 James A. Loehr
James A. Loehr Reem Abo-Zahrah
Reem Abo-Zahrah Rituraj Pal
Rituraj Pal George G. Rodney
George G. Rodney- Department of Molecular Physiology and Biophysics, Baylor College of Medicine, Houston, TX, USA
Elevated concentrations of sphingomyelinase (SMase) have been detected in a variety of diseases. SMase has been shown to increase muscle derived oxidants and decrease skeletal muscle force; however, the sub-cellular site of oxidant production has not been elucidated. Using redox sensitive biosensors targeted to the mitochondria and NADPH oxidase (Nox2), we demonstrate that SMase increased Nox2-dependent ROS and had no effect on mitochondrial ROS in isolated FDB fibers. Pharmacological inhibition and genetic knockdown of Nox2 activity prevented SMase induced ROS production and provided protection against decreased force production in the diaphragm. In contrast, genetic overexpression of superoxide dismutase within the mitochondria did not prevent increased ROS production and offered no protection against decreased diaphragm function in response to SMase. Our study shows that SMase induced ROS production occurs in specific sub-cellular regions of skeletal muscle; however, the increased ROS does not completely account for the decrease in muscle function.
Introduction
A variety of chronic diseases such as chronic heart failure (Doehner et al., 2007; Empinado et al., 2014), inflammatory disease (Wong et al., 2000), and sepsis (Okazaki et al., 2014) have been correlated with muscle weakness. Shingomyelinase (SMase), an enzyme involved in sphingolipid metabolism, has been shown to be elevated during these conditions (Wong et al., 2000; Claus et al., 2005; Empinado et al., 2014). Sphingolipids are abundant in skeletal muscle and their metabolites affect a wide variety of molecular processes (Nikolova-Karakashian and Reid, 2011). SMase induced sphingolipid metabolism results in an increase in ceramide production which has been shown to decrease force production and increase fatigue and muscle atrophy (Ferreira et al., 2010, 2012; De et al., 2012; Empinado et al., 2014).
Reactive oxygen species (ROS) have been shown to play a key role in modulating muscle function (Reid et al., 1993). In contracting skeletal muscle, mitochondria have traditionally been thought to be the major source of ROS; however, more recent evidence indicates that NADPH Oxidase (Nox2) may play a predominant role in ROS generation (Michaelson et al., 2010; Pal et al., 2013, 2014b; Sakellariou et al., 2013, 2014). In non-muscle cells, SMase has been shown to increase both mitochondrial and Nox2-dependent ROS (Sawada et al., 2004; Reinehr et al., 2005); while in skeletal muscle the mitochondria appear to be the primary source (Ferreira et al., 2012).
Until recently, there has been no reliable method to determine the sub-cellular origins of ROS. Traditionally, dichlorofluorescein (DCFH), in combination with various antioxidants, has been used to assess intracellular ROS production and extrapolate oxidant origin. While DCFH is useful for measuring general cytosolic ROS production, it is an irreversible dye that cannot differentiate between subcellular sources of ROS or determine redox state within the cell. Therefore, redox-sensitive green fluorescent probes (roGFP) with an artificial dithiol-disulfide pair inserted into the GFP (Hanson et al., 2004) were developed to function as reversible redox sensors and evaluate the local redox balance within the cell (Dooley et al., 2004; Hanson et al., 2004; Pal et al., 2013). We have shown that ro-GFPs specifically targeted to Nox2 (p47-roGFP) or the mitochondria (mito-roGFP) allow for measurement of site-specific ROS production within skeletal muscle (Michaelson et al., 2010; Pal et al., 2013, 2014a). Therefore, the aim of this project was to determine the specific subcellular site of ROS production in response to SMase stimulation. Our data indicate that Nox2 is a major contributor to ROS production in skeletal muscle (Michaelson et al., 2010; Pal et al., 2013, 2014b; Sakellariou et al., 2013, 2014); therefore,we hypothesized that SMase would induce Nox2-specific ROS and impair muscle function.
Materials and Methods
In-vivo Electroporation
C57Bl/6J wild type (WT) and Nox2 knockout (Nox2−/y) were purchased from Jackson Laboratories (Bar Harbor, ME) and bred following their breeding strategy. Mitochondrial SOD (MnSOD) transgenic mice were a kind gift from Dr. Pautler and were genotyped by PCR as previously described to verify transgene overexpression (Massaad et al., 2009). Redox sensitive GFP probes were transfected into the flexor digitorum brevis (FDB) of male WT, Nox2−/y, and MnSOD mice as previously described (Michaelson et al., 2010) and in accordance with National Institutes of Health guidelines and with the approval of the Institutional Animal Care and Use Committee of Baylor College of Medicine. Briefly, mice between 8 and 12 weeks of age were anesthetized with isoflurane (2%) and hyaluronidase (0.5 mg/ml) dissolved in sterile saline was injected subcutaneously into the foot pad of both hindlimb feet. Two hours later each foot was injected with 20–30 μg of rDNA in PBS; typically the right foot with p47-roGFP and the left foot with mito-roGFP. Two electrodes were placed subcutaneously at the proximal and distal FDB tendons to deliver 20 pulses of 150 V, 20 ms in duration at a frequency of 1 Hz with a square pulse stimulator (S48; Grass Technologies, West Warwick, RI). Mice were returned to their home cage and FDB muscle fibers were isolated 6–8 days later.
Isolation of FDB Fibers
Mice were deeply anesthetized by isofluorane (2%) inhalation and euthanized by rapid cervical dislocation. FDB muscles were surgically isolated and incubated at 37°C in minimal essential media containing 1% Pen Strep (Life Technologies, Grand Island, NY) and 0.4% Collagenase A (Roche Applied Science, Indianapolis, IN) for 2.0 h. FDB muscles were transferred to a serum containing media (10%, Atlanta Biologicals) without collagenase and triturated gently to release single fibers. Single fibers were then incubated in 21% O2/5% CO2 at 37°C until used, typically 12–36 h later.
