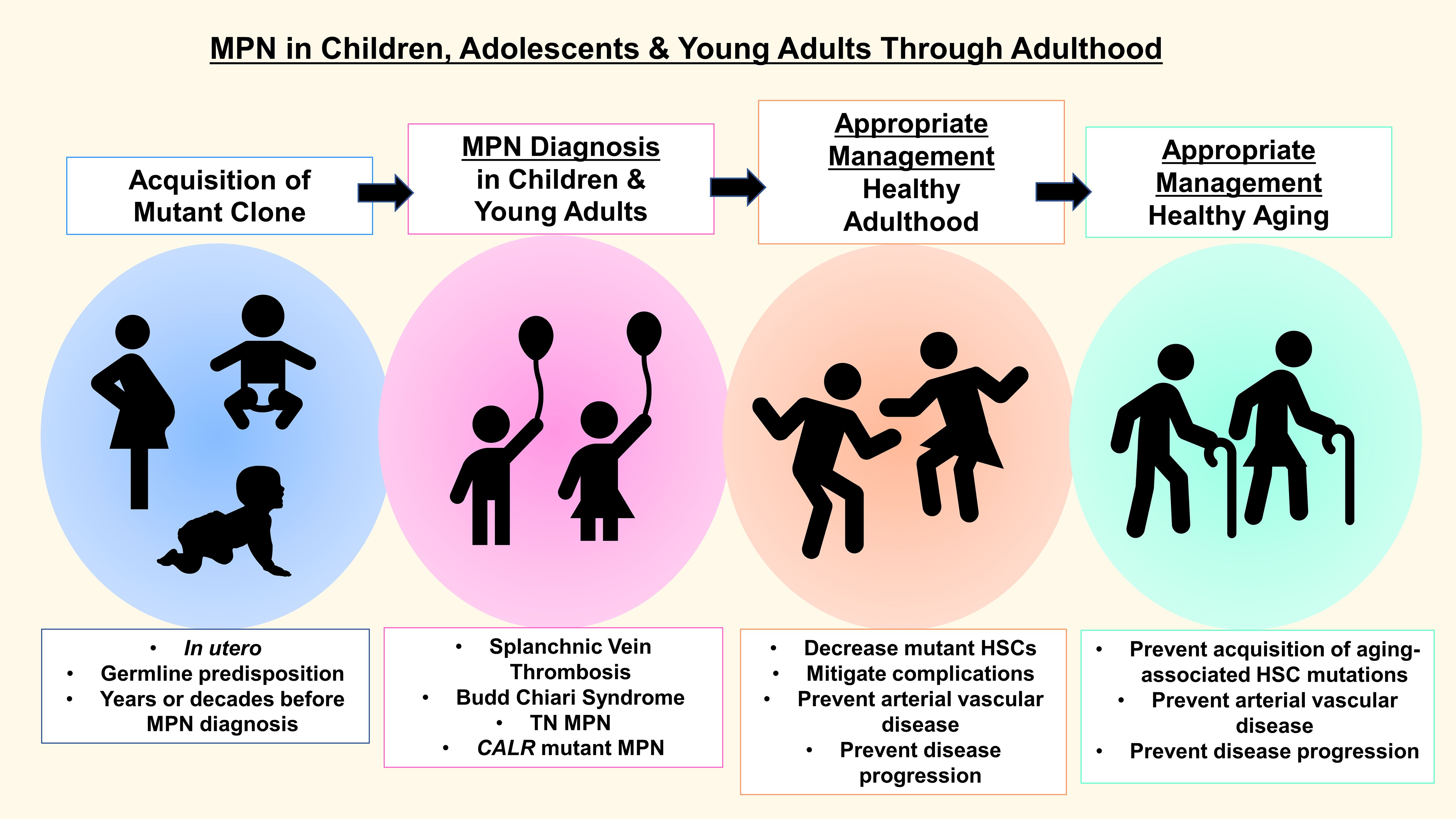
Figure 1. MPN in children, adolescents and young adults: diagnosis, management & treatment goals.
Special considerations for MPN in younger patients
While historically considered diseases of older adults, we now know that young patients develop MPN. Indeed, with the advent of more widely available genetic testing, MPN is being diagnosed more frequently in pediatric and young adults (16–37). Intriguingly, recent studies using mathematical modeling of clonal dynamics in adult-onset MPN suggest that driver mutations occur much earlier in life, and even in utero in some cases (33, 34). For example, clonal evolution studies traced the development of CALR driver mutations to fetal development in monozygotic twins with MPN, both of whom were diagnosed with MF in their third decade of life (34). In addition, newborn blood spots confirmed the presence of a JAK2V617F in neonatal blood in a PV patient who presented at age 34 years (34).
Why some patients present in childhood or early adulthood versus later in life remains a fundamental unanswered question in the field. Given the increasing numbers of children and young adults being diagnosed with MPN (16–34), recent studies have reviewed clinical and molecular features in younger patients, including a European consortium describing a large cohort of MPN patients diagnosed before 25 years and multiple studies from the United States (16–32). In these cohorts, children and AYA patients comprise anywhere from 8 to 27% of the total MPN population (18–23). Unfortunately, transformation to blast phase, MF, or frank AML occurs, albeit rarely, in MPN patients diagnosed as children or young adults (19–23). Thus, a critical goal in the field is to prevent evolution of more benign, chronic MPN to advanced diseases that will impact quality of life and shorten lifespans.
