 Mary Clare McKenna1
Mary Clare McKenna1 Philippe Corcia2,3,4
Philippe Corcia2,3,4 Philippe Couratier4,5
Philippe Couratier4,5 We Fong Siah1
We Fong Siah1 Pierre-Francois Pradat6
Pierre-Francois Pradat6 Peter Bede1,6*
Peter Bede1,6*- 1Computational Neuroimaging Group, Trinity College Dublin, Dublin, Ireland
- 2Department of Neurology-Neurophysiology, CRMR ALS, Tours, France
- 3UMR 1253 iBrain, University of Tours, Tours, France
- 4LITORALS, Federation of ALS Centres: Tours-Limoges, Limoges, France
- 5ALS Centre, Limoges University Hospital (CHU de Limoges), Limoges, France
- 6Pitié-Salpêtrière University Hospital, Sorbonne University, Paris, France
Frontotemporal involvement has been extensively investigated in amyotrophic lateral sclerosis (ALS) but remains relatively poorly characterized in other motor neuron disease (MND) phenotypes such as primary lateral sclerosis (PLS), progressive muscular atrophy (PMA), spinal muscular atrophy (SMA), spinal bulbar muscular atrophy (SBMA), post poliomyelitis syndrome (PPS), and hereditary spastic paraplegia (HSP). This review focuses on insights from structural, metabolic, and functional neuroimaging studies that have advanced our understanding of extra-motor disease burden in these phenotypes. The imaging literature is limited in the majority of these conditions and frontotemporal involvement has been primarily evaluated by neuropsychology and post mortem studies. Existing imaging studies reveal that frontotemporal degeneration can be readily detected in ALS and PLS, varying degree of frontotemporal pathology may be captured in PMA, SBMA, and HSP, SMA exhibits cerebral involvement without regional predilection, and there is limited evidence for cerebral changes in PPS. Our review confirms the heterogeneity extra-motor pathology across the spectrum of MNDs and highlights the role of neuroimaging in characterizing anatomical patterns of disease burden in vivo. Despite the contribution of neuroimaging to MND research, sample size limitations, inclusion bias, attrition rates in longitudinal studies, and methodological constraints need to be carefully considered. Frontotemporal involvement is a quintessential clinical facet of MND which has important implications for screening practices, individualized management strategies, participation in clinical trials, caregiver burden, and resource allocation. The academic relevance of imaging frontotemporal pathology in MND spans from the identification of genetic variants, through the ascertainment of presymptomatic changes to the design of future epidemiology studies.
Introduction
This review explores the role of neuroimaging in characterizing frontotemporal pathology in motor neuron diseases (MNDs). While frontotemporal involvement has been extensively investigated in amyotrophic lateral sclerosis (ALS) (Table 1), it is relatively under evaluated in other MND phenotypes, such as primary lateral sclerosis (PLS) (Table 2), progressive muscular atrophy (PMA), spinal bulbar muscular atrophy (SBMA), spinal muscular atrophy (SMA), hereditary spastic paraplegia (HSP), poliomyelitis and post poliomyelitis syndrome (PPS) (Table 3). Progressive muscular atrophy, SBMA, SMA, and poliomyelitis were once regarded as pure anterior horn cell disorders, but emerging data shows that the central nervous system is more widely involved than previously thought (48, 50). Primary lateral sclerosis was traditionally considered a pure UMN condition, but extra-motor manifestations are now gradually recognized (29) (Figure 1). The ALS-FTD continuum of neurodegenerative disorders share common clinical, radiological, genetic, and pathological features (80, 81). Similar cognitive and behavioral manifestations, however, have also been described in the non-ALS MND phenotypes (82, 83). The low incidence of these slowly progressive UMN or LMN predominant disorders coupled with heterogeneous frontotemporal manifestations are all factors that may contribute to delayed or mistaken diagnoses (84–87). Caregiver burden is not only heightened by diagnostic delay, but may be exacerbated by considerable behavioral challenges (88, 89). Frontotemporal involvement may impact on entry into clinical trials and decision to participate in research studies, potentially leading to participation bias. From an academic viewpoint, there are synergistic efforts to evaluate frontotemporal disease burden using computational imaging in combination with clinical instruments. In parallel, these advances help to advance our understanding of disease pathology, propagation patterns, and the dynamics of anatomical spread. The objective of this review is to collate evidence from robust neuroimaging studies, distill emerging research trends, identify pertinent gaps in the literature, highlight clinical implications, and postulate research priorities in the evaluation of frontotemporal pathology across the spectrum of MND phenotypes.
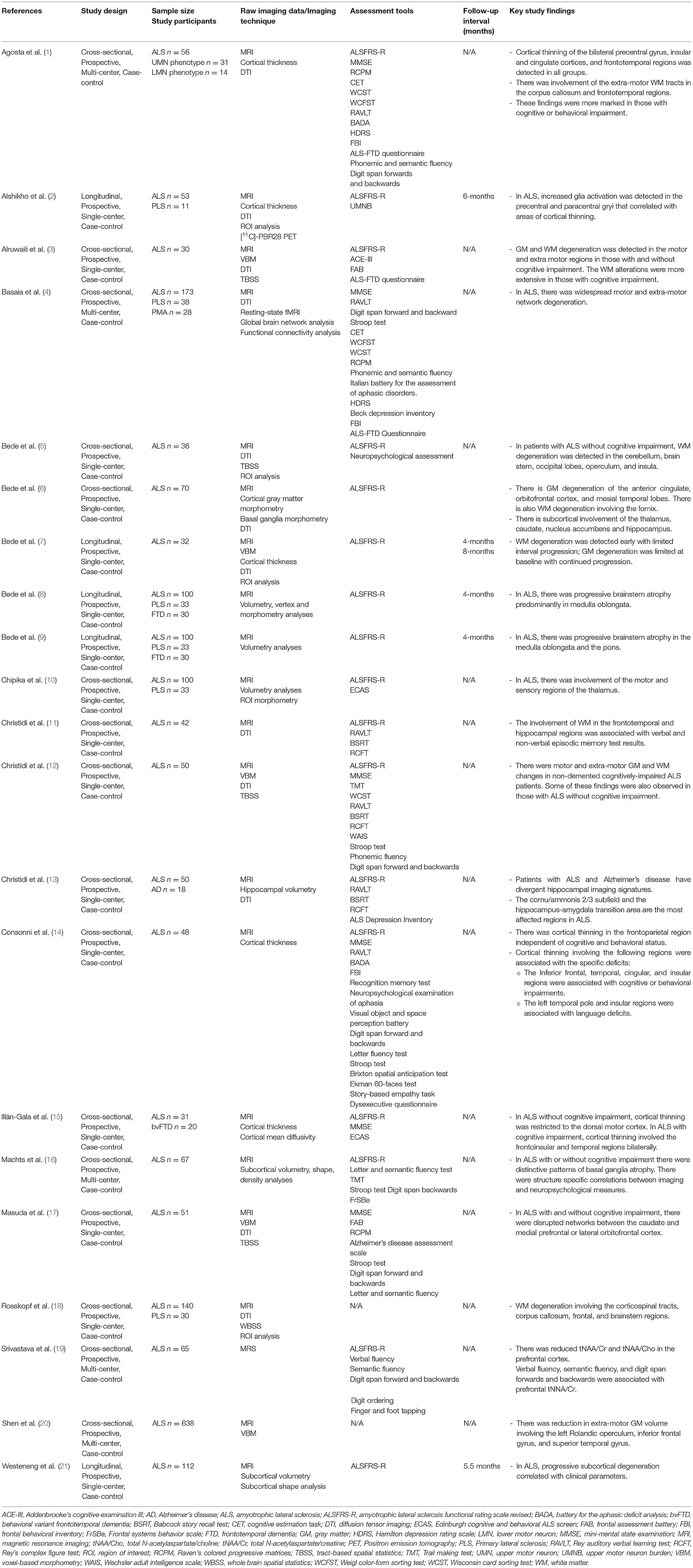
Table 1. Selection of original neuroimaging research articles in ALS since 2015 with more than 30 patients.
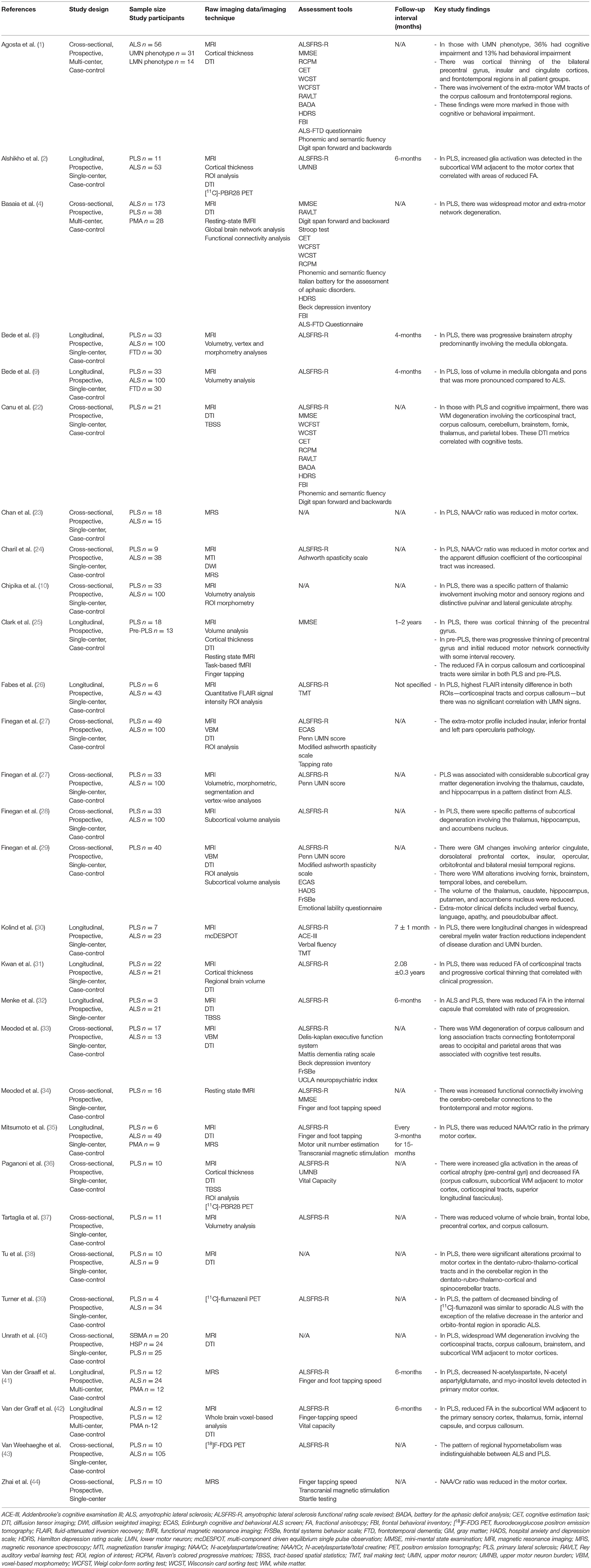
Table 2. Selection of original neuroimaging research articles in PLS.
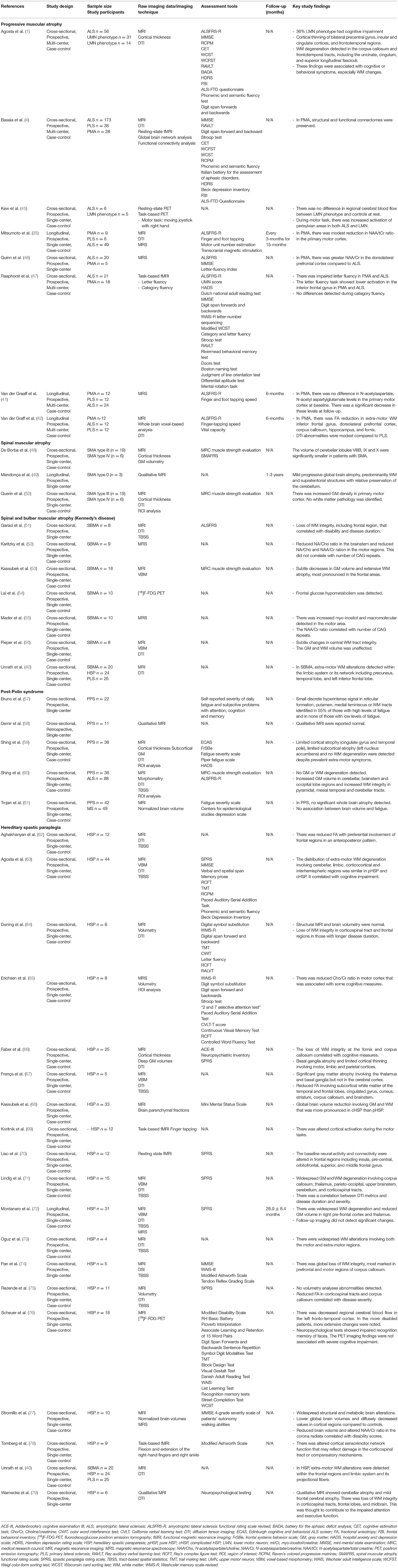
Table 3. Selection of original neuroimaging research articles in PMA, SMA, SBMA, PPS, and HSP.
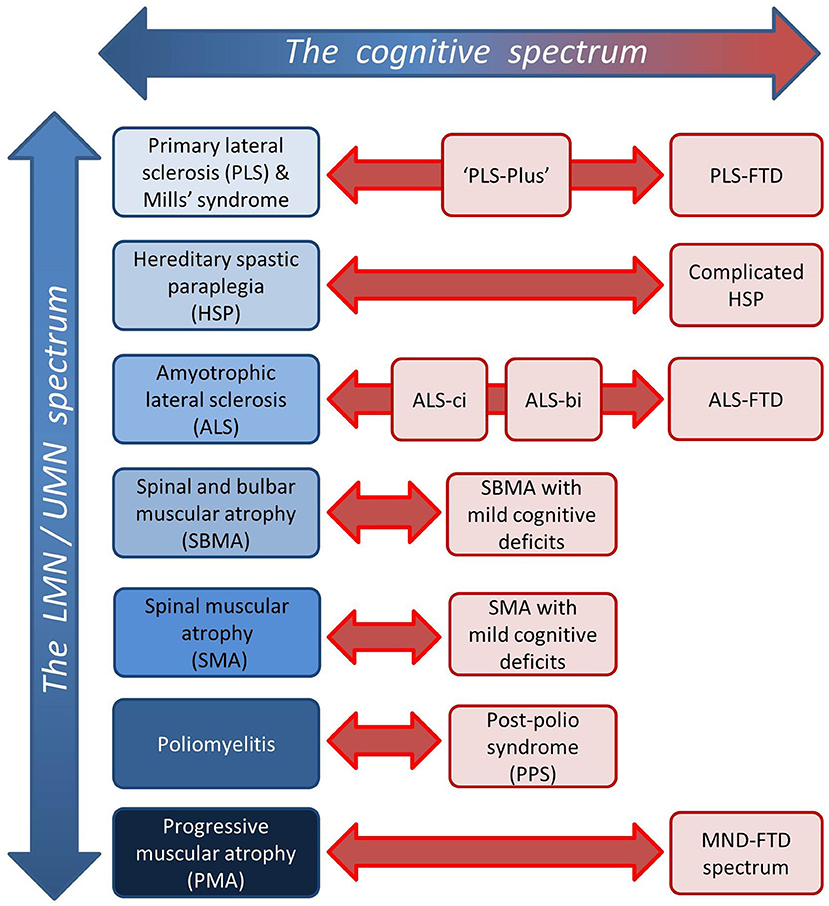
Figure 1. Dimensions of disease heterogeneity in MND; the spectrum of relative upper/lower motor neuron involvement and the spectrum of extra-motor manifestations.
Methods
This is a focused review of original neuroimaging studies that investigated frontotemporal pathology in the following MND phenotypes; PLS, PMA, SBMA, SMA, PPS, HSP, and ALS. The search engines PubMed and Google Scholar were used to identify key papers. Individual MND phenotypes were searched paired with keywords “MRI,” “PET,” “brain imaging,” “neuroimaging,” or “frontotemporal.” Only articles in English were reviewed. Editorials, opinion pieces and review articles were not selected. Additional papers were considered based on the reference list of reviewed publications. One hundred and forty-two original research neuroimaging studies were identified. Given the paucity of prospective neuroimaging studies in non-ALS MNDs, case series, neuropsychology, and post mortem studies were also reviewed in these conditions. The selected articles were systematically evaluated for cohort numbers, study design, clinical assessment, imaging methods, and anatomical focus.
Results
Primary Lateral Sclerosis
Primary Lateral Sclerosis is an upper motor neuron disorder that typically presents with insidious spino-bulbar spasticity in adulthood (27, 90). It is often associated with pseudobulbar affect that may trigger self-imposed social isolation. Extra-motor manifestations are increasingly recognized in PLS (83, 91), occurring in a similar behavioral and cognitive profile to ALS (83), and rarely fulfilling the diagnostic criteria for FTD (83, 91). Such deficits include impaired social cognition, executive function, verbal fluency, language, or apathy (29, 82, 83, 91). The reported cases of frank FTD evolved several years after the insidious onset of UMN signs and were associated with progressive radiological frontotemporal atrophy (91). This is in line with the mounting body of neuroimaging evidence that supports widespread frontotemporal involvement in PLS (22, 29, 33).
