 Carmen García-Rodríguez1,2*
Carmen García-Rodríguez1,2* Iván Parra-Izquierdo1
Iván Parra-Izquierdo1 Irene Castaños-Mollor1
Irene Castaños-Mollor1 Javier López2,3J. Alberto San Román2,3
Javier López2,3J. Alberto San Román2,3 Mariano Sánchez Crespo1
Mariano Sánchez Crespo1- 1Instituto de Biología y Genética Molecular, CSIC-Universidad de Valladolid, Valladolid, Spain
- 2Centro de Investigación Biomédica en Red de Enfermedades Cardiovasculares (CIBERCV), Madrid, Spain
- 3Hospital Clínico Universitario, Valladolid, Spain
Inflammation, the primary response of innate immunity, is essential to initiate the calcification process underlying calcific aortic valve disease (CAVD), the most prevalent valvulopathy in Western countries. The pathogenesis of CAVD is multifactorial and includes inflammation, hemodynamic factors, fibrosis, and active calcification. In the development of CAVD, both innate and adaptive immune responses are activated, and accumulating evidences show the central role of inflammation in the initiation and propagation phases of the disease, being the function of Toll-like receptors (TLR) particularly relevant. These receptors act as sentinels of the innate immune system by recognizing pattern molecules from both pathogens and host-derived molecules released after tissue damage. TLR mediate inflammation via NF-κB routes within and beyond the immune system, and play a crucial role in the control of infection and the maintenance of tissue homeostasis. This review outlines the current notions about the association between TLR signaling and the ensuing development of inflammation and fibrocalcific remodeling in the pathogenesis of CAVD. Recent data provide new insights into the inflammatory and osteogenic responses underlying the disease and further support the hypothesis that inflammation plays a mechanistic role in the initiation and progression of CAVD. These findings make TLR signaling a potential target for therapeutic intervention in CAVD.
Inflammation and Calcific Aortic Valve Stenosis
Calcific aortic stenosis is the final step of calcific aortic valve disease (CAVD), a slowly progressive complex process which begins with alterations in the valve leaflets that may lead to the development of left ventricular outflow obstruction (Miller et al., 2011; Rajamannan et al., 2011; Pawade et al., 2015). It has been related to atherosclerosis, inflammation, and hemodynamic factors, but active calcification is a feature characteristic (O'Brien, 2006; Yetkin and Waltenberger, 2009). Its prevalence is high and depends on age, affecting more than 2% of people older than 75 years and 8% of people older than 84 (Roberts and Ko, 2005; Nkomo et al., 2006; Go et al., 2014; Otto and Prendergast, 2014). Once symptoms appear the disease progresses rapidly, and most patients undergo either surgical aortic valve replacement or transcatheter aortic valve implantation. An estimated 42,000 and 65,000 valvular implants are performed per year in Europe and the USA, respectively (Bridgewater et al., 2011; Go et al., 2014; Otto and Prendergast, 2014), thus explaining the social impact and growing healthcare costs associated with CAVD.
CAVD is no longer considered a degenerative process related to aging, but an actively regulated process in which many pathogenetic factors remain unknown (O'Brien, 2006; Rajamannan et al., 2011; Towler, 2013). This change in the paradigm is based on three set of data: (1) epidemiological associations of risk factors and higher prevalence and faster progression of CAVD; (2) histopathologic identification in excised stenotic valves of chronic inflammation features, lipoprotein deposition, renin-angiotensin system components, and molecular mediators of calcification; and 3) elucidation of cell-signaling pathways and genetic factors linked to valve disease pathogenesis (O'Brien, 2006). From a clinical point of view, it would be very important to deepen in the knowledge of the disease and identifying potential novel therapeutic targets to delay its progression. Despite extensive research efforts including several randomized controlled trials (Cowell et al., 2005; Rossebø et al., 2008; Chan et al., 2010), no pharmacotherapy strategies currently exist to prevent or treat CAVD.
During the last decades, many studies have shown the relationship between classical atherogenic risk factors and valve calcification that might explain their frequent coexistence in clinical practice. Clinical factors associated with the development of CAVD found in the Cardiovascular Health Study are similar to the cardiovascular risk factors: older age, male gender, serum lipoprotein and LDL levels, height, hypertension, metabolic syndrome and smoking (Stewart et al., 1997). Even though CAVD and atherosclerosis share some ethiopathological features, recent reports have disclosed significant differences in calcification progression in the late stages and the lack of effect of statins in CAVD progression in the SALTIRE trial (Cowell et al., 2005; O'Brien, 2006; Hjortnaes et al., 2010; Dweck et al., 2013).
The inflammatory response is a hallmark of CAVD, which has been extensively demonstrated (O'Brien, 2006; Coté et al., 2013; Mathieu et al., 2015). In the 90s, some studies identified the presence in aortic valve lesions of macrophages and T lymphocytes and expression of chronic inflammation effector molecules like interleukins (IL)-1 and 2, class II human leukocyte antigen, and HLA-DR. More recently, mast cells, pro-inflammatory cytokines, IL-1β and tumor necrosis factor α have been linked to the disease (O'Brien, 2006; Hjortnaes et al., 2010; Dweck et al., 2013). Other mechanisms include the deposition of the atherogenic lipoproteins LDL and lipoprotein(a) in stenotic aortic valves and the activation of the renin-angiotensin system. Remarkably, the typical calcification during CAVD is considered an inflammation-dependent process (Coté et al., 2013; Dweck et al., 2013). New insights using novel imaging approaches that allow simultaneous visualization of inflammation and early mineralization support the paradigm of inflammation-dependent calcification and reveal that inflammation precedes calcification. This suggests three phases for CAVD progression (Otto, 2008; Aikawa and Otto, 2012; Pawade et al., 2015), inflammation being involved in the initiation and propagation phases, whereas in the last phase, calcification rather than inflammation is predominant.
