 Kevin A. Prestia1
Kevin A. Prestia1 Eugene A. Sosunov1 Evgeny P. Anyukhovsky1 Elena Dolmatova2
Eugene A. Sosunov1 Evgeny P. Anyukhovsky1 Elena Dolmatova2 Caitlin W. Kelly1
Caitlin W. Kelly1 Peter R. Brink3 Richard B. Robinson1 Michael R. Rosen1
Peter R. Brink3 Richard B. Robinson1 Michael R. Rosen1 Heather S. Duffy2*
Heather S. Duffy2*
- 1 Department of Pharmacology, College of Physicians and Surgeons, Columbia University, New York, NY, USA
- 2 Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, USA
- 3 Department of Physiology and Biophysics, State University of New York, Stonybrook, NY, USA
Increasing connexin43 (Cx43) gap junctional conductance as a means to improve cardiac conduction has been proposed as a novel antiarrhythmic modality. Yet, transmission of molecules via gap junctions may be associated with increased infarct size. To determine whether maintaining open gap junction channels impacts on infarct size and induction of ventricular tachycardia (VT) following coronary occlusion, we expressed the pH- and voltage-independent connexin isoform connexin32 (Cx32) in ventricle and confirmed Cx32 expression. Wild-type (WT) mice injected with adenovirus-Cx32 (Cx32inj) were examined following coronary occlusion to determine infarct size and inducibility of VT. There was an increased infarct size in Cx32inj hearts as compared to WT (WT 22.9 ± 4%; Cx32inj 44.3 ± 5%; p < 0.05). Programmed electrical stimulation showed no difference in VT inducibility in WT and Cx32inj mice (VT was reproducibly inducible in 55% of shams and 50% of Cx32inj mice (p > 0.05). Following coronary occlusion, improving cell–cell communication increased infarct size, and conferred no antiarrhythmic benefit.
Introduction
In the acute stage of a myocardial infarction loss of the gap junction protein connexin43 (Cx43) occurs contributing to slowed conduction and reentrant arrhythmias (Gutstein et al., 2001, 2003, 2005; Kieken et al., 2009). The mechanisms, by which Cx43 channels are lost, include the alteration of protein-partners of Cx43 (Kieken et al., 2009). These modifications result in the loss of channels previously localized to the intercalated disk (ID) in the canine heart (Kieken et al., 2009), as well as a decrease in new Cx43 trafficking to the ID in HeLa cells (Shaw et al., 2007). It has been hypothesized that restoring gap junctional communication may increase conduction velocity and thus be antiarrhythmic in settings where conduction is slow and reentry occurs (Roell et al., 2007; Hagen et al., 2009). The advantage of speeding conduction has received experimental validation in experiments in which a skeletal muscle Na+ channel was overexpressed in the canine subacute myocardial infarct model (Lau et al., 2009; Coronel et al., 2010). One concern regarding improving cell–cell connectivity comes from studies showing that a decrease in Cx43 (Kanno et al., 2003) causes a decrease in infarct size whereas a loss of the Cx43 regulatory carboxyl domain in the mouse (Maass et al., 2009) causes an increase in infarct size. This suggests that closing gap junctions may be cardioprotective. Based on these studies, we hypothesized that increasing cell coupling following coronary occlusion would produce larger infarcts. If this is the case then such an intervention might show limited or no antiarrhythmic efficacy or greater arrhythmogenesis while conferring the risks attendant on a larger infarct.
To test our hypothesis we overexpressed connexin32 (Cx32), which is primarily found in non-electrically excitable cells such hepatocytes, oligodendrocytes, and Schwann cells, in the heart. Cx43 channels are voltage dependent (closing under depolarizing conditions) and pH sensitive (closing when the tissue pH becomes lower than pH 6.5; Stergiopoulos et al., 1999) such that ischemia causes rapid channel closure. In contrast, Cx32 channels are non-voltage dependent and non-pH dependent. These channels should remain open in ischemic myocardium due to their decreased pH sensitivity. We examined infarct size in adenoviral-Cx32 injected wild-type (WT) mice (Cx32inj) and used program stimulation to determine if connexin addition was antiarrhythmic. We found that infarct size was increased with no change in ventricular tachycardia (VT) inducibility in Cx32inj mice, suggesting that the increase in infarct size may offset any potential antiarrhythmic benefit. This suggests that caution is warranted when considering using connexin replacement or connexin agonist therapies to decrease arrhythmias in the post-MI heart.
Materials and Methods
All protocols conformed to The Public Health Service Policy on Humane Care and Use of Laboratory Animals and were reviewed and approved by the Columbia University Animal Care and Use Committee.
Cx32 Adenovirus Preparation
We inserted murine Cx32 into the pDC516 shuttle vector (Microbix, Toronto, Canada) and prepared an adenovirus from this transgene using the Admax system (Microbix) and HEK293 cells. After plaque purification, total DNA was extracted from infected cells (Qiagen, Valencia, CA, USA). The cDNA transgene was amplified by polymerase chain reaction and sequenced to confirm the absence of mutations. The titer was determined using fluorescent focus assay with mouse anti-adenosine virus antiserum (Advanced ImmunoChemical, Long Beach, CA, USA) and goat anti-mouse antiserum (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Western Blot Analysis
As described previously (Ozgen et al., 2007), tissues were chopped and sonicated in two 15 s bursts in lysate buffer (mM) Tris–HCl 20 (pH 7.4), EDTA 10, sodium orthovanadate 0.04, benzamidine 3.2, phenylmethylsulfonyl fluoride 0.1, 1% Triton X-100, and complete proteinase inhibitor (Roche) and incubated on ice for 30 min. After centrifugation at 10,000 rcf for 10 min supernatants were collected and protein concentration was measured using Bio-Rad protein assay. Forty micrograms of whole cell lysate was separated on a 4–20% Tris–glycine gradient gel (Invitrogen) and transferred to PVDF membranes (Bio-Rad). Membranes were blocked for 1 h in 5% milk in phosphate buffered saline containing 0.1% Tween 20 (PBST) then incubated with anti-Cx43 rabbit polyclonal antibody (Zymed Lab., 1:1000) or anti-Cx32 rabbit polyclonal antibody (Sigma, 1:250) in 5% BSA/PBST overnight at 4°C. Membranes were washed in PBST three times for 5 min and anti-rabbit TrueBlot system (eBioscience, 1:2000) secondary antibody was used to eliminate interference of heavy and light chain endogenous IgG in 5% milk in PBST. Membranes were washed for three times for 5 min in PBST. Immunodetection was performed with an enhanced chemiluminescence method (Amersham Pharmacia Biotech) and developed using X-ray film (Amersham Pharmacia Biotech). Data were normalized to beta Actin as a loading control (Abcam Inc., 1:1000).
