 Joaquín Jerez
Joaquín Jerez Caterina Hernandez2,3
Caterina Hernandez2,3 Charlotte N. Hill
Charlotte N. Hill- 1Hematology Department, Fundación Arturo Lopez Perez, Santiago, Chile
- 2Centro de Investigación e Innovación Biomédica, Facultad de Medicina, Universidad de los Andes, Santiago, Chile
- 3IMPACT, Center of Interventional Medicine for Precision and Advanced Cellular Therapy, Santiago, Chile
Therapy-related acute myeloid leukemia (t-AML) is a complex clinical entity characterized by its association with prior exposure to cytotoxic agents or radiotherapy. Recent analyses have unveiled the intricate molecular landscape of t-AML, revealing a heterogeneous genetic profile marked by mutations in TP53, PPM1D, and other genes. While t-AML does not constitute a distinct molecular entity, its prognostic impact is integrated into current risk classifications, with particular emphasis on TP53 mutations. Treatment strategies should be guided by the underlying biology of the disease rather than solely by clinical history. The significance of t-AML lies in its role as a qualifying condition rather than an independent disease entity. Its association with germline mutations and clonal hematopoiesis of indeterminate potential represents an emerging and promising field for developing preventive and monitoring strategies. The standard therapeutic approach for t-AML has evolved, with promising alternatives emerging. The CPX-351 regimen has demonstrated superior outcomes compared to conventional 3 + 7 induction therapy in selected patients. The incorporation of Venetoclax, both in combination with hypomethylating agents and in high- or low-intensity chemotherapy regimens, has shown efficacy in high-risk AML, including t-AML cases; however, its validity must be confirmed in prospective studies. Allogeneic hematopoietic stem cell transplantation remains a crucial consolidation strategy. Participation in clinical trials is of paramount importance to optimize management strategies for this high-risk AML subset.
Introduction
From a biological standpoint, therapy-related acute myeloid leukemia (t-AML) refers to a form of AML that arises because of prior exposure to therapeutic treatments, primarily cytotoxic in nature. Although t-AML most commonly arises after treatment for solid or hematologic malignancies, it can also develop following therapy for non-malignant conditions, particularly inflammatory disorders (1–4). It has been reported that 5-10% of AML cases are related to previous chemotherapy. These treatments predispose to DNA damage and are frequently associated with high-risk cytogenetic alterations and an unfavorable prognosis (1, 5). However, the clinical conceptualization of this entity has evolved over time.
The 2016 World Health Organization (WHO) classification recognized therapy-related myeloid neoplasms as a distinct category, including both AML and myelodysplastic syndromes (MDS) (6). However, the following 2022 revision of the WHO Classification has retained a subset designated as “secondary myeloid neoplasms”, however, for AML the attribute “post-cytotoxic therapy” was instructed to be used as a new criterion for AML classification (7). As shown in Table 1, this category includes conventional chemotherapy (excluding methotrexate) and broad-field radiotherapy (though its use is decreasing in favor of conformational radiotherapy techniques). Notably, poly(ADP-ribose) polymerase (PARP) inhibitors have been added to this classification, while immunomodulators such as lenalidomide are excluded.
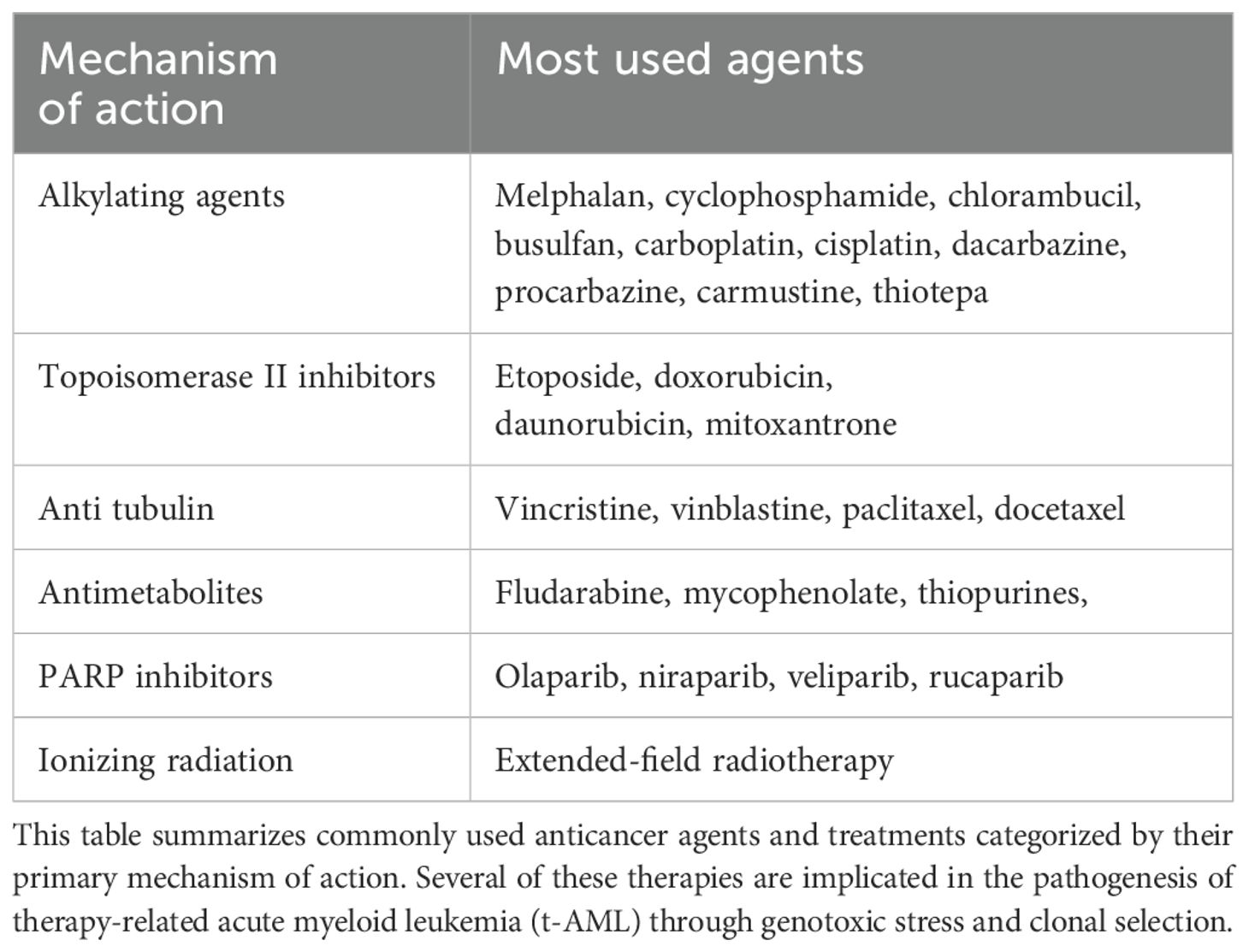
Table 1. Common cytotoxic and genotoxic therapies associated with therapy-related AML.
In 2022 the International Consensus Classification (ICC) eliminated the distinct category of therapy-related AML, instead considering it a qualifier (8). This decision is largely based on its frequent overlap with entities exhibiting myelodysplasia-related cytogenetic or molecular alterations, as well as with the newly proposed TP53-mutated entity. Similarly to the latest WHO classification, the ICC 2022 also distinguishes among prior exposures such as chemotherapy, radiotherapy, and immune interventions.