The radiological profile of PLS varies from limited extra-motor involvement to widespread pathology (27). Structural and diffusion data revealed degenerative changes in the fornix, body of the corpus callosum, anterior cingulate, dorsolateral prefrontal, insular, opercular, orbitofrontal, and temporal regions (22, 27, 29, 33, 37, 42). Some studies have explored associations with underlying structural abnormalities focusing on apathy, impaired executive function, language, and verbal fluency deficits (29, 33, 92). Longitudinal studies have yielded inconsistent findings with regards to progressive pathology (25, 26, 30–32, 35). A case report described progressive cortical atrophy over an 8.5-year timeframe (93). These observations would suggest that contrary to ALS, longer follow-up intervals may be required in PLS to characterize radiological trajectories. Extra-motor findings in PLS are also supported by metabolic and functional imaging studies. PET imaging studies have detected prefrontal and premotor areas of hypometabolism in PLS that are almost indistinguishable from the patterns seen in ALS (39, 43, 94). Whilst primarily used in a research setting, novel PET radioligand binding studies have also demonstrated alterations beyond the motor system, in the bilateral anterior cingulate gyri, and in left superior temporal lobe (2, 36, 39, 90, 95). MR Spectroscopy in PLS has mostly focused on the evaluation of the motor rather than extra-motor regions (24, 35). Similar to ALS, it shows reduced N-acetyl aspartate/creatinine ratios (23, 24, 35, 41, 44) and increased myo-inositol/creatinine ratios (41) in the motor cortex suggestive of neuronal dysfunction and gliosis, respectively. Resting-state fMRI studies report increased functional connectivity in frontotemporal networks (34, 92) which has been associated with executive dysfunction (4, 92). Similar to ALS, increased functional connectivity is typically considered a “compensatory response” to structural degeneration (92).
The few post-mortem studies are concordant with the extra-motor radiological profile of PLS (96–103). Frontotemporal lobar degeneration has been detected with some cases revealing ubiquitin- and TDP-43-immunoreactive neuronal cytoplasmic inclusion bodies in frontotemporal and hippocampal areas. Post-mortem studies seldom have accompanying comprehensive clinical information, but when available, features of nfvPPA or bvFTD have been described (96, 102).
Progressive Muscular Atrophy
Progressive Muscular Atrophy is a clinical diagnosis that is defined by a gradually progressive isolated lower motor neuron disorder, evolving over many years (104). Reports of extra-motor involvement are inconsistent which is further complicated by the debate on whether PMA is a distinct entity or embedded within the spectrum of ALS (105–108). There are undeniably shared clinical, radiological, and pathological features, albeit less severe compared to ALS. While the initial exclusive LMN clinical presentation distinguishes PMA from ALS, patients with PMA often later develop UMN signs (107). The cognitive profile is also strikingly similar to ALS, with varying levels of executive function, language, fluency, and memory affected (83, 109). In contrast, minimal behavioral impairment is observed, and very few patients with PMA fulfill the diagnostic criteria for FTD (83, 109).
Some imaging studies have identified radiological abnormalities in a distribution that may explain these cognitive deficits (1, 109). Structural analyses have reported loss of white matter integrity in inferior frontal, dorsolateral pre-frontal, and hippocampal regions (42). A task-based fMRI study utilizing a letter fluency task as a test of executive function showed impaired letter fluency and abnormal pre-frontal activation (47). As a counter-argument, a recent study in PMA reported preserved structural integrity with no functional connectivity alterations (4). Neither MRI spectroscopy nor PET imaging studies have identified radiological abnormalities in extra-motor regions (45, 46). It is noteworthy that a dedicated neuropsychological study failed to find a difference between patients with PMA compared to controls (110). Potential shortcomings of the study designs must be considered, including small numbers of patients and the lack of sensitivity of either the chosen task or the imaging modality (46).
The shared neuropathological hallmarks also lend support to the opinion that PMA is part of the ALS clinicopathological continuum (105–107). The pathological substrates of TDP-43 positive inclusions and occasional fused-in-sarcoma (FUS)-positive basophilic inclusions are observed in both conditions, but at a lesser burden and more limited distribution in PMA (108, 111). Post-mortem studies in PMA typically describe LMN degeneration, occasional pyramidal tract degeneration, and additional TDP-43 positive inclusions in the primary motor cortex and hippocampus even in the absence of UMN degeneration (108, 111). These findings raise the question, if in fact the results of PMA studies should be streamlined, interpreted, and analyzed under the umbrella of ALS.
Spinal Muscular Atrophy
Spinal Muscular Atrophy is an autosomal recessive disorder that is caused by either homozygous deletions or loss of function mutations in the survival motor neuron 1 (SMN1) gene resulting in a deficiency of survival motor neuron (SMN) protein (112). It typically manifests as a proximal, predominantly symmetrical motor weakness. The phenotype is stratified in levels of decreasing severity from type 0 to type IV, depending on age of symptom onset and achievement of developmental milestones (112). There are preliminary signals of cerebral involvement in the more severe phenotypes, but it is not yet clear if there is preferential involvement of frontotemporal regions.
The only two cross-sectional quantitative multimodal MRI brain studies evaluated the same 25 treatment naïve adults with type III or type IV SMA initially focusing on the cerebrum and then the cerebellum (48, 50). No supratentorial cortical atrophy was detected (50), but focal cerebellar changes were noted. In the more severe clinical phenotypes, qualitative MRI brain scans have captured more dramatic findings (49, 113–115). In type 0 SMA, widespread supratentorial, and sometimes infratentorial, brain atrophy has been reported. A longitudinal case series of patients with type 0 SMA showed interval radiological abnormalities involving the thalamus and basal ganglia (49, 114). Similar radiological findings have been described in type I SMA (115). For the most part, neuropsychological studies demonstrate preserved cognition (116–119). This is with the caveat that these studies are mostly limited to children and omit the more severe clinical phenotypes (120). Some aspects of childhood development are even deemed superior compared to healthy controls (121–125). The only neuropsychological study of adults with type II or type III SMA described normal rather than superior cognitive abilities. This study reported a possible adaptive mechanism of an inverse correlation between executive function and physical ability, but the level of executive function did not exceed healthy controls (116). In contrast, there are indications of attention and executive function deficits in children with type I SMA (120, 126).
The post mortem examination of the brain is often confounded by coexistent anoxic changes (127, 128). The more severe clinical phenotypes display more widespread features of degeneration involving the cerebral cortex, thalamus, brainstem, and some cranial nerve nuclei that are congruent with ante mortem radiological abnormalities (49, 129). Most of these regions seem spared in the milder phenotypes (127, 128, 130). This has been interpreted as selective neuronal network degeneration occurring below a threshold of SMN protein, although the true clinical significance of this is unknown (129). Overall, the radiological characterization of the more severe clinical phenotypes has proven challenging because of the rarity of the condition, significant disability, and limited life expectancy. In the advent of gene therapy, there may be opportunities for future research in this cohort.
Spinal and Bulbar Muscular Atrophy (Kennedy's Disease)
Spinal and Bulbar Muscular Atrophy, also known as Kennedy's disease, is an X-linked trinucleotide repeat disorder due to expansion of cytosine-adenine-guanine (CAG) repeat in the androgen receptor gene (131). It is a multisystem disorder that typically presents in men in their fourth decade of life with slowly progressive weakness, bulbar involvement, and muscle atrophy due to insidious lower motor neuron degeneration (132, 133). Relatively mild cognitive deficits have been consistently described (134–136). While it is a multi-system disorder, the involvement of the central nervous system has been relatively under-evaluated from a radiological viewpoint (131).
The few brain imaging studies indicate various degree of frontotemporal involvement (53, 54, 56). Quantitative MRI analyses demonstrate a spectrum of frontal gray and white matter abnormalities ranging from entirely unaffected to subtle gray matter atrophy and extensive white matter degeneration (53, 56). Widespread loss of white matter integrity has been reported in the brainstem, corticospinal tracts, and limbic system (40, 51, 56). A single PET imaging study showed hypometabolism in frontal areas (54). The results of conflicting MR spectroscopy studies highlight that subclinical neuronal dysfunction may not be detected by certain imaging protocols (52, 55). A long echo-time MR spectroscopy study demonstrated altered metabolite ratios in the brainstem and motor regions (52); however, a short echo-time MR spectroscopy study failed to reproduce these findings (55). The discrepancy in these results may be explained by the potential pitfall of artificial metabolite elevation because of either metabolite signal overlap or incorrect baseline determination in short echo-time MR spectroscopy (137). These radiological findings are complemented by consistent reports of neuropsychological dysfunction in this cohort albeit mostly at a subclinical level (134–136). Deficits may be so subtle that performance on standard tests of executive function can be normal (134, 135). Mild deficits in social cognition have also been recorded (135). In contrast, single cases of more severe frontal dysfunction have been repeatedly described (138, 139).
Most post-mortem studies in SBMA focus on cardinal spinal cord, peripheral nerve, and proximal muscle changes (133, 139, 140). The pathological examination of cerebral hemispheres is seldom reported. A post-mortem report of an SBMA patient with significant cognitive impairment demonstrated marked diffuse subcortical gliosis in the pre-frontal region, hippocampus, and the degeneration of fronto-bulbar fibers in the midbrain without accompanying cortical pathology (139). Immunohistochemical studies have shown that the pathogenic nuclear mutant AR protein is present in abundance in the central nervous system; supporting the rationale to systematically evaluate cerebral changes in future SBMA studies (141).
Poliomyelitis and Post-polio Syndrome
Post-polio syndrome is characterized by progressive muscular weakness with or without pain, fatigue, and muscle atrophy in patients who have recovered from a distant polio infection (61). Patients often report diverse cognitive symptoms, mostly deficits in attention or memory; however objective evidence is strongly confounded by comorbid factors such as fatigue (57, 142–144). The reportedly high prevalence of extra-motor symptoms is contrasted by the relative lack of cerebral radiological abnormalities in post-polio syndrome (59).
A quantitative MRI study detected minimal cortical and subcortical atrophy, involving the cingulate gyrus, temporal pole, and left nucleus accumbens (59). These subtle changes were not appreciated in other studies (60, 61). Qualitative MRI studies either identified no abnormalities or discrete subcortical hyperintensities that were hypothesized to contribute to the disabling comorbid fatigue (57, 58, 145). Patients with post-polio syndrome frequently exhibit high levels of self-reported fatigue, apathy, and verbal fluency deficits. In the absence of widespread frontotemporal imaging abnormalities, these extra-motor symptoms are postulated to be multifactorial in origin with factors such as low mood, poor sleep, and polypharmacy all playing an additive role (59).
These observations are corroborated by historical pathological studies that demonstrate preferential involvement of the brainstem rather than the cerebrum (57, 146, 147). This is further complicated by reports of patients with a history of polio, who later develop sporadic ALS and demonstrate mixed neuropathological features including the hallmarks of both diseases (148, 149).
Hereditary Spastic Paraplegia
Hereditary spastic paraplegias (HSPs) are a clinically and genetically heterogenous group of neurodegenerative disorders that present as progressive limb weakness and spasticity. They were traditionally divided into “pure” or “complicated” phenotypes based on the absence or presence of extra-motor involvement, respectively (63). In recent times, there has been a shift to stratify these cohorts in accordance with their genetic diagnoses (150). Interestingly, there are radiological indicators of frontotemporal dysfunction irrespective of the subgroup.
Brain imaging studies have shown a reduction in whole brain volume in both clinical phenotypes (68). In pure HSP the volume of gray matter volume is thought to be mostly preserved, whereas in complicated HSP the volume of cortical and deep gray matter may be reduced. The only longitudinal study detected no change in cerebral volume over a 2-year follow-up period (72). This is with the caveat that longer time intervals may be required to detect a significant change. Loss of white matter integrity has been identified in the corpus callosum, in the frontotemporal and parietal regions in both groups (40, 62–64, 72, 73). The severity of these findings correlate with the degree of cognitive impairment (63). Given the relative cortical sparing, cognitive deficits in these cohorts were postulated to be primarily subcortical in origin (64, 66, 67). This was supported by MR spectroscopy reports of abnormal metabolic ratios in the subcortical white matter (65, 72, 77, 151–155). PET imaging studies detected cortical hypometabolism, sometimes implicating the frontotemporal regions (76, 156–161). This was accompanied by clinical measures of frontal dysfunction (76, 161). Resting-state fMRI studies have shown altered functional connectivity involving the primary motor cortex, insula, and superior frontal gyrus (70). Task-based fMRI studies typically report abnormal activation patterns in sensorimotor areas whilst performing motor tasks (69, 78).
In the advent of genotyping, there has been a focused effort to define the radiological signatures of specific genotypes. Spastic paraplegia 4 (SPG 4) is the most common autosomal dominant HSP subtype that is characterized by widespread white matter degeneration with relatively preserved gray matter (64, 71, 75). Subclinical cognitive deficits have been described that later follow a more rapid trajectory of decline escalating in the eights decade of life (162–165). Spastic paraplegia 11 (SPG11) and spastic paraplegia 7 (SPG7) are rare autosomal recessive HSP subtypes that reveal white matter degeneration involving the frontotemporal regions amongst other features (67, 74, 79, 150, 152, 166–168). Varying degrees of cognitive deficits including attention, memory, and executive dysfunction have been described in these genotypes and others (66, 79, 169).
The few post-mortem studies corroborate the radiological descriptions of frontotemporal pathology. Autopsy reports of those with a clinical rather than genetic diagnosis must be interpreted with caution. In clinically defined cases, marked cerebral atrophy and severe gliosis of the cerebral white matter has been described sometimes preferentially involving prefrontal and frontal areas (170–172). In SPG11, widespread frontotemporal cortical degeneration has been described (85). Similar pathological observations have been reported in SPG4, in addition to widespread ubiquitin positivity (164).
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis is the most common form of MND that is characterized by progressive upper and lower motor neuron degeneration in the motor cortex, brainstem nuclei, and anterior horn of the spinal cord. It begins with progressive limb-onset or bulbar-onset muscle weakness that clinically manifests as cramps, fasciculations, muscle wasting, difficulty swallowing, or speaking before ultimately advancing to respiratory failure (173). Additional cognitive and/or behavioral impairment is universally recognized and a minority of patients with ALS also fulfill the diagnostic criteria for FTD (174).
