 Wei Li1*
Wei Li1* Andrew Q. Pucka2Lina Houran2Xiaoqing Huang3
Andrew Q. Pucka2Lina Houran2Xiaoqing Huang3 Candice Debats2
Candice Debats2 Brandon A. Reyes2Andrew R. W. O’Brien4Qigui Yu1*
Brandon A. Reyes2Andrew R. W. O’Brien4Qigui Yu1* Ying Wang2,4*
Ying Wang2,4*- 1Department of Microbiology and Immunology, Indiana University School of Medicine, Indianapolis, IN, United States
- 2Department of Anesthesia, Stark Neurosciences Research Institute, Indiana University School of Medicine, Indianapolis, IN, United States
- 3Department of Biostatistics and Health Data Science, Indiana University School of Medicine, Indianapolis, IN, United States
- 4Division of Hematology/Oncology, Department of Medicine, Indiana University School of Medicine, Indianapolis, IN, United States
Background: Sickle cell disease (SCD) is a chronic condition characterized by inflammation, immune dysregulation, and debilitating pain.
Aim: This study investigates soluble immune checkpoints (sICPs) and their associations with inflammatory mediators, immune cell profiles, autoantibodies, and clinical outcomes in SCD.
Method: Peripheral blood samples from 50 SCD patients and 40 demographic-matched healthy controls (HCs) were analyzed for 37 sICPs, 80 inflammatory mediators, and 18 autoantibodies using multiplex assays, alongside immune cell profiles via flow cytometry. Pain and quality of life (QoL) were assessed through patient-reported outcome measures (PROMs).
Results: Twenty-three sICPs, including arginase-1, BTLA, CD27, CD28, CD47, CD80, CD96, CD134, CD137, CD152, GITR, HVEM, IDO, LAG-3, MICA, MICB, Nectin-2, PD-1, Siglec-7, Siglec-9, TIM-3, TIMD-4, and VISTA, were significantly elevated in SCD patients compared to HCs. These sICPs correlated with multiple proinflammatory mediators (e.g., IL-18), autoantibodies (e.g., MPO), and immune cell activation markers (e.g., CD38/HLA-DR on CD8 T cells). Notably, CD28, CD152, HVEM, and VISTA were strongly associated with systemic inflammation and immune cell activation, while BTLA, LAG-3, PD-1, and CD80 correlated with pain and anxiety scores and QoL.
Conclusion: This study highlights complex interactions between sICPs, immune activation, inflammation, and clinical outcomes in SCD, underscoring their potential as biomarkers or therapeutic targets to alleviate inflammation and improve QoL in this challenging clinical population.
1 Introduction
Sickle cell disease (SCD) is a complex, lifelong condition characterized by systemic involvement and significant complications, including acute and chronic pain, anemia, stroke, pulmonary hypertension, and progressive organ damage (1, 2). Clinically, SCD alternates between two distinct states: the steady-state (StSt) phase, often presenting with mild symptoms related to chronic hemolysis and ongoing discomfort, and the acute vaso-occlusive crisis (VOC) phase, marked by severe pain episodes and potentially life-threatening complications such as acute chest syndrome and stroke due to blood vessel blockages by sickle-shaped red blood cells (3–5). Central to the pathology of SCD is a state of chronic inflammation and immune dysregulation, which underpins the disease’s clinical manifestations and contributes to its progression.
Chronic inflammation in SCD patients is driven by various mechanisms, including hemolysis, ischemia-reperfusion injury, oxidative stress, and immune dysregulation. Hemolysis releases hemoglobin and heme into circulation, triggering the activation of immune cells through NF-κB and TLR4 pathways, resulting in elevated levels of inflammatory cytokines such as IL-1β, IL-6, and TNF-α (6–8). Additionally, the ischemic and hypoxic conditions caused by vascular occlusion activate endothelial cells, promoting the expression of adhesion molecules and the recruitment of leukocytes, which further amplify the inflammatory cascade (9, 10). Autoimmune processes also play a role, with the production of autoantibodies driven by aberrant B-cell activation contributing to immune complex formation and tissue damage (11, 12). These sustained inflammatory responses are compounded by the oxidative stress inherent to SCD, further exacerbating endothelial dysfunction and immune cell activation.
Despite advances in understanding SCD pathophysiology, gaps remain in identifying the full spectrum of immune regulatory mechanisms involved in the disease. Recent advances have identified soluble immune checkpoints (sICPs) as critical modulators of immune regulation and inflammation (13, 14). Soluble forms of immune checkpoint (ICP) molecules, generated via protease-mediated shedding or alternative mRNA splicing, act as circulating regulators of immune responses. They play diverse roles in maintaining immune homeostasis, interacting with inflammatory pathways, and acting as biomarkers of disease severity or therapeutic response. While sICPs have been recently studied in conditions such as cancer, chronic viral infections, and autoimmune diseases, their role in SCD remains unexplored.
Given that chronic inflammation and immune dysregulation are prominent in SCD patients, studying sICPs could provide valuable insights into the disease’s inflammation and immune landscape. These molecules may represent novel biomarkers of disease severity or therapeutic targets, offering a new avenue for understanding and managing SCD. This study seeks to address this gap by utilizing baseline clinical samples from our clinical trial (NCT05045820) to investigate the levels and roles of sICPs in SCD for the first time. Alongside profiling inflammatory mediators, autoantibodies, and immune cell profiles, we aim to elucidate the associations between sICPs, inflammation, immune dysregulation, and clinical outcomes, including VOCs, pain severity, and quality of life (QoL) measures. By integrating this analysis, we provide insights into mechanisms underlying SCD pathophysiology and identify potential targets for innovative therapeutic interventions.
2 Materials and methods
2.1 Study participants
Eligible participants aged 14 to 80 were individuals diagnosed with SCD who experienced chronic pain within the last 6 months and/or at least one VOC in the past 12 months. Participants also met the following inclusion criteria: no recent changes in stimulant medication, willingness to continue ongoing treatments, and agreement to avoid new medications or pain management methods during the study. Major exclusion criteria included confirmed or suspected COVID-19, recent or ongoing acupuncture for pain management (within the last 6 months), presence of autoimmune or inflammatory diseases, and blood transfusion within 90 days before enrollment. Age-, gender-, and ethnicity-matched healthy controls (HCs) without SCD were also recruited. This study is part of our ongoing randomized clinical trial in SCD, and comprehensive criteria are available at ClinicalTrials.gov (NCT05045820).
Peripheral blood was collected from 50 SCD participants at steady-state (StSt) (58% female; median age: 31 years) and 40 HCs (60% female; median age: 33.5 years). All participants were Black/African American. Blood samples were collected between July 2021 and June 2024 from Indiana University Health hospitals, the Indiana Hemophilia & Thrombosis Center, community hospitals, and other external medical facilities. Plasma was separated and stored at -80°C, while peripheral blood mononuclear cells (PBMCs) were cryopreserved in liquid nitrogen or used directly. Detailed demographic, clinical, and laboratory characteristics are summarized in Table 1.
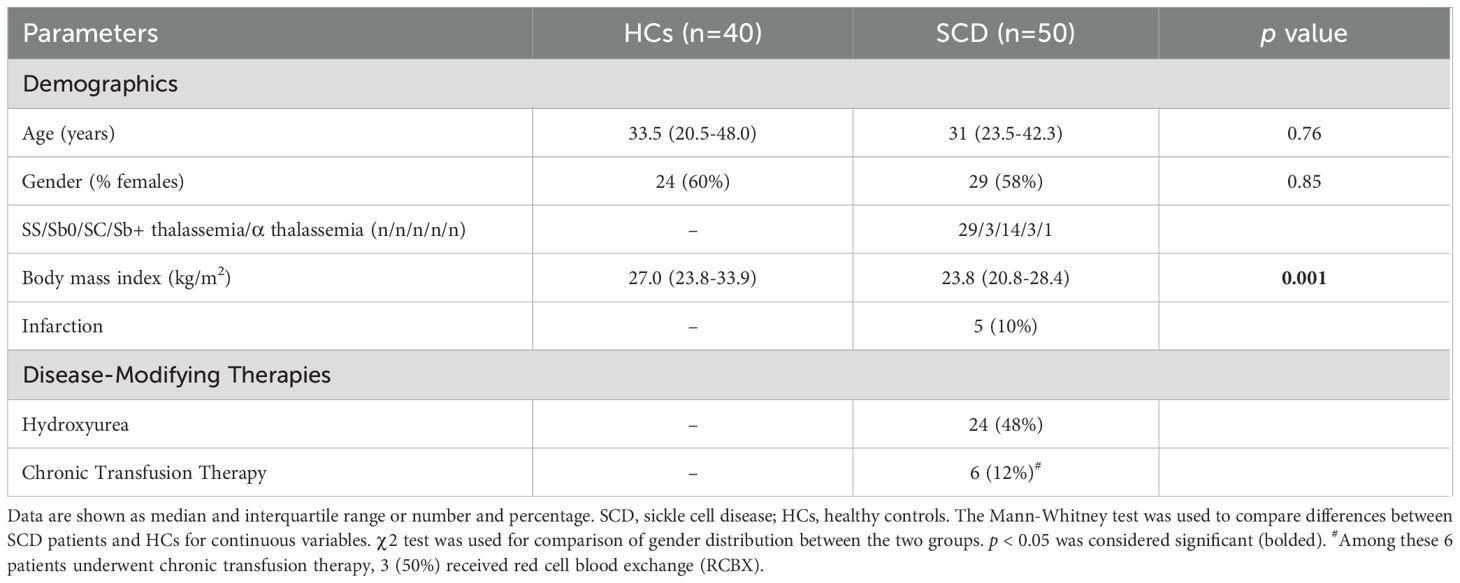
Table 1. Demographics and clinical characteristics of SCD patients.
