 Wen Zhou
Wen Zhou Xiaojia Guo
Xiaojia Guo Liansheng Zhang
Liansheng Zhang- Department of Hematology, Lanzhou University Second Hospital, Lanzhou University, Lanzhou, China
Intravascular large B-cell lymphoma (IVLBCL) presents with a wide range of clinical symptoms, making clinical diagnosis challenging. It is often misdiagnosed or overlooked, leading to delays in treatment for affected patients. We present a case of a patient exhibiting clinical symptoms such as chest tightness, dyspnea, fever, and edema, who was later diagnosed with secondary hemophagocytic syndrome (HPS). Laboratory tests indicated persistent hypoalbuminemia, significantly elevated lactate dehydrogenase levels, thrombocytopenia, and splenomegaly, with no evidence of lymphadenopathy. During the treatment for HPS, the patient developed a rash on both lower limbs and abdomen and was ultimately diagnosed as IVLBCL after a skin biopsy. Following four cycles of zanubrutinib in combination with the R-CHOP regimen (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone), the patient achieved complete resolution of both dermatological manifestations and systemic symptoms. Laboratory parameters, including complete blood count, serum albumin levels, and lactate dehydrogenase, were normalized. Additionally, ultrasonography demonstrated a marked reduction in splenic size. However, the patient exhibited suboptimal adherence to the prescribed treatment plan and did not complete the intended number of cycles. During a subsequent telephone follow-up, the patient was confirmed to be alive; however, the status of the disease could not be evaluated. As of the latest follow-up, the patient has survived for 2 years.
Introduction
It is estimated that 85% to 90% of intravascular large B-cell lymphoma (IVLBCL) cases are of B-cell origin, while NK/T-cell lymphomas are comparatively rare. Immunological marker analysis and T-cell receptor (TCR) gene rearrangement studies can aid in differential diagnosis (1). IVLBCL was first reported by Pfleger and Tappeiner in 1959. It is a rare subtype of non-Hodgkin lymphoma (NHL) of B-cell origin, characterized by the selective proliferation of tumor cells within the vascular lumen, particularly in the capillaries (2). The reported median age ranged from 60 to 70 years,with no statistically significant difference observed between genders (3).
IVLBCL may precede, occur secondarily to, or coexist concurrently with NHL, including large B-cell lymphoma, lymphocytic lymphoma, and follicular lymphoma (3). According to clinical manifestations and patterns of organ infiltration, IVLBCL is classified into three distinct entities: classical entity, hemophagocytic syndrome (HPS)-associated entity or Asian entity, and the cutaneous entity (4). Classical entity accounted for more than 75% of the cases, with approximately 40% of these presenting skin lesions. An HPS-associated entity, also referred to as an Asian entity, is more prevalent in Asian countries and is associated with the poorest prognosis. This entity is typically characterized by fever, pancytopenia, hepatosplenomegaly, and bone marrow infiltration (5). The skin entity accounts for approximately 20% of the cases and is typically confined to the skin, with no evidence of organ infiltration upon general examination (6).
Most patients with IVLBCL do not exhibit abnormal findings in the early bone marrow examination. Since IVLBCL typically lacks extravascular tumor masses, imaging studies often fail to reveal specific manifestations. Consequently, early diagnosis is extremely challenging, and misdiagnosis is a common occurrence. The case we present involves an HPS-related condition accompanied by skin lesions during disease progression and was ultimately confirmed as IVLBCL through skin biopsy.