MPN phenotypes in younger patients
In prior studies focused on pediatric and young adult patients, ET is the most common MPN type, followed by PV, with the lowest proportion of younger patients presenting with PMF (21, 23, 26–29). By contrast, in adult MPN patients, PV is the most common presenting diagnosis (21, 23, 26–29). In our Hopkins cohort of 630 patients enrolled between 2012-2022, older adults (≥ 40 years; n=459), presented most frequently with PV (45.5%), followed by ET (38.5%), and MF (15.9%) (21). In the younger patient cohort (<40 years old; n=171), ET was the most common diagnosis (67.6%), with PV next in frequency (26%), and MF in only 7% of cases (21). In the youngest subset of patients in the Hopkins study (< 20 years; n=21), 57% had ET and 43% had PV at diagnosis, while none presented with MF (21). In this Hopkins cohort, palpable splenomegaly was more common in older adults (≥ 40 years; 12.6%) compared to younger patients (< 40 years; 6.4%), although palpable splenomegaly was noted in almost half of the 21 patients who were < 20 years (47.6%; 10/21) (21). Platelet counts were not reported in this study. In a multi-institutional cohort of pediatric patients (< 20 years; n=58) in the United States (24), 64% had ET and 26% had PV, although 10% were diagnosed with either MF (7%) or pre-fibrotic MF (3%). Platelet counts were > 450,000/µL in 5 of 7 patients who developed thrombosis, although none with thromboses had platelet counts > 1 million/µL. Spleen sizes were not reported in this study (24). Other studies in the United States and Europe showed similar findings with ET being most prevalent among younger patients (19–32). A systematic review of European and American MPN cohorts (< 40 years) published in 2019 reported variable rates of splenomegaly in MPN patients < 20 years, ranging from 15.3% in PV to 54.7% in ET (27). Platelet counts in this review ranged from 476,000-799,000/µL for PV and 708,00 to 1,192,000/µL for ET (27). Thus, most young patients are diagnosed with ET, followed by PV, and rarely MF (21, 23, 26–29).
While PMF is rare in younger patients, a series from the Mayo Clinic reported clinical and molecular features of 63 young PMF patients (age range of 19-40 years) compared to older patients, who were divided into 2 cohorts (ages 41-60 or > 60 years) (29). In the young PMF cohort, the median age was 37 years and most were male (59%) (29). Compared to the older MPN patients, hemoglobin values were higher (12.1 versus 10.7 versus 10.0 g/dL) and transfusion requirements were lower in younger PMF patients (29). Constitutional symptoms were less frequent in younger patients and platelet counts, leukocytosis, circulating blasts, and lactate dehydrogenase concentrations were also lower (29). There were fewer arterial thrombotic events at diagnosis in the younger PMF patients (29). Most younger PMF patients (>68%) had enlarged spleens, although splenomegaly was slightly more common in older PMF patients (29). Unfortunately, 10% of the younger patients with PMF transformed to AML during the 7.1 year follow-up period, which is similar to the older patients in this cohort (10% and 9%). In the Hopkins cohort of 171 patients < 40 years, males were also more likely to present with PMF (23).
Distinct molecular features associated with MPN in young patients
While JAK2-V617F mutations comprise the most common mutations associated with MPN in young and older MPN patients, there are differences in mutational status based on age. ET in younger patients is associated with higher rates of “triple negative” (TN) status compared to older cohorts. In fact, younger children and teens have the highest rates of TN disease in most studies (16, 19–21, 23–31). In the Hopkins cohort, the presence of JAK2 somatic mutations was higher in older adults (67% in patients >40 years versus 39% in patients <40 years), although JAK2 mutations constitute the most frequent driver mutations in both young and older patients with MPN in most studies (21). In addition to TN, CALR mutations are more common in younger patients in studies comparing MPN in younger to older individuals based on available research (16, 19–21, 23–31). Similar to adults, over 90% of pediatric, adolescent, and young adults with PV harbor JAK2 mutations (16, 19–21, 23–31). Most younger cohorts have a slight female predominance, akin to older adult MPN cohorts. In the Mayo Clinic study which included young PMF patients (n=63, < 40 years), CALR mutations were also more common in the young PMF cohort (48%), with a lower frequency of JAK2 mutations (39%), whereas JAK2 was more prevalent in older patients with PMF (29). High risk mutations (ASXL1, SRSF2, U2AF1) were more common in the older cohorts (29). In the Hopkins cohort of 171 patients < 40 years, CALR mutations were more frequent than JAK2 in PMF among younger patients in our cohort (21). Across all published cohorts, younger patients have higher proportions of TN status and CALR mutations compared to older MPN patients. Nonetheless, JAK2-V617F mutations are the most common driver mutations in younger and older MPN patients. Intriguingly, a prior study also showed an increased incidence of family members with MPN among younger patients diagnosed with MPN (21).
MPN complications in younger patients
Thrombosis
Many pediatric and AYA patients are asymptomatic at diagnosis. However, in contrast to adults, prior studies report more frequent thrombotic events in younger patients (21, 23, 27, 35). In the Hopkins cohort (n=171, age < 40 years), 20% of young MPN patients developed VTE compared to 11% in older adults (n=459, age ≥ 40 years) (21). A retrospective analysis of predominantly European patients under 20 years (n= 471) found that 12% of all patients reported prior thrombosis while an additional 13% developed a new VTE after MPN diagnosis (27). A European retrospective review of young MPN patients (< 25 years) reported similar results with 11% of patients presenting with a history of VTE at MPN diagnosis and an additional 11% developing VTE after MPN diagnosis (23). A multi-institutional cohort across the United States of pediatric patients (< 21 years) reported similar rates of thrombosis (12%) (24). By contrast, a study of patients evaluated at Mayo Clinic (n=361, age ≤ 40 years) reported higher frequencies of VTE with 19% of PV and 16% of ET patients with VTE at MPN presentation and an additional 18% of PV and 21% of ET patients developing a VTE after diagnosis (29). Of note, this study included patients diagnosed as early as 1967. In more contemporary cohorts, approximately ¼ of all pediatric and young adult patients will develop venous thrombosis. In contrast to older adult patients, arterial thromboses are uncommon in pediatric and young adults (21, 23–24, 26–31, 35). However, as detailed next, splanchnic vein thrombosis (SVT), which refers to thrombosis in any of the vessels that drain the abdominal organs, including portal, splenic, mesenteric, or hepatic veins, is a particularly vexing thrombotic complication in younger patients, and unfortunately, often associated with significant sequelae (21, 23, 24–31). Similar to results with older MPN patients, prior studies of young MPN patients show no clear relationship between the higher platelet counts and thrombotic complications (21, 24, 27, 29, 35).
