 Sanda Despa
Sanda Despa- Department of Pharmacology and Nutritional Sciences, University of Kentucky, Lexington, KY, United States
By controlling the function of various sarcolemmal and mitochondrial ion transporters, intracellular Na+ concentration ([Na+]i) regulates Ca2+ cycling, electrical activity, the matching of energy supply and demand, and oxidative stress in cardiac myocytes. Thus, maintenance of myocyte Na+ homeostasis is vital for preserving the electrical and contractile activity of the heart. [Na+]i is set by the balance between the passive Na+ entry through numerous pathways and the pumping of Na+ out of the cell by the Na+/K+-ATPase. This equilibrium is perturbed in heart failure, resulting in higher [Na+]i. More recent studies have revealed that [Na+]i is also increased in myocytes from diabetic hearts. Elevated [Na+]i causes oxidative stress and augments the sarcoplasmic reticulum Ca2+ leak, thus amplifying the risk for arrhythmias and promoting heart dysfunction. This mini-review compares and contrasts the alterations in Na+ extrusion and/or Na+ uptake that underlie the [Na+]i increase in heart failure and diabetes, with a particular emphasis on the emerging role of Na+ - glucose cotransporters in the diabetic heart.
Maintenance of Myocyte Na+ Homeostasis is Vital for Preserving Heart Function
All mammalian cells maintain a low intracellular Na+ concentration ([Na+]i) by actively extruding Na+ through the Na+/K+-ATPase (NKA) at the expense of metabolic energy. The energy stored in the electrochemical Na+ gradient is then used for the transmembrane transport of other ions (e.g., Ca2+ through the Na+/Ca2+ exchanger, NCX, H+ via the Na+/H+ exchanger, NHE, etc.), uptake of energy substrates (glucose through the family of Na+-glucose cotransporters, SGLTs, and aminoacids through Na+-aminoacid cotransporter) and, in the case of excitable cells, generation of action potentials (via voltage-gated Na+ channels). Changes in [Na+]i critically affect the function of these transporters, therefore [Na+]i homeostasis is essential for numerous cellular processes.
In cardiac myocytes, NCX is the main route for Ca2+ extrusion from the cells (Bers, 2001), which intimately links Ca2+ cycling to [Na+]i. Even a small (few mM) increase in [Na+]i alters Ca2+ fluxes through NCX, resulting in higher Ca2+ levels in the cytosol and sarcoplasmic reticulum (SR) and consequently larger contractions (Figure 1). This mechanism is responsible for the inotropic effect of cardiac glycosides such as digoxin. However, as demonstrated clinically with digoxin, the beneficial effect of enhanced contractility is counteracted by a higher risk for ectopic arrhythmias, as larger SR Ca2+ load increases the incidence of spontaneous Ca2+ waves and delayed afterdepolarizations (Bers, 2014).
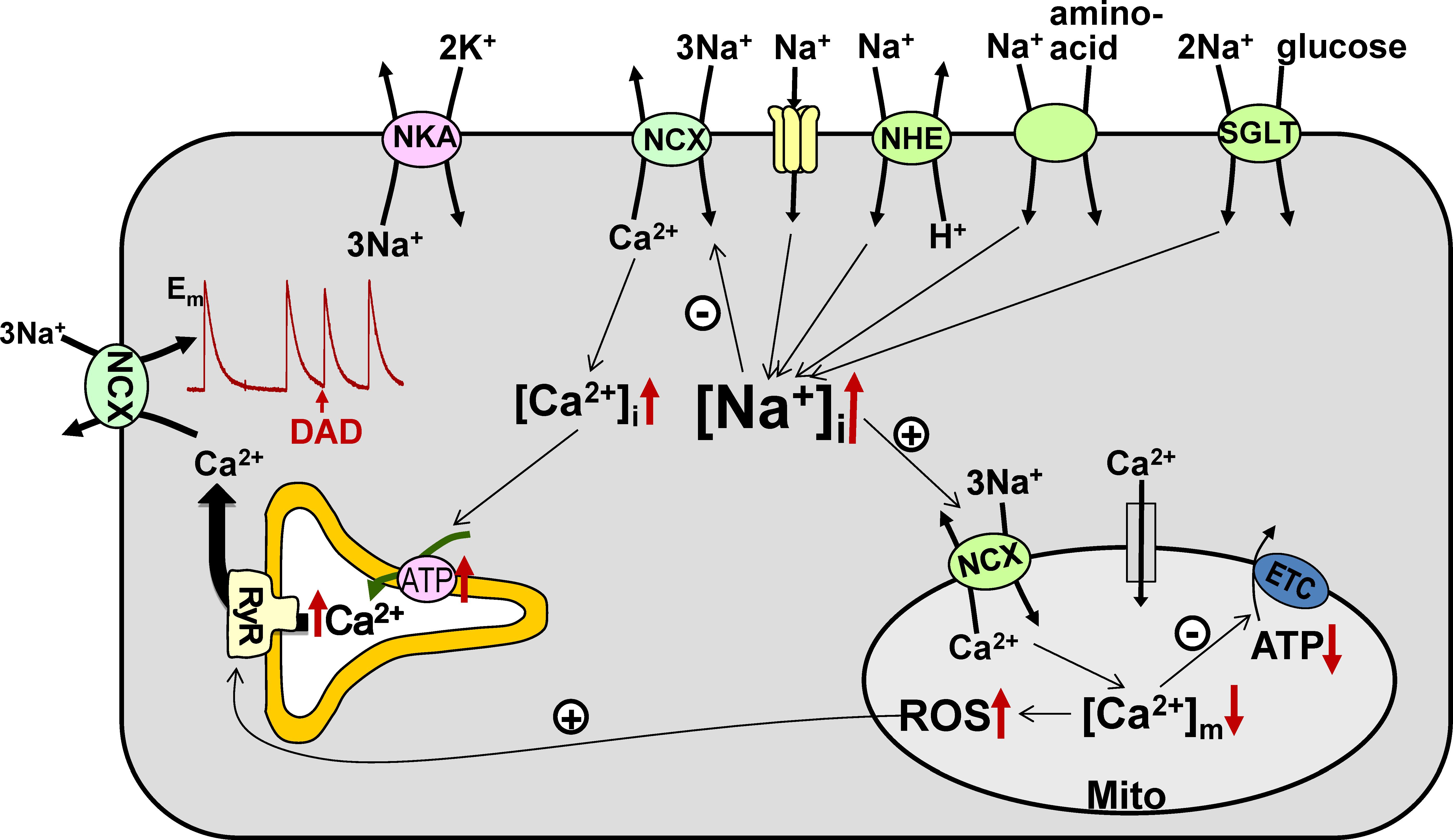
FIGURE 1. Na+ transport pathways and consequences of elevated [Na+]i in cardiac myocytes. Several pathways, including the Na+/Ca2+ exchanger (NCX), Na+ channels, Na+/H+ exchanger (NHE), Na+-glucose cotransporter (SGLT), and Na+-aminoacid cotransporter, contribute to the entry of Na+ into cardiac myocytes, while the Na+/K+-ATPase (NKA) is the essential route for Na+ efflux. Either enhanced Na+ influx or impaired Na+ extrusion result in higher [Na+]i. Elevated [Na+]i leads to (i) increased cellular and SR Ca2+ load, which augments the risk for the occurrence of delayed afterdepolarizations (DADs), and (ii) reduced mitochondrial Ca2+ levels, which leads to lower ATP production and causes oxidative stress. ETC – electron transport chain.