2.2 Patient-reported outcome measures
PROMs were collected using validated tools as used in our previous published work (15). The PainDETECT Questionnaire assessed neuropathic pain (higher scores indicated higher pain) (16, 17), while PROMIS-29 assessed pain intensity, interference, and physical dysfunction (18). The recency and frequency of VOCs were assessed by the Adult Sickle Cell Quality-of-Life Measurement Information System (ASCQ-Me) (19). Depression and anxiety were evaluated with the Hospital Anxiety and Depression Scale (HADS), and pain-related QoL was measured using the Pediatric Quality of Life Inventory (PedsQL, both peds and adult versions) (20). The Fibromyalgia Survey Questionnaire (FSQ), which includes the Widespread Pain Index, was used as a surrogate measure of nociplastic pain (21). Results from these assessments, including significant differences between SCD and HC groups, are presented in Table 2.
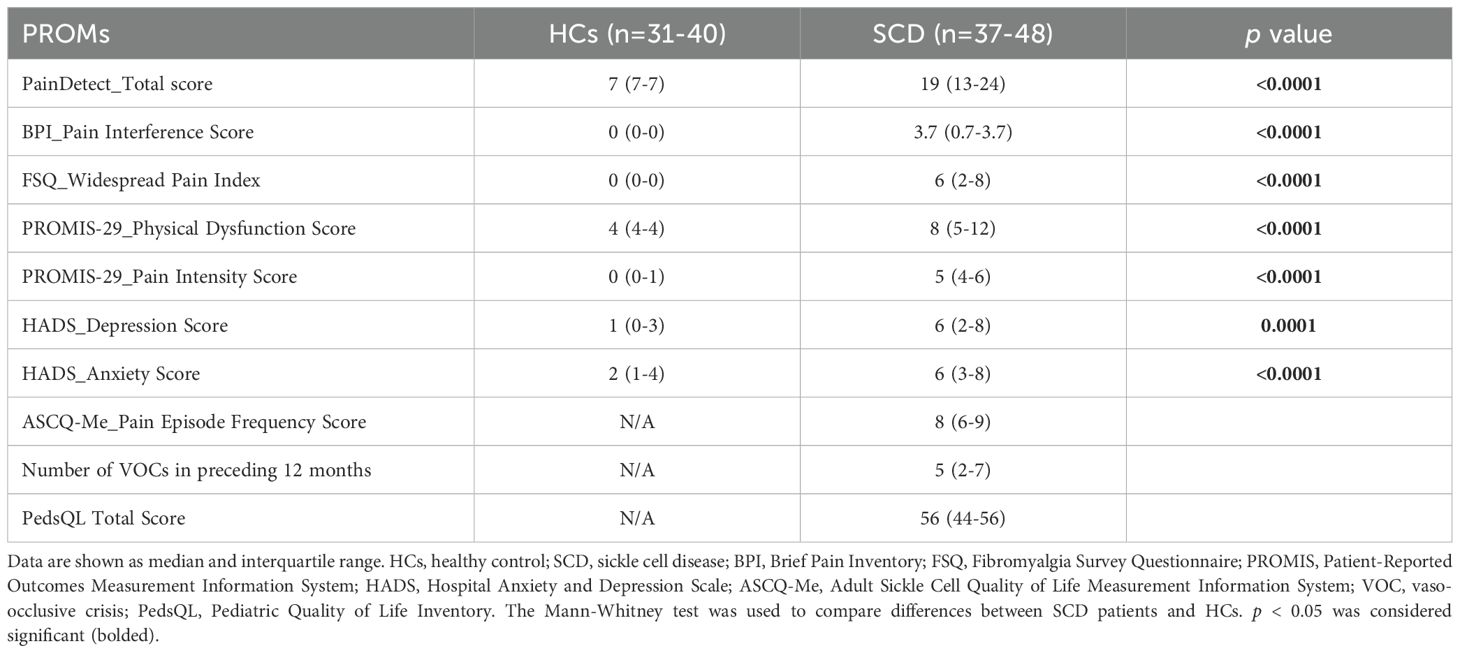
Table 2. Patient-reported outcome measures (PROMs).
2.3 Multiplex immunoassays
The ProcartaPlex Human Immune Checkpoint Panel 37plex (Cat. #: EPX370-15846-901, Invitrogen, Carlsbad, CA) was used to quantify plasma concentrations of thirty-seven sICPs, including twenty inhibitory ICPs (Arginase-1, B7-H6, BTLA, CD47, CD48, CD73, CD152, CD276, HVEM, IDO, LAG-3, PD-1, PD-L1, PD-L2, PVR, S100A8/A9, Siglec-7, Siglec-9, TIM-3, and VISTA) and seventeen stimulatory ICPs (CD27, CD28, CD80, CD96, CD134, CD137, E-Cadherin, GITR, ICOSL, MICA, MICB, Nectin-2, Perforin, TIMD-4, ULBP-1, ULBP-3, and ULBP-4). As the immune modulatory functions of the sICPs are not yet fully understood, we classified the sICPs as either inhibitory or stimulatory based on their membrane-bound counterparts. HVEM was originally described as an immunosuppressive molecule but has been found to exhibit dual functionality, depending on the receptors or ligands it engages. For simplicity, we classified HVEM as an inhibitory ICP. ProcartaPlex Human Immune Response Panel 80plex (Cat. #: EPX800-10080-901, Invitrogen) was used to quantify plasma concentrations of eighty inflammatory mediators, including twenty-seven inflammatory cytokines (IFN-α, IFN-γ, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12p70, IL-13, IL-15, IL-16, IL-17A, IL-18, IL-21, IL-22, IL-23, IL-27, IL-31, IL-37, MIF, TNF-α, TNF-β, and TSLP), twenty-seven chemokines (BLC, CCL1, CCL17, CCL21, CCL23, CCL25, CXCL6, ENA-78, Eotaxin, Eotaxin-2, Eotaxin-3, Fractalkine, Gro-α, IL-8, IP-10, I-TAC, MCP-1, MCP-2, MCP-3, MCP-4, MDC, MIG, MIP-1α, MIP-1β, MIP-2α, MIP-3α, and MIP-3β), twelve growth factors (bNGF, FGF-2, G-CSF, GM-CSF, HGF, IL-7, IL-20, IL-34, LIF, M-CSF, SCF, and VEGF-A), twelve soluble receptors/proteins (APRIL, BAFF, CD30, CD40L, Gal-3, IL-2R, MMP-1, PTX3, TNF-RII, TRAIL, TREM-1, and TWEAK), and two serine proteases (Granzyme A and Granzyme B). The MILLIPLEX MAP Human Autoimmune Autoantibody Panel (Cat. #: HAIAB-10K, MilliporeSigma, Burlington, MA) was used to quantify plasma concentrations of eighteen anti-human autoantibodies, including anti-C1q, anti-Centromere Protein A (CENP-A), anti-Centromere Protein B (CENP-B), anti-β 2-glycoprotein, anti-Ku, anti-Mi-2, anti-myeloperoxidase, anti-proliferating cell nuclear antigen A (PCNA), anti-PM/Scl-100, anti-proteinase 3, anti-Ribosomal P, anti-Ribonucleoprotein (RNP), anti-RNP/Smith (RNP/Sm), anti-Scl-70, anti-Sm, anti-Sjögren′s Syndrome-related antigen B/La (SSB/La), anti-anti-Sjögren′s Syndrome-related antigen A/Ro52 kDa (SSA/Ro52), and anti-Sjögren′s Syndrome-related antigen A/Ro60 kDa (SSA/Ro60). The beads were read on a BioPlex 200 system (Bio-Rad, Hercules, CA). The standards at 4-fold serial dilutions were run on each plate in duplicate and used to calculate the concentrations of sICPs and inflammatory mediators using the Bio-Plex Manager Software (Bio-Rad, Hercules, CA) as previously reported (15, 22). Plasma samples were diluted 100-fold for the autoantibody multiplex assay, and the levels of the autoantibodies were reported as mean fluorescence intensity (MFI) after background MFI subtraction. The inter-assay CV for the 37plex ICP kit and the 80plex kit is <15% and the inter-assay precision for the autoantibody kit is <20%. The lower limits of quantifications (LLOQ) for the 37plex ICP kit and the 80plex kit are provided by the manufacturer.
2.4 Flow cytometry
Frozen PBMCs were incubated with a fixable viability dye (Thermo Fisher Scientific, Cincinnati, OH, USA) to exclude dead cells prior to analysis. Cells were then stained with fluorochrome-conjugated antibodies against cell lineage markers (BV510-CD3 (clone; OKT3), BV605-CD4 (clone: SK3), Alexa Fluor 700-CD14 (clone: 63D3), BV650-CD16 (clone: 3G8), BV711-CD19 (clone: HIB19), BV785-CD56 (clone: 5.1H11), PE-Dazzle 594-CD161 (clone: HP-3G10), and PE-MR1-tetramer loaded with the ligand 5-OP-RU) and three immune cell activation markers (PE-Cy7-CD38 (clone: HIT2), FITC-CD69 (clone: FN50), and PerCp-Cy5.5-HLA-DR (clone: L243) and two membrane ICP (mICPs)/exhaustion markers (BV421-PD-1 (clone: EH12.2H7) and APC-TIM-3(F38-2E2)). PE-Cy7, FITC-, PerCp-Cy5.5-, BV421-, and APC-conjugated isotype control antibodies were used to set gates for positive expression for the activation markers and mICPs. All antibodies were purchased from BioLegend (San Diego, CA, USA). The MR1 tetramers were produced by the NIH Tetramer Core Facility as permitted to be distributed by the University of Melbourne. Stained cells were acquired using a BD LSRFortessa flow cytometer (BD Biosciences, San Jose, CA). Flow data were analyzed using FlowJo v10 software (Tree Star, San Carlos, CA). Immune cell types were gated as follows: Single live cells were divided into monocytes (MC) and lymphocytes (LYM) based on their forward scatter and side scatter characteristics. CD14 and CD16 expression were used to further divide MC into classic (CD14hiCD16-), non-classic (CD14loCD16hi), and transitional MC (CD14hiCD16+) subsets. Among LYM, B cells were defined as CD19+CD3-. Non-B LYM were further divided into T cells (CD3+) and NK cells (CD3-CD56+). T cells include the innate T cells subsets MAIT cells (CD161hiMR1-tetramer+) and NKT cells (CD3+CD56+) as well as CD4+ T cells (CD3+CD4+) and CD4- T cells (CD3+CD4-). As CD4- T cells are mostly CD8 T cells, they were defined as CD8 T cells.