Splanchnic vein thrombosis and Budd Chiari syndrome
In addition to an increased incidence of thrombosis overall, adolescent and young adult patients with MPN develop SVT at significantly higher rates than older patients (19, 21–31). In the Hopkins cohort, SVT accounted for the higher rates of thrombosis observed in younger patients (21). JAK2 mutation was significantly associated with thrombosis in the younger patients (< 40 years) but not in the older adults (≥ 40 years) in this cohort (21). Another group reported that 77% of all thromboses in younger MPN patients ranging from 18-45 years old were due to splanchnic events (28). A study examining clinical outcomes of 120 patients ≤ 45 years of age found higher SVT in younger patients compared to older individuals (13% versus 2%) (31). A multi-institutional cohort (n=58) of pediatric MPN patients (< 21 years old) across the United States also reported that 12% (n=7) developed VTE, primarily in patients who are JAK2V617F positive with a median age of 16 years (24). Strikingly, 5 of these 7 patients (71%) had SVT. Among the PV patients, 1/3 had thrombosis (24). Intriguingly, in 5 of 7 patients, VTE was their initial presenting manifestation of MPN highlighting VTE as a hallmark of MPN in younger patients (24).
Budd-Chiari Syndrome (BCS) is a special case of SVT caused by obstruction of the hepatic vein, most commonly by a hepatic vein thrombosis (36). BCS is rare, with estimates of about 1/1,000,000 people per year (36). Importantly, MPN is the most common cause for BCS (36). When hepatic vein thrombosis leads to increased intrahepatic pressure, venous congestion may ensue and cause hepatic cell injury, transaminitis, and direct hyperbilirubinemia. In severe cases, liver failure develops and patients require liver transplantation. The multi-institutional pediatric series from the United States (n=58) (24) reported that all patients with SVT had BCS, and one required liver transplantation for progressive hepatic failure associated with hyperammonemia, encephalopathy, and coagulopathy. The European cohort study also reported a high incidence of BCS (23). Most patients with BCS are managed with hepatic portosystemic shunts to relieve venous congestion in the liver, although hepatic failure may develop despite this intervention. Based on these findings, it is critical to consider the diagnosis of MPN in young patients presenting with hepatic vein thrombosis and BCS.
Hemorrhage
Paradoxically, MPN patients are also at increased risk for hemorrhage, which typically occurs in the setting of extreme thrombocytosis (> 1 million/µL) and acquired von Willebrand disease. Acquired von Willebrand disease occurs when excessive numbers of platelets consume and degrade von Willebrand factor, which interferes with platelet aggregation and normal clotting. Although hemorrhagic complications are reported in studies of younger MPN patients, the incidence is generally lower than that of thrombotic complications (21, 23–30). For example, a systemic review of published literature of patients < 20 years found that 3% and 4% of patients with ET or PV, respectively, had hemorrhage prior to MPN diagnosis, whereas 19% and 3% of patients with ET or PV, respectively, developed hemorrhage after MPN diagnosis (27).
Transformation to MF and AML in younger patients
In older adults with chronic, indolent MPN (ET, PV), transformation to MF is frequently associated with increasing symptom burden, worsening splenomegaly, anemia, and other blood count abnormalities, along with decreased survival (7–11). Progression to AML in adults with MPN is almost universally lethal. Unfortunately, children and young adults diagnosed with MPN also progress to more advanced disease, although this happens decades later and well into adulthood (21, 29). For example, in the Hopkins cohort, transformation from ET to MF in younger patients (< 40 years) occurred in 8.8% (10 of 114 patients) with a mean age of 52 years at MF diagnosis (21). The time to transformation ranged from 4-41 years, with a mean of 22 years. Transformation of PV to MF in younger patients occurred in 24.4% (11 of 45 patients) with a mean age of 54 at MF diagnosis and time to progression ranging from 10-53 years (mean 24 years) (21). When focusing on the youngest patients < 20 years, only 1 ET patient and 2 PV patients progressed to MF with a mean age of 62 at MF diagnosis (21). By contrast, in the older adults in this cohort (≥ 40 years), 25.4% of ET patients (177/459) and 21.5% of PV patients (209/459) progressed to MF at mean ages of 68 and 65 years, respectively (21). Years to transformation ranged from 1-32 years with a mean of 12 years for ET and 10 years for PV, which was about half the time to progression for the younger patients (21). Notably, the number of young patients who transformed to MF is relatively small (n=10) and therefore may not be generalizable to all younger MPN patients.
In the younger cohort (< 40 years), 1.8% (3 of 171 patients) progressed to AML, including 1 patient with ET who was diagnosed with AML at age 58, which was 25 years after the initial MPN diagnosis (21). Two patients developed AML at 27-28 years after their PV diagnosis, with AML diagnosed at age 58 or 65 years, respectively. None of the patients aged < 20 years old developed AML during the study interval in our Hopkins cohort. By contrast, in the older adult cohort (> 40 years), 3% (6 of 177) of patients with ET progressed to AML at a mean age of 76 with a mean time for progression of 20 years and 6% (13 of 209 patients) of patients with PV developed AML at a mean age of 74 with a mean time of progression of 10 years. While progression was relatively rare, particularly in our younger cohorts, these data suggest that the time to progression may be longer for younger patients, although the age at diagnosis for MF was slightly lower (52-54 for younger patients versus 65-68 for older patients) (21). For AML, the younger patients ages ranged from 58-62 at AML diagnosis, while ages for the older patients with AML ranged from 63-87 (21). As noted previously, the Mayo Clinic study reported that 10% of young PMF patients (n=63) transformed to AML over 7.1 years, although this included patients diagnosed between 1967-2017 (29). Importantly, the numbers of patients progressing to AML or MF in published cohorts are small and the time to progression and ages at progression may not be relevant to other cohorts or more contemporarily-treated patients (21, 29). In addition, our hope is that current therapeutic approaches will delay or prevent progression, particularly in our younger patients.