[Na+]i also controls the level of Ca2+ in the mitochondria ([Ca2+]m) through the mitochondrial Na+/Ca2+ exchanger (mitoNCX), which is the main route for mitochondrial Ca2+ extrusion in the heart (Griffiths, 2009). MitoNCX is half-maximally activated at [Na+]i in the physiological range (5–10 mM) (Boyman et al., 2013). Therefore, mitoNCX is very sensitive to changes in [Na+]i. An increase in [Na+]i accelerates mitochondrial Ca2+ efflux and thus reduces [Ca2+]m (Cox and Matlib, 1993; Maack et al., 2006). Because [Ca2+]m stimulates several dehydrogenases involved in the tricarboxylic acid cycle (McCormack et al., 1990), regeneration of NADH and NADPH from their oxidized forms slows down at lower [Ca2+]m. Slower restoration of the NADH pool limits the rate of electron transport and thus diminishes mitochondrial ATP production. Notably however, glycolytic ATP also drives cellular processes in the heart, particularly NKA (Glitsch and Tappe, 1993). NADPH is utilized to neutralize the H2O2 produced by the electron transport chain and lower NADPH levels may result in oxidative stress (Bertero and Maack, 2018). Abnormally low [Na+]i is likely to have the opposite effect and cause mitochondrial Ca2+ overload, which also has detrimental effects on myocyte function. This notion is supported by the recent finding that elimination of mitochondrial Ca2+ efflux through deletion of the gene encoding mitoNCX results in increased generation of superoxide and necrotic cell death leading to heart failure (Luongo et al., 2017).
In summary, [Na+]i regulates Ca2+ cycling, electrical activity, and oxidative stress in cardiac myocytes. Thus, maintenance of myocyte Na+ homeostasis is vital for preserving heart function.
Myocyte [Na+]i is Elevated in Heart Failure and Type-2 Diabetes
[Na+]i is in the 4–8 mM range in resting ventricular myocytes from healthy rabbit, guinea-pig, dog and human hearts (Harrison et al., 1992; Yao et al., 1998b; Gray et al., 2001; Despa et al., 2002a; Pieske et al., 2002; Gao et al., 2005) and somewhat higher (10–15 mM) in rat and mouse myocytes (Donoso et al., 1992; Yao et al., 1998a; Despa et al., 2002a, 2005). [Na+]i increases in a frequency dependent manner when myocytes are excited electrically. This [Na+]i rise is caused by enhanced Na+ entry due to the regular opening of the voltage-gated Na+ channels and activation of NCX during Ca2+ transients. As [Na+]i rises, NKA is activated to extrude more Na+ and a new steady-state is reached when the higher Na+ influx is balanced by an elevated Na+ efflux.
The balance between the passive Na+ entry and Na+ pumping out of the cell is perturbed in both humans and animal models with heart failure (HF), resulting in elevated [Na+]i. Pieske et al. (2002) found that [Na+]i is ∼4 mM higher in myocytes from failing human myocardium compared to non-failing hearts. A comparable [Na+]i increase was reported in myocytes from rabbits with heart failure induced by volume and pressure overload (Despa et al., 2002b; Baartscheer et al., 2003) or by rapid pacing (Schillinger et al., 2006). [Na+]i is also elevated in myocytes from mice with heart failure caused by conditional, cardiomyocyte-specific deletion of SERCA gene (Louch et al., 2010). These studies found that [Na+]i was elevated at all stimulation rates in the 0–3 Hz range. While a few mM rise in [Na+]i may seem modest, Despa et al. (2002b) showed that, compared to other HF-induced alterations such as smaller Ca2+ transients and longer action potentials, it has the greatest impact on NCX function, and thus on cellular and SR Ca2+ load.
Diabetes is a systemic disease that leads to structural, contractile and electrical abnormalities of the heart, even in the absence of coronary artery disease or hypertension (Taegtmeyer et al., 2002; Young et al., 2002, 2009; Guha et al., 2008; Boudina and Abel, 2010). Diabetic cardiomyopathy is characterized by diastolic dysfunction (>50% prevalence) that progresses to systolic dysfunction and heart failure at more advanced diabetic stages (Ingelsson et al., 2005; Kostis and Sanders, 2005; Masoudi and Inzucchi, 2007; Pataky et al., 2011; Chaudhary et al., 2015). Some studies reported alterations in myocyte Na+ transport consistent with elevated [Na+]i in animal models of both type-1 and type-2 diabetes (see below). However, whether or not [Na+]i is altered in diabetic hearts was largely unknown until we recently found higher [Na+]i in resting and contracting myocytes from rats with late-onset type-2 diabetes that display a cardiac phenotype that closely resembles the diabetic cardiomyopathy in humans (HIP rats) (Lambert et al., 2015). Interestingly, the [Na+]i rise that we measured in diabetic HIP rat hearts is comparable to the increase in [Na+]i that occurs in HF.
As discussed above, high [Na+]i may amplify the risk for arrhythmias and cause oxidative stress. Indeed, elevated [Na+]i was shown to cause oxidation of NAD(P)H and to increase the H2O2 level in myocytes from guinea-pigs with heart failure induced by aortic constriction (Liu and O’Rourke, 2008). These effects were prevented by pharmacological inhibition of mitoNCX (Liu and O’Rourke, 2008). In a guinea pig model of heart failure and sudden cardiac death (aortic constriction + daily β-adrenergic receptor stimulation), mitoNCX inhibition attenuated cardiac hypertrophic remodeling and prevented cardiac dysfunction and sudden cardiac death, likely through normalizing [Ca2+]m (Liu et al., 2014). Therefore, the increase in [Na+]i is an active contributor to heart dysfunction in HF. Similar mechanisms are likely also in play in diabetic cardiomyopathy. In support of this assertion, Babsky et al. (2001) found that higher [Na+] had a larger negative effect on state 3 respiration, rate of oxidative phosphorylation and ATP production in mitochondria isolated from rats with streptozotocin-induced diabetes compared to control.