In situ Scrape Loading
To determine if functional gap junction channels remained open in Cx32inj hearts following coronary occlusion we performed an in situ dye spread assay in the intact heart using Lucifer yellow (LY) and Texas red (TR). Scrape loading was performed on WT control hearts (n = 3), sham injected hearts following coronary occlusion (n = 4) and Cx32inj hearts following coronary occlusion (n = 5). LY readily passes through gap junctions thus the extent of LY spread can be used as an indication of gap junctional function. TR cannot pass through gap junction channels, and therefore only cells with punctured membranes are marked. For these experiments the hearts were removed and placed in warm saline and a 26-gage needle was used to puncture the peri-infarct region (as determined by tissue blanching) on the left ventricle. A mixture of 0.5% LY and 0.5% TR in 150 mM LiCl was infused into the hole. Following a 15-min incubation, the hearts were rinsed in Phosphate Buffered Saline, fixed in 4% paraformaldehyde for 30 min, and frozen in liquid nitrogen in OTC. Samples were sectioned to 10 μM (Microm HM505E), air dried, and fixed with 4% paraformaldehyde in PBS for 30 min at room temperature. Sections were imaged on a Leica DM5500 epifluorescence microscope. Dye spread was measured using NIH Image J. Degree of coupling was determined as the LY spread minus the TR spread (mm of LY spread − mm of TR spread).
Murine Infarct Preparation
Adult mice were anesthetized (ketamine 90 mg/kg plus xylazine 10 mg/kg i.p.) and fixed in the supine position on a heating pad. Intubation was attained by direct laryngoscopy and the outer sheath of a 24-gage intravenous catheter was placed into the distal trachea. The catheter was then connected to a rodent ventilator for continuous mechanical ventilation (respiratory rate 120/min with a tidal volume of 0.5 ml, Harvard Apparatus Respirator, model 707). Rectal temperature was maintained within 35–37°C. A left thoracotomy was performed via the fourth intercostal space. All muscles overlying the intercostal space were dissected free and retracted; only the intercostals muscles were transected. The heart was exposed and an 8-nylon suture was passed using a tapered needle around the left coronary artery 1–2 mm from the tip of the left atrium (Michael et al., 1995). Infarction was evident from discoloration of the ventricle. The chest and skin were closed with 6-0 sutures, the endotracheal tube was withdrawn, and the animals were allowed to recover (Lutgens et al., 1999). WT animals were injected with 25 μl of saline containing 1.6 × 109 ffu of either empty adenovirus (Sham) or adenoviral constructs expressing Cx32 at a single location into the anterior left ventricular wall approximately 5 min prior to the ligation of the coronary artery. Expression of Cx32 was observed following injection into the ventricle as confirmed in western blots following left coronary artery ligation. WT and Cx32inj experimental animals were re-anesthetized 4 days later and hearts were rapidly removed for scrape loading experiments or to determine the inducibility of VT.
Infarct Size Determination
Infarct sizing was performed as previously described (Lau et al., 2009). Briefly, hearts were removed and snap frozen in liquid nitrogen and placed in PBS cooled to 4°C. The heart was cut into transverse slices from apex to base using a vibratome (Leica VT 1000S) and incubated for 2 h in 1% tetrazolium red (pH, 7.4 buffer at 37°C). The tissue was immersed in 10% formalin for 15 min and pressed between two glass plates to obtain uniform thickness. Apical and basal sides of slices were photographed, and a digital image was obtained. Planimetry was used to determine overall infarct size (Image J, NIH Shareware). The volume of infarcted myocardium was expressed as percentage of total ventricular volume.
Electrophysiology Studies
Animals were re-anesthetized 4 days after surgery, and electrodes were placed to record the standard limb electrocardiogram throughout the experiments. An octapolar 1.1°F catheter (Scisense, Canada) was inserted into the right ventricle through a right external jugular vein cutdown (Sabada et al., 2000). Ventricular effective refractory period (ERP) was determined by introducing extrastimuli following a 10-beat S1-S1 train with a basic cycle length of 125 ms. To induce ventricular arrhythmias, three extrastimuli with a cycle length slightly longer than the ERP were applied after each train. Cycle length of extrastimuli was progressively shortened in steps of 1 ms. If VT (VT-five or more ventricular beats) was not inducible then the identical procedure was repeated with 6 and 10 extrastimuli.
Statistical Analysis
Infarct size and western blot results were compared using a Student’s t-test. Dye spread was compared using one-way ANOVA with Fishers test for least significant difference. Three separate sections from each animal were included in the dye spread analysis and data is represented as the mean, with error bars indicating standard error of the mean. Differences were considered to be significant at p < 0.05. VT inducibility was compared using Fisher’s exact test. Differences were considered to be significant at p < 0.05.
Results
Confirmation of the Presence of the Cx32 Construct in the Ventricular Myocardium
To determine if Cx32 was present in Cx32inj mice, we examined the ventricles from Cx32inj mice. Cx32 was absent in WT mice and was present in the ventricles of Cx32inj mice at 1.27 (AU; Figures 1A,B).