In alignment with ICC 2022, the European LeukemiaNet (ELN) 2022 recommendations adopted these guidelines, emphasizing that genetic characteristics hold greater significance in defining biologically distinct entities rather than focusing on clinical history (9). Conceptually, t-AML is now regarded as a qualifier providing valuable epidemiological and biological insights, which will be further explored in subsequent sections.
Epidemiology of t-AML
Various series estimate that approximately 10% of AML cases have a history of prior therapy (10). Moreover, after conventional chemotherapy, between 0.8% and 6.3% of patients will develop a myeloid neoplasm within a 20-year follow-up, with this probability increasing to 10-20% in those undergoing autologous transplantation (10). A combined analysis of the German Austrian AMLSG registry, including 3,026 patients, identified that 7% of cases corresponded to t-AML (n=200) (3). This patient subgroup tends to be older than de novo AML cases and exhibits a higher proportion of female patients, likely due to frequent chemotherapy exposure in breast cancer patients. Approximately 70% of primary neoplasms in this cohort comprised breast cancer and lymphomas, with a median latency period of four years. Latency duration varied by exposure type; patients treated with anthracyclines exhibited a shorter latency period (1.9 years) and a higher prevalence of KMT2A rearrangements. Similarly, a Danish registry of 3,055 patients detected a 6.6% prevalence of t-AML (n=203), with breast cancer and lymphomas accounting for approximately 70% of cases (5). The latency period was four to five years. Notably, this registry observed lower induction response rates compared to de novo AML, although early mortality did not increase.
The Swedish registry, comprising 3,363 patients, reported a 7.7% prevalence (n=259), again demonstrating a higher proportion of female patients (1). In this case, breast cancer and lymphomas represented approximately 50% of underlying causes. The Spanish PETHEMA registry, encompassing 8,521 patients, identified a 5.8% prevalence of t-AML (n=502), with a latency period of five years (11). Similarly to the Swedish registry, in the Spanish cohort breast cancer and lymphomas were the most frequent primary neoplasms, accounting for roughly 40% of cases.
For patients with t-AML secondary to isolated radiotherapy, a combined series from MD Anderson Cancer Center and Massachusetts General Hospital found that prostate and breast cancer were the primary etiologies (12). The relative (and absolute) proportion of t-AML cases is rising, likely due to increased survival and improved outcomes in solid tumor treatments such as breast and prostate cancer. The Swedish registry observed an increase in t-AML incidence, rising from approximately 8% to 16% of total AML cases in recent years (13). These findings are summarized in Table 2.
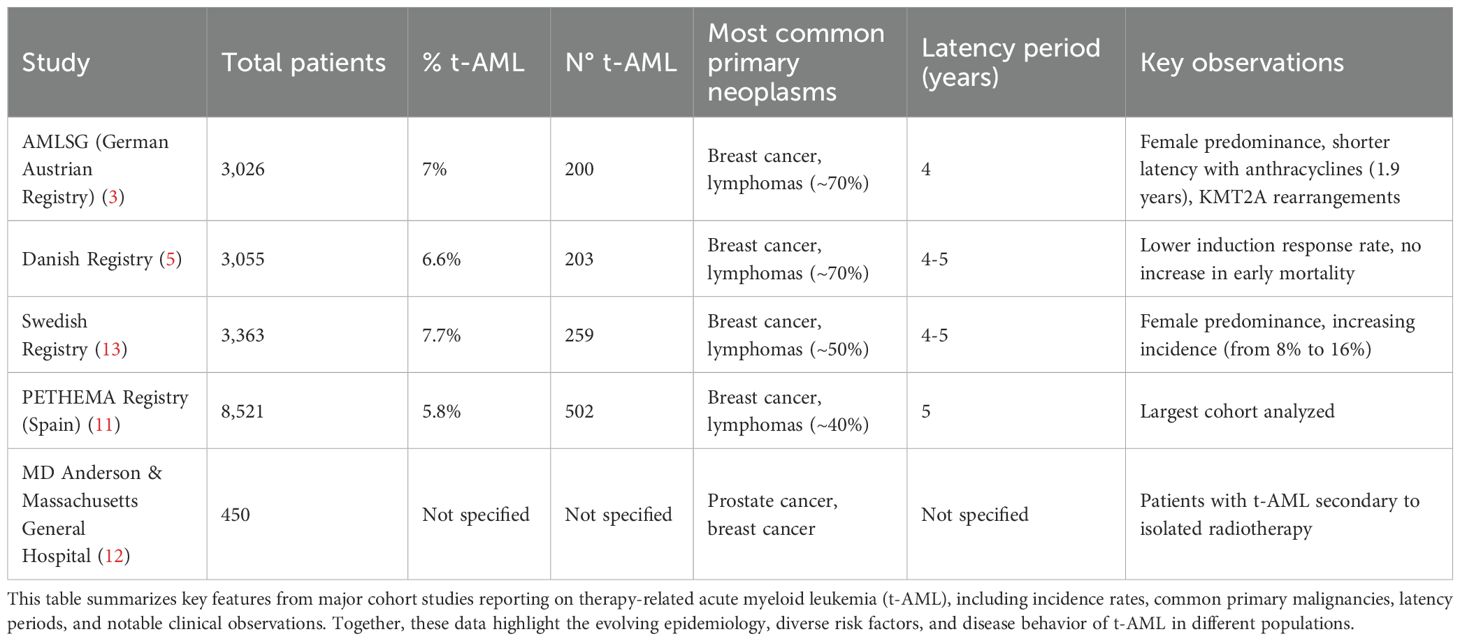
Table 2. Characteristics of therapy-related AML across large institutional and population-based cohorts.
Despite geographic variations, registries consistently report similar clinical characteristics among European populations. In Latin America, there is no specific cohort data focused on t-AML, however, there is a higher mortality compared to developed countries (14). Moreover, there are limited and restricted genomic studies to determine frequent mutations and their association with disease progression or treatment response (14).
Genetic landscape of t-AML
The first available records indicate that patients with therapy-related acute myeloid leukemia (t-AML) exhibit a higher prevalence of altered karyotypes compared to de novo AML cases (75% vs. 51%, respectively) (3). This increase in the frequency of cytogenetic alterations is of prognostic significance, as it is associated with high-risk modifications, such as complex karyotypes, chromosomal monosomies, alterations in chromosomes 5, 7, and 17, as well as rearrangements of the KMT2A gene. In contrast, core binding factor AML cases exhibit a similar proportion to those observed in de novo AML cases (3). In a series of patients with therapy-related myeloid neoplasms from MD Anderson Cancer Center (15), it was reported that 41% of cases had complex karyotypes, 25% showed alterations on chromosome 5q/-5, 20% on chromosome 7q/-7, 16% had alterations on chromosome 11q23, 13% on chromosome 17p/17, and only 6% exhibited core binding factor translocations. The Nordic registries and the PETHEMA registry also report a higher proportion of patients with adverse cytogenetics, accounting for 40-50% of cases (1, 5, 11). This suggests that conventional chemotherapy induces chromosomal instability, promoting both structural and numerical alterations.