Therapeutic considerations for younger MPN patients
Although most young patients with MPN are diagnosed with more indolent disease, such as ET or PV, younger patients experience symptoms and vascular complications which can impact quality of life and lead to shortened lifespans (16, 19–32). However, given the rarity of pediatric MPN, there are no studies defining optimal cytoreductive therapy for the younger patients. The foremost objective in caring for younger MPN patients is to limit disease-associated complications while minimizing toxicity from therapy. Another important goal particularly relevant to pediatric patients is to identify strategies to prevent disease progression, avoid the acquisition of high-risk mutations associated with transformation, and ideally, to eliminate the mutant clone, if this can be done without untoward toxicity. Cytoreductive medications used in published cohorts of pediatric MPN patients include hydroxyurea, interferon (IFN), anagrelide, and ruxolitnib (16, 19–32, 37). Given increasing evidence that hydroxyurea is associated with the acquisition of deleterious mutations in older patients, such as TP53, most MPN physicians prefer to avoid its use in younger patients (38). Experience with anagrelide is limited in younger patients. However, ruxolitinib, has been used at our institution and others in the United States, particularly in young adults who require liver transplantation for BCS since ruxolitinib controls counts effectively in most young patients. MAJIC-PV, a recent large multi-institutional trial of ruxolitinib in adults with MPN in England showed promising results marked by a lower incidence of progression in addition to decreases in mutant allele burdens in most patients compared to best available therapies (37).
Interferons are immunomodulating agents that have been shown to improve MPN-associated bone marrow abnormalities in some patients, lower blood counts, and induce hematologic remissions (22, 38–45). Perhaps the most exciting attribute of interferons for our youngest patients is their capacity to decrease the mutant JAK2 allele burden and induce molecular remissions (38–40). Significant decreases in allele burdens have been reported with IFN (38–45). While the underlying mechanisms continue to be explored, prior studies suggest that IFN may preferentially induce cell cycle progression in JAK2 mutant stem cells, leading to proliferation and ultimately exhaustion of mutant clones (46–49). Decreasing mutant allele burdens, and potentially eliminating them, is a particularly desirable goal for younger patients (22, 26). In one cohort, IFN treated patients with PV had the same overall survival as an age-matched US population (44). Discontinuation of IFN after achieving hematologic remission was also shown to be safe in several patients without negatively impacting survival (45). Compelling data from large studies using ropeginterferon alfa-2b (ROPEG), a more recent formulation of monopegylated IFN that can be given every 2−4 weeks, led to its approval as a first-line therapy for PV (43). In adults, ROPEG is administered every 2 weeks until hematologic remission, after which many patients do well with every 4-week dosing once sustained hematologic responses are achieved. The relatively infrequent dosing is advantageous for young adult patients who may be attending universities away from home and younger patients in general who may be resistant to taking medications.
Interferons are an appealing class of drug for younger women who become pregnant. IFNs have not been linked to mutagenicity or teratogenicity, which are the primary concerns for using hydroxyurea in younger patients (38). While prior studies are limited by small cohorts, results also suggest that the use of IFN in pregnant MPN patients led to increased odds of live births (50). IFN has a track record of use in children and teenagers (22, 25). Our group reported favorable outcomes with IFN in a small series of pediatric patients (n=13) with MPN, including six with ET and seven with PV (25). While generally well-tolerated, side effects reported in this cohort included psychiatric (depression) and elevations in transaminitis, which can limit its use particularly in patients who underwent liver transplantation for BCS (25). Autoimmune effects may also limit IFN use in younger patients (22, 25). In our prior study, five of 13 children discontinued IFN for mood-related effects, one for transaminitis post-liver transplant, and two because VTE events occurred while on IFN (25), although one of those patients subsequently restarted IFN without additional VTE events or side effects. Less significant side effects that did not result in stopping IFN in our study of younger MPN patients included dizziness and injection site reaction (25). Since this publication, we have treated additional young patients with IFN who have shown effective cytoreduction, elimination of phlebotomy, and resolution of iron deficiency. The effects on iron deficiency are particularly important in younger patients since iron is critical for neurocognitive function and brain development, which extends into the third decade of life (51). In the Low-PV study, phlebotomy and low dose aspirin (when not contraindicated) were compared to Ropeginterferon (ROPEG; 100 microgram subcutaneously every 2 weeks) plus phlebotomy and low dose aspirin in a multicenter, randomized trial across Italy. One-quarter of the ROPEG treatment arm were patients < 45.5 years, and ROPEG was superior to phlebotomy alone in maintaining the hematocrit <45%. There were no dose-limiting side effects or toxicities in all patients (52). A recent European study reported that IFN treatment yields significantly better myelosis-free survival compared to other cytoreductive therapies in young MPN patients (53). As recommended by a recently published consensus statement (22), future long-term studies in younger patients on IFN are warranted given the potential advantages of standard IFN and ROPEG for our youngest MPN patients.
Stem cell transplantation in younger patients
Despite improvements in available therapies, allogeneic hematopoietic stem cell transplantation (HSCT) remains the only cure for MPN (54, 55). Studies from primarily older adult patients with MF that incorporate poor prognostic disease-related factors with both clinical and molecular features recommend HSCT for patients with expected progression within 5 years (56–60). In the pediatric population, however, there are no established prognostic criteria, and consequently, evidence-based recommendations for HSCT are lacking. Since young patients with chronic MPN, such as ET and PV, have excellent long-term overall survivals, HSCT is currently reserved for those with advanced-stage disease. Fortunately, PMF is rare in younger patients, although when it does occur (12, 27, 29), optimal timing of HSCT is perhaps the most important consideration for younger patients with advanced-stage MPN. For example, high-risk disease features that warrant considering HSCT in older adults include somatic mutations associated with higher risk of progression (ASXL1, IDH1, IDH2, SETBP1, EZH2, U2AF1 Q157, TP53, KRAS, NRAS), cytopenias, especially those requiring transfusions which are often associated with triple-negative PMF in older patients, and accelerated or blast phase disease (57–60). While mostly anecdotal evidence is available on outcomes after HSCT in younger patients, one retrospective study from the European Society for Blood and Marrow Transplantation (EBMT) summarized outcomes of 35 children or adolescents of age ≤ 18 years who underwent HSCT for primary or post-ET/PV myelofibrosis between 2000-2022 in Europe (60). Most patients (n=32) underwent myeloablative conditioning, although the regimen varied across institutions and over time (61). At 6 years, non-relapse mortality was 18%, relapse incidence was 15.9%, progression-free survival was 66.1% and overall survival was 71.1% (61). Non-relapse mortality was <10% in patients transplanted between 2008-2022, although this was not statistically significant with the small patient numbers (n=19 from 2008-2022 and n=16 from 2000-2007) (61). However, overall survival was better when the stem cell source was bone marrow compared to peripheral blood or cord blood (61).