TLR, Inflammation, and Disease
The innate immune receptors termed Toll-like receptors (TLR) belong to a phylogenetically ancient system specialized in the recognition of conserved motifs present in pathogens, the so-called pathogen associated molecular patterns (PAMP), and of endogenous molecules released upon tissue injury (DAMP, also known as alarmins) (Kawai and Akira, 2010). The TLR family consists of membrane-spanned receptors recognizing a broad range of ligands, each member sensing a specific set of molecular patterns. So far, 10 TLRs have been identified in humans and 13 in mammalians (Kawai and Akira, 2010). TLR4, the first one described, is the receptor for lipopolysaccharide (LPS), a gram-negative bacterial toxin (Figure 1). TLR2 and its co-receptors TLR1/6 detect lipoproteins and lipopeptides, and TLR5 senses flagellin. TLR2/4 can also sense DAMPs, including heat-shock proteins, high-mobility group box 1 (HMGB1), reactive oxygen intermediates, and extracellular matrix breakdown products (Ionita et al., 2010). Finally, viral-derived molecules and host nucleic acids are sensed by nucleic acid-sensing TLRs localized in the endosomal compartment, i.e., TLR3, TLR7-9 (Takeuchi and Akira, 2009). Upon ligand binding, TLR dimerization activates common signaling pathways via the adaptor MyD88, except TLR3 that uses the adaptor TRIF, leading to the induction of either pro-inflammatory molecules through the activation of NF-κB or antiviral molecules via interferon regulatory factor (IRF) routes (Figure 1).
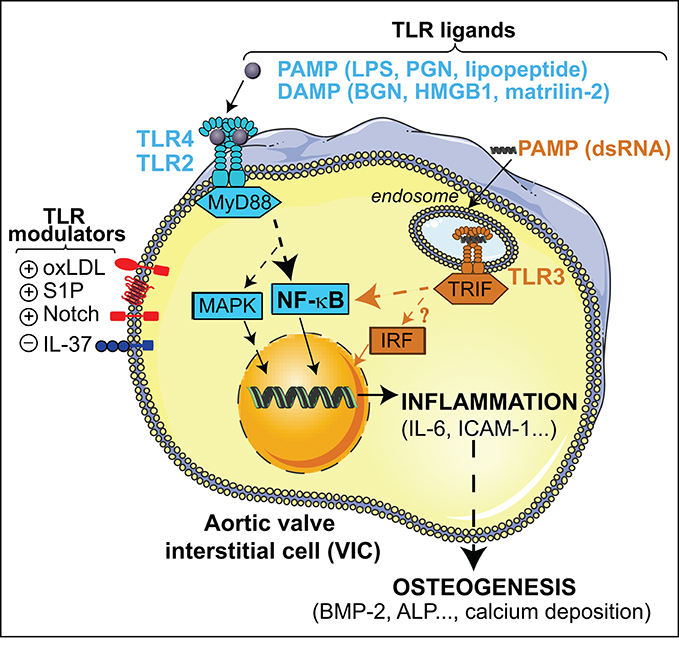
Figure 1. Overview of the TLR signaling pathways triggered by PAMPs and DAMPs in VIC. TLR2/4-MyD88 stimulation leads to NF-κB and MAP kinases activation and the subsequent induction of inflammatory mediators that promotes osteogenic reprogramming of VIC. Endosomal TLR3-TRIF stimulation triggers a non-canonical NF-κB signaling, and most likely IRF activation, thus promoting up-regulation of inflammatory and osteogenic mediators. Putative endosomal TLR4 signaling and TLR4-independent mechanisms are not depicted. ALP, alkaline phosphatase; ICAM-1, Intercellular Adhesion Molecule 1; PGN, peptidoglycan.
TLR-mediated inflammation is necessary for homeostasis and control of tissue damage; however, improper activation leads to chronic inflammation, thus promoting deleterious effects. A significant amount of evidence, including human genetic studies, supports the involvement of TLR in a variety of diseases such as sepsis, asthma and autoimmune diseases (Drexler and Foxwell, 2010; Netea et al., 2012). In the cardiovascular system, recent studies support the emerging role of TLR in inflammatory diseases like atherosclerosis and ischemia/reperfusion injury (Mann, 2011), and more recently CAVD (Mathieu et al., 2015).
TLR and CAVD Pathogenesis
The finding of pathogen cargo in stenotic aortic valve cusps prompted researchers to investigate the putative role of TLR in CAVD. Although a straightforward association between infective agents and valve calcification is lacking, Chlamydia pneumoniae and bacteria associated with chronic periodontal infection have been detected in stenotic valves at the level of the valvular fibrosa (Juvonen et al., 1997; Nakano et al., 2006; Skowasch et al., 2009; Edvinsson et al., 2010), and inoculation of oral bacteria has been found to cause aortic valve calcification in a rabbit model of recurrent low-grade endocarditis (Cohen et al., 2004). However, the potential pathogenic role of transient bacteremia associated with mucous membrane trauma and/or the ensuing cytokine response has not been translated into clinical practice.
Innate receptors have been posited as the molecular hubs between pathogen-derived molecules, inflammation and CAVD (Mathieu et al., 2015). Particularly, the association between TLR and CAVD is supported by around 30 articles conducted in cellular and animal models. This section summarizes current evidences supporting the key role of TLR in driving inflammation and valve calcification in response to several stimuli. In the aortic valve, TLR are present not only in infiltrating immune cells but also in resident cells, where TLR4 is the most abundant subtype (Meng et al., 2008; Yang et al., 2009; López et al., 2012). To note, most evidences are from aortic valve interstitial cells (VIC), a model used to study aortic valve inflammation and calcification (Mohler et al., 1999; Osman et al., 2006; Rutkovskiy et al., 2017) that shows a myofibroblast-like phenotype when cultured on plastic plates, a reason why they are termed activated VIC or aVIC (Liu et al., 2007; Wang et al., 2014).
TLR2/4 via MyD88/NF-κB Dependent Routes
The first members of the TLR family found to be expressed in aortic valve tissue and VIC were TLR4 and TLR2 (Meng et al., 2008), which are up-regulated in VIC explanted from calcified valves (Yang et al., 2009; López et al., 2012). In healthy human VICs, receptors are functional and mediate their effects via the MyD88 adaptor and the NF-κB route (Meng et al., 2008). Notably, the association of TLR with CAVD pathology has been established by using several pathogen-derived as well as endogenous molecules along the last decade (Figure 1).
PAMPs and Nonsterile Inflammation
Several highly conserved motifs present in pathogens but not in the host have been associated with inflammation and osteogenesis in the aortic valve via TLR.