Mechanisms Underlying the [Na+]i Rise in Heart Failure and Diabetes
Since [Na+]i is at steady-state when the active Na+ efflux through the Na+/K+-ATPase and the total Na+ influx through various pathways (Figure 1) are at equilibrium, the [Na+]i rise in HF and diabetic cardiomyopathy could be caused by both reduced Na+ extrusion and enhanced Na+ entry.
Na+/K+-ATPase Expression and Function in Heart Failure and Diabetes
Numerous studies in human myocardium and animal models reported lower protein expression of various NKA subunits in HF. Schwinger et al. (1999) found reduced expression of NKA α1-, α3-, and β1-subunits in failing human hearts. Protein expression of all three α-subunit isoforms is decreased in hearts from rabbits with pressure and volume overload-induced HF (Bossuyt et al., 2005). In contrast, expression of α1 isoform is unchanged while α2 is reduced and α3 is increased in most rat HF models (Verdonck et al., 2003a). These data suggest that NKA activity might be reduced in HF. However, NKA function depends strongly on regulation by various modulators, including the endogenous inhibitor phospholemman. Indeed, Boguslavskyi et al. (2014) reported hypophosphorylation of phospholemman with no change in NKA expression following aortic constriction in mice, which resulted in a progressive decline in NKA current and elevation of [Na+]i. Thus, functional measurements in live cells and intact beating hearts are needed in order to compare NKA activity in failing and control hearts. By measuring the rate of [Na+]i decline as a function of [Na+]i in live myocytes, we found no changes in either the maximal Na+ transport rate or the apparent affinity for internal Na+ in myocytes from failing rabbit hearts compared to controls (Despa et al., 2002b). Decreased maximal Na+ extrusion rate (mainly through NKA-α2 isoform) but unchanged [Na+]i-affinity were reported in myocytes from rats with HF following myocardial infarction (Semb et al., 1998; Swift et al., 2008) as well as in mice with end-stage HF following genetic deletion of SERCA2 (Louch et al., 2010). In contrast, myocytes from dogs with chronic atrioventricular block and hypertrophy have unaltered maximal NKA current but reduced NKA [Na+]i-affinity (Verdonck et al., 2003b). Overall, NKA activity is decreased in some but not all HF models investigated, which may contribute to the rise in [Na+]i.
There are significantly fewer studies of NKA expression and function in diabetic cardiomyopathy. NKA activity is reduced by 21% in the myocardium of rats with streptozocin-induced type-1 diabetes (Kjeldsen et al., 1987). Hansen et al. (2007) found decreased NKA current measured with 10 mM Na+ but not with 80 mM Na+ in the pipette solutions in myocytes from rabbits with alloxan-induced type-1 diabetes, which indicates a reduction in the affinity of NKA for internal Na+ but no change in the maximal NKA activity. Myocytes from type-2 diabetic HIP rats showed no change in NKA-mediated Na+ extrusion for [Na+]i in the physiological range (0–20 mM) compared to their control littermates (Lambert et al., 2015). In agreement with this result, we also found that NKA-α1 expression is unaltered, while there is ∼50% decrease in NKA-α2 (Lambert et al., 2015). Since NKA-α2 represents less than 25% of the total NKA in rat myocytes (Despa and Bers, 2007; Swift et al., 2007), a ∼50% reduction in its expression has only a minor effect on total NKA function. However, NKA-α2 has a preferential localization in the t-tubules (Despa and Bers, 2007; Swift et al., 2007, 2010; Despa et al., 2012a) and therefore reduced NKA-α2 expression might affect Ca2+ cycling and contractility through local, subcellular, effects.
Na+ Influx Pathways in Heart Failure and Diabetes
We (Despa et al., 2002b) and others (Baartscheer et al., 2003) found that higher [Na+]i is caused by enhanced Na+ influx rather than reduced NKA activity in hearts from rabbits with pressure and volume-overload induced HF. This is also the case in myocytes from type-2 diabetic HIP rats, where Na+ influx is increased by ∼40% compared to myocytes from control rats (Lambert et al., 2015). While not directly measuring the rate of Na+ entry, several other studies reported upregulation of membrane transporters that facilitate Na+ import in HF and diabetic cardiomyopathy (see below), supporting an essential role for Na+ influx in the [Na+]i rise that occurs in these pathological conditions.
Na+ Current in Heart Failure and Diabetes
We found that the excess Na+ influx in myocytes from rabbits with HF is TTX-sensitive, which suggests that it is carried by Na+ channels (Despa et al., 2002b). There are numerous reports of increased late Na+ current in HF (Maltsev et al., 1998; Valdivia et al., 2005; Mishra et al., 2015; Hegyi et al., 2018), a slowly inactivating current that may be carried by both cardiac and neuronal Na+ channels present in myocytes. While the amplitude of late Na+ current is small (∼0.1–0.5% of the amplitude of the peak Na+ current), the current is long lasting (hundreds of milliseconds) and thus may contribute to myocyte Na+ homeostasis. However, the role of late Na+ current in elevating Na+ influx and [Na+]i in HF is controversial. On one hand, ranolazine, a late Na+ current inhibitor, reduced [Na+]i, and diastolic Ca2+ overload in failing human hearts (Sossalla et al., 2008). Moreover, the CaMKII-dependent increase in late Na+ current produced by exogenous reactive oxygen species was associated with a TTX and ranolazine-dependent rise in [Na+]i (Wagner et al., 2011). On the other hand, computational modeling predicts that higher late Na+ current measured in myocytes from failing hearts generates only a modest increase in [Na+]i, smaller than measured experimentally (Wagner et al., 2011; Cardona et al., 2016). Alternatively, HF may also enhance a background Na+ channel conductance that is responsible for the higher rate of Na+ entry.
Independent of a potential effect on [Na+]i, increased late Na+ current contributes to the prolongation of the action potential in HF, which may result in early afterdepolarizations. Moreover, via NCX, longer action potentials favor Ca2+ loading of the myocyte, which increases the propensity for delayed afterdepolarizations. Indeed, ranolazine significantly abbreviated the action potential and prevented the occurrence of delayed afterdepolarizations in myocytes from mice with HF induced by aortic constriction (Toischer et al., 2013). Ranolazine prevented ventricular fibrillation in rabbits with pacing-induced HF (Frommeyer et al., 2012) Thus, while the contribution of increased late Na+ current to the rise in [Na+]i is not fully elucidated, late Na+ current inhibition has proven beneficial effects in HF.