In younger patients with MPN, specific issues related to HSCT are particularly relevant, such as fertility preservation. Planning for potential gonadal failure is important for younger patients, which can result from myeloablative conditioning (62, 63) In addition, late effects of HSCT, including chronic graft versus host disease and related manifestations, such as endocrine disorders, or secondary malignancies require monitoring. Since our younger patients are expected to have longer post-HSCT survival, survivorship care must be tailored to include monitoring for long-term effects (62–65). Thus, while there is no consensus in the field on when to consider HSCT in pediatric and young adult patients, the following clinical parameters are reasonable considerations based on available data: 1) Imminent or current transfusion dependence, 2) Increasing blast count, and 3) Acquisition of high-risk mutations. Intervening earlier may be advantageous, although precisely what parameters are needed for optimal outcomes in HSCT in younger patients will require further study.
Transitioning from a pediatrician to an adult MPN clinic
Another topic relevant to younger patients with MPN is how best to prepare adolescents for care in adult clinics where they will have more autonomy and responsibility with less parental involvement in most cases. Our group advocates use of a check list of topics to discuss with younger patients prior to transition. Importantly, we recommend that this process occur gradually, to allow the young person with MPN to learn more about the disease and management as well as to ask questions. Providers should ensure that young patients have the maturity and resources to pursue MPN follow-up in addition to an appropriate clinic with expertise in MPN prior to transitioning young MPN patients to adult care.
Summary and future directions
Despite being rare, MPN can lead to significant short- and long-term burdens in young patients and their families. Thus, there is an unmet need for further studies to focus on our youngest patients. Further work is warranted to fully characterize the scope of MPN in this population and to identify the best therapies for their diseases. Outcomes in young adults with acute lymphoblastic leukemia improved significantly when they were treated with therapies designed for younger, pediatric patients. Thus, it is possible that therapies and outcomes in younger patients will inform interventions for older MPN patients. Given the relatively low numbers of children with MPN, multi-institutional studies are needed. Clinical trials through cooperative groups such as the Children’s Oncology Group in the United States, or similar groups on other continents, will ensure that our youngest MPN patients have access to such opportunities. Although enrollment in trials will be limited by the low prevalence of MPN in pediatric and young adult patients, the Children’s Oncology Group has successfully enrolled children with rare cancers which ultimately led to better therapies. Endpoints for chronic diseases with low risk of progression in young patients represent an additional challenge, although studies such as the Low-PV trial used maintenance of hematocrit below 45% as a useful endpoint (52). While this endpoint may be relevant in older adolescents, age-specific hematocrits would be helpful for younger patients. In addition, further research to elucidate the mechanisms underlying MPN complications that occur primarily in children, adolescents, and young adults, such as thromboses and BCS, are needed to develop optimal therapies to prevent or mitigate these deleterious consequences of MPN. Importantly, identifying appropriate management and therapies for young patients with MPNs have the potential to modify their diseases and improve outcomes long-term which will optimally include many decades. While prior studies have begun to illuminate the scope of MPN in younger patients and identify promising therapies, further work will help to ensure that our youngest MPN patients can live long and healthy lives.
Author contributions
LR: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. TJ: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. LR reports: MPN Research Foundation to fund MPN research, R01 HL145780 to fund research investigating molecular mechanisms driving MPN progression, R01 HL143818 to study genetic drivers of clonal hematopoiesis, Maryland Stem Cell Discovery Award 2023-MSCRFD-6151 to investigate hematopoietic stem cell disorders such as MPN, and RALLY Foundation Consortium Award. Children’s Cancer Research Fund, B+ Foundation, American Cancer Society/St. Baldrick’s Foundation, and Alex’s Lemonade Stand Foundation to examine targetable mechanisms underlying leukemogenesis in children. TJ reports: Leukemia Research Foundation, MPN Research Foundation, Gabrielle’s angel Foundation, and Swim Across America foundation funding to support research on targetable mechanisms underlying MPN progression.