Case report
The patient was a 59-year-old man who presented with a chief complaint of intermittent chest tightness, dyspnea, fever, and edema lasting for 4 days. He was admitted to another department of our hospital in August 2023. Physical examination revealed stable vital signs, evidence of anemia, and absence of palpable lymphadenopathy. No hepatosplenomegaly was observed, and severe symmetrical pitting edema was noted in both lower extremities. The patient had no history of chronic illnesses and no family history of genetic disorders. Laboratory examinations demonstrated pancytopenia and hypoproteinemia, accompanied by elevated levels of lactate dehydrogenase (LDH), β2-microglobulin, blood creatinine, and erythrocyte sedimentation rate (Table 1). Furthermore, tumor markers, thyroid function tests, pituitary hormone assays, autoimmune disease examination, Epstein–Barr virus testing, and blood gas analysis all yielded negative results. Echocardiography revealed thickening of the interventricular septum and left ventricular wall, the presence of pericardial effusion, left ventricular diastolic dysfunction, and moderately elevated pulmonary artery pressure. Chest computed tomography (CT) demonstrated mild interstitial changes bilaterally, and the findings were consistent with chronic bronchitis. Abdominal ultrasound revealed splenomegaly (spleen index: 32 cm²), with the spleen not palpable below the costal margin. Bone marrow morphology revealed no evidence of neoplastic proliferation and exhibited prominent phagocytic activity (Figure 1). During the period, the patient presented with intermittent fever, and HPS was considered in the differential diagnosis. However, no specific tests for HPS were conducted. Following empirical anti-infective therapy and supportive care, the patient was discharged. One week after discharge, the patient was readmitted to our department due to progressive abdominal distension. Laboratory findings revealed reduced natural killer (NK) cell activity at 2.5% (reference range >4%) and elevated soluble CD25 (sCD25) levels at 4,089 U/mL (reference range 223–710 U/mL). Additional laboratory results showed the following abnormalities: WBC 2.3 × 109/L, HGB 74 g/L, PLT 81 × 109/L, LDH 813 U/L, ALB 24.5 g/L, and creatinine 93.2 μmol/L. Bone marrow biopsy demonstrated the absence of blast proliferation, megakaryocytic hypoplasia, and no evidence of dysplastic hematopoiesis. Flow cytometry did not detect any abnormal immunophenotypic cell populations. Serum interleukin-10 (IL-10) level was 2.3 pg/mL. CT imaging demonstrated interstitial changes in both lungs, splenomegaly, and no evidence of enlarged lymph nodes. Sequencing results for genes associated with familial hemophagocytic syndrome and related immunodeficiencies were negative. Chromosomal analysis showed a normal karyotype. Due to financial constraints, the patient declined to undergo the PET/CT examination. He was diagnosed with secondary HPS of undetermined etiology and was discharged following treatment with DEP (doxorubicin hydrochloride liposome, etoposide, methylprednisolone). Five days after discharge, the patient was readmitted due to bilateral lower extremity edema accompanied by fever. Laboratory tests revealed WBC 1.22 × 109/L, HGB 62 g/L, PLT 56 × 109/L, CREA 78.2 µmol/L, LDH 646 U/L, and ALB 24.2 g/L. Bone marrow morphology and biopsy findings were consistent with those from the initial admission, with no evidence of tumor cells identified. Serum IL-10 level was elevated at 270 pg/mL. The patient received the second and third cycles of DEP chemotherapy.