ROS Measurements
Prior to experiments, fibers were plated in 96 well dishes (Greiner Bio One, Monroe, NC) on ECM gel from Engelbreth-Holm-Swarm murine sarcoma (Sigma, St. Louis, MO). Intracellular ROS, in unstimulated FDB fibers, was measured using 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) (Invitrogen, Carlsbad, CA) or electroporated site-specific sub-cellular redox sensitive ROS probes targeted to Nox2 (p47-roGFP) or the mitochondria (mito-roGFP). All FDB fibers were imaged at baseline (no drug) and then incubated for 30 min at 37°C with either vehicle control (0.13% Glycerol final) or SMase (0.5 U/ml). Cells were imaged after 30 min, placed back in the incubator for another 30 min and imaged again at 60 min. To obtain maximal oxidation, p47-roGFP and mito-roGFP electroporated fibers were exposed to 100 μM H2O2 for 5 min followed by dithiothreitol (DTT, 10 mM) to obtain maximum reduction. Regions of interest (ROI) were drawn on the cell and in the background. Ratio images (403/470) were created by subtracting the mean background ROI from the mean cell ROI.
DCFH-DA Loading and Treatment of Single FDB Fibers
Plated FDB fibers were washed with 4-(2-Hydroxyethyl) piperazine-1-ethanesulfonic acid (HEPES) buffered Ringer's solution containing (in mM): 146 NaCl, 4.7 KCl, 0.6 MgSO4, 1.8 CaCl, 1.6, NaHCO3, 0.13 NaH2PO4, 7.8 glucose, 20.0 HEPES, pH 7.3 and incubated with DCFH-DA (5 μM) for 30 min at 37°C. The fibers were then washed with Ringer's solution and the dye was allowed to de-esterify for 20 min at 37°C prior to fluorescence microscopy. To prevent light induced oxidation of DCFH, all cell-loading and imaging was performed in the dark. In a subset of conditions, prior to DCFH-DA incubation, cells were incubated with a Nox2 peptide inhibitor, gp-91 ds (5 μM; Biosynthesis Inc., Lewisville, TX), for 45 min at 37°C.
Microscopy
A Sutter Lamda DG-5 Ultra high speed wavelength switcher was used to excite DCF (480 nm) and ro-GFP (403/12 nm and 470/20 nm) fluorescence. DCFH-DA and ro-GFP emission intensity was collected at 510 nm and 535/48 nm, respectively, on a charge coupled device (CCD) Camera (CoolSNAP MYO, Photometrics, Tucson, AZ) attached to an Axio Observer (Zeiss) inverted microscope (40× H2O objective, 1.2 NA) at a rate of 0.1 Hz.
Ex vivo Force Measurements
Diaphragm muscle was surgically dissected from mice and sectioned into diaphragm strips with one end attached to a fixed hook and the other to a force transducer (F30, Harvard Apparatus) using silk suture (4-0) in a physiological saline solution continuously gassed with 95% O2–5% CO2 at 25°C. Diaphragm strips were incubated at 37°C for 15 min and optimal muscle length (Lo) and voltage (Vmax) were adjusted to elicit maximum twitch force. SMase (0.5 U/ml) or vehicle control (0.13% glycerol final) was administered to the muscle bath and force at 300 Hz was monitored over time at 0, 30, and 60 min with pulse and train durations of 0.5 and 400 ms, respectively. After 60 min of incubation force-frequency characteristics were measured at stimulation frequencies of 1, 5, 10, 20, 40, 60, 80, 120, 150, 200, and 300-Hz every minute with pulse and train durations of 0.5 and 250 ms. Following a 5 min rest period fatigue was assessed using a matched force protocol in which frequency was adjusted so force generated by drug treated tissue matched the force of vehicle control at 40 Hz with pulse and train durations of 0.5 and 400 ms and a train rate of 0.5 Hz. ROS has been suggested to decrease force by altering Ca2+ sensitivity; therefore a frequency on the steep part of the force frequency relationship (40 Hz) was chosen to determine if inhibiting SMase induced ROS protected against decreased muscle force. At the end of the contractile protocol muscle length was measured using a hand-held electronic caliper, fiber bundles were removed from the bath and trimmed of excess bone and connective tissue, blotted dry, and weighed. Muscle weight and Lo were used to estimate cross-sectional area and absolute forces expressed as N/cm2 (Close, 1972).
Data Analysis
Data are reported as mean ± SEM, unless otherwise specified. A One-Way RM ANOVA was used to measure statistical differences within a group and a Two-Way RM ANOVA was used for between group differences. Tukey's post-hoc test was used when statistical differences were identified. Statistical analysis was performed in Origin Pro (OriginLab Corporation, Northhampton, MA) with significance set a priori at p ≤ 0.05.
Results
Effect of SMase Stimulation on General ROS Production
To assess the role of Nox2 in general non-specific intracellular ROS production, we measured changes in DCF fluorescence in FDB fibers in the presence or absence of the Nox2 specific peptide inhibitor gp91-ds (Rey et al., 2001; Csanyi et al., 2011; Pal et al., 2013). SMase resulted in an increase (p ≤ 0.05) in DCF fluorescence at 30 min and a further increase at 60min, which was inhibited by pre-incubation with gp91-ds (Figure 1). There was no change in control fibers over the same time. These data suggest that in skeletal muscle, SMase stimulates Nox2 dependent ROS production.
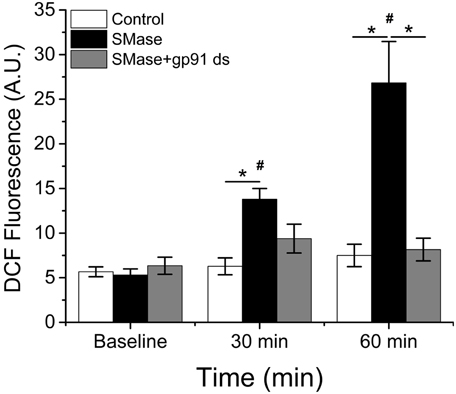
Figure 1. SMase increases ROS production through Nox2 in FDB fibers. SMase increased DCF fluorescence in WT FDB fibers (black bars), which could be completely inhibited upon pre-incubation with gp91-ds (gray bars). There was no time-dependent change in the absence of SMase (white bars). p ≤ 0.05 *significantly different from vehicle treated control, #significantly different from baseline in at least nanimals = 3, ncells = 31.