In patients exposed to therapy with isolated radiotherapy, the cytogenetic alterations resemble those observed in de novo AML cases (12). This finding is significant, as in the previously mentioned registries, approximately one-third of the patients were exclusively treated with radiotherapy. This initial data provided the first clue to address the question of whether isolated radiotherapy, in modern times, plays a pathogenic role in the pathogenesis of t-AML.
A comprehensive analysis of 352 patients with therapy-related myeloid neoplasms revealed that the most frequently detected mutations were TP53 (37%), followed by PPM1D (19%), TET2 (16%), DNMT3A (15%), and RUNX1 (13%) (15). In this context, it was observed that 61% of TP53 mutations presented multihit alterations, while PPM1D mutations tended to be subclonal, with small allele variant frequencies (VAFs). When comparing therapy-related acute myeloid leukemia (t-AML) with de novo AML, it was evident that the following molecular alterations were more prevalent in t-AML: PPM1D, TP53, and SETBP1 (15). In contrast, the following alterations were more frequently observed in de novo AML: STAG2, CEBPA, NPM1, IDH1/2, FLT3, ASXL1, NRAS, and DNMT3A. This higher prevalence of TP53 mutations has also been corroborated in other series (13, 14). The lower representation of NPM1 and FLT3 also aligns with data from the Nordic and PETHEMA registries (1, 5, 11), where the prevalence of NPM1 is around 20% and FLT3 around 10%.
The most prominent pivotal study in this area was conducted by Lindsley et al. (16). In this study, a cohort of patients from the ACCEDE trial with AML and a strict history of myelodysplastic syndrome/chronic myelomonocytic leukemia underwent massive sequencing. When comparing these results with those from de novo AML in the Cancer Genome Atlas cohort, three mutually exclusive mutation groups were generated: a) Mutations with high specificity for secondary AML: SRSF2, SF3B1, U2AF1, ZRSR2, ASXL1, EZH2, BCOR, and STAG2. b) Mutations in TP53. c) De novo/pan-AML alterations: Three alterations were strongly associated with de novo mutations: NPM1, nuclear binding factors, and rearrangements on 11q23. The remaining alterations were classified as “pan-AML.” Subsequently in the same study, a cohort of 101 patients from the ACCEDE trial with t-AML and mass sequencing data was analyzed. The most common mutations were TP53, DNMT3A, and TET2. What was novel in this study was the application of the classification previously described by the authors. It was found that 23% corresponded to TP53, 33% to secondary AML, and 47% to de novo/pan-AML. An analysis was conducted to determine whether t-AML constituted its own genetic group or fit into the proposed genetic classification. The conclusion was that prior therapy exposure does not define a genetically distinct group. Rather, t-AML can be assigned to the three proposed groups. Furthermore, it was observed that exposure to topoisomerase 2 inhibitors, which increase the risk of 11q23 rearrangements (considered de novo), is a contributing factor. Therefore, from a hierarchical perspective, mutational analysis encompasses the history of therapy.
Despite the initial classifications that grouped cases into two main categories: those related to alkylating agents or topoisomerase II inhibitors, this classification is somewhat impractical, as many cytotoxic therapy protocols use combinations of these drugs. In this regard, the previously mentioned series from MD Anderson provides essential data on the specific associations between therapies and molecular and cytogenetic elements (12) (Box 1).
Box 1. Associations between prior cancer therapy and genomic alterations in t-AML.
● Platinum-based drugs are cytogenetically associated with complex karyotypes, deletions on chromosome 17p/-17, deletions on chromosome 7q/-7, deletions on chromosome 5q/-5, inversion of chromosome 3 (inv(3)), and a lower likelihood of diploid karyotypes. From a molecular perspective, they are primarily associated with PPM1D.
● Alkylating agents increase the risk of alterations on 11q23 and decrease the risk of diploid karyotypes. Additionally, they significantly reduce the risk of mutations in NPM1, IDH2, and TET2.
● Anthracyclines and topoisomerase inhibitors increase the risk of alterations on 11q23 and inversion of chromosome 3 (inv(3)), while decreasing the risk of deletions on chromosome 5q/-5. They also reduce the risk of mutations in TP53, SRSF2, NRAS, and TET2.
● Vinca alkaloids increase the risk of deletions on chromosome 7q/-7, decrease the risk of alterations on 11q23, increase the risk of mutations in TP53, NRAS/KRAS, and EZH2, and decrease the risk of IDH2 mutations.
● Taxanes decrease the risk of mutations in NPM1 and splicing mutations.
● Immunomodulatory drugs (thalidomide analogs) increase the risk of deletions on chromosome 17p/-17, deletions on chromosome 7q/-7, and mutations in TP53, with the latter association representing the highest risk. Moreover, it was observed that this risk is time-dependent, with a greater risk of TP53 mutation associated with longer exposure.
In patients exposed to therapy with isolated radiotherapy, the cytogenetic alterations resemble those observed in de novo AML cases (12). This finding is significant, as in the previously mentioned registries, approximately one-third of the patients were exclusively treated with radiotherapy. This initial data provided the first clue to address the question of whether isolated radiotherapy, in modern times, plays a pathogenic role in the pathogenesis of t-AML.
Due to the nature of prior therapies, primary malignancies are associated with specific subsequent alterations, such as breast cancer with 11q23 abnormalities, lymphomas with chromosome 7q/-7 deletions, and multiple myeloma with TP53 mutations (15). It has been demonstrated that lenalidomide (but not pomalidomide) promotes the selection of hematopoietic stem cells harboring pre-existing TP53 mutations (15). The importance of genetic analysis, particularly regarding the negative association between chemotherapy use and mutations such as NPM1, IDH2, ASXL1, and splicing-related genes, could help differentiate cases where chemotherapy plays a true pathological role from those where its exposure represents a mere epiphenomenon (15).
PARP inhibitors warrant particular attention, given their explicit inclusion in the 2022 WHO classification. A meta-analysis of randomized controlled trials (RCTs) from the WHO pharmacovigilance program revealed a significant increase in the incidence of both AML and MDS associated with these agents (17). This phenomenon is predominantly observed in patients with a history of underlying ovarian cancer. Numerically, this translates to an incidence of 0.83%, with a latency period of approximately 20 months from exposure to myeloid neoplasia diagnosis. Notably, this risk is independent of the presence of BRCA1/2 mutations.
PARP inhibitors act by suppressing DNA repair pathways, thereby inducing apoptosis in cancer cells, particularly those incapable of homologous recombination repair (18). In a case series of patients who developed therapy-related AML, 100% exhibited adverse cytogenetic abnormalities (19). Of these, 95% harbored complex karyotypes, with 75% carrying TP53 mutations. In response to this observation, a study focusing on Clonal Hematopoiesis of Indeterminate Potential (CHIP) prior to PARP inhibitor therapy initiation revealed a 45% prevalence of TP53 mutations among patients who later developed therapy-related AML (20). This exposure correlated with clonal expansion, as evidenced by an increase in the variant allele frequency, a mechanism analogous to that observed with lenalidomide (15).
