 Maria Paulina Castelo Rueda1
Maria Paulina Castelo Rueda1 Athina Raftopoulou1,2
Athina Raftopoulou1,2 Martin Gögele1
Martin Gögele1 Max Borsche3
Max Borsche3 David Emmert1
David Emmert1 Christian Fuchsberger1
Christian Fuchsberger1 Essi M. Hantikainen1Vladimir Vukovic1,4
Essi M. Hantikainen1Vladimir Vukovic1,4 Christine Klein3
Christine Klein3 Peter P. Pramstaller1,5
Peter P. Pramstaller1,5 Irene Pichler1*†
Irene Pichler1*† Andrew A. Hicks1†
Andrew A. Hicks1†- 1Institute for Biomedicine, Eurac Research, Affiliated Institute of the University of Lübeck, Bolzano, Italy
- 2Department of Economics, University of Patras, Patras, Greece
- 3Institute of Neurogenetics, University of Lübeck, Lübeck, Germany
- 4Centre for Disease Control and Prevention, Institute of Public Health of Vojvodina, Novi Sad, Serbia
- 5Department of Neurology, University Medical Center Schleswig-Holstein, Lübeck, Germany
Mutations in the Parkin (PRKN) gene are the most frequent cause of autosomal recessive early-onset Parkinson's disease (PD). Heterozygous PRKN mutation carriers might also be at increased risk for developing clinical symptoms of PD. Given the high frequency of heterozygous mutations in the general population, it is essential to have better estimates of the penetrance of these variants, and to investigate, which clinical and biochemical markers are present in carriers and thus potentially useful for identifying those individuals at greater risk of developing clinical symptoms later in life. In the present study, we ascertained the frequency of heterozygous PRKN mutation carriers in a large population sample of the Cooperative Health Research in South Tyrol (CHRIS) study, and screened for reported PD risk markers. 164 confirmed heterozygous PRKN mutation carriers were compared with 2,582 controls. A higher number of heterozygous mutation carriers reported a detectable increase in an akinesia-related phenotype, and a higher percentage of carriers had manifested diabetes. We also observed lower resting heart rate in the PRKN mutation carriers. Extending our risk analyses to a larger number of potential carriers and non-carriers using genotype imputation (n = 299 carriers and n = 7,127 non-carriers), from previously published biomarkers we also observed a higher neutrophil-to-lymphocyte ratio (NLR) and lower serum albumin and sodium levels in the heterozygous PRKN variant carriers. These results identify a set of biomarkers that might be useful either individually or as an ensemble to identify variant carriers at greater risk of health issues due to carrier status.
Introduction
Parkinson's disease (PD) is one of the most common neurodegenerative diseases in aging, second only to Alzheimer's disease (1). Most PD cases are classified idiopathic (~90%), and about 10% of cases represent rare forms with Mendelian inheritance patterns, ascribable to rare mutations in specific genes, with more than 20 identified to date (2). However, many of these genes lack replication and their relevance is therefore still debated. In addition, genome-wide association studies have uncovered ~90 independent risk signals in 78 genomic loci at genome-wide significance, that contribute to genetic risk for sporadic PD (3). Mutations in PRKN (the gene that encodes Parkin) are the most common known cause of autosomal recessive early-onset PD, accounting for up to 42.2% of cases with an age of onset ≤20 years (4), and Parkin dysfunction represents a risk factor for sporadic PD (5). PRKN variants have been identified in numerous families with different genetic background (6) and include more than 130 mutations consisting of copy number variations (CNVs) (deletions or multiplications of exons), small deletions/insertions, as well as single nucleotide polymorphisms (SNPs) (missense, non-sense or splice side variations) (7, 8). Importantly, a large number of patients carry exon rearrangements that result in protein truncation (8, 9). The Parkin protein functions as an E3 ubiquitin ligase, which is implicated in the clearance of dysfunctional mitochondria by autophagy (mitophagy) and in several other cellular processes like mitochondrial biogenesis, free radical metabolism, and inflammation.
Homozygous or compound-heterozygous mutations in the PRKN gene result in highly penetrant symptom expression, while heterozygous mutations may predispose to disease symptoms with highly reduced penetrance (10, 11), with a similar pattern established for mutations in GBA (12, 13). Heterozygous PRKN mutations are found more frequently in PD patients than in controls (14), where they are detected with a frequency up to 3.1% (15). The presence of heterozygous PRKN mutations is one of the reported potential genetic risk factors for PD (10, 14, 15). Given the frequency (up to 3%) of heterozygous mutations in the population, it is essential to have better estimates of the penetrance of these variants and to investigate, which risk markers are manifesting in carriers and thus potentially useful for identifying those individuals at greater risk of clinical symptoms later in life. In the present study, we ascertained the frequency of heterozygous PRKN mutation carriers in a large population sample of the Cooperative Health Research in South Tyrol (CHRIS) study conducted in Northern Italy (16), in the same geographic region, where the largest pedigree to date with PRKN mutations was identified (17). We also screened for validated PD risk markers of the Movement Disorder Society (MDS) (18) and additional potential PD risk markers reported in the literature (19–22).
