 Nayoung Kim
Nayoung Kim Kiven Erique Lukong*
Kiven Erique Lukong*- Biochemistry, Microbiology and Immunology, College of Medicine, University of Saskatchewan, Saskatoon, SK, Canada
Breast cancer is one of the most significant causes of mortality among women and the second most prevalent cancer worldwide. Estrogen receptor (ER)-positive breast cancers are the most common molecular subtype of breast cancer, comprising about 70% of breast carcinoma diagnoses worldwide. Endocrine therapy is the foremost strategy for the treatment of ER-positive breast cancer. In the United States, the Food and Drug Administration (FDA) has approved endocrine therapies for ER-positive breast cancers that include selective estrogen receptor modulators (SERMs), selective estrogen receptor downregulators/degraders (SERDs) and aromatase inhibitors (AIs). The approved SERMS, tamoxifen, toremifene and raloxifene, are the gold-standard treatments. The only FDA-approved SERD available for treating ER and hormone-positive breast cancers is fulvestrant, and various generations of AIs, including exemestane, letrozole, and anastrozole, have also received FDA approval. Herein, we review the major FDA-approved SERMs and SERDs for treating ER-positive breast cancer, focusing on their mechanisms of action. We also explore molecular events that contribute to the resistance of these drugs to endocrine therapies and combinational strategies with drugs such as cyclin-dependant kinases 4/6 (CDK4/6) inhibitors in clinical trials to combat endocrine drug resistance.
1 Introduction
Breast cancer is currently the second most prevalent cancer worldwide, second only to lung cancer, as reported in Globocan 2022. There were approximately 2.3 million new cases of breast cancer diagnosed in 2022 alone, and over 660,000 deaths were attributed to these diagnoses (1). In the same year, Europe saw over half a million new cases of breast cancer, while 198,553 cases were reported in Africa. Furthermore, North America reported 306,307 newly diagnosed cases, making breast cancer the most prevalent cancer in both the United States of America and Canada (1) (Table 1).
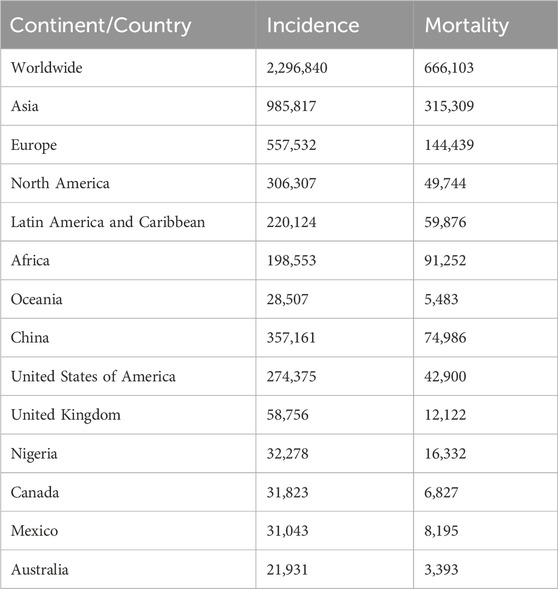
Table 1. Globocan 2022 – estimated breast cancer incidence and mortality. The table below lists the estimated number of new breast cancer cases and mortality for select continents and countries in 2022 (1). Adapted from (1).
Breast cancer encompasses a group of diseases originating from the breast and displays both biological and molecular heterogeneity (2, 3). Most breast tumours usually develop from the hyperproliferation of ductal epithelial cells before developing into in situ and invasive carcinomas, eventually resulting in metastatic disease (3–5). Histologically, breast cancer is divided into three broad categories: in situ carcinomas, invasive carcinomas, and metastatic breast carcinomas (2, 6, 7). In situ breast carcinoma is further subclassified into ductal in situ carcinoma and lobular in situ carcinoma (2, 7, 8). Additionally, the World Health Organization (WHO) Classification of Tumors of the Breast, fourth edition, acknowledges the most common histological subtypes of invasive breast carcinoma include invasive tubular, lobular, cribriform, metaplastic, mucinous, apocrine, papillary, and micropapillary carcinomas (9). Tumour grading for breast carcinomas is conducted using methods such as the Nottingham modification of the Scarff-Bloom-Richardson grading system, which grades the tumour on a scale from I-III based on the degree of variation from healthy breast epithelium for gland formation (tubularity) and nuclear size and shape (pleomorphism) (9, 10).
Breast carcinomas can be assessed for tumour stages based on the Tumour-Node-Metastasis (TNM) system, which is comprised of nine stages (0, IA, IB, IIA, IIB, IIIA, IIIB, IIIC, and IV) based on the different combinations for tumour status (T), regional lymph nodes status (N), and metastasis status (M) (9, 11). The American Joint Committee on Cancer (AJCC) Cancer Staging Manual, eighth edition, assigns the T0 category for tumour status when there is no evidence of primary tumour(s) and the Tis category for ductal carcinoma in situ. The T1 category is defined by tumours 2 cm in diameter or less; the T2 category of tumours are >2 cm but ≤5 cm in diameter; and the T3 category is defined as tumours greater than 5 cm in diameter. The T4 category is assigned when breast carcinoma cells invade neighbouring and distant tissues and organs (9, 11). The AJCC Cancer Staging Manual, eighth edition, separates regional lymph node status into pathologic N (pN) and clinical N (cN) categories (9, 11). The pN0 category is defined by the absence of regional lymph node metastasis, while the pN1, pN2, and pN3 categories display malignancy in 1-3 lymph nodes, 4-9 lymph nodes, and greater than 10 lymph nodes, respectively. Clinically, the cN classification comprises the cN, cN(f), and cN (sn) categories, which are assigned for confirmed regional lymph node metastasis by clinical findings, core biopsy or fine-needle aspiration, and sentinel node biopsy, respectively (9, 11). Furthermore, metastasis status is classified into two main categories: M0 and M1. M0 indicates the absence of distant metastasis based on radiographic or clinical evaluations. In contrast, M1 signifies the presence of distant metastasis, which includes metastases detected in distant organs or non-regional lymph nodes with a size greater than 0.2 mm (9, 11).
In addition to histological subtypes, breast cancer is classified into five molecular subtypes: luminal A, luminal B, HER2-positive, basal-like or triple-negative (TNBC), and normal-like breast cancers (12, 13). Both luminal A and luminal B subtypes exhibit the expression of estrogen receptors (ER) and are, therefore, considered to be ER-positive breast cancers. Luminal A breast cancers comprise approximately 30%–40% of all invasive breast cancers and are low in grade. Luminal A subtypes are also progesterone receptor (PR) positive and negative for HER2 receptor expression (12, 13). Luminal B breast cancers account for roughly 20%–30% of invasive breast carcinomas and are typically higher in grade. Luminal B subtypes can present as either PR positive or negative, and HER2-positive or HER2-negative with a higher proliferation (Ki-67) score than Luminal A (12–15). HER2-positive breast cancers constitute 10%–15% of invasive breast cancers and are characterized by the overexpression of the HER2 receptor and can be further subtyped based on the positive or negative expression of ER and/or PR (2, 12, 16). TNBC or basal-like subtypes, hallmarked as ER-negative, PR-negative and HER2-negative, represent approximately 15%–20% of invasive breast carcinomas and tend to be aggressive and high-grade. TNBC is further subtyped based on molecular characteristics, with basal-like being the most common subtype (8, 12, 17, 18). The normal-like subcategory of breast cancer makes up less than 10% of invasive breast cancers. The normal-like subtype is similar to luminal A and is characterized as ER and PR-positive, HER2-negative, and displays a low expression of the Ki-67 proliferation marker (2, 19, 20).
Currently, there are various therapeutic strategies for breast cancer, and treatment options predominantly depend on the subtype. Endocrine therapy is the first line of treatment for ER-positive and hormone-positive breast cancers, which account for approximately 70% of total breast cancer diagnoses. Endocrine therapy options comprise selective estrogen receptor modulators (SERMs), selective estrogen receptor degraders (SERDs), aromatase inhibitors (AIs) or combination therapy of two or more drugs (14, 21, 22). Given the similarity of normal-like breast cancer to luminal A subtypes, it is reasonable to infer that SERMs and SERDs provide therapeutic benefits in this setting. However, further research is needed to determine whether there are distinct responses to these agents within the normal-like subtype compared to other ER-positive breast cancers. Treatment for HER2-positive breast carcinoma is typically by targeting the HER2 receptor using anti-HER2 therapies (23–25). However, de novo or acquired resistance to anti-HER2 and hormone therapies is common, and therefore, combination therapy of anti-HER2 or hormone treatments with other agents like CDK4/6 inhibitors, PI3K/AKT/mTOR inhibitors, and immune checkpoint inhibitors are being extensively studied in clinical trials (24, 25). TNBC presents a great challenge for breast cancer treatment due to its heterogeneity, poor prognosis, and limited therapeutic options (26). Currently, the standard of care for treating TNBC is chemotherapy, although current research into PARP (Poly [ADP-ribose] polymerase) inhibitors and immunotherapy provides new and targeted treatments for TNBC (26–28). Metastatic breast cancer presents significant challenges due to its aggressive nature and poor prognosis. Unlike early-stage breast cancer, metastatic disease often lacks specific targeted treatment options and is, therefore, conventionally managed with systemic chemotherapy (29, 30). However, not all chemotherapy agents exhibit the same efficacy, and polychemotherapy—the use of multiple agents—generally yields better treatment responses and longer progression-free survival compared to single-agent chemotherapy. Unfortunately, this approach is frequently associated with increased toxicity and a higher risk of adverse effects, underscoring the need for a balanced treatment strategy that maximizes efficacy while minimizing patient burden (29–31).
The regulation of the development, production, marketing, and sales of pharmaceuticals and medical devices in the United States of America is the responsibility of the Food and Drug Administration (FDA). Established in 1906 with the passage of the Pure Food and Drug Act, the FDA is a federal agency responsible for ensuring the quality, safety, and efficacy of drugs, medical devices, food, cosmetics, and other consumer products in the United States. Through rigorous scientific evaluation and regulatory oversight, the FDA plays a critical role in protecting public health and advancing medical innovation (32, 33). The first drug approved by the FDA for treating breast cancer was the cytotoxic agent methotrexate in 1953. Since then, over 30 drugs have been approved for the treatment of both in situ and malignant breast carcinomas (34). Although ER-positive breast cancers are more common, less aggressive, and present a better prognosis than HER2-positive and triple-negative breast cancers, treatment for ER-positive breast carcinoma warrants further research as there are no definitive treatment strategies for therapeutic resistance (22, 35). Drug resistance develops in 30%–50% of ER-positive breast cancer patients treated with FDA-approved endocrine therapies (22). As previously mentioned, these endocrine therapies include SERMs and SERDs which function to target and modify estrogen receptor activity and to degrade and/or reduce the expression of the estrogen receptor (21, 22, 36). This review, therefore, examines the current FDA-approved treatments for ER-positive breast cancers and their limitations, with an emphasis on the mechanisms of action of SERMs and SERDs.
2 Estrogen receptor signaling
The ER-positive luminal cancers represent the most prevalent subtype of breast cancer, with nearly 70% of breast tumours overexpressing ER, with or without the progesterone receptor (37). Estrogen is the driving force behind mammary gland development and promotes the growth and survival of normal epithelial cells of the breast as well as mammary tumorigenesis. 17β-Estradiol (E2) is the predominant endogenous estrogen and ER ligand in humans (38). Aromatase (encoded by the Cyp19/CYP19 gene) is the rate-limiting enzyme responsible for the unidirectional conversion of androgens to E2 by aromatization in gonadal and extra-gonadal tissues, and it is essential throughout the lifespan in males and females (39, 40).
ERs are members of the nuclear hormone receptor family that include ERα and ERβ, which are encoded by the ESR1 and ESR2 genes and are composed of 595 amino acids and 530 amino acids, respectively (41) (Figure 1). ERα and ERβ share five functional domains: A/B, C, D, E and F (42). The A/B domain, also called the activation factor 1 (AF1) domain, is located in the amino-terminal and is involved in ligand-independent transcription and interactions with domain E. Domain C is the DNA-binding domain (DBD), while the D domain is a flexible hinge region that harbours a nuclear localization signal and heat shock proteins-binding domain. Domain E is the ligand-binding domain (LBD), and the activation factor 2 (AF2) domain is also involved in ligand-dependent transcriptional activation. The LBD also harbours an interface for homo-hetero-dimerization and a binding site for ligand-dependent co-regulator interaction (43). The C-terminal domain (domain F) regulates the transcriptional activation mediated by domains A/B and E (43).
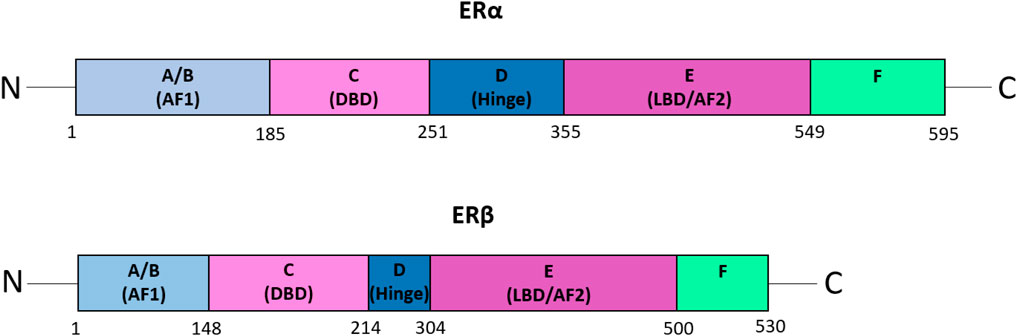
Figure 1. The functional domains of ERα and ERβ. A schematic representation of the domain structure of the estrogen receptors, ERα and ERβ. The five functional domains: A/B, C, D, E, and F, are shown. Domain A/B, also known as the activation factor 1 (AF1) domain, is involved in ligand-independent transcription and interactions with domain E. The C domain is the DNA-binding domain (DBD), whereas domain D is the flexible hinge region that harbours a nuclear localization signal and the domain for binding heat shock proteins. Domain E, also called the ligand-binding domain (LBD) or activation factor 2 (AF2) domain, is involved in ligand-dependent transcriptional activation. Furthermore, the LBD possesses a binding site for ligand-dependent co-regulator interaction as well as an interface for homo-hetero-dimerization. Domain F is the C-terminal domain, which regulates the transcriptional activation mediated by domains A/B and E (43). Figure adapted from (43).
ERs signal through various pathways, including i) the nuclear estrogen response element (ERE)-dependent pathway, ii) the nuclear ERE-independent pathway, and iii) the extranuclear/estrogen-independent pathway (44) (Figure 2). Without E2, ER monomers remain predominantly in the cytoplasm in an inactive (non-DNA-binding) state, sequestered in multiprotein chaperone complexes organized around heat shock proteins (HSPs), particularly HSP90 (45). It is important to note that while ERα plays a predominant role in driving cell proliferation and survival in ER-positive breast cancer, ERβ has been shown to exert opposing effects by inhibiting cell proliferation and promoting apoptosis (Murphy and Leygue, 2012). Furthermore, studies have demonstrated that ERβ antagonizes ERα-mediated transcriptional activity by competing for ERE binding, recruiting co-repressors, and modulating non-genomic signaling pathways (46).