2.5 Statistical analysis
Statistical analysis was performed using GraphPad Prism 10 and R (version 4.5.0). Data were expressed as median and interquartile range. Differences between 2 groups were calculated using the Mann-Whitney test for continuous variables. The χ2 test was used for comparison between two groups for gender distribution. Adjusted p-values were calculated for the 37plex, 80plex, and 18plex analytes using the Holm-Šídák correction for multiple comparisons.
Inflammatory mediators elevated in the SCD participants were used for subsequent Spearman correlation analyses performed in R using the pcor.test() function from the ppcor package, with age, gender, SCD genotype, hydroxyurea (HU) treatment, body mass index (BMI), and chronic transfusions included as covariables. SCD genotype was modeled as a binary covariate, distinguishing the classically more severe genotypes (HbSS and HbSβ°) from less severe forms (HbSC and HbSβ+) (23, 24). This binary stratification approach was chosen due to the limited number of participants in some diagnostic subgroups (e.g., Sβ° n = 3), which precludes stable estimation in more granular models and increases the risk of type II error.
As the sICP x 80-plex comparison generated 1,540 correlations, we controlled multiplicity by applying a false discovery rate using the Benjamini-Hochberg method. The full matrix of raw and false discovery rate (FDR)-adjusted p-values is provided in Supplementary Table S1. All partial correlation analyses were conducted using listwise deletion, such that any pairwise comparison was performed only in cases with complete data for both target variables and all covariates. Analytes in the ICP and 80plex multiplex immunoassays with values below the LLOQ were replaced with 0.1-fold of the LLOQ values provided by the manufacturer. In sICP analyses where over ≥50% of samples were below LLOQ, analysis of the marker was ignored. Significance was set at p < 0.05, except for the sICP × 80-plex matrix, where q < 0.05 applied.
We conducted a power analysis for Spearman correlation to evaluate the ability to detect statistically significant associations under a multiple-testing framework. Assuming a moderate correlation effect size (ρ = 0.4), a sample size of 90, and a total of 1,540 pairwise tests, statistical power was estimated using a 5% FDR threshold controlled via the Benjamini-Hochberg procedure. The analysis indicated statistical power exceeding 91.7% to detect true associations of this magnitude, demonstrating that the study is well-powered to identify moderate correlations while appropriately controlling for false positives. Power calculations were performed using PASS 2019 (v19.0.1).
3 Results
3.1 Characteristics of the SCD study cohort: demographics and patient-reported outcomes
The demographic and clinical characteristics of 50 SCD participants, alongside 40 healthy controls (HCs), are summarized in Table 1. There were no significant differences between SCD participants and HCs in terms of age (median: 31 vs. 33.5 years; p = 0.76) or gender distribution (58% vs. 60% females; p = 0.85). BMI was significantly lower in the SCD group compared to HCs (median: 23.8 vs. 27.0 kg/m²; p = 0.001). Five SCD participants (10%) had a history of infarction. Among SCD participants, 48% were on HU therapy, and 12% received chronic transfusion therapy.
PROMs indicated significantly greater pain burden in SCD participants compared to HCs, as detailed in Table 2. SCD participants reported higher PainDETECT scores (median 19 vs. 7; p < 0.0001), greater pain interference (median Brief Pain Inventory score: 3.7 vs. 0; p < 0.0001), and more widespread pain (FSQ Widespread Pain Index: median 6 vs. 0; p < 0.0001). Physical functioning and pain intensity scores were significantly worse among SCD participants (PROMIS-29 Physical Dysfunction Score: median 8 vs. 4; p < 0.0001 and PROMIS-29 Pain Intensity Score: median 5 vs. 0; p < 0.0001). Depression and anxiety levels were elevated (HADS Depression: median 6 vs. 1; p = 0.0001; HADS Anxiety: median 6 vs. 2; p < 0.0001). SCD participants reported multiple VOCs in the past 12 months (median: 5 events) and had a median PedsQL of 56.
3.2 Circulating sICPs were highly dysregulated in SCD patients
All sICPs, except for four (CD48, ULBP-1, ULBP-3, and ULBP-4), were detected in the plasma samples from both SCD patients and HCs. SCD patients exhibited significantly dysregulated levels of multiple sICPs compared to HCs (Table 3). Among the twenty inhibitory sICPs examined, twelve were elevated in SCD patients, including arginase-1, BTLA, CD47, CD152 (CTLA-4), HVEM, IDO, LAG-3, PD-1, Siglec-7, Siglec-9, TIM-3, and VISTA. After adjusting for multiple comparisons, seven (CD47 BTLA, CD152, HVEM, Siglec-7, TIM-3, and VISTA) remained significantly upregulated. Among the seventeen stimulatory sICPs examined, eleven of them, including CD27, CD28, CD80, CD96, CD134 (OX40), CD137 (4-1BB), GITR, MICA, MICB, Nectin-2, and TIMD-4, were markedly elevated in SCD patients, and all except CD96, GITR, and MICB remained significantly different after multiple comparison adjustment (Table 3). These findings highlight a dysregulated circulating immune profile.
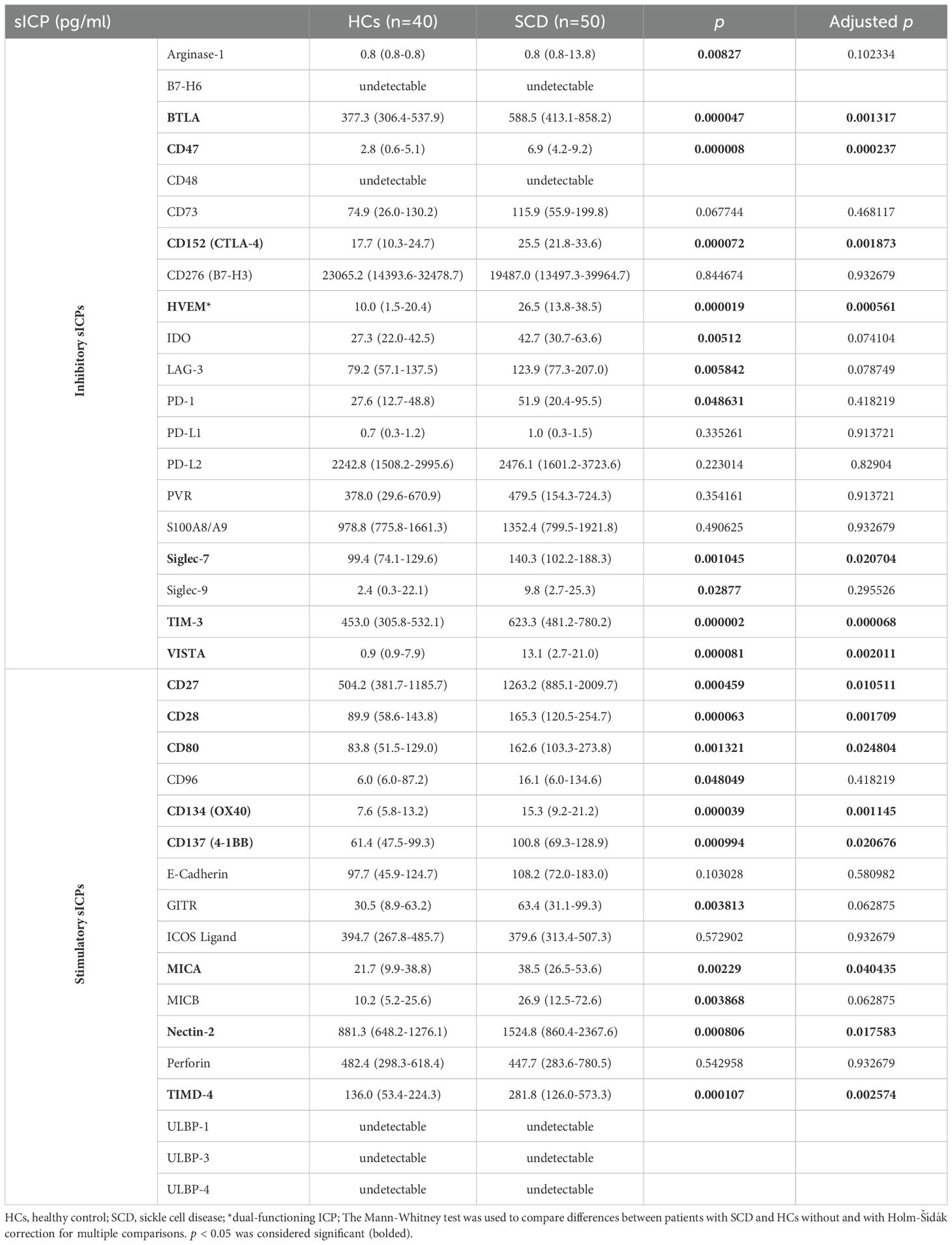
Table 3. Plasma levels of sICPs in SCD patients versus HCs.
