 Margaret V. Westfall
Margaret V. Westfall- Department of Cardiac Surgery, University of Michigan, Ann Arbor, MI, USA
Secondary shifts develop in post-translational phosphorylation of sarcomeric proteins in multiple animal models of inherited cardiomyopathy. These signaling alterations together with the primary mutation are predicted to contribute to the overall cardiac phenotype. As a result, identification and integration of post-translational myofilament signaling responses are identified as priorities for gaining insights into sarcomeric cardiomyopathies. However, significant questions remain about the nature and contribution of post-translational phosphorylation to structural remodeling and cardiac dysfunction in animal models and human patients. This perspective essay discusses specific goals for filling critical gaps about post-translational signaling in response to these inherited mutations, especially within sarcomeric proteins. The discussion focuses primarily on pre-clinical analysis of animal models and defines challenges and future directions in this field.
Introduction
More than 3800 gene mutations are linked to inherited cardiomyopathies (ICs) and identification of underlying gene mutations continues to expand (https://www.ncbi.nlm.nih.gov/clinvar/). Animal models expressing individual mutations have provided insight into the human disease and a better understanding of myofilament force transduction mechanisms (Tardiff, 2005, 2011). In these models, the pathophysiological response is often linked to a specific disease progression such as hypertrophic, dilated, restrictive, left ventricular noncompaction, and/or arrhythmogenic right ventricular phenotypes (Fatkin et al., 2014). However, understanding how a specific mutation leads to the cardiac phenotype remains a persistent question (van der Velden et al., 2015), and the factors contributing to disease variability in patients are only partially understood. One area which may provide insight into these issues, and therefore deserves further consideration, is dynamic local myofilament signaling and its impact on downstream networks and/or global signaling within cardiac myocytes. This Perspective focuses on the possibility that IC-linked mutations alter local myofilament signaling and contribute to downstream remodeling and disease progression. Our current understanding of dynamic post-translational myofilament signaling also is briefly summarized to lay the foundation for future work aimed at investigating relationships between IC-linked mutations and myofilament modulation.
First, it is important to point out that previous work in humans and animal models indicate IC-linked heart disease is complex. In patients, morbidity and mortality are often not easily explained by an identified mutation acting as a primary physiological insult or substrate (Ho et al., 2015). Instead, a temporal and spatial network of factors contributes to progressive cardiac structural and functional remodeling in IC patients, and can ultimately evolve into end stage heart failure. In addition to a primary mutation, factors known or suspected to increase the risk for disease include second hit and epigenetic mutations, polymorphisms, and other genetic modifiers, which include genes linked to cardiac remodeling, and environmental factors such as sex, aerobic activity levels, and risk factors such as hypertension (McNally et al., 2013; Månsson, 2014; Ho et al., 2015; van der Velden et al., 2015). In addition, cardiac remodeling and dysfunction is progressive on many levels and includes alterations in cellular morphology, signaling, and function, cell-cell architecture, plus organ-level electrical, and pump dysfunction. Signaling modulation is predicted to be an important focus for future work in recent reviews (Yar et al., 2014; van der Velden et al., 2015). Figure 1 illustrates the variety of signaling pathways known to phosphorylate myofilament proteins, and therefore, could contribute to modulation by targeting myofilament proteins for phosphorylation. The emphasis on myofilament modulation in this Perspective is based on the possibility that myofilament phosphorylation may be an early secondary response to IC-linked mutations, and therefore present prior to significant structural and functional remodeling within myocytes. Myofilament signaling may continue to contribute to adaptive functional responses and/or initiate one or more later compensatory behaviors associated with cardiac remodeling and disease, such as alterations in excitation-contraction (E-C) coupling, myocyte Ca2+ handling, and metabolism (Ashrafian et al., 2011).
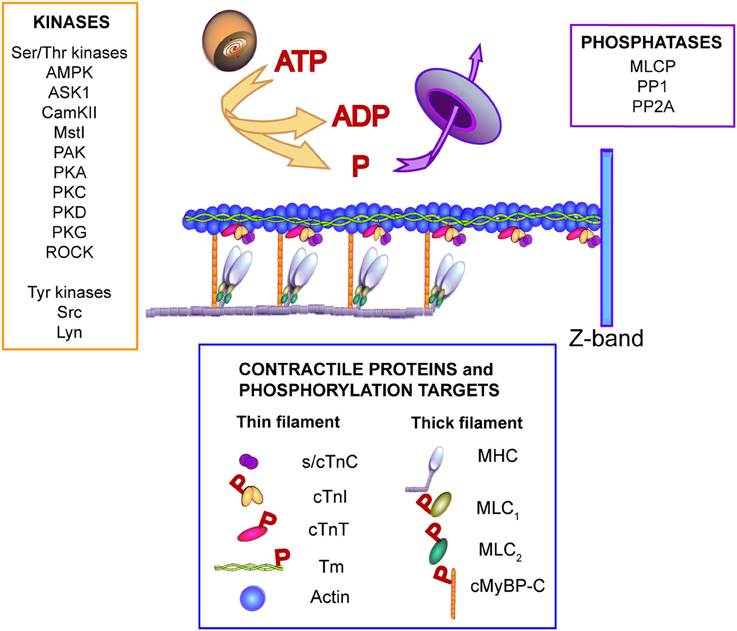
Figure 1. Illustration of signaling kinases and phosphatases known to modulate phosphorylation of contractile proteins. One or more of the kinases and/or phosphatases shown may contribute to secondary or “local” phosphorylation changes in response to IC-linked mutations. The Ser/Thr kinases shown to target myofilament proteins include adenosine monophosphate kinase (AMPK), apoptosis signal-regulating kinase 1 (ASK1), Ca2+/calmodulin protein kinase II (CamKII), sterile 20-like kinase 1 (Mst1), p21-activated kinase (PAK), protein kinase A (PKA), protein kinase C (PKC), protein kinase D (PKD), protein kinase G (PKG), and Rho-associated protein kinase (ROCK). Tyrosine kinases which target myofilament troponin I include non-receptor activated Src and the Lck/Yes novel (Lyn) kinase (Salhi et al., 2014). Phosphatases known to target myofilament proteins include myosin light chain phosphatase (MLCP), protein phosphatase I (PPI), protein phosphatase 2A (PP2A) (Solaro and Kobayashi, 2011). The contractile proteins shown in this illustration are slow/cardiac troponin C (s/cTnC), cardiac troponin I (cTnI), cardiac troponin T (cTnT), alpha-tropomyosin (Tm), and actin in the thin filament plus the myosin heavy chain (MHC), myosin light chains 1 and 2 (MLC1, MLC2, respectively), and cardiac myosin binding protein C (cMyBP-C). The proteins identified as phosphorylation targets include cTnI, cTnT, Tm, MLC1, MLC2, and cMyBP-C (indicated by red P in the legend). For further information see the following references: (He et al., 2003; Barefield and Sadayappan, 2010; Solaro and Kobayashi, 2011; Streng et al., 2013; Westfall, 2014; Huang and Szczesna-Cordary, 2015).