Na+/Ca2+ Exchanger in Heart Failure and Diabetes
NCX, which exchanges three Na+ ions for one Ca2+, is the main route for Ca2+ extrusion (Bers, 2001) and the most prominent contributor to Na+ influx (Despa et al., 2002a) in cardiac myocytes. Cardiac NCX expression is generally increased in both animal models of HF (O’Rourke et al., 1999; Pogwizd et al., 1999; Louch et al., 2010) and failing human hearts (Hasenfuss et al., 1999). However, higher NCX expression does not necessarily translate into higher rate of NCX-mediated Na+ entry. This is because the higher [Na+]i and smaller Ca2+ transients typically seen in HF shift the balance of fluxes through NCX to disfavor the Ca2+ out/Na+ in mode of function and may even cause the reversal of the exchanger during an action potential. In agreement with this reasoning, we found no change in the NCX-mediated Na+ influx in failing rabbit myocytes (Despa et al., 2002b) despite a 100% increase in NCX expression (Pogwizd et al., 1999).
In contrast to HF, NCX expression and function is decreased in rats with streptozotocin-induced type-1 diabetes (Chattou et al., 1999; Hattori et al., 2000) and in type-2 diabetic HIP rats (Despa et al., 2012b). It is thus unlikely that NCX contributes to the enhanced myocyte Na+ entry in diabetes.
Na+/H+ Exchanger in Heart Failure and Diabetes
NHE is markedly upregulated in HF (Yokoyama et al., 2000; Leineweber et al., 2007; Fliegel, 2009; Packer, 2017) and its inhibition improved heart function in various animal models of HF (Kusumoto et al., 2001; Engelhardt et al., 2002; Aker et al., 2004; Kilić et al., 2014). Baartscheer et al. (Baartscheer et al., 2003) reported that increased Na+/H+-exchange activity is responsible for elevated [Na+]i in myocytes from failing rabbit hearts. Moreover, chronic treatment with cariporide, an NHE inhibitor, prevented the onset of HF in rabbits with pressure and volume overload (Baartscheer et al., 2005). The activity of myocardial Na+/H+ exchanger (NHE) is enhanced and contributes to left ventricular hypertrophy in the Goto-Kakizaki rat model of T2D (Darmellah et al., 2007). Increased NHE activity leads to higher [Na+]i gain during ischemia-reperfusion in hearts from T2D db/db mice (Anzawa et al., 2006). Moreover, reducing [Na+]i gain during ischemia-reperfusion by NHE inhibition was associated with a lower incidence of ventricular tachycardia and fibrillation in db/db hearts (Anzawa et al., 2006). These data point to an important contribution of NHE to the excess cardiac Na+ influx in HF and diabetic cardiomyopathy.
Na+-Glucose Cotransporter and [Na+]i Dysregulation in Diabetic Hearts
One transporter known to be present in the heart but rarely discussed in the context of myocyte Na+ homeostasis is the Na+-glucose cotransporter (SGLT), which couples Na+ transport to glucose uptake and thus to energy substrate metabolism. The major SGLT isoforms, SGLT1, and SGLT2, have distinct tissue distribution and systemic role. SGLT1 is found predominantly in epithelial cells from the intestine, where it participates in dietary glucose absorption, whereas SGLT2 is the major isoform expressed in renal epithelial cells and is essential for glucose reabsorption from the forming urine. Highly specific SGLT2 inhibitors are the latest class of blood glucose lowering drugs. Recently, SGLT2 inhibitors were demonstrated to have beneficial cardiac effects in patients with type-2 diabetes and HF (Zinman et al., 2015). However, reports from several labs (Nishimura and Naito, 2005; Wright and Loo, 2011; Van Steenbergen et al., 2017) indicate that SGLT2 is not expressed in the heart. This suggests that the cardioprotection conferred by SGLT2 inhibitors is mediated by interaction with a different cardiac target and/or by effects in extracardiac tissues. Intriguingly, two recent studies (Baartscheer et al., 2017; Uthman et al., 2018) found that SGLT2 inhibitors (empagliflozin, dapagliglozin, and canagliflozin) block the Na+/H+ exchanger, possibly by binding to the Na+-binding site of NHE, and thus lower myocyte [Na+]i in multiple species. Furthermore, empagliflozin and canagliflozin also induced vasodilation (Uthman et al., 2018). While these measurements were performed in healthy hearts/myocytes, the effects uncovered are likely to play a part in the improvement of heart function by SGLT2 inhibitors that was observed clinically in patients with HF and type-2 diabetes.
In contrast to SGLT2, SGLT1 is highly expressed in the heart (Zhou et al., 2003; Banerjee et al., 2009, 2010; Wright and Loo, 2011). Recently, SGLT1 upregulation was causally linked to PRKAG2 cardiomyopathy that is caused by mutations in the gene encoding the γ2 subunit of AMP-activated protein kinase (Banerjee et al., 2010) and cardiac-specific SGLT1 deletion attenuated the cardiomyopathy (Ramratnam et al., 2014). Moreover, cardiac-specific overexpression of SGLT1 in mice causes hypertrophy and left-ventricular dysfunction (Ramratnam et al., 2014). Thus, there is increasing evidence that enhanced SGLT1 activity harms the heart.
Banerjee et al. (2009) reported that the mRNA level of SGLT1 is increased in hearts from humans and mice with type-2 diabetes and ischemic cardiomyopathy. In agreement with this result, we found higher SGLT1 protein expression in failing hearts from T2D patients compared to failing hearts from lean, non-diabetic individuals and in hearts from type-2 diabetic HIP rats vs. control rats (Lambert et al., 2015). Obesity, in the absence of type-2 diabetes, was also associated with elevated levels of cardiac SGLT1 in human hearts (Lambert et al., 2015). Moreover, the presence of HF alone resulted in higher cardiac SGLT1 protein expression in both lean and obese individuals (Lambert et al., 2015). Consistent with augmented SGLT1 expression, we found that the SGLT-mediated glucose uptake is significantly increased in hearts from diabetic vs. control rats (Lambert et al., 2015). Moreover, SGLT inhibition (both pharmacological and through omission of glucose from external solution) greatly reduced the rate of Na+ entry in myocytes from diabetic rats while it had no significant effect in myocytes from control rats. SGLT-mediated Na+ influx was ≈7 times higher in diabetic vs. control hearts (Lambert et al., 2015). Together, these results indicate that SGLT1 is upregulated in diabetic rat hearts and its activation is largely responsible for the excess Na+ influx that accounts for the increase in [Na+]i.