The adjusted analysis identified a subset of sICPs that were robustly increased, including BTLA, CD47, CD152, HVEM, TIM-3, VISTA, CD28, CD134, and TIMD-4, indicating their potential as biomarkers for immune dysfunction in SCD (Table 3). Interestingly, TIM-3, an inhibitory sICP linked to immune exhaustion, was among the most significantly elevated markers in SCD participants, emphasizing its role in immune dysregulation. The stimulatory checkpoint CD28, critical for T cell activation, showed pronounced upregulation in SCD participants. These data underscore the complex interplay between inhibitory and stimulatory sICPs in the immunopathogenesis of SCD, with a distinct shift towards heightened immune activation and exhaustion.
3.3 Comprehensive analysis revealed correlations of sICPs with inflammatory mediators and autoantibodies in SCD patients
To explore the interplay between immune regulation and inflammation in SCD patients, we first identified significantly dysregulated inflammatory mediators and autoantibodies by comparing SCD patients to HCs using Mann–Whitney tests (Supplementary Tables S2, S3); the significantly altered factors were then used in downstream Spearman correlation analyses with elevated sICPs (15). Notably, the associations varied depending on whether covariables (age, gender, SCD genotype, HU use, BMI, chronic transfusion) were included in the analysis. For simplicity, we only reported correlation results from multivariant analysis. Our analysis revealed significant associations between plasma levels of sICPs and inflammatory mediators, as depicted in an FDR-adjusted heatmap (Figure 1), where stronger correlations are represented by a higher intensity of color gradients (coefficient) and the number of stars (q-value). Specifically, twenty sICPs, including BTLA, CD47, CD152, HVEM, IDO, LAG-3, PD-1, Siglec-7, TIM-3, VISTA, CD27, CD28, CD80, CD134, CD137, GITR, MICA, MICB, Nectin-2, and TIMD-4, demonstrated positive correlations with various proinflammatory mediators, including multiple inflammatory cytokines (such as IFN-γ, IL-4, IL-6, IL-18, and TNF-α), chemokines (such as CCL21, IL-8, and MIP-1β), growth factors (such as IL-20, IL-34, and VEGF-A). A subset of the sICPs also positively correlated with two soluble receptors (PTX3 and TREM-1) and two serine proteases (granzyme A and granzyme B). Several upregulated sICPs, including CD152, HVEM, LAG-3, VISTA, CD28, CD80, GITR, and MICA, were highly associated with many inflammatory mediators. These results indicate a complex network of interactions between sICPs and inflammatory mediators, underscoring the potential role of specific sICPs in driving or modulating inflammation in SCD patients.
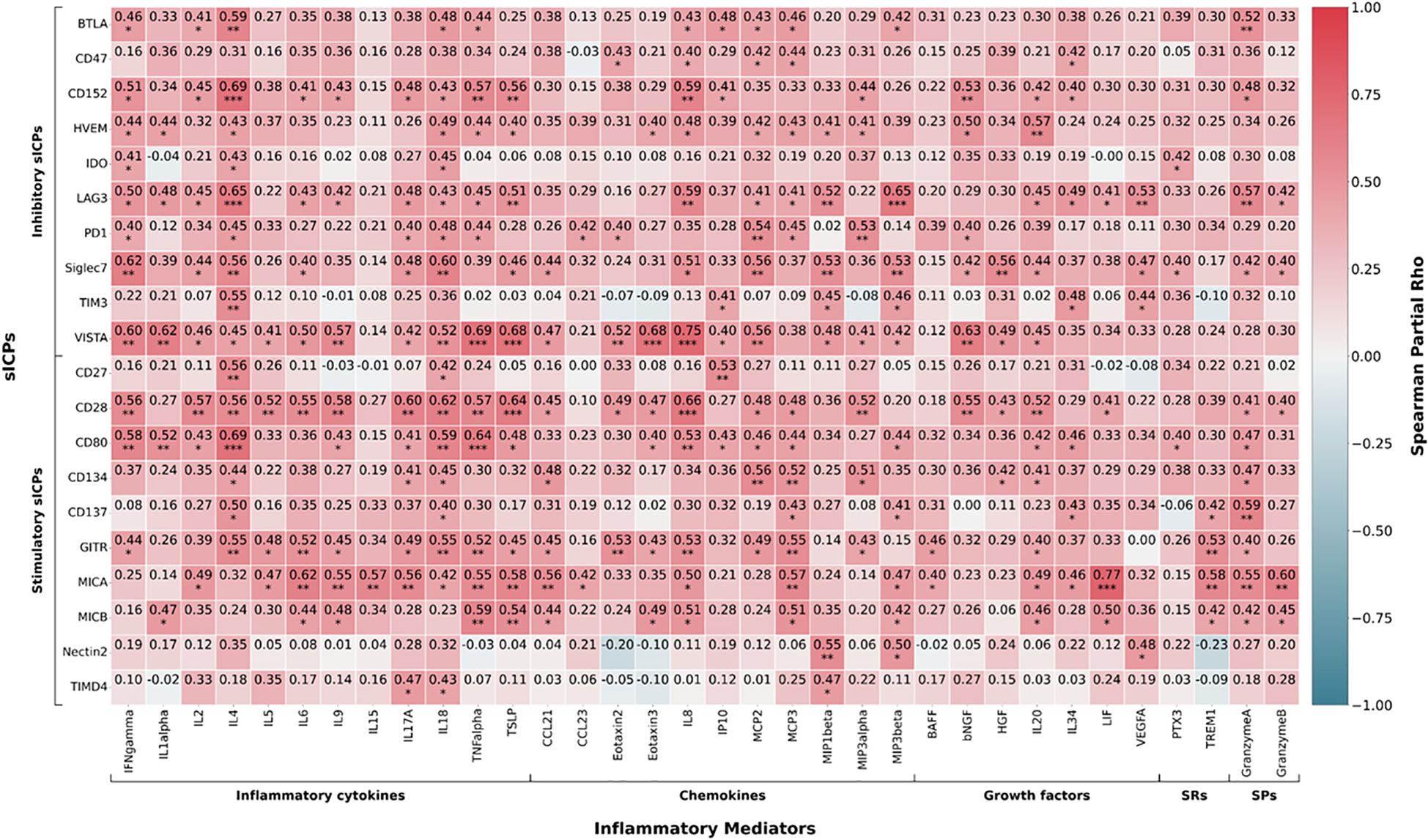
Figure 1. Correlations between altered plasma levels of sICPs and inflammatory mediators in participants with sickle cell disease. Heatmap showing Spearman correlation coefficient (color) and p values (stars). Covariables include age, gender SCD genotype, hydroxyurea use, body mass index, and chronic transfusion. sICPs, soluble immune checkpoints; SCD, sickle cell disease; SRs, soluble receptors; SPs, serine proteases. *p < 0.05; **p < 0.01; ***p < 0.001.
Similarly, we also performed a Spearman correlation analysis of the dysregulated sICPs with eight autoantibodies that were significantly upregulated in SCD patients relative to HCs (Ku, PM/Scl-100, ribosomal P, RNP/Sm, SSB/La, SSA/Ro60, myeloperoxidase, and proteinase 3) and one that trended higher (Smith antigens, Sm) (Supplementary Table S3). This analysis revealed significant associations between plasma levels of three sICPs and specific autoantibodies, represented by coefficient values and significance levels. The inhibitory sICPs CD47 and Siglec-7 demonstrated significant positive correlations with autoantibodies against myeloperoxidase and ribosomal P protein and Sm, respectively, while the stimulatory sICP CD80 exhibited significant positive correlations with the Ku and Sm autoantibodies (Table 4). These findings reveal intricate networks of interactions between sICPs and autoantibodies, emphasizing the dual roles of these molecules in both promoting and potentially mitigating autoimmunity in SCD patients.
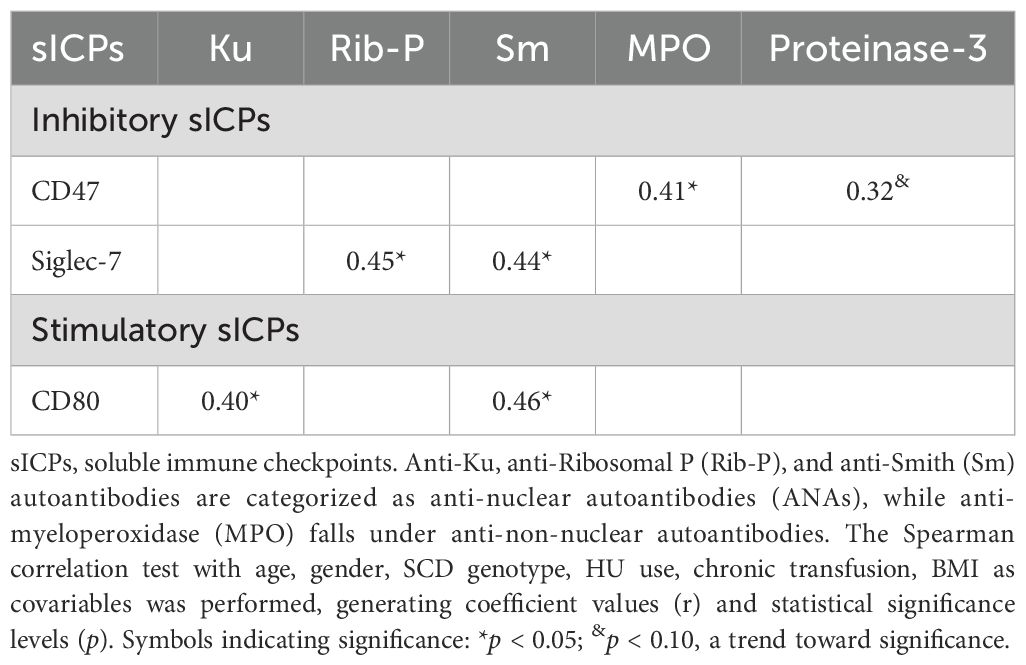
Table 4. Correlations between altered plasma levels of sICPs and autoantibodies.