Meng et al., first reported that the archetypal TLR4 agonist LPS acts as a proinflammatory factor in healthy human VIC by triggering NF-κB signaling and the subsequent induction of cytokines and adhesion molecules (Babu et al., 2008; Meng et al., 2008). These findings were later confirmed by our group and expanded to prostanoids (López et al., 2012). Moreover, VICs from calcified human valves are more responsive to LPS (Yang et al., 2009; Fernández-Pisonero et al., 2014). Remarkably, TLR4/2 ligands are also pro-calcific factors, as judged from several evidences emerged from bovine, porcine, and human VIC, as well as from mice models. Microarray and proteomic analysis revealed that LPS promotes a pro-calcific phenotype in human and clonal bovine VICs (Babu et al., 2008; Meng et al., 2008; Bertacco et al., 2010), where it functions as a pro-calcific factor by activating osteogenic mediators such as bone morphogenetic protein-2 (BMP-2), Runt-related transcription factor 2, and alkaline phosphatase. Moreover, LPS promoted in vitro calcification in studies using either β-glycerophosphate (Yang et al., 2009; López et al., 2012) or elevated phosphate levels (Rattazzi et al., 2008) as a phosphate source. In keeping with this, TLR2 ligands such as peptidoglycan, characteristic of the outer membrane of gram-positive bacteria, and synthetic peptides like Pam3CSK4 promote a pro-calcific phenotype in VIC (Yang et al., 2009; López et al., 2012). Even more, recent data highlight the in vivo role of TLR4/2 in osteogenesis since LPS promotes early aortic valve leaflet thickness increase in mice, and TLR4/2 deficiency abrogates high fat diet-induced aortic valve lesions. (Zeng et al., 2017). Interestingly, the response to TLR4/2 ligands differs according to cardiac valve site, being their effects stronger in aortic VICs as compared to pulmonary, mitral or tricuspid VICs (Yang et al., 2009; López et al., 2012; Venardos et al., 2014). Finally, a recent report hypothesizes that TLR-mediated effects are prevented in infants by a protective mechanism involving STAT3 activation that is absent in adults (Deng et al., 2015).
Together, evidences disclose a cardiac valve site-specific association between TLR4/2-NF-κB, inflammation and osteogenesis, and support an inflammation-driven calcification model in CAVD. Further studies on quiescent VIC cultured within 3D hydrogels (Hjortnaes et al., 2016) and on mice models to test the in vivo relevance are warranted. To note, additional LPS endosomal recognition via TRIF and TLR4-independent mechanisms cannot be ruled out (Tan and Kagan, 2014).
DAMPs and Sterile Inflammation
Endogenous molecules released upon tissue injury induce and perpetuate the so-called sterile inflammation (Ionita et al., 2010). Here, we describe several DAMPs recently linked to CAVD pathogenesis that mediate their effects via TLR.
Biglycan (BGN) is a small proteoglycan widely distributed within tissues that appears to be dysregulated in pathological conditions (Schaefer and Iozzo, 2008), including its overexpression in valves from CAVD patients (Derbali et al., 2010). Moreover, soluble BGN acts as a pro-inflammatory inducer through TLR pathways in human VICs by inducing lipid-modifying enzymes and cytokine expression via TLR2/4 (Derbali et al., 2010; Song et al., 2014). Song and colleagues further reported its pro-inflammatory and pro-osteogenic activities via a mechanism involving TLR2 and MAPK/ERK signaling (Song et al., 2012, 2014). A follow-up study demonstrated BGN-mediated pro-osteogenic reprogramming in VIC and identified BMP-2 and transforming growth factor-β1 as the molecular mediators (Song et al., 2015).
HMGB1 is a regulatory nuclear protein that when secreted extracellularly acts as a pro-inflammatory cytokine (Yang et al., 2005). Recent studies in patients and animal models have associated this protein with CAVD, since tissue and plasma levels of HMGB1 are increased in patients with CAVD (Wang et al., 2016a), and can be detected in the secretory granules of endothelial and interstitial cells explanted from diseased valves (Passmore et al., 2015). Additional evidences with recombinant HMGB1 show its pro-osteogenic activity by increasing osteogenic markers and calcium deposition in human VIC (Wang et al., 2016b). Moreover, the role of TLR4 and its transducers NF-κB and JNK, is supported by in vitro and in vivo studies, where HMGB1 pro-osteogenic activity was markedly decreased by gene silencing and TLR4-deficiency (Wang et al., 2016b; Shen et al., 2017).
Matrilin-2, an extracellular protein expressed in different tissues (Deák et al., 1999), accumulates in the calcific nodules of human aortic valves (Li et al., 2017). Moreover, matrilin-2 enhances osteogenic activity via TLR2/4 in VICs, as demonstrated by using both gene silencing and neutralizing antibodies, being the effects regulated by the NF-κB family of transcription factors and NFATc1 (Li et al., 2017).
Altogether, several DAMPs promote sterile inflammation and osteogenesis via TLR4/2-NF-κB routes, which warrants further investigation to test the in vivo relevance. Additional DAMPs associated to CAVD pathogenesis, i.e., galectin-3 and heat-shock protein (Skowasch et al., 2009; Sádaba et al., 2016) might mediate their effects through TLR signaling. It is plausible to think that the increased levels of DAMPs reported in calcified valves may act as a pro-calcific loop, thus contributing to disease progression.
TLR3 and the TRIF/IRF Dependent Routes
The TLR3 ligand dsRNA is present in virus and can also be produced under replication of positive-strand RNA viruses, dsRNA viruses, and DNA virus, i.e., poliovirus, coxsackievirus, and encephalomyocarditis virus, in the host. An endogenous source may be tissue damage or necrosis (Gantier and Williams, 2007). The first association of viral-derived molecules with inflammation and calcification of human VIC through TLR3 was reported using polyinosinic acid: polycytidylic acid to mimic dsRNA effects (López et al., 2012). Zhan et al., later confirmed dsRNA-mediated up-regulation of inflammatory mediators and pro-osteogenic activity, and further demonstrated by gene-silencing and neutralizing antibodies the involvement of TLR3-TRIF non canonical NF-κB signaling pathways as well as the ERK route (Zhan et al., 2015, 2017). Moreover, recent in vivo data presented at European Society of Cardiology Congress 2017 have associated TLR3 to the onset of calcific aortic valve disease by using TLR3- and ApoE-deficient mice models (Tepekoylu et al., 2017).