Methods
Study Participants
The CHRIS study (16) is a longitudinal population-based study to investigate the genetic and molecular basis of age-related common chronic conditions and their interaction with genes, lifestyle and environment in the general population. All adults of one valley in South Tyrol (Northern Italy) were invited, with 13,393 participants enrolled. Of these, 10,500 were genotyped with either the OmniExpressExome chip (Illumina), that contains 958,497 markers including over 273,000 functional exome markers, or with the Omni2.5Exome chip (Illumina) with 2,618,000 markers that include over 240,000 exonic markers. Genotypes were imputed (23) to 39.2 million variants using the Haplotype Reference Consortium panel (http://www.haplotype-reference-consortium.org). Exome sequencing was performed using the xGen® Exome Research Panel v1.0 in 3,603 individuals. Family participation was encouraged for complete pedigree reconstruction. The study was approved by the Ethics Committee of the Healthcare System of the Autonomous Province of Bozen/Bolzano (Italy).
Phenotypes
In this study, PD risk markers of the Movement Disorder Society (MDS) research criteria for prodromal PD were assessed (18). All risk markers, except for occupational solvent exposure and substantia nigra hyperechogenicity, were available for analysis. Significant pesticide exposure was assessed by a computer-assisted interviewer-administered question “Do you use pesticides? (insecticides, herbicides, fungicides).” The non-use of caffeine was defined as the response “rarely or never” (in the past 12 months) to the item “Coffee (mocha/espresso, filter coffee, instant)” of the GA2LEN food frequency questionnaire (24). Physical activity was assessed by using the International physical activity questionnaire (25), and life-course smoking was measured by the European Community Respiratory Health Survey (26). Diabetes was defined as either self-reported doctor-diagnosed and/or use of glucose-lowering drugs or fasting serum glucose ≥126 mg/dL or glycated hemoglobin (HbA1c) ≥6.5%. Additional potential PD risk markers were available for analysis: C-reactive protein (CRP) (21), Platelet count (PC), Mean platelet volume (MPV) (20), Platelet to lymphocyte ratio (PLR), Neutrophil to lymphocyte ratio (NLR) (22), heart rate, systolic blood pressure (BP), white blood cell (WBC), neutrophil counts, serum albumin, sodium (19). Blood samples were drawn after overnight fasting and blood parameters measured by standard methods (16). For evaluating PD symptoms, a validated screening questionnaire composed of nine symptom questions and two additional questions about the patient's diagnosis of parkinsonism and/or treatment was used (27).
Mutation Analysis
To search for predicted CNVs over the PRKN gene, we used the SNP & Variation Suite software (SVS version 8, Golden Helix) to examine the signal intensity information (LogR) from available chip genotyping data. To confirm predicted deletions or duplications of specific exons in the gene, DNA was further analyzed by Multiplex ligation-dependent probe amplification (MLPA) or digital PCR assays (ddPCR). In addition to the detection of deletions/duplications over exons of the PRKN gene, we used an in-house instance of the BRAVO (Browse All Variants Online) browser, which is a web-based browser for genetic variation determined in the whole exome sequencing dataset of 3,603 CHRIS participants. All variants were annotated with the Ensembl Variant Effect Predictor (VEP) and the Combined Annotation Dependent Depletion (CADD) algorithms (28, 29). We decided a CADD score cutoff of >20, which was more likely to capture predicted pathogenic variants by comparison with variants described in the MDSGene database (https://www.mdsgene.org/). We used these resources and databases to identify carriers of pathogenic or likely pathogenic point mutations and small deletions/insertions in our exome sequence and imputed chip genotyping datasets. Additionally, published or suspected pathogenic variants in 18 PD and dystonia-related genes other than PRKN were identified, and individuals carrying such variants were excluded from the control set used for the exome sequence dataset (resulting in n = 2,582 controls) and the imputed dataset (resulting in 7,127 controls).
Statistical Analysis
Mann-Whitney and Fisher's exact tests were used to compare non-normally distributed variables per heterozygous PRKN mutation carrier status. Analyses were conducted in Stata version 15.1 (StataCorp, College Station, TX, USA).