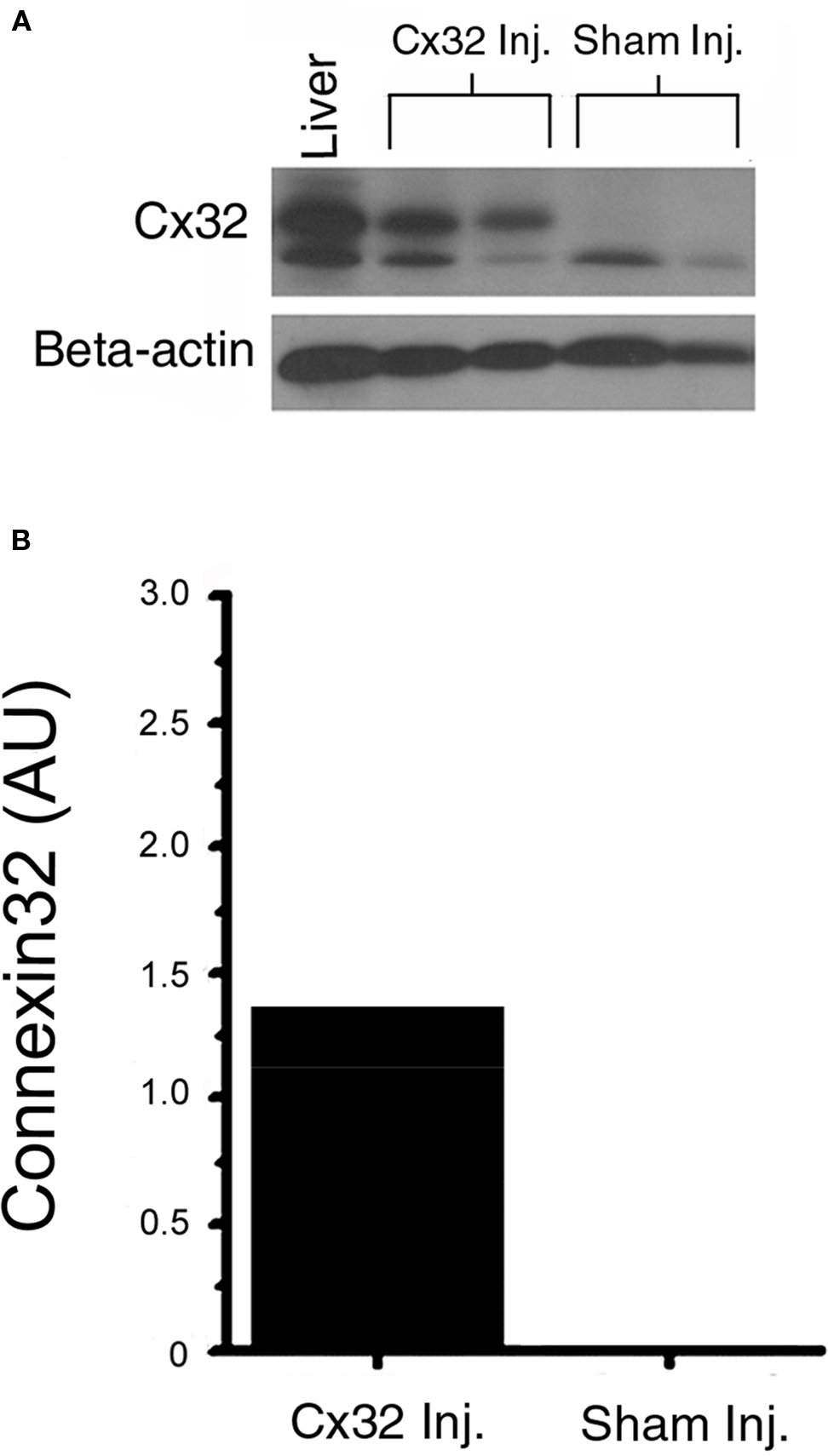
Figure 1. (A) Western blot analysis of Cx32 protein. Lysates of mouse ventricles from wild-type and Cx32 adenoviral vector-injected wild-type mice were run on 4–20% Tris–glycine gradient gels using mouse liver as a positive control for Cx32. (B) The data confirm the presence of Cx32 in injected wild type ventricles (Cx32 Inj), and the absence of Cx32 in the sham-injected ventricles (Sham Inj).
Determination of Cx32 Function in Cx32inj Mice Following Coronary Occlusion
Examination of LY spread in WT mice showed extensive dye spread within the left ventricle while TR labeling was only found in cells directly adjacent to the puncture site (dark core in the middle of the dye spread image). Quantification of the overlay of the two dyes indicated that LY spread extended several cells past the TR labeled cells indicating a high level of coupling between these myocytes (Figure 2, Control). In contrast, WT hearts following coronary occlusion showed very little dye spread of LY or of TR with almost complete colocalization indicating that in WT mice following coronary occlusion there is very little coupling between the myocytes (Figure 2, Sham Inj). The Cx32inj mice, on the other hand, showed extensive LY dye spread even following coronary occlusion well past the TR labeled cells. This is similar to what was seen in control WT mice (Figure 2, Cx32 Inj). These data show that the Cx32 channels in the hearts maintained coupling following coronary occlusion.