Given the evidence of positive selection for pre-mutated TP53 clones following lenalidomide and PARP inhibitor therapy, evaluating CHIP in patients undergoing cytotoxic treatment emerges as a pertinent clinical question. A study from Memorial Sloan Kettering Cancer Center (MSKCC) analyzing CHIP in patients with solid tumors found a 25% prevalence—significantly higher than that observed in the healthy population (21). The most frequently mutated genes were TET2, PPM1D, ASXL1, ATM, and TP53, collectively accounting for 60% of CHIP cases. Supporting this concept, a case-control study of over 50,000 patients demonstrated that those who developed therapy-related myeloid neoplasia had a 62% prevalence of CHIP prior to therapy, compared to 27% in controls (22). This was identified as a major risk factor, with a positive predictive value of 34% and a negative predictive value of 89%. Given the low overall incidence, the absence of CHIP significantly reduces the probability of progression to a myeloid neoplasm. These findings were corroborated with case-control study with external validation, reporting positive and negative predictive values of 26% and 98%, respectively (23).
The same MSKCC group, through a clonal evolution analysis, found that cytotoxic therapy selects clones with mutations in DNA damage repair genes (primarily TP53, PPM1D, and CHEK2). These clones exhibit lower competitive fitness compared to mutations in non-DNA repair genes in the absence of cytotoxic agents or radiotherapy (24). In this analysis, CHIP in TP53 was the most significant risk factor for therapy-related myeloid neoplasia. An independent group confirmed these findings, showing that most cases had pre-existing CHIP and that a mouse model demonstrated selective advantage for these clones upon chemotherapy exposure (25).
The management of CHIP in patients with solid tumors remains an emerging area, with numerous unresolved questions (26). Prospective validation in large cohorts, the assessment of high-risk mutations, and high-risk exposures are necessary to ultimately guide therapeutic decisions for solid tumors in a manner that reduces subsequent hematologic malignancies. In an analysis proposed by MSKCC, 96% of early-stage breast cancers were found to have a very low risk of subsequent myeloid neoplasia. However, 4% had an absolute risk of approximately 9% over ten years. In these cases, the benefit of adjuvant chemotherapy is outweighed by the high lethality of myeloid neoplasms (24). If validated, this could lead to the omission of adjuvant chemotherapy in such scenarios. Additionally, patients with solid tumors and CHIP with a variant allele frequency (VAF) >10% exhibited increased mortality due to the underlying cancer (26), likely impacting immune cells within the tumor microenvironment (27), making this a potential future prognostic factor.
In patients with therapy-related myeloid neoplasms, germline mutations have been detected in 10–30% of cases, predominantly in genes related to Fanconi anemia, BRCA1/2, TP53, and CHEK2 (28). A study of patients with acute leukemia following breast cancer treatment identified a 21% prevalence of germline mutations, mainly in BRCA1/2, TP53, CHEK2, and PALB2 (29). This finding suggests two potential pathways: one in which germline mutations predispose to both solid and hematologic malignancies, and another in which these mutations predispose to CHIP, which is then naturally selected under cytotoxic pressure. Emerging evidence suggests the existence of germline predisposition genes for CHIP, adding complexity to this model (30). Given these considerations and the potential need for allogeneic transplantation in affected patients with related donors, germline predisposition gene testing should be considered (31).
In therapy-related AML patients with a diploid karyotype, comparative mutational profiling with de novo AML cases of similar cytogenetic background reveals equivalent incidences of genetic alterations (6). The most frequently observed mutations include NPM1 (45%), DNMT3A (28%), and FLT3 (23%) (32). These findings, along with prior reports, indicate that while univariate analyses associate t-AML with diploid karyotype with worse prognosis compared to de novo AML, this difference is not sustained in multivariate analyses (32, 33). Furthermore, both groups exhibit comparable disease recurrence rates. This similarity in relapse rates suggests an increased mortality risk in remission among t-AML patients, possibly due to cumulative toxicities. These findings underscore the importance of risk stratification for treatment-related toxicities and, where appropriate, the need for comprehensive geriatric assessments in treatment selection.
We have proposed a hierarchical classification of therapy-related acute myeloid leukemia (t-AML) in Figure 1, which organizes various aspects of the disease based on its molecular ontogeny, associations with risk factors, and proposed pathophysiological mechanisms. The key categories identified include TP53 mutations, secondary AML, where a clear distinction must be made between cases due to cytogenetic alterations or specific mutations, as their causal relationships differ; and de novo AML with genetic profiles, where again, it is important to differentiate between those with causal mutations such as NPM1/FLT3/IDH1-2, and those with KMT2A rearrangements.
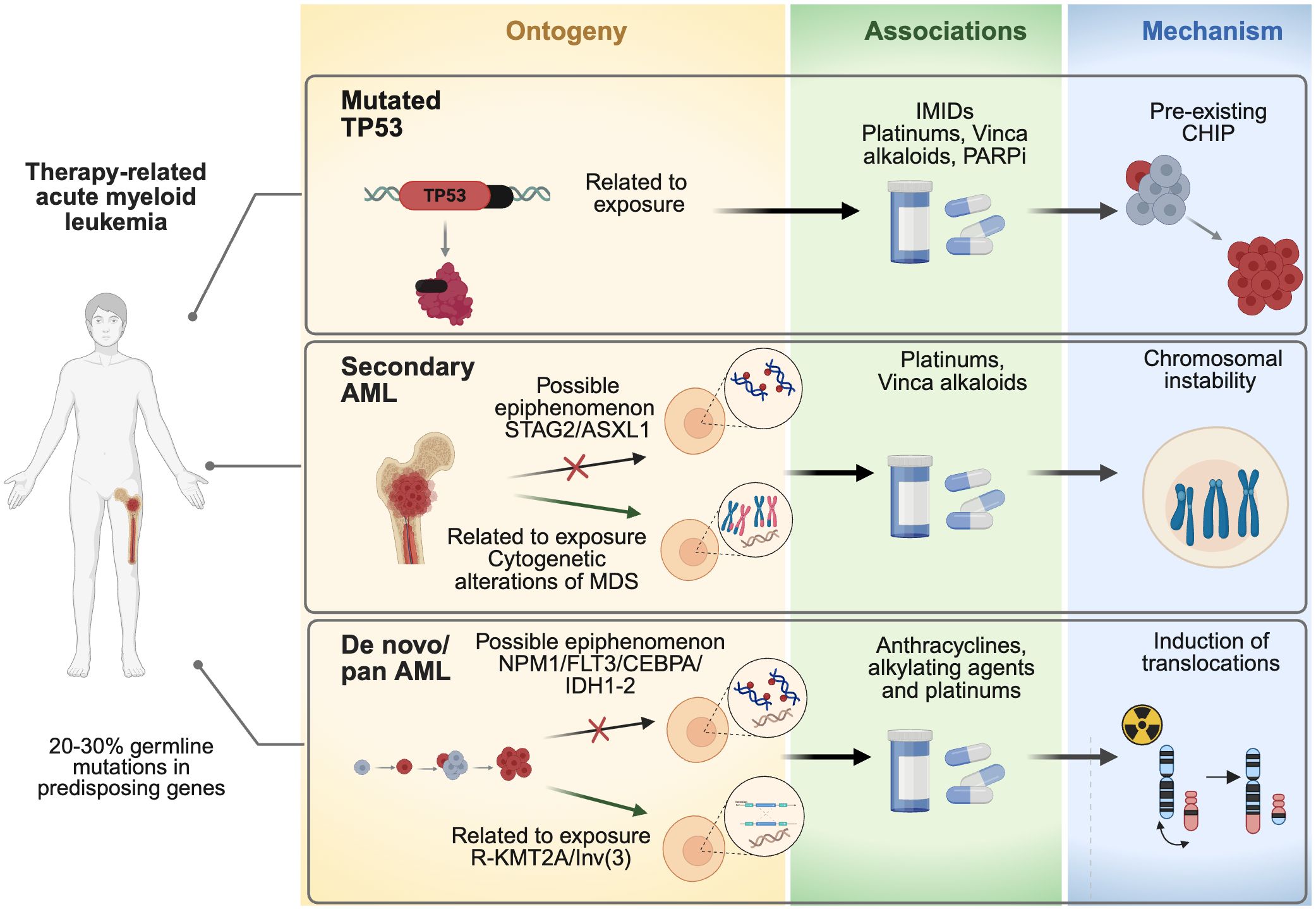
Figure 1. Hierarchical classification of therapy-related acute myeloid leukemia (t-AML). AML, Acute myeloid leukemia; ASXL1, ASXL Transcriptional Regulator 1; CEBPA, CCAAT Enhancer Binding Protein Alpha; CHIP, clonal hematopoiesis of indeterminate potential; IDH1, Isocitrate Dehydrogenase (NADP(+)) 1; IDH2, Isocitrate Dehydrogenase (NADP(+)) 2; IMIDs, Immunomodulatory drugs (IMiDs) like thalidomide, lenalidomide, and pomalidomide; FLT3, Fms Related Receptor Tyrosine Kinase 3; MDS, Myelodysplastic syndromes; NPM1, Nucleophosmin 1; TP53, tumor protein p53; STAG2, STAG2 Cohesin Complex Component; R-KMT2A Inv(3), an inversion of chromosome 3 (inv(3)), that leads to a rearrangement of the KMT2A gene (lysine methyltransferase 2A).