Results
In the subset of the CHRIS study, for which exome sequence data are available (n = 3,603 participants), 164 individuals carried one of 17 known or newly detected heterozygous mutations in PRKN resulting in a carrier frequency of 4.55%. Large exon rearrangements affecting exons 2, 3, 4, and 7 of the PRKN gene were identified and confirmed in 31 individuals (0.86%). Two small frameshift deletions (delA in exon 9, p.Val324AlafsTer111 - rs1562519380); del CT in exon 2, p.Gln34ArgfsTer5 - rs55777503) were detected in 35 individuals, and 98 individuals carried one out of 11 single nucleotide variations (eight known and three novel) resulting in a missense mutation. The most frequent mutation is the p.Arg275Trp missense mutation (rs34424986) with 1.64% (Table 1). Not surprisingly, a heterozygous deletion of exon 7 (n = 24 individuals, 0.67%) and the 1-bp deletion in exon 9 (n = 18 individuals, 0.50%), which are the two mutations identified in the previously described pedigree with PRKN mutations (17), are among the most frequent mutations identified. Heterozygous PRKN mutation carriers were clustered in eight multigenerational families and four small multiplex families. Most pedigrees with either the exon 7 deletion or the 1-bp deletion in exon 9 could be connected to the large pedigree with PRKN mutations, further pointing to a founder effect for these two mutations (6).
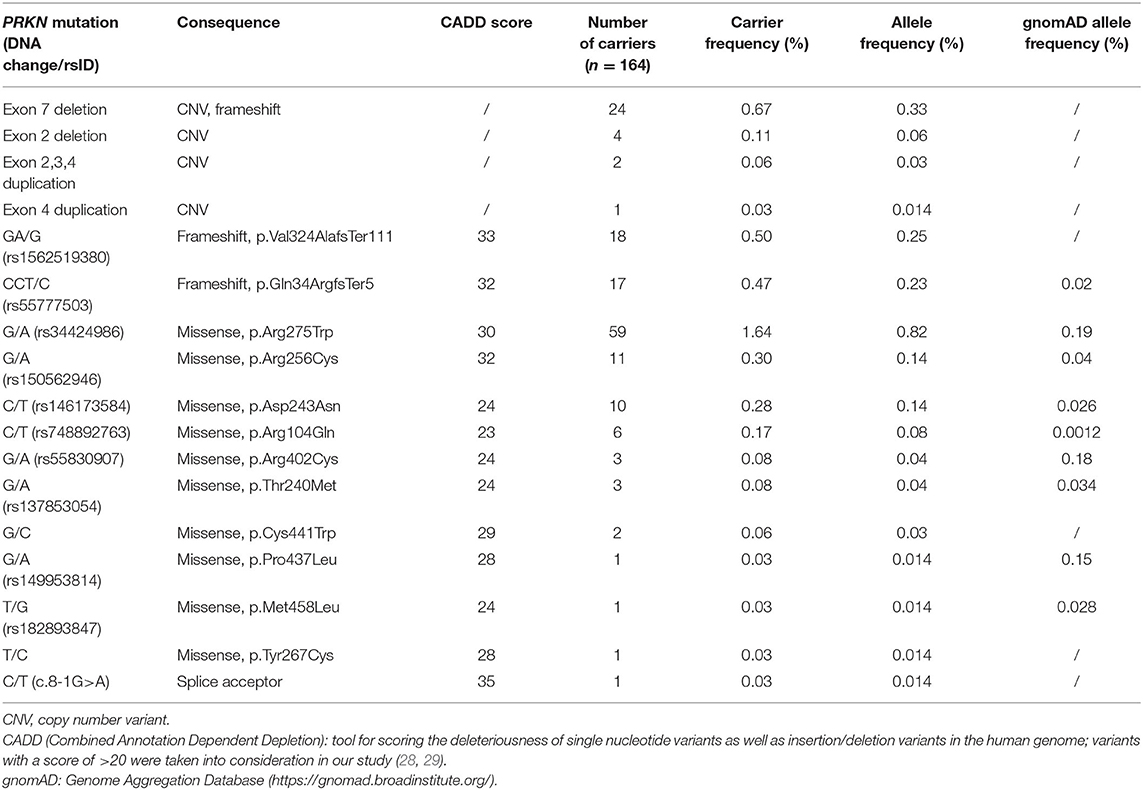
Table 1. Heterozygous mutations in the PRKN gene identified in the exome sequenced dataset of the CHRIS cohort (n = 3,603).
The analysis of the PD screening questionnaire revealed that a higher number of heterozygous carriers of a PRKN mutation reported that “their feet seem to get stuck to the floor” (symptom question 5) (p = 0.006, alpha set at 0.0045 after Bonferroni correction) as compared to non-carriers, indicating a detectable increase in an akinesia-related phenotype (Supplementary Table 1). The 164 heterozygous PRKN mutation carriers were further compared with 2,582 controls in terms of risk markers associated with PD. The descriptive analysis for all MDS risk markers (18), except for occupational solvent exposure and substantia nigra hyperechogenicity, is presented in Table 2, reporting percentages for the binary variables and means, medians, and inter-quartile range (IQR) for the continuous variables. Interestingly, a higher percentage of heterozygous PRKN mutation carriers manifest diabetes mellitus (p = 0.023), which is known to be associated with PD risk in multiple population-based prospective studies (18). In addition to the established PD risk markers, we included potential PD risk markers in our analyses, which we identified based on recent data in the literature. For these markers, we observed a trend toward a lower heart rate in the PRKN mutation carriers (59.01 vs. 60.6, p = 0.054) as compared to the controls. When extending our risk marker analyses to the imputed PRKN carrier dataset (n = 299 carriers and n = 7,127 non-carriers, Supplementary Table 2), we also found higher neutrophil-to-lymphocyte ratio (NLR) (4.91 vs. 4.71, p = 0.025) and lower serum albumin (4.45 vs. 4.49, p = 0.008) and sodium levels (140.22 vs. 140.44, p = 0.047) in the PRKN variant carriers (Supplementary Table 3).