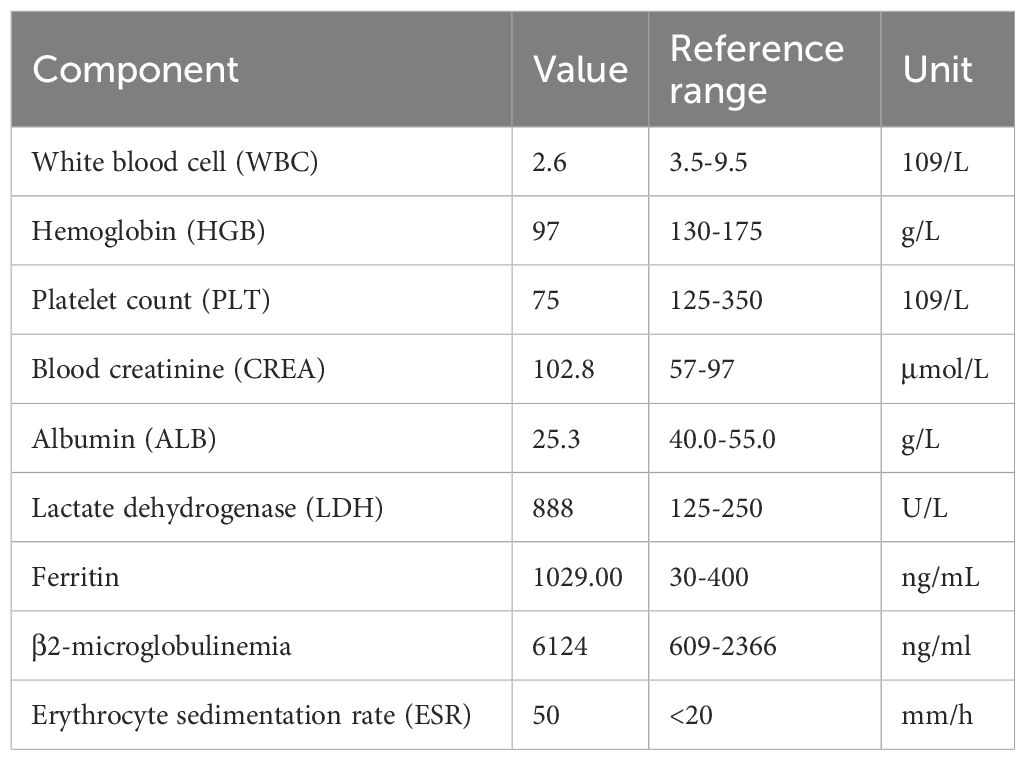
Table 1. Pertinent laboratory findings on hospitalization.
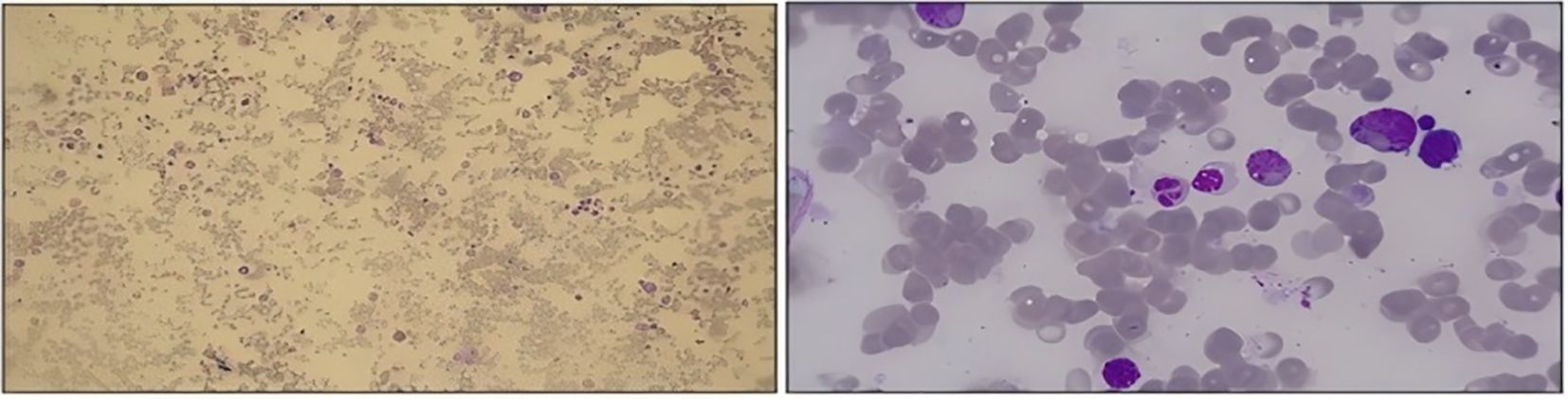
Figure 1. Bone marrow HPS.