This classification facilitates the understanding of how prior oncological treatments, such as chemotherapy or radiation, interact with specific genetic alterations, contributing to the development of t-AML. Furthermore, the figure highlights how these risk factors are stratified into different hierarchical levels, offering a more integrated and detailed approach to understanding and prognosticating the disease, considering both clinical history and underlying molecular mechanisms.
Standard and emerging therapeutic approaches in t-AML
A recent study including 734 patients concluded that the prognostic significance of a t-AML history diminishes when considering the 2022 ELN classification and TP53 mutational status (34). This finding underscores the importance of basing therapeutic approaches on the genetic landscape of the disease rather than prior therapy history. This observation is particularly relevant in favorable-risk acute leukemias, such as core-binding factor AML, NPM1-mutated AML, and acute promyelocytic leukemia (APL). Multivariate analyses of these entities demonstrate relapse rates comparable to those seen in de novo cases (35). A comparative study on NPM1-mutated AML revealed that patients with therapy-related NPM1 mutations share clinical characteristics, cytogenetic profiles, co-mutation patterns, and overall survival outcomes with de novo NPM1-mutated AML, suggesting that the presence of NPM1 mutations in therapy-exposed patients may reflect a de novo-like entity coinciding with prior cytotoxic exposure (36). For therapy-related APL, studies indicate comparable rates of complete remission, relapse, and overall survival when compared with de novo APL (37, 38). Collectively, these findings suggest that in patients with favorable cytogenetic risk and NPM1 mutations, leukemia-related mortality risk is comparable to de novo cases, supporting the use of standard treatment strategies in this setting (39).
The current standard treatment for therapy-related AML is the liposomal formulation of daunorubicin and cytarabine at a fixed molar ratio of 5:1, known as CPX-351. The pivotal CPX-351 trial included patients aged 60–75 years with t-AML or AML with high-risk cytogenetics and/or a history of myelodysplastic syndromes (MDS) or chronic myelomonocytic leukemia (CMML) (40). CPX-351 demonstrated superior efficacy compared to conventional chemotherapy. Among the 309 patients enrolled, 20% had t-AML. CPX-351 treatment resulted in higher complete remission (CR) or CR with incomplete hematologic recovery (CRi) rates (47% vs. 33%) and improved overall survival (median 9.5 vs. 5.9 months) compared to conventional chemotherapy. Subgroup analyses identified that patients deriving the greatest overall survival benefit were those with t-AML and a prior history of MDS who had not been previously exposed to hypomethylating agents. At a five-year follow-up, overall survival rates were 8% with standard 3 + 7 therapy versus 18% with CPX-351, highlighting a significant difference. The most substantial benefit was observed in patients who achieved CR/CRi and proceeded to allogeneic hematopoietic stem cell transplantation (allo-HSCT) (41). Specifically, the five-year overall survival rate in therapy-related AML patients reached 27% with CPX-351 versus 9% with conventional 3 + 7 induction.
A post hoc analysis suggested superior outcomes in patients achieving CR/CRi with CPX-351 compared to standard 3 + 7 therapy (42). This finding raises the hypothesis that CPX-351 may induce deeper remissions; however, this could not be confirmed due to the absence of measurable residual disease (MRD) assessment. Furthermore, as previously discussed, t-AML is a highly heterogeneous entity rather than a uniform biological group. Another post hoc analysis demonstrated that TP53-mutated patients did not exhibit differences in response rates (~33%) between CPX-351 and conventional 3 + 7 therapy, nor in overall survival (median ~5 months) (43). Given the high prevalence of TP53 mutations in this entity, these findings are significant. Despite their poor prognosis, these patients may still benefit from inclusion in clinical trials. Additionally, researchers from MD Anderson Cancer Center have suggested that patients with isolated TP53 mutations and a variant allele frequency (VAF) below 40% may achieve better outcomes with intensive chemotherapy compared to hypomethylating agents or Venetoclax-based therapy (44). Conversely, in cases where VAF exceeds 40%, overall survival outcomes appear comparable between both treatment strategies; however, lower response rates with intensive chemotherapy suggest that it may be a less effective bridge to allo-HSCT.
A recent Phase 3 study compared CPX-351 and FLAG-IDA in high-risk AML/MDS patients with adverse-risk karyotypes (45). This study included 189 patients, of whom 95% met ELN 2022 high-risk criteria, and 8% had therapy-related AML. No significant differences in response rates, overall survival, or progression-free survival were observed between treatment groups. This trial was prompted by an analysis of secondary AML patients (n=115) from the MRC AML 15 study (46), which demonstrated improved overall survival and progression-free survival with FLAG-IDA induction compared to anthracycline/cytarabine regimens. However, this study did not specify the proportion of t-AML patients or analyze this subgroup separately.
In summary, conventional 3 + 7 therapy yields poor outcomes in t-AML patients, who typically present with adverse prognostic features. Therefore, it is crucial to consider more effective induction strategies, such as CPX-351 or FLAG-IDA, and prioritize clinical trial enrollment to explore novel therapeutic options.
The use of Venetoclax in therapy-related AML is supported by emerging evidence. In a subgroup analysis of the VIALE-A study, which included 8% t-AML patients, secondary AML cases demonstrated a median overall survival of 16 months and a CR/CRi rate of 66% (47). A recent retrospective study also reported comparable outcomes between CPX-351 and Venetoclax plus hypomethylating agents, with 30% of patients having t-AML (48).