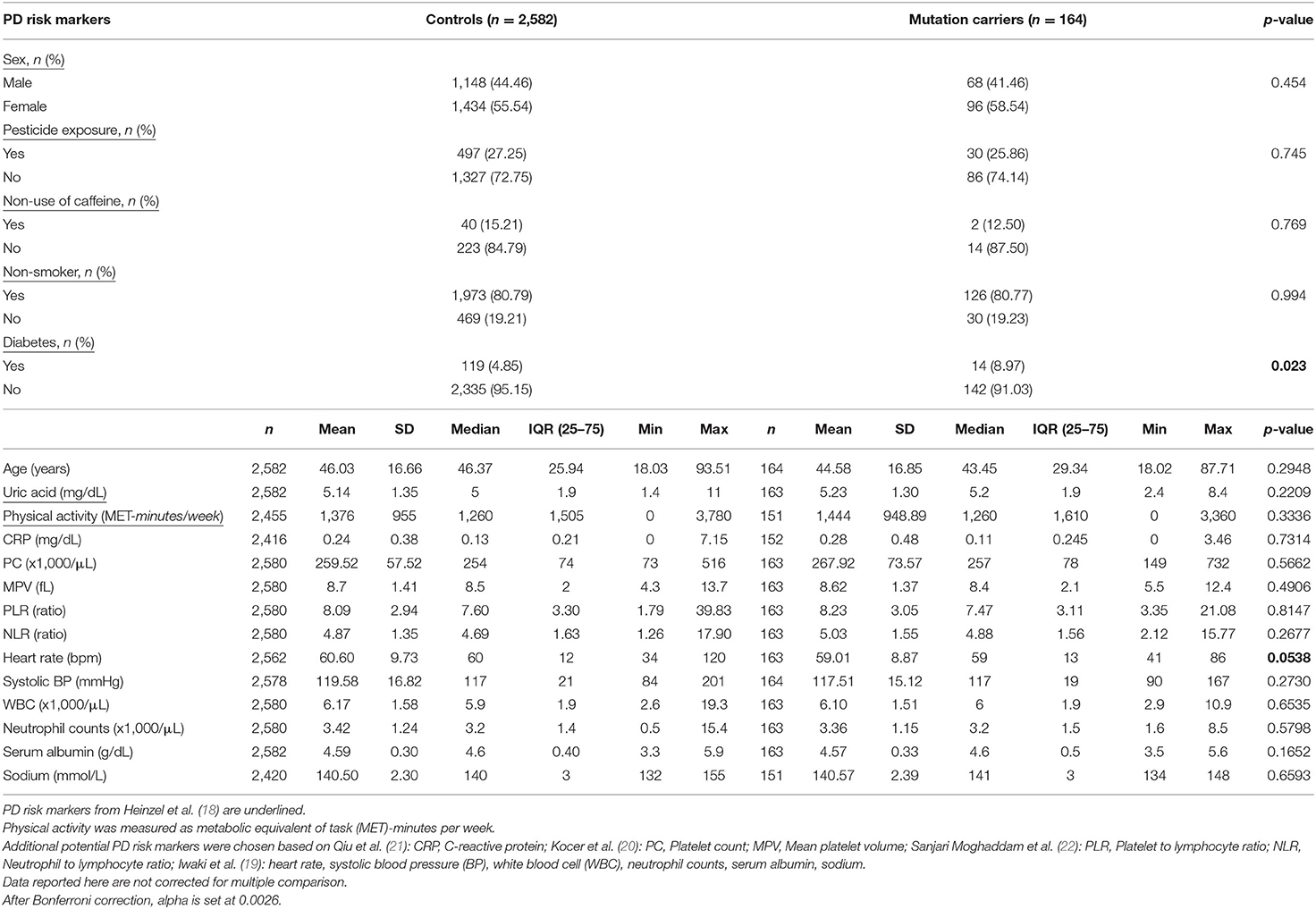
Table 2. Descriptive statistics of PD risk markers in heterozygous PRKN mutation carriers in the exome sequenced dataset of the CHRIS study.