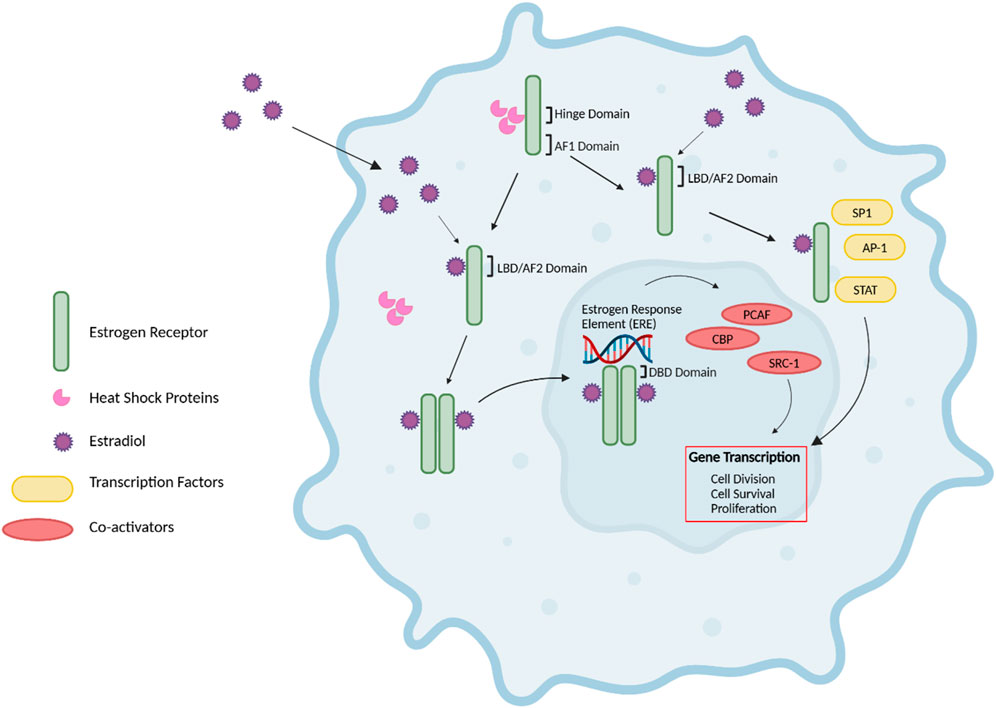
Figure 2. Estrogen Receptor Signaling. The nuclear estrogen response element (ERE) pathway is depicted. 17β-Estradiol (E2), the predominant endogenous estrogen and estrogen receptor (ER) ligand, binds to the ligand binding domain (LBD/AF2) domain of the ER to undergo a conformational change. This conformational change enables the dimerization, activation, and nuclear translocation of the ER to the nucleus, where the ER complex binds to EREs in the promoter region of the target gene via the DNA-binding domain (DBD). Subsequently, this facilitates the recruitment of co-activators such as PCAF and CBP histone acetyltransferases and steroid coactivator-1 (SRC-1), thus promoting various cellular activities, including cell division, survival and proliferation. Further, E2-bound ER can also result in non-classical genomic signaling where liganded ER regulates the expression of genes in an ERE-independent manner through the direct interaction with transcription factors, including specificity protein 1 (SP1), activator protein 1 (AP-1) transcription complex, and the signal transducers and activators of transcription (STAT) family of transcription factors. Without E2, ER monomers remain predominantly in the cytoplasm in an inactive (non-DNA-binding) state, sequestered in multiprotein chaperone complexes organized around heat shock proteins bound to the ER hinge domain. The activation factor 1 (AF1) domain primarily contributes to ligand-independent activation of the ER (43–45, 47, 48). Figure generated using (50).
The nuclear ERE-dependent pathway is the classic ER genomic pathway in which E2-activated ER undergoes a conformational change and dissociates from the multiprotein chaperone complexes, thus releasing HSP90. This conformational change enables the dimerization and activation of the ER. Activated ER translocates to the nucleus, where it binds to EREs in the promoter regions of target genes, facilitating the sequential recruitment of co-activators such as histone acetyltransferases (including CBP/p300 and PCAF) and steroid receptor coactivator-1 (SRC-1), which can robustly enhance the transcription of target genes and promote cellular activities such as cell survival, division, and proliferation (44).
Interactions between E2 and ER can also result in non-classical genomic signaling, whereby the liganded ER regulates the expression of genes in an ERE-independent manner via direct interaction with transcription factors such as the activator protein 1 (AP-1) or specificity protein 1 (SP1) transcription complex, NF-kappaB (NFkB), and the signal transducers and activators of transcription (STAT) family of transcription factors (47, 48).
In the extranuclear/estrogen-independent pathway, ERs may reside in or can be translocated to the cell membrane or cytoplasm, where they can rapidly initiate cellular signaling events by direct interaction with receptor tyrosine kinases such as HER2, EGFR, and insulin-like growth factor-1 receptor (IGF1R) [reviewed in (48)]. Crosstalk between the ER and these receptors can activate the mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) pathways, leading to gene expression changes that enhance cell growth [reviewed in (49)]. It is important to note the extent to which extranuclear/estrogen-independent pathway depends on classical ERs is complex. GPER (G protein-coupled estrogen receptor), an alternative membrane estrogen receptor, also binds 17β-estradiol and triggers overlapping signaling cascades, including activation of adenylate cyclase, ERK1/2, and calcium mobilization (50). Furthermore, Prossnitz and Barton describe how GPER functions independently of ERα and ERβ, yet there is evidence of some functional interplay between these receptors (49). Some studies suggest that classical ERs may even modulate GPER activity through direct or indirect interactions (51). The interplay between these pathways warrants further investigation, particularly in the context of endocrine resistance in ER-positive breast cancer.
The intricacy of ER-signaling in mammary gland development adds to the complexity of identifying a single effective therapeutic target for ER-positive breast cancer. However, the ER is targeted therapeutically directly by antiestrogen agents such as the SERM, tamoxifen, the SERD, fulvestrant, and indirectly by the use of aromatase inhibitors that block the production of estrogen (52, 53). Interestingly, fulvestrant and the SERMs tamoxifen and raloxifene have been found to activate the GPER, acting as a GPER agonist (50, 54–60). This information warrants additional research to fully understand the complex role of these drugs in the context and consideration of both the ER and GPER pathways for breast cancer therapy.
3 FDA-Approved drugs for ER-positive breast cancer
Current FDA-approved endocrine therapies for ER-positive breast cancers target to modify or decrease ER expression and activity or inhibit estrogen biosynthesis. As previously mentioned, these therapies fall under three main categories: i) selective ER modulators (SERMs), ii) selective ER downregulators/degraders (SERDs) and iii) aromatase inhibitors (AIs). SERMs function to target and modify ER activity, while SERDs degrade and/or decrease the expression of the ER. AIs function by blocking estrogen biosynthesis, thereby reducing the amount of estrogen circulating in the body (21, 22, 36). SERMs, such as tamoxifen, are approved endocrine therapies for premenopausal and postmenopausal women (61, 62). The SERD fulvestrant, warrants further treatment studies in premenopausal women, who are still producing estrogen via ovaries, and is more often used to treat breast cancers in postmenopausal women (34, 63, 64). Aromatase inhibitors on the other hand, are often utilized in postmenopausal women, as this treatment stops estrogen production in the breast of postmenopausal women via inhibition of the aromatase enzyme (62, 65, 66).
3.1 Selective estrogen receptor modulator (SERM)
SERMs have been used to treat various diseases and conditions, including breast cancer, osteoporosis and postmenopausal symptoms. Depending on the target tissue, SERMs have the potential to display characteristics of either estrogen agonists or antagonists (67). SERMs are able to interact with both ERα and ERβ to elicit either agonistic effects, such as growth and proliferation when targeting bone tissue, or opposing antagonistic effects in target tissues, such as the breast and mammary epithelia (68, 69). Additionally, selective estrogen receptor modulators have been found to increase the expression and activity of low-density lipoprotein (LDL) receptors, thus reducing LDL cholesterol levels (70, 71). Furthermore, SERMs have been found to inhibit the biosynthesis of cholesterol, resulting in a further contribution to the reduction of LDL cholesterol levels (60, 69, 72–75). Currently, three SERMs have been approved by the FDA for the treatment of ER-positive breast cancers. They include tamoxifen and toremifene, which are triphenylethylene derivatives differing only by the presence of a chlorine atom in the ethyl chain of toremifene, and raloxifene, a benzothiophene derivative (76).
3.1.1 Tamoxifen
3.1.1.1 Approval and use
Tamoxifen is a first-generation breast cancer drug that was approved by the FDA for the treatment of breast cancer in 1977. Originally synthesized as a method of contraception in 1962, tamoxifen is a non-steroidal derivative of triphenylethylene and is one of the most utilized endocrine therapies for ER-positive and hormone-positive breast cancers (77, 78). Various clinical trials have evaluated the efficacy of tamoxifen in the prevention and treatment of breast cancers, including the National Surgical Adjuvant Breast and Bowel Project (NSABP) Protocol B-14 trial in 1981. The Protocol B-14 trial was a randomized clinical study that evaluated the efficacy of tamoxifen in women with ER-positive breast cancer with negative axillary nodes. Participants received either 10 mg of tamoxifen twice daily or a placebo for 4 years following surgery. The results demonstrated that women treated with tamoxifen had a 75% higher likelihood of remaining disease-free compared to those who received the placebo (79). Furthermore, tamoxifen therapy was found to significantly reduce the rates of local and distant treatment failures. Although the trial was effective in prolonging the disease-free survival in those receiving tamoxifen treatment, adverse effects reported more frequently in the tamoxifen group compared to placebo include vaginal discharge, irregular menses, hot flashes, and thromboembolic events (79). Additional side effects include but are not limited to, headaches, dizziness, and depression (80). Further, treatment with tamoxifen has been found to increase the risk of endometrial cancer due to its estrogen agonist effects in the uterus (81–84). Tamoxifen treatment has also been shown to have effects on the ovaries in premenopausal women, such as an increased incidence of benign ovarian cysts, ovarian enlargement, stimulation of ovarian steroidogenesis, and induction of ovulation (85–89). Although tamoxifen displayed biological activity as a full antagonist in the mammary epithelium and is, therefore, an extremely effective treatment for breast cancer, the drug acts as an agonist in other organs, including the endometrium, where it can promote endometrial proliferative disorders, including hyperplasia (90). Tamoxifen is sold under the brand name Soltomax Oral Solution in the United States through pharmaceutical companies such as Fortovia Therapeutics Inc. Likewise, in Canada, AstraZeneca and several other companies sell brand names including Nolvadex-D among others. Tamoxifen is also marketed under various generic names, including tamoxifen citrate in the United States and supplied by companies such as Actavis Pharma, Inc. Additionally, Apo-Tamox tab 10/20 mg is one of the various generic brands of tamoxifen sold in Canada through companies such as Apotex Corporation. Standard tamoxifen dosing is typically 20 mg administered daily, either in tablet or solution form. Furthermore, drug interactions for tamoxifen include but are not limited to, warfarin, aromatase inhibitors such as anastrozole, inducers of CYP3A4, and strong inhibitors of CYP2D6 (80). According to the American Society of Clinical Oncology recommendations, newly diagnosed premenopausal and perimenopausal ER-positive patients take tamoxifen doses daily for 5 years as their first hormonal therapy.
3.1.1.2 Tamoxifen metabolism
Tamoxifen ((Z)-2-[p-(1,2-diphenyl-1-butenyl)phenoxy]N,N-dimethylethylamine) is a non-steroidal SERM exhibiting strong antiestrogen effects in mammary epithelia (91) (Figure 3). Tamoxifen is a prodrug possessing a low affinity for its target, the ER. Tamoxifen is extensively metabolized in the liver by the cytochrome P450 isoforms CYP2D6, CYP3A4, CYP3A5, CYP2C9, and CYP2C19 via two pathways to generate two of its most potent metabolites, afimoxifene (4-hydroxy tamoxifen, 4-OHT) and endoxifen (4-hydroxy, N-desmethyl tamoxifen) (92, 93) (Figure 4). The major pathway involves the cytochrome P450 isoenzyme (CYP) 3A4/5 (CYP3A4/5), which catalyzes the N-demethylation of tamoxifen to N-desmethyltamoxifen (NDM-tamoxifen), which is then 4-hydroxylated by the polymorphic CYP2D6 to produce endoxifen. In a separate metabolic pathway, tamoxifen is converted to 4-OHT by many enzymes, including CYP2D6 and CYP2C9 (93). Further, 4-hydroxy tamoxifen tends to lose a methyl group to yield endoxifen in a process catalyzed primarily by CYP3A (92). Although both 4-hydroxy tamoxifen and endoxifen exhibit 30–100-fold greater affinity for the estrogen receptor than the parent drug tamoxifen (94). Endoxifen, which is produced at five to ten times higher concentrations than 4-OHT, is considered the primary metabolite (95). The conversion of tamoxifen to NDM-tamoxifen constitutes about 92% of tamoxifen metabolism, while the pathway through 4-OHT represents only about 7% (96). However, tamoxifen, formulated as tamoxifen citrate, has a half-life of 5–7 days, whereas endoxifen, formulated as z-endoxifen hydrochloride, has a much shorter half-life between 49.0 and 68.1 h (98). Both 4-OHT and endoxifen are converted into excretable forms by the UDP-glucuronosyltransferase enzymes and sulfotransferase enzymes (99, 100). The ATP-binding cassette (ABC) transporters–ABCB1 (P-gp/MDR1), ABCC1 (BRCP), and ABCC2 (MRP2) are known to be elevated in multiple drug resistance and mediate the efflux of metabolites such as 4-OHT and endoxifen (101, 102). ABCB1, for example, is expressed in 28%–63% of breast tumours and has been shown to bind 4-OHT and endoxifen (102, 103).
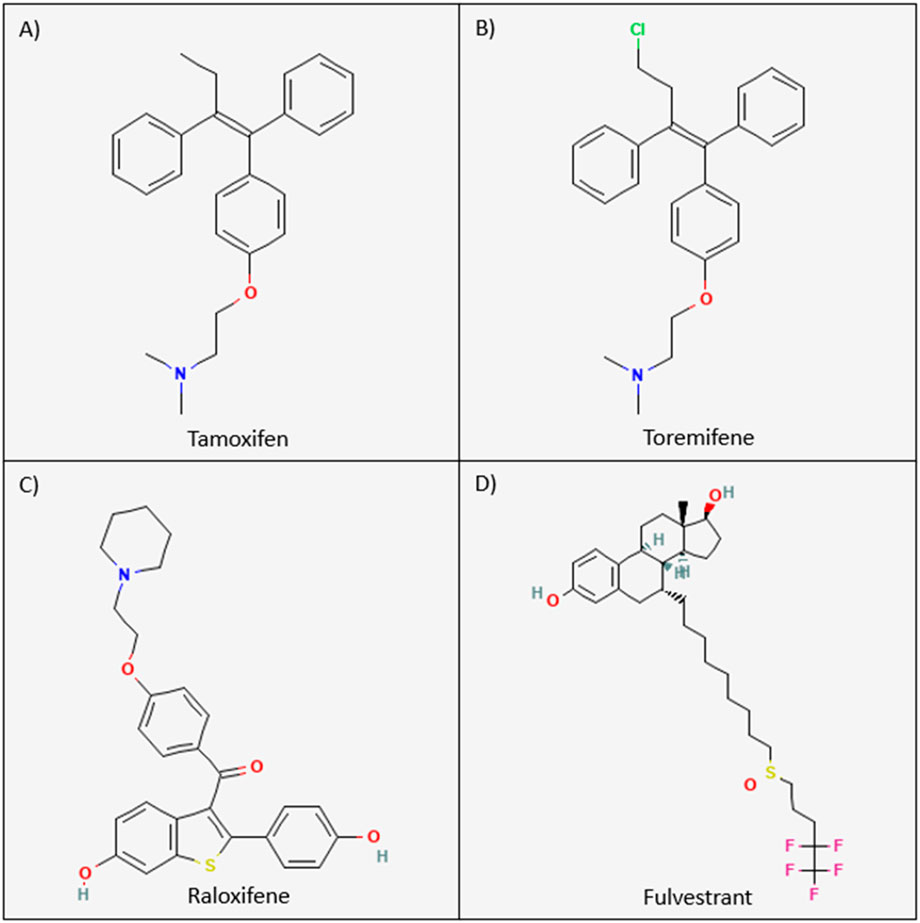
Figure 3. Chemical structures of the current FDA-approved SERMs and SERDs. The chemical structures for tamoxifen, toremifene, raloxifene, and fulvestrant are visualized above. (A) Tamoxifen: (Z)-2-[p-(1,2-diphenyl-1-butenyl)phenoxy]N,N-dimethylethylamine. (B) Toremifene: 2-[p-[(Z)-4-chloro-1,2¬diphenyl-1-butenyl]phenoxy]-N,N-dimethylethylamine. (C) Raloxifene: [6-hydroxy-2-(4-hydroxyphenyl)-1-benzothiophen-3-yl]-[4-(2-piperidin-1-ylethoxy)phenyl]methanone. (D) Fulvestrant: 7-alpha-[9-(4,4,5,5,5-penta fluoropentylsulphinyl)nonyl]estra-1,3,5-(10)-triene-3,17¬beta-diol (190).