Other members of the Na+-glucose cotransporter family may also play a role in the development of diabetic cardiomyopathy. Van Steenbergen et al. (2017) found that hyperglycemia stimulates the production of reactive oxygen species in the heart through activation of NADPH oxidase and this mechanism is prevented in myocytes from mice with genetic deletion of the sodium-myoinositol cotransporter-1 (SMIT1). This finding suggests that SMIT1 is activated by high glucose, which may also contribute to the [Na+]i rise in diabetic hearts.
In summary, [Na+]i is augmented in both HF and diabetic cardiomyopathy and higher [Na+]i further exacerbates heart dysfunction. An enhancement of Na+ influx is at least partially responsible for the [Na+]i rise in both conditions. However, the Na+ entry pathways that mediate the excess Na+ entry may be distinct in the two pathologies, with Na+ channels and NHE being activated in HF and SGLT isoforms and NHE having the main contribution in diabetic cardiomyopathy.
Author Contributions
SD reviewed the literature in the field, and wrote and edited the manuscript.
Funding
This work was supported by NIH (Grant R01HL135000).
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aker, S., Snabaitis, A. K., Konietzka, I., Van De Sand, A., Böngler, K., Avkiran, M., et al. (2004). Inhibition of the Na+/H+ exchanger attenuates the deterioration of ventricular function during pacing-induced heart failure in rabbits. Cardiovasc. Res. 63, 273–282. doi: 10.1016/j.cardiores.2004.04.014
Anzawa, R., Bernard, M., Tamareille, S., Baetz, D., Confort-Gouny, S., Gascard, J. P., et al. (2006). Intracellular sodium increase and susceptibility to ischaemia in hearts from type 2 diabetic db/db mice. Diabetologia 49, 598–606. doi: 10.1007/s00125-005-0091-5
Baartscheer, A., Schumacher, C. A., van Borren, M. M., Belterman, C. N., Coronel, R., and Fiolet, J. W. (2003). Increased Na+/H+-exchange activity is the cause of increased [Na+]i and underlies disturbed calcium handling in the rabbit pressure and volume overload heart failure model. Cardiovasc. Res. 57, 1015–1024. doi: 10.1016/S0008-6363(02)00809-X
Baartscheer, A., Schumacher, C. A., van Borren, M. M., Belterman, C. N., Coronel, R., Opthof, T., et al. (2005). Chronic inhibition of Na+/H+-exchanger attenuates cardiac hypertrophy and prevents cellular remodeling in heart failure. Cardiovasc. Res. 65, 83–92. doi: 10.1016/j.cardiores.2004.09.024
Baartscheer, A., Schumacher, C. A., Wüst, R. C., Fiolet, J. W., Stienen, G. J., Coronel, R., et al. (2017). Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia 60, 568–573. doi: 10.1007/s00125-016-4134-x
Babsky, A., Doliba, N., Doliba, N., Savchenko, A., Wehrli, S., and Osbakken, M. (2001). Na+ effects on mitochondrial respiration and oxidative phosphorylation in diabetic hearts. Exp. Biol. Med. 226, 543–551. doi: 10.1177/153537020122600606
Banerjee, S. K., McGaffin, K. R., Pastor-Soler, N. M., and Ahmad, F. (2009). SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc. Res. 84, 111–118. doi: 10.1093/cvr/cvp190
Banerjee, S. K., Wang, D. W., Alzamora, R., Huang, X. N., Pastor-Soler, N. M., Hallows, K. R., et al. (2010). SGLT1, a novel cardiac glucose transporter, mediates increased glucose uptake in PRKAG2 cardiomyopathy. J. Mol. Cell Cardiol. 49, 683–692. doi: 10.1016/j.yjmcc.2010.06.003
Bers, D. M. (2001). Excitation-Contraction Coupling and Cardiac Contractile Force, 2nd Edn. Dordrecht: Kluwer Academic. doi: 10.1007/978-94-010-0658-3
Bers, D. M. (2014). Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu. Rev. Physiol. 76, 107–127. doi: 10.1146/annurev-physiol-020911-153308
Bertero, E., and Maack, C. (2018). Calcium signaling and reactive oxygen species in mitochondria. Circ. Res. 122, 1460–1478. doi: 10.1161/CIRCRESAHA.118.310082
Boguslavskyi, A., Pavlovic, D., Aughton, K., Clark, J. E., Howie, J., Fuller, W., et al. (2014). Cardiac hypertrophy in mice expressing unphosphorylatable phospholemman. Cardiovasc. Res. 104, 72–82. doi: 10.1093/cvr/cvu182
Bossuyt, J., Ai, X., Moorman, R. J., Pogwizd, S. M., and Bers, D. M. (2005). Expression and phosphorylation of the Na+-pump regulatory subunit phospholemman in heart failure. Circ. Res. 97, 558–565. doi: 10.1161/01.RES.0000181172.27931.c3
Boudina, S., and Abel, E. D. (2010). Diabetic cardiomyopathy, causes and effects. Rev. Endocr. Metab. Disord. 11, 31–39. doi: 10.1007/s11154-010-9131-7
Boyman, L., Williams, G. S., Khananshvili, D., Sekler, I., and Lederer, W. J. (2013). NCLX: the mitochondrial sodium calcium exchanger. J. Mol. Cell Cardiol. 59, 205–213. doi: 10.1016/j.yjmcc.2013.03.012
Cardona, K., Trenor, B., and Giles, W. R. (2016). Changes in intracellular Na+ following enhancement of late Na+ current in virtual human ventricular myocytes. PLoS One 11:e0167060. doi: 10.1371/journal.pone.0167060
Chattou, S., Diacono, J., and Feuvray, D. (1999). Decrease in sodium-calcium exchange and calcium currents in diabetic rat ventricular myocytes. Acta Physiol. Scand. 166, 137–144. doi: 10.1046/j.1365-201x.1999.00547.x
Chaudhary, A. K., Aneja, G. K., Shukla, S., and Razi, S. M. (2015). Study on diastolic dysfunction in newly diagnosed type 2 diabetes mellitus and its correlation with glycosylated haemoglobin (HbA1C). J. Clin. Diagn. Res. 9, OC20–OC22. doi: 10.7860/JCDR/2015/13348.6376
Cox, D. A., and Matlib, M. A. (1993). A role for the mitochondrial Na+-Ca2+ exchanger in the regulation of oxidative phosphorylation in isolated heart mitochondria. J. Biol. Chem. 268, 938–947.
