 Rajvardhan Ravindra Patil
Rajvardhan Ravindra Patil Sourya Acharya
Sourya Acharya Vineet Karwa
Vineet Karwa- Department of Internal Medicine, Jawaharlal Nehru Medical College, Datta Meghe Institute Of Higher Education and Research, Wardha, India
Sickle cell disease is a prevalent haemoglobinopathy that is hereditary. People with sickle cell disease frequently experience vaso-occlusive crises, which lead to substantial morbidity from end-organ ischaemia and infarction. Numerous organs, including the bones, brain, lungs, spleen, retina, and penis, may be affected. As a result, symptoms may include painful bone crises, cerebrovascular accidents, acute chest syndrome, sequestration crises, retinal haemorrhage, and priapism. In sickle cell disease, subperiosteal orbital haemorrhage is a rare vaso-occlusive crisis (VOC). Roth spots (white-centred, flame-shaped retinal haemorrhages), occurring commonly in subacute bacterial endocarditis, can also be seen in leukaemia, pre-eclampsia, and hypertensive retinopathy. A 19-year-old male patient with sickle cell disease (HbSS) presented to the hospital with ptosis and swelling in his right periorbital region 3 days after being brought to the hospital for a sickle cell crisis. It was later determined to be an acute subperiosteal orbital hematoma along with Roth spots, an uncommon clinical manifestation of sickle cell disease, only after a thorough clinical and radiological assessment.
Introduction
Sickle cell disease is one of the most prevalent hereditary autosomal recessive monogenic haematological disorders that affect red blood cells (1–3). The gene-coding defect produces rigid, sticky, crescent-shaped red blood cells that cannot pass through the capillary bed, leading to ischaemia–reperfusion damage in organs (4, 5).
Vaso-occlusive crises are the predominant feature of sickle cell disease. All organ systems are impacted by vaso-occlusive crises, causing end-organ ischaemia and infarction, leading to high morbidity and early mortality. A diagnostic conundrum arises because the clinical presentation of the end-organ vaso-occlusive crisis may resemble that of several other clinical diseases. Splenic infarction (6), pulmonary involvement, acute chest syndrome (7), and orbital compression syndrome (8, 9) are among the recent instances of end-organ ischaemia.
Roth spots, characterised by white-centred retinal haemorrhages, can be caused by several factors. Still, the most common theory links them to retinal capillary rupture brought on by endothelial cell dysfunction. After the vessel ruptures, red blood cells spill out, causing fibrin–platelet plugs to develop on the injured endothelial cell lining and starting the clotting chain reaction. Sickle cell anaemia is a relatively rare but significant disorder among the many others linked to Roth spots (10, 11).
Orbital involvement in sickle cell disease is rare, but when it occurs, it can be made worse by orbital compression syndrome, which occurs when the bone marrow’s microvasculature is infarcted, leading to inflammation and necrosis, ultimately resulting in subperiosteal haemorrhage (12).
The rupture of diploic veins between the periosteum (periorbita) and the bony orbit results in subperiosteal orbital haemorrhage. A local haemorrhage from a rupture creates a haematoma between the bone and the periorbita. The periorbita peels away from the bone as a result of the pressure being raised by the blood, taking up space. Blood will not enter the orbit if the periorbita is intact, and imaging will show that the haematoma will have a bilenticular shape. If left untreated, a subperiosteal orbital haematoma can expand over time or resolve spontaneously. Generally, it is treated medically, but in cases of optic nerve dysfunction or extensive haematomas, it is treated surgically (13).
Case presentation
A 19 year old male patient with a known case of sickle cell disease (HbSS) rushed to the hospitals emergency room with a high grade fever that had been present for 3 days. The fever was insidious in onset, progressed over time, and was not resolved by medications. He also complained of pain and swelling over his right eyelid associated with frontal and occipital headaches.