In animal models expressing IC-linked mutations, E-C coupling and Ca2+ handling network alterations are often detected in parallel with in vivo evidence of cardiac performance compensation and/or dysfunction, and prior to end-stage heart failure (Ashrafian et al., 2011). These changes in Ca2+ increase the risk for developing arrhythmia and sudden cardiac death (Ashrafian et al., 2011; Yar et al., 2014), and the events responsible for initiating and/or causing remodeling of the Ca2+ signal may be critical for understanding IC-linked disease progression. Interventions to prevent or delay disease progression prior to the onset of Ca2+ remodeling would be desirable in high risk families and/or patients. However, little is known about the process or mechanism(s) responsible for the initiation of Ca2+ remodeling in these patients. IC-linked mutations may initiate changes in local myofilament signaling network(s) and the myofilament post-translational modification (PTM) pattern helps to maintain cardiac performance prior to any changes in Ca2+ handling. Evidence is accumulating that myofilament residues targeted by signaling pathways can initiate additional “secondary” or “adaptive” changes in the phosphorylation of other myofilament residues to modulate function (Montgomery et al., 2002; Scruggs et al., 2006; Lang et al., 2015), which appears to maintain steady state contractile function in the short term (Lang et al., 2015). Chronic activation of this secondary signaling within the myofilament may become inadequate and/or serve as a direct trigger for later structural and functional remodeling such as the IC-associated alterations in Ca2+ handling and E-C coupling described above. Although critical studies are still needed to prove this idea, future support for a direct role of local myofilament signaling in response to IC-linked mutations could lead to early diagnostic tests and/or therapeutic strategies to prevent or minimize IC disease progression in high risk patients.
IC-Linked Mutations and A Role for Local Myofilament Signaling
There are some general observations which are consistent with a role for local myofilament signaling responses in IC-linked structural and functional remodeling. First, a causative mutation is usually not identified in new probands until cardiac dysfunction develops, which is often during adolescence or later (Cirino and Ho, 2008). The known impact of an IC-linked mutation on myofilament function and/or Ca2+ remodeling also may not predict the cardiac phenotype in animal models or patients, especially at early time points (Jacques et al., 2008; Jensen et al., 2013). A recent developmental study also demonstrated that inhibition of IC-linked gene expression during the first 6 weeks of life markedly reduced cardiac remodeling at 40 weeks in an α–MHCR403Q mouse model, while more modest improvements developed if mutant protein expression was inhibited after 6 weeks of age (Cannon et al., 2015). Secondary modulatory mechanisms in the myofilament are consistent with these observations and could contribute to developmental lags and/or unexpected phenotypes. Myofilament modulatory networks also may undergo developmental transitions over the same perinatal period observed for many contractile proteins (Cummins, 1982; Lyons et al., 1990; Reiser et al., 1994, 2001; Suurmeijer et al., 2003). Impaired or altered myofilament signaling development could result in permanently sub-optimal myofilament modulation in adults with IC-linked mutations. Alternatively, this local signaling network modulation may be hard-wired to respond to myofilament perturbations such as IC-linked mutants, and either directly or indirectly trigger further adverse structural and functional remodeling of myocardium.
The local myofilament signaling concept also is supported by reported changes in the phosphorylation of multiple myofilament protein residues in response to IC-linked mutation expression, and alterations in additional phosphorylated residues linked to heart failure (Table 1). Altered myofilament phosphorylation develops in at least one IC-linked mutation for each contractile protein, and there is significant potential for myofilament phosphorylation to modulate contractile function based on the myofilament residues already identified as phosphorylation targets (see Table 1 and references). However, it is not known whether a given contractile protein or mutations clustered into a specific cardiomyopathy produce common spatial and/or temporal phosphorylation patterns. Thus, to test whether myofilament modulatory phosphorylation makes an early contribution to IC-linked phenotypes requires rigorous experimental testing in the future. As part of these studies, it is important to identify the dose-dependent spatial and temporal impact of each IC-linked mutation on myofilament phosphorylation and understand the modulatory impact of each phosphorylated contractile protein residue. Although not included here, phosphorylation of additional sarcomeric proteins, such as titin, also may contribute to this modulation. A few representative studies on cardiac troponin I (cTnI) mutations and phosphorylation are briefly presented below to illustrate our current understanding and the rationale for future directions on myofilament phosphorylation in response to IC-linked mutations.

Table 1. Contractile protein phosphorylation sites associated with inherited cardiomyopathies (IC) and heart failure (HF).
IC-Linked Mutations and β-AR Signaling in Myofilaments
Previous work on β-adrenergic receptor (β-AR) signaling provides direct support for a role of local myofilament signaling in IC-linked changes in cardiac function. Several IC-linked mutations directly influence myofilament phosphorylation and/or β-AR signal transduction, as illustrated by representative cTnI mutations. Protein kinase A (PKA)-induced cTnI-S23/24 phosphorylation significantly contributes to the positive β-AR-induced lusitropic response (Takimoto et al., 2004; Yasuda et al., 2007; note that residue numbering is based on Uniprot human protein accession numbers, see Table 1). Uncoupling between the β-AR receptor and this response often develops in IC-linked animal models (reviewed by Messer and Marston, 2014). Poor outcomes are associated with myofilament β-AR uncoupling in other types of human heart failure, and the ability of IC-linked mutations to cause this uncoupling is proposed to be a prognostic indicator in patients with IC-linked mutations (Messer and Marston, 2014).
