 Hua Zhou
Hua Zhou Jing Zhao*
Jing Zhao*- Department of Hematology and Oncology, Affiliated Changzhou Children’s Hospital of Nantong University, Changzhou, Jiangsu, China
Objective: To explore the clinical and genetic mutation characteristics of familial platelet disorder with acute myeloid leukemia tendency (FPD/AML) caused by RUNX1 germline mutations.
Methods: The clinical data and gene mutation results of a child with FPD/AML and family members admitted to Changzhou Children’s Hospital on 8 September 2022 were analyzed.
Results: A 1-year and 11-month-old male patient had recurrent thrombocytopenia for 1 year, with scattered bleeding points throughout the body. Blood routine tests showed thrombocytopenia, and peripheral blood smears and bone marrow cytology tests showed no significant abnormalities. The gene sequencing results showed that the patient carried a frameshift mutation in RUNX1: c.494delG (p.G165Vf*11). Sanger sequencing, which was used for family verification analysis, revealed that the mutation in the patient was inherited from his mother, and his grandmother had similar clinical symptoms.
Conclusions: These findings expand the spectrum of RUNX1 variants linked to familial platelet disorders. The familial platelet disorder (FPD) associated with this gene is clinically rare and has the potential to undergo malignant transformation. Continuous monitoring of biological evolution is essential, and early intervention and treatment may be warranted.
1 Introduction
The RUNX1 gene is located on chromosome 21q22.3 and spans approximately 261 kb. It is one of the members of the RUNX transcription factor family, which includes RUNX1, RUNX2, and RUNX3. This gene plays a crucial role in determining cell lineage differentiation, the formation of normal hematopoietic cells, and the proliferation of stem cells (1–3). Allelic mutations of the RUNX1 gene, including deletions, missense mutations, splicing alterations, frameshift mutations, and nonsense mutations, can lead to a rare autosomal recessive genetic disorder known as familial platelet disorder (FPD) (4). Mutations in RUNX1 are also significant in prognostic assessments of patients with AML. The World Health Organization (WHO) has categorized AML with RUNX1 mutations as a separate entity, currently termed “AML with RUNX1 mutations,” to highlight its clinical significance in AML (5). This study employed whole-exome sequencing (WES) to identify a RUNX1-FPD pedigree, which is reported herein.
2 Materials and methods
2.1 General information
The male patient, aged 1 year and 11 months, was the first child, born at term through a normal delivery, with a birth weight of 3.6 kg. He was admitted to the hospital after “noticing generalized petechiae for over a year.” Since birth, the child had exhibited scattered petechiae across his body that did not blanch on pressure and would resolve on their own; however, the family did not pay much attention to it. Four months prior to admission, the parents sought medical advice at a local hospital where a bone marrow cytology examination led to a diagnosis of “immune thrombocytopenia,” but no special treatment was administered, and the platelet count was maintained between 60 and 90 × 109/L. On physical examination, the child appeared well, with no palpable superficial lymph nodes, scattered pinhead-sized petechiae on the abdomen, and a few ecchymoses on the lower limbs. Examination of the heart, lungs, and abdomen showed no abnormalities. The results of the auxiliary examination revealed the following: blood routine test showed white blood cells at 6.35 × 109/L, hemoglobin at 111 g/L, and platelets at 69 × 109/L; and the other tests conducted (liver and kidney function, myocardiac zymogram, coagulation function, full set of antinuclear antibodies, immunoglobulins, and urine/feces) showed no significant abnormalities. Electrocardiogram and imaging of the liver, spleen, kidneys, and abdominal masses also showed no significant abnormalities.
2.2 Family information
The proband’s parents, brothers, sisters, and grandparents were interviewed regarding their clinical manifestations and medical history, including any occurrences of purpura, ecchymoses, gingival bleeding, epistaxis, and blood in stool. They also underwent blood routine tests, including platelet count and microscopic analysis of peripheral blood cell morphology, coagulation function, liver and kidney function, and autoantibodies, along with a rapid whole-exome sequencing test for the entire family.