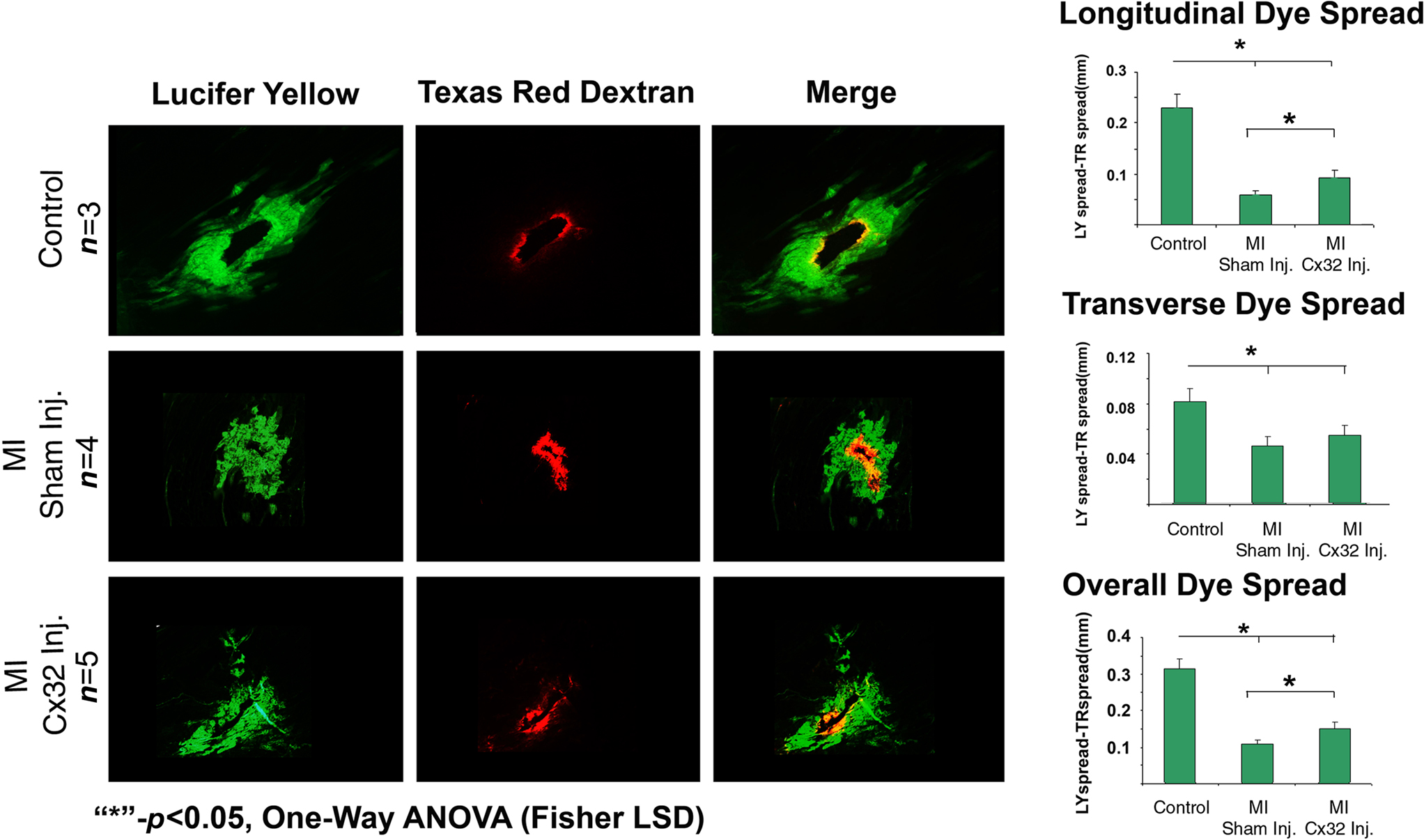
Figure 2. Determination of the function of Cx32 in the MICx32inj hearts. Dye spread studies show that wild-type hearts (Control) pass LY dye well past the RD markers (RD) indicating that these hearts exhibit high levels of coupling (Overlay). Following coronary occlusion LY is mostly confined to the cells which also contain RD indicating a total loss of gap junctional coupling. In contrast, the Cx32inj hearts still exhibit high levels of coupling even after coronary occlusion (MI Cx32 Inj). These data show that addition of Cx32 to the heart rescues the loss of coupling phenotype observed following coronary occlusion in wild-type mice.
Infarct Size
To determine the size of infarcts created by coronary ligation in WT and Cx32inj hearts, transverse slices of tetrazolium red stained hearts, which depict volume of infarcted myocardium, were measured and expressed as a percentage of volume of infarcted myocardium to total ventricular myocardial volume (Figure 3A). Examination of infarct size in Cx32inj hearts compared to WT showed that the presence of Cx32 correlated with an increase in infarct size. Infarct size was larger in Cx32inj mice vs WT mice (Cx32inj 44.3 ± 5%; WT 22.9 ± 4%; respectively; p < 0.05; Figure 3B).