The patient was readmitted to the hospital in December 2023. Physical examination revealed anemia and scattered patchy rashes on both lower limbs and the abdomen (Figure 2). No palpable enlargement of superficial lymph nodes or hepatosplenomegaly was noted, although pitting edema was present in both lower limbs. Laboratory findings included the following: WBC 0.67 × 109/L, HGB 82 g/L, PLT 87 × 109/L, CREA 70.8 μmol/L, ALB 23.1 g/L, and LDH 755 U/L. A skin biopsy revealed epidermal hyperkeratosis and the presence of atypical large lymphoid cells within the dermal capillaries of the right thigh skin. These cells exhibited morphological features consistent with centroblasts and immunoblasts, with occasional extravasation beyond the vascular boundaries and a high Ki-67 proliferation index. Immunohistochemical analysis demonstrated that the atypical cells were positive for CD20, Pax-5, Bcl-2 (90%+), Bcl-6 (90%+), MUM-1 (50%+), c-MYC (20%), CD10, CD34, vimentin, and CD163 (focal +), while being negative for CD3, CD5, CD43, pancytokeratin, Desmin, CD117, MPO, CD68, CD99, CD56, CD30, CD4, CD7, CD123, TdT, and p53 (wild-type expression). The Ki-67 labeling index was approximately 90%, indicating a highly proliferative activity within the lesion (Figure 3). Fluorescence in situ hybridization (FISH) studies for BCL-2/BCL-6/MYC rearrangements were all negative. Zanubrutinib combined with the R-CHOP regimen for a total of four cycles of chemotherapy resulted in complete resolution of the patient’s rash and normalization of laboratory parameters (Figure 4). Repeat CT scans demonstrated the disappearance of bilateral interstitial lung changes, reduced splenomegaly, and resolution of pulmonary hypertension. However, the patient exhibited poor treatment adherence and did not undergo regular follow-up or continued therapy thereafter. During a subsequent telephone follow-up, the patient was confirmed to be alive; however, the disease status could not be assessed.

Figure 2. Pertinent cutaneous lesion.
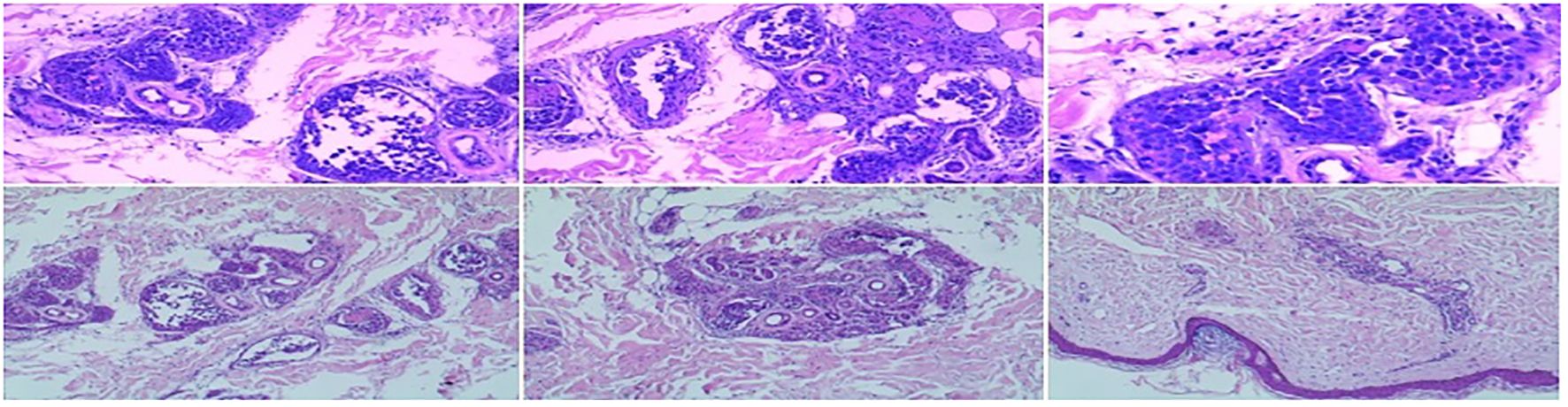
Figure 3. Histopathological characteristics of cutaneous CD20+ lymphoma include Pax-5+, Bcl-2 (90%+), Bcl-6 (90%+), MUM-1 (50%+), c-MYC (20%), CD10+, CD5−, and Ki-67 (90%).