On admission, the patient appeared pale, drowsy, and sick. The patient’s axillary temperature was 38.2°C, his heart rate was 115 beats per minute with regular rhythm and normal volume with no special character, and he had abdomino-thoracic breathing with a respiratory rate of 25 cycles per minute. His blood pressure was 100/60 mm Hg in the right brachial artery in the supine position, and his oxygen saturation was 96% on room air. The patient’s weight was 45 kg.
Upon general examination, the patient showed evident pallor without any icterus, cyanosis, clubbing, lymphadenopathy, or oedema. The ocular examination revealed peri-ocular soft tissue enlargement, right eyelid oedema, and restriction in the right eye’s elevation.
Using the colour plates, colour vision was assessed and found to be normal. The pupils were equal in size and were reactive to light. Both pupils reacted normally to the swinging light reflex. Afferent pupillary light reflex deficit (APD) was absent. Both eyes’ intraocular pressures were within normal limits. Fundus examination revealed round flame-shaped haemorrhages with a white/pale centre suggestive of Roth spots (Figure 1).
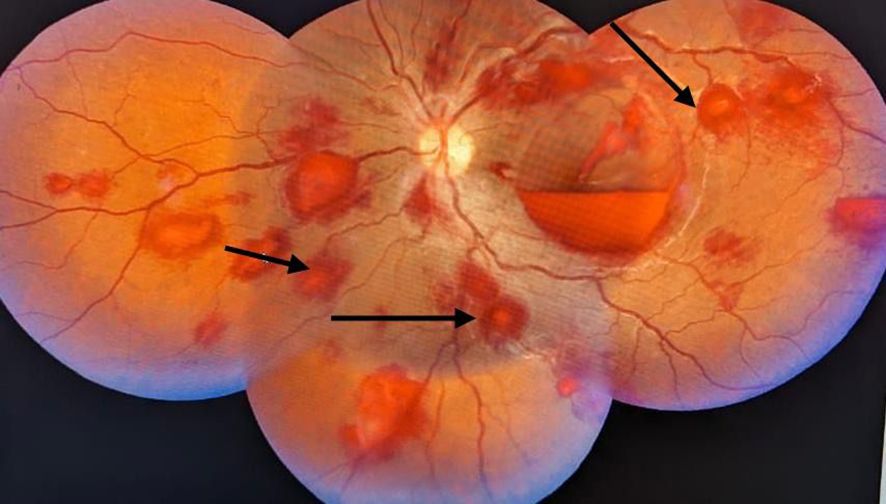
Figure 1. A fundus image of a right eye with round flame-shaped haemorrhages and a white/pale centre suggestive of Roth spots.