In summary, these findings posit the association between TLR and osteogenesis with no reported differences between sterile and non-sterile inflammation, and support an inflammation-driven calcification model in CAVD. Future studies are needed to analyze putative mechanistic differences associated to different TLR pathways and their in vivo relevance.
Modulation of TLR Signaling in the Aortic Valve: Crosstalk with Membrane Receptors
TLR activation relevant to CAVD can be modulated by several inflammatory mediators at various levels, but remarkably all converge on the NF-κB route. This section includes TLR modulators reported to date in the context of aortic valve physiopathology (Figure 1).
Lipoproteins and Lipid Components
Among CAVD risk factors are high levels of circulating oxidized LDL (ox-LDL), which also accumulate in diseased aortic valves (Côté et al., 2008; Yeang et al., 2016). oxLDL treatment of human VIC modulates LPS effects by synergistically increasing pro-osteogenic genes (Zeng et al., 2014). Pharmacological and gene silencing approaches revealed a mechanism involving the modulation of the Notch-NF-κB axis (Zeng et al., 2014). Remarkably, blocking TLR2/4 with neutralizing antibodies abolished oxLDL-induced activation of NF-κB and ERK1/2 (Zeng et al., 2017). Sphingosine 1-phosphate, a lipoprotein lipid component acting via G-protein coupled receptors, modulates TLR activation in human VICs, macrophages and aortic endothelial cells (Dueñas et al., 2008; Fernández-Pisonero et al., 2012, 2014). In healthy human VICs, TLR-sphingosine 1-phosphate receptor interplay leads to the potentiation of inflammatory, angiogenic, and osteogenic responses through NF-κB and p38/MAPK signaling (Fernández-Pisonero et al., 2014).
Notch
Genetic studies in humans and experiments in mice models suggest the association between the transmembrane receptor Notch and CAVD (Garg et al., 2005; Nigam and Srivastava, 2009). Recent studies provide evidence of the interplay between TLR and Notch pathways on the expression of inflammatory and osteogenic mediators and highlight the importance of their cross-talk in VICs from calcified valves, which have elevated levels of Notch1 (Zeng et al., 2012, 2013). Moreover, the mechanism of TLR4-Notch1 interplay includes the modulation of NF-κB and BMP-2 through a process dependent on Notch 1 cleavage and nuclear translocation (Zeng et al., 2012, 2013).
Cytokines
IL-37, an anti-inflammatory cytokine that mediates its effects through IL-18 receptor and suppression of NF-κB function (Dinarello et al., 2016), is expressed in human VIC and down-regulated in calcified valves (Zhan et al., 2017). Two recent studies uncovered its anti-inflammatory and anti-osteogenic functions by modulating LPS-induced responses in human VIC (Zeng et al., 2017; Zhan et al., 2017). IL-37 negatively modulates AVIC osteogenic responses to both PAMPs such as TLR4/2 agonists, and DAMPs like oxLDL, by inhibiting ERK1/2 and NF-κB activities. Remarkably, IL-37 transgenic mice are protected against early aortic valve lesions induced by prolonged exposure to proinflammatory agents such as LPS (Zeng et al., 2017). Interestingly, IL-37 anti-inflammatory effect is specific of TLR4/2 ligands, most likely by regulating the MyD88-mediated canonical activation of NF-κB (Zhan et al., 2017).
Collectively, evidences provide mechanistic insights into the crosstalk between TLR and membrane receptors relevant to CAVD. Whether this crosstalk occurs at the level of trafficking or signaling and how these multiple pathways integrate in vivo needs to be elucidated. Their regulation may lead to identify therapeutic targets for the suppression of valvular inflammation.
Conclusions and Perspectives
Recent data highlight a potentially important link between the TLR-NF-κB axis, inflammation, and CAVD pathogenesis. Accumulating evidences suggest that TLRs could be significant contributors to the pathogenesis of valvular inflammation, and the ensuing inflammation-driven calcification processes in aortic valves (Figure 2). The key question, whether TLR signaling is involved in the onset of clinically relevant calcification, needs to be investigated using in vivo models recapitulating hyperphosphatemic and/or inflammatory calcification triggers, with an emphasis on the analysis of cell phenotypes to elucidate osteogenic and/or dystrophic mechanisms (Hutcheson et al., 2017). Future studies will benefit from new approaches like optical molecular imaging for the detection of early-stage calcification and 3D cultures of VIC (New and Aikawa, 2011; Hjortnaes et al., 2016), and will help to clearly define the therapeutic potential of TLRs and their signaling transducers in the initial stages of CAVD. The major challenge of targeting this pathway will be minimizing harmful innate immune responses, while preserving appropriate innate immune defense mechanisms.
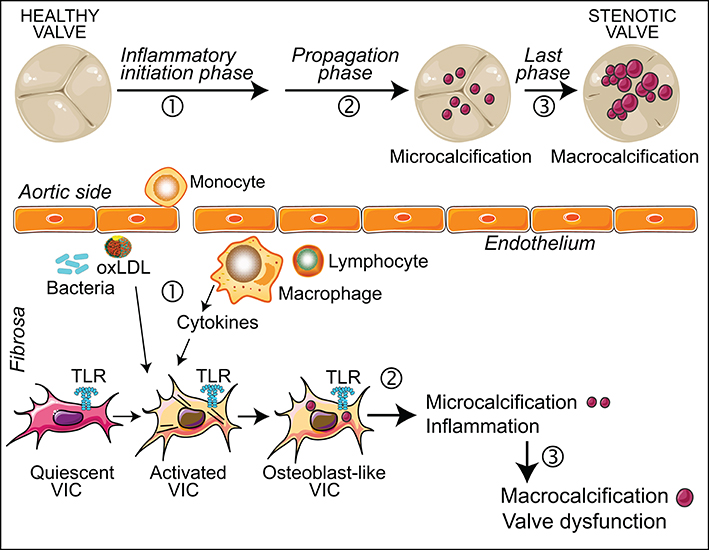
Figure 2. Inflammation in CAVD progression. The scheme shows the role of inflammation in the different stages of CAVD, at both valve and cellular levels. (1) Valve endothelial layer disruption leads to the recruitment of immune cells and oxLDL. This inflammatory milieu promotes the activation and differentiation of quiescent VIC. These cells express functional TLR, the activation of which induces inflammation and osteogenic reprogramming. (2) In the propagation phase, inflammation promotes the development of microcalcifications. (3) Large scale calcification leads to leaflet thickness and valve dysfunction.