Discussion
Previous studies have estimated the frequency of rare, heterozygous PRKN mutations in healthy control individuals up to 3.2%, with a previous South Tyrolean sample showing a frequency of 3.9%, and two studies that identified no mutation in healthy control individuals (30). We have screened 3,603 individuals of the population-based CHRIS study, free of overt clinical symptoms related to movement disorders, by exome sequencing and CNV analysis and have identified heterozygous mutations in 4.55% of the study participants (n = 164 individuals). Given the low residential mobility across generations in this restricted area of the Alps, rare variants might be overrepresented (31) as it can be seen also from the comparison with gnomAD allele frequencies (Table 1), and a founder effect can be assumed for some of the PRKN mutations identified in this study. This might explain the higher frequency compared to previous studies. However, the CNV carrier frequency of 0.86% detected in this study is in keeping with previously published reports (10, 32).
None of the mutation carriers identified fulfilled the clinical criteria for a PD diagnosis. However, a higher percentage of mutation carriers reported that their “feet seem to get stuck to the floor.” This specific symptom item might indicate subtle motor signs in an early phase of disease conversion. In support of this, we recently observed in a subset of 27 heterozygous PRKN mutation carriers and 24 non-mutation carriers from our study, who were recalled based on their genetic profile, that sensor-based quantification of movements allows discrimination between unaffected heterozygous mutation carriers and mutation-free controls. Thereby, it is crucial to challenge the motor system with more difficult tasks to unmask these subtle motor alterations (33). These results are further supported by a recent study, which found an increased risk of developing PD in heterozygous PRKN mutation carriers, although ~70% of putative heterozygous cases were not assessed for a second mutation (14). Pre-clinical changes identified by neuroimaging studies and a reduced fluorodopa uptake in the striatum previously observed in asymptomatic heterozygous PRKN mutation carriers also support an increased risk of developing PD symptoms in heterozygous PRKN mutation carriers (34, 35).
Diabetes has recently been defined as a novel PD risk marker as part of The MDS Research Criteria for Prodromal PD with a positive and negative likelihood ratio of 1.5 and 0.97, respectively (18). Furthermore, a recent meta-analysis found an association of type-2 diabetes with PD and some evidence for an increased progression of motor symptoms and cognitive decline (36). First studies showed a potential effect of anti-diabetic drugs on the progression of PD (37). Notably, diabetes was associated with the heterozygous PRKN mutation carrier status in our study. In this context, it has recently been shown that metformin, an anti-diabetic drug, which induces mild mitochondrial stress, phosphorylates and thereby activates endogenous Parkin in the liver, suggesting that Parkin plays an important role in mitochondrial homeostasis in this physiological context (38).
Furthermore, a previous study has found a higher heart rate in PD subjects (19), while we observed a tendency for a lower heart rate in our PRKN carriers. A recent study performed in cardiomyocytes and sera derived from PD patients with biallelic PRKN mutations found no alterations in Troponin T levels, suggesting that Parkin deficiency may not be associated with cardiac damage (39). However, animal studies suggested that Parkin might protect from cardiac damage (40, 41).
Moreover, when extending our analysis to the larger dataset with imputed genotypes, we also identified three molecular markers, which are different between putative mutation carriers and non-carrying controls. The peripheral immune biomarker NLR, which is increased in this group of mutation carriers, was previously found to be higher in idiopathic PD patients as compared to controls (42). Although there is one contradicting study for this association (43), it has been recently confirmed by Iwaki et al. (19). Interestingly, for NLR, a negative correlation to striatal binding ratios of DAT SPECT images in bilateral caudate and putamen nuclei of drug-naïve early PD patients was observed, and a positive association of NLR and motor severity was detected in the tremor-dominant subgroup of PD patients (22). Serum albumin and sodium are decreased in the PRKN mutation carriers of our enlarged dataset, in contrast to a previously found association with PD (19).
Carrying heterozygous PRKN mutations is one of the reported potential genetic risk factors for PD, and therefore identifying those individuals at possibly greater risk of disease development requires identification of biological markers that can be monitored in the prodromal phase. While data is lacking about conversion of individuals carrying such genetic risk factors to full disease, it is crucial to identify such markers, as these will be vital for testing potential neuroprotective therapies before progressive neurodegeneration advances, as it has been shown in individuals at high risk for type I diabetes (44). While we have identified associations for several risk markers, they cannot be used to predict an individual risk to develop PD. The prodromal phase of PD is clinically characterized not only by the presence of non-motor signs but also subtle motor disturbances as part of the phenotypic spectrum. To adequately understand individual disease conversion, there is a need for more longitudinal studies with individuals at greater risk and larger sample sizes using more in-depth phenotyping such as analysis of gait (33, 45). Here, we showed that heterozygous PRKN mutation carriers in the general population may self-report subtle motor signs (which can then be followed up by deeper phenotyping), have increased occurrence of diabetes, decreased heart rate, alterations of serum albumin and sodium, and increased NLR ratios, potentially further pointing to inflammatory activation as an early sign in the development of PRKN-related PD (46).