No apparent abnormalities were found after a thorough, comprehensive assessment of the cardiovascular, respiratory, central nervous, and gastrointestinal systems. The following are the results of routine and special laboratory investigations (Table 1). The blood culture results were negative, and the urine analysis was normal.
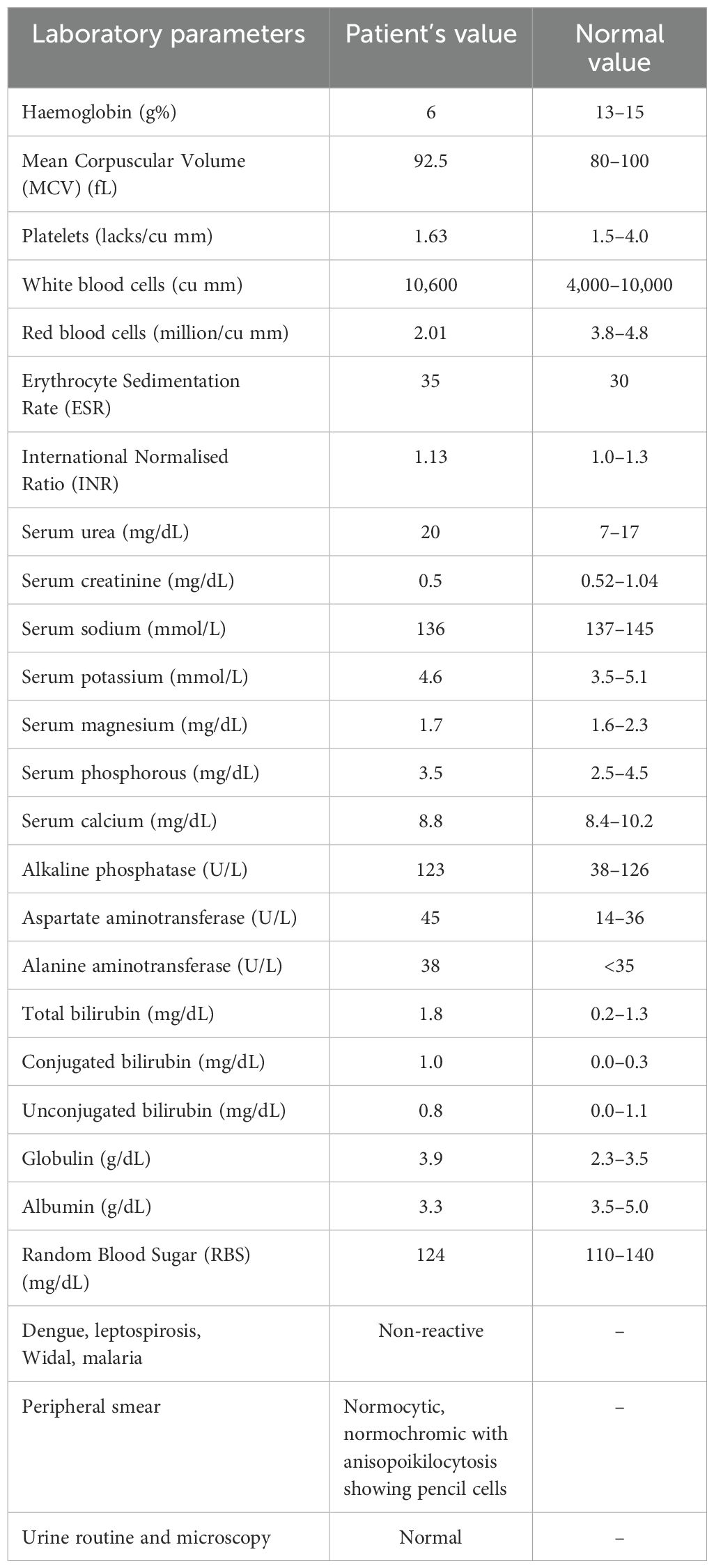
Table 1. Table showing laboratory parameters of the patient and the normal laboratory values.
Plain MRI and contrast MRI of the right orbit were conducted, showing well-defined elliptical altered intensity lesions noted in the extraconal space of the superior quadrant and appearing heterogeneously hyperintense on T1 Weighted Image (T1WI) (Figure 2) and hyperintense on T2 Weighted Image (T2WI)/Fluid Attenuated Inversion Recovery (FLAIR), causing mass effect over the superior oblique and superior rectus muscles, suggestive of subperiosteal orbital hematoma. A contrast MRI of the brain was conducted, revealing the heterogeneous signal intensity of the calvarium, suggestive of bone infarcts with subperiosteal collection involving the bilateral frontal and parietal bones, appearing hyperintense on T1WI and heterogeneously hyperintense on T2WI (Figure 3).