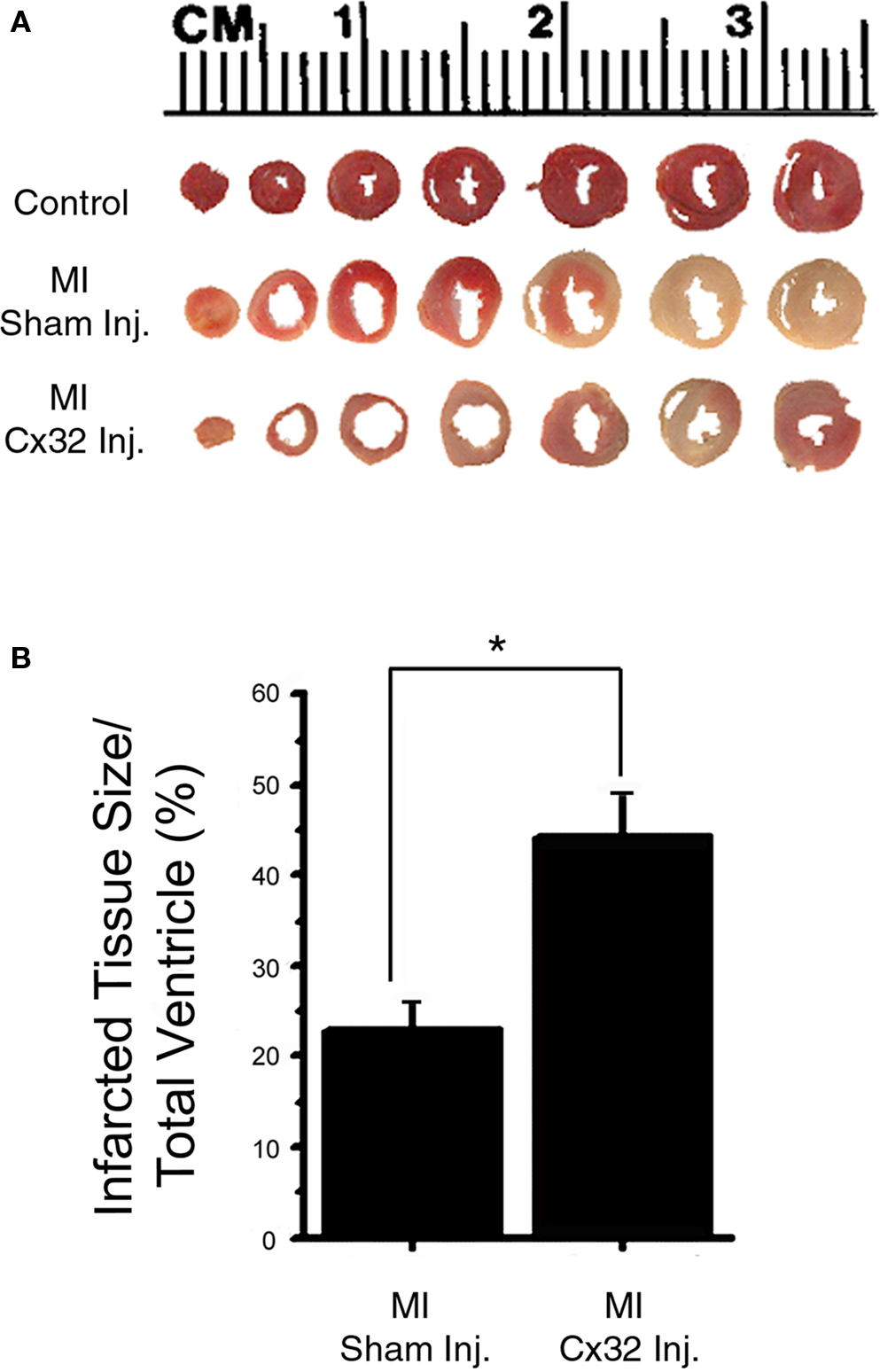
Figure 3. Infarct size analysis. (A) Transverse slices of tetrazolium red-stained wild-type (Control), wild-type with coronary occlusion (MI Sham-Inj), and Cx32 adenoviral vector-injected wild-type with coronary occlusion (MI Cx32 Inj) hearts, depicting volume of infarcted myocardium. Non-infarcted tissue stains red with tetrazolium, whereas infarcted tissue remains unstained. (B) Quantification of infarcted tissue volume shows an increase in infarct size in the Cx32 adenoviral vector-injected wild-type heart.
Electrophysiology Studies
Eleven shams and 10 Cx32inj mice were studied using programmed electrical stimulation following coronary occlusion. There was no significant difference in VT incidence: VT was reproducibly inducible in 55% of sham mice and 50% of the Cx32inj mice indicating that opening gap junctions was not antiarrhythmic in this setting. In those in which VT was induced, sham and Cx32inj mice required the same number of extra stimuli to induce VT. Ventricular fibrillation (VF) was not experimentally induced in the WT or Cx32inj mice. Experimental conditions for pacing-induced VF in mouse hearts vary among different studies. An important factor for VF inducibility is the size of the heart (so-called “critical mass”): the smaller the size of excitable myocardium the more difficult is VF induction. We used relatively small mice (∼20 g), whereas in the studies focused on VF induction 40–50 g mice were used (Chen et al., 2007). In addition, special interventions to facilitate VF induction have been employed, and include the use of isolated hearts (Vaidya et al., 1999), modification of ionic current, and application of pharmacological agents (Stables and Curtis, 2009). Even with particularly aggressive pacing protocols, VF incidence has been relatively low (Vaidya et al., 1999; Gehrmann et al., 2001).
Discussion
Following myocardial infarction gap junction channels close and Cx43 is lost, contributing to the formation of the arrhythmogenic substrate. Changes in localization and regulation of Cx43 during ischemic heart disease contribute to slowed conduction, unidirectional block, and formation of reentrant circuits (Peters et al., 1997; Kalcheva et al., 2007). Additionally, in a canine heart failure model, reduced Cx43 expression is associated with reductions in intercellular coupling leading to slower conduction and increased dispersion of repolarization, possibly contributing to an arrhythmic substrate (Poelzing and Rosenbaum, 2004). We now show that replacement of Cx43 with a non-voltage dependent, non-pH sensitive connexin isoform, Cx32 (Harris, 2001), increases cell coupling post-coronary occlusion while also producing larger infarcts. Because our data show that the Cx32 channels maintained coupling in hearts following coronary occlusion, we believe that the increase in infarct size is a result of the addition of Cx32. Additionally, in WT hearts LY dye spread was minimal indicating nominal coupling of myocytes. Mice expressing genetically engineered Cx43, that lack C-terminal regulation sites and maintain coupling during coronary occlusion, also have increased infarct size and enhanced susceptibility to arrhythmias (Maass et al., 2009). Cx32 channels do not pass apoptotic signals (see Discussion below) while Cx43 channels do, and therefore Cx32 could potentially provide antiarrhythmic effects without passage of cell death molecules which are believed to be the cause of increased infarct size.