Subcellular ROS Sensors Targeted to NADPH Oxidase (Nox2) and the Mitochondria
While our data indicate a role for Nox2 in SMase induced ROS production, others have indicated a potential role for mitochondrial ROS generation (Sawada et al., 2004; Nikolova-Karakashian and Reid, 2011; Ferreira et al., 2012). Therefore, we used redox sensitive GFP probes targeted to Nox2 (p47-roGFP) and the mitochondria (mito-roGFP) to determine the localization of subcellular ROS production. Similar to DCFH, in the absence of SMase, we saw no time dependent effects of ROS production using the p47-roGFP or mito-roGFP ROS sensors (data not shown). SMase exposure increased Nox2 derived ROS following 30 and 60 min (Figure 2C), while it had no effect on increasing mitochondrial oxidant burden (Figure 2D). Representative images demonstrate the change in fluorescence of both p47-roGFP (Figure 2A) and mito-roGFP (Figure 2B) in response to SMase. The use of site-specific redox probes further confirms ROS is produced through Nox2, with no additional mitochondrial contribution.
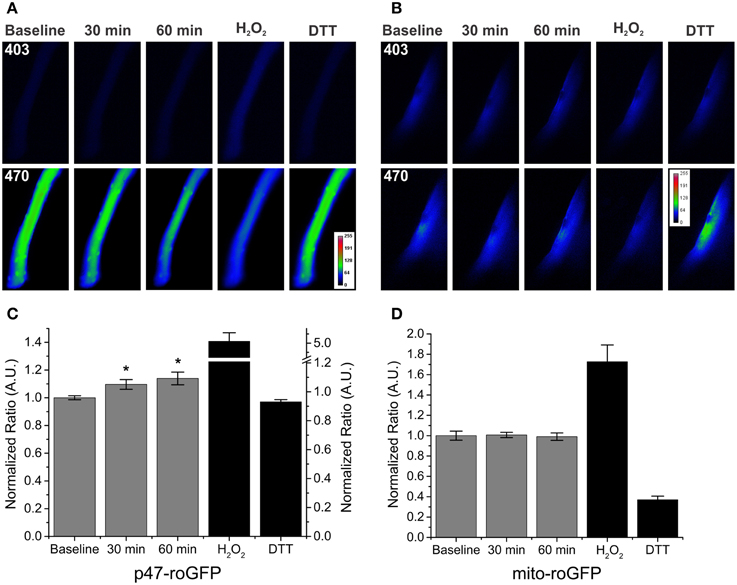
Figure 2. SMase increased Nox2 ROS production in WT FDB fibers with no effect on mitochondrial ROS. (A,B) Representative images of SMase induced changes in p47-roGFP and mito-roGFP fluorescence, respectively. (C) Nox2 ROS (p47-roGFP) in FDBs from WT animals was elevated following SMase at both 30 and 60 min. H2O2 and DTT (100 μM and 10 mM, black bars) resulted in further oxidation and reduction of p47-roGFP. (D) Mitochondrial ROS (mito-roGFP) did not change in response to SMase. H2O2 and DTT (100 μM and 10 mM, black bars) resulted in further oxidation and reduction of mito-roGFP. Fluorescence values were normalized to non-drug treated baseline measurements. p ≤ 0.05 *significantly different from baseline in at least nanimals = 4, ncells = 11.
Genetic Ablation of Nox2 Prevents Increased ROS Production
Our data indicate that Nox2 plays a central role in SMase induced ROS production in skeletal muscle. To further confirm the role of Nox2 we assessed the effects of SMase stimulated ROS production in FDB fibers from Nox2 deficient (Nox2−/y) mice. SMase induced oxidation of p47-roGFP was completely abolished (Figure 3A) and, consistent with our previous findings, there was no change in the oxidation of mito-roGFP (Figure 3B). The lack of ROS production in Nox2−/y animals further supports the role of Nox2 as the source of ROS production in adult skeletal muscle exposed to SMase.
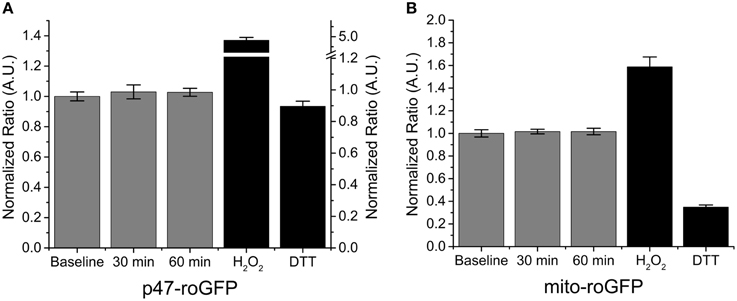
Figure 3. Genetic deletion of Nox2 prevents SMase induced ROS. (A) Nox2 ROS (p47-roGFP) was not altered with SMase in FDBs from Nox2−/y mice. (B) Mitochondrial ROS (mito-roGFP) did not change in response to SMase. H2O2 and DTT (100 μM and 10 mM, black bars) resulted in further oxidation and reduction of both p47-roGFP and mito-roGFP. Fluorescence values were normalized to non-drug treated baseline measurements in at least nanimals = 4, ncells = 12.
MnSOD Overexpression does not Prevent SMase Induced ROS Production
Previous data have shown increased mitochondrial ROS in response to SMase stimulation (Ferreira et al., 2012). Genetically overexpressing mitochondrial superoxide dismutase (MnSOD) in mice has been shown to reduce contraction induced mitochondrial ROS in skeletal muscle (McClung et al., 2010); therefore, we next wanted to determine if MnSOD overexpression had any effect on ROS production in response to SMase. A representative DNA gel image, verifying MnSOD transgene overexpression, is shown in Figure 4A. In FDBs from MnSOD overexpressing mice, SMase induced an increase in Nox2 (p47-roGFP) ROS production (Figure 4B), while no difference was detected in mitochondrial ROS production (Figure 4C). These results provide further support that SMase increases ROS production through Nox2, while mitochondrial ROS production is not altered.