3.4 Comprehensive analysis revealed significant correlations of sICPs with alterations of immune cell profiles in SCD patients
We first compared immune cell profiles in SCD patients and HCs using multiparametric flow cytometry (Supplementary Figure S1). PBMCs were immunophenotyped with cell lineage markers (CD3, CD4, CD14, CD16, CD19, CD56, CD161, and MR1-tetramer loaded with the ligand 5-OP-RU), three immune cell activation markers (CD38, CD69, and HLA-DR) and two membrane ICP/exhaustion markers (PD-1 and TIM-3). As shown in Supplementary Table S4, the immunophenotypes of both innate (monocytes and NK cells) and adaptive immune cells (various subsets of T cells, including MAIT, NKT, CD4, and CD8 T cells) were altered in SCD patients. In particular, NK cells and all subsets of T cells expressed higher levels of the cell activation marker CD69. NKT cells, CD8 T cells, and B cells also had higher levels of two other cell activation markers, CD38 and HLA-DR. On the other hand, the classic monocytes downregulated the inhibitory mICP TIM-3 and the monocyte lineage marker CD14, while the B cells trended towards having lower levels of TIM-3 expression (p = 0.07). Additionally, the frequency of NK cells trended higher (p = 0.06), while that of T cells was significantly lower in SCD patients. These results indicated dysregulation and hyperactivation of the immune system in SCD patients.
To investigate the interplay between dysregulated sICPs and altered immune cell profiles in SCD patients, Spearman correlation analyses were performed, controlling for covariables that included age, gender, SCD genotype, HU use, BMI, and chronic transfusion status (Table 5). Both inhibitory and stimulatory sICPs demonstrated significant associations with phenotypes of various immune cells, including subsets of monocytes, NK cells, T cells, MAIT cells, NKT cells, activated T cells, and B cells. Specifically, seven sICPs (CD152, IDO, TIM-3, CD28, CD80, GITR, and Nectin-2) displayed positive correlations with phenotypes of multiple immune cell types. Conversely, six sICPs (CD47, HVEM, LAG-3, Siglec-7, VISTA, and TIMD-4) showed both positive and negative correlations with immune cell phenotypes. Notably, seven sICPs (CD47, HVEM, LAG-3, Siglec-7, VISTA, MICB, and TIMD-4) exhibited significant negative correlations with the decreased percentage of T cells, while no sICPs were positively correlated with T cell percentages. Furthermore, CD152, HVEM, IDO, Siglec-7, VISTA, CD28, and CD80 correlated with multiple aspects of immune cell phenotypes. These findings highlight the intricate and multifaceted interactions between sICPs and immune cell profiles in SCD patients, underscoring the need for further investigation to elucidate their immunological and clinical implications.
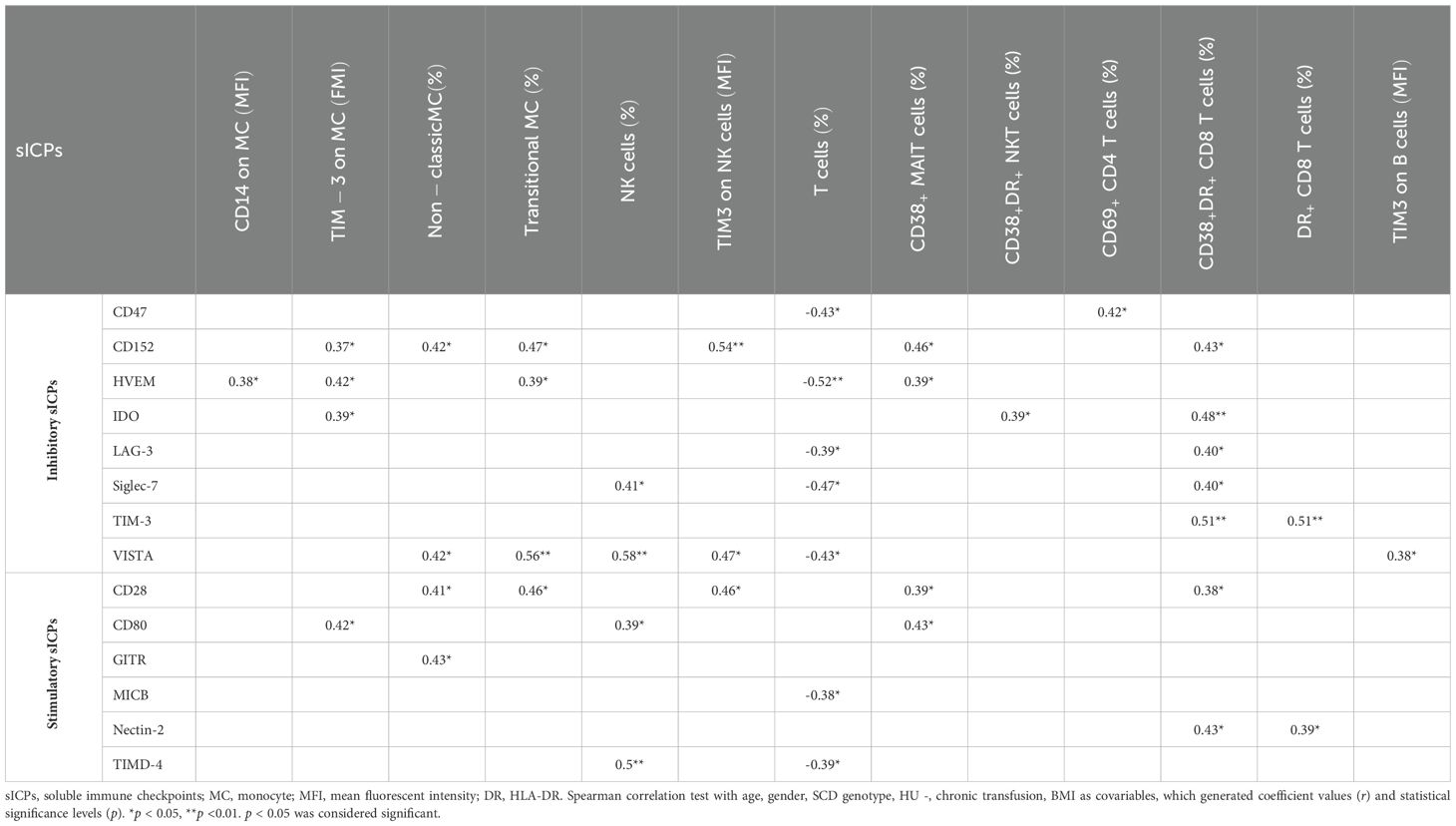
Table 5. Correlations between altered plasma levels of sICPs and immune cell profiles.
3.5 Correlation analysis revealed associations between altered plasma levels of sICPs and PROMs
To investigate the relationship between sICPs and PROMs in SCD patients, Spearman correlation analyses were performed with and without the inclusion of covariables (Table 6; Supplementary Table S5). Covariables included age, gender, SCD genotype, HU use, BMI, and chronic transfusion status.
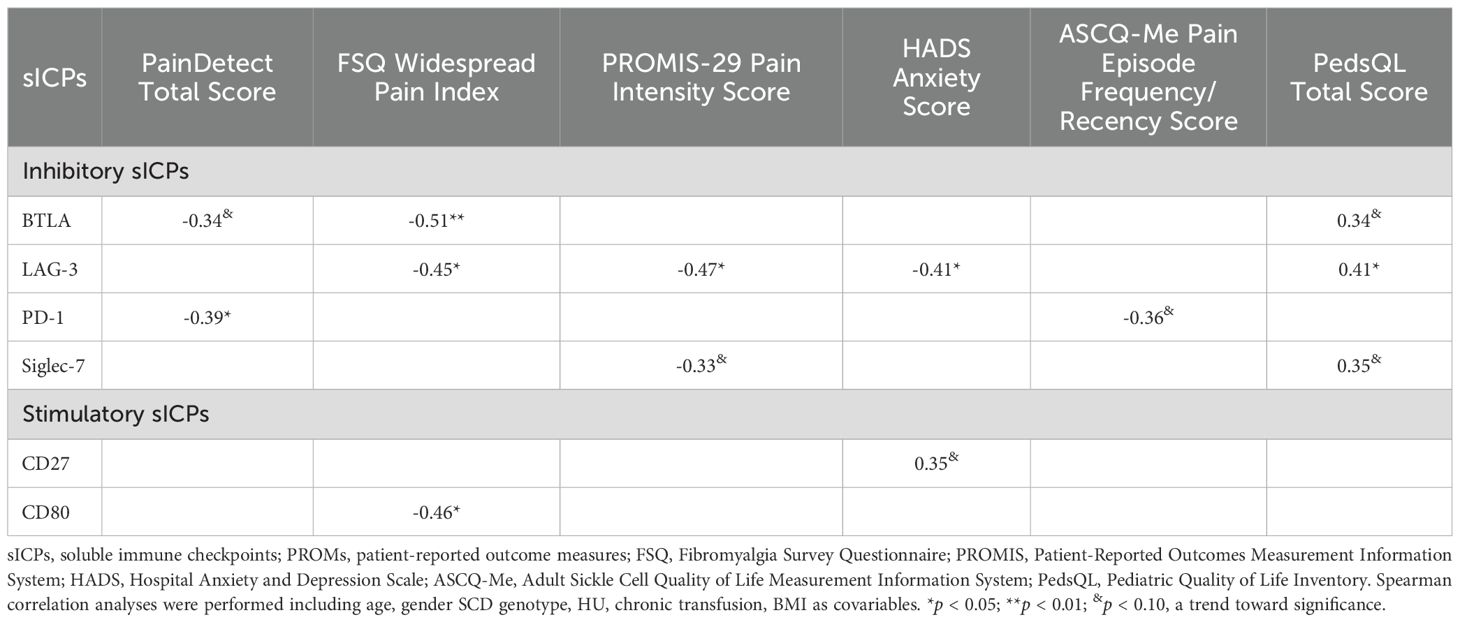
Table 6. Correlations between altered plasma levels of sICPs and PROMs.