Clinical observations are widely supported by extra-motor neuroimaging findings. Structural imaging consistently reveals frontotemporal gray and white matter degeneration (14, 15, 18, 175–188). Gray matter atrophy has been described in the anterior cingulate, insula, operculum, inferior frontal gyrus, superior temporal gyrus, cerebellum, parietal, and occipital cortex (1, 6, 7, 14, 15, 20, 179, 185, 187–191). White matter degeneration has been detected in the body of the corpus callosum, inferior longitudinal fasciculus, uncinate fasciculus, cerebellum, inferior frontal, middle temporal, superior temporal, orbitofrontal, occipital, and parietal regions (1, 5–7, 11, 14, 15, 18, 20, 175–191). These anatomical findings are often linked to structure-specific behavioral or cognitive deficits (1, 3, 11, 13–15, 186–188, 192–195), but similar patterns have been described in the absence of overt cognitive impairment (1, 17, 189, 191, 196). Extra-motor changes were initially considered to be more prominent in those with C9orf72 genotype compared to those with sporadic ALS (187, 196), but widespread frontotemporal involvement is not unique to C9orf72 (197). Subcortical gray matter involvement can also be readily detected in the hippocampus, amygdala, thalamus, caudate nucleus, putamen, nucleus accumbens, and globus pallidus (6, 10, 11, 13, 16, 21, 185, 189, 196, 198, 199). Progressive brainstem pathology has also been reported preferentially involving the pons and the medulla oblongata (8, 9). Structural and diffusion studies are complemented by robust metabolic and functional imaging studies. PET imaging studies have shown frontotemporal hypometabolism involving the dorsolateral prefrontal, orbitofrontal, anterior frontal, and anterior temporal areas (94, 200–202) and regional hypometabolism has been linked to cognitive deficits in ALS (201, 203, 204). PET imaging abnormalities may precede the detection of cortical atrophy (205). While in their infancy, novel PET radioligand studies highlight microglial activation in frontotemporal regions, suggestive of localized inflammatory processes (2, 206–210). MR spectroscopy detects extra-motor abnormalities, potentially before the emergence of clinical symptoms (211). It shows reduced N-acetylaspartate indicative of neuronal dysfunction in the mid-cingulate gyrus (212), dorsolateral (46, 213), ventrolateral (214), and mesial prefrontal cortices (211, 215). Sometimes these frontal lobe abnormalities are subtle (19) and may be associated with measures of executive dysfunction (19, 46, 213). Resting-state fMRI studies captured both increased and decreased functional connectivity within networks that mediate specific behavioral and cognitive functions (216–222). Task-based fMRI studies have linked these abnormal activation patterns with different facets of cognition, specifically executive function (223–225), social cognition (224, 226–230), memory (227, 231, 232), and language (233). Executive dysfunction is associated with increased activation of the right superior and inferior frontal areas (224), left superior and mid temporal gyrus and left anterior cingulate gyrus (223) and decreased activation in the left precentral gyrus (223), and dorsolateral pre-frontal cortex (223, 225, 233); impaired social cognition is associated with increased activation in the prefrontal cortex (224, 226, 228, 229), right supramarginal area (230), right posterior temporal sulcus, and decreased activation in the bilateral hippocampus (229); memory deficits are associated with increased activation in the hippocampus (231) and superior frontal gyrus (232), and decreased activation in the right pre-frontal cortex (232); and finally impaired language is associated with decreased activation patterns in the pre-frontal cortex, right cingulate gyrus, and left temporal lobes (233). For the most part there are increased (224, 226, 228–231) or co-existing (223, 225, 227, 229, 230, 232) activation patterns which suggests either loss of inhibitory dysfunction or partial compensation to overcome early functional impairment (234–236). Overall there does not seem to be a consistent compensatory or inhibitory effect which suggests that these patients may have been captured at different stages of disease. Functional studies have also been widely utilized to evaluate extra-pyramidal dysfunction in ALS (237, 238). Emerging functional modalities, such as magnetoencephalography or spectral EEG have also confirmed widespread extra-motor dysfunction and as these technologies develop they are likely to contribute important additional insights (239–243). The majority of imaging studies in ALS explored the underpinnings of the most commonly affected neuropsychological domains (233, 244), such as the substrate of verbal fluency deficits, executive dysfunction, and behavioral impairment, but with the recognition of the relatively high prevalence of impairments in social cognition, memory deficits, and of apathy, the focus of imaging studies is likely to gradually shift (12, 245–249). Imaging changes in ALS are typically solely interpreted based on genetic and clinical profiles, and seldom correlated with other markers such biofluid markers (250–252). The radiological patterns identified by various imaging studies are largely congruent with the distribution of pathological TDP-43 (pTDP-43) aggregates in extra-motor brain regions (253–257). Patients with ALS-FTD are thought to carry increased extra-motor pTDP-43 burden compared to patients without cognitive impairment (256). A study of patients with cognitive impairment revealed correlations between regional pTDP-43 load and executive, language and fluency deficits (253).
Discussion
This review collates evidence of radiological frontotemporal involvement in common MND phenotypes. Existing neuroimaging studies suggest that frontotemporal degeneration may be readily detected in ALS and PLS; a varying degree of frontotemporal pathology may be captured in PMA, SBMA, and HSP. Cerebral involvement without regional predilection may be exhibited in the more severe clinical phenotypes of SMA; and there is limited evidence for cerebral changes in PPS (Figure 2, Table 4). These radiological features may precede clinical symptoms, and longitudinal studies often capture gradual progression. Imaging studies in MND suffer from considerable inclusion bias because of disease-specific factors. Patients with significant apathy, motor disability, respiratory compromise, or sialorrhoea are less likely to participate or return for follow-up imaging.
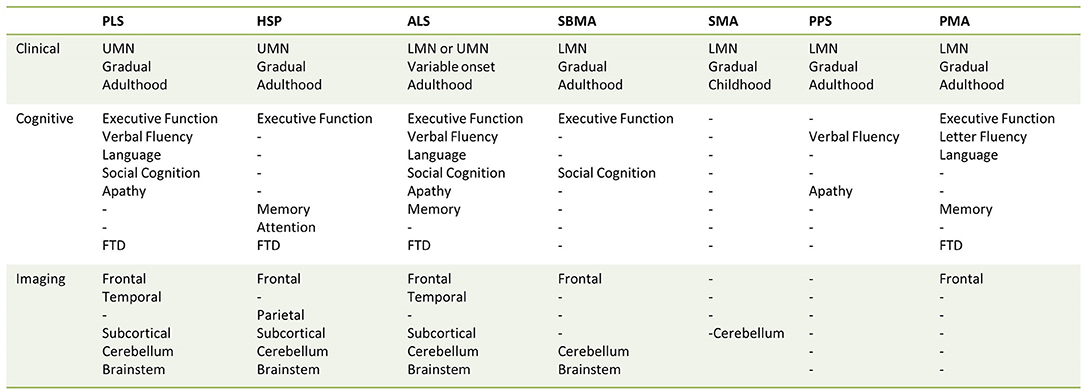
Figure 2. Anatomical vulnerability and clinical domains affected across the spectrum of motor neuron diseases.
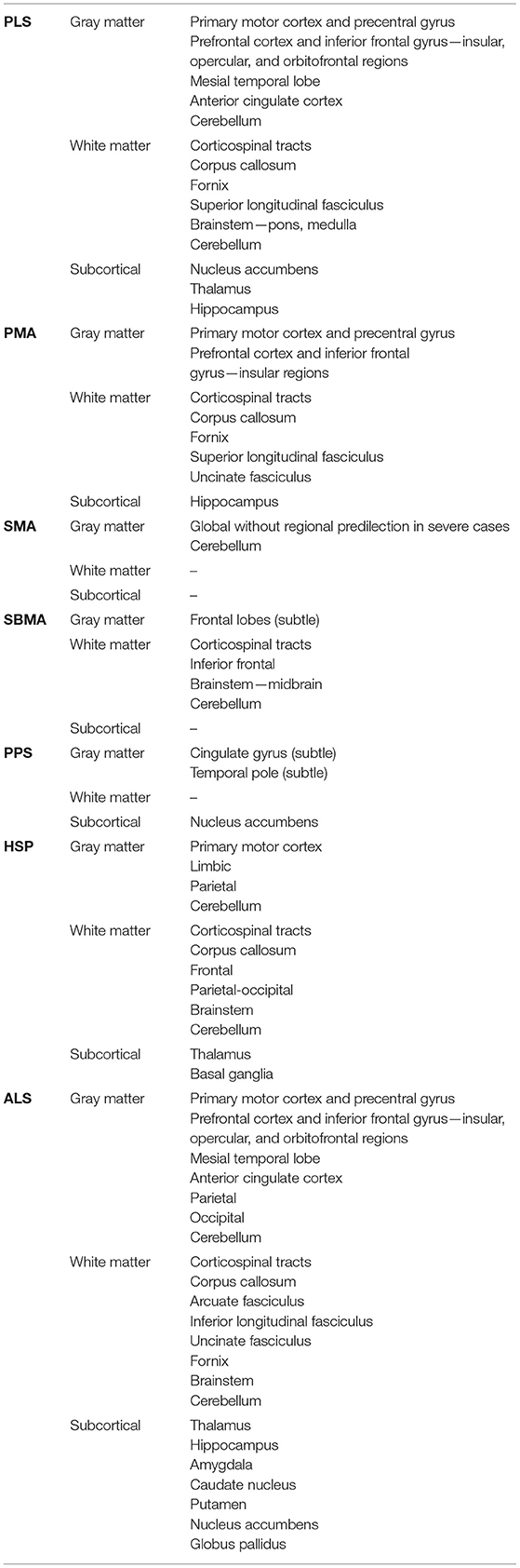
Table 4. An overview of preferential anatomical involvement in MND phenotypes.
In clinical practice, the wide spectrum of frontotemporal manifestations in ALS are already incorporated in the clinical diagnostic criteria (174). It is anticipated that these features will be a fundamental part of future revisions, in conjunction with supportive neuroimaging data (258). Despite implications for survival (259), clinical staging systems of ALS have omitted to include a cognitive facet thus far (260, 261). These observations have also not yet translated into the diagnostic criteria of other MND phenotypes. While such deficits are increasingly recognized in PLS, they are deemed too infrequent to be included in the core clinical features (262). The link between FTD and other rare MND phenotypes may have important implications for everyday clinical practice, particularly given that many non-ALS MND phenotypes are associated with longer survival than ALS (263). The awareness of possible frontotemporal dysfunction may prompt the use of neuropsychological screening tests in the routine evaluation of these patients. Validated, disease-specific screening tools are preferred to generic instruments, and these are available in ALS (264, 265). Several of these are adapted to motor disability and dysarthria, and interrogate domains commonly affected in ALS. It is worth noting that patients with predominant frontotemporal cognitive deficits should be screened for incipient motor deficits (266). The early recognition of neuropsychological deficits is crucial for individualized patient care including: the appraisal of decision-making capacity, caregiver support, resource allocation, and the anticipation of management challenges (89). It may also allow clinicians to consider pharmacological and non-pharmacological interventions such as cognitive or behavioral rehabilitation. In the context of FTD, this is primarily focused on developing compensatory skills for adapting to functional impairments with the lowest level of assistance required. For example, an electronic device calendar is a daily planning tool that can be used to establish routines and set reminders to initiate activities such as taking medications (267). There is also an evolving interest in early language therapy interventions (268, 269). The education of caregivers is crucial to identify unmet needs of the patient that may trigger behavioral problems. These measures have proven to be beneficial to both the patient and their caregivers (267).
In tandem, technological innovations have enriched the supportive radiological data. High-field MRI generates better quality images and acceleration techniques enable shorter data acquisition that may be better tolerated by patients. Quantitative MRI analyses using validated computational pipelines and reliance on robust comparative, correlative, and classifier models enhance the clinical interpretation of vast imaging datasets (270). The advent of structural and functional connectivity studies have ignited interest in the concept of disease-specific selective network degeneration rather than the emphasis on focal pathology (271). These methods have proven particularly useful to differentiate clinical phenotypes and map longitudinal changes in neurodegenerative disorders (7, 271). Novel MRI pulse sequences, non-Gaussian diffusion models such as DKI or NODDI, quantitative susceptibility mapping, and multi-voxel spectroscopy are just some of the promising new tools enriching our armamentarium of imaging tools (272–274). While these methods continue to be tested in the research community they have not been implemented in routine clinical radiology protocols (275).
Frontotemporal involvement across the spectrum of MND phenotypes has important implications for clinical trials. It invites the opportunity for the development of radiological biomarkers that quantify and track frontotemporal involvement (276). Pioneering gene therapy trials have primarily focused on clinical outcome measures such as motor milestones, requiring artificial ventilation, and survival (277–279). This is also relevant because there has been interest in developing adjunctive interventions such as transcranial or neuromuscular magnetic stimulation (280, 281). These methods are not only applicable to symptomatic patients but also to those in the pre-symptomatic stages of their disease. In ALS, genotype-specific radiological alterations have been detected in pre-symptomatic carriers of pathogenic mutations decades before the onset of clinical symptoms (282–284). Awareness of associated behavioral and cognitive impairment allows for due preparation and adaption of study designs if required (285).
As we begin to incorporate these developments into clinical practice and clinical trial designs, there are pressing academic questions to be elucidated. First and foremost, it is uncertain if the motor or extra-motor changes evolve in sequence or in parallel across all phenotypes. This topic is probably best explored in ALS where there is a unique opportunity to study the pre-symptomatic phase in carriers of pathogenic C9orf72 repeat expansions. In this cohort, radiological co-existence of motor and extra-motor involvement has been consistently described (282–286). Overall, the topography of radiological alterations is largely similar but less marked than what is described in symptomatic cases. It is unclear if the initial pattern dictates the ultimate clinical phenotype given that both FTD and ALS may have co-existent subclinical motor and extra-motor manifestations (284, 287, 288). It is also unclear whether these findings solely represent early neurodegenerative changes; some postulate that they capture a developmental abnormality (284). From a clinical perspective, early cognitive deficits have been described in pre-symptomatic carriers of C9orf72 expansion before the phenotype is defined (282–284, 286). The notion of cognitive reserve has been increasingly evaluated in ALS which may impact on the sequence of symptom manifestation. It suggests that those with a high level of cognitive reserve, often proxied with educational attainment, require a greater degree of brain pathology to meet the threshold for clinical symptoms (289). This concept has been investigated in greater detail in FTD, but similar themes are also emerging in ALS. The level of cognitive reserve appears to predict cognitive performance and the degree of brain imaging abnormalities (290–292). These observations suggest that patient-specific factors influence the chronology of clinically evident symptoms. Some argue that the debate of whether extra-motor or motor symptoms emerge first in ALS is antiquated and that cognition and motor function are inseparably intertwined (293). It is hypothesized that the selective deficit in action words and verb processing detected in patients with ALS is in fact a cognitive manifestation of motor dysfunction (293). Although some disagree and consider it to be a feature of executive dysfunction (294). Task-based fMRI studies in healthy controls have consistently shown that reading action words activates areas along the motor strip that were responsible for conducting these movements (295). In ALS, action observation and motor imagery are routinely utilized in fMRI studies to compensate for motor disability (296).
Cognitive deficits in specific domains have been linked to the degeneration of single structures in MND (1, 14, 16, 33, 186, 188, 244). Often there is frank dissociation between cognitive and radiological findings (59, 63), but a reporting bias for confirmed associations prevents the gauging of this occurrence. Correlation analyses in ALS linked apathy to anterior cingulate and accumbens nucleus degeneration (193–195), and memory impairment to hippocampal degeneration (13). Linking cognitive deficits to single structures however may be a reductionist approach, which overlooks the role of complex cortico-subcortical networks in mediating cognitive functions (297). Accordingly, the underpinnings of neuropsychological deficits are probably best evaluated at a circuitry-integrity level instead of seeking associations with focal structures (271). Traditional structural measures are increasingly complemented by connectivity metrics to appraise the integrity of functional circuits (240, 242). The selective vulnerability of functional networks is thought to drive cardinal manifestations of neurodegenerative conditions (271). It may or may not be associated with focal atrophy of crucial nodes within these networks (298).
There are stereotyped shortcomings in the current literature that remain to be addressed. First, the low incidence of these conditions leads to small sample size despite multi-center collaborations. Second, while case-control study designs are often used to evaluate these rare disorders, this cross-sectional approach is suboptimal to characterize dynamically evolving processes. Furthermore, the indolent progression of the non-ALS MND phenotypes may require relatively long follow-up intervals to detect progressive radiological changes (72). Third, co-existing neurodegenerative disorders are potential confounders, such as behavioral variant Alzheimer's dementia. To account for this, the use of serum or cerebrospinal fluid biomarkers should be considered in future study methods to enhance diagnostic certainty. Fourth, there is a scarcity of pre-symptomatic studies and often these cohorts are not followed longitudinally until phenoconversion. Fifth, the diagnostic criteria are not well-defined in some MNDs (104). The diagnosis of “definite PLS” requires a symptom duration of at least 4 years which may further limit the number of patients available for recruitment (28). Sixth, imaging studies often concentrate on supratentorial cortical regions, overlooking the contribution of subcortical and cerebellar pathology to cognitive and behavioral manifestations. The sensitivity limitations of single imaging modalities are seldom acknowledged. Subtle abnormalities may not be detected, considerable neuronal loss may ensue before it becomes radiologically evident. Seventh, the practical implications of cognitive deficits need to be specifically investigated. The presence of cognitive impairment in ALS is considered a negative prognostic indicator that is associated with increased caregiver burden, reduced quality of life, and reduced survival; whereas the implications of cognitive impairment in other MND phenotypes is woefully under-evaluated despite their markedly longer survival (259). Finally, there is a disappointing lack of post-mortem validation of radiological findings. This is further complicated by the inherent bias of the pathological literature to favor atypical cases that are unlikely to represent the true hallmarks of these conditions.
This paper offers an overview of imaging efforts across the spectrum of MNDs to investigate frontotemporal disease expansion. It highlights the disproportionate emphasis on ALS, which offers valuable lessons to conduct similar studies in other MND phenotypes. Radiological observations highlight the rationale for routine screening for frontotemporal dysfunction to inform individualized patient care. The quality of the data may be enhanced by using multiparametric imaging protocols, longitudinal study designs, and the inclusion of pre-symptomatic cohorts where possible. The opportunity for international collaborations through carefully harmonized protocols should be explored to maximize the number of study participants in low incidence phenotypes.
Conclusions
In contrast to ALS, the quantitative characterization of frontotemporal disease burden in non-ALS MND phenotypes remains under investigated. The nuanced evaluation of frontotemporal dysfunction across the entire spectrum of MNDs has important pragmatic implications for individualized clinical care, caregiver support, clinical trial designs, and more broadly, for our understanding of disease biology.