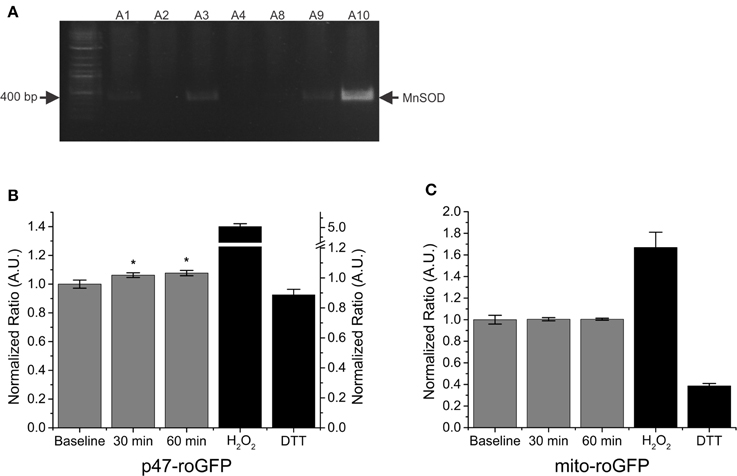
Figure 4. SMase increased Nox2 ROS production in FDB fibers from MnSOD overexpressing mice. (A) Representative DNA gel verifying MnSOD transgene overexpression. (B) SMase increased Nox2 ROS (p47-roGFP) in muscle from MnSOD overexpressing animals at both 30 and 60 min. (C) Mitochondrial ROS (mito-roGFP) did not change in response to SMase. H2O2 and DTT (100 μM and 10 mM, black bars) resulted in further oxidation and reduction of both p47-roGFP and mito-roGFP. Fluorescence values were normalized to non-drug treated baseline measurements. p ≤ 0.05 *significantly different from baseline in at least nanimals = 3, ncells = 10.
Genetic Deletion of Nox2 Partially Protects against SMase Induced Force Decrement
SMase induced ROS has been shown to decrease force in skeletal muscle (Ferreira et al., 2010, 2012). Our previous data indicate decreased ROS production in Nox2−/y animals in response to SMase; therefore, we wanted to determine whether genetically inhibiting Nox2-dependent ROS production would provide protection against SMase induced force depression in diaphragm strips. In the absence of SMase, there was a non-significant (<4%) time dependent change in force over 60 min (Figures 5A,B). SMase decreased tetanic force (p ≤ 0.05) in diaphragm strips from WT, Nox2−/y, and MnSOD overexpressing mice compared to their respective controls at 30 and 60 min (Figures 5A,B). Nox2 deficient animals conferred partial protection against the force decrement observed in the diaphragm of WT mice at 60 min (Figure 5A), while the MnSOD overexpressing mice offered no protection in force decrement (Figure 5B). While not statistically significant (p ≤ 0.08), there was a trend toward protection against force decrement in the Nox2−/y animals at 30 min.
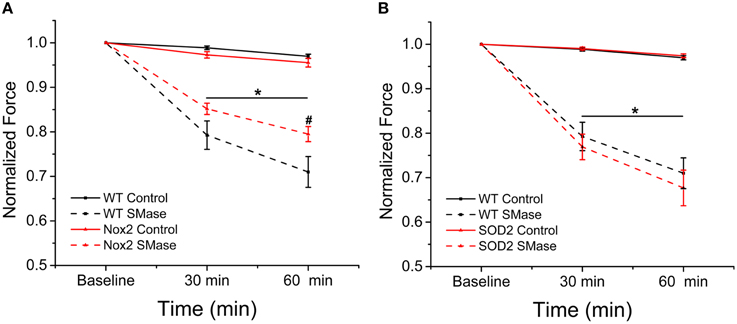
Figure 5. Genetic deletion of Nox2 protected against diaphragm force decrement over time. Peak tetanic force was decreased at both 30 and 60 min following SMase administration compared with respective controls. (A) Genetic deletion of Nox2 protected against SMase induced force loss compared with WT. Black solid = WT control, Black dash = WT SMase, Red solid = Nox2−/y control, Red dash = Nox2−/y SMase. (B) Overexpressing MnSOD did not affect force decrement compared to WT. Black solid = WT control, Black dash = WT SMase, Red solid = MnSOD control, Red dash = MnSOD SMase. Force values at 30 and 60 min were normalized to force measured in the absence of SMase (baseline). p ≤ 0.05 *All genotypes significantly different from respective control, #Nox2−/y SMase significantly different from WT SMase in at least nanimals = 6.
SMase induced force decrements were observed across all frequencies of stimulation (Figure 6) Genetic deletion of Nox2 protected against SMase induced force loss at all frequencies of stimulation, with an approximate 51 and 55% protection against twitch and peak tetanic force loss, respectively (Figure 6A). Interestingly, diaphragm from Nox2−/y mice displayed enhanced force production compared to WT animals at higher frequencies of stimulation (Figure 6A), suggesting that Nox2 specific ROS regulates muscle function even under basal conditions. Unlike the protection observed by eliminating Nox2 dependent ROS, no protection was evident in muscle from MnSOD overexpressing mice (Figure 6B), confirming our previous data that SMase does not result in a significant increase in ROS production from the mitochondria. Our data indicate that eliminating SMase stimulated Nox2-dependent ROS confers protection against force decrement and that Nox2-specific ROS may play a role in basal muscle function.