In analyses without covariables, multiple inhibitory and stimulatory sICPs demonstrated significant correlations with multiple PROMs (Supplementary Table S5). Nine (BTLA, LAG-3, PD-1, Siglec-7, CD28, CD80, CD137, GITR and Nectin-2) had significant negative correlations with various pain measurements, while CD80 and MICA inversely correlated with the PROMIS 29 Physical Dysfunction Score. On the other hand, ten sICPs (BTLA, LAG-3, Siglec-7, TIM-3, CD80, CD134, CD137, MICA, MICB, and Nectin-2) demonstrated positive correlations with PedsQL scores.
When covariables were included, three inhibitory (BTLA, LAG-3, and PD-1) and one stimulatory (CD80) sICPs remained significantly correlated with PROMs (Table 6). Key findings include: (1) BTLA showed negative correlations with the PainDetect Total Score (r = -0.34, p = 0.09) and FSQ Widespread Pain Index (r = -0.51, p = 0.006) while positively correlating with the PedsQL Total Score (r = 0.34, p = 0.09). (2) LAG-3 was negatively correlated with FSQ Widespread Pain Index (r = -0.45, p = 0.02), PROMIS-29 Pain Intensity Score (r = -0.47, p = 0.01) and HADS Anxiety Score (r = -0.41, p = 0.04), but positively correlated with the PedsQL Total Score (r = 0.41, p = 0.03), (3) PD-1 demonstrated negative correlations with PainDetect Total Score (r = -0.39, p = 0.05) and Pain Episode (r = -0.36, p = 0.07), and (4) CD80 demonstrated negative significance in correlation with FSQ Widespread Pain Index (r = -0.46, p = 0.02).
The analyses revealed significant and intricate associations between sICPs and PROMs, which varied with the inclusion of covariables. These findings underscore the multifaceted role of sICPs in modulating pain perception and QoL in SCD patients, warranting further exploration of their immunological and clinical implications.
3.6 Analysis revealed associations of sICPs, inflammatory mediators, autoantibodies, and immune cell phenotypes with VOCs
To identify potential underlying inflammatory traits contributing to VOCs, we performed Spearman correlation analysis of altered sICPs, inflammatory mediators, autoantibodies, and immune cell phenotypes with the time intervals from blood draw for biomarker analysis to the most recent VOC episode (Days after crisis) and the future crisis (Days before crisis). Age, gender, SCD genotype, HU use, BMI, and chronic transfusion were included as covariables. None of the sICPs and autoantibodies had significant correlations with Days after crisis and Days before crisis. As shown in Table 7, trends of negative correlations were observed between only one sICP, Nectin-2 (r = -0.34, p = 0.06) and Days post-crisis of SCD. Key inflammatory mediators, including IL-1α (r = -0.34, p = 0.07) and VEGF-A (r = -0.34, p = 0.07), showed trends towards significant negative correlations with post-crisis time intervals. Notably, the frequencies of HLA-DR+ (r = -0.72, p = 0.0001) or CD38+HLA-DR+ (r = -0.48, p = 0.02) activated CD8 T cells were highly inversely correlated with Days after crisis. Regarding correlations with Days before crisis, a positive correlation was noted for CCL23 (r = 0.33, p = 0.08), while IL-18 (r = -0.38, p = 0.04) and TIM-3 expression on B cells (r = -0.45, p = 0.03) displayed significant negative correlations. Lastly, the autoantibodies to Sm trended to negatively correlate with Days before crisis.
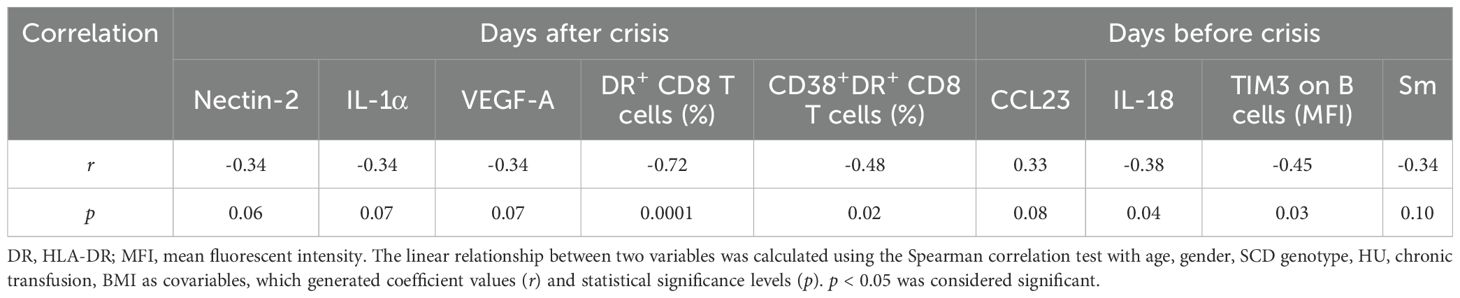
Table 7. Correlations between altered plasma levels of inflammatory factors and time interval from after and before VOC occurrence.
Taken together, these results indicate that levels of IL-18, T cell activation, and B cell immune exhaustion are linked with VOCs and that levels of circulating IL-18 and TIM-3 expression on B cells may predict VOC occurrence.
4 Discussion
This study investigates the profile of sICPs in SCD and their associations with inflammatory mediators, autoantibodies, immune cell profiles, and clinical outcomes. ICPs consist of paired receptor-ligand molecules that exert inhibitory, stimulatory, or dual effects on immune regulation, surveillance, defense, and self-tolerance (25–29). ICP molecules exist in both membrane and soluble forms in vivo and in vitro (30–41). Similar to membrane-bound ICPs (mICPs), sICPs are also present in normal physiological conditions and highly dysregulated in patients with cancer, viral infections, or alcohol-associated liver disease (ALD) (30–42). Soluble ICPs can be generated through either the secretion of protein isoforms encoded by alternative mRNA splicing or protease-mediated shedding from mICPs, mediated by the actions of matrix metalloproteinases (MMPs) (41, 43). Since sICPs are paired receptor-ligand molecules and circulate in the bloodstream, they likely form a circulating immune regulatory system. In addition, increasing evidence has shown that sICPs interact with their mICP compartments to positively or negatively regulate immune responses (40). Furthermore, sICPs can compete with their mICP compartments for binding to ICP-blocking antibodies, thereby interrupting the efficacy of ICP blockade therapies. Thus, there is an urgent need to study the role of sICPs in immune regulation in health and disease. Given that sICPs have not been studied in patients with SCD, we utilized clinical samples from our ongoing randomized clinical trial in SCD (ClinicalTrials.gov, NCT05045820) to study the profile of sICPs in SCD and their associations with inflammatory mediators, autoantibodies, immune cell profiles, and clinical outcomes. Our findings highlight the significant dysregulation of sICPs in SCD patients and their potential as biomarkers or therapeutic targets.
In comparison to HCs, SCD patients exhibited significantly elevated levels of multiple sICPs (Table 3). Among the 20 inhibitory sICPs analyzed, 7 (BTLA, CD47, CD152, HVEM, Siglec-7, TIM-3, and VISTA) were markedly elevated in SCD patients. Similarly, 8 of the 17 stimulatory sICPs (CD27, CD28, CD80, CD134, CD137, MICA, Nectin-2, and TIMD-4) showed significant increases in the SCD cohort (Table 3). Notably, some ICPs, such as HVEM, exhibit dual functionality, acting as either coinhibitory or costimulatory molecules depending on the receptors or ligands they engage. Soluble HVEM has also been shown to regulate immune response, such as the production of proinflammatory cytokines IFN-γ and TNF-α by T cells and NK cells (42, 44). Consistent with this, we found sHVEM levels in the SCD patients positively correlated with multiple inflammatory cytokines (including IFN-γ and TNF-α), chemokines, and growth factors (Figure 1). Understanding the context-dependent roles of ICPs like HVEM in SCD is vital for addressing the chronic inflammation, immune dysregulation, and pain experienced by these patients. These insights could pave the way for targeted therapeutic strategies to modulate ICP pathways, ultimately improving clinical outcomes for individuals with SCD.
We have recently reported that inflammation and autoimmunity are interrelated in SCD patients in this study cohort (15). Following this path, we analyzed the association of sICPs with inflammatory mediators and autoantibodies. Our analysis revealed significant correlations between sICPs and various inflammatory mediators (Figure 1). Specifically, 20 sICPs, including BTLA, CD47, CD152, HVEM, IDO, LAG-3, PD-1, Siglec-7, TIM-3, VISTA, CD27, CD28, CD80, CD134, CD137, GITR, MICA, MICB, Nectin-2, and TIMD-4, positively correlated with various proinflammatory mediators. Notably, all except CD47, TIM-3, MICB, and Nectin-2 positively correlated with the proinflammatory cytokine IL-18, a critical proinflammatory regulator of both innate and adaptive immune responses, which was significantly upregulated in SCD compared to healthy controls in our recently published results (15). These findings suggest that sICPs play a crucial role in modulating inflammation in SCD. In turn, it is possible that inflammatory mediators reciprocally regulate sICPs, forming a positive feedback loop.