Data Availability Statement
The datasets presented in this report are not readily available because the data are part of a large population-based study, and the informed consent provided by the study participants does not allow the upload of individual-level genetic data to public repositories. Requests to access the datasets (including individual-level genetic data) should be directed to the Eurac data access committee (http://chris.eurac.edu).
Ethics Statement
The study was approved by the Ethics Committee of the Healthcare System of the Autonomous Province of Bozen/Bolzano (Italy). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
MPCR performed ddPCR experiments. AR performed the statistical analyses. MG organized the clinical data collection. MB performed MLPA experiments. DE and CF performed the genotype imputation. EMH extracted the genotypes. VV elaborated the clinical data. CK and PP provided critical review of the manuscript. IP wrote the manuscript and organized the study. AH wrote the manuscript, designed, and organized the study. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the Deutsche Forschungsgemeinschaft to AH, CK, and PP (FOR2488) and by the Department of Educational Assistance, University and Research of the Autonomous Province of Bolzano, Italy, through a core funding initiative to the Institute for Biomedicine.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are very grateful to the study participants of the CHRIS study. We thank Heike Pawlack for technical support of the MLPA measurements. The authors thank the Department of Innovation, Research, University and Museums of the Autonomous Province of Bozen/Bolzano for covering the Open Access publication costs.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.706145/full#supplementary-material
References
1. Bloem BR, Okun MS, Klein C. Parkinson's disease. Lancet. (2021) 12:2284–303. doi: 10.1016/S0140-6736(21)00218-X
2. Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson's disease. Lancet Neurol. (2020) 19:170–8. doi: 10.1016/S1474-4422(19)30287-X
3. Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet Neurol. (2019) 18:1091–102. doi: 10.1016/S1474-4422(19)30320-5
4. Lesage S, Lunati A, Houot M, Romdhan SB, Clot F, Tesson C, et al. Characterization of recessive Parkinson's disease in a large multicenter study. Ann Neurol. (2020) 88:843–50. doi: 10.1002/ana.25787
5. Dawson TM, Dawson VL. Parkin plays a role in sporadic Parkinson's disease. Neurodegener Dis. (2014) 13:69–71. doi: 10.1159/000354307
6. Hedrich K, Eskelson C, Wilmot B, Marder K, Harris J, Garrels J, et al. Distribution, type, and origin of Parkin mutations: review and case studies. Mov Disord. (2004) 19:1146–57. doi: 10.1002/mds.20234
7. Corti O, Lesage S, Brice A. What genetics tells us about the causes and mechanisms of Parkinson's disease. Physiol Rev. (2011) 91:1161–218. doi: 10.1152/physrev.00022.2010
8. Kasten M, Hartmann C, Hampf J, Schaake S, Westenberger A, Vollstedt EJ, et al. Genotype-phenotype relations for the Parkinson's disease genes Parkin, PINK1, DJ1: MDSGene systematic review. Mov Disord. (2018) 33:730–41. doi: 10.1002/mds.27352
9. Hedrich K, Kann M, Lanthaler AJ, Dalski A, Eskelson C, Landt O, et al. The importance of gene dosage studies: mutational analysis of the parkin gene in early-onset parkinsonism. Hum Mol Genet. (2001) 10:1649–56. doi: 10.1093/hmg/10.16.1649
10. Huttenlocher J, Stefansson H, Steinberg S, Helgadottir HT, Sveinbjornsdottir S, Riess O, et al. Heterozygote carriers for CNVs in PARK2 are at increased risk of Parkinson's disease. Hum Mol Genet. (2015) 24:5637–43. doi: 10.1093/hmg/ddv277
11. Weissbach A, Konig IR, Huckelheim K, Pramstaller PP, Werner E, Bruggemann N, et al. Influence of L-dopa on subtle motor signs in heterozygous Parkin- and PINK1 mutation carriers. Parkinsonism Relat Disord. (2017) 42:95–9. doi: 10.1016/j.parkreldis.2017.07.003
12. Avenali M, Blandini F, Cerri S. Glucocerebrosidase defects as a major risk factor for Parkinson's Disease. Front Aging Neurosci. (2020) 12:97. doi: 10.3389/fnagi.2020.00097
13. Klein C, Krainc D. Glucocerebrosidase mutations: tipping point toward Parkinson disease and dementia? JAMA Neurol. (2013) 70:686–8. doi: 10.1001/jamaneurol.2013.87