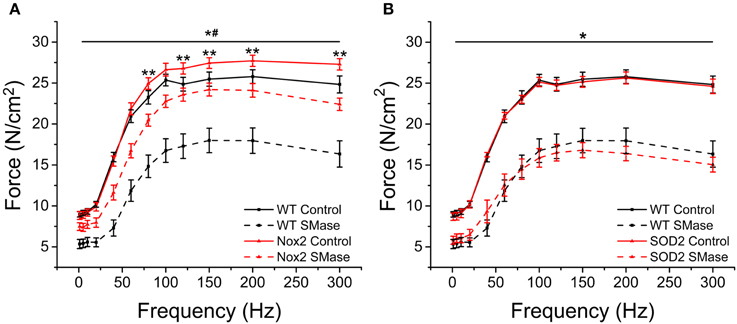
Figure 6. Nox2 deletion protected against SMase induced decreased force. Force was decreased at all frequencies following SMase administration compared with respective controls. (A) SMase induced force loss was prevented in diaphragm muscle from Nox2−/y mice compared to WT at all frequencies. Nox2−/y controls produced greater force at higher frequencies compared with WT control. Black solid = WT control, Black dash = WT SMase, Red solid = Nox2−/y control, Red dash = Nox2−/y SMase. (B) MnSOD overexpression did not prevent force loss at any frequency compared to WT SMase. Black solid = WT control, Black dash = WT SMase, Red solid = MnSOD control, Red dash = MnSOD SMase. P ≤ 0.05 *All genotypes significantly different from respective control, #Nox2−/y SMase significantly different from WT SMase, **Nox2 control significantly different from WT control in at least nanimals = 4.
Nox2 does not Confer Protection Against SMase Induced Fatigue
Oxidants have been shown to promote skeletal muscle fatigue (Moopanar and Allen, 2005; Bruton et al., 2008) and SMase has been shown to increase fatigue in an oxidant dependent manner (Ferreira et al., 2010). Our results show that Nox2 deficiency decreases SMase induced oxidant production and offers partial protection against the decrease in force observed in WT animals; therefore, we wanted to determine if it also would protect against SMase induced fatigue. In the vehicle treated control groups, neither diaphragm from the Nox2−/y nor the MnSOD overexpressing mice showed altered fatigue compared with WT. Following SMase administration, there was an increase in the rate of fatigue for diaphragm from WT, Nox2−/y and MnSOD overexpressing mice (Figures 7A,B). Neither the Nox2 knockout nor the MnSOD overexpressing mice conferred any protection against increased rate of fatigue (Figures 7A,B). The fatigue index, calculated as the last tetanus divided by the first tetanus, indicated diaphragm strips from all animals fatigued approximately 75–80%. These data suggest that fatigue observed in the presence of SMase is likely independent of ROS.
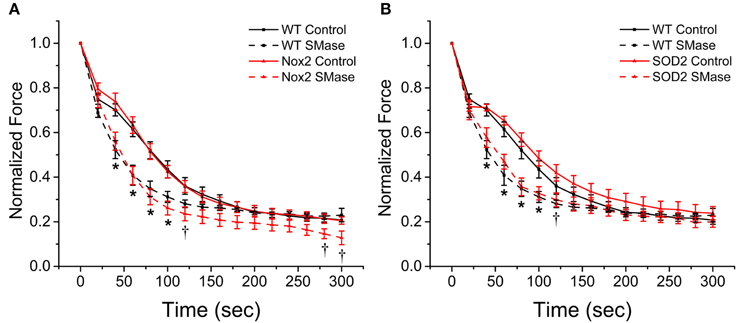
Figure 7. Genetic ablation of Nox2 or MnSOD overexpression does not protect against SMase induced fatigue. All genotypes fatigued approximately 75–80%. SMase induced an increased rate of fatigue in diaphragm from WT (A,B, black dashed), Nox2−/y (A, red dashed), and MnSOD (B, red dashed) compared with their respective controls (solid lines). All force values were normalized to initial force. p ≤ 0.05 *All genotypes significantly different from respective control, †Nox2−/y and MnSOD significantly different from respective controls in at least nanimals = 6.
Discussion
SMase, an enzyme which catalyzes the production of metabolites that affect a number of cellular functions (Nikolova-Karakashian and Reid, 2011), has been shown to be elevated in response to a variety of diseases (Wong et al., 2000; Claus et al., 2005; Empinado et al., 2014). Increased SMase results in increased ROS production, which has been thought to negatively impact muscle force and promote fatigue (Ferreira et al., 2010, 2012). Previous research has shown that SMase can induce either mitochondrial or Nox2 ROS in non-muscle cells (Sawada et al., 2004; Reinehr et al., 2005) while recent indirect evidence suggests SMase induced ROS in skeletal muscle was generated by the mitochondria (Ferreira et al., 2012). The majority of ROS production in skeletal muscle has generally been assumed to be mitochondrial; however, more recent evidence by our lab and others implicate Nox2 as a major source of ROS in skeletal muscle (Michaelson et al., 2010; Pal et al., 2013, 2014b; Sakellariou et al., 2013, 2014). Using site-specific subcellular redox sensors and various genetic animal models, we have shown that SMase induces ROS through Nox2 and eliminating Nox2-dependent ROS partially recovers muscle function.
Using the general ROS probe, DCFH, and a Nox2-specific peptide inhibitor (gp91-ds), we show that Nox2 plays a significant role in SMase induced ROS production. However, since DCFH is a non-specific, irreversible probe that does not allow for the evaluation of cellular redox homeostasis, there is still uncertainty as to the source of ROS within the cell and whether there is a true shift in the redox balance upon SMase exposure. The recent development of reversible, site-specific subcellular redox probes allows for a more thorough evaluation of the source of ROS production and redox balance in response to various stimuli or in response to different disease states. Using ROS sensors specifically targeted to Nox2 (p47-roGFP) and the mitochondria (mito-roGFP), we were able to show that SMase increased Nox2 ROS production, but had no effect on mitochondrial ROS in mouse FDB muscle. The lack of response of mito-roGFP to SMase cannot be attributed to the failure of the probe to respond, as the fluorescence ratio increased approximately two-fold upon oxidation (100 μM H2O2). These findings are in disagreement with other reports, which suggest SMase induces mitochondrial ROS production in the mouse diaphragm. Using an antioxidant peptide targeted to the inner mitochondrial membrane (SS-31) Reid and colleagues have suggested that SMase induces ROS generation from the mitochondria in mouse diaphragm muscle, which then diffuses into the cytosol where it is detected by the non-specific ROS probe DCFH (Ferreira et al., 2012). While SS-31 has been shown to localize to the mitochondria Szeto and colleagues have also shown that it also localizes to the cytosol (Zhao et al., 2004). This would allow SS-31 to act as a cytosolic ROS scavenger; reducing SMase induced ROS production generated by Nox2. Using pharmacological inhibition of Nox2 (gp91-ds) and genetic deletion of Nox2 (Nox2−/y), our results confirm that SMase induces ROS production by activating Nox2 in skeletal muscle. Furthermore, MnSOD overexpressing mice did not confer protection against SMase induced oxidation of our Nox2 redox biosensor.