Several mechanisms can produce β-AR uncoupling in response to IC-linked mutations. Some IC-linked mutations directly disrupt post-translational cTnI-S23/24 phosphorylation. For example, the IC-linked cTnI-R21C mutation directly blocks PKA-induced phosphorylation of the adjacent S23/24 residues (Gomes et al., 2005; Wang et al., 2012; Dweck et al., 2014; Cheng et al., 2015). IC-linked mutations in more distant proteins also modify this PKA-targeted cTnI phosphorylation (Najafi et al., 2016). Alternatively, PKA continues to phosphorylate myofilament targets, such as cTnI-S23/24, in the presence of other IC-linked mutations. Representative mutations such as cTnI-R145G and -P82S, disrupt signal transduction within cTnI to cause β-AR uncoupling (Deng et al., 2001; Messer and Marston, 2014; Ramirez-Correa et al., 2015; Cheng et al., 2016). The cTnI-P82S mutation is noteworthy because the diastolic dysfunction and late-onset of disease in humans associated with this mutation is postulated to be a long-term consequence of secondary alterations in PKA-related myofilament signaling (Nimura et al., 2002; Mogensen et al., 2004; Frazier et al., 2008; Ramirez-Correa et al., 2015). In addition, IC-linked mutations may indirectly cause β-AR uncoupling due to changes in the overall myofilament phosphorylation status (Kooij et al., 2010), which could result from differences in other myofilament associated kinase and phosphatase activities (Figure 1).
IC-Linked Mutations and Additional Myofilament Signaling Pathways
While β-AR induced PKA modulation is among the most studied signaling pathways targeting myofilaments, a number of additional signaling pathways also modulate myofilament function (Figure 1). A few studies also indicate that IC-linked mutations modify both kinase and phosphatase signaling pathways and downstream target residues other than β-AR/PKA-targeted sites. Mutation-related alterations associated with the protein kinase C (PKC) second messenger serve as a representative example. First, progressive increases in cardiac PKC expression and increased PKC affinity for sarcomeric proteins are associated with IC-linked mutations (Arimura et al., 2004; Sfichi-Duke et al., 2010). Modification of end-targets, such as PKC phosphorylation of cTnI-S42/44 provides some initial support. Myofilament function is similarly modified by either PKC-induced phosphorylation or phospho-mimetic cTnI-S42/44 substitutions (Noland et al., 1996; Burkart et al., 2003b). Interestingly, the myofilament response to phospho-mimetic cTnI-S42/44 is significantly greater in myofilaments expressing IC-linked tropomyosin (Tm)-E180G compared to controls (Burkart et al., 2003a).
Multiple neurohormones activate receptor-induced PKC signaling in myocytes, such as angiotensin II (AgII), endothelin, and catecholamine activation of α-adrenergic receptors (Dorn and Force, 2005). Accelerated and/or exaggerated cardiac remodeling and dysfunction develop in response to one or more of these neurohormones in mice with IC-linked mutations (Maass et al., 2004; Gramlich et al., 2009). This severe response has been interpreted as a stress response, but myofilament-associated PKC activity may already be modified in myofilaments expressing IC-linked mutations independent from receptor activation. Thus, further neurohormone stimulation of PKC may accelerate additional remodeling and produce progressive deterioration in cardiac function. This interpretation is supported by evidence of more severe remodeling in endothelin-treated, cardiac-derived stem cells from patients with IC-linked mutations (Tanaka et al., 2014). Pro-left ventricular polymorphisms present in the renin-angiotensin-aldosterone (RAA) axis also are associated with higher morbidity and mortality in patients with IC-linked mutations (Ortlepp et al., 2002; Kaufman et al., 2007). In addition, environmental stressors known to activate the RAA axis, such as pressure overload, further exacerbate IC-associated contractile dysfunction (Chen et al., 2013). While angiotensin receptor inhibitors failed to reverse fibrosis (Axelsson et al., 2015), some tangential evidence in Duchenne's muscular dystrophy (DMD) patients indicates earlier treatment with these types of inhibitors may be beneficial in treating DMD patients with cardiomyopathy (Duboc et al., 2007; Kamdar and Garry, 2016). DMD is caused by mutations in dystrophin, a crucial component of the costamere, which anchors sarcomeres to the sarcolemma.
Many of the potential signaling networks associated with myofilaments have the ability to produce a range of outputs via multi-layer signaling cascades capable of targeting both kinases and phosphatases, multiple sarcomeric protein targets, and multiple residues targeted within a single myofilament protein (Figure 1). The complexity of local myofilament signaling contributes to difficulties in defining the modulatory role for a given signal in myofilament function (Angeli et al., 2004). As a result, these signaling networks may not be easily recognized as contributors to IC-linked remodeling and/or disease. However, these types of pathways also are noteworthy because they are either predicted or known to act as oscillators capable of flexible outputs. Oscillatory signals have the potential to provide highly dynamic modulation to maintain steady state structure/function (Angeli et al., 2004). An IC-linked mutation could disrupt or alter one or more signals in an oscillatory pathway to produce subtle shifts in phosphorylation turnover at multiple target residues. These types of pathways may have little initial impact, but lead to bi- or multi-phasic temporal alterations in one or more target PTMs (Angeli et al., 2004). An IC-linked mutation which chronically induces secondary myofilament signaling to modulate function may either become inadequate to maintain myofilament structure and function, or directly trigger further adaptations beyond the myofilament, such as the myocyte Ca2+ handling modifications described earlier.
While a secondary reduction in myofilament phosphorylation can coincide with cardiac dysfunction (Bayliss et al., 2013; Alves et al., 2014), no published reports prove bifurcative/ oscillatory signaling and/or altered PTM levels develop in multiple myofilament proteins prior to detectable morphological and/or functional remodeling in animal models with IC-linked mutations. Short-term expression of cardiac troponin T (cTnT)-R92Q in bigenic mice provides some indirect evidence. These mice develop early alterations in a range of signaling pathways associated with structural remodeling, which returned to baseline after turning off mutant expression (Lutucuta et al., 2004). There is also some indirect support for IC-induced oscillatory changes based on changes in Ca2+ wave frequencies in cardiomyocytes expressing gain-of-function SHP-2/PTPN11 mutations, which are linked to Noonan's syndrome (Uhlén et al., 2006). In addition, myofilament PTMs and functional responses observed in adult myocytes after PKC gene transfer are consistent with bifurcative myofilament signaling (Hwang et al., 2013). These data alone do not provide adequate proof that local myofilament modulation contributes to IC-linked remodeling, but suggest that local myofilament signaling in IC-linked mice is worthy of analysis. Specifically, studies are needed to determine whether local myofilament modulation precedes and/or works in parallel with other adaptive responses associated with structural and functional remodeling observed in IC animal models.