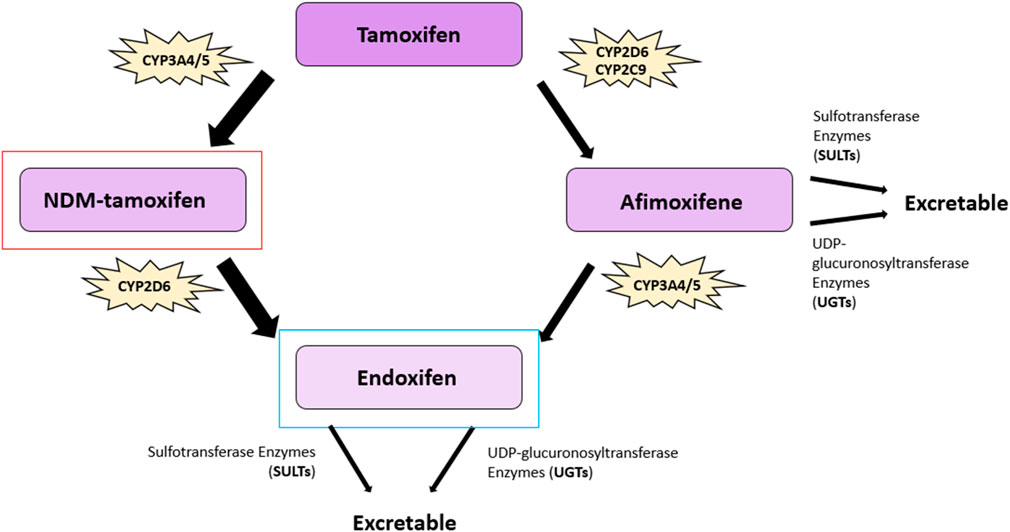
Figure 4. Tamoxifen metabolic pathway. The major metabolites of tamoxifen: 4-hydroxy tamoxifen (afimoxifene), and 4-hydroxy, N-desmethyl tamoxifen (endoxifen), are shown, as well as the metabolite N-desmethyltamoxifen (NDM-tamoxifen). The primary active tamoxifen metabolite, endoxifen, is highlighted in blue and NDM-tamoxifen, the major circulating metabolite, is highlighted in red. The conversion of tamoxifen to NDM-tamoxifen constitutes approximately 92% of tamoxifen metabolism, whereas the pathway through 4-OHT represents about 7%. The key cytochrome P450 (CYP) enzymes involved in bioconversion are highlighted alongside the other cytochrome P450 isoenzymes. CYP3A4/5 catalyzes the N-demethylation of tamoxifen to produce NDM-tamoxifen, which is subsequently 4-hydroxylated by CYP2D6 to generate endoxifen. Tamoxifen is converted to afimoxifene by enzymes such as CYP2D6 and CYP2C9 and is further metabolized into endoxifen primarily via CYP3A enzymes. Afimoxifene and endoxifen are converted into excretable forms via sulfotransferase and UDP-glucuronosyltransferase enzymes (92, 93, 99, 100). Figure adapted from (97).
3.1.1.3 Mechanism of action
Tamoxifen, a non-steroidal ER antagonist, produces metabolites which competitively bind to the ER and displace estrogen to inhibit its proliferative effects in breast tissue (91) (Figure 5). Like E2, tamoxifen also binds to the ER, albeit with lower affinity, and induces a distinct conformational change that promotes the release of HSP90 and ER dimerization. The ER dimer translocates to the nucleus where it will first promote the activation of the activation factor 1 (AF1) domain and inhibit the activation factor 2 (AF2) domain or ligand-binding, and second, bind to ERE on promotors of target genes (104). Through these processes, the Tamoxifen-ER dimer attenuates the transcription of the E2-responsive genes since the ligand-dependent AF2 domain is inactivated and ER co-activator binding is reduced. Further, tamoxifen-induced ER dimers recruit corepressors such as the HDACs (histone deacetylases) and SMRT (silencing mediator of retinoid acid and thyroid hormone receptor), also known as N-CoR2 (105–107). HDACs, for instance, deacetylate histones that subsequently lead to the inhibition of transcription. The recruitment of the co-repressor proteins is, therefore, pivotal to the antiestrogen effects of tamoxifen in mammary epithelia (105–107).
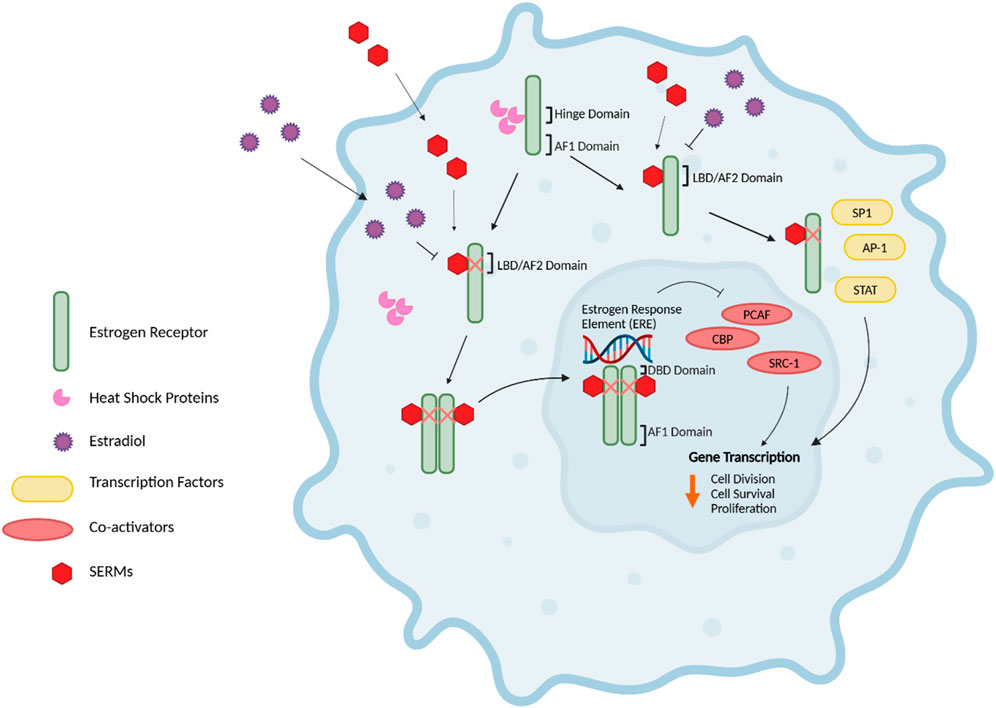
Figure 5. Mechanism of action of SERMs. The selective estrogen receptor modulators (SERMs), tamoxifen, toremifene, and raloxifene bind to the ER like 17β-Estradiol (E2) to induce conformational change. Heat shock proteins (HSP) chaperones, such as HSP90 are released as part of ER activation and nuclear translocation from the ER hinge domain. This results in the dimerization of ER and translocation to the nucleus, preferentially activating the activation factor 1 (AF1) domain while suppressing the activation factor 2 (AF2)/ligand-binding domain (LDB). Secondly, SERM-bound ER will bind to the Estrogen Response Element (ERE) on promotors of target genes, thus resulting in transcription attenuation of E2-responsive genes due to the inactivation of the ligand-dependent AF2 domain and reduced ER co-activator binding. Additionally, SERMs can influence transcription factor pathways involving proteins such as STAT and AP-1 pathways via meditation through altered ER-cofactor interactions (22, 35, 104). Figure generated using (106).
3.1.2 Toremifene
3.1.2.1 Approval and use
Toremifene is a selective estrogen receptor modulator and a chlorinated derivative of tamoxifen. Toremifene was therefore approved by the FDA in 1997 for the treatment of metastatic breast cancers of ER-positive origin or tumours of unknown ER expression (108, 109). The drug is widely used for the treatment of both early and late stages of metastatic breast cancer (109). Toremifene was approved with the hope that it would display a better safety profile than tamoxifen (108). A study conducted by the International Breast Cancer Study Group found that 76% of patients with ER-positive tumours demonstrated a 5-year disease-free survival after receiving 60 mg of toremifene daily following chemotherapy on day 15 over a 5-year period. Additionally, the 5-year survival of patients with ER-positive breast cancer who received toremifene treatment over a 5-year duration was 90% (110). In the United States, toremifene is sold as tablets under the brand name Fareston and as the generic product, toremifene citrate, through suppliers such as Kyowa Kirin, Inc. and Rising Pharmaceuticals, Inc., respectively. The FDA-recommended dose of toremifene for the treatment of metastatic breast cancer in postmenopausal women is 60 mg once daily. However, doses as high as 120 mg have been approved in countries such as Japan and shown to be effective in treating metastatic breast cancer in patients who have relapsed on aromatase inhibitors (111, 112). Toremifene toxicity of grade 3 or higher was experienced in 7% of patients taking 60 mg of toremifene daily in the study for the International Breast Cancer Study Group (n = 499). Grade 3 toxicity symptoms include vascular events such as deep vein thrombosis and phlebitis pulmonary embolism, myocardial infarction, and congestive heart failure, among others (110). Drug interactions listed by the FDA for toremifene include warfarin, agents that prolong the QT interval, drugs that decrease the excretion of renal calcium, and strong inducers or inhibitors of CYP3A4 (113).
3.1.2.2 Toremifene metabolism
Toremifene (2-[p-[(Z)-4-chloro-1,2diphenyl-1-butenyl]phenoxy]-N,N-dimethylethylamine) is structurally similar to tamoxifen, differing in just one chlorine atom substituting a hydrogen atom in the ethyl side chain, but it is equally as effective as tamoxifen for treating breast cancer (114–116) (Figure 3). Toremifene is lipophilic and over 99% bound to plasma proteins such as albumin (117, 118). Like tamoxifen, toremifene is metabolized in the liver to form metabolites, including N-desmethyl (NDM) toremifene, 4-hydroxy (4OH) toremifene and 4-hydroxy-N-desmethyl (4OH-NDM) toremifene (119) (Figure 6). The major circulating metabolite of toremifene is NDM-toremifene, while levels of 4OH-toremifene, the primary active metabolite, and 4OH-NDM-toremifene are much lower in human plasma (119, 120). In breast cancer cell lines, the 4OH- and 4OH-NDM-toremifene metabolites are approximately 100-fold higher in activity than both toremifene and NDM-toremifene (119). The metabolism of toremifene into its metabolites occurs via two pathways. The first pathway is the conversion of toremifene into NDM-toremifene mainly through the cytochrome P450 isoenzyme CYP3A4, although CYP3A5 and CYP2D6 are also contributors (119, 121). The conversion of NDM-toremifene into its secondary metabolite, 4OH-NDM-toremifene, occurs via the CYP2D6 and CYP2C9 isozymes. The second pathway of toremifene metabolism involves the conversion of toremifene into 4OH-toremifene primarily through the CYP2C9 isoenzyme, with CYP2D6 playing a minor role. The bioconversion of 4OH-toremifene into 4OH-NDM-toremifene occurs mainly through CYP3A4, although CYP2D6 and CYP2C9 are also involved to a lesser degree (119, 121). Due to its similarities to tamoxifen, toremifene metabolites such as 4-OH-toremifene, are also converted into excretable forms through sulfotransferase and UDP-glucuronosyltransferase enzymes (100, 122, 123). The half-life of toremifene and NDM-toremifene is typically 5 days, whereas the half-life for 4OH-toremifene is approximately 6 days (124). Additionally, the concentration of the major metabolite, NDM-toremifene, is greater than toremifene 8 hours after administration (119, 125).
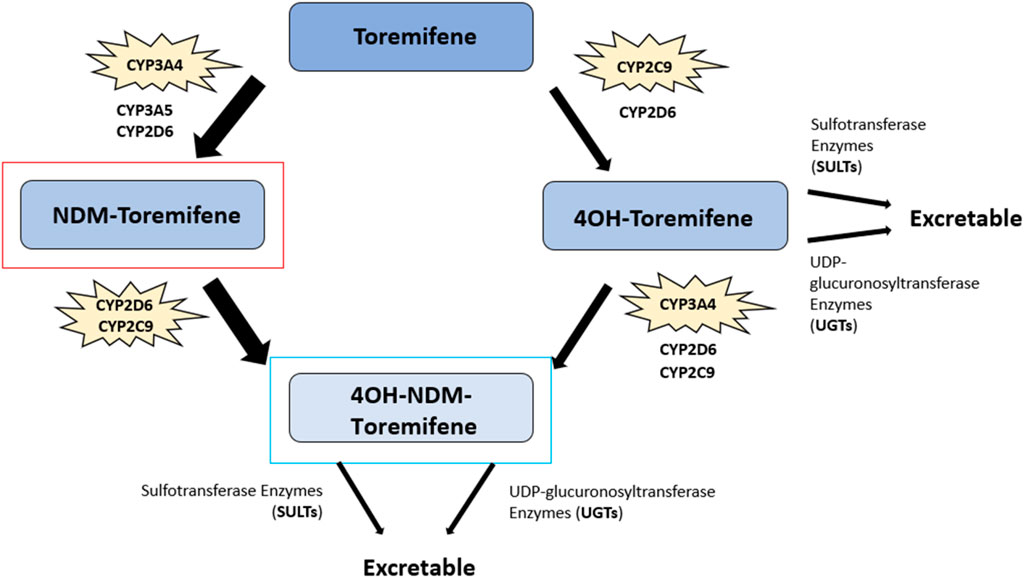
Figure 6. Toremifene metabolic pathway. The major circulating metabolite of toremifene, N-desmethyl (NDM) toremifene, is highlighted in red while the primary active metabolite of tamoxifen, 4OH-NDM-toremifene, is highlighted in blue. 4-hydroxy (4-OH) toremifene, another metabolite, is also shown. The key cytochrome P450 (CYP) enzymes involved in toremifene metabolism are noted alongside other cytochrome P450 isoenzymes. The conversion of toremifene into NDM-toremifene occurs primarily via CYP3A4, with CYP3A5 and CYP2D6 also contributing to a lesser extent. NDM-toremifene is converted into its secondary metabolite, 4OH-NDM-toremifene, through the CYP2D6 and CYP2C9 isozymes. Further, toremifene is converted to 4OH-toremifene mainly via CYP2C9, with CYP2D6 contributing a minor role. The CYP3A4 is the main isozyme in converting 4OH-toremifene into 4OH-NDM-toremifene, with CYP2D6 and CYP2C9 also involved to a lesser degree. 4OH-toremifene and 4OH-NDM-toremifene are converted into excretable forms through sulfotransferase and UDP-glucuronosyltransferase enzymes (100, 117, 118, 122, 123).