Author Contributions
Manuscript drafted by MM and PB. Review for intellectual content by PCor, PCou, WS, and P-FP. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the Spastic Paraplegia Foundation (SPF). PB and the Computational Neuroimaging Group are also supported by the Health Research Board (HRB EIA-2017-019), the EU Joint Programme-Neurodegenerative Disease Research (JPND), the Irish Institute of Clinical Neuroscience (IICN), and the Iris O'Brien Foundation. PCou was supported by the Inter-Universities Limoges—Tours federation LiTorals and by the ERN-NMD. P-FP was supported by the French charity for ALS research (ARSLA), the Thierry Latran Foundation, and the EU Joint Programme—Neurodegenerative Disease Research (JPND).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Agosta F, Ferraro PM, Riva N, Spinelli EG, Chiò A, Canu E, et al. Structural brain correlates of cognitive and behavioral impairment in MND. Hum Brain Mapp. (2016) 37:1614–26. doi: 10.1002/hbm.23124
2. Alshikho MJ, Zurcher NR, Loggia ML, Cernasov P, Reynolds B, Pijanowski O, et al. Integrated magnetic resonance imaging and ((11) C)-PBR28 positron emission tomographic imaging in amyotrophic lateral sclerosis. Ann Neurol. (2018) 83:1186–97. doi: 10.1002/ana.25251
3. Alruwaili AR, Pannek K, Coulthard A, Henderson R, Kurniawan ND, McCombe P. A combined tract-based spatial statistics and voxel-based morphometry study of the first MRI scan after diagnosis of amyotrophic lateral sclerosis with subgroup analysis. J Neuroradiol. (2018) 45:41–8. doi: 10.1016/j.neurad.2017.03.007
4. Basaia S, Agosta F, Cividini C, Trojsi F, Riva N, Spinelli EG, et al. Structural and functional brain connectome in motor neuron diseases. A multicenter MRI study. Neurlogy. (2020) 95:e2552–64. doi: 10.1212/WNL.0000000000010731
5. Bede P, Elamin M, Byrne S, McLaughlin RL, Kenna K, Vajda A, et al. Patterns of cerebral and cerebellar white matter degeneration in ALS. J Neurol Neurosurg Psychiatry. (2015) 86:468–70. doi: 10.1136/jnnp-2014-308172
6. Bede P, Iyer PM, Schuster C, Elamin M, McLaughlin RL, Kenna K, et al. The selective anatomical vulnerability of ALS: 'disease-defining' and 'disease-defying' brain regions. Amyotroph Lateral Scler Frontotemporal Degener. (2016) 17:561–70. doi: 10.3109/21678421.2016.1173702
7. Bede P, Hardiman O. Longitudinal structural changes in ALS: a three time-point imaging study of white and gray matter degeneration. Amyotroph Lateral Scler Frontotemporal Degener. (2018) 19:232–41. doi: 10.1080/21678421.2017.1407795
8. Bede P, Chipika RH, Finegan E, Li Hi Shing S, Doherty MA, Hengeveld JC, et al. Brainstem pathology in amyotrophic lateral sclerosis and primary lateral sclerosis: a longitudinal neuroimaging study. NeuroImage Clin. (2019) 24:102054. doi: 10.1016/j.nicl.2019.102054
9. Bede P, Chipika RH, Finegan E, Li Hi Shing S, Chang KM, Doherty MA, et al. Progressive brainstem pathology in motor neuron diseases: imaging data from amyotrophic lateral sclerosis and primary lateral sclerosis. Data Brief. (2020) 29:105229. doi: 10.1016/j.dib.2020.105229
10. Chipika RH, Finegan E, Li Hi Shing S, McKenna MC, Christidi F, Chang KM, et al. “Switchboard” malfunction in motor neuron diseases: selective pathology of thalamic nuclei in amyotrophic lateral sclerosis and primary lateral sclerosis. NeuroImage Clin. (2020) 27:102300. doi: 10.1016/j.nicl.2020.102300
11. Christidi F, Karavasilis E, Zalonis I, Ferentinos P, Giavri Z, Wilde EA, et al. Memory-related white matter tract integrity in amyotrophic lateral sclerosis: an advanced neuroimaging and neuropsychological study. Neurobiol Aging. (2017) 49:69–78. doi: 10.1016/j.neurobiolaging.2016.09.014
12. Christidi F, Karavasilis E, Velonakis G, Ferentinos P, Rentzos M, Kelekis N, et al. The clinical and radiological spectrum of hippocampal pathology in amyotrophic lateral sclerosis. Front Neurol. (2018) 9:523. doi: 10.3389/fneur.2018.00523
13. Christidi F, Karavasilis E, Rentzos M, Velonakis G, Zouvelou V, Xirou S, et al. Hippocampal pathology in amyotrophic lateral sclerosis: selective vulnerability of subfields and their associated projections. Neurobiol Aging. (2019) 84:178–88. doi: 10.1016/j.neurobiolaging.2019.07.019
14. Consonni M, Contarino VE, Catricalà E, Dalla Bella E, Pensato V, Gellera C, et al. Cortical markers of cognitive syndromes in amyotrophic lateral sclerosis. Neuroimage Clin. (2018) 19:675–82. doi: 10.1016/j.nicl.2018.05.020
15. llán-Gala I, Montal V, Pegueroles J, Vilaplana E, Alcolea D, Dols-Icardo O, et al. Cortical microstructure in the amyotrophic lateral sclerosisifrontotemporal dementia continuum. Neurology. (2020) 95:e2565–76. doi: 10.1212/WNL.0000000000010727
16. Machts J, Loewe K, Kaufmann J, Jakubiczka S, Abdulla S, Petri S, et al. Basal ganglia pathology in ALS is associated with neuropsychological deficits. Neurology. (2015) 85:1301–9. doi: 10.1212/WNL.0000000000002017
17. Masuda M, Senda J, Watanabe H, Epifanio B, Tanaka Y, Imai K, et al. Involvement of the caudate nucleus head and its networks in sporadic amyotrophic lateral sclerosis-frontotemporal dementia continuum. Amyotroph Lateral Scler Frontotemporal Degener. (2016) 17:571–9. doi: 10.1080/21678421.2016.1211151
18. Rosskopf J, Muller HP, Dreyhaupt J, Gorges M, Ludolph AC, Kassubek J. Ex post facto assessment of diffusion tensor imaging metrics from different MRI protocols: preparing for multicentre studies in ALS. Amyotroph Lateral Scler Frontotemporal Degener. (2015) 16:92–101. doi: 10.3109/21678421.2014.977297
19. Srivastava O, Hanstock C, Chenji S, Mah D, Eurich D, Ta D, et al. Cerebral degeneration in amyotrophic lateral sclerosis: a prospective multicenter magnetic resonance spectroscopy study. Neurol Clin Pract. (2019) 9:400–7. doi: 10.1212/CPJ.0000000000000674
20. Shen D, Cui L, Fang J, Cui B, Li D, Tai H. Voxel-wise meta-analysis of gray matter changes in amyotrophic lateral sclerosis. Front Aging Neurosci. (2016) 8:64. doi: 10.3389/fnagi.2016.00064
21. Westeneng HJ, Verstraete E, Walhout R, Schmidt R, Hendrikse J, Veldink JH, et al. Subcortical structures in amyotrophic lateral sclerosis. Neurobiol Aging. (2015) 36:1075–82. doi: 10.1016/j.neurobiolaging.2014.09.002
22. Canu E, Agosta F, Galantucci S, Chio A, Riva N, Silani V, et al. Extramotor damage is associated with cognition in primary lateral sclerosis patients. PLoS ONE. (2013) 8:e82017. doi: 10.1371/journal.pone.0082017
23. Chan S, Shungu DC, Douglas-Akinwande A, Lange DJ, Rowland LP. Motor neuron diseases: comparison of single-voxel proton MR spectroscopy of the motor cortex with MR imaging of the brain. Radiology. (1999) 212:763–9. doi: 10.1148/radiology.212.3.r99au35763
24. Charil A, Corbo M, Filippi M, Kesavadas C, Agosta F, Munerati E, et al. Structural and metabolic changes in the brain of patients with upper motor neuron disorders: a multiparametric MRI study. Amyotroph Lateral Scler. (2009) 10:269–79. doi: 10.3109/17482960902777339
25. Clark MG, Smallwood Shoukry R, Huang CJ, Danielian LE, Bageac D, et al. Loss of functional connectivity is an early imaging marker in primary lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2018) 19:562–9. doi: 10.1080/21678421.2018.1517180
26. Fabes J, Matthews L, Filippini N, Talbot K, Jenkinson M, Turner MR. Quantitative FLAIR MRI in amyotrophic lateral sclerosis. Acad. Radiol. (2017) 24:1187–94. doi: 10.1016/j.acra.2017.04.008
27. Finegan E, Chipika RH, Shing SLH, Doherty MA, Hengeveld JC, Vajda A, et al. The clinical and radiological profile of primary lateral sclerosis: a population-based study. J Neurol. (2019) 266:2718–33. doi: 10.1007/s00415-019-09473-z
28. Finegan E, Li Hi Shing S, Siah WF, Chipika RH, Chang KM, McKenna MC, et al. Evolving diagnostic criteria in primary lateral sclerosis: the clinical and radiological basis of “probable PLS”. J Neurol Sci. (2020) 417:117052. doi: 10.1016/j.jns.2020.117052
29. Finegan E, Shing SLH, Chipika RH, Chang KM, McKenna MC, Doherty MA, et al. Extra-motor cerebral changes and manifestations in primary lateral sclerosis. Brain Imaging Behav. (2021). doi: 10.1007/s11682-020-00421-4
30. Kolind S, Sharma R, Knight S, Johansen-Berg H, Talbot K, Turner MR. Myelin imaging in amyotrophic and primary lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2013) 14:562–73. doi: 10.3109/21678421.2013.794843
31. Kwan JY, Meoded A, Danielian LE, Wu T, Floeter MK. Structural imaging differences and longitudinal changes in primary lateral sclerosis and amyotrophic lateral sclerosis. NeuroImage Clin. (2013) 2:151–60. doi: 10.1016/j.nicl.2012.12.003
32. Menke RA, Abraham I, Thiel CS, Filippini N, Knight S, Talbot K, et al. Fractional anisotropy in the posterior limb of the internal capsule and prognosis in amyotrophic lateral sclerosis. Arch Neurol. (2012) 69:1493–9. doi: 10.1001/archneurol.2012.1122
33. Meoded A, Kwan JY, Peters TL, Huey ED, Danielian LE, Wiggs E, et al. Imaging findings associated with cognitive performance in primary lateral sclerosis and amyotrophic lateral sclerosis. Dement Geriatr Cogn Disord Extra. (2013) 3:233–50. doi: 10.1159/000353456
34. Meoded A, Morrissette AE, Katipally R, Schanz O, Gotts SJ, Floeter MK. Cerebro-cerebellar connectivity is increased in primary lateral sclerosis. NeuroImage Clin. (2015) 7: 288–96. doi: 10.1016/j.nicl.2014.12.009
35. Mitsumoto H, Ulug AM, Pullman SL, Gooch CL, Chan S, Tang MX, et al. Quantitative objective markers for upper and lower motor neuron dysfunction in ALS. Neurology. (2007) 68:1402–10. doi: 10.1212/01.wnl.0000260065.57832.87
36. Paganoni S, Alshikho MJ, Zürcher NR, Cernasov P, Babu S, Loggia ML, et al. Imaging of glia activation in people with primary lateral sclerosis. NeuroImage Clin. (2018) 17:347–53. doi: 10.1016/j.nicl.2017.10.024
37. Tartaglia MC, Laluz V, Rowe A, Findlater K, Lee DH, Kennedy K, et al. Brain atrophy in primary lateral sclerosis. Neurology. (2009) 72:1236–41. doi: 10.1212/01.wnl.0000345665.75512.f9
38. Tu S, Menke RAL, Talbot K, Kiernan MC, Turner MR. Cerebellar tract alterations in PLS and ALS. Amyotroph Lateral Scler Frontotemporal Degener. (2019) 20:281–4. doi: 10.1080/21678421.2018.1562554
39. Turner MR, Hammers A, Al-Chalabi A, Shaw CE, Andersen PM, Brooks DJ, et al. Cortical involvement in four cases of primary lateral sclerosis using. (11C)-flumazenil PET. J Neurol. (2007) 254:1033–36. doi: 10.1007/s00415-006-0482-7
40. Unrath A, Müller HP, Riecker A, Ludolph AC, Sperfeld AD, Kassubek J. Whole brain-based analysis of regional white matter tract alterations in rare motor neuron diseases by diffusion tensor imaging. Hum Brain Mapp. (2010) 31:1727–40. doi: 10.1002/hbm.20971
41. van der Graaff MM, Lavini C, Akkerman EM, Majoie Ch B, Nederveen AJ, Zwinderman AH, et al. MR spectroscopy findings in early stages of motor neuron disease. AJNR Am J Neuroradiol. (2010) 31:1799–806. doi: 10.3174/ajnr.A2217
42. van der Graaff MM, Sage CA, Caan MWA, Akkerman EM, Lavini C, Majoie CB, et al. Upper and extra-motoneuron involvement in early motoneuron disease: a diffusion tensor imaging study. Brain. (2011) 134:1211–28. doi: 10.1093/brain/awr016
43. Van Weehaeghe D, Ceccarini J, Delva A, Robberecht W, Van Damme P, Van Laere K. Prospective validation of 18F-FDG brain PET discriminant analysis methods in the diagnosis of amyotrophic lateral sclerosis. J Nucl Med. (2016) 57:1238–43. doi: 10.2967/jnumed.115.166272
44. Zhai P, Pagan F, Statland J, Butman JA, Floeter MK. Primary lateral sclerosis. Neurology. (2003) 60:1258. doi: 10.1212/01.WNL.0000058900.02672.D2
45. Kew JJ, Brooks DJ, Passingham RE, Rothwell JC, Frackowiak RS, Leigh PN. Cortical function in progressive lower motor neuron disorders and amyotrophic lateral sclerosis: a comparative PET study. Neurology. (1994) 44:1101–10. doi: 10.1212/WNL.44.6.1101
46. Quinn C, Elman L, McCluskey L, Hoskins K, Karam C, Woo JH, et al. Frontal lobe abnormalities on MRS correlate with poor letter fluency in ALS. Neurology. (2012) 79: 583–8. doi: 10.1212/WNL.0b013e3182635720
47. Raaphorst J, van Tol MJ, Groot PF, Altena E, van der Werf YD, Majoie CB, et al. Prefrontal involvement related to cognitive impairment in progressive muscular atrophy. Neurology. (2014) 83:818–25. doi: 10.1212/WNL.0000000000000745
48. de Borba FC, Querin G, França MC, Pradat PF. Cerebellar degeneration in adult spinal muscular atrophy patients. J Neurol. (2020) 267:2625–31. doi: 10.1007/s00415-020-09875-4
49. Mendonça RH, Rocha AJ, Lozano-Arango A, Diaz AB, Castiglioni C, Silva AMS, et al. Severe brain involvement in 5q spinal muscular atrophy type 0. Ann Neurol. (2019) 86:458–62. doi: 10.1002/ana.25549
50. Querin G, El Mendili MM, Lenglet T, Behin A, Stojkovic T, et al. The spinal and cerebral profile of adult spinal-muscular atrophy: a multimodal imaging study. NeuroImage Clin. (2019) 21:101618. doi: 10.1016/j.nicl.2018.101618
51. Garaci F, Toschi N, Lanzafame S, Marfia GA, Marziali S, Meschini A, et al. Brain MR diffusion tensor imaging in Kennedy's disease. Neuroradiol J. (2015) 28:126–32. doi: 10.1177/1971400915581740
52. Karitzky J, Block W, Mellies JK, Träber F, Sperfeld A, Schild HH, et al. Proton magnetic resonance spectroscopy in Kennedy syndrome. Arch Neurol. (1999) 56:1465–71. doi: 10.1001/archneur.56.12.1465
53. Kassubek J, Juengling FD, Sperfeld AD. Widespread white matter changes in Kennedy disease: a voxel based morphometry study. J Neurol Neurosurg Psychiatry. (2007) 78:1209–12. doi: 10.1136/jnnp.2006.112532
54. Lai TH, Liu RS, Yang BH, Wang PS, Lin KP, Lee YC, Soong BW. Cerebral involvement in spinal and bulbar muscular atrophy (Kennedy's disease): a pilot study of PET. J Neurol Sci. (2013) 335:139–44. doi: 10.1016/j.jns.2013.09.016
55. Mader I, Karitzky J, Klose U, Seeger U, Sperfeld A, Naegele T, et al. Proton MRS in Kennedy disease: absolute metabolite and macromolecular concentrations. J Magn Reson Imaging. (2002) 16:160–7. doi: 10.1002/jmri.10141
56. Pieper CC, Konrad C, Sommer J, Teismann I, Schiffbauer H. Structural changes of central white matter tracts in Kennedy's disease – a diffusion tensor imaging and voxel-based morphometry study. Acta Neurol Scand. (2013) 127:323–8. doi: 10.1111/ane.12018
57. Bruno RL, Cohen JM, Galski T, Frick NM. The neuroanatomy of post-polio fatigue. Arch Phys Med Rehabil. (1994) 75:498–504.
58. Demir CF, Berilgen MS, Mungen B, Bulut S. Do polio survivors have a higher risk of epilepsy? Epilepsy Res. (2012) 98:72–5. doi: 10.1016/j.eplepsyres.2011.08.019
59. Shing SLH, Lope J, Chipika RH, Hardiman O, Bede P. Extra-motor manifestations in post-polio syndrome (PPS): fatigue, cognitive symptoms and radiological features. Neurol Sci. (2021). doi: 10.1007/s10072-021-05130-4. [Epub ahead of print].