Darmellah, A., Baetz, D., Prunier, F., Tamareille, S., Rücker-Martin, C., and Feuvray, D. (2007). Enhanced activity of the myocardial Na+/H+ exchanger contributes to left ventricular hypertrophy in the Goto-Kakizaki rat model of type 2 diabetes: critical role of Akt. Diabetologia 50, 1335–1344. doi: 10.1007/s00125-007-0628-x
Despa, S., and Bers, D. M. (2007). Functional analysis of Na/K-ATPase isoform distribution in rat ventricular myocytes. Am. J. Physiol. Cell Physiol. 293, C321–C327. doi: 10.1152/ajpcell.00597.2006
Despa, S., Bossuyt, J., Han, F., Ginsburg, K. S., Jia, L. G., Kutchai, H., et al. (2005). Phospholemman -phosphorylation mediates the β-adrenergic effects on Na/K pump function in cardiac myocytes. Circ. Res. 97, 252–259. doi: 10.1161/01.RES.0000176532.97731.e5
Despa, S., Islam, M. A., Pogwizd, S. M., and Bers, D. M. (2002a). Intracellular [Na+]i and Na+-pump rate in rat and rabbit ventricular myocytes. J. Physiol. 539, 133–143. doi: 10.1113/jphysiol.2001.012940
Despa, S., Islam, M. A., Weber, C. R., Pogwizd, S. M., and Bers, D. M. (2002b). Intracellular Na+ concentration is elevated in heart failure, but Na/K-pump function is unchanged. Circulation 105, 2543–2548.
Despa, S., Lingrel, J. B., and Bers, D. M. (2012a). Na/K-ATPase (2-subunit preferentially affects sarcoplasmic reticulum Ca release in mouse cardiac myocytes. Cardiovasc. Res. 95, 480–486.
Despa, S., Margulies, K. B., Chen, L., Knowlton, A. A., Havel, P. J., Taegtmeyer, H., et al. (2012b). Hyperamylinemia contributes to heart dysfunction in obesity and diabetes, a study in humans and rats. Circ. Res. 110, 598–608. doi: 10.1161/CIRCRESAHA.111.258285
Donoso, P., Mill, J. G., O’Neil, S. C., and Eisner, D. A. (1992). Fluorescence measurements of cytoplasmic and mitochondrial sodium concentration in rat ventricular myocytes. J. Physiol. 448, 493–509. doi: 10.1113/jphysiol.1992.sp019053
Engelhardt, S., Hein, L., Keller, U., Klämbt, K., and Lohse, M. J. (2002). Inhibition of Na+/H+ exchange prevents hypertrophy, fibrosis, and heart failure in β1-adrenergic receptor transgenic mice. Circ. Res. 90, 814–819. doi: 10.1161/01.RES.0000014966.97486.C0
Fliegel, L. (2009). Regulation of the Na+/H+ exchanger in the healthy and diseased myocardium. Expert Opin. Ther. Targets 13, 55–68. doi: 10.1517/14728220802600707
Frommeyer, G., Rajamani, S., Grundmann, F., Stypmann, J., Osada, N., Breithardt, G., et al. (2012). New insights into the beneficial electrophysiologic profile of ranolazine in heart failure: prevention of ventricular fibrillation with increased postrepolarization refractoriness and without drug-induced proarrhythmia. J. Card. Fail. 18, 939–949. doi: 10.1016/j.cardfail.2012.10.017
Gao, J., Wang, W., Cohen, I. S., and Mathias, R. T. (2005). Transmural gradients in Na/K pump activity and [Na+]i in canine ventricle. Biophys. J. 89, 1700–1709. doi: 10.1529/biophysj.105.062406
Glitsch, H. G., and Tappe, A. (1993). The Na+/K+ pump of cardiac Purkinje cells is preferentially fuelled by glycolytic ATP production. Pflugers Arch. 422, 380–385. doi: 10.1007/BF00374294
Gray, R. P., McIntyre, H., Sheridan, D. S., and Fry, C. H. (2001). Intracellular sodium and contractile function in hypertrophied human and guinea-pig myocardium. Pflugers Arch. 442, 117–123. doi: 10.1007/s004240000512
Griffiths, E. J. (2009). Mitochondrial calcium transport in the heart: physiological and pathological roles. J. Mol. Cell Cardiol. 46, 789–803. doi: 10.1016/j.yjmcc.2009.03.001
Guha, A., Harmancey, R., and Taegtmeyer, H. (2008). Nonischemic heart failure in diabetes mellitus. Curr. Opin. Cardiol. 23, 241–248. doi: 10.1097/HCO.0b013e3282fcc2fa
Hansen, P. S., Clarke, R. J., Buhagiar, K. A., Hamilton, E., Garcia, A., White, C., et al. (2007). Alloxan-induced diabetes reduces sarcolemmal Na+-K+ pump function in rabbit ventricular myocytes. Am. J. Physiol. Cell Physiol. 292, C1070–C1077. doi: 10.1152/ajpcell.00288.2006
Harrison, S. M., McCall, E., and Boyet, M. R. (1992). The relationship between contraction and intracellular sodium in rat and guinea-pig ventricular myocytes. J. Physiol. 449, 517–550. doi: 10.1113/jphysiol.1992.sp019100
Hasenfuss, G., Schillinger, W., Lehnart, S. E., Preuss, M., Pieske, B., Maier, L. S., et al. (1999). Relationship between Na+-Ca2+-exchanger protein levels and diastolic function of failing human myocardium. Circulation 99, 641–648. doi: 10.1161/01.CIR.99.5.641
Hattori, Y., Matsuda, N., Kimura, J., Ishitani, T., Tamada, A., Gando, S., et al. (2000). Diminished function and expression of the cardiac Na+-Ca2+ exchanger in diabetic rats: implication in Ca2+ overload. J. Physiol. 527, 85–94. doi: 10.1111/j.1469-7793.2000.00085.x
Hegyi, B., Bossuyt, J., Griffiths, L. G., Shimkunas, R., Coulibaly, Z., Jian, Z., et al. (2018). Complex electrophysiological remodeling in postinfarction ischemic heart failure. Proc. Natl. Acad. Sci. U.S.A. 115, E3036–E3044. doi: 10.1073/pnas.1718211115
Ingelsson, E., Sundstrom, J., Arnlov, J., Zethelius, B., and Lind, L. (2005). Insulin resistance and risk of congestive heart failure. J. Am. Med. Assoc. 294, 334–341. doi: 10.1001/jama.294.3.334
Kilić, A., Huang, C. X., Rajapurohitam, V., Madwed, J. B., and Karmazyn, M. (2014). Early and transient sodium-hydrogen exchanger isoform 1 inhibition attenuates subsequent cardiac hypertrophy and heart failure following coronary artery ligation. J. Pharmacol. Exp. Ther. 351, 492–499. doi: 10.1124/jpet.114.217091
Kjeldsen, K., Braendgaard, H., Sidenius, P., Larsen, J. S., and Nørgaard, A. (1987). Diabetes decreases Na+-K+ pump concentration in skeletal muscles, heart ventricular muscle, and peripheral nerves of rat. Diabetes Metab. Res. Rev. 36, 842–848.