Conflict of interest
The authors declare investigator-initiated grant funding administered through Johns Hopkins University from PharmaEssentia for an unrelated project.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dameshek W. Some speculations on the myeloproliferative syndromes. Blood. (1951) 6:372–5. doi: 10.1182/blood-2015-12-686402
2. Pardanani A, Fridley BL, Lasho TL, Gilliland DG, and Tefferi A. Host genetic variation contributes to phenotypic diversity in myeloproliferative disorders. Blood. (2008) 111:2785–9. doi: 10.1182/blood-2007-06-095703
3. Tam CS, Nussenzveig RM, Popat U, Bueso-Ramos CE, Thomas DA, Cortes JA, et al. The natural history and treatment outcome of blast phase BCR-ABL- myeloproliferative neoplasms. Blood. (2008) 112:1628–37. doi: 10.1182/blood-2008-02-138230
4. Skoda RC, Duek A, and Grisouard J. Pathogenesis of myeloproliferative neoplasms. Exp Hematol. (2015) 43:599–608. doi: 10.1016/j.exphem.2015.06.007
5. Lundberg P, Karow A, Nienhold R, Looser R, Hao-Shen H, Nissen I, et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood. (2014) 123:2220–8. doi: 10.1182/blood-2013-11-537167
6. Tefferi A, Guglielmelli P, Larson DR, Finke C, Wassie EA, Pieri L, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood. (2014) 124:2507–13. doi: 10.1182/blood-2014-05-579136
7. Spivak JL, Considine M, Williams DM, Albot CC Jr, Rogers O, Moliterno AR, et al. Two clinical phenotypes in polycythemia vera. N Engl J Med. (2014) 371:808–17. doi: 10.1056/NEJMoa1403141
8. Spivak JL. Myeloproliferative neoplasms. N Engl J Med. (2017) 376:2168–81. doi: 10.1056/NEJMra1406186
9. Grinfeld J, Nangalia J, and Green AR. Molecular determinants of pathogenesis and clinical phenotype in myeloproliferative neoplasms. Haematologica. (2017) 102:7–17. doi: 10.3324/haematol.2014.113845
10. Tefferi A and Vannucchi AM. Genetic risk assessment in myeloproliferative neoplasms. Mayo Clin Proc. (2017) 92:1283–90. doi: 10.1016/j.mayocp.2017.06.002
11. Dunbar AJ, Rampal RK, and Levine R. Leukemia secondary to myeloproliferative neoplasms. Blood. (2020) 136:61–70. doi: 10.1182/blood.2019000943
12. Barbui T, Thiele J, Passamonti F, Rumi E, Boveri E, Ruggeri M, et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: an international study. J Clin Oncol. (2011) 29:3179–84. doi: 10.1200/JCO.2010.34.5298
13. Cervantes F, Tassies D, Salgado C, Rovira M, Pereira A, and Rozman C. Acute transformation in nonleukemic chronic myeloproliferative disorders: actuarial probability and main characteristics in a series of 218 patients. Acta Haematol. (1991) 85:124–7. doi: 10.1159/000204873
14. Tefferi A, Rumi E, Finazzi G, Gisslinger H, Vannucchi AM, Rodeghiero F, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. (2013) 27:1874–81. doi: 10.1038/leu.2013.163
15. Chim CS, Kwong YL, Lie AK, Ma SK, Chan CC, Wong LG, et al. Long-term outcome of 231 patients with essential thrombocythemia: prognostic factors for thrombosis, bleeding, myelofibrosis, and leukemia. Arch Intern Med. (2005) 165:2651–8. doi: 10.1001/archinte.165.22.2651
16. Chamseddine RS, Savenkov O, Rana S, Khalid M, Silver RT, Kucine N, et al. Cytoreductive therapy in younger adults with polycythemia vera: a meta-analysis of safety and outcomes. Blood Adv. (2024) 8:2520–6. doi: 10.1182/bloodadvances.2023012459
17. Cordua S, Kjaer L, Skov V, Pallisgaard N, Hasselbalch HC, and Ellervik C. Prevalence and phenotypes of JAK2 V617F and calreticulin mutations in a Danish general population. Blood. (2019) 134:469–79. doi: 10.1182/blood.2019001113
18. Hinds DA, Barnholt KE, Mesa RA, Kiefer AK, Do CB, Eriksson N, et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood. (2016) 128:1121–8. doi: 10.1182/blood-2015-06-652941
19. Goulart H, Masarova L, Mesa R, Harrison C, Kiladjian JJ, and Pemmaraju N. Myeloproliferative neoplasms in the adolescent and young adult population: A comprehensive review of the literature. Br J Haematol. (2024) 205:48–60. doi: 10.1111/bjh.19557
20. England JT, Szuber N, Sirhan S, Dunne T, Cerquozzi S, Hill M, et al. Clinical features and long-term outcomes of a pan-canadian cohort of adolescents and young adults with myeloproliferative neoplasms: A Canadian MPN group study. Leukemia. (2024) 38:570–8. doi: 10.1038/s41375-024-02155-4
21. Harris Z, Kaizer H, Wei A, Karantanos T, Williams DM, Chaturvedi S, et al. Characterization of myeloproliferative neoplasms in the paediatric and young adult population. Br J Haematol. (2023) 201:449–58. doi: 10.1111/bjh.18650
22. Kucine N, Jessup JA, Cooper TM, Urbanski RW, Kolb EA, and Resar LMS. Position paper: The time for cooperative group study of ropeginterferon alfa-2b in young patients with myeloproliferative neoplasms is now. Pediatr Blood Cancer. (2023) 70:e30559. doi: 10.1002/pbc.30559
23. Sobas M, Kiladjian JJ, Beauvard Y, Curto-Garcia N, Sadjadian P, Shih LY, et al. Real-world study of children and young adults with myeloproliferative neoplasms: identifying risks and unmet needs. Blood Adv. (2022) 6:5171–83. doi: 10.1182/bloodadvances.2022007201
24. Shimano KA, Vanderpoel V, Stone H, Resar L, and Kucine N. Clinical features associated with thrombotic events in children with myeloproliferative neoplasms. Am J Hematol. (2022) 97:E353–5. doi: 10.1002/ajh.26646