Similarly, significant associations were found between sICPs and specific autoantibodies (Table 4). With covariables included, three sICPs (CD47, Siglec-7, and CD80), demonstrated positive correlations with autoantibodies (Table 4). Notably, the associations between sICPs and different types of autoantibodies highlight distinct pathways in autoimmunity. Siglec-7 and CD80 were significantly correlated with anti-nuclear autoantibodies (ANAs), such as anti-Sm, anti-Ribosomal P (Rib-P), and anti-Ku (Table 4), suggesting a role for these sICPs in pathways related to nuclear antigen response and immune tolerance breakdown. In contrast, CD47 showed a positive correlation with autoantibody against myeloperoxidase (MPO) (Table 4), a hallmark of small-vessel vasculitis and other systemic autoimmune diseases. This distinction between the associations of sICPs with ANAs and anti-non-nuclear autoantibodies emphasizes the diverse roles these sICPs may play in modulating specific immune responses. ANAs are primarily associated with systemic autoimmune conditions targeting nuclear components, whereas anti-non-nuclear autoantibodies are often linked to organ-specific damage and systemic inflammation. These findings provide new insights into the potential mechanistic roles of sICPs in shaping the immune landscape of autoimmune diseases, paving the way for targeted therapeutic strategies based on sICP modulation.
Both inhibitory and stimulatory sICPs demonstrated significant associations with various immune cell phenotypes, including monocytes, NK cells, T cells, MAIT cells, NKT cells, activated T cells, and B cells (Table 5). Notably, Four sICPs (HVEM, CD47, LAG-3, and Siglec-7) exhibited significant negative correlations with the percentage of T cells but positive correlations with T cell activation (Table 5). The results suggest that sICPs are linked to the regulation of T cell dynamics and activation in the immune system of SCD patients. A significant negative correlation between sICPs and the percentage of T cells implies that higher levels of sICPs likely play a role in reducing T cell abundance, potentially by promoting T cell exhaustion, apoptosis, or suppression of T cell proliferation. These mechanisms are often observed in chronic infections (e.g., HIV) or cancer and may also hold truth in SCD. A significant positive correlation between sICPs and T cell activation further suggests that higher levels of sICPs are associated with excessive activation and exhaustion of T cells in SCD. Alternatively, it could mean that sICPs are being released into circulation in response to T cell activation signals, serving as markers of immune system engagement.
Our analyses also demonstrated the correlations of PROMs in pain and QoL with inhibitory (BTLA, LAG-3, PD-1, and Siglec-7) and/or stimulatory (CD27 and CD80) sICPs, respectively (Table 6). Interestingly, the widespread pain index showed a strong correlation with BTLA, LAG-3, and CD80, suggesting the correlation between nociplastic pain and circulating sICPs. Consistent with our previous finding (15), IL-18 is elevated prior to VOC onset (Table 7). The changes in sICPs, inflammatory mediators, autoantibodies, and immune cell phenotypes were observed in relation to SCD crisis timing (Table 7), providing insights into potential immune and inflammatory dynamics surrounding VOC onset. A longitudinal extension of the same trial is now collecting sequential samples at StSt, during defined time points of VOCs and through recovery. Once adequately powered, these data will reveal whether the sICPs highlighted here fluctuate dynamically with the crisis stage.
In SCD, chronic inflammation and sustained immune activation contribute to the dysfunction and exhaustion of immune cells, particularly among T cells (45–47). Membrane-bound ICPs, including PD-1, PD-L1, TIM-3, and LAG-3, play central roles in regulating T cell responses and serve as markers of exhaustion in immune cells. Their engagement with ligands suppresses T cell proliferation, cytokine secretion, and cytotoxic activity (48). Soluble ICPs, released through alternative mRNA splicing or proteolytic cleavage, can modulate immunity in parallel or in opposition to their membrane-bound counterparts (49, 50). Some sICPs, such as sPD-1 and sTIM-3, function as decoy receptors, sequestering ligands and reducing inhibitory signaling (51), while others, including PD-L1 and LAG-3, can bind to their inhibitory receptors to transmit immunosuppressive signals, similar to their membrane-bound forms (52, 53). Thus, sICPs likely represent circulating immune regulators that dynamically influence systemic immune responses. While immune dysregulation in SCD is well documented, the contribution of ICP pathways remains unexplored. Our study is the first to characterize the sICP landscape in SCD and demonstrates that these molecules are linked to systemic inflammation, autoimmunity, and pain in SCD patients.
In conclusion, this study highlights the intricate relationship between sICPs, inflammatory mediators, autoantibodies, immune cell profiles, and clinical outcomes in SCD. The significant associations between sICPs and various clinical outcome measures in pain and QoL highlight their potential as biomarkers and therapeutic targets in SCD pain management. Future research should focus on elucidating the mechanistic roles of sICPs in modulating immune activation, autoimmunity, and inflammation in SCD, which may lead to novel strategies for managing VOCs and improving the QoL in SCD patients.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Indiana University Institutional Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
WL: Writing – original draft, Writing – review & editing. AP: Writing – review & editing. LH: Writing – review & editing. XH: Writing – review & editing. CD: Writing – review & editing. BR: Writing – review & editing. AO’B: Writing – review & editing. QY: Writing – original draft, Writing – review & editing. YW: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by NIH K99/R00 award (Grant # 5R00AT010012) and Indiana University Health – Indiana University School of Medicine Strategic Research Initiative funding to YW. This work was also supported by the Bill & Melinda Gates Foundation (OPP1035237 to QY).
Acknowledgments
The authors would like to thank Tyler James Barret, Nayana Dutt, Payton Mittman, Bea Paras, Ramat Gbemisola Suleiman-Oba, Amy Gao and Yongqi Yu for assisting with experimental performance, and clinical team staff at Indiana University Clinical Research Center for patient scheduling and blood draw performance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frhem.2025.1580009/full#supplementary-material
Abbreviations
HADS, Hospital Anxiety and Depression Scale; HC, healthy control; MFI, median fluorescent intensity; PBMC, peripheral blood mononuclear cell; PedsQL, Pediatric Quality of Life Inventory; PROMIS, Patient-Reported Outcomes Measurement Information System; ASCQ-Me, Adult Sickle Cell Quality of Life Measurement Information System; PROMs, patient-reported outcome measures; QoL, quality of life; SCD, sickle cell disease; StSt, steady-state condition; VOC, vaso-occlusive crisis.
References
1. Ware RE, de Montalembert M, Tshilolo L, and Abboud MR. Sickle cell disease. Lancet. (2017) 390:311–23. doi: 10.1016/S0140-6736(17)30193-9
2. Rees DC, Williams TN, and Gladwin MT. Sickle-cell disease. Lancet. (2010) 376:2018–31. doi: 10.1016/S0140-6736(10)61029-X
3. Ilesanmi OO. Pathological basis of symptoms and crises in sickle cell disorder: implications for counseling and psychotherapy. Hematol Rep. (2010) 2:e2. doi: 10.4081/hr.2010.e2
4. Novelli EM and Gladwin MT. Crises in sickle cell disease. Chest. (2016) 149:1082–93. doi: 10.1016/j.chest.2015.12.016
5. Darbari DS, Sheehan VA, and Ballas SK. The vaso-occlusive pain crisis in sickle cell disease: definition, pathophysiology, and management. Eur J Haematol. (2020) 105:237–46. doi: 10.1111/ejh.13430
6. Allali S, Maciel TT, Hermine O, and de Montalembert M. Innate immune cells, major protagonists of sickle cell disease pathophysiology. Haematologica. (2020) 105:273–83. doi: 10.3324/haematol.2019.229989
7. Sesti-Costa R, Borges MD, Lanaro C, de Albuquerque DM, Saad STO, and Costa FF. Inflammatory dendritic cells contribute to regulate the immune response in sickle cell disease. Front Immunol. (2020) 11:617962. doi: 10.3389/fimmu.2020.617962
8. Adisa OA, Hu Y, Ghosh S, Aryee D, Osunkwo I, and Ofori-Acquah SF. Association between plasma free haem and incidence of vaso-occlusive episodes and acute chest syndrome in children with sickle cell disease. Br J Haematol. (2013) 162:702–5. doi: 10.1111/bjh.12445
9. Al-Habboubi HH, Mahdi N, Abu-Hijleh TM, Abu-Hijleh FM, Sater MS, and Almawi WY. The relation of vascular endothelial growth factor (VEGF) gene polymorphisms on VEGF levels and the risk of vasoocclusive crisis in sickle cell disease. Eur J Haematol. (2012) 89:403–9. doi: 10.1111/ejh.12003
10. Redha NA, Mahdi N, Al-Habboubi HH, and Almawi WY. Impact of VEGFA -583C > T polymorphism on serum VEGF levels and the susceptibility to acute chest syndrome in pediatric patients with sickle cell disease. Pediatr Blood Cancer. (2014) 61:2310–2. doi: 10.1002/pbc.25158
11. Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity. (2004) 20:441–53. doi: 10.1016/s1074-7613(04)00079-2
12. Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, et al. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. (1999) 190:1697–710. doi: 10.1084/jem.190.11.1697