Chordates have over 20 isoforms of connexin proteins with an approximate 40% sequence identity, with even greater homology in the transmembrane and extracellular domains. The different isoforms compose channels with diverse unitary conductances, permeabilities, voltage sensitivities, and sensitivities to modulatory substrates (Yan and Kléber, 1992). Cx43 has a unitary conductance of 100 pS, and is less charge selective than Cx32 (60 pS), although selectivity to small molecules is dependent on the size and hydration of the molecule in addition to its charge (Goldberg et al., 2002). One of the most relevant differences between Cx43 and Cx32 in the context of tissue ischemia is the difference in the effect of intracellular pH on junctional conductance. Following myocardial infarction the intracellular pH in papillary muscles of the heart has been reported to be 6.93 (Liu et al., 1993). Cx43 channel conductance is more sensitive to intracellular acidification than Cx32 with KD values around pH 6.7 as opposed to pH 5.9 for Cx32. Therefore, channels formed from Cx43 are likely to close under the pH conditions found in the post-ischemic heart, while Cx32 channels are likely to remain open. It has been theorized that the mechanism by which coupling increases infarct size is by passage of apoptotic signals through open channels (Cusato et al., 2003). Cx32 is unable to pass apoptotic signals and has a reduced permeability to several intracellular messengers such as: AMP, ADP, and ATP (Goldberg et al., 2002). Nonetheless Cx32 can replace the function of Cx43 regarding propagation of electrical impulses regardless of Cx43 and Cx32 protein-partners (Rodríguez-Sinovas et al., 2010). Our studies now suggest that passage of apoptotic signals is not necessary to cause increased infarct size; rather the presence of any open gap junction channel suffices. One possibility is that Cx32 expression rescues Cx43 channels. Based on our immunostaining, which shows no Cx43 at the IDs in the heterozygous mouse after MI we do not think that this is likely. However, if it did occur in very low levels it would be undetectable by our methods. Because our dye spread studies show that following MI Cx43 channels are closed, any small remaining rescue of Cx43 by Cx32 is unlikely to produce coupling. Cx32 is a low conducting pore as compared to Cx43 (60 vs 100 pS). Therefore less dye spread would be expected even if the number of coupled Cx32 channels were identical to the number of Cx43 channels. Additionally, we expressed Cx32 in levels that are lower than endogenous Cx43, and the combination of these two events would lead to less dye spread via Cx32 channels. With regard to electrophysiology, this would limit ion flux but not action potential propagation slowing, as the threshold for activation of a cardiac myocyte is lower than the Cx32 permeability. Additionally there were no differences in the arrhythmogenicity of these hearts indicating that addition of a connexin is not enough to rescue the arrhythmic phenotype. Hence, adding connexin proteins to the heart following coronary occlusion is not antiarrhythmic at this stage, and may actually increase the risk for later stage arrhythmias by aiding in formation of a larger scar.
We speculate that there are several potential explanations for the increase in infarct size with the presence of open Cx32 channels. It may be that, as opposed to what has been previously shown (Rodríguez-Sinovas et al., 2010) Cx32 does indeed pass apoptotic signals under the conditions of myocardial ischemia. While our studies cannot rule this out definitively we suggest this is unlikely. Studies in heterologous cell systems and in other tissues have shown that connexin channel properties are not altered by the system in which they are introduced (Schiller et al., 2001; Hanstein et al., 2009). The low Cx32 conductance to apoptotic signals in cells in culture (Andrade-Rozental et al., 2000) suggests Cx32 gap junctions cannot pass these signals in whole tissues. The second possibility is that the presence of Cx32 at the ID helps maintain Cx43 at the disk as well; Thus it would be the passage of apoptotic signals through these channels that causes the increase in infarct size. Our scrape loading data are not suggestive of maintenance of Cx43 protein at the ID, and moreover, the intracellular acidification of cells within the infarct zone would ensure that the channels would remain closed. In addition, Xenopus oocyte preparations expressing Cx43 fail to become electrically coupled to cells expressing Cx32 (White et al., 1995) indicating that this type of heteromerism is unlikely to occur. Being that Cx32 caused increased infarct size and we believe that the apoptotic signals are not passed via these channels the likely mechanism by which the spread of cell death occurs is via the electrical current. We believe the significant finding is that a connexin isoform that cannot spread apoptotic signals still increases infarct size. This leads to the new theory that electrical signals may cause cellular damage that then initiates cell death far from the initial site of injury.
Another possible mechanism for the spread of cell death is via maintenance of Ca2+ signaling between myocytes following coronary occlusion. Normal cardiac myocytes release calcium from intracellular stores leading to a calcium spike and activation of excitation–contraction (EC) coupling (Kranias and Bers, 2007). At the end of the EC cycle, excess calcium undergoes reuptake into the sarcoplasmic reticulum (SR) decreasing cytosolic calcium and allowing the cell to relax prior to the next contraction. This process is regulated by the SERCA system which involves a tightly regulated cascade of signals which switch the SERCA pumps off and on in response to intracellular calcium levels (White et al., 1995). In ischemic myocardium levels of SERCA are decreased, leading to incomplete calcium reuptake into the SR (White et al., 1995) and a concomitant increase in the level of intracellular calcium. This high calcium level then stimulates the activation of apoptotic pathways including opening of the mitochondrial permeability transition pore, and is followed by cell death (Moens et al., 2005). Based on this study we speculate that when gap junctions are closed in the infarct region, electrical signals are unable to pass from cell to cell thus limiting the spread of calcium-dependent cell death. The addition of a gap junction channel which does not close under ischemic conditions is likely to increase infarct size by increasing depolarization- dependent calcium release and subsequent cell death in areas that would have survived otherwise.