Author Contributions
CG-R: Contributed to the design and the writing of the manuscript, and to figure preparation; IP-I: Drafted the manuscript and designed figures; IC-M, JL, JAS, and MS: Drafted and revised the manuscript.
Funding
Sources of funding include grants from the Instituto de Salud Carlos III (Spanish Ministry of Health): PI14/00022, CIBERCV; the Ministry of Economy and Competitivity of Spain, cofunded by the European Social Fund: SAF2013-44521-R; Junta de Castilla y León: GRS 1432/A/16, BIO/VA47/14, BIO/VA36/15, CSI035P17; Fundación Domingo Martínez. IP-I was a fellow from the University of Valladolid co-funded by Banco de Santander; IC-M was supported by a pre-doctoral fellowship from Regional Government of Castilla y León and the European Social Fund co-funded by CSIC.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
BGN, Biglycan; BMP-2, Bone morphogenetic protein-2; CAVD, calcific aortic valve disease; DAMP, danger/damage associated molecular patterns; HMGB1, high-mobility group box 1; IL, interleukin; IRF, interferon regulatory factor; LPS, Lipopolysaccharide; oxLDL, oxidized low-density lipoprotein; PAMP, pathogen associated molecular patterns; TLR, Toll-like receptor; VIC, aortic valve interstitial cell.
References
Aikawa, E., and Otto, C. M. (2012). Look more closely at the valve: imaging calcific aortic valve disease. Circulation 125, 9–11. doi: 10.1161/CIRCULATIONAHA.111.073452
Babu, A. N., Meng, X., Zou, N., Yang, X., Wang, M., Song, Y., et al. (2008). Lipopolysaccharide stimulation of human aortic valve interstitial cells activates inflammation and osteogenesis. Ann. Thorac. Surg. 86, 71–76. doi: 10.1016/j.athoracsur.2008.03.008
Bertacco, E., Millioni, R., Arrigoni, G., Faggin, E., Iop, L., Puato, M., et al. (2010). Proteomic analysis of clonal interstitial aortic valve cells acquiring a pro-calcific profile. J. Proteome Res. 9, 5913–5921. doi: 10.1021/pr100682g
Bridgewater, B., Kinsman, R., Walton, P., Gummert, J., and Kappetein, A. P. (2011). The 4th European Association for cardio-thoracic surgery adult cardiac surgery database report. Interact. Cardiovasc. Thorac. Surg. 12, 4–5. doi: 10.1510/icvts.2010.251744
Chan, K. L., Teo, K., Dumesnil, J. G., Ni, A., and Tam, J. (2010). Effect of lipid lowering with rosuvastatin on progression of aortic stenosis: results of the aortic stenosis progression observation: measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation 121, 306–314. doi: 10.1161/CIRCULATIONAHA.109.900027
Cohen, D. J., Malave, D., Ghidoni, J. J., Iakovidis, P., Everett, M. M., You, S., et al. (2004). Role of oral bacterial flora in calcific aortic stenosis: an animal model. Ann. Thorac. Surg. 77, 537–543. doi: 10.1016/S0003-4975(03)01454-1
Côté, C., Pibarot, P., Després, J. P., Mohty, D., Cartier, A., Arsenault, B. J., et al. (2008). Association between circulating oxidised low-density lipoprotein and fibrocalcific remodelling of the aortic valve in aortic stenosis. Heart 94, 1175–1180. doi: 10.1136/hrt.2007.125740
Coté, N., Mahmut, A., Bosse, Y., Couture, C., Pagé, S., Trahan, S., et al. (2013). Inflammation is associated with the remodeling of calcific aortic valve disease. Inflammation 36, 573–581. doi: 10.1007/s10753-012-9579-6
Cowell, S. J., Newby, D. E., Prescott, R. J., Bloomfield, P., Reid, J., Northridge, D. B., et al. (2005). A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N. Engl. J. Med. 352, 2389–2397. doi: 10.1056/NEJMoa043876
Deák, F., Wagener, R., Kiss, I., and Paulsson, M. (1999). The matrilins: a novel family of oligomeric extracellular matrix proteins. Matrix Biol. 18, 55–64. doi: 10.1016/S0945-053X(98)00006-7
Deng, X. S., Meng, X., Zeng, Q., Fullerton, D., Mitchell, M., and Jaggers, J. (2015). Adult aortic valve interstitial cells have greater responses to toll-like receptor 4 stimulation. Ann. Thorac. Surg. 99, 62–71. doi: 10.1016/j.athoracsur.2014.07.027
Derbali, H., Bossé, Y., Côté, N., Pibarot, P., Audet, A., Pepin, A., et al. (2010). Increased biglycan in aortic valve stenosis leads to the overexpression of phospholipid transfer protein via Toll-like receptor 2. Am. J. Pathol. 176, 2638–2645. doi: 10.2353/ajpath.2010.090541
Dinarello, C. A., Nold-Petry, C., Nold, M., Fujita, M., Li, S., Kim, S., et al. (2016). Suppression of innate inflammation and immunity by interleukin-37. Eur. J. Immunol. 46, 1067–1081. doi: 10.1002/eji.201545828
Drexler, S. K., and Foxwell, B. M. (2010). The role of toll-like receptors in chronic inflammation. Int. J. Biochem. Cell Biol. 42, 506–518. doi: 10.1016/j.biocel.2009.10.009
Dueñas, A. I., Aceves, M., Fernández-Pisonero, I., Gómez, C., Orduña, A., Crespo, M. S., et al. (2008). Selective attenuation of Toll-like receptor 2 signalling may explain the atheroprotective effect of sphingosine 1-phosphate. Cardiovasc. Res. 79, 537–544. doi: 10.1093/cvr/cvn087