3 Results
3.1 Laboratory tests
The investigation revealed that both the proband’s mother and maternal grandmother exhibited varying degrees of bleeding tendencies, including frequent skin bruising and heavy menstrual flow. The mother had a healthy pregnancy without any history of spontaneous miscarriage. There was no history of malignancy or congenital hereditary diseases reported. Blood routine tests showed that both the proband’s mother and grandmother had thrombocytopenia, while the proband’s father and grandfather had normal platelet counts and no notable abnormalities in platelet morphology under microscopy. Coagulation profiles, liver and kidney functions, and autoantibody panels were all normal for all four individuals.
Genetic testing was conducted using high-throughput sequencing (NGS) followed by Sanger sequencing for confirmation. Figure 1 shows that the proband, his mother, and his grandmother all possess the RUNX1 gene frameshift mutation c.494delG/p.G165Vfs*11. This mutation results in premature termination of the polypeptide chain synthesis at a highly conserved RUNT domain (shown in Figure 2), lacking subsequent amino acid sequences and affecting the gene’s function. The protein structure prediction diagram was generated using the SWISS-MODEL, showcasing the three-dimensional structures of both the wild-type and mutant proteins. Due to a frameshift mutation (highlighted in red) in the mutant protein, the C-terminal sequence has undergone significant alterations, which may affect the protein’s folding and function. The C-terminal structure of the mutant protein exhibits an irregular conformation, potentially associated with loss of function or decreased protein stability. In the diagram, the mutant protein is represented in yellow, the wild-type protein in green, and the frameshift mutation region of the mutant protein in red. Although the structure prediction tool provides valuable structural insights, the predicted results still require further experimental validation (shown in Figure 3). According to the ACMG guidelines, this variant is classified as pathogenic (PVS1+PS4_Supporting+PM2_Supporting). PVS1: This variant may result in the premature termination of polypeptide chain synthesis, thereby affecting gene function. Loss of function (LOF) serves as the pathogenic mechanism underlying the disease. PS4_Supporting: The variant demonstrated phenotypic enrichment in at least two patients, consistent with a dominant disease model, and was concordant with the disease phenotype associated with the implicated gene. PM2_Supporting: This variant was not