3.1.2.3 Mechanism of action
Toremifene is a non-steroidal SERM exhibiting antiestrogen effects on target tissues such as the mammary epithelia and partial agonist effects in uterine and bone tissues (109, 118). Although equivalent in its estrogen binding and anti-tumour properties as tamoxifen, toremifene may be less genotoxic, as research conducted in rat hepatocytes found lower toremifene DNA adducts compared to tamoxifen (110, 118, 126, 127). Toremifene and its metabolites competitively bind to the ER to displace estrogen, thereby inhibiting the growth and proliferative effects of estrogen on breast tissue (118, 128). The mechanism of action of toremifene is similar to that of tamoxifen (Figure 5). Upon binding to the ER, toremifene and its metabolites initiate ER dimerization in the same fashion as E2. The ER dimer is translocated to the nucleus, leading to the activation of the AF1 domain, the inactivation of the AF2 domain, and the binding of the dimer to the ERE of target gene promoters. The toremifene-ER complex results in a decrease in the binding of ER co-activators in addition to the already inactivated AF2 domain, resulting in the reduced transcription of estrogen-responsive genes (118, 128).
3.1.3 Raloxifene
3.1.3.1 Approval and use
Raloxifene is a second-generation, selective estrogen receptor modulator approved by the FDA for the prevention of postmenopausal osteoporosis in 1997 and for the treatment of osteoporosis in postmenopausal women in 1999. In 2007, the FDA approved raloxifene for the risk reduction of invasive breast carcinoma in postmenopausal women (34, 63). Clinical trials such as the Multiple Outcomes of Raloxifene Evaluation (MORE) were conducted to observe the effectiveness of raloxifene in the risk reduction of breast cancer in postmenopausal women with osteoporosis. The MORE trial, a randomized, multicenter, double-blind trial, was conducted between 1994 and 1998, where 7705 postmenopausal women with osteoporosis across 25 countries were administered raloxifene or placebo and followed up after a median of 40 months. The trial found that the risk of invasive breast carcinoma decreased by 76% during the 3 years that patients received raloxifene treatment. Furthermore, the risk of ER-positive breast cancer was decreased by 90% during the raloxifene treatment (129). In general, raloxifene is well tolerated, although an increase in the rate of hot flashes and leg cramps was reported. Other adverse effects reported in the trial include peripheral edema, endometrial cavity fluid and influenza-like syndromes (129).
Raloxifene is sold in the United States under the brand name Evista by companies such as Physicians Total Care, Inc. and as the generic raloxifene hydrochloride through companies like Actavis Pharma Company. Furthermore, raloxifene is sold in other countries, such as Canada, for the prevention of breast cancer. For breast cancer risk reduction, the standard dosing of raloxifene is 60 mg taken orally, once daily. Interactions listed by the FDA for raloxifene include cholestyramine, warfarin, systemic estrogens, and other highly protein-bound drugs, among others (130).
3.1.3.2 Raloxifene metabolism
Raloxifene ([6-hydroxy-2-(4-hydroxyphenyl)benzo [b]thien-3-yl]-[4-[2-(1-piperidinyl) ethoxy]phenyl]methanone) is a selective estrogen receptor modulator belonging to a class of compounds known as the benzothiophenes (Figure 3). Like tamoxifen and toremifene, raloxifene also exhibits estrogen agonist or antagonist effects on differing target tissues (63). Raloxifene is rapidly absorbed after oral administration, with up to 60% absorbed following administration of a treatment dose. Additionally, the drug does not undergo significant P450-dependent oxidation. Instead, raloxifene undergoes extensive first-pass glucuronidation upon absorption into the gastrointestinal tract, resulting in less than 2% bioavailability (131–133). Upon first pass glucuronidation, raloxifene is converted into the metabolites raloxifene-6-β-glucuronide (raloxifene-6-gluc) and raloxifene-4′-β-glucuronide (raloxifene-4′-gluc) (131, 134) (Figure 7). Raloxifene-4′-gluc is the major metabolite found in human plasma, with an approximate ratio of 8:1 for raloxifene-4′-gluc:raloxifene-6-gluc. Further, only less than 1% of unconjugated raloxifene is found in human plasma (131, 132). Previous research has found the 1A UDP-glucuronosyltransferase (UGT) enzyme family to be involved in the metabolism of raloxifene (135). UGTs are membrane-bound enzymes found in the endoplasmic reticulum and are involved in catalyzing the transfer of glucuronic acid to substrates. The resulting conjugates possess an increase in water solubility (136, 137). The conversion of raloxifene to the raloxifene-4′-gluc metabolite occurs primarily by intestinal UGT1A10 and UGT1A8 enzymes, while the bioconversion of raloxifene to raloxifene-6-gluc occurs mainly through hepatic UGT1A1. Additionally, UGT1A9 was shown to play a minor role in catalyzing the formation of raloxifene-4′-gluc and raloxifene-6-gluc (135). Raloxifene possesses an elimination half-life of approximately 28 h and an apparent oral clearance of 44 L/kg per hour (131, 133).
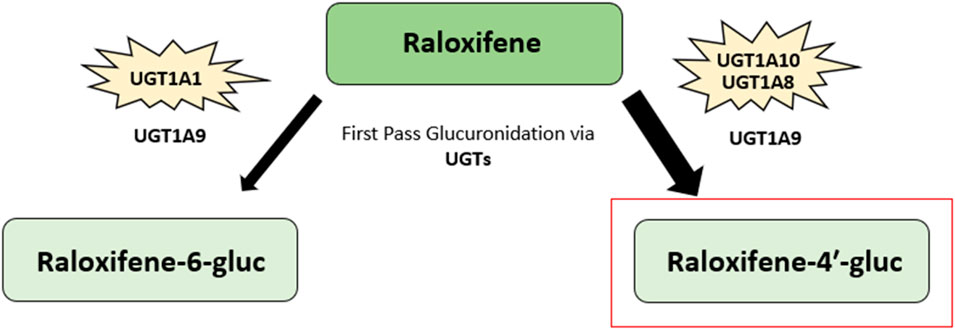
Figure 7. Raloxifene metabolic pathway. The major metabolite of raloxifene, raloxifene-4′-β-glucuronide (raloxifene-4′-gluc), is highlighted in red. Another metabolite, raloxifene-6-β-glucuronide (raloxifene-6-gluc), is also shown. Raloxifene undergoes extensive first-pass glucuronidation into its metabolites upon gastrointestinal tract absorption. The 1A UDP-glucuronosyltransferase (UGT) enzyme family are involved in raloxifene metabolism. Raloxifene is converted to raloxifene-4′-gluc metabolite mainly through intestinal UGT1A10 and UGT1A8 enzymes. The conversion of raloxifene to raloxifene-6-gluc occurs primarily via hepatic UGT1A1. The UGT1A9 enzyme has been found to play a minor role in catalyzing the formation of both raloxifene-4′-gluc and raloxifene-6-gluc (131–135).
3.1.3.3 Mechanism of action
Raloxifene is a non-steroidal SERM exhibiting estrogen agonist or antagonist effects depending on the target tissue (63). Estrogenic effects of raloxifene occur in bone and lipid metabolism, whereas antiestrogen effects are exhibited in breast tissue, and neutral effects are observed in the endometrium (63, 67, 138, 139). Raloxifene exhibits a higher affinity for the ER than its glucuronide conjugate metabolites (140). Raloxifene has a similar affinity to the ER as E2 and can initiate ER dimerization and translocation to the nucleus once bound to the ER via its benzothiophene ring (Figure 5). The binding of raloxifene to the ER results in a spatial change in the configuration of the ER, resulting in the activation of the AF1 domain and inactivation of the AF2 domain of the ER. Subsequently, the raloxifene-ER complex binds to the ERE of target gene promoters (141–143). Furthermore, the raloxifene-ER complex results in a decrease in the binding of ER co-activators, thus resulting in the reduced transcription of estrogen responsive genes). Additionally, once raloxifene binds to the ER, the raloxifene-ER complex then recruits coregulators to further enhance the antiestrogen effects of raloxifene (63, 144). However, raloxifene exerts a different effect on other tissues, such as bone. The raloxifene-ER complex, with the aid of various helping, activating, and/or adapting (HP) proteins, is able to bind and activate a specific DNA sequence known as the Raloxifene Responding Element (RRE). The binding of the raloxifene-ER complex to the RRE results in the transcription of genes involved in the synthesis of specific cell proteins responsible for the estrogen growth and proliferative effects of raloxifene on these tissues (141–143).
3.2 Selective estrogen receptor downregulators/degraders (SERDs)
As discussed earlier, anti-endocrine therapies such as tamoxifen, a selective ER modulator, and aromatase inhibitors such as anastrozole were FDA-approved in 1977 and 1995, respectively (65, 145). Tamoxifen quickly became the gold standard therapy for advanced and early-stage estrogen-sensitive cancers. However, the potentially serious side effects and other presentations following tamoxifen treatment, such as relapses, exposed the need for other anti-endocrine therapies with fewer or less severe side effects. The advent of selective estrogen receptor downregulators/degraders (SERDs) was, therefore, a welcome addition to antiestrogen therapeutic armamentarium. SERDs are estrogen receptor antagonists employed for treating ER and hormone-positive breast cancers. SERDs exert their anti-tumour effects through the inhibition, downregulation, and degradation of the ER, therefore abrogating the proliferative effects of estrogen in breast cancer cells (21, 146, 147). Fulvestrant is the only SERD approved by the FDA for the treatment of hormone-positive breast carcinomas. The drug is an effective and well-tolerated drug for the treatment of metastatic ER-positive breast cancer.
3.2.1 Fulvestrant
3.2.1.1 Approval and use
Fulvestrant was approved by the FDA in 2002 for the treatment of ER and hormone-positive, metastatic breast cancers in postmenopausal women (34, 148). Fulvestrant is sold in the United States as the brand name Faslodex by companies such as AstraZeneca Pharmaceuticals LP and as the generic fulvestrant through companies like Amneal Pharmaceuticals LLC. Furthermore, fulvestrant is sold in various countries including Canada, France, and Spain. The FDA lists the standard dosing of fulvestrant for the treatment of breast cancer as 500 mg as two intramuscular injections, one for each buttock, administered on days 1, 15, and 29 and subsequent once-monthly doses (149). Additionally, there are no known drug interactions for fulvestrant (150). Clinical trials have been conducted to evaluate the efficacy of fulvestrant for use in hormone-positive breast cancers. Such trials include the FALCON study, a randomized, double-blind, international, phase III clinical trial where 524 patients with hormone-positive, locally advanced or metastatic breast cancer were enrolled between 2012 and 2014 (151). The FALCON trial aimed to evaluate the efficacy of fulvestrant compared to the aromatase inhibitor, anastrozole, in patients who had not received prior endocrine therapy. The trial demonstrated that patients receiving a 500 mg dose of fulvestrant had a longer median progression-free survival than those treated with 1 mg of anastrozole daily, with medians of 16.6 months and 13.8 months, respectively. Therefore, fulvestrant demonstrates comparable, if not superior, efficacy in extending progression-free survival in patients with hormone receptor-positive, locally advanced, or metastatic breast cancer when compared to aromatase inhibitors like anastrozole (151). Although fulvestrant is an effective and generally well-tolerated treatment option, common adverse effects reported in trials such as the FALCON include arthralgia, hot flashes and gastrointestinal disturbances (151, 152).
3.2.1.2 Fulvestrant metabolism
Fulvestrant (7-alpha-[9-(4,4,5,5,5-penta fluoropentylsulphinyl)nonyl]estra-1,3,5-(10)-triene-3,17beta-diol) is a 7α-alkylsulphinyl analogue of estradiol that competes with E2 for binding to the ER (148) (Figure 3). Fulvestrant is metabolized in the body via rapid glucuronidation at its −3 and −17 positions and through sulfate conjugation to produce sulfated-fulvestrant (153, 154) (Figure 8). The glucuronidation of fulvestrant is catalyzed by the A1 UDP-glucuronosyltransferase (UGT) family enzymes, namely UGT1A1, UGT1A3, UGT1A4, and UGT1A8 (154). The majority of fulvestrant is metabolized by UGT1A3 and UGT1A4, the main enzymes which catalyze the glucuronidation of fulvestrant at the 3-hydroxyl position, although UGT1A1 and UGT1A8 were also found to also play a minor role. Furthermore, UGT1A8 can convert fulvestrant into fulvestrant-17-glucuronide, although this metabolite only accounts for 5%–10% of total fulvestrant glucuronidation (154). Previous research suggests that fulvestrant may be inactivated in both the liver and intestine due to the high expression levels of the UGT1A3 and UGT1A4 enzymes (154).
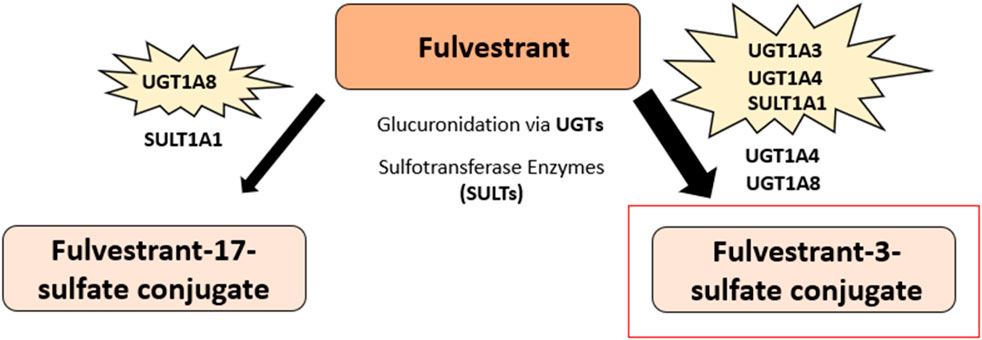
Figure 8. Fulvestrant predominant metabolic pathway. The major metabolite of fulvestrant, fulvestrant-3-sulfate conjugate, is highlighted in red. Fulvestrant glucuronidation at the 3-hydroxyl position and sulfate conjugation produces sulfated-fulvestrant, specifically the metabolite fulvestrant-3-sulfate conjugate. Another metabolite of fulvestrant, fulvestrant-17-sulfate conjugate, is also shown and is produced via fulvestrant glucuronidation at the −17 position and through sulfate conjugation. The 1A UDP-glucuronosyltransferase (UGT) enzyme family are involved in the metabolism of fulvestrant. The majority of fulvestrant is metabolized via UGT1A3 and UGT1A4 enzymes, which catalyze the glucuronidation of fulvestrant at the 3-hydroxyl position. UGT1A1 and UGT1A8 were also found to also play a minor role in the glucuronidation of fulvestrant. Further, fulvestrant can be converted to fulvestrant-17-glucuronide through the UGT1A8 enzyme. Fulvestrant is sulfated by sulfotransferase enzymes, namely SULT1A1, to produce sulfated-fulvestrant (153–155).
Additionally, the cytochrome p450 isoenzymes, CYP1A2, CYP2C9 and CYPA4, may also be involved in the metabolism of fulvestrant, although the sulfate conjugation of fulvestrant is suggested to be the more predominant pathway in comparison (154, 155). Fulvestrant is sulfated via sulfotransferase enzymes, namely SULT1A1, to produce sulfated-fulvestrant, particularly the fulvestrant-3-sulfate conjugate (153, 154). Due to its low bioavailability and pre-systemic metabolism, fulvestrant was designed for administration via intramuscular injection over oral ingestion (156). Fulvestrant has an elimination half-life of approximately 40 days after a 250 mg dose, with an estimated apparent volume of distribution at steady state ranging from 3 to 5 L/kg (155).