60. Shing SLH, Lope J, McKenna MC, Chipika RH, Hardiman O, Bede P. Increased cerebral integrity metrics in poliomyelitis survivors: putative adaptation to longstanding lower motor neuron degeneration. J Neurol Sci. (2021) 424:117361. doi: 10.1016/j.jns.2021.117361
61. Trojan DA, Narayanan S, Francis SJ, Caramanos Z, Robinson A, Cardoso M, et al. Brain volume and fatigue in patients with postpoliomyelitis syndrome. PM R. (2014) 6:215–20. doi: 10.1016/j.pmrj.2013.09.009
62. Aghakhanyan G, Martinuzzi A, Frijia F, Vavla M, Hlavata H, Baratto A, et al. Brain white matter involvement in hereditary spastic paraplegias: analysis with multiple diffusion tensor indices. Amer J Neuroradiol. (2014) 35:1533–8. doi: 10.3174/ajnr.A3897
63. Agosta F, Scarlato M, Spinelli EG, Canu E, Benedetti S, Bassi MT, et al. Hereditary spastic paraplegia: beyond clinical phenotypes toward a unified pattern of central nervous system damage. Radiology. (2015) 276:207–18. doi: 10.1148/radiol.14141715
64. Duning T, Warnecke T, Schirmacher A, Schiffbauer H, Lohmann H, Mohammadi S, et al. Specific pattern of early white-matter changes in pure hereditary spastic paraplegia. Mov Disord. (2010) 25:1986–92. doi: 10.1002/mds.23211
65. Erichsen AK, Server A, Landrø NI, Sandvik L, Tallaksen CM. Proton magnetic resonance spectroscopy and cognition in patients with spastin mutations. J Neurol Sci. (2009) 277:124–9. doi: 10.1016/j.jns.2008.10.030
66. Faber I, Martinez ARM, de Rezende TJR, Martins CR Jr, Martins MP, Lourenço CM, et al. SPG11 mutations cause widespread white matter and basal ganglia abnormalities, but restricted cortical damage. Neuroimage Clin. (2018) 19:848–57. doi: 10.1016/j.nicl.2018.05.031
67. França MC, Yasuda CL, Pereira FRS, D'Abreu A, Lopes-Ramos CM, Rosa MV, et al. White and grey matter abnormalities in patients with SPG11 mutations. J Neurol Neurosurg Psychiatry. (2012) 83:828–33. doi: 10.1136/jnnp-2011-300129
68. Kassubek J, Sperfeld AD, Baumgartner A, Huppertz HJ, Riecker A, Juengling FD. Brain atrophy in pure and complicated hereditary spastic paraparesis: a quantitative 3D MRI study. Eur J Neurol. (2006) 13:880–6. doi: 10.1111/j.1468-1331.2006.01380.x
69. Koritnik B, Azam S, Knific J, Zidar J. Functional changes of the cortical motor system in hereditary spastic paraparesis. Acta Neurol Scand. (2009) 120:182–90. doi: 10.1111/j.1600-0404.2008.01143.x
70. Liao X, Huang M, Xing W, Wu X, Liao W, Wang X, et al. Resting state fMRI studies in SPG4-linked hereditary spastic paraplegia. J Neurol Sci. (2018) 384:1–6. doi: 10.1016/j.jns.2017.10.048
71. Lindig T, Bender B, Hauser TK, Mang S, Schweikardt D, Klose U, et al. Gray and white matter alterations in hereditary spastic paraplegia type SPG4 and clinical correlations. J Neurol. (2015) 262:1961–71. doi: 10.1007/s00415-015-7791-7
72. Montanaro D, Vavla M, Frijia F, Aghakhanyan G, Baratto A, Coi A, et al. Multimodal MRI longitudinal assessment of white and gray matter in different SPG types of hereditary spastic paraparesis. Front Neurosci. (2020) 14:325. doi: 10.3389/fnins.2020.00325
73. Oguz KK, Sanverdi E, Has A, Temuçin Ç, Türk S, Doerschner K. Tract-based spatial statistics of diffusion tensor imaging in hereditary spastic paraplegia with thin corpus callosum reveals widespread white matter changes. Diagn Interv Radiol. (2013) 19:181–6. doi: 10.5152/dir.2013.046
74. Pan MK, Huang SC, Lo YC, Yang CC, Cheng TW, Yang CC, et al. Microstructural integrity of cerebral fiber tracts in hereditary spastic paraparesis with SPG11 mutation. AJNR Am J Neuroradiol. (2013) 34:990–6. doi: 10.3174/ajnr.A3330
75. Rezende TJR, de Albuquerque M, Lamas GM, Martinez ARM, Campos BM, Casseb RF, et al. Multimodal MRI-based study in patients with SPG4 mutations. PLoS ONE. (2015) 10:e0117666. doi: 10.1371/journal.pone.0117666
76. Scheuer KH, Nielsen JE, Krabbe K, Simonsen C, Koefoed P, Sørensen SA, et al. Reduced regional cerebral blood flow in SPG4-linked hereditary spastic paraplegia. J Neurol Sci. (2005) 235:23–32. doi: 10.1016/j.jns.2005.03.051
77. Stromillo ML, Malandrini A, Dotti MT, Battaglini M, Borgogni F, Tessa A, et al. Structural and metabolic damage in brains of patients with SPG11-related spastic paraplegia as detected by quantitative MRI. J Neurol. (2011) 258:2240–7. doi: 10.1007/s00415-011-6106-x
78. Tomberg T, Braschinsky M, Rannikmns K, Kepler J, Kepler K, K,pl J, et al. Functional MRI of the cortical sensorimotor system in patients with hereditary spastic paraplegia. Spinal Cord. (2012) 50:885–90. doi: 10.1038/sc.2012.70
79. Warnecke T, Duning T, Schwan A, Lohmann H, Epplen JT, Young P. A novel form of autosomal recessive hereditary spastic paraplegia caused by a new SPG7 mutation. Neurology. (2007) 69:368–75. doi: 10.1212/01.wnl.0000266667.91074.fe
80. Montuschi A, Iazzolino B, Calvo A, Moglia C, Lopiano L, Restagno G, et al. Cognitive correlates in amyotrophic lateral sclerosis: a population-based study in Italy. J Neurol Neurosurg Psychiatry. (2015) 86:168–73. doi: 10.1136/jnnp-2013-307223
81. Phukan J, Elamin M, Bede P, Jordan N, Gallagher L, Byrne S, et al. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry. (2012) 83:102–8. doi: 10.1136/jnnp-2011-300188
82. de Vries BS, Spreij LA, Rustemeijer LMM, Bakker LA, Veldink JH, van den Berg LH, et al. A neuropsychological and behavioral study of PLS. Amyotroph Lateral Scler Frontotemporal Degener. (2019) 20:376–84. doi: 10.1080/21678421.2019.1620284
83. de Vries BS, Rustemeijer LMM, Bakker LA, Schröder CD, Veldink JH, van den Berg LH, et al. Cognitive and behavioural changes in PLS and PMA: challenging the concept of restricted phenotypes. J Neurol Neurosurg Psychiatry. (2019) 90:141–7. doi: 10.1136/jnnp-2018-318788
84. Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman O. Amyotrophic lateral sclerosis mimic syndromes: a population-based study. Arch Neurol. (2000) 57:109–13. doi: 10.1001/archneur.57.1.109
85. Denora PS, Smets K, Zolfanelli F, Ceuterick-de Groote C, Casali C, Deconinck T, et al. Motor neuron degeneration in spastic paraplegia 11 mimics amyotrophic lateral sclerosis lesions. Brain. (2016) 139:1723–34. doi: 10.1093/brain/aww061
86. Romagnolo A, Masera S, Mattioda A, Superti G, Santorelli FM, Mongini T, et al. Atypical hereditary spastic paraplegia mimicking multiple sclerosis associated with a novel SPG11 mutation. Eur J Neurol. (2014) 21:e14–5. doi: 10.1111/ene.12297
87. Visser J, van den Berg-Vos RM, Franssen H, van den Berg LH, Vogels OJ, Wokke JHJ, et al. Mimic syndromes in sporadic cases of progressive spinal muscular atrophy. Neurology. (2002) 58:1593. doi: 10.1212/WNL.58.11.1593
88. Rasmussen H, Hellzen O, Stordal E, Enmarker I. Family caregivers experiences of the pre-diagnostic stage in frontotemporal dementia. Geriatric Nursing. (2019) 40:246–51. doi: 10.1016/j.gerinurse.2018.10.006
89. Diehl-Schmid J, Schmidt EM, Nunnemann S, Riedl L, Kurz A, Förstl H, et al. Caregiver burden and needs in frontotemporal dementia. J Geriatr Psychiatry Neurol. (2013) 26:221–9. doi: 10.1177/0891988713498467
90. Le Forestier N, Maisonobe T, Piquard A, Rivaud S, Crevier-Buchman L, Salachas F, et al. Does primary lateral sclerosis exist? A study of 20 patients and a review of the literature. Brain. (2001) 124:1989–99. doi: 10.1093/brain/124.10.1989
91. de Vries BS, Rustemeijer LMM, van der Kooi AJ, Raaphorst J, Schröder CD, Nijboer TCW, et al. A case series of PLS patients with frontotemporal dementia and overview of the literature. Amyotroph Lateral Scler Frontotemporal Degener. (2017) 18:534–48. doi: 10.1080/21678421.2017.1354996
92. Agosta F, Canu E, Inuggi A, Chio A, Riva N, Silani V, et al. Resting state functional connectivity alterations in primary lateral sclerosis. Neurobiol Aging. (2014) 35:916–25. doi: 10.1016/j.neurobiolaging.2013.09.041
93. Smith CD. Serial MRI findings in a case of primary lateral sclerosis. Neurology. (2002) 58:647–9. doi: 10.1212/WNL.58.4.647
94. Van Laere K, Vanhee A, Verschueren J, De Coster L, Driesen A, Dupont P, et al. Value of 18fluorodeoxyglucose-positron-emission tomography in amyotrophic lateral sclerosis: a prospective study. JAMA Neurol. (2014) 71:553–61. doi: 10.1001/jamaneurol.2014.62
95. Turner MR, Gerhard A, Al-Chalabi A, Shaw CE, Hughes RAC, Banati RB, et al. Mills' and other isolated upper motor neurone syndromes: in vivo study with 11C-(R)-PK11195 PET. J Neurol Neurosurg Psychiatry. (2005) 76:871–4. doi: 10.1136/jnnp.2004.047902
96. Gazulla J, Ferrer I, Izquierdo-Alvarez S, Alvarez S, Sánchez-Alcudia R, Bestué-Cardiel M, et al. Hereditary primary lateral sclerosis and progressive nonfluent aphasia. J Neurol. (2019) 266:1079–90. doi: 10.1007/s00415-019-09235-x
97. Kobayashi Z, Tsuchiya K, Arai T, Yokota O, Yoshida M, Shimomura Y, et al. Clinicopathological characteristics of FTLD-TDP showing corticospinal tract degeneration but lacking lower motor neuron loss. J Neurol Sci. (2010) 298:70–7. doi: 10.1016/j.jns.2010.08.013
98. Konagaya M, Sakai M, Matsuoka Y, Konagaya Y, Hashizume Y. Upper motor neuron predominant degeneration with frontal and temporal lobe atrophy. Acta Neuropathol. (1998) 96:532–6. doi: 10.1007/s004010050930
99. Mochizuki A, Komatsuzaki Y, Iwamoto H, Shoji S. Frontotemporal dementia with ubiquitinated neuronal inclusions presenting with primary lateral sclerosis and parkinsonism: clinicopathological report of an autopsy case. Acta Neuropathol. (2004) 107:377–80. doi: 10.1007/s00401-003-0818-7
100. Tan CF, Kakita A, Piao YS, Kikugawa K, Endo K, Tanaka M, et al. Primary lateral sclerosis: a rare upper-motor-predominant form of amyotrophic lateral sclerosis often accompanied by frontotemporal lobar degeneration with ubiquitinated neuronal inclusions? Report of an autopsy case and a review of the literature. Acta Neuropathol. (2003) 105:615–20. doi: 10.1007/s00401-003-0687-0
101. Kosaka T, Fu YJ, Shiga A, Ishidaira H, Tan CF, Tani T, et al. Primary lateral sclerosis: upper-motor-predominant amyotrophic lateral sclerosis with frontotemporal lobar degeneration–immunohistochemical and biochemical analyses of TDP-43. Neuropathology. (2012) 32:373–84. doi: 10.1111/j.1440-1789.2011.01271.x
102. Hirsch-Reinshagen V, Alfaify OA, Hsiung GR, Pottier C, Baker M, Perkerson RB III, et al. Clinicopathologic correlations in a family with a TBK1 mutation presenting as primary progressive aphasia and primary lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2019) 20:568–75. doi: 10.1080/21678421.2019.1632347
103. Mackenzie IRA, Briemberg H. TDP-43 pathology in primary lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2020) 21:52–8. doi: 10.1080/21678421.2020.1790607
104. Liewluck T, Saperstein DS. Progressive muscular atrophy. Neurol Clin. (2015) 33:761–73. doi: 10.1016/j.ncl.2015.07.005
105. Nishihira Y, Tan CF, Hoshi Y, Iwanaga K, Yamada M, Kawachi I, et al. Sporadic amyotrophic lateral sclerosis of long duration is associated with relatively mild TDP-43 pathology. Acta Neuropathol. (2008) 117:45–53. doi: 10.1007/s00401-008-0443-6
106. Geser F, Stein B, Partain M, Elman LB, McCluskey LF, Xie SX, et al. Motor neuron disease clinically limited to the lower motor neuron is a diffuse TDP-43 proteinopathy. Acta Neuropathol. (2011) 121:509–17. doi: 10.1007/s00401-011-0797-z
107. Kim WK, Liu X, Sandner J, Pasmantier M, Andrews J, Rowland LP, et al. Study of 962 patients indicates progressive muscular atrophy is a form of ALS. Neurology. (2009) 73:1686–92. doi: 10.1212/WNL.0b013e3181c1dea3
108. Ince PG, Evans J, Knopp M, Forster G, Hamdalla HHM, Wharton SB, et al. Corticospinal tract degeneration in the progressive muscular atrophy variant of ALS. Neurology. (2003) 60:1252–8. doi: 10.1212/01.WNL.0000058901.75728.4E
109. Raaphorst J, de Visser M, van Tol MJ, Linssen WHJP, van der Kooi AJ, de Haan RJ, et al. Cognitive dysfunction in lower motor neuron disease: executive and memory deficits in progressive muscular atrophy. J Neurol Neurosurg Psychiatry. (2011) 82:170–5. doi: 10.1136/jnnp.2009.204446
110. Wicks P, Abrahams S, Leigh PN, Williams T, Goldstein LH. Absence of cognitive, behavioral, or emotional dysfunction in progressive muscular atrophy. Neurology. (2006) 67:1718–19. doi: 10.1212/01.wnl.0000242726.36625.f3
111. Riku Y, Atsuta N, Yoshida M, Tatsumi S, Iwasaki Y, Mimuro M, et al. Differential motor neuron involvement in progressive muscular atrophy: a comparative study with amyotrophic lateral sclerosis. BMJ Open. (2014) 4:e005213. doi: 10.1136/bmjopen-2014-005213
112. D'Amico A, Mercuri E, Tiziano FD, Bertini E. Spinal muscular atrophy. Orph J Rare Dis. (2011) 6:71. doi: 10.1186/1750-1172-6-71
113. Losito L, Gennaro L, Lucarelli E, Trabacca A. Brain MRI abnormalities in a child with spinal muscular atrophy type II. Acta Neurol Belg. (2020). doi: 10.1007/s13760-020-01524-x. [Epub ahead of print].