Another potential confounding variable is that mitochondrial content varies in different muscles based on fiber type composition. Ferreira et al. (2012) used mouse diaphragm, which is comprised of 42–49% oxidative (Type I and IIA) fibers with the mitochondria encompassing approximately 34% of the fiber volume (Gamboa and Andrade, 2010; Guido et al., 2010; Pal et al., 2014b). We evaluated ROS production in FDB fibers, which have been reported to contain approximately 30% oxidative fibers (Gonzalez et al., 2003) with a mitochondrial volume between 10 and 36% of the fiber volume (Bruton et al., 2003; Laker et al., 2014). However, if SMase was able to induce a shift in the mitochondrial redox balance in muscle with a higher mitochondrial content (i.e., diaphgram), one would predict that overexpressing MnSOD would protect against SMase induced dysfunction, which was not observed in our studies. Therefore, we believe that differences in fiber type or mitochondrial content do not affect SMase induced mitochondrial ROS production.
ROS has been shown to be a potent modifier of skeletal muscle contractility and fatigue by modulating myofilament calcium sensitivity (Reid et al., 1993; Andrade et al., 1998; Callahan et al., 2001; Moopanar and Allen, 2005; Bruton et al., 2008). In the present study, eliminating SMase induced ROS provided partial protection against decreased force and had no effect on fatigue. It is unlikely that reactive nitrogen species, another source of oxidants in skeletal muscle, could account for the continued force decrement, as SMase had no effect on NO activity and blocking NO activity had no effect on SMase induced force decrement (Ferreira et al., 2010). Sarcoplasmic reticulum (SR) calcium release has been shown to play an important role in skeletal muscle function (Ortenblad et al., 2000; Allen et al., 2008). The direct metabolite of SMase activity, ceramide, can be further metabolized by ceramidase into sphingosine. Sphingosine has been shown to decrease SR calcium release (Betto et al., 1992; Sabbadini et al., 1992; McDonough et al., 1994) by decreasing ryanodine receptor open probability (Needleman et al., 1997; Sabbadini et al., 1999). Increased levels of sphingosines have also been shown to decrease myocyte calcium transients, cell shortening, and contractile function, (Webster et al., 1994; Oral et al., 1997; Friedrichs et al., 2002; Favory et al., 2004) in addition to promoting muscle fatigue (Danieli-Betto et al., 2000). Therefore, SMase may affect muscle function through a non-ROS dependent mechanism. We are currently investigating the role of these potential mechanisms in skeletal muscle force depression and fatigue.
In summary, we have shown that SMase increases Nox2 dependent ROS and has no effect on mitochondrial ROS production. Genetically deleting or pharmacologically inhibiting Nox2 reduced SMase stimulated ROS production to control levels. In addition, Nox2 inhibition provided protection against decreased force production induced by SMase; however, there was no effect on muscle fatigue. This indicates that SMase, through its downstream metabolites, may affect contractile function in a ROS independent manner, potentially through modifying SR calcium release.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Robia Pautler and Deric Griffin for providing and genotyping the MnSOD overexpression mice. Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number R01 AR061370 to George G. Rodney, the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under Award Number R21 NS085208 to George G. Rodney, the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Number T32 HL007676 to James A. Loehr, and a Gillson Longenbaugh Foundation Award to George G. Rodney.
References
Allen, D. G., Lamb, G. D., and Westerblad, H. (2008). Impaired calcium release during fatigue. J. Appl. Physiol. (1985) 104, 296–305. doi: 10.1152/japplphysiol.00908.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Andrade, F. H., Reid, M. B., Allen, D. G., and Westerblad, H. (1998). Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J. Physiol. 509(Pt 2), 565–575. doi: 10.1111/j.1469-7793.1998.565bn.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Betto, R., Teresi, A., Fachechi-Cassano, G., Salviati, G., and Sabbadini, R. A. (1992). Sphingosine inhibits calcium release from sarcoplasmic reticulum membranes. Adv. Exp. Med. Biol. 311, 403–404. doi: 10.1007/978-1-4615-3362-7_55
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bruton, J. D., Dahlstedt, A. J., Abbate, F., and Westerblad, H. (2003). Mitochondrial function in intact skeletal muscle fibres of creatine kinase deficient mice. J. Physiol. 552, 393–402. doi: 10.1113/jphysiol.2003.050732
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bruton, J. D., Place, N., Yamada, T., Silva, J. P., Andrade, F. H., Dahlstedt, A. J., et al. (2008). Reactive oxygen species and fatigue-induced prolonged low-frequency force depression in skeletal muscle fibres of rats, mice and SOD2 overexpressing mice. J. Physiol. 586, 175–184. doi: 10.1113/jphysiol.2007.147470
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Callahan, L. A., She, Z. W., and Nosek, T. M. (2001). Superoxide, hydroxyl radical, and hydrogen peroxide effects on single-diaphragm fiber contractile apparatus. J. Appl. Physiol. (1985) 90, 45–54.
Claus, R. A., Bunck, A. C., Bockmeyer, C. L., Brunkhorst, F. M., Losche, W., Kinscherf, R., et al. (2005). Role of increased sphingomyelinase activity in apoptosis and organ failure of patients with severe sepsis. FASEB J. 19, 1719–1721. doi: 10.1096/fj.04-2842fje
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Csanyi, G., Cifuentes-Pagano, E., Al Ghouleh, I., Ranayhossaini, D. J., Egana, L., Lopes, L. R., et al. (2011). Nox2 B-loop peptide, Nox2ds, specifically inhibits the NADPH oxidase Nox2. Free Radic. Biol. Med. 51, 1116–1125. doi: 10.1016/j.freeradbiomed.2011.04.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Danieli-Betto, D., Germinario, E., Esposito, A., Biral, D., and Betto, R. (2000). Effects of fatigue on sarcoplasmic reticulum and myofibrillar properties of rat single muscle fibers. J. Appl. Physiol. (1985) 89, 891–898.