Dynamic Signaling Modulation in Cardiac Myofilaments
IC-linked mutations also may trigger a secondary signaling response, which may arise from structural changes imposed by a mutation. This secondary response could involve one or more signaling pathways known to target myofilament proteins (Figure 1). However, in contrast to the typical receptor-based signaling activation discussed earlier, only signaling networks localized to the myofilament undergo changes in activity. This localized signaling also may undergo dynamic changes in response to structural alterations produced during contraction and relaxation. Signaling studies utilizing phospho-mimetic and -null substitutions at myofilament target residues provide some initial support for the presence and role of dynamic, local myofilament signaling modulation (Lang et al., 2013, 2015). Specifically, alterations in the phosphorylation of other myofilament residues develops in myocytes expressing phospho-mimetic or non-phosphorylatable substitutions at one or more kinase-targeted myofilament residues (Montgomery et al., 2002; Scruggs et al., 2006, 2009; Lang et al., 2013; Nixon et al., 2014; Lang et al., 2015). This secondary phosphorylation also is associated with altered functional responses (Lang et al., 2013, 2015). Secondary adaptations in myofilament phosphorylation are reported in both thick and thin filaments using a variety of approaches. For example, a cTnI-S150 phospho-mimetic blunts the β-AR/PKA myofilament response (Nixon et al., 2014) and elevated myofilament phosphorylation develops in mice expressing non-phosphorylatable ventricular myosin light chain (MLC; Scruggs et al., 2009). Other post-translational modifications also trigger local myofilament phospho-modulation, as indicated by alterations in cardiac myosin binding protein C (cMyBP-C) phosphorylation after S-glutathiolation increases during heart failure (Stathopoulou et al., 2016). Taken together, these results are consistent with local myofilament modulatory signaling changes during sustained structural or functional perturbations in the sarcomere.
Other approaches, such as proteomic analysis of myofilament proteins during heart failure, also hint at dynamic PTM modulation within myofilaments. Heart failure is associated with altered phosphorylation residues in several contractile proteins (see Table 1; Dubois et al., 2011; Zhang et al., 2012; Kooij et al., 2013; Walker et al., 2013). Secondary signaling also is reported in some, but not all myofilament phospho-mimetic and -null animal models. These local signaling changes may contribute to phenotypic differences among these models, such as the significant differences reported in cTnI models with phosho-substitutions at PKC-targeted sites (Pi et al., 2002; Sakthivel et al., 2005; Bilchick et al., 2007; Kirk et al., 2009). While differences in genetic approach, mouse strain, age, and mutant expressivity may factor into these differences, highly organized signaling network(s) which locally modulate myofilament structure and function also may contribute to divergent phenotypes.
Future Directions
Future work needs to establish the circuitry, physiological functions, and temporal response of local myofilament modulatory signaling, and test whether this local modulation is an early or longer-term contributor to IC-linked remodeling and/or dysfunction. A parallel, translational goal for this work is the development of diagnostic tools, improved clinical management, and therapies to prevent and/or delay disease progression in IC patients. The integration of computational modeling, myofilament, cellular, and in vivo genetic model work is critical for achieving these goals. As a result, significant advancements are likely to depend on an unusually high level of cooperativity and resource sharing among investigators.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alves, M. L., Dias, F. A., Gaffin, R. D., Simon, J. N., Montminy, E. M., Biesiadecki, B. J., et al. (2014). Desensitization of myofilaments to Ca2+ as a therapeutic target for hypertrophic cardio-myopathy with mutations in thin filament proteins. Circ. Cardiovasc. Genet. 7, 132–143. doi: 10.1161/CIRCGENETICS.113.000324
Angeli, D., Ferrell, J. E. Jr., and Sontag, E. D. (2004). Detection of multistability, bifurcations, and hysteresis in a large class of biological positive-feedback systems. Proc. Natl. Acad. Sci. U.S.A. 101, 1822–1827. doi: 10.1073/pnas.0308265100
Arimura, T., Hayashi, T., Terada, H., Lee, S.-Y., Zhou, Q., Takahashi, M., et al. (2004). A Cypher/ZASP mutation associated with dilated cardiomyopathy alters the binding affinity to protein kinase C. J. Biol. Chem. 279, 6746–6752. doi: 10.1074/jbc.M311849200
Ashrafian, H., McKenna, W. J., and Watkins, H. (2011). Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ. Res. 209, 86–96. doi: 10.1161/CIRCRESAHA.111.242974
Axelsson, A., Iversen, K., Vejlstrup, N., Ho, C., Langhoff, L., Ahtarovski, K., et al. (2015). Efficacy and safety of the angiotensin II receptor blocker losartan for hypertrophic cardiomyopathy: the INHERIT randomized, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 3, 123–131. doi: 10.1016/S2213-8587(14)70241-4
Barefield, D., and Sadayappan, S. (2010). Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J. Mol. Cell. Cardiol. 48, 866–875. doi: 10.1016/j.yjmcc.2009.11.014