Kostis, J. B., and Sanders, M. (2005). The association of heart failure with insulin resistance and the development of type 2 diabetes. Am. J. Hypertens. 18, 731–737. doi: 10.1016/j.amjhyper.2004.11.038
Kusumoto, K., Haist, J. V., and Karmazyn, M. (2001). Na+/H+ exchange inhibition reduces hypertrophy and heart failure after myocardial infarction in rats. Am. J. Physiol. Heart Circ. Physiol. 280, H738–H745. doi: 10.1152/ajpheart.2001.280.2.H738
Lambert, R., Srodulski, S., Peng, X., Margulies, K. B., Despa, F., and Despa, S. (2015). Intracellular Na+ concentration ([Na+]i) is elevated in diabetic hearts due to enhanced Na+-glucose cotransport. J. Am. Heart Assoc. 4:e002183. doi: 10.1161/JAHA.115.002183
Leineweber, K., Heusch, G., and Schulz, R. (2007). Regulation and role of the presynaptic and myocardial Na+/H+ exchanger NHE1: effects on the sympathetic nervous system in heart failure. Cardiovasc. Drug Rev. 25, 123–131. doi: 10.1111/j.1527-3466.2007.00010.x
Liu, T., and O’Rourke, B. (2008). Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply, and demand matching. Circ. Res. 103, 279–288. doi: 10.1161/CIRCRESAHA.108.175919
Liu, T., Takimoto, E., Dimaano, V. L., DeMazumder, D., Kettlewell, S., Smith, G., et al. (2014). Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ. Res. 115, 44–54. doi: 10.1161/CIRCRESAHA.115.303062
Louch, W. E., Hougen, K., Mørk, H. K., Swift, F., Aronsen, J. M., Sjaastad, I., et al. (2010). Sodium accumulation promotes diastolic dysfunction in end-stage heart failure following Serca2 knockout. J. Physiol. 588, 465–478. doi: 10.1113/jphysiol.2009.183517
Luongo, T. S., Lambert, J. P., Gross, P., Nwokedi, M., Lombardi, A. A., Shanmughapriya, S., et al. (2017). The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 545, 93–97. doi: 10.1038/nature22082
Maack, C., Cortassa, S., Aon, M. A., Ganesan, A. N., Liu, T., and O’Rourke, B. (2006). Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. 99, 172–182. doi: 10.1161/01.RES.0000232546.92777.05
Maltsev, V. A., Sabbah, H. N., Higgins, R. S., Silverman, N., Lesch, M., and Undrovinas, A. I. (1998). Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation 98, 2545–2552. doi: 10.1161/01.CIR.98.23.2545
Masoudi, F. A., and Inzucchi, S. E. (2007). Diabetes mellitus and heart failure: epidemiology, mechanisms, and pharmacotherapy. Am. J. Cardiol. 99,113B–132B. doi: 10.1016/j.amjcard.2006.11.013
McCormack, J. G., Halestrap, A. P., and Denton, R. M. (1990). Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 70, 391–425. doi: 10.1152/physrev.1990.70.2.391
Mishra, S., Reznikov, V., Maltsev, V. A., Undrovinas, N. A., Sabbah, H. N., and Undrovinas, A. (2015). Contribution of sodium channel neuronal isoform Nav1.1 to late sodium current in ventricular myocytes from failing hearts. J. Physiol. 593, 1409–1427. doi: 10.1113/jphysiol.2014.278259
Nishimura, M., and Naito, S. (2005). Tissue-specific mRNA expression profiles of human ATP-binding cassette and solute carrier transporter superfamilies. Drug Metab. Pharmacokinet. 20, 452–477. doi: 10.2133/dmpk.20.452
O’Rourke, B., Kass, D. A., Tomaselli, G. F., Kääb, S., Tunin, R., and Marbán, E. (1999). Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure. Circ. Res. 84, 562–570. doi: 10.1161/01.RES.84.5.562
Packer, M. (2017). Activation and inhibition of sodium-hydrogen exchanger is a mechanism that links the pathophysiology and treatment of diabetes mellitus with that of heart failure. Circulation 136, 1548–1559. doi: 10.1161/CIRCULATIONAHA.117.030418
Pataky, Z., Makoundou, V., Nilsson, P., Gabriel, R. S., Lalic, K., Muscelli, E., et al. (2011). Metabolic normality in overweight and obese subjects. Which parameters? Which risks? Int. J. Obes. 35, 1208–1215. doi: 10.1038/ijo.2010.264
Pieske, B., Maier, L. S., Piacentino, V. I. I. I., Weisser, J., Hasenfuss, G., and Houser, S. (2002). Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation 106, 447–453. doi: 10.1161/01.CIR.0000023042.50192.F4
Pogwizd, S. M., Qi, M., Yuan, W., Samarel, A. M., and Bers, D. M. (1999). Upregulation of Na+/Ca2+ exchanger expression and function in an arrh ythmogenic rabbit model of heart failure. Circ. Res. 85, 1009–1019. doi: 10.1161/01.RES.85.11.1009
Ramratnam, M., Sharma, R. K., D’Auria, S., Lee, S. J., Wang, D., Huang, X. Y., et al. (2014). Transgenic Knockdown of Cardiac Sodium/Glucose Cotransporter 1 (SGLT1) Attenuates PRKAG2 Cardiomyopathy, Whereas Transgenic Overexpression of Cardiac SGLT1 Causes Pathologic Hypertrophy and Dysfunction in Mice. J. Am. Heart Assoc. 3:e000899. doi: 10.1161/JAHA.114.000899
Schillinger, W., Teucher, N., Christians, C., Kohlhaas, M., Sossalla, S., Van Nguyen, P., et al. (2006). High intracellular Na+ preserves myocardial function at low heart rates in isolated myocardium from failing hearts. Eur. J. Heart Fail. 8, 673–680. doi: 10.1016/j.ejheart.2006.01.013
Schwinger, R. H., Wang, J., Frank, K., Müller-Ehmsen, J., Brixus, K., McDonough, A. A., et al. (1999). Reduced sodium pump (1, (3 and β1-isoform protein levels and Na+,K+-ATPase activity but unchanged Na+-Ca2+ exchanger protein levels in human heart failure. Circulation 99, 2105–2112. doi: 10.1161/01.CIR.99.16.2105
Semb, S. O., Lunde, P. K., Holt, E., Tonnessen, T., Christensen, G., and Sejersted, O. M. (1998). Reduced myocardial Na-K pump capacity in congestive heart failure following myocardial infarction in rats. J. Mol. Cell Cardiol. 30, 1311–1328. doi: 10.1006/jmcc.1998.0696