De, L. J., Zufferli, A., Serra, F., Isidori, A. M., Naro, F., Dessalle, K., et al. (2012). TNF-alpha- and tumor-induced skeletal muscle atrophy involves sphingolipid metabolism. Skelet. Muscle 2:2. doi: 10.1186/2044-5040-2-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Doehner, W., Bunck, A. C., Rauchhaus, M., von, H. S., Brunkhorst, F. M., Cicoira, M., et al. (2007). Secretory sphingomyelinase is upregulated in chronic heart failure: a second messenger system of immune activation relates to body composition, muscular functional capacity, and peripheral blood flow. Eur. Heart J. 28, 821–828. doi: 10.1093/eurheartj/ehl541
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dooley, C. T., Dore, T. M., Hanson, G. T., Jackson, W. C., Remington, S. J., and Tsien, R. Y. (2004). Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J. Biol. Chem. 279, 22284–22293. doi: 10.1074/jbc.M312847200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Empinado, H. M., Deevska, G. M., Nikolova-Karakashian, M., Yoo, J. K., Christou, D. D., and Ferreira, L. F. (2014). Diaphragm dysfunction in heart failure is accompanied by increases in neutral sphingomyelinase activity and ceramide content. Eur. J. Heart Fail. 16, 519–525. doi: 10.1002/ejhf.73
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Favory, R., Lancel, S., Marchetti, P., Mordon, S., Chopin, C., Formstecher, P., et al. (2004). Endotoxin-induced myocardial dysfunction: evidence for a role of sphingosine production. Crit. Care Med. 32, 495–501. doi: 10.1097/01.CCM.0000109452.36271.FA
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferreira, L. F., Moylan, J. S., Gilliam, L. A., Smith, J. D., Nikolova-Karakashian, M., and Reid, M. B. (2010). Sphingomyelinase stimulates oxidant signaling to weaken skeletal muscle and promote fatigue. Am. J. Physiol. Cell Physiol. 299, C552–C560. doi: 10.1152/ajpcell.00065.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferreira, L. F., Moylan, J. S., Stasko, S., Smith, J. D., Campbell, K. S., and Reid, M. B. (2012). Sphingomyelinase depresses force and calcium sensitivity of the contractile apparatus in mouse diaphragm muscle fibers. J. Appl. Physiol. (1985) 112, 1538–1545. doi: 10.1152/japplphysiol.01269.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Friedrichs, G. S., Swillo, R. E., Jow, B., Bridal, T., Numann, R., Warner, L. M., et al. (2002). Sphingosine modulates myocyte electrophysiology, induces negative inotropy, and decreases survival after myocardial ischemia. J. Cardiovasc. Pharmacol. 39, 18–28. doi: 10.1097/00005344-200201000-00003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gamboa, J. L., and Andrade, F. H. (2010). Mitochondrial content and distribution changes specific to mouse diaphragm after chronic normobaric hypoxia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R575–R583. doi: 10.1152/ajpregu.00320.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gonzalez, E., Messi, M. L., Zheng, Z., and Delbono, O. (2003). Insulin-like growth factor-1 prevents age-related decrease in specific force and intracellular Ca2+ in single intact muscle fibres from transgenic mice. J. Physiol. 552, 833–844. doi: 10.1113/jphysiol.2003.048165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guido, A. N., Campos, G. E., Neto, H. S., Marques, M. J., and Minatel, E. (2010). Fiber type composition of the sternomastoid and diaphragm muscles of dystrophin-deficient mdx mice. Anat. Rec. (Hoboken.) 293, 1722–1728. doi: 10.1002/ar.21224
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hanson, G. T., Aggeler, R., Oglesbee, D., Cannon, M., Capaldi, R. A., Tsien, R. Y., et al. (2004). Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J. Biol. Chem. 279, 13044–13053. doi: 10.1074/jbc.M312846200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Laker, R. C., Xu, P., Ryall, K. A., Sujkowski, A., Kenwood, B. M., Chain, K. H., et al. (2014). A novel MitoTimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J. Biol. Chem. 289, 12005–12015. doi: 10.1074/jbc.M113.530527
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Massaad, C. A., Washington, T. M., Pautler, R. G., and Klann, E. (2009). Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 106, 13576–13581. doi: 10.1073/pnas.0902714106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McClung, J. M., Deruisseau, K. C., Whidden, M. A., Van, R. H., Richardson, A., Song, W., et al. (2010). Overexpression of antioxidant enzymes in diaphragm muscle does not alter contraction-induced fatigue or recovery. Exp. Physiol. 95, 222–231. doi: 10.1113/expphysiol.2009.049650
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McDonough, P. M., Yasui, K., Betto, R., Salviati, G., Glembotski, C. C., Palade, P. T., et al. (1994). Control of cardiac Ca2+ levels. Inhibitory actions of sphingosine on Ca2+ transients and L-type Ca2+ channel conductance. Circ. Res. 75, 981–989. doi: 10.1161/01.RES.75.6.981
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Michaelson, L. P., Shi, G., Ward, C. W., and Rodney, G. G. (2010). Mitochondrial redox potential during contraction in single intact muscle fibers. Muscle Nerve 42, 522–529. doi: 10.1002/mus.21724
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moopanar, T. R., and Allen, D. G. (2005). Reactive oxygen species reduce myofibrillar Ca2+ sensitivity in fatiguing mouse skeletal muscle at 37 degrees C. J. Physiol. 564, 189–199. doi: 10.1113/jphysiol.2005.083519
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Needleman, D. H., Aghdasi, B., Seryshev, A. B., Schroepfer, G. J. Jr., and Hamilton, S. L. (1997). Modulation of skeletal muscle Ca2(+)-release channel activity by sphingosine. Am. J. Physiol. 272, C1465–C1474.