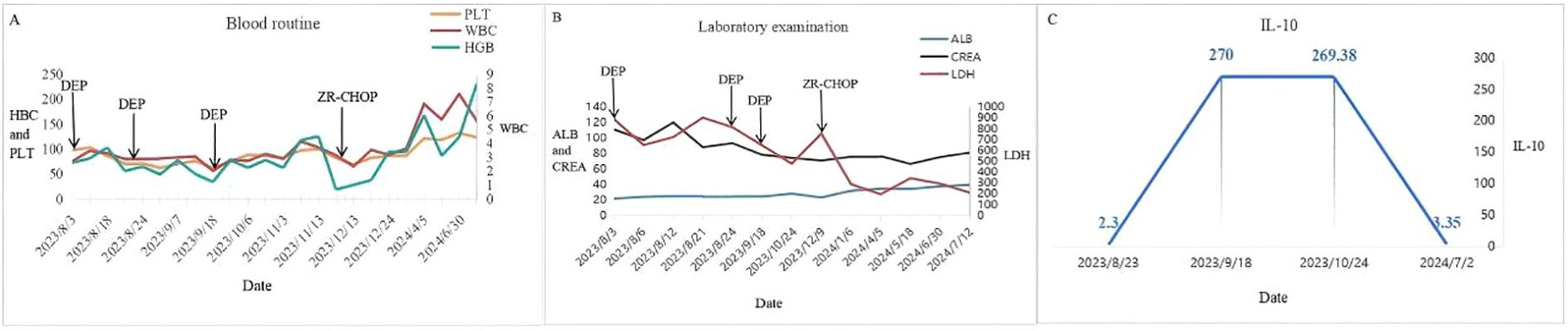
Figure 4. (A) Recovery of blood routine. (B) Recovery of laboratory examinations. (C) Change of IL-10.
Discussion
IVLBCL is a rare form of extranodal large B-cell lymphoma characterized by the selective proliferation of lymphoma cells within the lumina of capillaries and small blood vessels. The exact pathogenic mechanism remains unclear; however, evidence suggests that IVLBCL is associated with interactions between tumor cells and endothelial cells, as well as alterations in adhesion molecules and chemokines involved in tumor cell motility (7, 8). Studies have shown that IVLBCL cells often lack certain key molecules essential for lymphocyte infiltration, such as the surface proteins CD29 (β-integrin) and CD54 (ICAM-1). Additionally, these cells do not express matrix metalloproteinases 2 and 9, which are crucial for parenchymal invasion (7, 9). Multiple chromosomal translocations have been implicated in the pathogenesis of IVLBCL, including those involving the B-cell lymphoma 2 (BCL2) and B-cell lymphoma 6 (BCL6) genes. Overexpression of these genes has been linked to the inhibition of apoptosis and the subsequent development of lymphoma (10, 11). The high frequency of PD-L1 and PD-L2 gene rearrangements in IVLBCL suggests that immune escape may play a role in disease pathogenesis and indicates a potential therapeutic benefit of immune checkpoint inhibitors in IVLBCL (12). Frequent mutations in NF-κB pathway genes have been observed in IVLBCL patients, particularly in PIM1, MYD88 (L265P), CD79B, TMEM30A, and IRF4. Additionally, alterations in several driver genes, including IRF4 translocation, NOTCH2, CCND3, and GNA13, have been identified (13). These genetic aberrations may contribute to the underlying pathogenesis of IVLBCL and offer potential targets for therapeutic intervention. IVLBCL typically shows positive expression of CD79a (100%), CD20 (96%), MUM1/IRF4 (95%), CD5 (38%), CD10 (12%), and MYC in lymphoma cells. Its characteristic feature is that lymphoma cells selectively proliferate within the lumens of capillaries and small blood vessels. This is distinct from other types of lymphoma.