3.2.1.3 Mechanism of action
Unlike SERMs, which exert both estrogen agonist and antagonist effects depending on the target tissue, fulvestrant exhibits purely antiestrogen effects. Fulvestrant has a high affinity for the ER, with an approximate 89% binding affinity to that of estradiol (157). The attenuation of ER dimerization via fulvestrant, results in the subsequent impairment and inhibition of the fulvestrant-ER complex for translocation to the nucleus (158) (Figure 9). Furthermore, the impaired dimerization inhibits the activation of the ER, as receptor dimerization is crucial for ER function and nuclear localization (158, 159). While tamoxifen blocks the E2-mediated activity of the AF2 domain, resulting in ER-antagonistic activity, the fulvestrant-induced conformational change of the ER disrupts both AF2- and AF1-related transcriptional activity (158, 159). Since fulvestrant hinders the ER’s ability for nuclear translocation, the fulvestrant-ER complex thus cannot localize and bind to the ERE of target gene promoters. However, if any fulvestrant-ER complexes are successful in nuclear localization, the complex is still rendered transcriptionally inactive due to the hindered AF1 and AF2 domains (158). Additionally, the fulvestrant-ER complex is unstable and fragile, thus resulting in the accelerated proteasomal degradation of the ER compared to the ER bound with estradiol or tamoxifen (160). The accumulation of the fulvestrant-ER complex in the cytoplasm may also promote its degradation, as the ER is not shuttled to the nucleus as per usual in its functioning state (161). The reduction in cellular ER protein caused by fulvestrant is not due to the downregulation of ER mRNA levels, but rather to the accelerated degradation of the receptor, driven by the instability of the fulvestrant-ER complex. Therefore, fulvestrant exerts its antiestrogen effects through various mechanisms, including the impairment of ER dimerization, the inhibition of ER activity, and the accelerated proteasomal degradation of the ER (21, 158, 160).
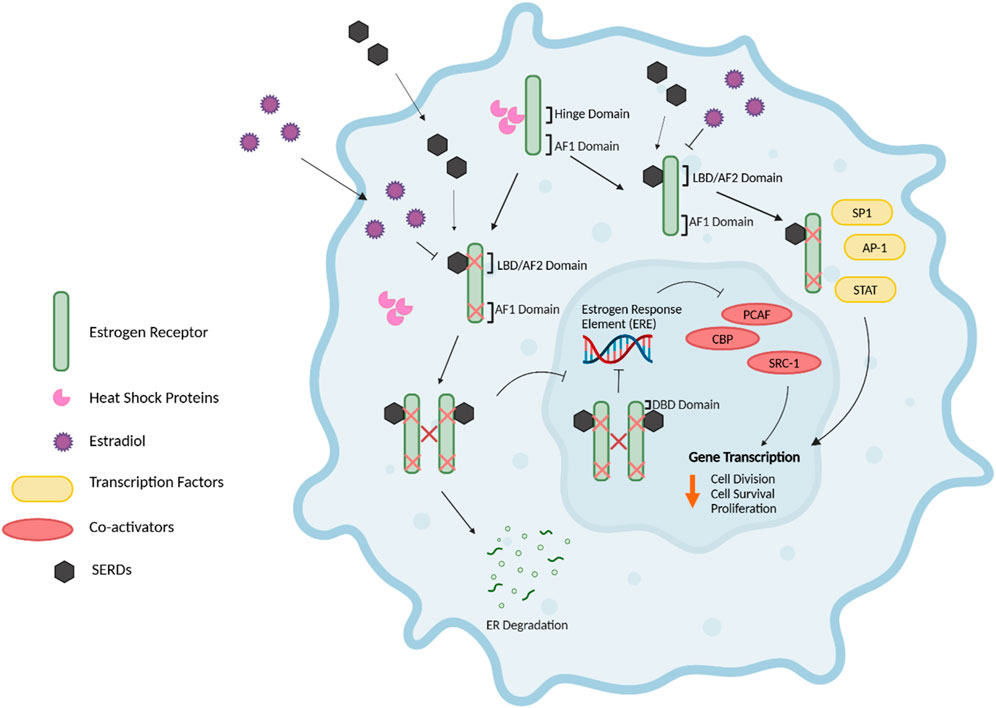
Figure 9. Mechanism of action of SERDs. The selective estrogen receptor downregulator/degrader (SERD), fulvestrant, exerts anti-tumour effects via the inhibition, downregulation, and degradation of the ER, therefore hindering the proliferative effects of estrogen in breast cancer cells. The attenuation of ER dimerization via fulvestrant competitively binding to the ligand binding domain (LBD)/AF2 domain of the ER results in the impairment and inhibition of the fulvestrant-ER complex for nuclear translocation, thus preventing the fulvestrant-ER complex from binding to the estrogen response element (ERE) of target gene promoters. The impaired dimerization of the ER inhibits its activation, as receptor dimerization is critical for ER function and nuclear localization. Additionally, the conformational change of the ER via fulvestrant disrupts both the Activation Factor 1 and 2 (AF2)- and (AF1)-related transcriptional activity. The fulvestrant-ER complex is fragile and unstable, therefore resulting in the accelerated proteasomal degradation of the ER in comparison to the ER bound with estradiol or SERMs such as tamoxifen. Further, SERDs can influence the pathways involving transcription factors such as STAT and AP-1 through the mediation of altered ER-cofactor interactions (21, 158–161, 191). Figure generated using (50).
4 Current progress and future directions
4.1 Current progress in tamoxifen treatment
Although there have been numerous clinical trials conducted for tamoxifen use in breast cancer endocrine therapy, a more recent trial known as MONALEESA 7 was conducted with a focus on combination therapy in breast cancer treatment. The MONALEESA 7 trial is a randomized, double-blind, placebo-controlled, international, phase III clinical trial comparing the CDK 4/6 inhibitor, ribociclib, with a placebo or in addition to endocrine therapy such as tamoxifen. Premenopausal and perimenopausal women (n = 672) with HER2-negative, hormone receptor-positive, advanced breast cancer were enrolled in the study between 2014 and 2016 (162). The trial found an estimated overall survival among those who received tamoxifen treatment in the ribociclib group was 71.2% versus 54.5% in the placebo group at 42 months. Furthermore, there was approximately 29% lower risk of death in those receiving ribociclib compared to those receiving endocrine therapy alone (162). Additionally, an update to the study showed that ribociclib plus endocrine therapies, such as tamoxifen, displayed a persistent and significantly longer overall survival compared to endocrine therapy alone when observing the 58.7 vs. 48.0 months timeframes. These results demonstrate a reduction in the relative risk of death by 24% and were consistent with the final analysis for overall survival (163). Therefore, utilizing endocrine therapies such as tamoxifen in combination with other drugs like CDK 4/6 inhibitors are a promising future for the prevention, disease-free maintenance, and treatment of hormone-positive breast carcinomas.
4.2 Current progress in toremifene therapy
Several clinical trials for high dose-toremifene treatment have been conducted in Japan and found to be effective as part of “hormone rotation therapy” for the treatment of metastatic breast cancer. Although clinical trials are yet to be conducted regarding the effectiveness of high-dose toremifene therapy against recurrent breast cancer or postmenopausal hormone-sensitive progressive breast cancer, Fushima et al. show promising research in their study of high dose-toremifene for hormone receptor-positive metastatic breast carcinoma with secondary resistance to aromatase inhibitors (112).
4.3 Current progress in raloxifene therapy
Various clinical trials, including the MONA trial and the National Surgical Adjuvant Breast and Bowel Project Study (NSABP) of Tamoxifen and Raloxifene (STAR) P-2 trial, have been undertaken regarding the effectiveness and safety of raloxifene in the risk reduction of breast carcinoma in postmenopausal women. However, due to its low bioavailability, the generation of a raloxifene-like drug with an increase in pharmacokinetics was developed (164, 165). This benzothiophene analog of raloxifene is a prodrug known as arzoxifene. However, a phase III trial comparing arzoxifene to tamoxifen as a first-line treatment was terminated when data suggested that arzoxifene was inferior to tamoxifen for the treatment of locally advanced or metastatic breast cancer with respect to a time to progression endpoint (166). Currently, raloxifene is the sole benzothiophene SERM approved by the FDA for use in breast cancer risk reduction. Upon comparing and reviewing data collected from clinical trials, raloxifene is a generally well-tolerated and an effective drug in the risk reduction of breast carcinoma in postmenopausal women with osteoporosis (129, 167).
4.4 Current progress in fulvestrant therapy
Recently, the FDA has approved the use of inavolisib, a potent and selective inhibitor of the p110α catalytic subunit of phosphatidylinositol 3-kinase (PIK3CA), plus the CDK4/6 inhibitor palbociclib and the sole FDA-approved SERD, fulvestrant. Inavolisib promotes the degradation of mutated p110α and has been shown to display synergistic activity in combination treatment with palbociclib and fulvestrant for PIK3CA-mutated, hormone receptor (HR) positive, HER2-negative, locally advanced or metastatic breast cancer following relapse on or after the completion of adjuvant endocrine therapy (168, 169). Treatment with inavolisib plus palbociclib-fulvestrant resulted in a longer progression-free survival for patients than those placed on placebo plus palbociclib-fulvestrant, although with greater incidence of toxic effects. Overall, this combination therapy demonstrated good tolerability with a manageable safety profile (168, 169).
4.5 Future directions
Alongside the extensive research for the treatment of ER-positive breast cancer with SERMs, SERDs, and AIs, new drug therapies are currently under research and progress in clinical trials. These include the Selective Estrogen Receptor Antagonist/Degrader, Giredestrant, and the Selective Androgen Receptor Modulator (SARM), Enobosarm (170–172). Giredestrant, a non-steroidal, oral, selective ER antagonist and SERD, has shown promise in phase II trials as a single-agent drug therapy to treat locally advanced or metastatic breast cancer with an ER-positive, HER-negative profile (171, 173). In the phase II acelERA BC study, daily 30 mg giredestrant was administered orally to patients until disease progression or unacceptable toxicity for 28-day cycles (171). Following the treatment course, giredestrant displayed a numerical improvement compared to the physician’s choice of endocrine monotherapy. Additionally, in the overall study population, there was an approximate 20% relative reduction in the risk of disease progression or death, which was a favourable benefit for patients with ESR1 m tumours (171). This indicates that giredestrant can target mutant ER-reversing progesterone hypersensitivity more effectively than fulvestrant or AIs, as it is a potent antagonist for targeting this mechanism of endocrine resistance (171, 174). Other novel SERDs that are in varying phases of clinical studies include elacestrant, camizestrant, amcenestrant, imlunestrant, and rintodestrant (173, 175).
Additionally, SARMs like enobosarm show great promise as a novel class of endocrine therapy. Like SERMs, SARMs also exhibit both tissue-dependent agonist and antagonist effects (172). Enobosarm is a first-in-class oral SARM that targets the Androgen Receptor (AR), thus inhibiting the growth of AR-positive, ER-positive breast carcinoma cells (172). As the AR is expressed in approximately 80%–90% of ER-positive breast carcinomas, SARMs serve as alternative means of endocrine therapy for those patients with AR-positive, ER-positive, metastatic breast cancer (176–182). A phase II clinical trial conducted showed that 9 mg or 18 mg, once daily treatment with enobosarm showed a clinical benefit rate of 32% in the 9-mg cohort and 29% in the 18-mg cohort for those enrolled in the study (170, 176). The phase III ARTEST trial is currently underway which will observe enobosarm monotherapy versus an active control of a SERM or exemestane, in patients with metastatic breast cancer of ER-positive, HER2-negative, and AR-positive (≥40% nuclei staining) origin who had progressed on previous therapy with a non-steroidal AI, fulvestrant, and a CDK4/6 inhibitor (172).
Although resistance to hormone therapy develops in 30%–50% of ER-positive breast cancer patients, the utilization of combination therapies provides insight into the treatment of resistant and relapse cases (22). The development of tamoxifen resistance is the result of various underlying mechanisms that are still being explored. These include the activation of the signaling pathways via receptor tyrosine kinases, as well as more recent studies which have found the involvement of cell cycle regulators and transcription factors in tamoxifen treatment resistance (183–188). The activation of the PI3K-PTEN/AKT/mTOR pathway via receptor tyrosine kinases overexpression is believed to be closely related to tamoxifen resistance (185, 186). Additionally, Breast Tumour Kinase (BRK), a non-receptor type tyrosine kinase, has been found to confer resistance to tamoxifen treatment in breast cancer through the regulation of CDK1 tyrosine phosphorylation (187). Further, cell cycle regulators, such as LEM4, have been found to render ER + breast cancer cells resistant to tamoxifen when overexpressed through ERα signaling and activation of the cyclin D-CDK4/6 axis (184). Furthermore, the transcription factor KLF4, has been shown to overcome tamoxifen resistance via suppression of the MAPK signaling pathway (183).
5 Conclusion
ER-positive breast cancers are the most common molecular subtype of breast cancer, but this does not imply a dismal prognosis. The use of SERMs and SERDs offers hope and improved survival for those diagnosed with ER-positive breast cancers. However, given the opposing cellular effects of ERα and Erβ, the Erα/ERβ ratio is, therefore, an important determinant of breast cancer behavior and response to endocrine therapy. A higher ERα/ERβ ratio has been linked to increased resistance to SERMs and endocrine therapy, whereas a higher ERβ expression correlates with improved responsiveness to treatment (189). These findings highlight the therapeutic potential of ERβ agonists or strategies that restore ERβ expression to counteract ERα-driven malignancy. Additionally, the recent advancements in expanding the scope of endocrine therapies through the use of SARMs and combination therapies provide further inspiration and optimism for those currently battling ER-positive breast cancers.
Author contributions
NK: Conceptualization, Writing – original draft, Writing – review and editing. KL: Conceptualization, Funding acquisition, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bray, F, Laversanne, M, Sung, H, Ferlay, J, Siegel, RL, Soerjomataram, I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J Clinicians (2024) 74(3):229–63. doi:10.3322/caac.21834
2. Feng, Y, Spezia, M, Huang, S, Yuan, C, Zeng, Z, Zhang, L, et al. Breast cancer development and progression: risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes and Dis (2018) 5(2):77–106. doi:10.1016/j.gendis.2018.05.001
3. Polyak, K. Breast cancer: origins and evolution. J Clin Invest (2007) 117(11):3155–63. doi:10.1172/jci33295
4. Burstein, HJ, Polyak, K, Wong, JS, Lester, SC, and Kaelin, CM. Ductal carcinoma in situ of the breast. N Engl J Med (2004) 350(14):1430–41. doi:10.1056/nejmra031301
5. Sun, YS, Zhao, Z, Yang, ZN, Xu, F, Lu, HJ, Zhu, ZY, et al. Risk factors and preventions of breast cancer. Int J Biol Sci (2017) 13(11):1387–97. doi:10.7150/ijbs.21635
6. Makki, J. Diversity of breast carcinoma: histological subtypes and clinical relevance. Clin Med Insights Pathol (2015) 8:23–31. doi:10.4137/cpath.s31563
7. Malhotra, GK, Zhao, X, Band, H, and Band, V. Histological, molecular and functional subtypes of breast cancers. Cancer Biol and Ther (2010) 10(10):955–60. doi:10.4161/cbt.10.10.13879
8. Li, CI, Uribe, DJ, and Daling, JR. Clinical characteristics of different histologic types of breast cancer. Br J Cancer (2005) 93(9):1046–52. doi:10.1038/sj.bjc.6602787
9. Oluogun, WA, Adedokun, KA, Oyenike, MA, and Adeyeba, OA. Histological classification, grading, staging, and prognostic indexing of female breast cancer in an African population: a 10-year retrospective study. Int J Health Sci (Qassim) (2019) 13(4):3–9.