identified in the dbSNP, 1000 Genomes, or gnomAD databases. The presence of the same heterozygous RUNX1 mutation across two generations, with no other known pathogenic variants associated with FPD identified, is consistent with an autosomal dominant inheritance pattern. The diagnosis of FPD with a predisposition to acute myeloid leukemia (FPD/AML) is confirmed due to the RUNX1 gene mutation.
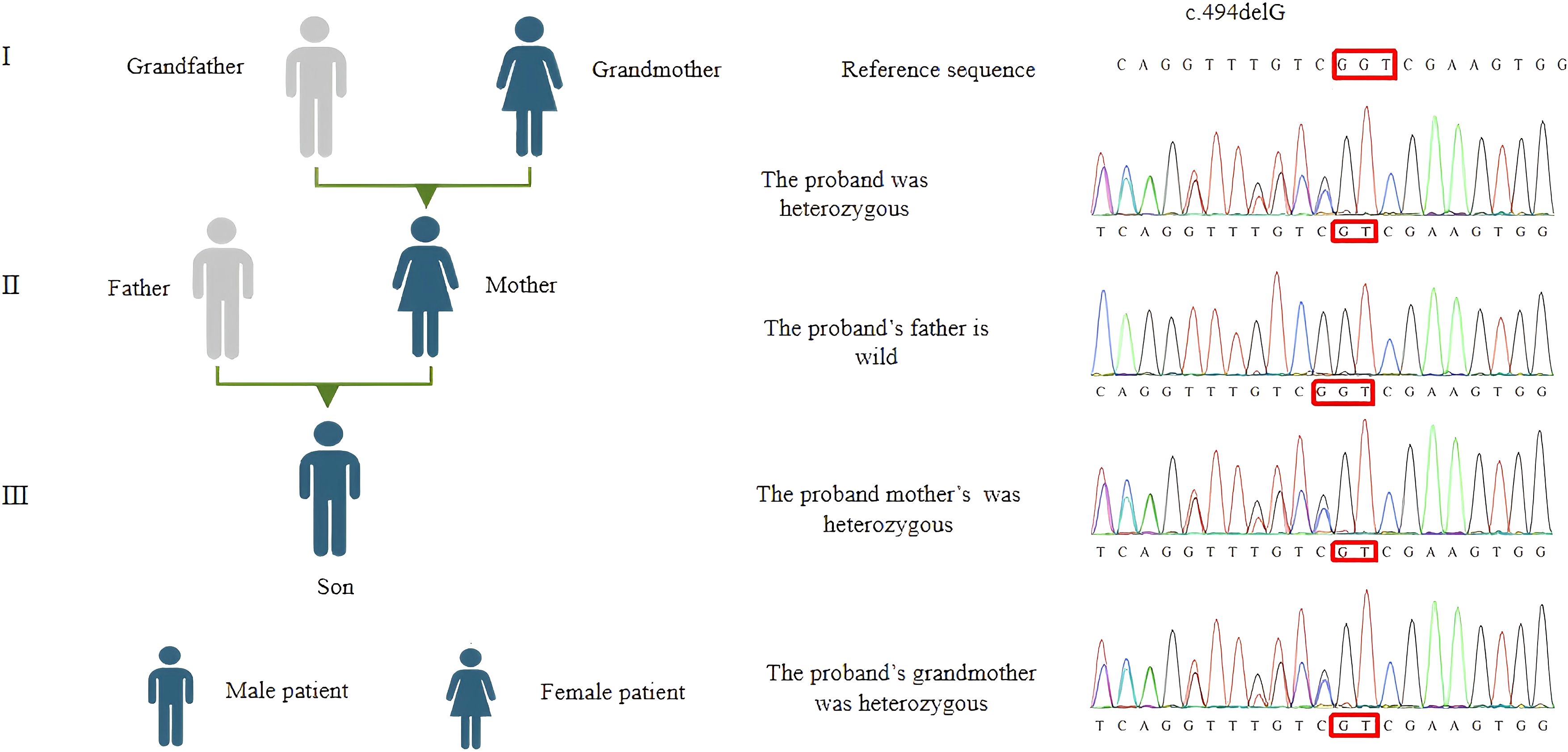
Figure 1. The proband’s family tree and RUNX1 gene sequencing results.
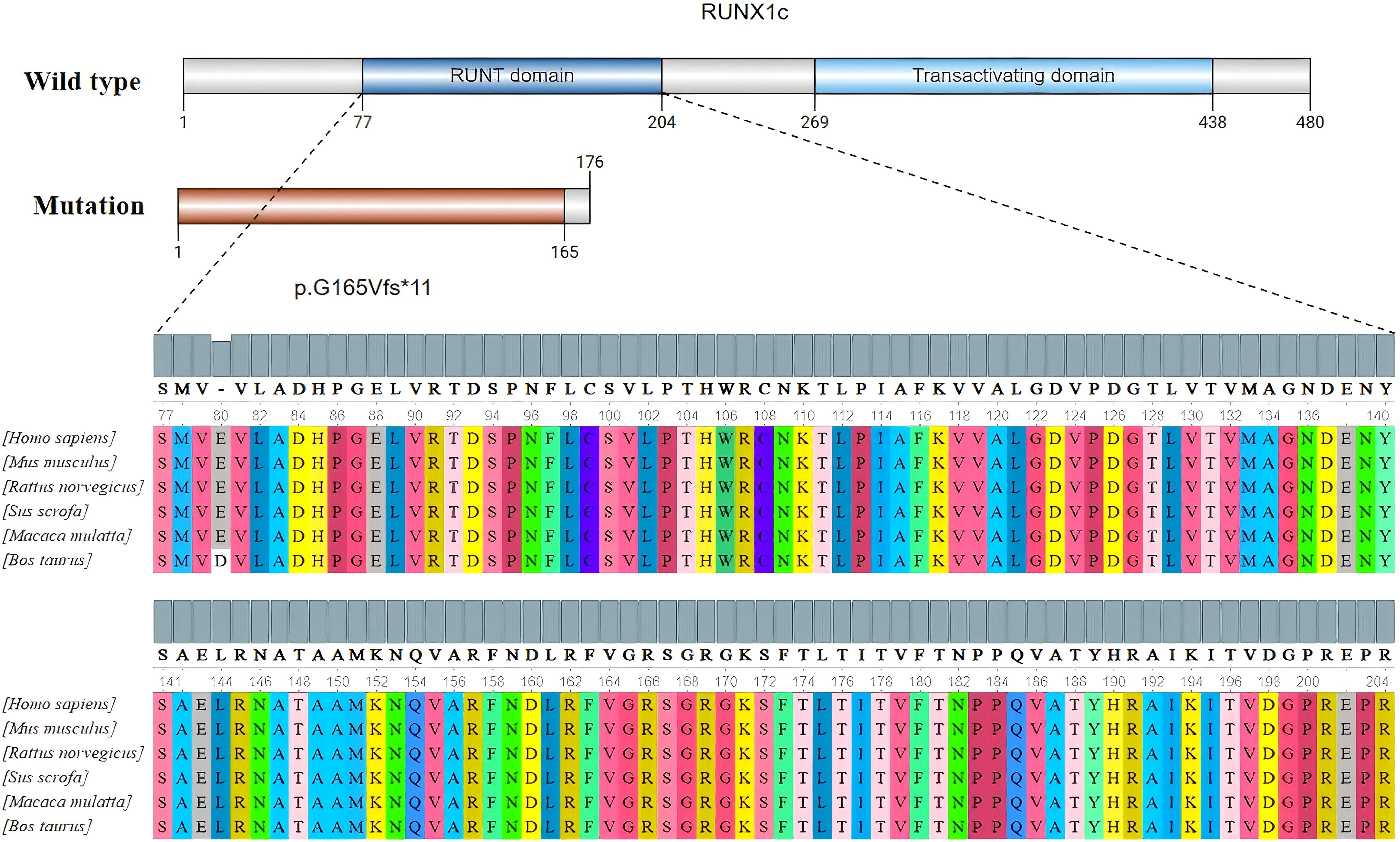
Figure 2. RUNX1 secondary structure and RUNT domain conservation analysis.
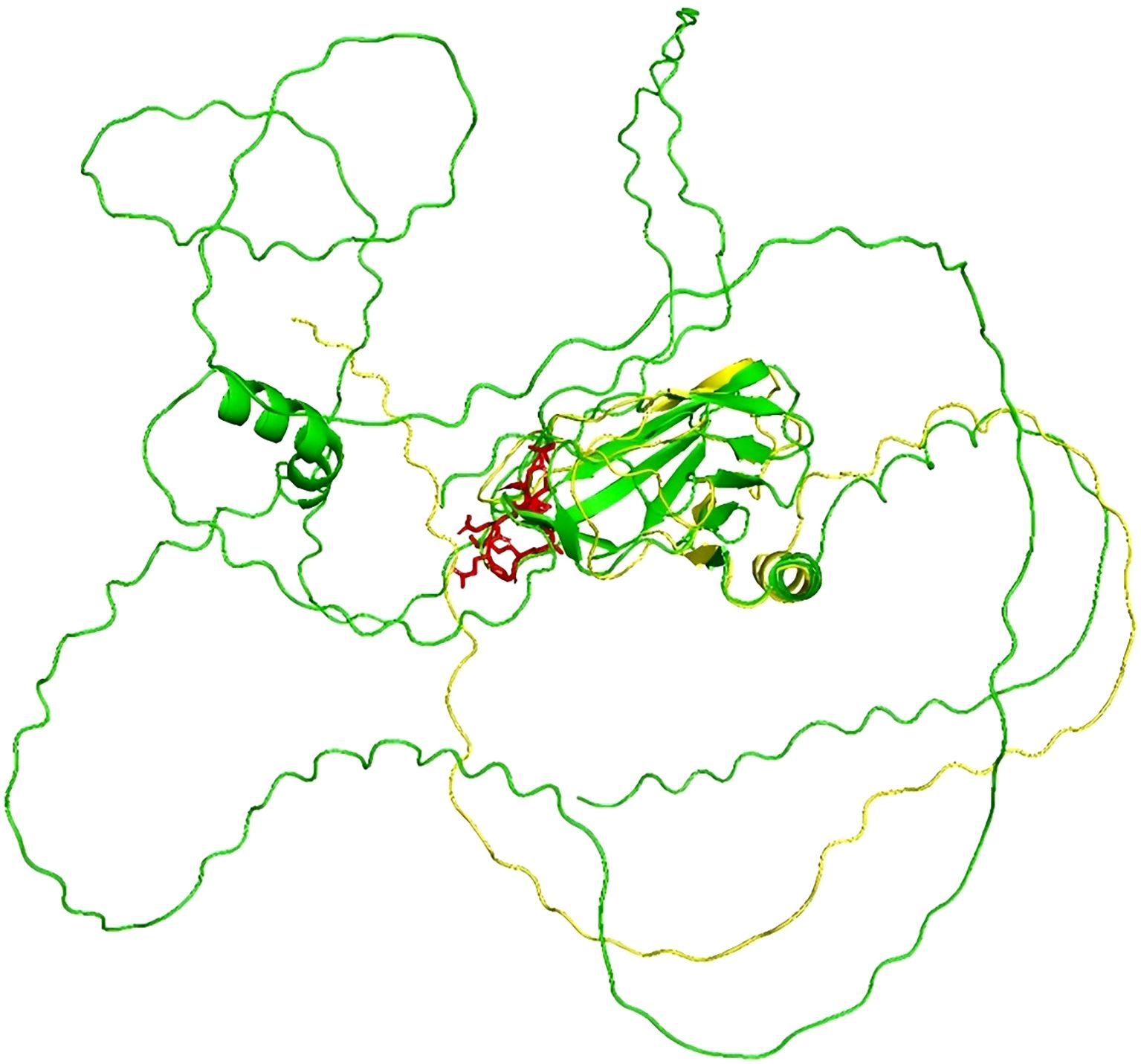
Figure 3. The protein structure prediction diagram.
4 Discussion
Familial platelet disorder with a predisposition to acute myeloid leukemia (FPD/AML; OMIM #601399) is an autosomal dominant disorder caused by mutations in the RUNX1 gene. This condition is characterized by thrombocytopenia, with or without functional impairment, and carries a significant risk of progression to hematological malignancies, particularly AML (6, 7). The earliest report of FPD/AML was in 1978 by Luddy et al., who described a family where early-onset thrombocytopenia and platelet dysfunction were observed, with three siblings eventually succumbing to myeloproliferative tumors (8). In 1999, Song and colleagues first described germline heterozygous mutations in RUNX1 across six families, each carrying different mutations (9). To date, more than 130 families with one to four different germline RUNX1 mutations have been reported (10).
The RUNX protein family comprises highly conserved transcription factors crucial for lineage-specific gene expression during development and essential for cell proliferation and differentiation. In mammals, this family has three members, namely, RUNX1, RUNX2, and RUNX3, in which RUNX1 is a key regulator in hematopoiesis and a common target of various chromosomal translocations in human leukemia.
The type of RUNX1 mutation (haploinsufficiency and dominant negative effects) or the location of the mutation influences the tendency to transform into leukemia (11), with most mutations causing haploinsufficiency. Frameshift mutations and large deletions typically result in haploinsufficiency, which often leads to thrombocytopenia, whereas gene deletions are more likely to lead to leukemia. The family in question carries the RUNX1 gene c.494delC (p.G165Vfs*11) frameshift mutation, displaying clinical manifestations of thrombocytopenia and skin ecchymosis, consistent with patient phenotypes reported in the literature.