Dweck, M. R., Khaw, H. J., Sng, G. K., Luo, E. L., Baird, A., Williams, M. C., et al. (2013). Aortic stenosis, atherosclerosis, and skeletal bone: is there a common link with calcification and inflammation? Eur. Heart J. 34, 1567–1574. doi: 10.1093/eurheartj/eht034
Edvinsson, M., Hjelm, E., Thelin, S., Friman, G., and Nystrom-Rosander, C. (2010). Presence of Chlamydophila pneumoniae DNA but not mRNA in stenotic aortic heart valves. Int. J. Cardiol. 143, 57–62. doi: 10.1016/j.ijcard.2009.01.052
Fernández-Pisonero, I., Dueñas, A. I., Barreiro, O., Montero, O., Sánchez-Madrid, F., and García-Rodríguez, C. (2012). Lipopolysaccharide and sphingosine-1-phosphate cooperate to induce inflammatory molecules and leukocyte adhesion in endothelial cells. J. Immunol. 189, 5402–5410. doi: 10.4049/jimmunol.1201309
Fernández-Pisonero, I., López, J., Onecha, E., Dueñas, A. I., Maeso, P., Crespo, M. S., et al. (2014). Synergy between sphingosine 1-phosphate and lipopolysaccharide signaling promotes an inflammatory, angiogenic and osteogenic response in human aortic valve interstitial cells. PLoS ONE 9:e109081. doi: 10.1371/journal.pone.0109081
Gantier, M. P., and Williams, B. R. (2007). The response of mammalian cells to double-stranded RNA. Cytokine Growth Factor Rev. 18, 363–371. doi: 10.1016/j.cytogfr.2007.06.016
Garg, V., Muth, A. N., Ransom, J. F., Schluterman, M. K., Barnes, R., King, I. N., et al. (2005). Mutations in NOTCH1 cause aortic valve disease. Nature 437, 270–274. doi: 10.1038/nature03940
Go, A. S., Mozaffarian, D., Roger, V. L., Benjamin, E. J., Berry, J. D., Blaha, M. J., et al. (2014). Heart disease and stroke statistics−2014 update: a report from the American Heart Association. Circulation 129, e28–e292. doi: 10.1161/01.cir.0000442015.53336.12
Hjortnaes, J., Butcher, J., Figueiredo, J. L., Riccio, M., Kohler, R. H., Kozloff, K. M., et al. (2010). Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodelling: a role for inflammation. Eur. Heart J. 31, 1975–1984. doi: 10.1093/eurheartj/ehq237
Hjortnaes, J., Goettsch, C., Hutcheson, J. D., Camci-Unal, G., Lax, L., Scherer, K., et al. (2016). Simulation of early calcific aortic valve disease in a 3D platform: a role for myofibroblast differentiation. J. Mol. Cell. Cardiol. 94, 13–20. doi: 10.1016/j.yjmcc.2016.03.004
Hutcheson, J. D., Blaser, M. C., and Aikawa, E. (2017). Giving calcification its due: recognition of a diverse disease: a first attempt to standardize the field. Circ. Res. 120, 270–273. doi: 10.1161/CIRCRESAHA.116.310060
Ionita, M. G., Arslan, F., de Kleijn, D. P., and Pasterkamp, G. (2010). Endogenous inflammatory molecules engage Toll-like receptors in cardiovascular disease. J. Innate Immun. 2, 307–315. doi: 10.1159/000314270
Juvonen, J., Laurila, A., Juvonen, T., Aläkarppä, H., Surcel, H. M., Lounatmaa, K., et al. (1997). Detection of Chlamydia pneumoniae in human nonrheumatic stenotic aortic valves. J. Am. Coll. Cardiol. 29, 1054–1059. doi: 10.1016/S0735-1097(97)00003-X
Kawai, T., and Akira, S. (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384. doi: 10.1038/ni.1863
Li, F., Song, R., Ao, L., Reece, T. B., Cleveland, J. C. Jr., Dong, N., et al. (2017). ADAMTS5 deficiency in calcified aortic valves is associated with elevated pro-osteogenic activity in valvular interstitial cells. Arterioscler. Thromb. Vasc. Biol. 37, 1339–1351. doi: 10.1161/ATVBAHA.117.309021
Liu, A. C., Joag, V. R., and Gotlieb, A. I. (2007). The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am. J. Pathol. 171, 1407–1418. doi: 10.2353/ajpath.2007.070251
López, J., Fernández-Pisonero, I., Dueñas, A. I., Maeso, P., Román, J. A., Crespo, M. S., et al. (2012). Viral and bacterial patterns induce TLR-mediated sustained inflammation and calcification in aortic valve interstitial cells. Int. J. Cardiol. 158, 18–25. doi: 10.1016/j.ijcard.2010.12.089
Mann, D. L. (2011). The emerging role of innate immunity in the heart and vascular system: for whom the cell tolls. Circ. Res. 108, 1133–1145. doi: 10.1161/CIRCRESAHA.110.226936
Mathieu, P., Bouchareb, R., and Boulanger, M. C. (2015). Innate and adaptive immunity in calcific aortic valve disease. J. Immunol. Res. 2015:851945. doi: 10.1155/2015/851945
Meng, X., Ao, L., Song, Y., Babu, A., Yang, X., Wang, M., et al. (2008). Expression of functional Toll-like receptors 2 and 4 in human aortic valve interstitial cells: potential roles in aortic valve inflammation and stenosis. Am. J. Physiol. Cell Physiol. 294, C29–C35. doi: 10.1152/ajpcell.00137.2007
Miller, J. D., Weiss, R. M., and Heistad, D. D. (2011). Calcific aortic valve stenosis: methods, models, and mechanisms. Circ. Res. 108, 1392–1412. doi: 10.1161/CIRCRESAHA.110.234138
Mohler, E. R. III., Chawla, M. K., Chang, A. W., Vyavahare, N., Levy, R. J., Graham, L., et al. (1999). Identification and characterization of calcifying valve cells from human and canine aortic valves. J. Heart Valve Dis. 8, 254–260.