10. Meyer, JS, Alvarez, C, Milikowski, C, Olson, N, Russo, I, Russo, J, et al. Breast carcinoma malignancy grading by Bloom-Richardson system vs proliferation index: reproducibility of grade and advantages of proliferation index. Mod Pathol (2005) 18(8):1067–78. doi:10.1038/modpathol.3800388
11. Koh, J, and Kim, MJ. Introduction of a new staging system of breast cancer for radiologists: an emphasis on the prognostic stage. Korean J Radiol (2019) 20(1):69–82. doi:10.3348/kjr.2018.0231
12. Fragomeni, SM, Sciallis, A, and Jeruss, JS. Molecular subtypes and local-regional control of breast cancer. Surg Oncol Clin North America (2018) 27(1):95–120. doi:10.1016/j.soc.2017.08.005
13. Vuong, D, Simpson, PT, Green, B, Cummings, MC, and Lakhani, SR. Molecular classification of breast cancer. Virchows Arch (2014) 465(1):1–14. doi:10.1007/s00428-014-1593-7
14. Johnson, KS, Conant, EF, and Soo, MS. Molecular subtypes of breast cancer: a review for breast radiologists. J Breast Imaging (2021) 3(1):12–24. doi:10.1093/jbi/wbaa110
15. Cheang, MC, Chia, SK, Voduc, D, Gao, D, Leung, S, Snider, J, et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. JNCI: J Natl Cancer Inst (2009) 101(10):736–50. doi:10.1093/jnci/djp082
16. Staaf, J, Ringner, M, Vallon-Christersson, J, Jonsson, G, Bendahl, PO, Holm, K, et al. Identification of subtypes in human epidermal growth factor receptor 2--positive breast cancer reveals a gene signature prognostic of outcome. J Clin Oncol (2010) 28(11):1813–20. doi:10.1200/jco.2009.22.8775
17. Badve, S, Dabbs, DJ, Schnitt, SJ, Baehner, FL, Decker, T, Eusebi, V, et al. Basal-like and triple-negative breast cancers: a critical review with an emphasis on the implications for pathologists and oncologists. Mod Pathol (2011) 24(2):157–67. doi:10.1038/modpathol.2010.200
18. Nielsen, TO, Hsu, FD, Jensen, K, Cheang, M, Karaca, G, Hu, Z, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res (2004) 10(16):5367–74. doi:10.1158/1078-0432.ccr-04-0220
19. Dai, X, Li, T, Bai, Z, Yang, Y, Liu, X, Zhan, J, et al. Breast cancer intrinsic subtype classification, clinical use and future trends. Am J Cancer Res (2015) 5(10):2929–43.
20. Perou, CM, Sorlie, T, Eisen, MB, van de Rijn, M, Jeffrey, SS, Rees, CA, et al. Molecular portraits of human breast tumours. Nature (2000) 406(6797):747–52. doi:10.1038/35021093
21. Lumachi, F, Santeufemia, DA, and Basso, SM. Current medical treatment of estrogen receptor-positive breast cancer. World J Biol Chem (2015) 6(3):231–9. doi:10.4331/wjbc.v6.i3.231
22. Rozeboom, B, Dey, N, and De, P. ER+ metastatic breast cancer: past, present, and a prescription for an apoptosis-targeted future. Am J Cancer Res (2019) 9(12):2821–31.
23. Loibl, S, and Gianni, L. HER2-positive breast cancer. The Lancet (2017) 389(10087):2415–29. doi:10.1016/s0140-6736(16)32417-5
24. Montagna, E, and Colleoni, M. Hormonal treatment combined with targeted therapies in endocrine-responsive and HER2-positive metastatic breast cancer. Ther Adv Med Oncol (2019) 11:1758835919894105. doi:10.1177/1758835919894105
25. Pernas, S, and Tolaney, SM. HER2-positive breast cancer: new therapeutic frontiers and overcoming resistance. Ther Adv Med Oncol (2019) 11:1758835919833519. doi:10.1177/1758835919833519
26. Mehanna, J, Haddad, FG, Eid, R, Lambertini, M, and Kourie, HR. Triple-negative breast cancer: current perspective on the evolving therapeutic landscape. Int J Women's Health (2019) 11:431–7. doi:10.2147/ijwh.s178349
27. Mandapati, A, and Lukong, KE. Triple negative breast cancer: approved treatment options and their mechanisms of action. J Cancer Res Clin Oncol (2023) 149(7):3701–19. doi:10.1007/s00432-022-04189-6
28. Lebert, JM, Lester, R, Powell, E, Seal, M, and McCarthy, J. Advances in the systemic treatment of triple-negative breast cancer. Curr Oncol (2018) 25(Suppl. 1):S142–150. doi:10.3747/co.25.3954
29. Cortazar, P, Justice, R, Johnson, J, Sridhara, R, Keegan, P, and Pazdur, R. US Food and Drug Administration approval overview in metastatic breast cancer. J Clin Oncol (2012) 30(14):1705–11. doi:10.1200/jco.2011.39.2613
30. Westphal, T, Gampenrieder, SP, Rinnerthaler, G, and Greil, R. Cure in metastatic breast cancer. Memo (2018) 11(3):172–9. doi:10.1007/s12254-018-0426-9
31. Carrick, S, Parker, S, Thornton, CE, Ghersi, D, Simes, J, and Wilcken, N. Single agent versus combination chemotherapy for metastatic breast cancer. The Cochrane database Syst Rev (2009) 2009(2):CD003372. doi:10.1002/14651858.cd003372.pub3
32. Jazowski, SA, and Winn, AN. The role of the FDA and regulatory approval of new devices for diabetes care. Curr Diab Rep (2017) 17(6):40. doi:10.1007/s11892-017-0871-6
33. Van Norman, GA. Drugs, devices, and the FDA: Part 2. JACC: Basic Translational Sci (2016) 1(4):277–87. doi:10.1016/j.jacbts.2016.03.009
34. Leo, CP, Leo, C, and Szucs, TD. Publisher Correction: breast cancer drug approvals by the US FDA from 1949 to 2018. Nat Rev Drug Discov (2020) 19(4):291. doi:10.1038/s41573-020-0060-1
35. Fallahpour, S, Navaneelan, T, De, P, and Borgo, A. Breast cancer survival by molecular subtype: a population-based analysis of cancer registry data. CMAJ Open (2017) 5(3):E734–E739. doi:10.9778/cmajo.20170030
36. Jordan, VC, and Brodie, AM. Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of breast cancer. Steroids (2007) 72(1):7–25. doi:10.1016/j.steroids.2006.10.009
37. Musgrove, EA, and Sutherland, RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer (2009) 9(9):631–43. doi:10.1038/nrc2713
38. Platet, N, Cathiard, AM, Gleizes, M, and Garcia, M. Estrogens and their receptors in breast cancer progression: a dual role in cancer proliferation and invasion. Crit Rev Oncology/Hematology (2004) 51(1):55–67. doi:10.1016/j.critrevonc.2004.02.001
39. Blakemore, J, and Naftolin, F. Aromatase: contributions to physiology and disease in women and men. Physiology (Bethesda) (2016) 31(4):258–69. doi:10.1152/physiol.00054.2015
40. Simpson, ER, Clyne, C, Rubin, G, Boon, WC, Robertson, K, Britt, K, et al. Aromatase--a brief overview. Annu Rev Physiol (2002) 64:93–127. doi:10.1146/annurev.physiol.64.081601.142703
41. Murphy, LC, and Leygue, E. The role of estrogen receptor-beta in breast cancer. Semin Reprod Med (2012) 30(1):05–13. doi:10.1055/s-0031-1299592
42. Rondon-Lagos, M, Villegas, VE, Rangel, N, Sanchez, MC, and Zaphiropoulos, PG. Tamoxifen resistance: emerging molecular targets. Int J Mol Sci (2016) 17(8):1357. doi:10.3390/ijms17081357
43. Kumar, V, Green, S, Stack, G, Berry, M, Jin, JR, and Chambon, P. Functional domains of the human estrogen receptor. Cell (1987) 51(6):941–51. doi:10.1016/0092-8674(87)90581-2
44. Welboren, WJ, Stunnenberg, HG, Sweep, FC, and Span, PN. Identifying estrogen receptor target genes. Mol Oncol (2007) 1(2):138–43. doi:10.1016/j.molonc.2007.04.001
45. Yager, JD, and Davidson, NE. Estrogen carcinogenesis in breast cancer. N Engl J Med (2006) 354(3):270–82. doi:10.1056/nejmra050776
46. Paplomata, E, and O'Regan, R. New and emerging treatments for estrogen receptor-positive breast cancer: focus on everolimus. Ther Clin Risk Management (2013) 9:27–36. doi:10.2147/tcrm.s30349
47. Safe, S, and Kim, K. Non-classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein-1 signaling pathways. J Mol Endocrinol (2008) 41(5):263–75. doi:10.1677/jme-08-0103
48. Glidewell-Kenney, C, Weiss, J, Lee, EJ, Pillai, S, Ishikawa, T, Ariazi, EA, et al. ERE-independent ERα target genes differentially expressed in human breast tumors. Mol Cell Endocrinol (2005) 245(1-2):53–9. doi:10.1016/j.mce.2005.10.003
49. Prossnitz, ER, and Barton, M. The G protein-coupled oestrogen receptor GPER in health and disease: an update. Nat Rev Endocrinol (2023) 19(7):407–24. doi:10.1038/s41574-023-00822-7
50. Revankar, CM, Cimino, DF, Sklar, LA, Arterburn, JB, and Prossnitz, ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science (2005) 307(5715):1625–30. doi:10.1126/science.1106943
51. Filardo, EJ, and Thomas, P. GPR30: a seven-transmembrane-spanning estrogen receptor that triggers EGF release. Trends Endocrinol and Metab (2005) 16(8):362–7. doi:10.1016/j.tem.2005.08.005
52. Wakeling, AE. Similarities and distinctions in the mode of action of different classes of antioestrogens. Endocrine-related cancer (2000) 7(1):17–28. doi:10.1677/erc.0.0070017
53. Smith, IE, and Dowsett, M. Aromatase inhibitors in breast cancer. N Engl J Med (2003) 348(24):2431–42. doi:10.1056/nejmra023246
54. Prossnitz, ER, and Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol (2011) 7(12):715–26. doi:10.1038/nrendo.2011.122
55. Prossnitz, ER, and Barton, M. Estrogen biology: new insights into GPER function and clinical opportunities. Mol Cell Endocrinol (2014) 389(1-2):71–83. doi:10.1016/j.mce.2014.02.002
56. Vergote, I, and Abram, P. Fulvestrant, a new treatment option for advanced breast cancer: tolerability versus existing agents. Ann Oncol (2006) 17(2):200–4. doi:10.1093/annonc/mdj047
57. Petrie, WK, Dennis, MK, Hu, C, Dai, D, Arterburn, JB, Smith, HO, et al. G protein-coupled estrogen receptor-selective ligands modulate endometrial tumor growth. Obstet Gynecol Int (2013) 2013:1–17. doi:10.1155/2013/472720
58. Filardo, EJ, Quinn, JA, Bland, KI, and Frackelton, AR. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol (2000) 14(10):1649–60. doi:10.1210/mend.14.10.0532
59. Prossnitz, ER, and Arterburn, JB. International union of basic and clinical pharmacology. XCVII. G protein-coupled estrogen receptor and its pharmacologic modulators. Pharmacol Rev (2015) 67(3):505–40. doi:10.1124/pr.114.009712
60. Barton, M. Not lost in translation: emerging clinical importance of the G protein-coupled estrogen receptor GPER. Steroids (2016) 111:37–45. doi:10.1016/j.steroids.2016.02.016
61. Rugo, HS, Rumble, RB, Macrae, E, Barton, DL, Connolly, HK, Dickler, MN, et al. Endocrine therapy for hormone receptor-positive metastatic breast cancer: American society of clinical Oncology guideline. J Clin Oncol (2016) 34(25):3069–103. doi:10.1200/jco.2016.67.1487
62. Gradishar, WJ, Anderson, BO, Balassanian, R, Blair, SL, Burstein, HJ, Cyr, A, et al. NCCN guidelines insights: breast cancer, version 1.2017. J Natl Compr Canc Netw (2017) 15(4):433–51. doi:10.6004/jnccn.2017.0044
63. Provinciali, N, Suen, C, Dunn, BK, and DeCensi, A. Raloxifene hydrochloride for breast cancer risk reduction in postmenopausal women. Expert Rev Clin Pharmacol (2016) 9(10):1263–72. doi:10.1080/17512433.2016.1231575
64. Pistelli, M, Mora, AD, Ballatore, Z, and Berardi, R. Aromatase inhibitors in premenopausal women with breast cancer: the state of the art and future prospects. Curr Oncol (2018) 25(2):e168–e175. doi:10.3747/co.25.3735
65. Chumsri, S, Howes, T, Bao, T, Sabnis, G, and Brodie, A. Aromatase, aromatase inhibitors, and breast cancer. The J Steroid Biochem Mol Biol (2011) 125(1-2):13–22. doi:10.1016/j.jsbmb.2011.02.001
66. Bradley, R, Braybrooke, J, Gray, R, Hills, RK, Liu, Z, Pan, H, et al. Aromatase inhibitors versus tamoxifen in premenopausal women with oestrogen receptor-positive early-stage breast cancer treated with ovarian suppression: a patient-level meta-analysis of 7030 women from four randomised trials. The Lancet Oncol (2022) 23(3):382–92. doi:10.1016/s1470-2045(21)00758-0
67. An, KC. Selective estrogen receptor modulators. Asian Spine J (2016) 10(4):787–91. doi:10.4184/asj.2016.10.4.787
68. Nelson, ER, Wardell, SE, and McDonnell, DP. The molecular mechanisms underlying the pharmacological actions of estrogens, SERMs and oxysterols: implications for the treatment and prevention of osteoporosis. Bone (2013) 53(1):42–50. doi:10.1016/j.bone.2012.11.011
69. Maximov, PY, Lee, TM, and Jordan, VC. The discovery and development of selective estrogen receptor modulators (SERMs) for clinical practice. Curr Clin Pharmacol (2013) 8(2):135–55. doi:10.2174/1574884711308020006
70. Suarez, Y, Fernandez, C, Gomez-Coronado, D, Ferruelo, AJ, Davalos, A, Martinez-Botas, J, et al. Synergistic upregulation of low-density lipoprotein receptor activity by tamoxifen and lovastatin. Cardiovasc Res (2004) 64(2):346–55. doi:10.1016/j.cardiores.2004.06.024
71. Cerrato, F, Fernández-Suárez, ME, Alonso, R, Alonso, M, Vázquez, C, Pastor, O, et al. Clinically used selective oestrogen receptor modulators increase LDL receptor activity in primary human lymphocytes. Br J Pharmacol (2015) 172(5):1379–94. doi:10.1111/bph.13016
72. Gylling, H, Pyrhonen, S, Mantyla, E, Maenpaa, H, Kangas, L, and Miettinen, TA. Tamoxifen and toremifene lower serum cholesterol by inhibition of delta 8-cholesterol conversion to lathosterol in women with breast cancer. J Clin Oncol (1995) 13(12):2900–5. doi:10.1200/jco.1995.13.12.2900
73. Gylling, H, Mantyla, E, and Miettinen, TA. Tamoxifen decreases serum cholesterol by inhibiting cholesterol synthesis. Atherosclerosis (1992) 96(2-3):245–7. doi:10.1016/0021-9150(92)90071-n
74. Cypriani, B, Tabacik, C, Descomps, B, and de Paulet, A. Role of estrogen receptors and antiestrogen binding sites in an early effect of antiestrogens, the inhibition of cholesterol biosynthesis. J Steroid Biochem (1988) 31(5):763–71. doi:10.1016/0022-4731(88)90284-1
75. Gomez-Coronado, D, Lasuncion, MA, Martinez-Botas, J, and Fernandez-Suarez, ME. Role of cholesterol metabolism in the anticancer pharmacology of selective estrogen receptor modulators. Semin Cancer Biol (2021) 73:101–15. doi:10.1016/j.semcancer.2020.08.015
76. Pickar, JH, MacNeil, T, and Ohleth, K. SERMs: progress and future perspectives. Maturitas (2010) 67(2):129–38. doi:10.1016/j.maturitas.2010.05.009
77. Quirke, VM. Tamoxifen from failed contraceptive pill to best-selling breast cancer medicine: a case-study in pharmaceutical innovation. Front Pharmacol (2017) 8:620. doi:10.3389/fphar.2017.00620
78. Jordan, VC. Long-term tamoxifen therapy to control or to prevent breast cancer: laboratory concept to clinical trials. Prog Clin Biol Res (1988) 262:105–23.