Furthermore, high-intensity chemotherapy regimens combined with Venetoclax have significantly improved response rates and overall survival in patients with intermediate- and high-risk AML (49–51). In these studies, approximately 8% of patients had t-AML. Additionally, in older patients (>60 years), lower-intensity chemotherapy combined with Venetoclax has achieved response rates exceeding 90%, presenting a promising bridge to allo-HSCT (52). Notably, retrospective studies have indicated that splicing factor mutations lose their adverse prognostic impact in patients receiving Venetoclax-based regimens (53). However, these findings require validation in prospective trials.
Regarding allo-HSCT in therapy-related AML, prior cytotoxic exposure loses prognostic relevance, while cytogenetics and ELN risk classification emerge as more determinant factors (54, 55). A study from the CIBMTR registry involving 868 patients with therapy-related myeloid neoplasms reported a five-year overall survival rate of 22% (56). Multivariate analysis identified four prognostic variables (age >35 years, adverse cytogenetics, active disease at transplant, and non-matched related donors), which were used to develop a predictive score. Patients without any risk factors had an estimated five-year overall survival of 50%, while those with one or more risk factors had a reduced probability of around 25%.
A COMMAND registry series analyzing TP53-mutated patients found that 18% underwent allo-HSCT, with a median overall survival of 24 months (57). Multivariate analysis identified key predictors of survival post-transplant, including achievement of complete marrow response at day +100 and the presence of chronic graft-versus-host disease, whereas pre-transplant response was not a significant predictor. Given the inherent chemoresistance of this entity, sequential conditioning regimens warrant further exploration as a strategy to optimize transplant outcomes (58).
Conclusions
In summary, therapy-related acute myeloid leukemia (t-AML) does not constitute a distinct molecular entity; rather, its prognostic value is integrated into current risk classifications, particularly in relation to TP53 mutations. Consequently, treatment decisions should be guided by the underlying disease biology rather than the clinical history of prior cytotoxic exposure. Nonetheless, the presence of t-AML is frequently associated with adverse-risk classifications.
Based on the pivotal Phase 3 CPX-351 study, the conventional 3 + 7 regimen should no longer be considered the standard of care or the optimal therapeutic approach for this patient population. Instead, prioritizing enrollment in clinical trials for high-risk patients is imperative (Box 2). In cases where trial participation is not feasible, alternative therapeutic strategies should be considered, including intensive chemotherapy regimens such as CPX-351 or FLAG-IDA, as well as Venetoclax-based protocols in combination with hypomethylating agents or chemotherapy. Furthermore, the critical role of allogeneic hematopoietic stem cell transplantation as a key component of consolidation therapy should be emphasized in the majority of cases.
Box 2. Key clinical considerations for therapy-related AML.
● Approximately 10% of AML cases are therapy-related AML (t-AML), with an increasing incidence trend.
● t-AML exhibits a marked tendency to cluster high-risk cytogenetic abnormalities and TP53 mutations.
● The pathogenic role of radiotherapy using current conformal techniques remains uncertain.
● Mutational analysis via next-generation sequencing (NGS) is essential for characterizing this entity.
● High-risk exposures include platinum-based agents, vinca alkaloids, and immunomodulatory drugs.
● An emerging area of research explores the role of clonal hematopoiesis of indeterminate potential (CHIP) and germline predisposition mutations in the pathogenesis of this disease.
● The prognosis of t-AML is encompassed within current risk classifications; therefore, treatment should be guided by the mutational profile.
● The conventional 3 + 7 chemotherapy regimen is unsatisfactory for this high-risk AML subgroup. Priority should be given to enrollment in clinical trials.
Author contributions
JJ: Conceptualization, Funding acquisition, Investigation, Writing – original draft, Writing – review & editing. CaH: Writing – original draft, Writing – review & editing. ChH: Funding acquisition, Investigation, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by ANID-Basal funding for Scientific and Technological Center of Excellence, IMPACT, #FB210024, and Fundación Arturo López Pérez.
Acknowledgments
We gratefully acknowledge the support of the IMPACT Research Center, Fundación Arturo López Pérez (FALP), and the PhD Program in Ciencias Biomedicas at Universidad de los Andes. Figures were created with Biorender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hulegårdh E, Nilsson C, Lazarevic V, Garelius H, Antunovic P, Rangert Derolf Å, et al. Characterization and prognostic features of secondary acute myeloid leukemia in a population-based setting: a report from the Swedish Acute Leukemia Registry. Am J Hematol. (2015) 90:208–14. doi: 10.1002/ajh.23908
2. Smith SM, Le Beau MM, Huo D, Karrison T, Sobecks RM, Anastasi J, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood. (2003) 102:43–52. doi: 10.1182/blood-2002-11-3343
3. Kayser S, Döhner K, Krauter J, Köhne CH, Horst HA, Held G, et al. The impact of therapy-related acute myeloid leukemia (AML) on outcome in 2853 adult patients with newly diagnosed AML. Blood. (2011) 117:2137–45. doi: 10.1182/blood-2010-08-301713
4. Fianchi L, Pagano L, Piciocchi A, Candoni A, Gaidano G, Breccia M, et al. Characteristics and outcome of therapy-related myeloid neoplasms: Report from the Italian network on secondary leukemias. Am J Hematol. (2015) 90:E80–5. doi: 10.1002/ajh.23966
5. Granfeldt Østgård LS, Medeiros BC, Sengeløv H, Nørgaard M, Andersen MK, Dufva IH, et al. Epidemiology and clinical significance of secondary and therapy-related acute myeloid leukemia: A national population-based cohort study. J Clin Oncol. (2015) 33:3641–9. doi: 10.1200/JCO.2014.60.0890
6. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
7. Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the World Health Organization classification of Haematolymphoid Tumours: Myeloid and histiocytic/dendritic neoplasms. Leukemia. (2022) 36:1703–19. doi: 10.1038/s41375-022-01613-1
8. Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. (2022) 140:1200–28. doi: 10.1182/blood.2022015850