Nakano, K., Inaba, H., Nomura, R., Nemoto, H., Takeda, M., Yoshioka, H., et al. (2006). Detection of cariogenic Streptococcus mutans in extirpated heart valve and atheromatous plaque specimens. J. Clin. Microbiol. 44, 3313–3317. doi: 10.1128/JCM.00377-06
Netea, M. G., Wijmenga, C., and O'neill, L. A. (2012). Genetic variation in Toll-like receptors and disease susceptibility. Nat. Immunol. 13, 535–542. doi: 10.1038/ni.2284
New, S. E., and Aikawa, E. (2011). Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ. Res. 108, 1381–1391. doi: 10.1161/CIRCRESAHA.110.234146
Nigam, V., and Srivastava, D. (2009). Notch1 represses osteogenic pathways in aortic valve cells. J. Mol. Cell. Cardiol. 47, 828–834. doi: 10.1016/j.yjmcc.2009.08.008
Nkomo, V. T., Gardin, J. M., Skelton, T. N., Gottdiener, J. S., Scott, C. G., and Enriquez-Sarano, M. (2006). Burden of valvular heart diseases: a population-based study. Lancet 368, 1005–1011. doi: 10.1016/S0140-6736(06)69208-8
O'Brien, K. D. (2006). Pathogenesis of calcific aortic valve disease: a disease process comes of age (and a good deal more). Arterioscler. Thromb. Vasc. Biol. 26, 1721–1728. doi: 10.1161/01.ATV.0000227513.13697.ac
Osman, L., Yacoub, M. H., Latif, N., Amrani, M., and Chester, A. H. (2006). Role of human valve interstitial cells in valve calcification and their response to atorvastatin. Circulation 114, I547–552. doi: 10.1161/CIRCULATIONAHA.105.001115
Otto, C. M. (2008). Calcific aortic stenosis–time to look more closely at the valve. N. Engl. J. Med. 359, 1395–1398. doi: 10.1056/NEJMe0807001
Otto, C. M., and Prendergast, B. (2014). Aortic-valve stenosis–from patients at risk to severe valve obstruction. N. Engl. J. Med. 371, 744–756. doi: 10.1056/NEJMra1313875
Passmore, M., Nataatmadja, M., Fung, Y. L., Pearse, B., Gabriel, S., Tesar, P., et al. (2015). Osteopontin alters endothelial and valvular interstitial cell behaviour in calcific aortic valve stenosis through HMGB1 regulation. Eur. J. Cardiothorac. Surg. 48, e20–e29. doi: 10.1093/ejcts/ezv244
Pawade, T. A., Newby, D. E., and Dweck, M. R. (2015). Calcification in aortic stenosis: the skeleton key. J. Am. Coll. Cardiol. 66, 561–577. doi: 10.1016/j.jacc.2015.05.066
Rajamannan, N. M., Evans, F. J., Aikawa, E., Grande-Allen, K. J., Demer, L. L., Heistad, D. D., et al. (2011). Calcific aortic valve disease: not simply a degenerative process: a review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: calcific aortic valve disease-2011 update. Circulation 124, 1783–1791. doi: 10.1161/CIRCULATIONAHA.110.006767
Rattazzi, M., Iop, L., Faggin, E., Bertacco, E., Zoppellaro, G., Baesso, I., et al. (2008). Clones of interstitial cells from bovine aortic valve exhibit different calcifying potential when exposed to endotoxin and phosphate. Arterioscler. Thromb. Vasc. Biol. 28, 2165–2172. doi: 10.1161/ATVBAHA.108.174342
Roberts, W. C., and Ko, J. M. (2005). Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation 111, 920–925. doi: 10.1161/01.CIR.0000155623.48408.C5
Rossebø, A. B., Pedersen, T. R., Boman, K., Brudi, P., Chambers, J. B., Egstrup, K., et al. (2008). Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N. Engl. J. Med. 359, 1343–1356. doi: 10.1056/NEJMoa0804602
Rutkovskiy, A., Malashicheva, A., Sullivan, G., Bogdanova, M., Kostareva, A., Stensløkken, K. O., et al. (2017). Valve interstitial cells: the key to understanding the pathophysiology of heart valve calcification. J. Am. Heart Assoc. 6:e006339. doi: 10.1161/JAHA.117.006339
Sádaba, J. R., Martínez-Martínez, E., Arrieta, V., Álvarez, V., Fernández-Celis, A., Ibarrola, J., et al. (2016). Role for galectin-3 in calcific aortic valve stenosis. J. Am. Heart. Assoc. 5:e004360. doi: 10.1161/JAHA.116.004360
Schaefer, L., and Iozzo, R. V. (2008). Biological functions of the small leucine-rich proteoglycans: from genetics to signal transduction. J. Biol. Chem. 283, 21305–21309. doi: 10.1074/jbc.R800020200
Shen, W., Zhou, J., Wang, C., Xu, G., Wu, Y., and Hu, Z. (2017). High mobility group box 1 induces calcification of aortic valve interstitial cells via toll-like receptor 4. Mol. Med. Rep. 15, 2530–2536. doi: 10.3892/mmr.2017.6287
Skowasch, D., Tuleta, I., Steinmetz, M., Pabst, S., Preusse, C. J., Welz, A., et al. (2009). Pathogen burden in degenerative aortic valves is associated with inflammatory and immune reactions. J. Heart Valve Dis. 18, 411–417. Available online at: https://www.icr-heart.com/?cid=2098
Song, R., Ao, L., Zhao, K. S., Zheng, D., Venardos, N., Fullerton, D. A., et al. (2014). Soluble biglycan induces the production of ICAM-1 and MCP-1 in human aortic valve interstitial cells through TLR2/4 and the ERK1/2 pathway. Inflamm. Res. 63, 703–710. doi: 10.1007/s00011-014-0743-3
Song, R., Fullerton, D. A., Ao, L., Zheng, D., Zhao, K. S., and Meng, X. (2015). BMP-2 and TGF-beta1 mediate biglycan-induced pro-osteogenic reprogramming in aortic valve interstitial cells. J. Mol. Med. 93, 403–412. doi: 10.1007/s00109-014-1229-z
Song, R., Zeng, Q., Ao, L., Yu, J. A., Cleveland, J. C., Zhao, K. S., et al. (2012). Biglycan induces the expression of osteogenic factors in human aortic valve interstitial cells via Toll-like receptor-2. Arterioscler. Thromb. Vasc. Biol. 32, 2711–2720. doi: 10.1161/ATVBAHA.112.300116