79. Fisher, B, Costantino, J, Redmond, C, Poisson, R, Bowman, D, Couture, J, et al. A randomized clinical trial evaluating tamoxifen in the treatment of patients with node-negative breast cancer who have estrogen-receptor-positive tumors. N Engl J Med (1989) 320(8):479–84. doi:10.1056/nejm198902233200802
80. FDA U.S. Food and Drug Administration (2019). Prescribing information for SOLTAMOX® (tamoxifen citrate). Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/021807s006lbl.pdf (Accessed March 2027).
81. Fisher, B, Costantino, JP, Redmond, CK, Fisher, ER, Wickerham, DL, and Cronin, WM. Endometrial cancer in tamoxifen-treated breast cancer patients: findings from the national surgical adjuvant breast and Bowel Project (NSABP) B-14. JNCI: J Natl Cancer Inst (1994) 86(7):527–37. doi:10.1093/jnci/86.7.527
82. van Leeuwen, FE, Benraadt, J, Coebergh, JW, Kiemeney, LA, Gimbrère, CH, Otter, R, et al. Risk of endometrial cancer after tamoxifen treatment of breast cancer. Lancet (1994) 343(8895):448–52. doi:10.1016/s0140-6736(94)92692-1
83. Rutqvist, LE, Johansson, H, Signomklao, T, Johansson, U, Fornander, T, and Wilking, N. Adjuvant tamoxifen therapy for early stage breast cancer and second primary malignancies. JNCI J Natl Cancer Inst (1995) 87(9):645–51. doi:10.1093/jnci/87.9.645
84. National Toxicology Program. 15th report on carcinogens. Rep Carcinog (2021) 15:roc15. doi:10.22427/NTP-OTHER-1003
85. Messinis, IE, and Nillius, SJ. Comparison between tamoxifen and clomiphene for induction of ovulation. Acta Obstet Gynecol Scand (1982) 61(4):377–9.
86. Jordan, VC, Fritz, NF, Langan-Fahey, S, Thompson, M, and Tormey, DC. Alteration of endocrine parameters in premenopausal women with breast cancer during long-term adjuvant therapy with tamoxifen as the single agent. JNCI J Natl Cancer Inst (1991) 83(20):1488–91. doi:10.1093/jnci/83.20.1488
87. Gerhard, I, and Runnebaum, B. Comparison between tamoxifen and clomiphene therapy in women with anovulation. Arch Gynecol (1979) 227(4):279–88. doi:10.1007/bf02109916
88. Powles, TJ, Jones, AL, Ashley, SE, O'Brien, ME, Tidy, VA, Treleavan, J, et al. The Royal Marsden Hospital pilot tamoxifen chemoprevention trial. Breast Cancer Res Treat (1994) 31(1):73–82. doi:10.1007/bf00689678
89. Swerdlow, AJ, and Jones, ME. Ovarian cancer risk in premenopausal and perimenopausal women treated with Tamoxifen: a case-control study. Br J Cancer (2007) 96(5):850–5. doi:10.1038/sj.bjc.6603605
90. Adamo, V, Iorfida, M, Montalto, E, Festa, V, Garipoli, C, Scimone, A, et al. Overview and new strategies in metastatic breast cancer (MBC) for treatment of tamoxifen-resistant patients. Ann Oncol (2007) 18(Suppl. 6):vi53–7. doi:10.1093/annonc/mdm225
91. Johnston, SR. Endocrinology and hormone therapy in breast cancer: selective oestrogen receptor modulators and downregulators for breast cancer - have they lost their way? Breast Cancer Res (2005) 7(3):119–30. doi:10.1186/bcr1023
92. Desta, Z, Ward, BA, Soukhova, NV, and Flockhart, DA. Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: prominent roles for CYP3A and CYP2D6. The J Pharmacol Exp Ther (2004) 310(3):1062–75. doi:10.1124/jpet.104.065607
93. Coller, JK, Krebsfaenger, N, Klein, K, Endrizzi, K, Wolbold, R, Lang, T, et al. The influence of CYP2B6, CYP2C9 and CYP2D6 genotypes on the formation of the potent antioestrogen Z-4-hydroxy-tamoxifen in human liver. Br J Clin Pharmacol (2002) 54(2):157–67. doi:10.1046/j.1365-2125.2002.01614.x
94. Lien, EA, Solheim, E, Lea, OA, Lundgren, S, Kvinnsland, S, and Ueland, PM. Distribution of 4-hydroxy-N-desmethyltamoxifen and other tamoxifen metabolites in human biological fluids during tamoxifen treatment. Cancer Res (1989) 49(8):2175–83.
95. Stearns, V, Johnson, MD, Rae, JM, Morocho, A, Novielli, A, Bhargava, P, et al. Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J Natl Cancer Inst (2003) 95(23):1758–64. doi:10.1093/jnci/djg108
96. Klein, DJ, Thorn, CF, Desta, Z, Flockhart, DA, Altman, RB, and Klein, TE. PharmGKB summary: tamoxifen pathway, pharmacokinetics. Pharmacogenetics and Genomics (2013) 23(11):643–7. doi:10.1097/fpc.0b013e3283656bc1
97. Jin, Y, Desta, Z, Stearns, V, Ward, B, Ho, H, Lee, KH, et al. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. JNCI J Natl Cancer Inst (2005) 97(1):30–9. doi:10.1093/jnci/dji005
98. Goetz, MP, Suman, VJ, Reid, JM, Northfelt, DW, Mahr, MA, Ralya, AT, et al. First-in-Human phase I study of the tamoxifen metabolite Z-endoxifen in women with endocrine-refractory metastatic breast cancer. J Clin Oncol (2017) 35(30):3391–400. doi:10.1200/jco.2017.73.3246
99. Ahern, TP, Christensen, M, Cronin-Fenton, DP, Lunetta, KL, Soiland, H, Gjerde, J, et al. Functional polymorphisms in UDP-glucuronosyl transferases and recurrence in tamoxifen-treated breast cancer survivors. Cancer Epidemiol Biomarkers and Prev (2011) 20(9):1937–43. doi:10.1158/1055-9965.epi-11-0419
100. Lazarus, P, Blevins-Primeau, AS, Zheng, Y, and Sun, D. Potential role of UGT pharmacogenetics in cancer treatment and prevention: focus on tamoxifen. Ann N Y Acad Sci (2009) 1155:99–111. doi:10.1111/j.1749-6632.2009.04114.x
101. Mooij, MG, Nies, AT, Knibbe, CA, Schaeffeler, E, Tibboel, D, Schwab, M, et al. Development of human membrane transporters: drug disposition and pharmacogenetics. Clin Pharmacokinet (2016) 55(5):507–24. doi:10.1007/s40262-015-0328-5
102. Teft, WA, Mansell, SE, and Kim, RB. Endoxifen, the active metabolite of tamoxifen, is a substrate of the efflux transporter P-glycoprotein (multidrug resistance 1). Drug Metab Disposition (2011) 39(3):558–62. doi:10.1124/dmd.110.036160
103. Clarke, R, Leonessa, F, and Trock, B. Multidrug resistance/P-glycoprotein and breast cancer: review and meta-analysis. Semin Oncol (2005) 32(6 Suppl. 7):S9–15. doi:10.1053/j.seminoncol.2005.09.009
104. Martinkovich, S, Shah, D, Planey, SL, and Arnott, JA. Selective estrogen receptor modulators: tissue specificity and clinical utility. Clin Interventions Aging (2014) 9:1437–52. doi:10.2147/cia.s66690
105. Wong, MM, Guo, C, and Zhang, J. Nuclear receptor corepressor complexes in cancer: mechanism, function and regulation. Am J Clin Exp Urol (2014) 2(3):169–87.
106. Shang, Y, and Brown, M. Molecular determinants for the tissue specificity of SERMs. Science (2002) 295(5564):2465–8. doi:10.1126/science.1068537
107. Ali, S, Rasool, M, Chaoudhry, H, Pushparaj, PN, Jha, P, Hafiz, A, et al. Molecular mechanisms and mode of tamoxifen resistance in breast cancer. Bioinformation (2016) 12(3):135–9. doi:10.6026/97320630012135
108. Vogel, CL, Johnston, MA, Capers, C, and Braccia, D. Toremifene for breast cancer: a review of 20 years of data. Clin Breast Cancer (2014) 14(1):1–9. doi:10.1016/j.clbc.2013.10.014
109. Mustonen, MV, Pyrhönen, S, and Kellokumpu-Lehtinen, PL. Toremifene in the treatment of breast cancer. World J Clin Oncol (2014) 5(3):393–405. doi:10.5306/wjco.v5.i3.393
110. International Breast Cancer Study, G, Pagani, O, Gelber, S, Price, K, Zahrieh, D, Gelber, R, et al. Toremifene and tamoxifen are equally effective for early-stage breast cancer: first results of International Breast Cancer Study Group Trials 12-93 and 14-93. Ann Oncol (2004) 15(12):1749–59.
111. Yamamoto, Y, Masuda, N, Ohtake, T, Yamashita, H, Saji, S, Kimijima, I, et al. Clinical usefulness of high-dose toremifene in patients relapsed on treatment with an aromatase inhibitor. Breast Cancer (2010) 17(4):254–60. doi:10.1007/s12282-009-0148-2
112. Fushimi, A, Tabei, I, Fuke, A, Okamoto, T, and Takeyama, H. High-dose toremifene as a promising candidate therapy for hormone receptor-positive metastatic breast cancer with secondary resistance to aromatase inhibitors. Int J Breast Cancer (2020) 2020:1–7. doi:10.1155/2020/7156574
113. FDA U.S. Food and Drug Administration (2024). Prescribing information for FARESTON® (toremifene citrate). Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/020497s018lbl.pdf (Accessed March 2027).
114. Zhou, WB, Ding, Q, Chen, L, Liu, XA, and Wang, S. Toremifene is an effective and safe alternative to tamoxifen in adjuvant endocrine therapy for breast cancer: results of four randomized trials. Breast Cancer Res Treat (2011) 128(3):625–31. doi:10.1007/s10549-011-1556-5
115. Kuramochi, H. Conformational studies and electronic structures of tamoxifen and toremifene and their allylic carbocations proposed as reactive intermediates leading to DNA adduct formation. J Med Chem (1996) 39(15):2877–86. doi:10.1021/jm960255g
116. Gams, R. Phase III trials of toremifene vs tamoxifen. Oncology (Williston Park) (1997) 11(5 Suppl. 4):23–8.
117. Lønning, PE, and Lien, EA. Pharmacokinetics of anti-endocrine agents. Cancer Surv (1993) 17:343–70.
118. Wiseman, LR, and Goa, KL. Toremifene. A review of its pharmacological properties and clinical efficacy in the management of advanced breast cancer. Drugs (1997) 54(1):141–60. doi:10.2165/00003495-199754010-00014
119. Watanabe, M, Watanabe, N, Maruyama, S, and Kawashiro, T. Comparative metabolic study between two selective estrogen receptor modulators, toremifene and tamoxifen, in human liver microsomes. Drug Metab Pharmacokinet (2015) 30(5):325–33. doi:10.1016/j.dmpk.2015.05.004
120. Berthou, F, Dreano, Y, Belloc, C, Kangas, L, Gautier, JC, and Beaune, P. Involvement of cytochrome P450 3A enzyme family in the major metabolic pathways of toremifene in human liver microsomes. Biochem Pharmacol (1994) 47(10):1883–95. doi:10.1016/0006-2952(94)90319-0
121. Kim, J, Coss, CC, Barrett, CM, Mohler, ML, Bohl, CE, Li, CM, et al. Role and pharmacologic significance of cytochrome P-450 2D6 in oxidative metabolism of toremifene and tamoxifen. Int J Cancer (2013) 132(6):1475–85. doi:10.1002/ijc.27794
122. Mazzarino, M, de la Torre, X, and Botre, F. Urinary excretion profiles of toremifene metabolites by liquid chromatography-mass spectrometry. Towards targeted analysis to relevant metabolites in doping control. Anal Bioanal Chem (2011) 401(2):529–41. doi:10.1007/s00216-011-4695-y
123. Edavana, VK, Dhakal, IB, Yu, X, Williams, S, and Kadlubar, S. Sulfation of 4-hydroxy toremifene: individual variability, isoform specificity, and contribution to toremifene pharmacogenomics. Drug Metab Disposition (2012) 40(6):1210–5. doi:10.1124/dmd.111.044040
124. Wiebe, VJ, Benz, CC, Shemano, I, Cadman, TB, and DeGregorio, MW. Pharmacokinetics of toremifene and its metabolites in patients with advanced breast cancer. Cancer Chemother Pharmacol (1990) 25(4):247–51. doi:10.1007/bf00684880
125. Anttila, M, Valavaara, R, Kivinen, S, and Maenpaa, J. Pharmacokinetics of toremifene. J Steroid Biochem (1990) 36(3):249–52. doi:10.1016/0022-4731(90)90019-o
126. Shibutani, S, Ravindernath, A, Terashima, I, Suzuki, N, Laxmi, YR, Kanno, Y, et al. Mechanism of lower genotoxicity of toremifene compared with tamoxifen. Cancer Res (2001) 61(10):3925–31.
127. Brown, K. Is tamoxifen a genotoxic carcinogen in women? Mutagenesis (2009) 24(5):391–404. doi:10.1093/mutage/gep022
128. Kangas, L. Biochemical and pharmacological effects of toremifene metabolites. Cancer Chemother Pharmacol (1990) 27(1):8–12. doi:10.1007/bf00689269
129. Cummings, SR, Eckert, S, Krueger, KA, Grady, D, Powles, TJ, Cauley, JA, et al. The effect of raloxifene on risk of breast cancer in postmenopausal women: results from the MORE randomized trial. Multiple Outcomes of Raloxifene Evaluation. JAMA (1999) 281(23):2189–97. doi:10.1001/jama.281.23.2189
130. FDA U.S. Food and Drug Administration (2019). Prescribing information for EVISTA® (raloxifene hydrochloride). Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/020815s034lbl.pdf (Accessed March 2027).