FPD/AML is a distinct subtype within IT (12). First described by Wessi and colleagues in 1969, it is characterized by mild to moderate reduction in platelet counts, abnormal platelet aggregation, easy bleeding, and a high tendency to progress to hematological malignancies (13). In 1998, germline mutations in the core transcription factor RUNX1 were identified as the cause of FPD/AML (12). Besides germline mutations, somatic mutations in RUNX1 are also seen in patients with isolated thrombocytopenia without a family history of FPD/AML (14). These patients are often misdiagnosed or overlooked as having immune thrombocytopenia (ITP), showing poor response to steroids and immunomodulatory treatments, and even undergoing splenectomy.
Currently, there is no uniform treatment protocol for RUNX1-FPD, with very few reports on treatment strategies and prognosis. Allogeneic hematopoietic stem cell transplantation (allo-HSCT) offers a potential cure for the disease. DiFilippo and colleagues reported three cases of RUNX1-FPD-associated myeloid tumors achieving sustained complete remission (CR) after HSCT (15). Christopher et al. conducted experiments on mice and found that knocking out or inhibiting RUNX1 can induce more apoptosis in AML cells expressing mtRUNX1, thereby improving the survival rate of AML mice expressing mtRUNX1 (16). Lachowies and others reported that among 12 RUNX1-FPD patients, five who underwent allo-HSCT had a median survival of approximately 2.9 years, with significant benefits seen in MDS patients (17). Limited data suggest that hematopoietic stem cell transplantation may benefit such patients, although the number of cases is too small and may be biased. The child in this case has been followed for more than 6 months, occasionally showing bleeding spots on the trunk that resolve on their own, with platelet counts maintained at 80 × 10 (9)/L and no significant abnormalities in other lineages, without special treatment. It is estimated that germline RUNX1 mutations result in the well-described autosomal-dominant familial platelet disorder with predisposition to hematologic malignancies (RUNX1-FPD, FPD/AML, FPDMM); ∼44% of affected individuals progress to AML or myelodysplastic syndromes (18). Given that neither the patient’s mother nor grandmother exhibited progression to leukemia, this may be indicative of a potential protective factor.
In summary, RUNX1 mutations are highly prevalent in thrombocytopenia (including recurrent refractory ITP), and thrombocytopenia carrying these mutations has a high risk of transforming into malignant hematological diseases. Therefore, it is necessary to perform NGS testing for recurrent refractory ITP and closely monitor patients positive for RUNX1 mutations. Particularly for patients who have already experienced thrombocytopenia, there should be high vigilance for malignant hematological diseases, especially the potential transformation into MDS and AML myeloid tumors.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Changzhou Children’s Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
HZ: Conceptualization, Writing – original draft. JZ: Writing – review & editing. XG: Methodology, Resources, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by Nantong University Special Research Fund for Clinical Medicine (Grant No. 2024LY027).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Miyoshi H, Shimizu K, Kozu T, Maseki N, Kaneko Y, Ohki M, et al. t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proc Natl Acad Sci U S A. (1991) 88:10431–4. doi: 10.1073/pnas.88.23.10431
2. Link KA, Chou FS, and Mulloy JC Core binding factor at the crossroads: determining the fate of the HSC. J Cell Physiol. (2010) 222:50–6. doi: 10.1002/jcp.21950