In the Asian entity of IVLBCL, HPS is frequently associated and can manifest with a range of clinical symptoms and laboratory abnormalities. However, the most commonly observed features include fever, pancytopenia, hepatosplenomegaly, and elevated ferritin levels (14). We report a case of a patient presenting with recurrent chest tightness, dyspnea, fever, and peripheral edema. Laboratory examinations revealed pancytopenia, splenomegaly, refractory hypoproteinemia, transient renal dysfunction, and significantly elevated LDH levels. Phagocytosis was identified in both bone marrow aspiration and biopsy, while no lymphomatous cells were detected. A diagnosis of HPS was made, and the patient was treated with the DEP regimen; however, there was no notable improvement in either clinical symptoms or laboratory parameters. During the course of treatment, the patient developed a rash on the thighs and abdomen, prompting a skin biopsy that confirmed the diagnosis of IVLBCL. Subsequently, the patient was treated with zanubrutinib combined with the R-CHOP regimen, leading to complete resolution of symptoms and normalization of laboratory findings. The patient has remained in remission for 2 years post-treatment. The patient we reported had HPS and skin simultaneously. Imaging findings revealed interstitial lung changes and a hypoechoic lesion measuring approximately 0.8 cm × 0.6 cm in the middle of the right lobe of the thyroid gland, as well as at the posterior peritoneum, which resolved after treatment. This suggests possible lymphomatous involvement of these organs. The patient experienced transient renal insufficiency; however, renal function normalized prior to the initiation of treatment for IVLBCL, making it uncertain whether the kidneys were truly involved. Therefore, this case exhibited clinical features consistent with IVLBCL-associated HPS, along with skin and multi-organ involvement, indicating a presentation involving multiple overlapping manifestations.
IVLBCL can affect any organ system, with the central nervous system (CNS) being the most commonly involved organ (60%), followed by bone marrow and spleen (11%), skin (8%), and lungs (7%). It may also involve the kidneys, adrenal glands, reproductive organs, thyroid gland, gallbladder, and other organs (15). CT scans of the lungs in affected patients may reveal ground-glass opacities or interstitial lesions, reduced pulmonary diffusion capacity, and the presence of pulmonary hypertension (16). In the case we reported, pulmonary hypertension was observed, although pulmonary function tests were not performed. Following treatment, the pulmonary artery pressure normalized. When the CNS is involved, the most common manifestations are those related to ischemia and infarction. Brain imaging findings are typically non-specific. Cerebrospinal fluid (CSF) analysis often reveals elevated protein levels, although lymphomatous infiltration is rare (17). The cranial imaging examination revealed no obvious abnormalities, and there were no clinical manifestations suggestive of central nervous system infiltration. Therefore, CNS involvement was not considered in this case. A marked elevation of IL-10 has been recognized as a potential indicator of IVLBCL (18). In the patient we described, IL-10 levels were not significantly elevated during the early phase of the disease but increased notably during disease progression (Figure 4). A higher success rate of skin biopsy has been associated with elevated IL-10 levels (19). Whether IL-10 levels consistently rise significantly in all IVLBCL cases as the disease progresses remains to be determined and requires further clinical data for validation. Additionally, the diagnostic yield of skin biopsy under conditions of low IL-10 levels is still unclear. Given the patient’s unwillingness to undergo a skin biopsy in the absence of a rash, this diagnostic procedure was not performed during the early stage of the disease.