131. Snyder, KR, Sparano, N, and Malinowski, JM. Raloxifene hydrochloride. Am J Health-System Pharm (2000) 57(18):1669–75. doi:10.1093/ajhp/57.18.1669
132. Morello, KC, Wurz, GT, and DeGregorio, MW. Pharmacokinetics of selective estrogen receptor modulators. Clin Pharmacokinet (2003) 42(4):361–72. doi:10.2165/00003088-200342040-00004
133. Hochner-Celnikier, D. Pharmacokinetics of raloxifene and its clinical application. Eur J Obstet and Gynecol Reprod Biol (1999) 85(1):23–9. doi:10.1016/s0301-2115(98)00278-4
134. Sun, D, Jones, NR, Manni, A, and Lazarus, P. Characterization of raloxifene glucuronidation: potential role of UGT1A8 genotype on raloxifene metabolism in vivo. Cancer Prev Res (2013) 6(7):719–30. doi:10.1158/1940-6207.capr-12-0448
135. Kemp, DC, Fan, PW, and Stevens, JC. Characterization of raloxifene glucuronidation in vitro: contribution of intestinal metabolism to presystemic clearance. Drug Metab Disposition (2002) 30(6):694–700. doi:10.1124/dmd.30.6.694
136. Tukey, RH, and Strassburg, CP. Human UDP-glucuronosyltransferases: metabolism, expression, and disease. Annu Rev Pharmacol Toxicol (2000) 40:581–616. doi:10.1146/annurev.pharmtox.40.1.581
137. Meech, R, and Mackenzie, PI. Structure and function of uridine diphosphate glucuronosyltransferases. Clin Exp Pharmacol Physiol (1997) 24(12):907–15. doi:10.1111/j.1440-1681.1997.tb02718.x
138. McDonnell, DP, and Wardell, SE. The molecular mechanisms underlying the pharmacological actions of ER modulators: implications for new drug discovery in breast cancer. Curr Opin Pharmacol (2010) 10(6):620–8. doi:10.1016/j.coph.2010.09.007
139. Maeda, SS, and Lazaretti-Castro, M. An overview on the treatment of postmenopausal osteoporosis. Arquivos Brasileiros de Endocrinologia and Metabologia (2014) 58(2):162–71. doi:10.1590/0004-2730000003039
140. Dodge, JA, Lugar, CW, Cho, S, Short, LL, Sato, M, Yang, NN, et al. Evaluation of the major metabolites of raloxifene as modulators of tissue selectivity. The J Steroid Biochem Mol Biol (1997) 61(1-2):97–106. doi:10.1016/s0960-0760(97)00008-3
141. Rey, JR, Cervino, EV, Rentero, ML, Crespo, EC, Alvaro, AO, and Casillas, M. Raloxifene: mechanism of action, effects on bone tissue, and applicability in clinical traumatology practice. The Open Orthopaedics J (2009) 3:14–21. doi:10.2174/1874325000903010014
142. Grese, TA, Sluka, JP, Bryant, HU, Cole, HW, Kim, JR, Magee, DE, et al. Benzopyran selective estrogen receptor modulators (SERMs): pharmacological effects and structural correlation with raloxifene. Bioorg and Med Chem Lett (1996) 6(7):903–8. doi:10.1016/0960-894x(96)00141-2
143. Bryant, HU, Glasebrook, AL, Yang, NN, and Sato, M. A pharmacological review of raloxifene. J Bone Mineral Metab (1996) 14(1):1–9. doi:10.1007/bf01771666
144. Ma, G, Ren, Y, Wang, K, and He, J. SRC-3 has a role in cancer other than as a nuclear receptor coactivator. Int J Biol Sci (2011) 7(5):664–72. doi:10.7150/ijbs.7.664
145. Jordan, VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov (2003) 2(3):205–13. doi:10.1038/nrd1031
146. Yeh, WL, Shioda, K, Coser, KR, Rivizzigno, D, McSweeney, KR, and Shioda, T. Fulvestrant-induced cell death and proteasomal degradation of estrogen receptor alpha protein in MCF-7 cells require the CSK c-Src tyrosine kinase. PLoS One (2013) 8(4):e60889. doi:10.1371/journal.pone.0060889
147. Lumachi, F, Luisetto, G, Basso, SM, Basso, U, Brunello, A, and Camozzi, V. Endocrine therapy of breast cancer. Curr Med Chem (2011) 18(4):513–22. doi:10.2174/092986711794480177
148. Curran, M, and Wiseman, L. Fulvestrant. Drugs (2001) 61(6):807–13. doi:10.2165/00003495-200161060-00013
149. FDA U.S. Food and Drug Administration (2019). Prescribing information for FASLODEX® (fulvestrant). Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/021344s044lbl.pdf.
150. Shafaee, MN, and Ellis, MJ. Fulvestrant in management of hormone receptor-positive metastatic breast cancer. Future Oncol (2018) 14(18):1789–800. doi:10.2217/fon-2017-0489
151. Robertson, JFR, Bondarenko, IM, Trishkina, E, Dvorkin, M, Panasci, L, Manikhas, A, et al. Fulvestrant 500 mg versus anastrozole 1 mg for hormone receptor-positive advanced breast cancer (FALCON): an international, randomised, double-blind, phase 3 trial. The Lancet (2016) 388(10063):2997–3005. doi:10.1016/s0140-6736(16)32389-3
152. Vergote, I, and Robertson, JF. Fulvestrant is an effective and well-tolerated endocrine therapy for postmenopausal women with advanced breast cancer: results from clinical trials. Br J Cancer (2004) 90(Suppl. 1):S11–4. doi:10.1038/sj.bjc.6601631
153. Hui, Y, Luo, L, Zhang, L, Kurogi, K, Zhou, C, Sakakibara, Y, et al. Sulfation of afimoxifene, endoxifen, raloxifene, and fulvestrant by the human cytosolic sulfotransferases (SULTs): a systematic analysis. J Pharmacol Sci (2015) 128(3):144–9. doi:10.1016/j.jphs.2015.06.004
154. Chouinard, S, Tessier, M, Vernouillet, G, Gauthier, S, Labrie, F, Barbier, O, et al. Inactivation of the pure antiestrogen fulvestrant and other synthetic estrogen molecules by UDP-glucuronosyltransferase 1A enzymes expressed in breast tissue. Mol Pharmacol (2006) 69(3):908–20. doi:10.1124/mol.105.015891
155. Robertson, JF, and Harrison, M. Fulvestrant: pharmacokinetics and pharmacology. Br J Cancer (2004) 90(Suppl. 1):S7–10. doi:10.1038/sj.bjc.6601630
156. M Harrison, A Laight, D Clarke, P Giles, and R Yates, editors. Pharmacokinetics and metabolism of fulvestrant after oral, intravenous and intramuscular administration in healthy volunteers (2003).Proc ASCO
157. Wakeling, AE, Dukes, M, and Bowler, J. A potent specific pure antiestrogen with clinical potential. Cancer Res (1991) 51(15):3867–73.
158. Fawell, SE, White, R, Hoare, S, Sydenham, M, Page, M, and Parker, MG. Inhibition of estrogen receptor-DNA binding by the “pure” antiestrogen ICI 164,384 appears to be mediated by impaired receptor dimerization. Proc Natl Acad Sci U S A (1990) 87(17):6883–7. doi:10.1073/pnas.87.17.6883
159. Lees, JA, Fawell, SE, White, R, and Parker, MG. A 22-amino-acid peptide restores DNA-binding activity to dimerization-defective mutants of the estrogen receptor. Mol Cell Biol (1990) 10(10):5529–31. doi:10.1128/mcb.10.10.5529
160. Nicholson, RI, Gee, JM, Manning, DL, Wakeling, AE, Montano, MM, and Katzenellenbogen, BS. Responses to pure antiestrogens (ICI 164384, ICI182780) in estrogen-sensitive and-resistant experimental and clinical breast cancera. Ann New York Acad Sci (1995) 761:148–63. doi:10.1111/j.1749-6632.1995.tb31376.x
161. Dauvois, S, White, R, and Parker, MG. The antiestrogen ICI 182780 disrupts estrogen receptor nucleocytoplasmic shuttling. J Cell Sci (1993) 106(Pt 4):1377–88. doi:10.1242/jcs.106.4.1377
162. Im, SA, Lu, YS, Bardia, A, Harbeck, N, Colleoni, M, Franke, F, et al. Overall survival with ribociclib plus endocrine therapy in breast cancer. N Engl J Med (2019) 381(4):307–16. doi:10.1056/nejmoa1903765
163. Lu, YS, Im, SA, Colleoni, M, Franke, F, Bardia, A, Cardoso, F, et al. Updated overall survival of ribociclib plus endocrine therapy versus endocrine therapy alone in pre- and perimenopausal patients with hr+/HER2- advanced breast cancer in MONALEESA-7: a phase III randomized clinical trial. Clin Cancer Res (2022) 28(5):851–9. doi:10.1158/1078-0432.ccr-21-3032
164. Palkowitz, AD, Glasebrook, AL, Thrasher, KJ, Hauser, KL, Short, LL, Phillips, DL, et al. Discovery and synthesis of [6-hydroxy-3-[4-[2-(1-piperidinyl)ethoxy]phenoxy]-2-(4-hydroxyphenyl)]b enzo[b]thiophene: a novel, highly potent, selective estrogen receptor modulator. J Med Chem (1997) 40(10):1407–16. doi:10.1021/jm970167b
165. Patel, HK, and Bihani, T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol and Ther (2018) 186:1–24. doi:10.1016/j.pharmthera.2017.12.012
166. Deshmane, V, Krishnamurthy, S, Melemed, AS, Peterson, P, and Buzdar, AU. Phase III double-blind trial of arzoxifene compared with tamoxifen for locally advanced or metastatic breast cancer. J Clin Oncol (2007) 25(31):4967–73. doi:10.1200/jco.2006.09.5992
167. Vogel, VG, Costantino, JP, Wickerham, DL, Cronin, WM, Cecchini, RS, Atkins, JN, et al. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA (2006) 295(23):2727–41. doi:10.1001/jama.295.23.joc60074
168. Jhaveri, KL, Accordino, MK, Bedard, PL, Cervantes, A, Gambardella, V, Hamilton, E, et al. Phase I/ib trial of inavolisib plus palbociclib and endocrine therapy for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced or metastatic breast cancer. J Clin Oncol (2024) 42(33):3947–56. doi:10.1200/jco.24.00110
169. Turner, NC, Im, SA, Saura, C, Juric, D, Loibl, S, Kalinsky, K, et al. Inavolisib-based therapy in PIK3CA-mutated advanced breast cancer. N Engl J Med (2024) 391(17):1584–96. doi:10.1056/nejmoa2404625
170. Palmieri, C, Linden, H, Birrell, SN, Wheelwright, S, Lim, E, Schwartzberg, LS, et al. Activity and safety of enobosarm, a novel, oral, selective androgen receptor modulator, in androgen receptor-positive, oestrogen receptor-positive, and HER2-negative advanced breast cancer (Study G200802): a randomised, open-label, multicentre, multinational, parallel design, phase 2 trial. The Lancet Oncol (2024) 25(3):317–25. doi:10.1016/s1470-2045(24)00004-4
171. Martin, M, Lim, E, Chavez-MacGregor, M, Bardia, A, Wu, J, Zhang, Q, et al. Giredestrant for estrogen receptor-positive, HER2-negative, previously treated advanced breast cancer: results from the randomized, phase II acelERA breast cancer study. J Clin Oncol (2024) 42(18):2149–60. doi:10.1200/jco.23.01500
172. Author anonymous, Novel agents show promise against acquired endocrine resistance in ER+ advanced breast cancer. Oncologist. (2021). 26 3:S15–S6.
173. Chen, YC, Yu, J, Metcalfe, C, De Bruyn, T, Gelzleichter, T, Malhi, V, et al. Latest generation estrogen receptor degraders for the treatment of hormone receptor-positive breast cancer. Expert Opin Investig Drugs (2022) 31(6):515–29. doi:10.1080/13543784.2021.1983542
174. Liang, J, Ingalla, ER, Yao, X, Wang, BE, Tai, L, Giltnane, J, et al. Giredestrant reverses progesterone hypersensitivity driven by estrogen receptor mutations in breast cancer. Sci Transl Med (2022) 14(663):eabo5959. doi:10.1126/scitranslmed.abo5959
175. Patel, R, Klein, P, Tiersten, A, and Sparano, JA. An emerging generation of endocrine therapies in breast cancer: a clinical perspective. NPJ Breast Cancer (2023) 9(1):20. doi:10.1038/s41523-023-00523-4
176. Palmieri, C, Linden, HM, Birrell, S, Lim, E, Schwartzberg, LS, Rugo, HS, et al. Efficacy of enobosarm, a selective androgen receptor (AR) targeting agent, correlates with the degree of AR positivity in advanced AR+/estrogen receptor (ER)+ breast cancer in an international phase 2 clinical study. J Clin Oncol (2021) 39(15_Suppl. l):1020. doi:10.1200/jco.2021.39.15_suppl.1020
177. Ponnusamy, S, Asemota, S, Schwartzberg, LS, Guestini, F, McNamara, KM, Pierobon, M, et al. Androgen receptor is a non-canonical inhibitor of wild-type and mutant estrogen receptors in hormone receptor-positive breast cancers. iScience (2019) 21:341–58. doi:10.1016/j.isci.2019.10.038
178. Niemeier, LA, Dabbs, DJ, Beriwal, S, Striebel, JM, and Bhargava, R. Androgen receptor in breast cancer: expression in estrogen receptor-positive tumors and in estrogen receptor-negative tumors with apocrine differentiation. Mod Pathol (2010) 23(2):205–12. doi:10.1038/modpathol.2009.159
179. Narita, D, Raica, M, Suciu, C, Cîmpean, A, and Anghel, A. Immunohistochemical expression of androgen receptor and prostate-specific antigen in breast cancer. Folia Histochem Cytobiol (2006) 44(3):165–72.
180. Hu, R, Dawood, S, Holmes, MD, Collins, LC, Schnitt, SJ, Cole, K, et al. Androgen receptor expression and breast cancer survival in postmenopausal women. Clin Cancer Res (2011) 17(7):1867–74. doi:10.1158/1078-0432.ccr-10-2021
181. Garay, JP, and Park, BH. Androgen receptor as a targeted therapy for breast cancer. Am J Cancer Res (2012) 2(4):434–45.
182. Collins, LC, Cole, KS, Marotti, JD, Hu, R, Schnitt, SJ, and Tamimi, RM. Androgen receptor expression in breast cancer in relation to molecular phenotype: results from the Nurses' Health Study. Mod Pathol (2011) 24(7):924–31. doi:10.1038/modpathol.2011.54
183. Jia, Y, Zhou, J, Luo, X, Chen, M, Chen, Y, Wang, J, et al. KLF4 overcomes tamoxifen resistance by suppressing MAPK signaling pathway and predicts good prognosis in breast cancer. Cell Signal (2018) 42:165–75. doi:10.1016/j.cellsig.2017.09.025
184. Gao, A, Sun, T, Ma, G, Cao, J, Hu, Q, Chen, L, et al. LEM4 confers tamoxifen resistance to breast cancer cells by activating cyclin D-CDK4/6-Rb and ERα pathway. Nat Commun (2018) 9(1):4180. doi:10.1038/s41467-018-06309-8
185. Yin, L, Zhang, XT, Bian, XW, Guo, YM, and Wang, ZY. Disruption of the ER-α36-EGFR/HER2 positive regulatory loops restores tamoxifen sensitivity in tamoxifen resistance breast cancer cells. PLoS One (2014) 9(9):e107369. doi:10.1371/journal.pone.0107369
186. Hosford, SR, and Miller, TW. Clinical potential of novel therapeutic targets in breast cancer: CDK4/6, Src, JAK/STAT, PARP, HDAC, and PI3K/AKT/mTOR pathways. Pharmacogenomics Personalized Med (2014) 7:203–15. doi:10.2147/pgpm.s52762
187. Mandapati, A, Ning, Z, Baharani, A, and Lukong, KE. BRK confers tamoxifen-resistance in breast cancer via regulation of tyrosine phosphorylation of CDK1. Cell Signal (2023) 108:110723. doi:10.1016/j.cellsig.2023.110723
188. Yao, J, Deng, K, Huang, J, Zeng, R, and Zuo, J. Progress in the understanding of the mechanism of tamoxifen resistance in breast cancer. Front Pharmacol (2020) 11:592912. doi:10.3389/fphar.2020.592912
189. Massarweh, S, and Schiff, R. Resistance to endocrine therapy in breast cancer: exploiting estrogen receptor/growth factor signaling crosstalk. Endocr Relat Cancer (2006) 13(Suppl. 1):S15–24. doi:10.1677/erc.1.01273
190. Kim, S, Chen, J, Cheng, T, Gindulyte, A, He, J, He, S, et al. PubChem 2025 update. Nucleic Acids Res (2025) 53(D1):D1516–D1525. doi:10.1093/nar/gkae1059
Keywords: breast cancer, estrogen receptor, SERMs, SERDS, tamoxifen, toremifene, raloxifene, fulvestrant
Citation: Kim N and Lukong KE (2025) Treating ER-positive breast cancer: a review of the current FDA-approved SERMs and SERDs and their mechanisms of action. Oncol. Rev. 19:1564642. doi: 10.3389/or.2025.1564642
Received: 21 January 2025; Accepted: 31 March 2025;
Published: 10 April 2025.
Edited by:
Daniel P. Bezerra, Oswaldo Cruz Foudantion (FIOCRUZ), BrazilReviewed by:
Diego Gomez-Coronado, Ramón y Cajal University Hospital, SpainKeenan Flynn, New Mexico State University, United States
Copyright © 2025 Kim and Lukong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kiven Erique Lukong, a2l2ZW4ubHVrb25nQHVzYXNrLmNh