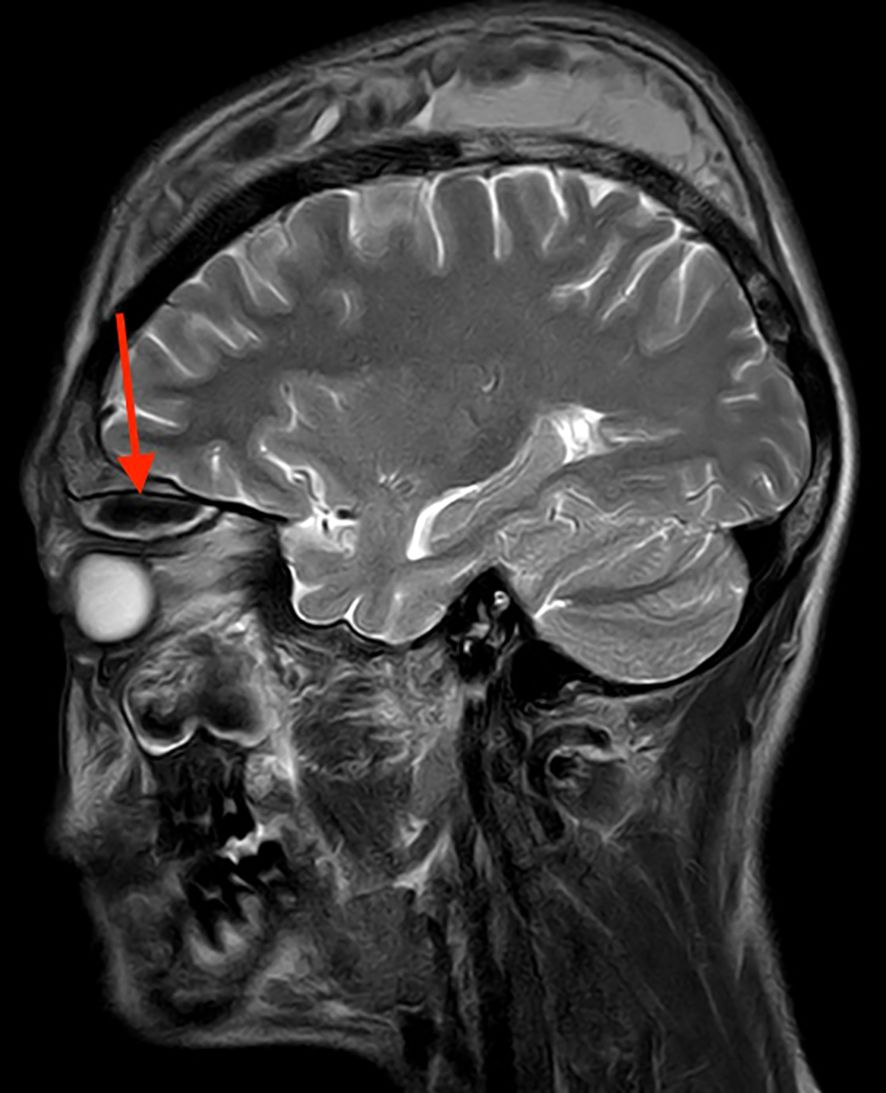
Figure 2. MRI orbit (plain) T2WI in sagittal view having well-defined peripherally enhancing elliptical altered signal intensity lesion measuring 27 × 10 mm in extraconal space of superior quadrant of the right orbit, appearing hyperintense, causing mass effect over superior oblique and superior rectus muscles.

Figure 3. MRI brain (contrast) T1WI in coronal view with the heterogeneous signal intensity of the calvarium, suggestive of bone infarcts with subperiosteal collection involving bilateral frontal and parietal bones measuring approximately 7.8 × 4.4 × 1.4 cm on the right side and 4.4 × 1.3 × 6.4 cm on the left side, appearing hyperintense.
The patient was promptly given injectable methylprednisolone pulse therapy, intravenous fluids, analgesics, wide-spectrum antibiotics, hydroxyurea, and two units of packed red blood cell transfusions. Following a favourable response to medical treatment and full recovery, the patient was discharged once his condition was stabilised.
Discussion
Ocular manifestations of sickle cell disease have been reported; when these manifestations are interconnected to the retina, they are called sickle cell retinopathy. Sickled red blood cells cause vaso-occlusion of the retinal microvasculature, which results in ischaemia and subsequent retinal tissue destruction. This is the pathogenesis of sickle cell retinopathy. Clinical manifestations of sickle cell retinopathies are often neovascularisation, cotton-wool spots, macular ischaemia, peripheral retinal vascular occlusion, and retinal haemorrhages (14).