25. Kucine N, Bergmann S, Krichevsky S, Jones D, Rytting M, Jain J, et al. Use of pegylated interferon in young patients with polycythemia vera and essential thrombocythemia. Pediatr Blood Cancer. (2021) 68:e28888. doi: 10.1002/pbc.28888
26. Kucine N. Myeloproliferative neoplasms in children, adolescents, and young adults. Curr Hematol Malig Rep. (2020) 15:141–8. doi: 10.1007/s11899-020-00571-8
27. Ianotto JC, Curto-Garcia N, Lauermanova M, Radia D, Kiladjian JJ, and Harrison CN. Characteristics and outcomes of patients with essential thrombocythemia or polycythemia vera diagnosed before 20 years of age: a systematic review. Haematologica. (2019) 104:1580–8. doi: 10.3324/haematol.2018.200832
28. Barzilai M, Kirgner I, Avivi I, Ellis M, Dally N, Rozovski U, et al. Characteristics and outcomes of young adults with Philadelphia-negative myeloproliferative neoplasms. Eur J Haematol. (2019) 102:504–8. doi: 10.1111/ejh.13232
29. Szuber N, Vallapureddy RR, Penna D, Lasho TL, Finke C, Hanson CA, et al. Myeloproliferative neoplasms in the young: Mayo Clinic experience with 361 patients age 40 years or younger. Am J Hematol. (2018) 93:1474–84. doi: 10.1002/ajh.25270
30. Boddu P, Masarova L, Verstovsek S, Strati P, Kantarjian H, Cortes J, et al. Patient characteristics and outcomes in adolescents and young adults with classical Philadelphia chromosome-negative myeloproliferative neoplasms. Ann Hematol. (2018) 97:109–12. doi: 10.1007/s00277-017-3165-9
31. Stein BL, Saraf S, Sobol U, Halpern A, Shammo J, Rondelli D, et al. Age-related differences in disease characteristics and clinical outcomes in polycythemia vera. Leuk Lymph. (2013) 54:1989–95. doi: 10.3109/10428194.2012.759656
32. Barbui T. How to manage children and young adults with myeloproliferative neoplasms. Leukemia. (2012) 26:1452–7. doi: 10.1038/leu.2012.12
33. Williams N, Lee J, Mitchell E, Moore L, Baxter EJ, Hewinson J, et al. Life histories of myeloproliferative neoplasms inferred from phylogenies. Nature. (2022) 602:162–8. doi: 10.1038/s41586-021-04312-6
34. Sousos N, Ní Leathlobhair M, Simoglou Karali C, Louka E, Bienz N, Royston D, et al. In utero origin of myelofibrosis presenting in adult monozygotic twins. Nat Med. (2022) 28:1207–11. doi: 10.1038/s41591-022-01793-4
35. Moliterno AR, Ginzburg YZ, and Hoffman R. Clinical insights into the origins of thrombosis in myeloproliferative neoplasms. Blood. (2021) 137:1145–53. doi: 10.1182/blood.2020008043
36. Garcia-Pagán JC and Valla DC. Primary budd-chiari syndrome. N Engl J Med. (2023) 388:1307–16. doi: 10.1056/NEJMra2207738
37. Harrison CN, Nangalia J, Boucher R, Jackson A, Yap C, O’Sullivan J, et al. Ruxolitinib versus best available therapy for polycythemia vera intolerant or resistant to hydroxycarbamide in a randomized trial. J Clin Oncol. (2023) 41:3534–44. doi: 10.1200/JCO.22.01935
38. Knudsen TA, Skov V, Stevenson K, Werner L, Duke W, Laurore C, et al. Genomic profiling of a randomized trial of interferon-α vs hydroxyurea in MPN reveals mutation-specific responses. Blood Adv. (2022) 6:2107–19. doi: 10.1182/bloodadvances.2021004856
39. Kiladjian JJ, Cassinat B, Turlure P, Cambier N, Roussel M, Bellucci S, et al. High molecular response rate of polycythemia vera patients treated with pegylated interferon alpha-2a. Blood. (2006) 108:2037–40. doi: 10.1182/blood-2006-03-009860
40. Silver RT, Kiladjian JJ, and Hasselbalch HC. Interferon and the treatment of polycythemia vera, essential thrombocythemia and myelofibrosis. Expert Rev Hematol. (2013) 6:49–58. doi: 10.1586/ehm.12.69
41. Gisslinger H, Klade C, Georgiev P, Krochmalczyk D, Gercheva-Kyuchukova L, Egyed M, et al. Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): a randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol. (2020) 7:e196–208. doi: 10.1016/S2352-3026(19)30236-4
42. Abu-Zeinah G, Krichevsky S, Cruz T, Hoberman G, Jaber D, Savage N, et al. Interferon-alpha for treating polycythemia vera yields improved myelofibrosis-free and overall survival. Leukemia. (2021) 35:2592–601. doi: 10.1038/s41375-021-01183-8
43. Kiladjian JJ, Klade C, Georgiev P, Krochmalczyk D, Gercheva-Kyuchukova L, Egyed M, et al. Long-term outcomes of polycythemia vera patients treated with ropeginterferon Alfa-2b. Leukemia. (2022) 36:1408–11. doi: 10.1038/s41375-022-01528-x
44. Abu-Zeinah G, Silver RT, Abu-Zeinah K, and Scandura JM. Normal life expectancy for polycythemia vera (PV) patients is possible. Leukemia. (2022) 36:569–72. doi: 10.1038/s41375-021-01447-3
45. De Oliveira DR, Soret-Dulphy J, Zhao LP, Marcault C, Gauthier N, Verger E, et al. Interferon-alpha (IFN) therapy discontinuation is feasible in myeloproliferative neoplasm (MPN) patients with complete hematological remission. Blood. (2020) 136:35–6. doi: 10.1182/blood-2020-141223
46. Mullally A, Bruedigam C, Poveromo L, Heidel FH, Purdon A, Vu T, et al. Depletion of Jak2V617F myeloproliferative neoplasm-propagating stem cells by interferon-α in a murine model of polycythemia vera. Blood. (2013) 121:3692–702. doi: 10.1182/blood-2012-05-432989
47. Verger E, Soret-Dulphy J, Maslah N, et al. Ropeginterferon alpha-2b targets JAK2V617F-positive polycythemia vera cells in vitro and in vivo. Blood Cancer J. (2018) 8:94. doi: 10.1038/s41408-018-0133-0
48. Austin RJ, Straube J, Bruedigam C, Pali G, Jacquelin S, Vu T, et al. Distinct effects of ruxolitinib and interferon-alpha on murine JAK2V617F myeloproliferative neoplasm hematopoietic stem cell populations. Leukemia. (2020) 34:1075–89. doi: 10.1038/s41375-019-0638-y