Bayliss, C. R., Jacques, A. M., Leung, M.-C., Ward, D. G., Redwood, C. S., Gallon, C. E., et al. (2013). Myofibrillar Ca2+ sensitivity is uncoupled from troponin I phosphorylation in hypertrophic obstructive cardiomyopathy due to abnormal troponin T. Cardiovasc. Res. 97, 500–508. doi: 10.1093/cvr/cvs322
Bilchick, K. C., Duncan, J. G., Ravi, R., Takimoto, E., Champion, H. C., Gao, W. D., et al. (2007). Heart failure-associated alterations in troponin I phosphorylation impair ventricular relaxation-afterload and force-frequency responses and systolic function. Am. J. Physiol. Heart Circ. Physiol. 292, H318–H325. doi: 10.1152/ajpheart.00283.2006
Burkart, E. M., Arteaga, G. M., Sumandea, M. P., Prabhakar, R., Wieczorek, D. F., and Solaro, R. J. (2003a). Altered signaling surrounding the C-lobe of cardiac troponin C in myofilaments containing an α-tropomyosin mutation linked to familial hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 35, 1285–1293. doi: 10.1016/S0022-2828(03)00240-2
Burkart, E. M., Sumandea, M. P., Kobayashi, T., Nili, M., Martin, A. F., Homsher, E., et al. (2003b). Phosphorylation or glutamic acid substitution at protein kinase C sites on cardiac troponin I differentially depress myofilament tension and shortening velocity. J. Biol. Chem. 278, 11265–11272. doi: 10.1074/jbc.M210712200
Cannon, L., Yu, Z.-Y., Marciniec, T., Waardenberg, A. J., Iismaa, S. E., Nikolova-Krstevski, V., et al. (2015). Irreversible triggers for hypertrophic cardiomyopathy are established in the early postnatal period. J. Am. Coll. Cardiol. 65, 560–569. doi: 10.1016/j.jacc.2014.10.069
Chen, H., Hwang, H., McKee, L. A. K., Perez, J. N., Regan, J. A., Constantopoulos, E., et al. (2013). Temporal and morphological impact of pressure overload in transgenic FHC mice. Front. Physiol. 4:205. doi: 10.3389/fphys.2013.00205
Cheng, Y., Lindert, S., Oxenford, L., Tu, A-y., McCulloch, A. D., and Regnier, M. (2016). Effects of cardiac troponin I mutation P83S on contractile properties and the modulation by PKA-mediated phosphorylation. J. Phys. Chem. B. 120, 8238–8253. doi: 10.1021/acs.jpcb.6b01859
Cheng, Y., Rao, V., Tu, A., Lindert, S., Wang, D., Oxenford, L., et al. (2015). Troponin I mutations R146G and R21C alter cardiac troponin function, contractile properties, and modulation by protein kinase A (PKA)-mediated phosphorylation. J. Biol. Chem. 290, 27749–27776. doi: 10.1074/jbc.M115.683045
Cirino, A. L., and Ho, C. (2008). “Hypertrophic cardiomyopathy overview,” in GeneReviews, eds R. A. Pagon, M. P. Adam, H. H. Ardinger, S. E. Wallace, A. Amemiya, L. J. H., Bean, T. D. Bird, C. T. Fong, H. C. Mefford, R. J. H. Smith, and K. Stephens (Seattle, WA: University of Washington).
Cummins, P. (1982). Transitions in human atrial and ventricular light-chain isoenzymes in response to cardiac-pressure-overload-induced hypertrophy. Biochem. J. 205, 195–204. doi: 10.1042/bj2050195
Deng, Y., Schmidtmann, A., Redlich, A., Westerdorf, B., Jaquet, K., and Thieleczek, R. (2001). Effects of phosphorylation and mutation R145G on human cardiac troponin I function. Biochemistry 40, 14593–14602. doi: 10.1021/bi0115232
Dorn, G. W. I. I., and Force, T. (2005). Protein kinase cascades in the regulation of cardiac hypertrophy. J. Clin. Invest. 115, 527–537. doi: 10.1172/JCI200524178
Duboc, D., Meune, C., Pierre, B., Wahbi, K., Eymard, B., Toutain, A., et al. (2007). Perindolpril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years' follow-up. Am. Heart J. 154, 596–602. doi: 10.1016/j.ahj.2007.05.014
Dubois, E., Richard, V., Mulder, P., Lamblin, N., Drobecq, H., Henry, J. P., et al. (2011). Decreased serine 207 phosphorylation of troponin T as a biomarker of left ventricular myocardial infarction. Eur. Heart J. 32, 115–123. doi: 10.1093/eurheartj/ehq108
Dweck, D., Sanchez-Gonzalez, M. A., Chang, A. N., Dulce, R. A., Badger, C. D., Koutnik, A. P., et al. (2014). Long term ablation of protein kinase A (PKA)-mediated cardiac troponin I phosphorylation leads to excitation-contraction uncoupling and diastolic dysfunction in a knock-in mouse model of hypertrophic cardiomyopathy. J. Biol. Chem. 289, 23097–23111. doi: 10.1074/jbc.M114.561472
Fatkin, D., Seidman, C. E., and Seidman, J. G. (2014). Genetics and disease of ventricular muscle. Cold Spring Harb. Perspect. Med. 4:a021063. doi: 10.1101/cshperspect.a021063
Frazier, A., Judge, D. P., Schulman, S. P., Johnson, N., Holmes, K. W., and Murphy, A. M. (2008). Familial hypertrophic cardiomyopathy associated with cardiac beta-myosin heavy chain and troponin I mutations. Pediatr. Cardiol. 29, 846–850. doi: 10.1007/s00246-007-9177-9
Gomes, A. V., Harada, K., and Potter, J. D. (2005). A mutation in the N-terimus of troponin I that is associated with hypertrophic cardiomyopathy affects the Ca2+ sensitivity, phosphorylation kinetics and proteolytic susceptibility of troponin. J. Mol. Cell. Cardiol. 39, 754–765. doi: 10.1016/j.yjmcc.2005.05.013
Gramlich, M., Michely, B., Krohne, C., Heuser, A., Erdmann, B., Klaassen, S., et al. (2009). Stress-induced dilated cardiomyopathy in a knock-in mouse model mimicking human titin-based disease. J. Mol. Cell. Cardiol. 47, 352–358. doi: 10.1016/j.yjmcc.2009.04.014
He, X., Liu, Y., Sharma, V., Dirsken, R. T., Waugh, R., Sheu, S.-S., et al. (2003). ASK1 associates with troponin T and induces troponin T phosphorylation and contractile dysfunction in cardiomyocytes. Am. J. Pathol. 163, 243–251. doi: 10.1016/S0002-9440(10)63647-4