Stewart, B. F., Siscovick, D., Lind, B. K., Gardin, J. M., Gottdiener, J. S., Smith, V. E., et al. (1997). Clinical factors associated with calcific aortic valve disease. Cardiovascular health study. J. Am. Coll. Cardiol. 29, 630–634. doi: 10.1016/S0735-1097(96)00563-3
Takeuchi, O., and Akira, S. (2009). Innate immunity to virus infection. Immunol. Rev. 227, 75–86. doi: 10.1111/j.1600-065X.2008.00737.x
Tan, Y., and Kagan, J. C. (2014). A cross-disciplinary perspective on the innate immune responses to bacterial lipopolysaccharide. Mol. Cell 54, 212–223. doi: 10.1016/j.molcel.2014.03.012
Tepekoylu, C. G. M., Poelzl, L., Hirsch, J., Kirchmair, E., Degenhart, G., et al. (2017). Toll-like receptor 3 mediates the onset of calcific aortic valve disease. Eur. Heart J. 38:ehx501.39. doi: 10.1093/eurheartj/ehx501.39
Towler, D. A. (2013). Molecular and cellular aspects of calcific aortic valve disease. Circ. Res. 113, 198–208. doi: 10.1161/CIRCRESAHA.113.300155
Venardos, N., Nadlonek, N. A., Zhan, Q., Weyant, M. J., Reece, T. B., Meng, X., et al. (2014). Aortic valve calcification is mediated by a differential response of aortic valve interstitial cells to inflammation. J. Surg. Res. 190, 1–8. doi: 10.1016/j.jss.2014.03.051
Wang, B., Wei, G., Liu, B., Zhou, X., Xiao, H., Dong, N., et al. (2016a). The role of high mobility group box 1 protein in interleukin-18-induced myofibroblastic transition of valvular interstitial cells. Cardiology 135, 168–178. doi: 10.1159/000447483
Wang, B., Li, F., Zhang, C., Wei, G., Liao, P., and Dong, N. (2016b). High-mobility group box-1 protein induces osteogenic phenotype changes in aortic valve interstitial cells. J. Thorac. Cardiovasc. Surg. 151, 255–262. doi: 10.1016/j.jtcvs.2015.09.077
Wang, H., Leinwand, L. A., and Anseth, K. S. (2014). Cardiac valve cells and their microenvironment–insights from in vitro studies. Nat. Rev. Cardiol. 11, 715–727. doi: 10.1038/nrcardio.2014.162
Yang, H., Wang, H., Czura, C. J., and Tracey, K. J. (2005). The cytokine activity of HMGB1. J. Leukoc. Biol. 78, 1–8. doi: 10.1189/jlb.1104648
Yang, X., Fullerton, D. A., Su, X., Ao, L., Cleveland, J. C. Jr., and Meng, X. (2009). Pro-osteogenic phenotype of human aortic valve interstitial cells is associated with higher levels of Toll-like receptors 2 and 4 and enhanced expression of bone morphogenetic protein 2. J. Am. Coll. Cardiol. 53, 491–500. doi: 10.1016/j.jacc.2008.09.052
Yeang, C., Wilkinson, M. J., and Tsimikas, S. (2016). Lipoprotein(a) and oxidized phospholipids in calcific aortic valve stenosis. Curr. Opin. Cardiol. 31, 440–450. doi: 10.1097/HCO.0000000000000300
Yetkin, E., and Waltenberger, J. (2009). Molecular and cellular mechanisms of aortic stenosis. Int. J. Cardiol. 135, 4–13. doi: 10.1016/j.ijcard.2009.03.108
Zeng, Q., Jin, C., Ao, L., Cleveland, J. C. Jr., Song, R., Xu, D., et al. (2012). Cross-talk between the Toll-like receptor 4 and Notch1 pathways augments the inflammatory response in the interstitial cells of stenotic human aortic valves. Circulation 126, S222–230. doi: 10.1161/CIRCULATIONAHA.111.083675
Zeng, Q., Song, R., Ao, L., Weyant, M. J., Lee, J., Xu, D., et al. (2013). Notch1 promotes the pro-osteogenic response of human aortic valve interstitial cells via modulation of ERK1/2 and nuclear factor-kappaB activation. Arterioscler. Thromb. Vasc. Biol. 33, 1580–1590. doi: 10.1161/ATVBAHA.112.300912
Zeng, Q., Song, R., Ao, L., Xu, D., Venardos, N., Fullerton, D. A., et al. (2014). Augmented osteogenic responses in human aortic valve cells exposed to oxLDL and TLR4 agonist: a mechanistic role of Notch1 and NF-kappaB interaction. PLoS ONE 9:e95400. doi: 10.1371/journal.pone.0095400
Zeng, Q., Song, R., Fullerton, D. A., Ao, L., Zhai, Y., Li, S., et al. (2017). Interleukin-37 suppresses the osteogenic responses of human aortic valve interstitial cells in vitro and alleviates valve lesions in mice. Proc. Natl. Acad. Sci. U.S.A. 114, 1631–1636. doi: 10.1073/pnas.1619667114
Zhan, Q., Song, R., Zeng, Q., Yao, Q., Ao, L., Xu, D., et al. (2015). Activation of TLR3 induces osteogenic responses in human aortic valve interstitial cells through the NF-kappaB and ERK1/2 pathways. Int. J. Biol. Sci. 11, 482–493. doi: 10.7150/ijbs.10905
Keywords: Toll-like receptor (TLR), inflammation, NF-κB, osteogenesis, aortic valve interstitial cell (VIC), calcific aortic valve disease (CAVD)
Citation: García-Rodríguez C, Parra-Izquierdo I, Castaños-Mollor I, López J, San Román JA and Sánchez Crespo M (2018) Toll-Like Receptors, Inflammation, and Calcific Aortic Valve Disease. Front. Physiol. 9:201. doi: 10.3389/fphys.2018.00201
Received: 08 January 2018; Accepted: 23 February 2018;
Published: 12 March 2018.
Edited by:
Alexandrina Ferreira Mendes, University of Coimbra, PortugalReviewed by:
Joshua D. Hutcheson, Florida International University, United StatesCynthia St. Hilaire, University of Pittsburgh, United States
Copyright © 2018 García-Rodríguez, Parra-Izquierdo, Castaños-Mollor, López, San Román and Sánchez Crespo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carmen García-Rodríguez, Y2dhcmNpYUBpYmdtLnV2YS5lcw==