14. Lubbe SJ, Bustos BI, Hu J, Krainc D, Joseph T, Hehir J, et al. Assessing the relationship between monoallelic PRKN mutations and Parkinson's risk. Hum Mol Genet. (2021) 30:78–86. doi: 10.1093/hmg/ddaa273
15. Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. (2007) 6:652–62. doi: 10.1016/S1474-4422(07)70174-6
16. Pattaro C, Gogele M, Mascalzoni D, Melotti R, Schwienbacher C, De Grandi A, et al. The Cooperative Health Research in South Tyrol (CHRIS) study: rationale, objectives, preliminary results. J Transl Med. (2015) 13:348. doi: 10.1186/s12967-015-0704-9
17. Pramstaller PP, Schlossmacher MG, Jacques TS, Scaravilli F, Eskelson C, Pepivani I, et al. Lewy body Parkinson's disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol. (2005) 58:411–422. doi: 10.1002/ana.20587
18. Heinzel S, Berg D, Gasser T, Chen H, Yao C, Postuma RB, et al. Update of the MDS research criteria for prodromal Parkinson's disease. Mov Disord. (2019) 34:1464–70. doi: 10.1002/mds.27802
19. Iwaki H, Leonard H, Bandrés-Ciga S, Blauwendraat C, Scholz SW, Faghri F, et al. Biomarkers of Parkinson's Disease: screening vital signs and routine blood tests. medRxiV [Preprint]. (2020). doi: 10.1101/2020.05.18.20103085
20. Kocer A, Yaman A, Niftaliyev E, Duruyen H, Eryilmaz M, Kocer E. Assessment of platelet indices in patients with neurodegenerative diseases: mean platelet volume was increased in patients with Parkinson's disease. Curr Gerontol Geriatr Res. (2013) 2013:986254. doi: 10.1155/2013/986254
21. Qiu X, Xiao Y, Wu J, Gan L, Huang Y, Wang J. C-reactive protein and risk of Parkinson's Disease: a systematic review and meta-analysis. Front Neurol. (2019) 10:384. doi: 10.3389/fneur.2019.00384
22. Sanjari Moghaddam H, Ghazi Sherbaf F, Mojtahed Zadeh M, Ashraf-Ganjouei A, Aarabi MH. Association between peripheral inflammation and datscan data of the striatal nuclei in different motor subtypes of Parkinson Disease. Front Neurol. (2018) 9:234. doi: 10.3389/fneur.2018.00234
23. Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A, et al. Next-generation genotype imputation service and methods. Nat Genet. (2016) 48:1284–7. doi: 10.1038/ng.3656
24. Garcia-Larsen V, Luczynska M, Kowalski ML, Voutilainen H, Ahlstrom M, Haahtela T, et al. Use of a common food frequency questionnaire (FFQ) to assess dietary patterns and their relation to allergy and asthma in Europe: pilot study of the GA2LEN FFQ. Eur J Clin Nutr. (2011) 65:750–6. doi: 10.1038/ejcn.2011.15
25. Craig CL, Marshall AL, Sjostrom M, Bauman AE, Booth ML, Ainsworth BE, et al. International physical activity questionnaire: 12-country reliability and validity. Med Sci Sports Exerc. (2003) 35:1381–95. doi: 10.1249/01.MSS.0000078924.61453.FB
26. Janson C, Anto J, Burney P, Chinn S, de Marco R, Heinrich J, et al. The European Community Respiratory Health Survey: what are the main results so far? European Community Respiratory Health Survey II. Eur Respir J. (2001) 18:598–611. doi: 10.1183/09031936.01.00205801
27. Pramstaller PP, Falk M, Schoenhuber R, Poewe W. Validation of a mail questionnaire for parkinsonism in two languages (German and Italian). J Neurol. (1999) 246:79–86. doi: 10.1007/s004150050312
28. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. (2014) 46:310–5. doi: 10.1038/ng.2892
29. McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The ensembl variant effect predictor. Genome Biol. (2016) 17:122. doi: 10.1186/s13059-016-0974-4
30. Bruggemann N, Mitterer M, Lanthaler AJ, Djarmati A, Hagenah J, Wiegers K, et al. Frequency of heterozygous Parkin mutations in healthy subjects: need for careful prospective follow-up examination of mutation carriers. Parkinsonism Relat Disord. (2009) 15:425–9. doi: 10.1016/j.parkreldis.2008.11.014
31. Pattaro C, Marroni F, Riegler A, Mascalzoni D, Pichler I, Volpato CB, et al. The genetic study of three population microisolates in South Tyrol (MICROS): study design and epidemiological perspectives. BMC Med Genet. (2007) 8:29. doi: 10.1186/1471-2350-8-29