Ho, C. Y., Charron, P., Richard, P., Girolami, F., Van Spaendonck-Zwarts, K. Y., and Pinto, Y. (2015). Genetic advances in sarcomeric cardiomyopathies: state of the art. Cardiovasc. Res.105, 397–408. doi: 10.1093/cvr/cvv025
Huang, W., and Szczesna-Cordary, D. (2015). Molecular mechanisms of cardiomyopathy phenotypes associated with myosin light chain mutations. J. Muscle Res. Cell Motil. 36, 433–445. doi: 10.1007/s10974-015-9423-3
Hwang, H., Robinson, D., Rogers, J. B., Stevenson, T. K., Lang, S. E., Sadayappan, S., et al. (2013). Agonist activated PKCβII translocation and modulation of cardiac myocyte contractile function. Sci. Rep. 3:1971. doi: 10.1038/srep01971
Jacques, A., Hoskins, A. C., Kentish, J. C., and Marston, S. B. (2008). From genotype to phenotype: a longitudinal study of a patient with hypertrophic cardiomyopathy due to a mutation in the MYBPC3 gene. J. Muscle Res. Cell Motil. 29, 239–246. doi: 10.1007/s10974-009-9174-0
Jensen, M. K., Havndrup, O., Christiansen, M., Andersen, P. S., Diness, B., Axelsson, A., et al. (2013). Penetrance of hypertrophic cardiomyopathy in children and adolescents. Circulation 127, 48–54. doi: 10.1161/CIRCULATIONAHA.111.090514
Jia, W., Shaffer, J. F., Harris, S. P., and Leary, J. A. (2010). Identification of novel protein kinase A phosphorylation sites in the M-domain of human and murine cardiac myosin binding protein-C using mass spectrometry analysis. J. Proteome Res. 9, 1843–1853. doi: 10.1021/pr901006h
Kamdar, F., and Garry, D. J. (2016). Dystrophin-deficient cardiomyopathy. J. Am. Coll. Cardiol. 67, 2533–2546. doi: 10.1016/j.jacc.2016.02.081
Kaufman, B. D., Auerbach, S., Reddy, S., Manlhiot, C., Deng, L., Prakash, A., et al. (2007). RAAS gene polymorphisms influence progression of pediatric hypertrophic cardiomyopathy. Hum. Genet. 122, 515–523. doi: 10.1007/s00439-007-0429-9
Kirk, J. A., MacGowan, G. A., Evans, C., Smith, S. H., Warren, C. M., Mamidi, R., et al. (2009). Left ventricular and myocardial function in mice expressing constitutively pseudophosphorylated cardiac troponin I. Circ. Res. 105, 1232–1239. doi: 10.1161/CIRCRESAHA.109.205427
Kobayashi, T., Dong, W.-J., Burkart, E. M., Cheung, H. C., and Solaro, R. J. (2004). Effects of protein kinase C dependent phosphorylation and a familial hypertrophic cardiomyopathy-related mutation of cardiac troponin I on structural transition of troponin C and myofilament activation. Biochemistry 43, 5996–6004. doi: 10.1021/bi036073n
Kooij, V., Holewinski, J., Murphy, A. M., and Van Eyk, J. E. (2013). Characterization of the cardiac myosin binding protein phosphoproteome in healthy and failing human hearts. J. Mol. Cell. Cardiol. 60, 116–120. doi: 10.1016/j.yjmcc.2013.04.012
Kooij, V., Saes, M., Jaquet, K., Zaremba, R., Foster, D. B., Murphy, A. M., et al. (2010). Effect of troponin I Ser23/24 phosphorylation on Ca2+-sensitivity in human myocardium depends on the phosphorylation background. J. Mol. Cell. Cardiol. 48, 954–963. doi: 10.1016/j.yjmcc.2010.01.002
Lang, S. E., Robinson, D. A., Wu, H. C., Herron, T. J., Wahr, P. A., and Westfall, M. V. (2013). Myofilament incorporation and contractile function after gene transfer of cardiac troponin I Ser43/45Ala. Arch. Biochem. Biophys. 535, 49–55. doi: 10.1016/j.abb.2012.12.021
Lang, S. E., Schwank, J., Stevenson, T. K., Jensen, M. A., and Westfall, M. V. (2015). Independent modulation of contractile performance by cardiac troponin I Ser43 and Ser45 in the dynamic sarcomere. J. Mol. Cell. Cardiol. 79, 264–274. doi: 10.1016/j.yjmcc.2014.11.022
Lutucuta, S., Tsybouleva, N., Ishiyama, M., DeFreitas, G., Wei, L., Carabello, B., et al. (2004). Induction and reversal of cardiac phenotype of human hypertrophic cardiomyopathy mmutation cardiac troponin T-Q92 in switch on-switch off bigenic mice. J. Am. Coll. Cardiol. 44, 2221–2230. doi: 10.1016/j.jacc.2004.09.005
Lyons, G. E., Schiaffino, S., Sassoon, D., Barton, P., and Buckingham, M. (1990). Developmental regulation of myosin gene expression in mouse cardiac muscle. J. Cell Biol. 111, 2427–2436. doi: 10.1083/jcb.111.6.2427
Maass, A. H., Ikeda, K., Oberdorf-Maass, S., Maler, S. K. G., and Leinwand, L. A. (2004). Hypertrophy, fibrosis, and sudden cardiac death in response to pathological stimuli in mice with mutations in cardiac troponin T. Circulation 110, 2102–2109. doi: 10.1161/01.CIR.0000144460.84795.E3
Månsson, A. (2014). Hypothesis and theory: mechanical instabilities and non-uniformities in hereditary sarcomere myopathies. Front. Physiol. 5:350. doi: 10.3389/fphys.2014.00350
Marston, S. B., Copeland, O., Messer, A. E., MacNamara, E., Nowak, K., Zampronio, C. G., et al. (2013). Tropomyosin isoform expression and phosphorylation in the human heart in health and disease. J. Muscle Res. Cell Motil. 34, 189–197. doi: 10.1007/s10974-013-9347-8
McNally, E. M., Golbus, J. R., and Puckelwartz, M. J. (2013). Genetic mutations and mechanisms in dilated cardiomyopathy. J. Clin. Invest. 123, 19–26. doi: 10.1172/JCI62862
Messer, A. E., and Marston, S. B. (2014). Investigating the role of uncoupling of troponin I phosphorylation from changes in myofibrillar Ca2+-sensitivity in the pathogenesis of cardiomyopathy. Front. Physiol. 5:315. doi: 10.3389/fphys.2014.00315