Orbital bone infarction, followed by an inflammatory reaction that can quickly spread to the orbit and cause orbital pain and proptosis, is the primary mechanism for developing orbital compression syndrome (9), an acute illness. The hallmark features of orbital compression syndrome (OCS) include eyelid oedema, proptosis, periorbital pain, and restriction of extra-ocular movement, with or without reduced visual acuity. The development of haematomas, which can be intracranial (epidural) or orbital (sub-periosteal), is a distinctive characteristic of orbital wall infarction in SCD (15). Subperiosteal haemorrhage has been attributed to several processes, including mild trauma, underlying bleeding diathesis, and blood extravasation from necrotised vessel walls (16). In this instance, orbital infarction was thought to be the aetiology of the orbital haematoma, as evidenced by aberrant frontal bone and bone marrow heterogeneous signals adjacent to the hematoma formation.
Since children’s orbital bones have greater marrow space than adults’, orbital wall infarction usually affects young patients (16). The youngest recorded age was 2 years, while the average age of presentation in the instances that were documented was 13 years. Nearly all cases had a fever as a presenting symptom, and over two-thirds of the patients had associated pain crises elsewhere (17).
Intravenous corticosteroid administration may alleviate orbital pressure brought on by the inflammatory component of orbital bone infarction. Since osteomyelitis and bone infarction are sometimes difficult to distinguish clinically, concurrent antibiotic coverage is advised. If there are signs of optic nerve dysfunction, surgical exploration and haematoma evacuation are necessary to avoid vision loss and ensure prompt recovery. Approximately one-fifth of the patients reported needing surgical procedures, whereas the majority of patients recovered with medical therapy (15). Orbital infarction is a rare manifestation of sickle cell disease, which may also be considered a vaso-occlusive crisis. The management is the same as for other crises: potent analgesics, cautious yet vigorous hydration, and a comprehensive assessment of the underlying cause (18).
Conclusion
Sickle cell disease is known to have ocular manifestations, known as sickle cell retinopathy, which includes neovascularisation, ischaemia, and occlusion of retinal vessels. However, Roth spots are an uncommon finding in sickle cell disease, which indicates complex ocular manifestations. Orbital infarction leading to subperiosteal orbital hematoma is another very rare vaso-occlusive crisis of sickle cell disease. Therefore, any patient with a known sickle cell disease presenting to the hospital with complaints of unilateral orbital swelling should be further evaluated with magnetic resonance imaging to rule out orbital hematoma and fundus examination to look for Roth spots. Careful assessment, diagnosis, and the timely initiation of the proper supportive treatment are highly recommended to avoid irreversible vision loss. The majority of ocular involvement cases do not need specialised treatment; instead, they respond well to conservative treatment, including adequate hydration, analgesics, and hydroxyurea.
In conclusion, even though they are uncommon, orbital infarction, hematoma, and retinal haemorrhage should be suspected in all sickle cell disease individuals who present with eyelid swelling.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Datta Meghe Institute of Higher Education and Research Institutional Ethics Committee. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
RP: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. SA: Formal Analysis, Supervision, Writing – review & editing. VK: Formal Analysis, Writing – review & editing. RD: Formal Analysis, Writing – review & editing. SS: Formal Analysis, Writing – review & editing. MK: Formal Analysis, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bello-Manga H, DeBaun MR, and Kassim AA. Epidemiology and treatment of relative anemia in children with sickle cell disease in sub-Saharan Africa. Expert Rev Hematol. (2016) 9:1031–42. doi: 10.1080/17474086.2016.1240612
2. Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, et al. Sickle cell disease. Nat Rev Dis Primers. (2018) 4:18010. doi: 10.1038/nrdp.2018.10
3. Rees DC, Williams TN, and Gladwin MT. Sickle-cell disease. Lancet. (2010) 11:2018–31. doi: 10.1016/S0140-6736(10)61029-X
4. Inusa BP, Hsu LL, Kohli N, Patel A, Ominu-Evbota K, Anie KA, et al. Sickle cell disease-genetics, pathophysiology, clinical presentation and treatment. Int J Neonatal Screen. (2019) 5. doi: 10.3390/ijns5020020
5. Panepinto JA, Brandow A, Mucalo L, Yusuf F, Singh A, Taylor B, et al. Coronavirus disease among persons with sickle cell disease, United States, March 20-May 21, 2020. Emerg Infect Dis. (2020) 26(10):2473–6. doi: 10.3201/eid2610.202792
6. Frisancho OE and Ichiyanagui RC. Spleen infarction and S hemoglobinopathies S in the high altitude lands. Rev Gastroenterol Peru. (2012) 32(1):68–78.