The 2023 European Hematology Association Congress presented findings from a prospective single-arm phase II clinical trial confirming the therapeutic efficacy and safety profile of zanubrutinib combined with R-CHOP as a first-line treatment for IVLBCL. All 13 enrolled patients received the ZR-CHOP regimen and completed eight cycles of treatment, achieving 12 complete remissions and one partial remission. At the mid-term evaluation, which included 19 patients, 18 achieved complete remission and one achieved partial remission. With a median follow-up of 439 days, only one patient died due to bacteremia during the follow-up period, and there was no disease recurrence. Neither median progression-free survival nor overall survival was reached, suggesting promising long-term outcomes. Notably, in this study, one patient completed only the first four cycles of treatment and achieved a favorable initial response, but did not complete the remaining four cycles due to poor adherence. Despite this, the patient still achieved a prolonged survival time.
Conclusion
Through this case, when physicians encounter patients presenting with idiopathic HPS, intractable hypoproteinemia, elevated LDH, pancytopenia, multi-organ dysfunction, and increased IL-10 levels, IVLBCL should be considered as a differential diagnosis. Performing random skin biopsies, even in the absence of cutaneous manifestations, may facilitate early diagnosis within the patient’s acceptable limits. This case report serves as a reminder to clinicians to maintain a high index of suspicion for IVLBCL, aiming to promote timely diagnosis and treatment.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Written informed consent was obtained from the individuals for the publication of any potentially identifiable images or data included in this article.
Author contributions
WZ: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. XG: Data curation, Formal Analysis, Investigation, Software, Writing – review & editing. YL: Conceptualization, Data curation, Methodology, Writing – review & editing. XW: Investigation, Validation, Writing – review & editing. HZ: Data curation, Writing – review & editing. LZ: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing. LL: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The authors declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Gu L, Li M, Tong G, Wei W, Fan Y, and Zhao Y. Prostate primary intravascular large B-cell lymphoma: A case report. Urol Case Rep. (2022) 45:102276. doi: 10.1016/j.eucr.2022.102276
2. Pfleger L and Tappeiner J. On the recognition of systematized endotheliomatosis of the cutaneous blood vessels (reticuloendotheliosis. Hautarzt. (1959) 10:359–63.
3. Ponzoni M, Campo E, and Nakamura S. Intravascular large B-cell lymphoma: a chameleon with multiple faces and many masks. Blood. (2018) 132:1561–7. doi: 10.1182/blood-2017-04-737445
4. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. (2016) 127:2375–90. doi: 10.1182/blood-2016-01-643569
5. Yoon SE, Kim WS, and Kim SJ. Asian variant of intravascular large B-cell lymphoma: a comparison of clinical features based on involvement of the central nervous system. Korean J Intern Med. (2020) 35:946–56. doi: 10.3904/kjim.2018.396
6. Willemze R, Cerroni L, Kempf W, Berti E, Facchetti F, Swerdlow SH, et al. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood. (2019) 133:1703–14. doi: 10.1182/blood-2018-11-881268
7. Ponzoni M, Arrigoni G, Gould VE, Del Curto B, Maggioni M, Scapinello A, et al. Lack of CD 29 (beta1 integrin) and CD 54 (ICAM-1) adhesion molecules in intravascular lymphomatosis. Hum Pathol. (2000) 31:220–6. doi: 10.1016/S0046-8177(00)80223-3
8. Fonkem E, Lok E, Robison D, Gautam S, and Wong ET. The natural history of intravascular lymphomatosis. Cancer Med. (2014) 3:1010–24. doi: 10.1002/cam4.2014.3.issue-4
9. Ferry JA, Harris NL, Picker LJ, Weinberg DS, Rosales RK, Tapia J, et al. Intravascular lymphomatosis (malignant angioendotheliomatosis). A B-cell neoplasm expressing surface homing receptors. Mod Pathol. (1988) 1:444–52.