Michael, J. J., and Chandra, M. (2016). Interplay between the effects of dilated cardiomyopathy mutation R206L and the protein kinase C phosphomimic (T204E) of rat cardiac troponin T are differently modulated by a- and b-myosin heavy chain isoforms. J. Am. Heart Assoc. 5:e002777. doi: 10.1161/JAHA.115.002777
Mogensen, J., Murphy, R. T., Shaw, T., Bahl, A., Redwood, C., Watkins, H., et al. (2004). Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 44, 2033–2040. doi: 10.1016/j.jacc.2004.08.027
Montgomery, D. E., Wolska, B. M., Pyle, W. G., Roman, B. B., Dowell, J. C., Buttrick, P. M., et al. (2002). α-Adrenergic response and myofilament activity in mouse hearts lacking PKC phosphorylation sites on cardiac TnI. Am. J. Physiol. Heart Circ. Physiol. 282, H2397–H2405. doi: 10.1152/ajpheart.00714.2001
Najafi, A., Sequeira, V., Helmes, M., Bollen, I. A., Goebel, M., Regan, J. A., et al. (2016). Selective phosphorylation of PKA targets after β-adrenergic receptor stimulation impairs myofilament function in Mybpc3-targeted HCM mouse model. Cardiovasc. Res. 110, 200–214. doi: 10.1093/cvr/cvw026
Nimura, H., Patton, K. K., McKenna, W. J., Soults, J., Maron, B. J., Seidman, J. G., et al. (2002). Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation 105, 446–451. doi: 10.1161/hc0402.102990
Nixon, B. R., Thawornkalwong, A., Jin, J., Brundage, E. A., Little, S. C., Davis, J. P., et al. (2012). AMP-activated protein kinase phosphorylates cardiac troponin I at Ser-150 to increase myofilament calcium sensitivity and blunt PKA-dependent function. J. Biol. Chem. 287, 19136–19147. doi: 10.1074/jbc.M111.323048
Nixon, B. R., Walton, S. D., Zhang, B., Brundage, E. A., Little, S. C., Ziolo, M. T., et al. (2014). Combined troponin I Ser-150 and Ser-23/24 phosphorylation sustains thin filament Ca2+ sensitivity and accelerates deactivation in an acidic environment. J. Mol. Cell Cardiol. 72, 177–185. doi: 10.1016/j.yjmcc.2014.03.010
Noland, T. A. Jr., Raynor, R. L., Jideama, N. M., Guo, X., Kazanietz, M. G., Blumberg, P. M., et al. (1996). Differential regulation of cardiac actomyosin S-1 MgATPase by protein kinase C isozyme-specific phosphorylation of specific sites in cardiac troponin I and its phosphorylation site mutants. Biochemistry 35, 14923–14931.
Oliveira, S. M., Zhang, Y. H., Sancho Soli, R., Isackson, H., Bellahcene, M., Yavari, A., et al. (2012). AMP-activated protein kinase phosphorylates cardiac troponin I and alters contractility of murine ventricular myocytes. Circ. Res. 110, 1192–1201. doi: 10.1161/CIRCRESAHA.111.259952
Ortlepp, J. R., Vosberg, H. P., Reith, S., Ohme, F., Mahon, N. G., Schröder, D., et al. (2002). Genetic polymorphisms in the renin-angiotensin-aldosterone system associated with expression of left ventricular hypertrophy in hypertrophic cardiomyopathy: a study of five polymorphic genes in a family with a disease causing mutation in the myosin binding protein C gene. Heart 87, 270–275. doi: 10.1136/heart.87.3.270
Pi, Y. Q., Kemnitz, K. R., Zhang, D., Kranias, E. G., and Walker, J. W. (2002). Phosphorylation of troponin I controls cardiac twitch dynamics. Circ. Res. 90, 649–656. doi: 10.1161/01.RES.0000014080.82861.5F
Ramirez-Correa, G. A., Frazier, A. H., Zhu, G., Zhang, P., Rappod, T., Kooij, V., et al. (2015). Cardiac troponin I Pro82Ser variant induces diastolic dysfunction, blunts β-adrenergic response, and impairs myofilament cooperativity. J. Appl. Physiol. 118, 212–223. doi: 10.1152/japplphysiol.00463.2014
Reiser, P. J., Portman, M. A., Ning, X.-H., and Moravec, C. S. (2001). Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. Am. J. Physiol. Heart Circ. Physiol. 280, H1814–H1820.
Reiser, P. J., Westfall, M. V., Schiaffino, S., and Solaro, R. J. (1994). Tension production and thin-filament protein isoforms in developing rat myocardium. Am. J. Physiol. Heart Circ. Physiol. 267, H1589–H1596.
Sakthivel, S., Finley, N. L., Rosevear, P. R., Lorenz, J. N., Gulick, J., Kim, S., et al. (2005). In vivo and in vitro analysis of cardiac troponin I phosphorylation. J. Biol. Chem. 280, 703–714. doi: 10.1074/jbc.M409513200
Salhi, H. E., Walton, S. D., Hassel, N. C., Brundage, E. A., de Tombe, P. P., Janssen, P. M. L., et al. (2014). Cardiac troponin I tyrosine 26 phosphorylation decreases myofilament Ca2+ sensitivity and accelerates deactivation. J. Mol. Cell. Cardiol. 76, 257–264. doi: 10.1016/j.yjmcc.2014.09.013
Sanbe, A., Fewell, J. G., Gulick, J., Osinska, H., Lorenz, J., Hall, D. G., et al. (1999). Abnormal cardiac structure and function in mice expressing nonphosphorylatable cardiac regulatory myosin light chain 2. J. Biol. Chem. 274, 21085–21094. doi: 10.1074/jbc.274.30.21085
Schulz, E. M., Wilder, T., Chowdhury, S. A. K., Sheikh, H. N., Wolska, B. M., Solaro, R. J., et al. (2013). Decreasing tropomyosin phosphorylation rescues tropomyosin induced familial hypertrophic cardiomyopathy. J. Biol. Chem. 288, 28925–28935. doi: 10.1074/jbc.M113.466466
Scruggs, S. B., Hinken, A. C., Thawornkalwong, A., Robbins, J., Walker, L. A., de Tombe, P. P., et al. (2009). Ablation of ventricular regulatory light chain phosphorylation in mice causes cardiac dysfunction in situ and affects neighboring myofilament protein phosphorylation. J. Biol. Chem. 284, 5097–5106. doi: 10.1074/jbc.M807414200
Scruggs, S. B., Walker, L. A., Lyu, T., Geenen, D. L., Solaro, R. J., Buttrick, P. M., et al. (2006). Partial replacement of cardiac troponin I with a non-phosphorylatable mutant at serines 43/45 attenuates the contractile dysfunction associated with PKCepsilon phosphorylation. J. Mol. Cell. Cardiol. 40, 465–473. doi: 10.1016/j.yjmcc.2005.12.009