Nikolova-Karakashian, M. N., and Reid, M. B. (2011). Sphingolipid metabolism, oxidant signaling, and contractile function of skeletal muscle. Antioxid Redox Signal. 15, 2501–2517. doi: 10.1089/ars.2011.3940
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Okazaki, T., Liang, F., Li, T., Lemaire, C., Danialou, G., Shoelson, S. E., et al. (2014). Muscle-specific inhibition of the classical nuclear factor-kappaB pathway is protective against diaphragmatic weakness in murine endotoxemia. Crit. Care Med. 42, e501–e509. doi: 10.1097/CCM.0000000000000407
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oral, H., Dorn, G. W., and Mann, D. L. (1997). Sphingosine mediates the immediate negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian cardiac myocyte. J. Biol. Chem. 272, 4836–4842. doi: 10.1074/jbc.272.8.4836
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ortenblad, N., Sjogaard, G., and Madsen, K. (2000). Impaired sarcoplasmic reticulum Ca(2+) release rate after fatiguing stimulation in rat skeletal muscle. J. Appl. Physiol. 89, 210–217.
Pal, R., Basu, T. P., Li, S., Minard, C., and Rodney, G. G. (2013). Real-time imaging of NADPH oxidase activity in living cells using a novel fluorescent protein reporter. PLoS ONE 8:e63989. doi: 10.1371/journal.pone.0063989
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pal, R., Monroe, T. O., Palmieri, M., Sardiello, M., and Rodney, G. G. (2014a). Rotenone induces neurotoxicity through Rac1-dependent activation of NADPH oxidase in SHSY-5Y cells. FEBS Lett. 588, 472–481. doi: 10.1016/j.febslet.2013.12.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pal, R., Palmieri, M., Loehr, J. A., Li, S., Abo-Zahrah, R., Monroe, T. O., et al. (2014b). Src-dependent impairment of autophagy by oxidative stress in a mouse model of Duchenne muscular dystrophy. Nat. Commun. 5:4425. doi: 10.1038/ncomms5425
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reid, M. B., Khawli, F. A., and Moody, M. R. (1993). Reactive oxygen in skeletal muscle. III. Contractility of unfatigued muscle. J. Appl. Physiol. (1985) 75, 1081–1087.
Reinehr, R., Becker, S., Eberle, A., Grether-Beck, S., and Haussinger, D. (2005). Involvement of NADPH oxidase isoforms and Src family kinases in CD95-dependent hepatocyte apoptosis. J. Biol. Chem. 280, 27179–27194. doi: 10.1074/jbc.M414361200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rey, F. E., Cifuentes, M. E., Kiarash, A., Quinn, M. T., and Pagano, P. J. (2001). Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ. Res. 89, 408–414. doi: 10.1161/hh1701.096037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sabbadini, R. A., Betto, R., Teresi, A., Fachechi-Cassano, G., and Salviati, G. (1992). The effects of sphingosine on sarcoplasmic reticulum membrane calcium release. J. Biol. Chem. 267, 15475–15484.
Sabbadini, R. A., Danieli-Betto, D., and Betto, R. (1999). The role of sphingolipids in the control of skeletal muscle function: a review. Ital. J. Neurol. Sci. 20, 423–430. doi: 10.1007/s100720050062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sakellariou, G. K., Jackson, M. J., and Vasilaki, A. (2014). Redefining the major contributors to superoxide production in contracting skeletal muscle. The role of NAD(P)H oxidases. Free Radic. Res. 48, 12–29. doi: 10.3109/10715762.2013.830718
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sakellariou, G. K., Vasilaki, A., Palomero, J., Kayani, A., Zibrik, L., McArdle, A., et al. (2013). Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid Redox Signal. 18, 603–621. doi: 10.1089/ars.2012.4623
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sawada, M., Kiyono, T., Nakashima, S., Shinoda, J., Naganawa, T., Hara, S., et al. (2004). Molecular mechanisms of TNF-alpha-induced ceramide formation in human glioma cells: P53-mediated oxidant stress-dependent and -independent pathways. Cell Death Differ. 11, 997–1008. doi: 10.1038/sj.cdd.4401438
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Webster, R. J., Sabbadini, R. A., Dettbarn, C. A., and Paolini, P. J. (1994). Sphingosine effects on the contractile behavior of skinned cardiac myocytes. J. Mol. Cell Cardiol. 26, 1273–1290. doi: 10.1006/jmcc.1994.1147
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wong, M. L., Xie, B., Beatini, N., Phu, P., Marathe, S., Johns, A., et al. (2000). Acute systemic inflammation up-regulates secretory sphingomyelinase in vivo: a possible link between inflammatory cytokines and atherogenesis. Proc. Natl. Acad. Sci. U.S.A. 97, 8681–8686. doi: 10.1073/pnas.150098097
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhao, K., Zhao, G. M., Wu, D., Soong, Y., Birk, A. V., Schiller, P. W., et al. (2004). Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J. Biol. Chem. 279, 34682–34690. doi: 10.1074/jbc.M402999200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: ROS, skeletal muscle, Nox2, sphingomyelinase, force
Citation: Loehr JA, Abo-Zahrah R, Pal R and Rodney GG (2015) Sphingomyelinase promotes oxidant production and skeletal muscle contractile dysfunction through activation of NADPH oxidase. Front. Physiol. 5:530. doi: 10.3389/fphys.2014.00530
Received: 31 October 2014; Accepted: 27 December 2014;
Published online: 21 January 2015.
Edited by:
Brian McDonagh, University of Liverpool, UKReviewed by:
Francisco H. Andrade, University of Kentucky College of Medicine, USAGeorge K. Sakellariou, University of Liverpool, UK
Copyright © 2015 Loehr, Abo-Zahrah, Pal and Rodney. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George G. Rodney, Department of Molecular Physiology and Biophysics, Baylor College of Medicine, One Baylor Plaza-BCM335, Houston, TX 77030, USA e-mail:cm9kbmV5QGJjbS5lZHU=