3. Kim W, Barron DA, San Martin R, Chan KS, Tran LL, Yang F, et al. RUNX1 is essential for mesenchymal stem cell proliferation and myofibroblast differentiation. Proc Natl Acad Sci U S A. (2014) 111:16389–94. doi: 10.1073/pnas.1407097111
4. Gaidzik VI, Teleanu V, Papaemmanuil E, Weber D, Paschka P, Hahn J, et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia. (2016) 30:2282. doi: 10.1038/leu.2016.207
5. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
6. Dowton SB, Beardsley D, Jamison D, Blattner S, and Li FP Studies of a familial platelet disorder. Blood. (1985) 65:557–63. doi: 10.1182/blood.V65.3.557.557
7. Minelli A, Maserati E, Rossi G, Bernardo ME, De Stefano P, Cecchini MP, et al. Familial platelet disorder with propensity to acute myelogenous leukemia: genetic heterogeneity and progression to leukemia via acquisition of clonal chromosome anomalies. Genes Chromosomes Cancer. (2004) 40:165–71. doi: 10.1002/gcc.20030
8. Luddy RE, Champion LA, and Schwartz AD A fatal myeloproliferative syndrome in a family with thrombocytopenia and platelet dysfunction. Cancer. (1978) 41:1959–63. doi: 10.1002/1097-0142(197805)41:5<1959::AID-CNCR2820410540>3.0.CO;2-8
9. Song WJ, Sullivan MG, Legare RD, Hutchings S, Tan X, Kufrin D, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. (1999) 23:166–75. doi: 10.1038/13793
10. Brown AL, Arts P, Carmichael CL, Babic M, Dobbins J, Chong CE, et al. RUNX1-mutated families show phenotype heterogeneity and a somatic mutation profile unique to germline predisposed AML. Blood Adv. (2020) 4:1131–44. doi: 10.1182/bloodadvances.2019000901
11. Antony-Debré I, Manchev VT, and Balayn N Level of RUNX1 activity is critical for leukemic predisposition but not for thrombocytopenia. Blood. (2015) 125:930–40. doi: 10.1182/blood-2014-06-585513
12. Michaud J, Wu F, Osato M, Cottles GM, Yanagida M, Asou N, et al. In vitro analyses of known and novel RUNX1/AML1 mutations in dominant familial platelet disorder with predisposition to acute myelogenous leukemia: implications for mechanisms of pathogenesis. Blood. (2002) 99:1364–72. doi: 10.1182/blood.v99.4.1364
13. Weiss HJ, Chervenick PA, Zalusky R, and Factor A A familial defect in platelet function associated with impaired release of adenosine diphosphate. N Engl J Med. (1969) 281:1264–70. doi: 10.1056/NEJM196912042812303
14. Sood R, Kamikubo Y, and Liu P Role of RUNX1 in hematological Malignancies. Blood: J Am Soc Hematol. (2018) 3):131.
15. DiFilippo EC, Coltro G, Carr RM, Mangaonkar AA, Binder M, Khan SP, et al. Spectrum of abnormalities and clonal transformation in germline RUNX1 familial platelet disorder and a genomic comparative analysis with somatic RUNX1 mutations in MDS/MPN overlap neoplasms. Leukemia. (2020) 34:2519–24. doi: 10.1038/s41375-020-0752-x
16. Mill CP, Fiskus W, DiNardo CD, Qian Y, Raina K, Rajapakshe K, et al. RUNX1-targeted therapy for AML expressing somatic or germline mutation in RUNX1. Blood. (2019) 134:59–73. doi: 10.1182/blood.2018893982
17. Lachowiez C, Bannon S, Loghavi S, Wang F, Kanagal-Shamanna R, Mehta R, et al. Clonal evolution and treatment outcomes in hematopoietic neoplasms arising in patients with germline RUNX1 mutations. Am J Hematol. (2020) 95:E313–5. doi: 10.1002/ajh.25965
Keywords: thrombocytopenia, RUNX1, FPD/AML, frameshift mutation, sequencing
Citation: Zhou H, Zhao J and Gu X (2025) Case report: Clinical characteristics and genetic analysis of a familial platelet disorder associated with RUNX1 gene mutation. Front. Hematol. 4:1608314. doi: 10.3389/frhem.2025.1608314
Received: 08 April 2025; Accepted: 28 May 2025;
Published: 23 June 2025.
Edited by:
John Strouboulis, King’s College London, United KingdomReviewed by:
Richard Oscar Francis, Columbia University, United StatesRoy Drissen, University of Oxford, United Kingdom
Nehal Joshi, Imperial College London, United Kingdom
Copyright © 2025 Zhou, Zhao and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Zhao, MTMzMzk0MzZAcXEuY29t