Sossalla, S., Wagner, S., Rasenack, E. C. L., Ruff, H., Weber, S. L., Schondube, F. A., et al. (2008). Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts - Role of late sodium current and intracellular ion accumulation. J. Mol. Cell Cardiol. 45, 32–43. doi: 10.1016/j.yjmcc.2008.03.006
Swift, F., Birkeland, J. A., Tovsrud, N., Enger, U. H., Aronsen, J. M., Louch, W. E., et al. (2008). Altered Na+/Ca2+-exchanger activity due to downregulation of Na+/K+-ATPase α2-isoform in heart failure. Cardiovasc. Res. 78, 71–78. doi: 10.1093/cvr/cvn013
Swift, F., Tovsrud, N., Enger, U. H., Sjaastad, I., and Sejersted, O. M. (2007). The Na+/K+-ATPase (2-isoform regulates cardiac contractility in rat cardiomyocytes. Cardiovasc. Res. 75, 109–117. doi: 10.1016/j.cardiores.2007.03.017
Swift, F., Tovsrud, N., Sjaastad, I., Sejersted, O. M., Niggli, E., and Egger, M. (2010). Functional coupling of (2-isoform Na+/K+-ATPase and Ca2+ extrusion through the Na+/Ca2+-exchanger in cardiomyocytes. Cell Calcium 48, 54–60. doi: 10.1016/j.ceca.2010.06.006
Taegtmeyer, H., McNulty, P., and Young, M. E. (2002). Adaptation and maladaptation of the heart in diabetes: part I: general concepts. Circulation 105, 1727–1733. doi: 10.1161/01.CIR.0000012466.50373.E8
Toischer, K., Hartmann, N., Wagner, S., Fischer, T. H., Herting, J., Danner, B. C., et al. (2013). Role of late sodium current as a potential arrhythmogenic mechanism in the progression of pressure-induced heart disease. J. Mol. Cell Cardiol. 61, 111–122. doi: 10.1016/j.yjmcc.2013.03.021
Uthman, L., Baartscheer, A., Bleijlevens, B., Schumacher, C. A., Fiolet, J. W. T., Koeman, A., et al. (2018). Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na+/H+ exchanger, lowering of cytosolic Na+ and vasodilation. Diabetologia 61, 722–726. doi: 10.1007/s00125-017-4509-7
Valdivia, C. R., Chu, W. W., Pu, J., Foell, J. D., Haworth, R. A., Wolff, M. R., et al. (2005). Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J. Mol. Cell Cardiol. 38, 475–483. doi: 10.1016/j.yjmcc.2004.12.012
Van Steenbergen, A., Balteau, M., Ginion, A., Ferté, L., Battault, S., Ravenstein, C. M., et al. (2017). Sodium-myoinositol cotransporter-1, SMIT1, mediates the production of reactive oxygen species induced by hyperglycemia in the heart. Sci. Rep. 7:41166. doi: 10.1038/srep41166
Verdonck, F., Volders, P. G. A., Vos, M. A., and Sipido, K. R. (2003a). Intracellular Na+ and altered Na+ transport mechanisms in cardiac hyperhtrophy and failure. J. Mol. Cell Cardiol. 35, 5–25.
Verdonck, F., Volders, P. G., Vos, M. A., and Sipido, K. R. (2003b). Increased Na+ concentration and altered Na+/K+ pump activity in hypertrophied canine ventricular cells. Cardiovasc. Res. 57, 1035–1043.
Wagner, S., Ruff, H. M., Weber, S. L., Bellmann, S., Sowa, T., Schulte, T., et al. (2011). Reactive oxygen species-activated Ca/calmodulin kinase IIδ is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ. Res. 108, 555–565. doi: 10.1161/CIRCRESAHA.110.221911
Wright, E. M., and Loo, D. D. (2011). Hirayama BA. Biology of human sodium glucose transporters. Physiol. Rev. 91, 733–794. doi: 10.1152/physrev.00055.2009
Yao, A., Su, Z., Nonaka, A., Zubair, I., Lu, L., Philipson, K. D., et al. (1998a). Effects of overexpression of the Na+-Ca2+ exchanger on [Ca2+] transients in murine ventricular myocytes. Circ. Res. 82, 657–665.
Yao, A., Su, Z., Nonaka, A., Zubair, I., Spitzer, K. W., Bridge, J. H., et al. (1998b). Abnormal myocyte Ca2+ homeostasis in rabbits with pacing-induced heart failure. Am. J. Physiol. 275, H1441–H1448.
Yokoyama, H., Gunasegaram, S., Harding, S. E., and Avkiran, M. (2000). Sarcolemmal Na+/H+ exchanger activity and expression in human ventricular myocardium. J. Am. Coll. Cardiol. 36, 534–540. doi: 10.1016/S0735-1097(00)00730-0
Young, L. H., Wackers, F. J., Chyun, D. A., Davey, J. A., Barrett, E. J., Taillefer, R., et al. (2009). DIAD Investigators. Cardiac outcomes after screening for asymptomatic coronary artery disease in patients with type 2 diabetes: the DIAD study: a randomized controlled trial. JAMA 301, 1547–1555. doi: 10.1001/jama.2009.476
Young, M. E., McNulty, P., and Taegtmeyer, H. (2002). Adaptation and maladaptation of the heart in diabetes: Part II: potential mechanisms. Circulation 105, 1861–1870. doi: 10.1161/01.CIR.0000012467.61045.87
Zhou, L., Cryan, E. V., D’Andrea, M. R., Belkowski, S., Conway, B. R., and Demarest, K. T. (2003). Human cardiomyocytes express high level of Na+/glucose cotransporter 1 (SGLT1). J. Cell. Biochem. 90, 339–346. doi: 10.1002/jcb.10631
Keywords: heart failure, type-2 diabetes, Na+-glucose cotransporter, Na+/H+ exchanger, Na+/K+-ATPase, Na+/Ca2+ exchanger
Citation: Despa S (2018) Myocyte [Na+]i Dysregulation in Heart Failure and Diabetic Cardiomyopathy. Front. Physiol. 9:1303. doi: 10.3389/fphys.2018.01303
Received: 13 July 2018; Accepted: 29 August 2018;
Published: 12 September 2018.
Edited by:
Coert J. Zuurbier, Academic Medical Center (AMC), NetherlandsReviewed by:
Thomas Hund, The Ohio State University, United StatesDunja Aksentijevic, Queen Mary University of London, United Kingdom
Copyright © 2018 Despa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sanda Despa, cy5kZXNwYUB1a3kuZWR1