9. Döhner H, Wei AH, Appelbaum FR, Craddock C, DiNardo CD, Dombret H, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. (2022) 140:1345–77. doi: 10.1182/blood.2022016867
10. Bhatia S. Therapy-related myelodysplasia and acute myeloid leukemia. Semin Oncol. (2013) 40:666–75. doi: 10.1053/j.seminoncol.2013.09.013
11. Martínez-Cuadrón D, Megías-Vericat JE, Serrano J, Martínez-Sánchez P, Rodríguez-Arbolí E, Gil C, et al. Treatment patterns and outcomes of 2310 patients with secondary acute myeloid leukemia: a PETHEMA registry study. Blood Adv. (2022) 6:1278–95. doi: 10.1182/bloodadvances.2021005335
12. Nardi V, Winkfield KM, Ok CY, Niemierko A, Kluk MJ, Attar EC, et al. Acute myeloid leukemia and myelodysplastic syndromes after radiation therapy are similar to de novo disease and differ from other therapy-related myeloid neoplasms. J Clin Oncol. (2012) 30:2340–7. doi: 10.1200/JCO.2011.38.7340
13. Nilsson C, Linde F, Hulegårdh E, Garelius H, Lazarevic V, Antunovic P, et al. Characterization of therapy-related acute myeloid leukemia: increasing incidence and prognostic implications. Haematologica. (2023) 108:1015–25. doi: 10.3324/haematol.2022.281233
14. Gómez-De León A, Demichelis-Gómez R, da Costa-Neto A, Gómez-Almaguer D, and Rego EM. Acute myeloid leukemia: challenges for diagnosis and treatment in Latin America. Hematology. (2023) 28:2158015. doi: 10.1080/16078454.2022.2158015
15. Sperling AS, Guerra VA, Kennedy JA, Yan Y, Hsu JI, Wang F, et al. Lenalidomide promotes the development of TP53-mutated therapy-related myeloid neoplasms. Blood. (2022) 140:1753–63. doi: 10.1182/blood.2021014956
16. Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. (2015) 125:1367–76. doi: 10.1182/blood-2014-11-610543
17. Morice PM, Leary A, Dolladille C, Chrétien B, Poulain L, González-Martín A, et al. Myelodysplastic syndrome and acute myeloid leukaemia in patients treated with PARP inhibitors: a safety meta-analysis of randomised controlled trials and a retrospective study of the WHO pharmacovigilance database. Lancet Haematol. (2021) 8:e122–34. doi: 10.1016/S2352-3026(20)30360-4
18. Csizmar CM, Saliba AN, Swisher EM, and Kaufmann SH. PARP inhibitors and myeloid neoplasms: A double-edged sword. Cancers (Basel). (2021) 13:6385. doi: 10.3390/cancers13246385
19. Martin JE, Khalife-Hachem S, Grinda T, Kfoury M, Garciaz S, Pasquier F, et al. Therapy-related myeloid neoplasms following treatment with PARP inhibitors: new molecular insights. Ann Oncol. (2021) 32:1046–8. doi: 10.1016/j.annonc.2021.04.015
20. Kwan TT, Oza AM, Tinker AV, Ray-Coquard I, Oaknin A, Aghajanian C, et al. Preexisting TP53-variant clonal hematopoiesis and risk of secondary myeloid neoplasms in patients with high-grade ovarian cancer treated with rucaparib. JAMA Oncol. (2021) 7:1772–81. doi: 10.1001/jamaoncol.2021.4664
21. Coombs CC, Zehir A, Devlin SM, Kishtagari A, Syed A, Jonsson P, et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell. (2017) 21:374–82. doi: 10.1016/j.stem.2017.07.010
22. Gillis NK, Ball M, Zhang Q, Ma Z, Zhao Y, Yoder SJ, et al. Clonal haemopoiesis and therapy-related myeloid Malignancies in elderly patients: a proof-of-concept, case-control study. Lancet Oncol. (2017) 18:112–21. doi: 10.1016/S1470-2045(16)30627-1
23. Takahashi K, Wang F, Kantarjian H, Doss D, Khanna K, Thompson E, et al. Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid neoplasms: a case-control study. Lancet Oncol. (2017) 18:100–11. doi: 10.1016/S1470-2045(16)30626-X
24. Bolton KL, Ptashkin RN, Gao T, Braunstein L, Devlin SM, Kelly D, et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet. (2020) 52:1219–26. doi: 10.1038/s41588-020-00710-0
25. Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. (2015) 518:552–5. doi: 10.1038/nature13968
26. Bolton KL, Gillis NK, Coombs CC, Takahashi K, Zehir A, Bejar R, et al. Managing clonal hematopoiesis in patients with solid tumors. J Clin Oncol. (2019) 37:7–11. doi: 10.1200/JCO.18.00331
27. Buttigieg MM and Rauh MJ. Clonal hematopoiesis: Updates and implications at the solid tumor-immune interface. JCO Precis Oncol. (2023) 7:e2300132. doi: 10.1200/PO.23.00132
28. Baranwal A, Hahn CN, Shah MV, and Hiwase DK. Role of germline predisposition to therapy-related myeloid neoplasms. Curr Hematol Malig Rep. (2022) 17:254–65. doi: 10.1007/s11899-022-00676-2
29. Churpek JE, Marquez R, Neistadt B, Claussen K, Lee MK, Churpek MM, et al. Inherited mutations in cancer susceptibility genes are common among survivors of breast cancer who develop therapy-related leukemia. Cancer. (2016) 122:304–11. doi: 10.1002/cncr.v122.2
30. Liu J, Osman AEG, Bolton K, and Godley LA. Germline predisposition to clonal hematopoiesis. Leuk Res. (2023) 132:107344. doi: 10.1016/j.leukres.2023.107344
31. Voso MT, Falconi G, and Fabiani E. What’s new in the pathogenesis and treatment of therapy-related myeloid neoplasms. Blood. (2021) 138:749–57. doi: 10.1182/blood.2021010764
32. Samra B, Richard-Carpentier G, Kadia TM, Ravandi F, Daver N, DiNardo CD, et al. Characteristics and outcomes of patients with therapy-related acute myeloid leukemia with normal karyotype. Blood Cancer J. (2020) 10:47. doi: 10.1038/s41408-020-0316-3
33. Cantu MD, Kanagal-Shamanna R, Wang SA, Kadia T, Bueso-Ramos CE, Patel SS, et al. Clinicopathologic and molecular analysis of normal karyotype therapy-related and DE Novo acute myeloid leukemia: A multi-institutional study by the Bone Marrow Pathology Group. JCO Precis Oncol. (2023) 7:e2200400. doi: 10.1200/PO.22.00400
34. Hochman MJ, Othus M, Hasserjian RP, Ambinder A, Brunner A, Percival MEM, et al. Prognostic impact of secondary versus de novo ontogeny in acute myeloid leukemia is accounted for by the European LeukemiaNet 2022 risk classification. Leukemia. (2023) 37:1915–8. doi: 10.1038/s41375-023-01985-y
35. Gross S, Ihlow J, Busack L, Adamiak K, Schrezenmeier J, Jesse J, et al. Therapy-related AML: long-term outcome in a large cohort of AML-patients with intensive and non-intensive therapy. Blood Cancer J. (2024) 14:160. doi: 10.1038/s41408-024-01140-5
36. Othman J, Meggendorfer M, Tiacci E, Thiede C, Schlenk R, Dillon R, et al. Overlapping features of therapy-related and de novo NPM1-mutated AML. Blood. (2023) 141:1846–57. doi: 10.1182/blood.2022018108
37. Kayser S, Krzykalla J, Elliott MA, Norsworthy K, Gonzales P, Hills RK, et al. Characteristics and outcome of patients with therapy-related acute promyelocytic leukemia front-line treated with or without arsenic trioxide. Leukemia. (2017) 31:2347–54. doi: 10.1038/leu.2017.92