114. Maeda K, Chong PF, Yamashita F, Akamine S, Kawakami S, Saito K, et al. Global central nervous system atrophy in spinal muscular atrophy type 0. Ann Neurol. (2019) 86:801–2. doi: 10.1002/ana.25596
115. Ito Y, Kumada S, Uchiyama A, Saito K, Osawa M, Yagishita A, et al. Thalamic lesions in a long-surviving child with spinal muscular atrophy type I: MRI and EEG findings. Brain Dev. (2004) 26:53–6. doi: 10.1016/S0387-7604(03)00075-5
116. Mix L, Schreiber-Katz O, Wurster CD, Uzelac Z, Platen S, Gipperich C, et al. Executive function is inversely correlated with physical function: the cognitive profile of adult Spinal Muscular Atrophy (SMA). Orphanet J Rare Dis. (2021) 16:10. doi: 10.1186/s13023-020-01661-9
117. Billard C, Gillet P, Signoret JL, Uicaut E, Bertrand P, Fardeau M, et al. Cognitive functions in duchenne muscular dystrophy: a reappraisal and comparison with spinal muscular atrophy. Neuromuscul Disord. (1992) 2:371–8. doi: 10.1016/S0960-8966(06)80008-8
118. Billard C, Gillet P, Barthez MA, Hommet C, Bertrand P. Reading ability and processing in Duchenne muscular dystrophy and spinal muscular atrophy. Dev Med Child Neurol. (1998) 40:12–20. doi: 10.1111/j.1469-8749.1998.tb15351.x
119. Oudgenoeg-Paz O, Rivieno J. Self-locomotion and spatial language and spatial cognition: insights from typical and atypical development. Front Psychol. (2014) 5:521. doi: 10.3389/fpsyg.2014.00521
120. Polido GJ, de Miranda MMV, Carvas N, Mendonça RDH, Caromano FA, Reed UC, et al. Cognitive performance of children with spinal muscular atrophy: a systematic review. Dement Neuropsychol. (2019) 13:436–43. doi: 10.1590/1980-57642018dn13-040011
121. Bénony C, Bénony H. Precocity of the acquisition of language and type II spinal muscular atrophy in 3-4-year-old children: a study of 12 cases. Eur J Paediatr Neurol. (2005) 9:71–6. doi: 10.1016/j.ejpn.2005.02.001
122. Rivière J, Lécuyer R. Spatial cognition in young children with spinal muscular atrophy. Dev Neuropsychol. (2002) 21:273–83. doi: 10.1207/S15326942DN2103_4
123. Riviere J, Lecuyer R. The C-not-B error: a comparative study. Cogn Dev. (2003) 18:285–97. doi: 10.1016/S0885-2014(03)00003-0
124. Leaffer E, Hinton V, Salazar R, Montes J, Dunaway Young S, Holuba LaMarca N, et al. Pediatrics-1 spinal muscular atrophy type I: cases of normal cognitive function despite having limited motor function and physical-environmental interaction. Arch Clin Neuropsychol. (2015) 30: 481. doi: 10.1093/arclin/acv046.17
125. von Gontard A, Zerres K, Backes M, Laufersweiler-Plass C, Wendland C, Melchers P, et al. Intelligence and cognitive function in children and adolescents with spinal muscular atrophy. Neuromuscul Disord. (2002) 12:130–6. doi: 10.1016/S0960-8966(01)00274-7
126. Polido GJ, Barbosa AF, Morimoto CH, Caromano FA, Favero FM, Zanoteli E, et al. Matching pairs difficulty in children with spinal muscular atrophy type I. Neuromuscul Disord. (2017) 27:419–27. doi: 10.1016/j.nmd.2017.01.017
127. Thirunavukkarasu B, Gupta K, Bansal A, Dhanasekaran N, Baranwal A. Spinal muscular atrophy: autopsy based neuropathological demonstration. Neurol India. (2020) 68:882–5. doi: 10.4103/0028-3886.293477
128. Kuru S, Sakai M, Konagaya M, Yoshida M, Hashizume Y, Saito K. An autopsy case of spinal muscular atrophy type III (Kugelberg-Welander disease). Neuropathology. (2009) 29:63–7. doi: 10.1111/j.1440-1789.2008.00910.x
129. Harding BN, Kariya S, Monani UR, Chung WK, Benton M, Yum SW, et al. Spectrum of neuropathophysiology in spinal muscular atrophy type I. J Neuropathol Exp Neurol. (2015) 74:15–24. doi: 10.1097/NEN.0000000000000144
130. Araki S, Hayashi M, Tamagawa K, Saito M, Kato S, Komori T, et al. Neuropathological analysis in spinal muscular atrophy type II. Acta Neuropathol. (2003) 106:441–8. doi: 10.1007/s00401-003-0743-9
131. Finsterer J, Scorza FA. Central nervous system abnormalities in spinal and bulbar muscular atrophy (Kennedy's disease). Clin Neurol Neurosurg. (2019) 184:105426. doi: 10.1016/j.clineuro.2019.105426
132. Manzano R, Sorarn G, Grunseich C, Fratta P, Zuccaro E, Pennuto M, et al. Beyond motor neurons: expanding the clinical spectrum in Kennedymg disease. J Neurol Neurosurg Psychiatry. (2018) 89:808–12. doi: 10.1136/jnnp-2017-316961
133. Kennedy WR, Alter M, Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology. (1998) 50:583. doi: 10.1212/WNL.50.3.583
134. Soukup GR, Sperfeld AD, Uttner I, Karitzky J, Ludolph AC, Kassubek J, et al. Frontotemporal cognitive function in X-linked spinal and bulbar muscular atrophy (SBMA): a controlled neuropsychological study of 20 patients. J Neurol. (2009) 256:1869–75. doi: 10.1007/s00415-009-5212-5
135. Di Rosa E, Sorarù G, Kleinbub JR, Calvo V, Vallesi A, Querin G, et al. Theory of mind, empathy and neuropsychological functioning in X-linked spinal and bulbar muscular atrophy: a controlled study of 20 patients. J Neurol. (2015) 262:394–401. doi: 10.1007/s00415-014-7567-5
136. Kasper E, Wegrzyn M, Marx I, Korp C, Kress W, Benecke R, et al. Minor cognitive disturbances in X-linked spinal and bulbar muscular atrophy, Kennedy's disease. Amyotroph Lateral Scler Frontotemporal Degener. (2014) 15:15–20. doi: 10.3109/21678421.2013.837927
137. Cianfoni A, Law M, Re TJ, Dubowitz DJ, Rumboldt Z, Imbesi SG. Clinical pitfalls related to short and long echo times in cerebral MR spectroscopy. J Neuroradiol. (2011) 38:69–75. doi: 10.1016/j.neurad.2010.10.001
138. Kessler H, Prudlo J, Kraft S, Supprian T. Dementia of frontal lobe type in Kennedy's disease. Amyotroph Lateral Scler. (2005) 6:250–3. doi: 10.1080/14660820510036558
139. Shaw PJ, Thagesen H, Tomkins J, Slade JY, Usher P, Jackson A, et al. Kennedy's disease: unusual molecular pathologic and clinical features. Neurology. (1998) 51:252–5. doi: 10.1212/WNL.51.1.252
140. Sobue GEN, Hashizume Y, Mukai E, Hirayama M, Mitsuma T, Takahashi A. X-linked recessive bulbospinal neuronopathy: a clinicopathological study. Brain. (1989) 112:209–32. doi: 10.1093/brain/112.1.209
141. Adachi H, Katsuno M, Minamiyama M, Waza M, Sang C, Nakagomi Y, et al. Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain. (2005) 128:659-70. doi: 10.1093/brain/awh381
142. Bruno RL, Zimmerman JR. Word finding difficulty as a post-polio sequelae. Am J Phys Med Rehabil. (2000) 79:343–8. doi: 10.1097/00002060-200007000-00005
143. Ostlund G, Borg K, Wahlin A. Cognitive functioning in post-polio patients with and without general fatigue. J Rehabil Med. (2005) 37:147–51. doi: 10.1080/16501970410024172
144. Hazendonk KM, Crowe SF. A neuropsychological study of the postpolio syndrome: support for depression without neuropsychological impairment. Neuropsychiatry Neuropsychol Behav Neurol. (2000) 13:112–8.
145. França MC, Schmutzler KMR, Garibaldi SG, Zanardi VA, Nucci A. Bilateral substantia nigra involvement in vaccine-associated poliomyelitis. Neurology. (2006) 66:1597–8. doi: 10.1212/01.wnl.0000216264.69966.50
146. Matzke HA, Baker AB. Poliomyelitis: IV. A study of the midbrain. AMA Arch Neurol Psychiatry. (1951) 65:1–15. doi: 10.1001/archneurpsyc.1951.02320010007001
147. Miller DC. Post-polio syndrome spinal cord pathology. Case report with immunopathology. Ann N Y Acad Sci. (1995) 753:186–93. doi: 10.1111/j.1749-6632.1995.tb27544.x
148. Shimada A, Lange DJ, Hays AP. Amyotrophic lateral sclerosis in an adult following acute paralytic poliomyelitis in early childhood. Acta Neuropathol. (1999) 97:317–21. doi: 10.1007/s004010050991
149. Casula M, Steentjes K, Aronica E, van Geel BM, Troost D. Concomitant CNS pathology in a patient with amyotropic lateral sclerosis following poliomyelitis in childhood. Clin Neuropathol. (2011) 30:111–7. doi: 10.5414/NPP30111
150. da Graça FF, de Rezende TJR, Vasconcellos LFR, Pedroso JL, Barsottini OGP, França MC Jr. Neuroimaging in hereditary spastic paraplegias: current use and future perspectives. Front Neurol. (2019) 9:1117. doi: 10.3389/fneur.2018.01117
151. Pizzini F, Fatemi AS, Barker PB, Nagae-Poetscher LM, Horská A, Zimmerman AW, et al. Proton MR spectroscopic imaging in Pelizaeus-Merzbacher disease. AJNR Am J Neuroradiol. (2003) 24:1683–9.
152. Fraidakis MJ, Brunetti M, Blackstone C, Filippi M, Chiò A. Novel compound heterozygous spatacsin mutations in a greek kindred with hereditary spastic paraplegia SPG11 and dementia. Neurodegener Dis. (2016) 16:373–81. doi: 10.1159/000444715
153. Roos P, Svenstrup K, Danielsen ER, Thomsen C, Nielsen JE. CYP7B1: novel mutations and magnetic resonance spectroscopy abnormalities in hereditary spastic paraplegia type 5A. Acta Neurol Scand. (2014) 129:330–4. doi: 10.1111/ane.12188
154. Dreha-Kulaczewski S, Dechent P, Helms G, Frahm J, Gärtner J, Brockmann K, et al. Cerebral metabolic and structural alterations in hereditary spastic paraplegia with thin corpus callosum assessed by MRS and DTI. Neuroradiology 48 (2006) 89–8. doi: 10.1007/s00234-006-0148-2
155. Svenstrup K, Giraud G, Boespflug-Tanguy O, Danielsen ER, Thomsen C, Rasmussen K, et al. Hereditary spastic paraplegia caused by the PLP1 'rumpshaker mutation'. J Neurol Neurosurg Psychiatry. (2010) 81:666–72. doi: 10.1136/jnnp.2009.180315
156. Orlén H, Melberg A, Raininko R, Kumlien E, Entesarian M, Söderberg P, et al. SPG11 mutations cause Kjellin syndrome, a hereditary spastic paraplegia with thin corpus callosum and central retinal degeneration. Am J Med Genet B Neuropsychiatr Genet. (2009) 150:984–92. doi: 10.1002/ajmg.b.30928
157. Hehr U, Bauer P, Winner B, Schule R, Olmez A, Koehler W, et al., Long-term course and mutational spectrum of spatacsin-linked spastic paraplegia. Ann Neurol. (2007) 62:656–65. doi: 10.1002/ana.21310
158. Ma J, Xiong L, Chang Y, Jing X, Huang W, Hu B, et al. Novel mutations c.(5121_5122insAG) + (6859C>T) of the SPG11 gene associated with cerebellum hypometabolism in a Chinese case of hereditary spastic paraplegia with thin corpus callosum. Parkinsonism Relat Disord. (2014) 20:256–9. doi: 10.1016/j.parkreldis.2013.11.004
159. Nielsen JE, Johnsen B, Koefoed P, Scheuer KH, Grønbech-Jensen M, Law I, et al. Hereditary spastic paraplegia with cerebellar ataxia: a complex phenotype associated with a new SPG4 gene mutation. Eur J Neurol. (2004) 11:817–24. doi: 10.1111/j.1468-1331.2004.00888.x
160. Samaranch L, Riverol M, Masdeu JC, Lorenzo E, Vidal-Taboada JM, Irigoyen J, et al. SPG11 compound mutations in spastic paraparesis with thin corpus callosum. Neurology. (2008) 71:332-6. doi: 10.1212/01.wnl.0000319646.23052.d1
161. Terada T, Kono S, Ouchi Y, Yoshida K, Hamaya Y, Kanaoka S, et al. SPG3A-linked hereditary spastic paraplegia associated with cerebral glucose hypometabolism. Ann Nucl Med. (2013) 27:303–6. doi: 10.1007/s12149-012-0673-5
162. Tallaksen CME, Guichart-Gomez E, Verpillat P, Hahn-Barma V, Ruberg M, Fontaine B, et al. Subtle cognitive impairment but no dementia in patients with spastin mutations. Arch Neurol. (2003) 60:1113–8. doi: 10.1001/archneur.60.8.1113
163. Byrne PC, McMonagle P, Webb S, Fitzgerald B, Parfrey NA, Hutchinson M. Age-related cognitive decline in hereditary spastic paraparesis linked to chromosome 2p. Neurology. (2000) 54:1510–7. doi: 10.1212/WNL.54.7.1510
164. Murphy S, Gorman G, Beetz C, Byrne P, Dytko M, McMonagle P, et al. Dementia in SPG4 hereditary spastic paraplegia: clinical, genetic, and neuropathologic evidence. Neurology. (2009) 73:378–84. doi: 10.1212/WNL.0b013e3181b04c6c
165. McMonagle P, Byrne P, Hutchinson M. Further evidence of dementia in SPG4-linked autosomal dominant hereditary spastic paraplegia. Neurology. (2004) 62:407–10. doi: 10.1212/01.WNL.0000108629.04434.05
166. Goizet C, Boukhris A, Maltete D, Guyant-Maréchal L, Truchetto J, Mundwiller E, et al. SPG15 is the second most common cause of hereditary spastic paraplegia with thin corpus callosum. Neurology. (2009) 73:1111–9. doi: 10.1212/WNL.0b013e3181bacf59
167. Chen Q, Lui S, Wang JG, Ou-Yang L, Zhou D, Burgunder JM, et al. Diffusion tensor imaging of two unrelated Chinese men with hereditary spastic paraplegia associated with thin corpus callosum. Neurosci Lett. (2008) 441:21–4. doi: 10.1016/j.neulet.2008.05.114
168. Schneider-Gold C, Dekomien G, Regensburger M, Schneider R, Trampe N, Krogias C, et al. Monozygotic twins with a new compound heterozygous SPG11 mutation and different disease expression. J Neurol Sci. (2017) 381:265–8. doi: 10.1016/j.jns.2017.09.005