13. Clements DM, Crumley B, Chew GM, Davis E, Bruhn R, Murphy EL, et al. Phenotypic and functional analyses guiding combination immune checkpoint immunotherapeutic strategies in HTLV-1 infection. Front Immunol. (2021) 12:608890. doi: 10.3389/fimmu.2021.608890
14. Joseph J, Rahmani B, Cole Y, Puttagunta N, Lin E, Khan ZK, et al. Can soluble immune checkpoint molecules on exosomes mediate inflammation? J Neuroimmune Pharmacol. (2022) 17:381–97. doi: 10.1007/s11481-021-10018-3
15. Li W, Pucka AQ, Debats C, Reyes BA, Syed F, O’Brien ARW, et al. Inflammation and autoimmunity are interrelated in patients with sickle cell disease at a steady-state condition: implications for vaso-occlusive crisis, pain, and sensory sensitivity. Front Immunol. (2024) 15:1288187. doi: 10.3389/fimmu.2024.1288187
16. Brandow AM, Farley RA, and Panepinto JA. Neuropathic pain in patients with sickle cell disease. Pediatr Blood Cancer. (2014) 61:512–7. doi: 10.1002/pbc.24838
17. Freynhagen R, Baron R, Gockel U, and Tolle TR. Paindetect: A new screening questionnaire to identify neuropathic components in patients with back pain. Curr Med Res Opin. (2006) 22:1911–20. doi: 10.1185/030079906X132488
18. Curtis S and Brandow AM. Responsiveness of patient-reported outcome measurement information system (Promis) pain domains and disease-specific patient-reported outcome measures in children and adults with sickle cell disease. Hematol Am Soc Hematol Educ Program. (2017) 2017:542–5. doi: 10.1182/asheducation-2017.1.542
19. Treadwell MJ, Hassell K, Levine R, and Keller S. Adult sickle cell quality-of-life measurement information system (ASCQ-me): conceptual model based on review of the literature and formative research. Clin J Pain. (2014) 30:902–14. doi: 10.1097/AJP.0000000000000054
20. Panepinto JA, Torres S, Bendo CB, McCavit TL, Dinu B, Sherman-Bien S, et al. Pedsql sickle cell disease module: feasibility, reliability, and validity. Pediatr Blood Cancer. (2013) 60:1338–44. doi: 10.1002/pbc.24491
21. Dudeney J, Law EF, Meyyappan A, Palermo TM, and Rabbitts JA. Evaluating the psychometric properties of the widespread pain index and the symptom severity scale in youth with painful conditions. Can J Pain. (2019) 3:137–47. doi: 10.1080/24740527.2019.1620097
22. Syed F, Li W, Relich RF, Russell PM, Zhang S, Zimmerman MK, et al. Excessive matrix metalloproteinase-1 and hyperactivation of endothelial cells occurred in covid-19 patients and were associated with the severity of covid-19. J Infect Dis. (2021) 224:60–9. doi: 10.1093/infdis/jiab167
23. Njoku F, Zhang X, Shah BN, MaChado RF, Han J, Saraf SL, et al. Biomarkers of clinical severity in treated and untreated sickle cell disease: A comparison by genotypes of a single center cohort and African Americans in the NHANES study. Br J Haematol. (2021) 194:767–78. doi: 10.1111/bjh.17682
24. Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. (1994) 330:1639–44. doi: 10.1056/NEJM199406093302303
25. Sanmamed MF and Chen L. A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell. (2018) 175:313–26. doi: 10.1016/j.cell.2018.09.035
26. Chen L and Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. (2013) 13:227–42. doi: 10.1038/nri3405
27. Sharma P and Allison JP. The future of immune checkpoint therapy. Science. (2015) 348:56–61. doi: 10.1126/science.aaa8172
28. Ishida Y, Agata Y, Shibahara K, and Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. (1992) 11:3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x
29. Wei SC, Duffy CR, and Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. (2018) 8:1069–86. doi: 10.1158/2159-8290.CD-18-0367
30. Riva A and Chokshi S. Immune checkpoint receptors: homeostatic regulators of immunity. Hepatol Int. (2018) 12:223–36. doi: 10.1007/s12072-018-9867-9
31. Li YM, Shi YY, Li Y, Yan L, Tang JT, Bai YJ, et al. Soluble Tim-3 and Gal-9 are associated with renal allograft dysfunction in kidney transplant recipients: A cross-sectional study. Int Immunopharmacol. (2018) 55:330–5. doi: 10.1016/j.intimp.2018.01.008
32. Chen D, Peng W, Jiang H, Yang H, Wu J, Wang H, et al. Noninvasive detection of acute renal allograft rejection by measurement of soluble Tim-3 in urine. Mol Med Rep. (2017) 16:915–21. doi: 10.3892/mmr.2017.6670
33. Dahal LN, Basu N, Youssef H, Khanolkar RC, Barker RN, Erwig LP, et al. Immunoregulatory soluble CTLA-4 modifies effector T-cell responses in systemic lupus erythematosus. Arthritis Res Ther. (2016) 18:180. doi: 10.1186/s13075-016-1075-1
34. Ren F, Li J, Jiang X, Xiao K, Zhang D, Zhao Z, et al. Plasma soluble Tim-3 emerges as an inhibitor in sepsis: sepsis contrary to membrane Tim-3 on monocytes. Tissue Antigens. (2015) 86:325–32. doi: 10.1111/tan.12653
35. Simone R, Pesce G, Antola P, Rumbullaku M, Bagnasco M, Bizzaro N, et al. The soluble form of CTLA-4 from serum of patients with autoimmune diseases regulates T-cell responses. BioMed Res Int. (2014) 2014:215763. doi: 10.1155/2014/215763
36. Casati C, Camisaschi C, Rini F, Arienti F, Rivoltini L, Triebel F, et al. Soluble human LAG-3 molecule amplifies the in vitro generation of type 1 tumor-specific immunity. Cancer Res. (2006) 66:4450–60. doi: 10.1158/0008-5472.CAN-05-2728
37. Haile ST, Horn LA, and Ostrand-Rosenberg S. A soluble form of CD80 enhances antitumor immunity by neutralizing programmed death ligand-1 and simultaneously providing costimulation. Cancer Immunol Res. (2014) 2:610–5. doi: 10.1158/2326-6066.CIR-13-0204
38. Zhu X and Lang J. Soluble PD-1 and PD-L1: predictive and prognostic significance in cancer. Oncotarget. (2017) 8:97671–82. doi: 10.18632/oncotarget.18311
39. Heo SK, Ju SA, Kim GY, Park SM, Back SH, Park NH, et al. The presence of high level soluble herpes virus entry mediator in sera of gastric cancer patients. Exp Mol Med. (2012) 44:149–58. doi: 10.3858/emm.2012.44.2.010
40. Gu D, Ao X, Yang Y, Chen Z, and Xu X. Soluble immune checkpoints in cancer: production, function and biological significance. J Immunother Cancer. (2018) 6:132. doi: 10.1186/s40425-018-0449-0
41. Oaks MK and Hallett KM. Cutting edge: A soluble form of CTLA-4 in patients with autoimmune thyroid disease. J Immunol. (2000) 164:5015–8. doi: 10.4049/jimmunol.164.10.5015
42. Li W, Xia Y, Yang J, Guo H, Sun G, Sanyal AJ, et al. Immune checkpoint axes are dysregulated in patients with alcoholic hepatitis. Hepatol Commun. (2020) 4:588–605. doi: 10.1002/hep4.1475
43. Oaks MK, Hallett KM, Penwell RT, Stauber EC, Warren SJ, and Tector AJ. A native soluble form of CTLA-4. Cell Immunol. (2000) 201:144–53. doi: 10.1006/cimm.2000.1649
44. Meng Q, Zaidi AK, Sedy J, Bensussan A, and Popkin DL. Soluble Fc-disabled herpes virus entry mediator augments activation and cytotoxicity of NK cells by promoting cross-talk between NK cells and monocytes. J Immunol. (2019) 202:2057–68. doi: 10.4049/jimmunol.1801449
45. Aboderin FI, Oduola T, Davison GM, and Oguntibeju OO. A review of the relationship between the immune response, inflammation, oxidative stress, and the pathogenesis of sickle cell anaemia. Biomedicines. (2023) 11. doi: 10.3390/biomedicines11092413
46. Marchesani S, Bertaina V, Marini O, Cossutta M, Di Mauro M, Rotulo GA, et al. Inflammatory status in pediatric sickle cell disease: unravelling the role of immune cell subsets. Front Mol Biosci. (2022) 9:1075686. doi: 10.3389/fmolb.2022.1075686
47. Nickel RS, Osunkwo I, Garrett A, Robertson J, Archer DR, Promislow DE, et al. Immune parameter analysis of children with sickle cell disease on hydroxycarbamide or chronic transfusion therapy. Br J Haematol. (2015) 169:574–83. doi: 10.1111/bjh.13326
48. Wherry EJ and Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
49. Gato-Canas M, Zuazo M, Arasanz H, Ibanez-Vea M, Lorenzo L, Fernandez-Hinojal G, et al. Pdl1 signals through conserved sequence motifs to overcome interferon-mediated cytotoxicity. Cell Rep. (2017) 20:1818–29. doi: 10.1016/j.celrep.2017.07.075
50. Zhou L, Chong MM, and Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. (2009) 30:646–55. doi: 10.1016/j.immuni.2009.05.001
51. Wang J, Singh M, Sun C, Besser D, Prigione A, Ivics Z, et al. Author correction: isolation and cultivation of naive-like human pluripotent stem cells based on HERVH expression. Nat Protoc. (2019) 14:2596. doi: 10.1038/s41596-018-0086-6
52. Gorgulho J, Roderburg C, Beier F, Bokemeyer C, Brummendorf TH, Loosen SH, et al. Soluble lymphocyte activation gene-3 (sLAG3) and CD4/CD8 ratio dynamics as predictive biomarkers in patients undergoing immune checkpoint blockade for solid Malignancies. Br J Cancer. (2024) 130:1013–22. doi: 10.1038/s41416-023-02558-7
Keywords: sickle cell disease, soluble immune checkpoints, inflammation, autoantibodies, steady-state condition, vaso-occlusive crisis
Citation: Li W, Pucka AQ, Houran L, Huang X, Debats C, Reyes BA, O’Brien ARW, Yu Q and Wang Y (2025) Soluble immune checkpoint landscape in sickle cell disease links systemic inflammation, autoimmunity, and pain. Front. Hematol. 4:1580009. doi: 10.3389/frhem.2025.1580009
Received: 26 February 2025; Accepted: 18 June 2025;
Published: 14 July 2025.
Edited by:
Mariasanta Napolitano, University of Palermo, ItalyReviewed by:
Roland Fiskesund, Karolinska Institutet (KI), SwedenRosario Hernández Armengol, Cedars Sinai Medical Center, United States
Copyright © 2025 Li, Pucka, Houran, Huang, Debats, Reyes, O’Brien, Yu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Wang, eXdhMTJAaXUuZWR1; Wei Li, d2wxQGl1LmVkdQ==; Qigui Yu, YW5keXVAaXUuZWR1