38. Braun T, Cereja S, Chevret S, Raffoux E, Beaumont M, Detourmignies L, et al. Evolving characteristics and outcome of secondary acute promyelocytic leukemia (APL): A prospective analysis by the French-Belgian-Swiss APL group. Cancer. (2015) 121:2393–9. doi: 10.1002/cncr.v121.14
39. Aldoss I and Pullarkat V. Therapy-related acute myeloid leukemia with favorable cytogenetics: still favorable? Leuk Res. (2012) 36:1547–51. doi: 10.1016/j.leukres.2012.09.008
40. Lancet JE, Uy GL, Cortes JE, Newell LF, Lin TL, Ritchie EK, et al. CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J Clin Oncol. (2018) 36:2684–92. doi: 10.1200/JCO.2017.77.6112
41. Lancet JE, Uy GL, Newell LF, Lin TL, Ritchie EK, Stuart RK, et al. CPX-351 versus 7 + 3 cytarabine and daunorubicin chemotherapy in older adults with newly diagnosed high-risk or secondary acute myeloid leukaemia: 5-year results of a randomised, open-label, multicentre, phase 3 trial. Lancet Haematol. (2021) 8:e481–91. doi: 10.1016/S2352-3026(21)00134-4
42. Lin TL, Rizzieri DA, Ryan DH, Schiller GJ, Kolitz JE, Uy GL, et al. Older adults with newly diagnosed high-risk/secondary AML who achieved remission with CPX-351: phase 3 post hoc analyses. Blood Adv. (2021) 5:1719–28. doi: 10.1182/bloodadvances.2020003510
43. Cortes JE, Lin TL, Asubonteng K, Faderl S, Lancet JE, and Prebet T. Efficacy and safety of CPX-351 versus 7 + 3 chemotherapy by European LeukemiaNet 2017 risk subgroups in older adults with newly diagnosed, high-risk/secondary AML: post hoc analysis of a randomized, phase 3 trial. J Hematol Oncol. (2022) 15:155. doi: 10.1186/s13045-022-01361-w
44. Short NJ, Montalban-Bravo G, Hwang H, Ning J, Franquiz MJ, Kanagal-Shamanna R, et al. Prognostic and therapeutic impacts of mutant TP53 variant allelic frequency in newly diagnosed acute myeloid leukemia. Blood Adv. (2020) 4:5681–9. doi: 10.1182/bloodadvances.2020003120
45. Othman J, Wilhelm-Benartzi C, Dillon R, Knapper S, Freeman SD, Batten LM, et al. A randomized comparison of CPX-351 and FLAG-Ida in adverse karyotype AML and high-risk MDS: the UK NCRI AML19 trial. Blood Adv. (2023) 7:4539–49. doi: 10.1182/bloodadvances.2023010276
46. Russell N, Hills R, Kjeldsen L, Dennis M, and Burnett A. Treatment intensification with FLAG-Ida may improve disease control in younger patients with secondary acute myeloid leukaemia: long-term follow up of the MRC AML15 trial. Br J Haematol. (2022) 196:1344–7. doi: 10.1111/bjh.v196.6
47. DiNardo CD, Jonas BA, Pullarkat V, Thirman MJ, Garcia JS, Wei AH, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. (2020) 383:617–29. doi: 10.1056/NEJMoa2012971
48. Salhotra A, Aribi A, Ngo D, Zhang J, Sandhu K, Al-Malki M, et al. Outcome of secondary acute myeloid leukemia treated with hypomethylating agent plus venetoclax (HMA-Ven) or liposomal daunorubicin-cytarabine (CPX-351). Am J Hematol. (2021) 96:E196–200. doi: 10.1002/ajh.26157
49. DiNardo CD, Lachowiez CA, Takahashi K, Loghavi S, Kadia T, Daver N, et al. Venetoclax combined with FLAG-IDA induction and consolidation in newly diagnosed acute myeloid leukemia. Am J Hematol. (2022) 97:1035–43. doi: 10.1002/ajh.26601
50. Kadia TM, Reville PK, Borthakur G, Yilmaz M, Kornblau S, Alvarado Y, et al. Venetoclax plus intensive chemotherapy with cladribine, idarubicin, and cytarabine in patients with newly diagnosed acute myeloid leukaemia or high-risk myelodysplastic syndrome: a cohort from a single-centre, single-arm, phase 2 trial. Lancet Haematol. (2021) 8:e552–61. doi: 10.1016/S2352-3026(21)00192-7
51. Lachowiez CA, Reville PK, Kantarjian H, Jabbour E, Borthakur G, Daver N, et al. Venetoclax combined with induction chemotherapy in patients with newly diagnosed acute myeloid leukaemia: a post-hoc, propensity score-matched, cohort study. Lancet Haematol. (2022) 9:e350–60. doi: 10.1016/S2352-3026(22)00076-X
52. Kadia TM, Reville PK, Wang X, Rausch CR, Borthakur G, Pemmaraju N, et al. Phase II study of venetoclax added to cladribine plus low-dose cytarabine alternating with 5-azacitidine in older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol. (2022) 40:3848–57. doi: 10.1200/JCO.21.02823
53. Senapati J, Urrutia S, Loghavi S, Short NJ, Issa GC, Maiti A, et al. Venetoclax abrogates the prognostic impact of splicing factor gene mutations in newly diagnosed acute myeloid leukemia. Blood. (2023) 142:1647–57. doi: 10.1182/blood.2023020649
54. Armand P, Kim HT, DeAngelo DJ, Ho VT, Cutler CS, Stone RM, et al. Impact of cytogenetics on outcome of de novo and therapy-related AML and MDS after allogeneic transplantation. Biol Blood Marrow Transpl. (2007) 13:655–64. doi: 10.1016/j.bbmt.2007.01.079
55. Jentzsch M, Grimm J, Bill M, Brauer D, Backhaus D, Goldmann K, et al. ELN risk stratification and outcomes in secondary and therapy-related AML patients consolidated with allogeneic stem cell transplantation. Bone Marrow Transpl. (2021) 56:936–45. doi: 10.1038/s41409-020-01129-1
56. Litzow MR, Tarima S, Pérez WS, Bolwell BJ, Cairo MS, Camitta BM, et al. Allogeneic transplantation for therapy-related myelodysplastic syndrome and acute myeloid leukemia. Blood. (2010) 115:1850–7. doi: 10.1182/blood-2009-10-249128
57. Badar T, Atallah E, Shallis R, Saliba AN, Patel A, Bewersdorf JP, et al. Survival of TP53-mutated acute myeloid leukemia patients receiving allogeneic stem cell transplantation after first induction or salvage therapy: results from the Consortium on Myeloid Malignancies and Neoplastic Diseases (COMMAND). Leukemia. (2023) 37:799–806. doi: 10.1038/s41375-023-01847-7
58. Stelljes M, Middeke JM, Bug G, Wagner-Drouet EM, Müller LP, Schmid C, et al. Remission induction versus immediate allogeneic haematopoietic stem cell transplantation for patients with relapsed or poor responsive acute myeloid leukaemia (ASAP): a randomised, open-label, phase 3, non-inferiority trial. Lancet Haematol. (2024) 11:e324–35. doi: 10.1016/S2352-3026(24)00065-6
Keywords: therapy-related AML, acute myeloid leukemia, clonal hematopoiesis, target therapy, genetic risk classifications
Citation: Jerez J, Hernandez C and Hill CN (2025) Understanding therapy-related AML: genetic insights and emerging strategies for high-risk patients. Front. Hematol. 4:1609642. doi: 10.3389/frhem.2025.1609642
Received: 10 April 2025; Accepted: 26 May 2025;
Published: 19 June 2025.
Edited by:
Giuseppe Gaetano Loscocco, University of Florence, ItalyReviewed by:
Francesco La Rocca, Madonna delle Grazie Hospital, ItalyCopyright © 2025 Jerez, Hernandez and Hill. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joaquín Jerez, am9hcXVpbi5qZXJlekBmYWxwLm9yZw==; Charlotte N. Hill, aGlsbG1hY2hhZG8uY0BnbWFpbC5jb20=