Initial identification of gap junction closure as a major mechanism for induction of life threatening arrhythmias suggested that therapeutically targeting Cx43 would decrease the incidence of arrhythmias. These studies were confounded by findings that lower levels of Cx43 corresponded to smaller infarct size post-MI (Gutstein et al., 2001) leading to the hypothesis that increasing Cx43 would increase infarct size. The study described here shows directly that a chronic increase in coupling increases infarct size, although the results do not rule out the possibility that temporary increases in coupling may provide protection from arrhythmias without increasing infarct size.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported in part by USPHS-NHLBI grants to Michael R. Rosen (HL-67101 and HL-094410) and to Heather S. Duffy (AHA SDG 0535084; NIH HL-083205 and HL-066140). We would also like to acknowledge Nancy Mutsaers for assistance in preparing the Cx32 adenovirus.
References
Andrade-Rozental, A. F., Rozental, R., Hopperstad, M. G., Wu, J. K., Vrionis, F. D., and Spray, D. C. (2000). Gap junctions: the “kiss of death” and the “kiss of life.” Brain Res. Rev. 32, 308–315.
Chen, M. H., Liu, T. W., Xie L., Song, F.Q., He, T., Mo, S. R., and Zeng Z. Y. (2007). A simpler cardiac arrest model in the mouse. Resuscitation 75, 372–379.
Coronel, R., Lau, D. H., Sosunov, E. A., Janse, M. J., Danilo, P. Jr., Anyukhovsky, E. P., Wilms-Schopman, F. J., Opthof, T., Shlapakova, I. N., Ozgen, N., Prestia, K., Kryukova, Y., Cohen, I. S., Robinson, R. B., and Rosen, M. R. (2010). Cardiac expression of skeletal muscle sodium channels increases longitudinal conduction velocity in the canine 1-week myocardial infarction. Heart Rhythm 7, 1104–1110.
Cusato, K., Bosco, A., Rozental, R., Guimarães, C. A., Reese, B. E., Linden, R., and Spray, D. C. (2003). Gap junctions mediate bystander cell death in developing retina. J. Neurosci. 23, 6413–6422.
Gehrmann, J., Frantz, S., Maguire, C. T., Vargas, M., Ducharme, A., Wakimoto, H., Lee, R. T., and Berul, C. I. (2001). Electrophysiological characterization of murine myocardial ischemia and infarction. Basic Res. Cardiol. 96, 237–250.
Goldberg, G. S., Moreno, A. P., and Lampe, P. D. (2002). Gap junctions between cells expressing connexin 43 or 32 show inverse permselectivity to adenosine and ATP. J. Biol. Chem. 277, 36725–36730.
Gutstein, D. E., Danik, S. B., Lewitton, S., France, D., Liu, F., Chen, F. L., Zhang, J., Ghodsi, N., Morley, G. E., and Fishman, G. I. (2005). Focal gap junction uncoupling and spontaneous ventricular ectopy. Am. J. Physiol. Heart Circ. Physiol. 289, 1091–1098.
Gutstein, D. E., Danik, S. B., Sereysky, J. B., Morley, G. E., and Fishman, G. I. (2003). Subdiaphragmatic murine electrophysiological studies: sequential determination of ventricular refractoriness and arrhythmia induction. Am. J. Physiol. Heart Circ. Physiol. 285, 1091–1096.
Gutstein, D. E., Morley, G. E., and Fishman, G. I. (2001). Conditional gene targeting of connexin43: exploring the consequences of gap junction remodeling in the heart. Cell Commun. Adhes. 8, 345–348.
Gutstein, D. E., Morley, G. E., Tamaddon, H., Vaidya, D., Schneider, M. D., Chen, J., Chien, K. R., Stuhlmann, H., and Fishman, G. I. (2001). Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ. Res. 88, 333–339.
Gutstein, D. E., Morley, G. E., Vaidya, D., Liu, F., Chen, F. L., Stuhlmann, H., and Fishman, G. I. (2001). Heterogeneous expression of gap junction channels in the heart leads to conduction defects and ventricular dysfunction. Circulation 104, 1194–1199.
Hagen, A., Dietze, A., and Dhein, S. (2009). Human cardiac gap-junction coupling: effects of antiarrhythmic peptide AAP10. Cardiovasc. Res. 83, 405–415.
Hanstein, R., Trotter, J., Behl, C., and Clement, A. B. (2009). Increased connexin 43 expression as a potential mediator of the neuroprotective activity of the corticotropin-releasing hormone. Mol. Endocrinol. 23, 1479–1493.
Harris, A. L. (2001). Emerging issues of connexin channels: biophysics fills the gap. Q. Rev. Biophys. 34, 325–472.
Kalcheva, N., Qu, J., Sandeep, N., Garcia, L., Zhang, J., Wang, Z., Lampe, P. D., Suadicani, S. O., Spray, D. C., and Fishman, G. I. (2007). Gap junction remodeling and cardiac arrhythmogenesis in a murine model of oculodentodigital dysplasia. Proc. Natl. Acad. Sci. U.S.A. 104, 20512–20516.
Kanno, S., Kovacs, A., Yamada, K. A., and Saffitz, J. E. (2003). Connexin43 as a determinant of myocardial infarct size following coronary occlusion in mice. J. Am. Coll. Cardiol. 41, 681–686.
Kieken, F., Mutsaers, N., Dolmatova, E., Virgil, K., Wit, A. L., Kellezi, A., Hirst-Jensen, B. J., Duffy, H. S., and Sorgen, P. L. (2009). Structural and molecular mechanisms of gap junction remodeling in epicardial border zone myocytes following myocardial infarction. Circ. Res. 104, 1103–1112.
Kranias, E. G., and Bers, D. M. (2007). Calcium and cardiomyopathies. Subcell. Biochem. 45, 523–537.
Lau, D. H., Clausen, C., Sosunov, E. A., Shlapakova, I. N., Anyukhovsky, E. P., Danilo, P. Jr., Rosen, T. S., Kelly, C., Duffy, H. S., Szabolcs, M. J., Chen, M., Robinson, R. B., Lu, J., Kumari, S., Cohen, I. S., and Rosen, M. R. (2009). Epicardial border zone overexpression of skeletal muscle sodium channel SkM1 normalizes activation, preserves conduction, and suppresses ventricular arrhythmia: an in silico, in vivo, in vitro study. Circulation 119, 19–27.