49. Rao TN, Hansen N, Stetka J, Luque Paz D, Kalmer M, Hilfiker J, et al. JAK2-V617F and interferon-α induce megakaryocyte-biased stem cells characterized by decreased long-term functionality. Blood. (2021) 137:2139–51. doi: 10.1182/blood.2020005563
50. Maze D, Kazi S, Gupta V, Malinowski AK, Fazelzad R, Shah PS, et al. Association of treatments for myeloproliferative neoplasms during pregnancy with birth rates and maternal outcomes: A systematic review and meta-analysis. JAMA Netw Open. (2019) 2:e1912666. doi: 10.1001/jamanetworkopen.2019.12666
51. Sowell E, Peterson B, Thompson P, Welcome SE, Henkenius AL, Toga AW, et al. Mapping cortical change across the human life span. Nat Neurosci. (2003) 6:309–15. doi: 10.1038/nn1008
52. Barbui T, Vannucchi AM, De Stefano V, Masciulli A, Carobbio A, Ferrari A, et al. Ropeginterferon alfa-2b versus phlebotomy in low-risk patients with polycythaemia vera (Low-PV study): a multicentre, randomised phase 2 trial. Lancet Haematol. (2021) 8(3):e175–84. doi: 10.1016/S2352-3026(20)30373-2
53. Yan B, Ianotto J-C, Thaw KH, Sobas M, Sadjadian P, Curto-Garcia N, et al. Impact of cytoreductive drugs upon outcomes in a contemporary cohort of adolescent and young adults with essential thrombocythemia and polycythemia vera. Blood. (2023) 142:748. doi: 10.1182/blood-2023-185108
54. Gowin K, Ballen K, Ahn KW, Hu ZH, Ali H, Arcasoy MO, et al. Survival following allogeneic transplant in patients with myelofibrosis. Blood Adv. (2020) 4:1965–73. doi: 10.1182/bloodadvances.2019001084
55. Jain T, Estrada-Merly N, Salas MQ, Kim S, DeVos J, Chen M, et al. Donor types and outcomes of transplantation in myelofibrosis: a CIBMTR study. Blood Adv. (2024) 8:4281–93. doi: 10.1182/bloodadvances.2024013451
56. Kröger N, Bacigalupo A, Barbui T, Ditschkowski M, Gagelmann N, Griesshammer M, et al. Indication and management of allogeneic haematopoietic stem-cell transplantation in myelofibrosis: updated recommendations by the EBMT/ELN International Working Group. Lancet Haematol. (2024) 11:e62–74. doi: 10.1016/S2352-3026(23)00305-8
57. Gangat N, Caramazza D, Vaidya R, George G, Begna K, Schwager S, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. (2011) 29:392–7. doi: 10.1200/JCO.2010.32.2446
58. Passamonti F, Cervantes F, Vannucchi AM, Morra E, Rumi E, Cazzola M, et al. Dynamic International Prognostic Scoring System (DIPSS) predicts progression to acute myeloid leukemia in primary myelofibrosis. Blood. (2010) 116:2857–8. doi: 10.1182/blood-2010-06-293415
59. Vannucchi AM, Lasho TL, Guglielmelli P, Biamonte F, Pardanani A, Pereira A, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. (2013) 27:1861–9. doi: 10.1038/leu.2013.119
60. Tefferi A, Finke CM, Lasho TL, Hanson CA, Ketterling RP, Gangat N, et al. U2AF1 mutation types in primary myelofibrosis: phenotypic and prognostic distinctions. Leukemia. (2018) 32:2274–8. doi: 10.1038/s41375-018-0078-0
61. Bender JD, Oquendo-Del Toro H, Benoit J, Howell JC, Badia P, Davies SM, et al. Reduced-intensity conditioning mitigates risk for primary ovarian insufficiency but does not decrease risk for infertility in pediatric and young adult survivors of hematopoietic stem cell transplantation. Transplant Cell Ther. (2023) 29:130.e1–8. doi: 10.1016/j.jtct.2022.10.018
62. Wachowiak J, Galimard JE, Dalissier A, Rihani R, AlSaedi H, Wynn RF, et al. Outcomes of allogeneic haematopoietic cell transplantation for myelofibrosis in children and adolescents: the retrospective study of the EBMT Paediatric Diseases WP. Bone Marrow Transpl. (2024) 59:1057–69. doi: 10.1038/s41409-024-02286-3
63. Rotz SJ, Hamilton BK, Wei W, Ahmed I, Winston SA, Ballard S, et al. Fertility potential and gonadal function in survivors of reduced-intensity hematopoietic stem cell transplantation. Transplant Cell Ther. (2024) 30:534.e1–534.e13. doi: 10.1016/j.jtct.2024.02.002
64. Chow EJ, Anderson L, Baker KS, Bhatia S, Guilcher GM, Huang JT, et al. Late effects surveillance recommendations among survivors of childhood hematopoietic cell transplantation: A children’s oncology group report. Biol Blood Marrow Transpl. (2016) 22:782–95. doi: 10.1016/j.bbmt.2016.01.023
65. Shenoy S, Gaziev J, Angelucci E, King A, Bhatia M, Smith A, et al. Late Effects Screening Guidelines after Hematopoietic Cell Transplantation (HCT) for Hemoglobinopathy: Consensus Statement from the Second Pediatric Blood and Marrow Transplant Consortium International Conference on Late Effects after Pediatric HCT. Biol Blood Marrow Transpl. (2018) 24:1313–21. doi: 10.1016/j.bbmt.2018.04.002
Keywords: MPN, myeloproliferative neoplasm, children, adolescents and young adults, therapy, Thrombosis in young MPN patients, MPN in young patients
Citation: Resar L and Jain T (2025) Not just for mature audiences: myeloproliferative neoplasms in children, adolescents, and young adults. Front. Hematol. 3:1483322. doi: 10.3389/frhem.2024.1483322
Received: 19 August 2024; Accepted: 05 November 2024;
Published: 13 May 2025.
Edited by:
Hans Carl Hasselbalch, Zealand University Hospital, DenmarkReviewed by:
Danijela Lekovic, University of Belgrade, SerbiaGhaith Abu-Zeinah, New York-Presbyterian, United States
Copyright © 2025 Resar and Jain. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linda Resar, bHJlc2FyQGpobWkuZWR1