Sfichi-Duke, L., Garcia-Cazarin, M. L., Sumandea, C. A., Sievert, G. A., Balke, C. W., Zhan, D. Y., et al. (2010). Cardiomyopathy-causing deletion K210 in cardiac troponin T alters phosphorylation propensity of sarcomeric proteins. J. Mol. Cell. Cardiol. 48, 934–942. doi: 10.1016/j.yjmcc.2010.01.005
Solaro, R. J., and Kobayashi, T. (2011). Protein phosphorylation and signal transduction in cardiac thin filaments. J. Biol. Chem. 286, 9935–9940. doi: 10.1074/jbc.R110.197731
Stathopoulou, K., Wittig, I., Heidler, J., Piasecki, A., Richter, F., Diering, S., et al. (2016). S-glutathiolation impairs phosphoregulation and function of cardiac myosin-binding protein C in human heart failure. FASEB J. 30, 1849–1864. doi: 10.1096/fj.201500048
Streng, A. S., de Boer, D., van der Velden, J., van Dieijen-Visser, M. P., and Wodzig, W. K. (2013). Posttranslational modifications of cardiac troponin T: an overview. J. Mol. Cell. Cardiol. 63, 47–56. doi: 10.1016/j.yjmcc.2013.07.004
Suurmeijer, A. J. H., Clément, S., Francesconi, A., Bocchi, L., Angelini, A., Van Veldhuisen, D. J., et al. (2003). α-Actin isoform distribution in normal and failing human heart: a morphological, morphometric and biochemical study. J. Pathol. 199, 387–397. doi: 10.1002/path.1311
Takimoto, E., Soergel, D. G., Janssen, P. M., Stull, L. B., Kass, D. A., and Murphy, A. M. (2004). Frequency- and afterload-dependent cardiac modulation in vivo by troponin I with constitutively active protein kinase A phosphorylation sites. Circ. Res. 94, 496–504. doi: 10.1161/01.RES.0000117307.57798.F5
Tanaka, A., Yuasa, S., Mearini, G., Egashira, T., Seki, T., Kodaira, M., et al. (2014). Endothelin-1 induces myofibrillar disarray and contractile vector variability in hypertrophic cardiomyopathy-induced pluripotent stem cell-derived cardiomyocytes. JAHA 3:e001263. doi: 10.1161/JAHA.114.001263
Tardiff, J. C. (2005). Sarcomeric proteins and familial hypertrophic cardiomyopathy: linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Fail. Rev. 10, 237–248. doi: 10.1007/s10741-005-5253-5
Tardiff, J. C. (2011). Thin filament mutations. Circ. Res. 108, 765–782. doi: 10.1161/CIRCRESAHA.110.224170
Uhlén, P., Burch, P. M., Zito, C. I., Estrada, M., and Ehrlich BE Bennett, A. M. (2006). Gain-of-function/Noonan syndrome SHP-2/Ptpn11 mutants enhance calcium oscillations and impair NFAT signaling. PNAS 103, 2160–2165. doi: 10.1073/pnas.0510876103
van der Velden, J., Ho, C. Y., Tardiff, J. C., Olivotto, I., and Knollmann BC Carrier, L. (2015). Research priorities in sarcomeric cardiomyopathy. Cardiovasc. Res. 105, 449–456. doi: 10.1093/cvr/cvv019
van Dijk, S. J., Paalberends, R., Najafi, A., Michels, M., Sadayappan, S., Carrier, L., et al. (2012). Contractile dysfunction irrespective of the mutant protein in human hypertrophic cardiomyopathy with normal systolic function. Circ. Heart Fail. 5, 36–46. doi: 10.1161/CIRCHEARTFAILURE.111.963702
Walker, L. A., Fullerton, D. A., and Buttrick, P. M. (2013). Contractile protein phosphorylation predicts human heart disease phenotypes. Am. J. Physiol. Heart Circ. Physiol. 304, H1644–H1650. doi: 10.1152/ajpheart.00957.2012
Wang, Y., Pinto, J. R., Solis, R. S., Dweck, D., Liang, J., Diaz-Perez, Z., et al. (2012). Generation and functional characterization of knock-in mice harboring the cardiac troponin I-R21C mutation associated with hypertrophic cardiomyopathy. J. Biol. Chem. 287, 2156–12167. doi: 10.1074/jbc.M111.294306
Warren, C. M., Arteaga, G. M., Rajan, S., Ahmed, R. P. H., Wieczorek, D. F., and Solaro, R. J. (2008). Use of 2-D DIGE analysis reveals altered phosphorylation in a tropomyosin mutant (Flu54Lys) linked to dilated cardiomyopathy. Proteomics 8, 100–105. doi: 10.1002/pmic.200700772
Wei, B., and Jin, J. P. (2011). Troponin T isoforms and post-transcriptional modifications: Evolution, regulation and function. Arch. Biochem. Biophys. 505, 144–154. doi: 10.1016/j.abb.2010.10.013
Westfall, M. V. (2014). “Post-translational modifications of troponin,” in Troponin: Regulator of Muscle Contraction, ed J.-P. Jin (New York, NY: Nova), 163–202.
Yar, S., Monasky, M. M., and Solaro, R. J. (2014). Maladaptive modifications in myofilament proteins and triggers in the progression to heart failure and sudden death. Pflugers Arch. 466, 1189–1197. doi: 10.1007/s00424-014-1457-7
Yasuda, S., Coutu, P., Sadayappan, S., Robbins, J., and Metzger, J. M. (2007). Cardiac transgenic and gene transfer strategies converge to support an important role for troponin I in regulating relaxation in cardiac myocytes. Circ. Res. 101, 377–386. doi: 10.1161/CIRCRESAHA.106.145557
Keywords: myofilament, post-translational modification, signaling, cardiomyopathy, phosphorylation
Citation: Westfall MV (2016) Contribution of Post-translational Phosphorylation to Sarcomere-Linked Cardiomyopathy Phenotypes. Front. Physiol. 7:407. doi: 10.3389/fphys.2016.00407
Received: 19 July 2016; Accepted: 30 August 2016;
Published: 14 September 2016.
Edited by:
P. Bryant Chase, Florida State University, USAReviewed by:
Aldrin V. Gomes, University of California, Davis, USAYuanhua Cheng, University of Washington, USA
Copyright © 2016 Westfall. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margaret V. Westfall, d2ZhbGxAbWVkLnVtaWNoLmVkdQ==