169. Warnecke T, Duning T, Schirmacher A, Mohammadi S, Schwindt W, Lohmann H, et al. A novel splice site mutation in the SPG7 gene causing widespread fiber damage in homozygous and heterozygous subjects. Mov Disord. (2010) 25:413–20. doi: 10.1002/mds.22949
170. Kuru S, Sakai M, Konagaya M, Yoshida M, Hashizume Y. Autopsy case of hereditary spastic paraplegia with thin corpus callosum showing severe gliosis in the cerebral white matter. Neuropathology. (2005) 25:346–52. doi: 10.1111/j.1440-1789.2005.00620.x
171. Nomura H, Koike F, Tsuruta Y, Iwaki A, Iwaki T. Autopsy case of autosomal recessive hereditary spastic paraplegia with reference to the muscular pathology. Neuropathology. (2001) 21:212–7. doi: 10.1046/j.1440-1789.2001.00388.x
172. Ferrer I, Olive M, Rivera R, Pou A, Narberhaust B, Ugartet A. Hereditary spastic paraparesis with dementia, amyotrophy and peripheral neuropathy. A neuropathological study. Neuropathol Appl Neurobiol. (1995) 21:255–61. doi: 10.1111/j.1365-2990.1995.tb01057.x
173. Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, et al. Amyotrophic lateral sclerosis. Lancet. (2011) 377:942–55. doi: 10.1016/S0140-6736(10)61156-7
174. Strong MJ, Abrahams S, Goldstein LH, Woolley S, McLaughlin P, Snowden J, et al. Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener. (2017) 18:153–74. doi: 10.1080/21678421.2016.1267768
175. Ciccarelli O, Behrens TE, Johansen-Berg H, Talbot K, Orrell RW, Howard RS, et al. Investigation of white matter pathology in ALS and PLS using tract-based spatial statistics. Hum Brain Mapp. (2009) 30:615–24. doi: 10.1002/hbm.20527
176. Sato K, Aoki S, Iwata NK, Masutani Y, Watadani T, Nakata Y, et al. Diffusion tensor tract-specific analysis of the uncinate fasciculus in patients with amyotrophic lateral sclerosis. Neuroradiology. (2010) 52:729–33. doi: 10.1007/s00234-010-0653-1
177. Prell T, Peschel T, Hartung V, Kaufmann J, Klauschies R, Bodammer N, et al. Diffusion tensor imaging patterns differ in bulbar and limb onset amyotrophic lateral sclerosis. Clin Neurol Neurosurg. (2013) 115:1281–7. doi: 10.1016/j.clineuro.2012.11.031
178. Abe O, Yamada H, Masutani Y, Aoki S, Kunimatsu A, Yamasue H, et al. Amyotrophic lateral sclerosis: diffusion tensor tractography and voxel-based analysis. NMR Biomed. (2004) 17:411–6. doi: 10.1002/nbm.907
179. Agosta F, Pagani E, Rocca MA, Caputo D, Perini M, Salvi F, et al. Voxel-based morphometry study of brain volumetry and diffusivity in amyotrophic lateral sclerosis patients with mild disability. Hum Brain Mapp. (2007) 28:1430–8. doi: 10.1002/hbm.20364
180. Filippini N, Douaud G, Mackay CE, Knight S, Talbot K, Turner MR. Corpus callosum involvement is a consistent feature of amyotrophic lateral sclerosis. Neurology. (2010) 75:1645–52. doi: 10.1212/WNL.0b013e3181fb84d1
181. Keil C, Prell T, Peschel T, Hartung V, Dengler R, Grosskreutz J. Longitudinal diffusion tensor imaging in amyotrophic lateral sclerosis. BMC Neurosci. (2012) 13:141. doi: 10.1186/1471-2202-13-141
182. Sach M, Winkler G, Glauche V, Liepert J, Heimbach B, Koch MA, et al. Diffusion tensor MRI of early upper motor neuron involvement in amyotrophic lateral sclerosis. Brain. (2004) 127:340–50. doi: 10.1093/brain/awh041
183. Sage CA, Peeters RR, Gorner A, Robberecht W, Sunaert S. Quantitative diffusion tensor imaging in amyotrophic lateral sclerosis. NeuroImage. (2007) 34:486–99. doi: 10.1016/j.neuroimage.2006.09.025
184. Sage CA, Van Hecke W, Peeters R, Sijbers J, Robberecht W, Parizel P, et al. Quantitative diffusion tensor imaging in amyotrophic lateral sclerosis: revisited. Hum Brain Mapp. (2009) 30:3657–75. doi: 10.1002/hbm.20794
185. Thivard L, Pradat PF, Lehericy S, Lacomblez L, Dormont D, Chiras J, et al. Diffusion tensor imaging and voxel based morphometry study in amyotrophic lateral sclerosis: relationships with motor disability. J Neurol Neurosurg Psychiatry. (2007) 78:889–92. doi: 10.1136/jnnp.2006.101758
186. Sarro L, Agosta F, Canu E, Riva N, Prelle A, Copetti M, et al. Cognitive functions and white matter tract damage in amyotrophic lateral sclerosis: a diffusion tensor tractography study. AJNR Am J Neuroradiol. (2011) 32:1866–72. doi: 10.3174/ajnr.A2658
187. Floeter MK, Bageac D, Danielian LE, Braun LE, Traynor BJ, Kwan JY. Longitudinal imaging in C9orf72 mutation carriers: relationship to phenotype. Neuroimage Clin. (2016) 12:1035–43. doi: 10.1016/j.nicl.2016.10.014
188. Schuster C, Kasper E, Dyrba M, Machts J, Bittner D, Kaufmann J, et al. Cortical thinning and its relation to cognition in amyotrophic lateral sclerosis. Neurobiol Aging. (2014) 35:240–6. doi: 10.1016/j.neurobiolaging.2013.07.020
189. Bede P, Bokde A, Elamin M, Byrne S, McLaughlin RL, Jordan N, et al. Grey matter correlates of clinical variables in amyotrophic lateral sclerosis (ALS): a neuroimaging study of ALS motor phenotype heterogeneity and cortical focality. J Neurol Neurosurg Psychiatry. (2013) 84:766–73. doi: 10.1136/jnnp-2012-302674
190. Chang JL, Lomen-Hoerth C, Murphy J, Henry RG, Kramer JH, Miller BL, et al. A voxel-based morphometry study of patterns of brain atrophy in ALS and ALS/FTLD. Neurology. (2005) 65:75–80. doi: 10.1212/01.wnl.0000167602.38643.29
191. Christidi F, Karavasilis E, Riederer F, Zalonis I, Ferentinos P, Velonakis G, et al. Gray matter and white matter changes in non-demented amyotrophic lateral sclerosis patients with or without cognitive impairment: a combined voxel-based morphometry and tract-based spatial statistics whole-brain analysis. Brain Imaging Behav. (2018) 12:547–63. doi: 10.1007/s11682-017-9722-y
192. Murphy JM, Henry RG, Langmore S, Kramer JH, Miller BL, Lomen-Hoerth C. Continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch Neurol. (2007) 64:530–4. doi: 10.1001/archneur.64.4.530
193. Tsujimoto M, Senda J, Ishihara T, Niimi Y, Kawai Y, Atsuta N, et al. Behavioral changes in early ALS correlate with voxel-based morphometry and diffusion tensor imaging. J Neurol Sci. (2011) 307:34–40. doi: 10.1016/j.jns.2011.05.025
194. Woolley SC, Zhang Y, Schuff N, Weiner MW, Katz JS. Neuroanatomical correlates of apathy in ALS using 4 Tesla diffusion tensor MRI. Amyotroph Lateral Scler. (2011) 12:52–8. doi: 10.3109/17482968.2010.521842
195. Femiano C, Trojsi F, Caiazzo G, Siciliano M, Passaniti C, Russo A, et al. Apathy is correlated with widespread diffusion tensor imaging (DTI) impairment in amyotrophic lateral sclerosis. Behav Neurol. (2018) 2018:2635202. doi: 10.1155/2018/2635202
196. Bede P, Bokde AL, Byrne S, Elamin M, McLaughlin RL, Kenna K, et al. Multiparametric MRI study of ALS stratified for the C9orf72 genotype. Neurology. (2013) 81:361–9. doi: 10.1212/WNL.0b013e31829c5eee
197. Westeneng HJ, Walhout R, Straathof M, Schmidt R, Hendrikse J, Veldink JH, et al. Widespread structural brain involvement in ALS is not limited to the C9orf72 repeat expansion. J Neurol Neurosurg Psychiatry. (2016) 87:1354–60. doi: 10.1136/jnnp-2016-313959
198. Bede P, Elamin M, Byrne S, McLaughlin RL, Kenna K, Vajda A, et al. Basal ganglia involvement in amyotrophic lateral sclerosis. Neurology. (2013) 81:2107–15. doi: 10.1212/01.wnl.0000437313.80913.2c
199. Pinkhardt EH, van Elst LT, Ludolph AC, Kassubek J. Amygdala size in amyotrophic lateral sclerosis without dementia: an in vivo study using MRI volumetry. BMC Neurol. (2006) 6:48. doi: 10.1186/1471-2377-6-48
200. Pagani M, Chio A, Valentini MC, Oberg J, Nobili F, Calvo A, et al. Functional pattern of brain FDG-PET in amyotrophic lateral sclerosis. Neurology. (2014) 83:1067–74. doi: 10.1212/WNL.0000000000000792
201. Renard D, Collombier L, Castelnovo G, Fourcade G, Kotzki PO, LaBauge P. Brain FDG-PET changes in ALS and ALS-FTD. Acta Neurol Belg. (2011) 111:306–9.
202. Cistaro A, Valentini MC, Chio A, Nobili F, Calvo A, Moglia C, et al. Brain hypermetabolism in amyotrophic lateral sclerosis: a FDG PET study in ALS of spinal and bulbar onset. Eur J Nucl Med Mol Imaging. (2012) 39:251–9. doi: 10.1007/s00259-011-1979-6
203. Ludolph AC, Langen KJ, Regard M, Herzog H, Kemper B, Kuwert T, et al. Frontal lobe function in amyotrophic lateral sclerosis: a neuropsychologic and positron emission tomography study. Acta Neurol Scand. (1992) 85:81–9. doi: 10.1111/j.1600-0404.1992.tb04003.x
204. Abrahams S, Leigh PN, Kew JJ, Goldstein LH, Lloyd CM, Brooks DJ. A positron emission tomography study of frontal lobe function (verbal fluency) in amyotrophic lateral sclerosis. J Neurol Sci. (1995) 129(Suppl.): 44–6. doi: 10.1016/0022-510X(95)00060-F
205. Rajagopalan V, Pioro EP Comparing brain structural MRI and metabolic FDG-PET changes in patients with ALS-FTD: 'the chicken or the egg?' question. J Neurol Neurosurg Psychiatry. (2015) 86:952–8. doi: 10.1136/jnnp-2014-308239
206. Cistaro A, Cuccurullo V, Quartuccio N, Pagani M, Valentini MC, Mansi L. Role of PET and SPECT in the study of amyotrophic lateral sclerosis. Biomed Res Int. (2014). 2014:237437. doi: 10.1155/2014/237437
207. Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an. (11C)(R)-PK11195 positron emission tomography study. Neurobiol Dis. (2004) 15:601–9. doi: 10.1016/j.nbd.2003.12.012
208. Corcia P, Tauber C, Vercoullie J, Arlicot N, Prunier C, Praline J, et al. Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS ONE. (2012) 7:e52941. doi: 10.1371/journal.pone.0052941
209. Zurcher NR, Loggia ML, Lawson R, Chonde DB, Izquierdo-Garcia D, Yasek JE, et al. Increased in vivo glial activation in patients with amyotrophic lateral sclerosis: assessed with ((11)C)-PBR28. NeuroImage Clin. (2015) 7:409–14. doi: 10.1016/j.nicl.2015.01.009
210. Johansson A, Engler H, Blomquist G, Scott B, Wall A, Aquilonius SM, et al. Evidence for astrocytosis in ALS demonstrated by (11C)(L)-deprenyl-D2 PET. J Neurol Sci. (2007) 255:17–22. doi: 10.1016/j.jns.2007.01.057
211. Usman U, Choi C, Camicioli R, Seres P, Lynch M, Sekhon R, et al. Mesial prefrontal cortex degeneration in amyotrophic lateral sclerosis: a high-field proton MR spectroscopy study. AJNR Am J Neuroradiol. (2011) 32:1677–80. doi: 10.3174/ajnr.A2590
212. Sudharshan N, Hanstock C, Hui B, Pyra T, Johnston W, Kalra S. Degeneration of the mid-cingulate cortex in amyotrophic lateral sclerosis detected in vivo with MR spectroscopy. Amer J Neuroradiol. (2011) 32:403–407. doi: 10.3174/ajnr.A2289
213. Abe K, Takanashi M, Watanabe Y, Tanaka H, Fujita N, Hirabuki N, et al. Decrease in N-acetylaspartate/creatine ratio in the motor area and the frontal lobe in amyotrophic lateral sclerosis. Neuroradiology. (2001) 43:537–41. doi: 10.1007/s002340000521
214. Verma G, Woo JH, Chawla S, Wang S, Sheriff S, Elman LB, et al. Whole-brain analysis of amyotrophic lateral sclerosis by using echo-planar spectroscopic imaging. Radiology. (2013) 267:851–7. doi: 10.1148/radiol.13121148
215. Strong MJ, Grace GM, Orange JB, Leeper HA, Menon RS, Aere C. A prospective study of cognitive impairment in ALS. Neurology. (1999) 53:1665–70. doi: 10.1212/WNL.53.8.1665
216. Mohammadi B, Kollewe K, Samii A, Krampfl K, Dengler R, Munte TF. Changes of resting state brain networks in amyotrophic lateral sclerosis. Exp Neurol. (2009) 217:147–53. doi: 10.1016/j.expneurol.2009.01.025
217. Fekete T, Zach N, Mujica-Parodi LR, Turner MR. Multiple kernel learning captures a systems-level functional connectivity biomarker signature in amyotrophic lateral sclerosis. PLoS ONE. (2013) 8:e85190. doi: 10.1371/journal.pone.0085190
218. Agosta F, Canu E, Valsasina P, Riva N, Prelle A, Comi G, et al. Divergent brain network connectivity in amyotrophic lateral sclerosis. Neurobiol Aging. (2013) 34:419–27. doi: 10.1016/j.neurobiolaging.2012.04.015
219. Luo C, Chen Q, Huang R, Chen X, Chen K, Huang X, et al. Patterns of spontaneous brain activity in amyotrophic lateral sclerosis: a resting-state fMRI study. PLoS ONE. (2012) 7:e45470. doi: 10.1371/journal.pone.0045470
220. Trojsi F, Esposito F, de Stefano M, Buonanno D, Conforti FL, Corbo D, et al. Functional overlap and divergence between ALS and bvFTD. Neurobiol Aging. (2015) 36:413–23. doi: 10.1016/j.neurobiolaging.2014.06.025
221. V H, Tietz F, Prell T, Penzlin S, Ilse B, Bokemeyer M, et al. The resting state default mode network (DMN) is pathologically hyperactive in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2012) 13:171.