7. Pincez T, Calamy L, Germont Z, Lemoine A, Lopes AA, Massiot A, et al. Pulmonary complications of sickle cell disease in children. Arch Pediatr. (2016) 23:1094–106. doi: 10.1016/j.arcped.2016.06.014
8. Emerson GG and Lutty GA. Effects of sickle cell disease on the eye: clinical features and treatment. Hematol Oncol Clin North Am. (2005) 19:957–73. doi: 10.1016/j.hoc.2005.07.005
9. Sokol JA, Baron E, Lantos G, and Kazim M. Orbital compression syndrome in sickle cell disease. Ophthal Plast Reconstr Surg. (2008) 24:181–4. doi: 10.1097/iop.0b013e31816b960e
10. Shah D, Reddy H, Kumar S, and Acharya S. Sickle cell disease presenting as extradural hematoma: an extremely rare fatal crisis. Cureus. (2022) 14:e27004. doi: 10.7759/cureus.27004
11. Piel FB, Patil AP, Howes RE, et al. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun. (2010) 1:104. doi: 10.1038/ncomms1104
12. Khawaji A and Alsini H. Acute orbitopathy manifesting as periorbital cellulitis in sickle cell disease. Case Rep Clin Med. (2020) 9:137–43. doi: 10.4236/crcm.2020.96020
13. Available online at: https://eyewiki.org/w/index.php?title=Subperiosteal_Hematoma&action=edit (Accessed December 17, 2024).
14. Kothari M, Chiwhane A, Kumar S, Wanjari A, and Reddy H. Roth spots: A rare finding in sickle cell anemia. Cureus. (2024) 16:e59047. doi: 10.7759/cureus.59047
15. Ghafouri RH, Lee I, Freitag SK, and Pira TN. Bilateral orbital bone infarction in sickle-cell disease. Ophthal Plast Reconstr Surg. (2011) 27:26–7. doi: 10.1097/iop.0b013e3181c70b65
16. Janssens C, Claeys L, Maes P, Boiy T, and Wojciechowski M. Orbital wall infarction in child with sickle cell disease. Acta Clin Belg. (2015) 70:451–2. doi: 10.1179/2295333715y.0000000053
17. Alghamdi A. Recurrent orbital bone sub-periosteal hematoma in sickle cell disease: a case study. BMC Ophthalmol. (2018) 18. doi: 10.1186/s12886-018-0884-1
Keywords: sickle cell disease, vaso-occlusive crisis, subperiosteal orbital hematoma, Roth spots, magnetic resonance imaging
Citation: Patil RR, Acharya S, Karwa V, Dhondge R, Shaikh S and Kothari M (2025) Case Report: Expanding the ocular horizon: subperiosteal orbital hematoma and Roth spots in known case of sickle cell disease. Front. Hematol. 4:1558509. doi: 10.3389/frhem.2025.1558509
Received: 10 January 2025; Accepted: 25 April 2025;
Published: 16 June 2025.
Edited by:
Arturo J. Martí-Carvajal, Universidad de Carabobo, VenezuelaReviewed by:
Anna Rita Migliaccio, Campus Bio-Medico University, ItalyRamesh C. Nayak, University of Cincinnati, United States
Copyright © 2025 Patil, Acharya, Karwa, Dhondge, Shaikh and Kothari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajvardhan Ravindra Patil, bWUucmFqcGF0aWwxOTk3QGdtYWlsLmNvbQ==