10. Vieites B, Fraga M, Lopez-Presas E, Pintos E, Garcia-Rivero A, and Forteza J. Detection of t(14;18) translocation in a case of intravascular large B-cell lymphoma: a germinal centre cell origin in a subset of these lymphomas? Histopathology. (2005) 46:466–8. doi: 10.1111/j.1365-2559.2005.02013.x
11. Khoury H, Lestou VS, Gascoyne RD, Bruyere H, Li CH, Nantel SH, et al. Multicolor karyotyping and clinicopathological analysis of three intravascular lymphoma cases. Mod Pathol. (2003) 16:716–24. doi: 10.1097/01.MP.0000077515.68734.85
12. Shimada K, Yoshida K, Suzuki Y, Iriyama C, Inoue Y, Sanada M, et al. Frequent genetic alterations in immune checkpoint-related genes in intravascular large B-cell lymphoma. Blood. (2021) 137:1491–502. doi: 10.1182/blood.2020007245
13. Gonzalez-Farre B, Ramis-Zaldivar JE, Castrejón de Anta N, Rivas-Delgado A, Nadeu F, Salmeron-Villalobos J, et al. Intravascular large B-cell lymphoma genomic profile is characterized by alterations in genes regulating NF-kappaB and immune checkpoints. Am J Surg Pathol. (2023) 47:202–11. doi: 10.1097/PAS.0000000000001978
14. Lee H, Kim HS, Lee JM, Park KH, Choi AR, Yoon JH, et al. Natural killer cell function tests by flowcytometry-based cytotoxicity and IFN-gamma production for the diagnosis of adult hemophagocytic lymphohistiocytosis. Int J Mol Sci. (2019) 20:5413. doi: 10.3390/ijms20215413
15. Roy AM, Pandey Y, Middleton D, Broadfoot B, Sasapu A, and Veeraputhiran M. Intravascular large B-cell lymphoma: A diagnostic dilemma. Cureus. (2021) 13:e16459. doi: 10.7759/cureus.16459
16. Nguyen TT, Sekiguchi H, Yi ES, and Ryu JH. Occult diffuse neoplasm in the lungs: intravascular large B-cell lymphoma. Am J Med. (2021) 134:926–9. doi: 10.1016/j.amjmed.2020.12.028
17. Cheng JW and Li JH. Intravascular large B-cell lymphoma. N Engl J Med. (2023) 389:2188. doi: 10.1056/NEJMicm2307122
18. Li J, Li YJ, Zhao DQ, Jia CW, Xu N, and Zeng XJ. Intravascular large B-cell lymphoma presenting with fever and dyspnea: a case report. Zhonghua Nei Ke Za Zhi. (2024) 63:499–501. doi: 10.3760/cma.j.cn112138-20231123-00334
19. Lymphoid Disease Group, C.S.o.H.C.M.A, and O. Lymphoma Expert Committee of Chinese Society of Clinical. Chinese expert consensus on the diagnosis and management of intravascular large B cell lymphoma (2023). Zhonghua Xue Ye Xue Za Zhi. (2023) 44:177–81. doi: 10.3760/cma.j.issn.0253-2727.2023.03.001
Keywords: intravascular large B-cell lymphoma, hemophagocytic syndrome, skin biopsy, zanubrutinib, case report
Citation: Zhou W, Guo X, Liu Y, Wang X, Zhang H, Zhang L and Li L (2025) Case report: A case of intravascular large B-cell lymphoma and related literature review. Front. Hematol. 4:1639726. doi: 10.3389/frhem.2025.1639726
Received: 02 June 2025; Accepted: 28 July 2025;
Published: 22 August 2025.
Edited by:
Tayfur Toptas, Marmara University, TürkiyeReviewed by:
Jinming Song, Moffitt Cancer Center, United StatesKarthik Kumar, Madras Medical College, India
Copyright © 2025 Zhou, Guo, Liu, Wang, Zhang, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liansheng Zhang, ZG9jdG9yemhhbmdsc2hAc2luYS5jb20=; Lijuan Li, ZG9jdG9yanVhbkBzaW5hLmNvbQ==