222. Tedeschi G, Trojsi F, Tessitore A, Corbo D, Sagnelli A, Paccone A, et al. Interaction between aging and neurodegeneration in amyotrophic lateral sclerosis. Neurobiol Aging. (2012) 33:886–98. doi: 10.1016/j.neurobiolaging.2010.07.011
223. Goldstein LH, Newsom-Davis IC, Bryant V, Brammer M, Leigh PN, Simmons A. Altered patterns of cortical activation in ALS patients during attention and cognitive response inhibition tasks. J Neurol. (2011) 258:2186–98. doi: 10.1007/s00415-011-6088-8
224. Keller J, Böhm S, Aho-Özhan HEA, Loose M, Gorges M, Kassubek J, et al. Functional reorganization during cognitive function tasks in patients with amyotrophic lateral sclerosis. Brain Imaging Behav. (2018) 12:771–84. doi: 10.1007/s11682-017-9738-3
225. Witiuk K, Fernandez-Ruiz J, McKee R, Alahyane N, Coe BC, Melanson M, et al. Cognitive deterioration and functional compensation in ALS measured with fMRI using an inhibitory task. J Neurosci. (2014) 34:14260–71. doi: 10.1523/JNEUROSCI.1111-14.2014
226. Jelsone-Swain L, Persad C, Burkard D, Welsh RC Action processing and mirror neuron function in patients with amyotrophic lateral sclerosis: an fMRI study. PLoS ONE. (2015) 10:e0119862. doi: 10.1371/journal.pone.0119862
227. Palmieri A, Naccarato M, Abrahams S, Bonato M, D'Ascenzo C, Balestreri S, et al. Right hemisphere dysfunction and emotional processing in ALS: an fMRI study. J Neurol. (2010) 257:1970–8. doi: 10.1007/s00415-010-5640-2
228. Passamonti L, Fera F, Tessitore A, Russo A, Cerasa A, Gioia CM, et al. Dysfunctions within limbic-motor networks in amyotrophic lateral sclerosis. Neurobiol Aging. (2013) 34:2499–509. doi: 10.1016/j.neurobiolaging.2013.05.016
229. Aho-Özhan HE, Keller J, Heimrath J, Uttner I, Kassubek J, Birbaumer N, et al. Perception of emotional facial expressions in Amyotrophic Lateral Sclerosis (ALS) at behavioural and brain metabolic level. PLoS ONE. (2016) 11:e0164655. doi: 10.1371/journal.pone.0164655
230. Lule D, Diekmann V, Anders S, Kassubek J, Kubler A, Ludolph AC, et al. Brain responses to emotional stimuli in patients with amyotrophic lateral sclerosis (ALS). J Neurol. (2007) 254:519–27. doi: 10.1007/s00415-006-0409-3
231. Stoppel CM, Vielhaber S, Eckart C, Machts J, Kaufmann J, Heinze HJ, et al. Structural and functional hallmarks of amyotrophic lateral sclerosis progression in motor- and memory-related brain regions. Neuroimage Clin. (2014) 5:277–90. doi: 10.1016/j.nicl.2014.07.007
232. Vellage AK, Veit M, Kobeleva X, Petri S, Vielhaber S, Müller NG. Working memory network changes in ALS: an fMRI study. Front Neurosci. (2016) 10:158. doi: 10.3389/fnins.2016.00158
233. Abrahams S, Goldstein LH, Simmons A, Brammer M, Williams SC, Giampietro V, et al. Word retrieval in amyotrophic lateral sclerosis: a functional magnetic resonance imaging study. Brain. (2004) 127:1507–17. doi: 10.1093/brain/awh170
234. Turner MR, Kiernan MC. Does interneuronal dysfunction contribute to neurodegeneration in amyotrophic lateral sclerosis? Amyotroph Lateral Scler. (2012) 13:245–50. doi: 10.3109/17482968.2011.636050
235. Douaud G, Filippini N, Knight S, Talbot K, Turner MR. Integration of structural and functional magnetic resonance imaging in amyotrophic lateral sclerosis. Brain. (2011) 134:3470–9. doi: 10.1093/brain/awr279
236. Bede P, Bogdahn U, Lope J, Chang KM, Xirou S, Christidi F. Degenerative and regenerative processes in amyotrophic lateral sclerosis: motor reserve, adaptation and putative compensatory changes. Neural Regen Res. (2021) 16:1208–9. doi: 10.4103/1673-5374.300440
237. Abidi M, de Marco G, Couillandre A, Feron M, Mseddi E, Termoz N, et al. Adaptive functional reorganization in amyotrophic lateral sclerosis: coexisting degenerative and compensatory changes. Eur J Neurol. (2020) 27:121–8. doi: 10.1111/ene.14042
238. Feron M, Couillandre A, Mseddi E, Termoz N, Abidi M, Bardinet E, et al. Extrapyramidal deficits in ALS: a combined biomechanical and neuroimaging study. J Neurol. (2018) 265:2125–36. doi: 10.1007/s00415-018-8964-y
239. Proudfoot M, Rohenkohl G, Quinn A, Colclough GL, Wuu J, Talbot K, et al. Altered cortical beta-band oscillations reflect motor system degeneration in amyotrophic lateral sclerosis. Hum Brain Mapp. (2017) 38:237–54. doi: 10.1002/hbm.23357
240. Dukic S, McMackin R, Buxo T, Fasano A, Chipika R, Pinto-Grau M, et al. Patterned functional network disruption in amyotrophic lateral sclerosis. Hum Brain Mapp. (2019) 40:4827–42. doi: 10.1002/hbm.24740
241. McMackin R, Dukic S, Broderick M, Iyer PM, Pinto-Grau M, Mohr K, et al. Dysfunction of attention switching networks in amyotrophic lateral sclerosis. NeuroImage Clin. (2019) 22:101707. doi: 10.1016/j.nicl.2019.101707
242. Nasseroleslami B, Dukic S, Broderick M, Mohr K, Schuster C, Gavin B, McLaughlin R, Heverin M, Vajda A, Iyer PM, Pender N, Bede P, et al. Characteristic increases in EEG connectivity correlate with changes of structural MRI in AMYOTROPHIC LATERAL SCLEROSIS. Cereb Cortex. (2019) 29:27–41. doi: 10.1093/cercor/bhx301
243. McMackin R, Dukic S, Costello E, Pinto-Grau M, McManus L, Broderick M, et al. Cognitive network hyperactivation and motor cortex decline correlate with ALS prognosis. Neurobiol Aging. (2021) 104:57–70. doi: 10.1016/j.neurobiolaging.2021.03.002
244. Pettit LD, Bastin ME, Smith C, Bak TH, Gillingwater TH, Abrahams S. Executive deficits, not processing speed relates to abnormalities in distinct prefrontal tracts in amyotrophic lateral sclerosis. Brain. (2013) 136:3290–304. doi: 10.1093/brain/awt243
245. Burke T, Pinto-Grau M, Lonergan K, Elamin M, Bede P, Costello E, et al. Measurement of social cognition in amyotrophic lateral sclerosis: a population based study. PLoS ONE. (2016) 11:e0160850. doi: 10.1371/journal.pone.0160850
246. Burke T, Pinto-Grau M, Lonergan K, Bede P, O'Sullivan M, Heverin M, et al. A cross-sectional population-based investigation into behavioral change in amyotrophic lateral sclerosis: subphenotypes, staging, cognitive predictors, and survival. Ann Clin Transl Neurol. (2017) 4:305–17. doi: 10.1002/acn3.407
247. Crockford C, Newton J, Lonergan K, Chiwera T, Booth T, Chandran S, et al. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology. (2018) 91:e1370–80. doi: 10.1212/WNL.0000000000006317
248. Radakovic R, Gray D, Dudley K, Mioshi E, Dick D, Melchiorre G, et al. Reliability and validity of the brief dimensional apathy scale. Arch Clin Neuropsychol. (2020) 35:539–44. doi: 10.1093/arclin/acaa002
249. Burke T, Elamin M, Bede P, Pinto-Grau M, Lonergan K, Hardiman O, et al. Discordant performance on the 'Reading the Mind in the Eyes' Test, based on disease onset in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2016) 17:467–72. doi: 10.1080/21678421.2016.1177088
250. De Vocht J, Blommaert J, Devrome M, Radwan A, Van Weehaeghe D, De Schaepdryver M, et al. Use of multimodal imaging and clinical biomarkers in presymptomatic carriers of C9orf72 repeat expansion. JAMA Neurol. (2020) 77:1–10. doi: 10.1001/jamaneurol.2020.1087
251. Blasco H, Patin F, Descat A, Garcon G, Corcia P, Gele P, et al. A pharmaco-metabolomics approach in a clinical trial of ALS: identification of predictive markers of progression. PLoS ONE. (2018) 13:e0198116. doi: 10.1371/journal.pone.0198116
252. Devos D, Moreau C, Kyheng M, Garcon G, Rolland AS, Blasco H, et al. A ferroptosis-based panel of prognostic biomarkers for amyotrophic lateral sclerosis. Sci Rep. (2019) 9:2918. doi: 10.1038/s41598-019-39739-5
253. Gregory JM, McDade K, Bak TH, Pal S, Chandran S, Smith C, et al. Executive, language and fluency dysfunction are markers of localised TDP-43 cerebral pathology in non-demented ALS. J Neurol Neurosurg Psychiatry. (2020) 91:149–57. doi: 10.1136/jnnp-2019-320807
254. Geser F, Martinez-Lage M, Robinson J, Uryu K, Neumann M, Brandmeir NJ, et al. Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch Neurol. (2009) 66:180–9. doi: 10.1001/archneurol.2008.558
255. Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M, et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol. (2013) 74:20–38. doi: 10.1002/ana.23937
256. Prudlo J, König J, Schuster C, Kasper E, Büttner A, Teipel S, et al. TDP-43 pathology and cognition in ALS: a prospective clinicopathologic correlation study. Neurology. (2016) 87:1019–23. doi: 10.1212/WNL.0000000000003062
257. Mollink J, Hiemstra M, Miller KL, Huszar IN, Jenkinson M, Raaphorst J, et al. White matter changes in the perforant path area in patients with amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol. (2019) 45:570–85. doi: 10.1111/nan.12555
258. Shefner JM, Al-Chalabi A, Baker MR, Cui LY, de Carvalho M, Eisen A, et al. A proposal for new diagnostic criteria for ALS. Clin Neurophysiol. (2020) 131:1975–8. doi: 10.1016/j.clinph.2020.04.005
259. Elamin M, Bede P, Byrne S, Jordan N, Gallagher L, Wynne B, et al. Cognitive changes predict functional decline in ALS: a population-based longitudinal study. Neurology. (2013) 80:1590–7. doi: 10.1212/WNL.0b013e31828f18ac
260. Roche JC, Rojas-Garcia R, Scott KM, Scotton W, Ellis CE, Burman R, et al. A proposed staging system for amyotrophic lateral sclerosis. Brain. (2012) 135:847–52. doi: 10.1093/brain/awr351
261. Chiò A, Hammond ER, Mora G, Bonito V, Filippini G. Development and evaluation of a clinical staging system for amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. (2015) 86:38–44. doi: 10.1136/jnnp-2013-306589
262. Turner MR, Barohn RJ, Corcia P, Fink JK, Harms MB, Kiernan MC, et al. Primary lateral sclerosis: consensus diagnostic criteria. J Neurol Neurosurg Psychiatry. (2020) 91:373–7. doi: 10.1136/jnnp-2019-322541
263. Almeida V, de Carvalho M, Scotto M, Pinto S, Pinto A, Ohana B, et al. Primary lateral sclerosis: predicting functional outcome. Amyotroph Lateral Scler Frontotemporal Degener. (2013) 14:141–5. doi: 10.3109/17482968.2012.719237
264. Abrahams S, Newton J, Niven E, Foley J, Bak TH Screening for cognition and behaviour changes in ALS. Amyotroph Lateral Scler Frontotemporal Degener. (2014) 15:9–14. doi: 10.3109/21678421.2013.805784
265. Radakovic R, Stephenson L, Colville S, Swingler R, Chandran S, Abrahams S. Multidimensional apathy in ALS: validation of the Dimensional Apathy Scale. J Neurol Neurosurg Psychiatry. (2016) 87:663–9. doi: 10.1136/jnnp-2015-310772
266. Burrell JR, Kiernan MC, Vucic S, Hodges JR. Motor Neuron dysfunction in frontotemporal dementia. Brain. (2011) 134:2582–94. doi: 10.1093/brain/awr195
267. Kortte KB Rogalski EJ. Behavioural interventions for enhancing life participation in behavioural variant frontotemporal dementia and primary progressive aphasia. Int Rev Psychiatry. (2013) 25:237–45. doi: 10.3109/09540261.2012.751017
268. Henry ML, Meese MV, Truong S, Babiak MC, Miller BL, Gorno-Tempini ML. Treatment for apraxia of speech in nonfluent variant primary progressive aphasia. Behav Neurol. (2013) 26:77–88. doi: 10.1155/2013/824302
269. Jokel R, Anderson ND. Quest for the best: effects of errorless and active encoding on word re-learning in semantic dementia. Neuropsychol Rehabil. (2012) 22:187–214. doi: 10.1080/09602011.2011.639626
270. Grollemund V, Chat GL, Secchi-Buhour MS, Delbot F, Pradat-Peyre JF, Bede P, et al. Development and validation of a 1-year survival prognosis estimation model for Amyotrophic Lateral Sclerosis using manifold learning algorithm UMAP. Sci Rep. (2020) 10:13378. doi: 10.1038/s41598-020-70125-8
271. Filippi M, Basaia S, Canu E, Imperiale F, Meani A, Caso F, et al. Brain network connectivity differs in early-onset neurodegenerative dementia. Neurology. (2017) 89:1764. doi: 10.1212/WNL.0000000000004577
272. Ravanfar P, Loi SM, Syeda WT, Van Rheenen TE, Bush AI, Desmond P, et al. Systematic Review: Quantitative Susceptibility Mapping (QSM) of brain iron profile in neurodegenerative diseases. Front Neurosci. (2021) 15:618435. doi: 10.3389/fnins.2021.618435
273. Cheng JX, Zhang HY, Peng ZK, Xu Y, Tang H, Wu JT, et al. Divergent topological networks in Alzheimer's disease: a diffusion kurtosis imaging analysis. Transl Neurodegener. (2018) 7:10. doi: 10.1186/s40035-018-0115-y
274. Broad RJ, Gabel MC, Dowell NG, Schwartzman DJ, Seth AK, Zhang H, et al. Neurite orientation and dispersion density imaging (NODDI) detects cortical and corticospinal tract degeneration in ALS. J Neurol Neurosurg Psychiatry. (2019) 90:404–11. doi: 10.1136/jnnp-2018-318830
275. Feis RA, Bouts MJRJ, Panman JL, Jiskoot LC, Dopper EGP, Schouten TM, et al. Single-subject classification of presymptomatic frontotemporal dementia mutation carriers using multimodal MRI. NeuroImage Clin. (2019) 22:101718. doi: 10.1016/j.nicl.2019.101718
276. Mitsumoto H, Brooks BR, Silani V. Clinical trials in amyotrophic lateral sclerosis: why so many negative trials and how can trials be improved? Lancet Neurol. (2014) 13:1127–38. doi: 10.1016/S1474-4422(14)70129-2
277. Miller T, Cudkowicz M, Shaw PJ, Andersen PM, Atassi N, Bucelli RC, et al. Phase 1–2 trial of antisense oligonucleotide tofersen for SOD1 ALS. N Eng J Med. (2020) 383:109–19. doi: 10.1056/NEJMoa2003715
278. Baranello G, Darras BT, Day JW, Deconinck N, Klein A, Masson R, et al. Risdiplam in type 1 spinal muscular atrophy. N Eng J Med. (2021) 384:915–23. doi: 10.1056/NEJMoa2009965
279. Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, et al. Nusinersen versus Sham control in later-onset spinal muscular atrophy. N Eng J Med. (2018) 378:625–35. doi: 10.1056/NEJMoa1710504
280. Musarò A, Dobrowolny G, Cambieri C, Onesti E, Ceccanti M, Frasca V, et al. Neuromuscular magnetic stimulation counteracts muscle decline in ALS patients: results of a randomized, double-blind, controlled study. Sci Rep. (2019) 9:2837. doi: 10.1038/s41598-019-39313-z
281. Fang J, Zhou M, Yang M, Zhu C, He L. Repetitive transcranial magnetic stimulation for the treatment of amyotrophic lateral sclerosis or motor neuron disease. Cochrane Database Syst Rev. (2013) 2013:Cd008554. doi: 10.1002/14651858.CD008554.pub3
282. Rohrer JD, Nicholas JM, Cash DM, van Swieten J, Dopper E, Jiskoot L, et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol. (2015) 14:253–62. doi: 10.1016/S1474-4422(14)70324-2
283. Bertrand A, Wen J, Rinaldi D, Houot M, Sayah S, Camuzat A, et al. Early cognitive, structural, and microstructural changes in presymptomatic C9orf72 carriers younger than 40 years. JAMA Neurol. (2018) 75:236–245. doi: 10.1001/jamaneurol.2017.4266
284. Lee SE, Sias AC, Mandelli ML, Brown JA, Brown AB, Khazenzon AM, et al. Network degeneration and dysfunction in presymptomatic C9ORF72 expansion carriers. Neuroimage Clin. (2016) 14:286–97. doi: 10.1016/j.nicl.2016.12.006
285. Meyer S, Mueller K, Stuke K, Bisenius S, Diehl-Schmid J, Jessen F, et al. Predicting behavioral variant frontotemporal dementia with pattern classification in multi-center structural MRI data. NeuroImage Clin. (2017) 14:656–62. doi: 10.1016/j.nicl.2017.02.001
286. Papma JM, Jiskoot LC, Panman JL, Dopper EG, den Heijer T, Donker Kaat L, et al. Cognition and gray and white matter characteristics of presymptomatic C9orf72 repeat expansion. Neurology. (2017) 89:1256–64. doi: 10.1212/WNL.0000000000004393
287. Crespi C, Dodich A, Iannaccone S, Marcone A, Falini A, Cappa SF, et al. Diffusion tensor imaging evidence of corticospinal pathway involvement in frontotemporal lobar degeneration. Cortex. (2020) 125:1–11. doi: 10.1016/j.cortex.2019.11.022
288. Häkkinen S, Chu SA, Lee SE. Neuroimaging in genetic frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol Dis. (2020) 145:105063. doi: 10.1016/j.nbd.2020.105063
289. Meng X, D,ng01 C. Education and dementia in the context of the cognitive reserve hypothesis: a systematic review with meta-analyses and qualitative analyses. PLoS ONE. (2012) 7:e38268. doi: 10.1371/journal.pone.0038268
290. Beyer L, Meyer-Wilmes J, Scher-Wilm S, Schnabel J, Sauerbeck J, Scheifele M, et al. Cognitive reserve hypothesis in frontotemporal dementia: a FDG-PET study. Neuroimage Clin. (2021) 29:102535. doi: 10.1016/j.nicl.2020.102535
291. Temp AGM, Prudlo J, Vielhaber S, Machts J, Hermann A, Teipel SJ, et al. Cognitive reserve and regional brain volume in amyotrophic lateral sclerosis. Cortex. (2021) 139:240–8. doi: 10.1016/j.cortex.2021.03.005
292. Canosa A, Palumbo F, Iazzolino B, Peotta L, Di Pede F, Manera U, et al. The interplay among education, brain metabolism, and cognitive impairment suggests a role of cognitive reserve in Amyotrophic Lateral Sclerosis. Neurobiol Aging. (2021) 98:205–13. doi: 10.1016/j.neurobiolaging.2020.11.010
293. Bak TH, Chandran S. What wires together dies together: verbs, actions and neurodegeneration in motor neuron disease. Cortex. (2012) 48:936–44. doi: 10.1016/j.cortex.2011.07.008
294. Papeo L, Cecchetto C, Mazzon G, Granello G, Cattaruzza T, Verriello L, et al. The processing of actions and action-words in amyotrophic lateral sclerosis patients. Cortex. (2015) 64:136–47. doi: 10.1016/j.cortex.2014.10.007
295. Hauk O, Johnsrude I, Pulvermde.co F. Somatotopic representation of action words in human motor and premotor cortex. Neuron. (2004) 41:301–7. doi: 10.1016/S0896-6273(03)00838-9
296. Lule D, Diekmann V, Kassubek J, Kurt A, Birbaumer N, Ludolph AC, et al. Cortical plasticity in amyotrophic lateral sclerosis: motor imagery and function. Neurorehabil Neural Repair. (2007) 21:518–26. doi: 10.1177/1545968307300698
297. O'Callaghan C, Bertoux M, Hornberger M. Beyond and below the cortex: the contribution of striatal dysfunction to cognition and behaviour in neurodegeneration. J Neurol Neurosurg Psychiatry. (2013) 85:371–8. doi: 10.1136/jnnp-2012-304558
Keywords: ALS, MND, PLS, MRI, PET, SMA, PMA, SBMA
Citation: McKenna MC, Corcia P, Couratier P, Siah WF, Pradat P-F and Bede P (2021) Frontotemporal Pathology in Motor Neuron Disease Phenotypes: Insights From Neuroimaging. Front. Neurol. 12:723450. doi: 10.3389/fneur.2021.723450
Received: 10 June 2021; Accepted: 22 July 2021;
Published: 16 August 2021.
Edited by:
William Huynh, The University of Sydney, AustraliaReviewed by:
Sicong Tu, The University of Sydney, AustraliaDeepak Menon, University Health Network (UHN), Canada
Copyright © 2021 McKenna, Corcia, Couratier, Siah, Pradat and Bede. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Bede, bedep@tcd.ie