32. Kay DM, Stevens CF, Hamza TH, Montimurro JS, Zabetian CP, Factor SA, et al. A comprehensive analysis of deletions, multiplications, and copy number variations in PARK2. Neurology. (2010) 75:1189–94. doi: 10.1212/WNL.0b013e3181f4d832
33. Prasuhn J, Borsche M, Hicks AA, Gögele M, Egger C, Pichler I, et al. Task matters - challenging the motor system allows distinguishing unaffected Parkin mutation carriers from mutation-free controls. Parkinsonism Relat Disord. (2021) 86:101–4. doi: 10.1016/j.parkreldis.2021.03.028
34. Hilker R, Klein C, Ghaemi M, Kis B, Strotmann T, Ozelius LJ, et al. Positron emission tomographic analysis of the nigrostriatal dopaminergic system in familial parkinsonism associated with mutations in the parkin gene. Ann Neurol. (2001) 49:367–76. doi: 10.1002/ana.74
35. Khan NL, Scherfler C, Graham E, Bhatia KP, Quinn N, Lees AJ, et al. Dopaminergic dysfunction in unrelated, asymptomatic carriers of a single parkin mutation. Neurology. (2005) 64:134–6. doi: 10.1212/01.WNL.0000148725.48740.6D
36. Chohan H, Senkevich K, Patel RK, Bestwick JP, Jacobs BM, Bandres Ciga S, et al. Type 2 diabetes as a determinant of Parkinson's disease risk and progression. Mov Disord. (2021) 36:1420–9. doi: 10.1002/mds.28551
37. Sportelli C, Urso D, Jenner P, Chaudhuri KR. Metformin as a potential neuroprotective agent in prodromal Parkinson's Disease-viewpoint. Front Neurol. (2020) 11:556. doi: 10.3389/fneur.2020.00556
38. Hung CM, Lombardo PS, Malik N, Brun SN, Hellberg K, Van Nostrand JL, et al. AMPK/ULK1-mediated phosphorylation of Parkin ACT domain mediates an early step in mitophagy. Sci Adv. (2021) 7:eabg4544. doi: 10.1126/sciadv.abg4544
39. Trilck-Winkler M, Borsche M, Konig IR, Balck A, Lenz I, Kasten M, et al. Parkin deficiency appears not to be associated with cardiac damage in Parkinson's disease. Mov Disord. (2021) 36:271–3. doi: 10.1002/mds.28422
40. Shao D, Kolwicz SC Jr, Wang P, Roe ND, Villet O, Nishi K, et al. Increasing fatty acid oxidation prevents high-fat diet-induced cardiomyopathy through regulating Parkin-mediated mitophagy. Circulation. (2020) 142:983–97. doi: 10.1161/CIRCULATIONAHA.119.043319
41. Sun Y, Yao X, Zhang QJ, Zhu M, Liu ZP, Ci B, et al. Beclin-1-dependent autophagy protects the heart during sepsis. Circulation. (2018) 138:2247–62. doi: 10.1161/CIRCULATIONAHA.117.032821
42. Akil E, Bulut A, Kaplan I, Ozdemir HH, Arslan D, Aluclu MU. The increase of carcinoembryonic antigen (CEA), high-sensitivity C-reactive protein, and neutrophil/lymphocyte ratio in Parkinson's disease. Neurol Sci. (2015) 36:423–8. doi: 10.1007/s10072-014-1976-1
43. Atac Ucar C, Gokce Cokal B, Unal Artik HA, Inan LE, Yoldas TK. Comparison of neutrophil-lymphocyte ratio (NLR) in Parkinson's disease subtypes. Neurol Sci. (2017) 38:287–93. doi: 10.1007/s10072-016-2758-8
44. Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ, et al. An Anti-CD3 Antibody, Teplizumab, in relatives at risk for type 1 diabetes. N Engl J Med. (2019) 381:603–13. doi: 10.1056/NEJMoa1902226
45. Del Din S, Elshehabi M, Galna B, Hobert MA, Warmerdam E, Suenkel U, et al. Gait analysis with wearables predicts conversion to parkinson disease. Ann Neurol. (2019) 86:357–67. doi: 10.1002/ana.25548
Keywords: Parkin, heterozygote mutation, Parkinson's disease, risk markers, penetrance, population
Citation: Castelo Rueda MP, Raftopoulou A, Gögele M, Borsche M, Emmert D, Fuchsberger C, Hantikainen EM, Vukovic V, Klein C, Pramstaller PP, Pichler I and Hicks AA (2021) Frequency of Heterozygous Parkin (PRKN) Variants and Penetrance of Parkinson's Disease Risk Markers in the Population-Based CHRIS Cohort. Front. Neurol. 12:706145. doi: 10.3389/fneur.2021.706145
Received: 06 May 2021; Accepted: 02 July 2021;
Published: 09 August 2021.
Edited by:
Jie Shen, Brigham and Women's Hospital and Harvard Medical School, United StatesReviewed by:
Suzanne Lesage, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceMarie Ynez Davis, University of Washington, United States
Copyright © 2021 Castelo Rueda, Raftopoulou, Gögele, Borsche, Emmert, Fuchsberger, Hantikainen, Vukovic, Klein, Pramstaller, Pichler and Hicks. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Irene Pichler, irene.pichler@eurac.edu
†These authors have contributed equally to this work