Liu, S., Taffet, S., Stoner, L., Delmar, M., Vallano, M. L., and Jalife, J. (1993). A structural basis for the unequal sensitivity of the major cardiac and liver gap junctions to intracellular acidification: the carboxyl tail length. Biophys. J. 64, 1422–1433.
Lutgens, E., Daemen, M. J., de Muinck, E. D., Debets, J., Leenders, P., and Smits, J. F. (1999). Chronic myocardial infarction in the mouse: cardiac structural and functional changes. Cardiovasc. Res. 41, 586–593.
Maass, K., Chase, S. E., Lin, X., and Delmar, M. (2009). Cx43 CT domain influences infarct size and susceptibility to ventricular tachyarrhythmias in acute myocardial infarction. Cardiovasc. Res. 84, 361–367.
Michael, L. H., Entman, M. L., Hartley, C. J., Youker, K. A., Zhu, J., Hall, S. R., Hawkins, H. K., Berens, K., and Ballantyne, C. M. (1995). Myocardial ischemia and reperfusion: a murine model. Am. J. Physiol. 269, 2147–2154.
Moens, A. L., Claeys, M. J., Timmermans, J. P., and Vrints, C. J. (2005). Myocardial ischemia/reperfusion-injury, a clinical view on a complex pathophysiological process. Int. J. Cardiol. 100, 179–190.
Ozgen, N., Dun, W., Sosunov, E. A., Anyukhovsky, E. P., Hirose, M., Duffy, H. S., Boyden, P. A., and Rosen, M. R. (2007). Early electrical remodeling in rabbit pulmonary vein results from trafficking of intracellular SK2 channels to membrane sites. Cardiovasc. Res. 75, 758–769.
Peters, N. S., Coromilas, J., Severs, N. J., and Wit, A. L. (1997). Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation 95, 988–996.
Poelzing, S., and Rosenbaum, D. S. (2004). Altered connexin43 expression produces arrhythmia substrate in heart failure. Am. J. Physiol. Heart Circ. Physiol. 287, 762–770.
Rodríguez-Sinovas, A., Sánchez, J. A., González-Loyola, A., Barba, I, Morente, M., Aguilar, R., Agulló, E., Miró-Casas, E., Esquerda, N., Ruiz-Meana, M., and García-Dorado, D. (2010). Effects of substitution of Cx43 by Cx32 on myocardial energy metabolism, tolerance to ischaemia and preconditioning protection. J. Physiol. 588, 1139–1151.
Roell, W., Lewalter, T., Sasse, P., Tallini, Y. N., Choi, B. R., Breitbach, M., Doran, R., Becher, U. M., Hwang, S. M., Bostani, T., von Maltzahn, J., Hofmann, A., Reining, S., Eiberger, B., Gabris, B., Pfeifer, A., Welz, A., Willecke, K., Salama, G., Schrickel, J. W., Kotlikoff, M. I., and Fleischmann, B. K. (2007). Engraftment of connexin 43-expressing cells prevents post-infarct arrhythmia. Nature 450, 819–824.
Sabada, S., Wang, P. J., and Estes, N. A. (2000). Invasive cardiac electrophysiology in the mouse: techniques and applications. Trends Cardiovasc. Med. 10, 122–132.
Schiller, P. C., D’Ippolito, G., Balkan, W., Roos, B. A., and Howard, G. A. (2001). Gap-junctional communication mediated parathyroid hormone stimulation of mineralization in osteoblastic cultures. Bone 28, 38–44.
Shaw, R. M., Fay, A. J., Puthenveedu, M. A., von Zastrow, M., Jan, Y. N., and Jan, L. Y. (2007). Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell 128, 547–560.
Stables, C. L., and Curtis, M. J. (2009). Development and characterization of a mouse in vitro model of ischaemia-induced ventricular fibrillation. Cardiovasc. Res. 83, 397–404.
Stergiopoulos, K., Alvarado, J. L., Mastroianni, M., Ek-Vitorin, J. F., Taffet, S. M., and Delmar, M. (1999). Hetero-domain interactions as a mechanism for the regulation of connexin channels. Circ. Res. 84, 1144–1155.
Vaidya, D., Morley, G. E., Samie, F. H., and Jalife, J. (1999). Reentry and fibrillation in the mouse heart. A challenge to the critical mass hypothesis. Circ Res. 85, 174–181.
White, T. W., Paul, D. L., Goodenough, D. A., and Bruzzone, R. (1995). Functional analysis of selective interactions among rodent connexins. Mol. Biol. Cell 6, 459–470.
Keywords: connexin43, connexin32, myocardial infarction, infarct size, antiarrhythmic therapy, coronary occlusion
Citation: Prestia KA, Sosunov EA, Anyukhovsky EP, Dolmatova E, Kelly CW, Brink PR, Robinson RB, Rosen MR and Duffy HS (2011) Increased cell–cell coupling increases infarct size and does not decrease incidence of ventricular tachycardia in mice. Front. Physio. 2:1. doi: 10.3389/fphys.2011.00001
Received: 22 October 2010;
Accepted: 04 January 2011;
Published online: 31 January 2011.
Edited by:
Jiashin Wu, University of South Florida, USAReviewed by:
Tobias Opthof, Academic Medical Center, NetherlandsCraig Doupnik, University of South Florida College of Medicine, USA
Copyright: © 2011 Prestia, Sosunov, Anyukhovsky, Dolmatova, Kelly, Brink, Robinson, Rosen and Duffy. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Heather S. Duffy, Beth Israel Deaconess Medical Center, Harvard Medical School, Center for Life Sciences, CLS 913, 3 Blackfan Circle, Boston, MA 02115, USA. e-mail:aGR1ZmZ5QGJpZG1jLmhhcnZhcmQuZWR1