 Zeki Ilkan
Zeki Ilkan Fadi G. Akar
Fadi G. Akar- Cardiovascular Research Center, Icahn School of Medicine at Mount Sinai, New York, NY, United States
The mitochondrial translocator protein (TSPO) is a key outer mitochondrial membrane protein that regulates the activity of energy-dissipating mitochondrial channels in response to oxidative stress. In this article, we provide an overview of the role of TSPO in the systematic amplification of reactive oxygen species (ROS) through an autocatalytic process known as ROS-induced ROS-release (RIRR). We describe how this TSPO-driven process destabilizes the mitochondrial membrane potential leading to electrical instability at the cellular and whole heart levels. Finally, we provide our perspective on the role of TSPO in the pathophysiology of diabetes, in general and diabetes-related arrhythmias, in particular.
Introduction
Diabetes mellitus is a global public health epidemic that continues to expand, in both its incidence and prevalence. Diabetic patients are predisposed to an increasing number of debilitating cardiovascular disorders such as stroke and myocardial infarction (Aune et al., 2018; Yang et al., 2018). This metabolic disease is also an important risk factor in the development of cardiac rhythm disorders (Movahed et al., 2005; Huxley et al., 2012; Lau et al., 2017). In addition to predisposing to atrial fibrillation (Huxley et al., 2011), diabetes along with its numerous complications, has been linked to increased prevalence of ventricular arrhythmias leading to sudden cardiac death (Stahn et al., 2014; Pistrosch et al., 2015; Xie et al., 2015; Agarwal and Singh, 2017). Importantly, oxidative stress, a major factor in the pathophysiology of diabetes, has been linked to arrhythmias either directly or through exacerbation of atherogenic risk factors (Van Wagoner, 2008; Gutierrez and Van Wagoner, 2015). Oxidative stress arises from enhanced production of free radicals and defective antioxidant defense mechanisms in the diabetic heart (Bashan et al., 2009; Bajaj and Khan, 2012). This, in turn, contributes to the pathogenesis of numerous diabetes-related cardiovascular complications, including endothelial dysfunction (Higashi et al., 2009), atherosclerosis (Harrison et al., 2003), myocardial infarction (Misra et al., 2009), and diabetic cardiomyopathy (Jia et al., 2018), all of which as illustrated in Figure 1 can lead to sudden cardiac death (Anderson et al., 2009; Duicu et al., 2016; Peiro et al., 2016; Zhang et al., 2017). In this article, we focus on a key outer mitochondrial membrane protein known as the mitochondrial translocator protein (TSPO) as a source of oxidative stress-related cardiac dysfunction. We begin by highlighting its role in linking mitochondrial instability to arrhythmias in the heart through a regenerative process known as reactive oxygen species (ROS) -induced ROS-release (RIRR). We then provide a new perspective on its potential importance to the pathophysiology of diabetes, in general and diabetes-related arrhythmias, in particular.
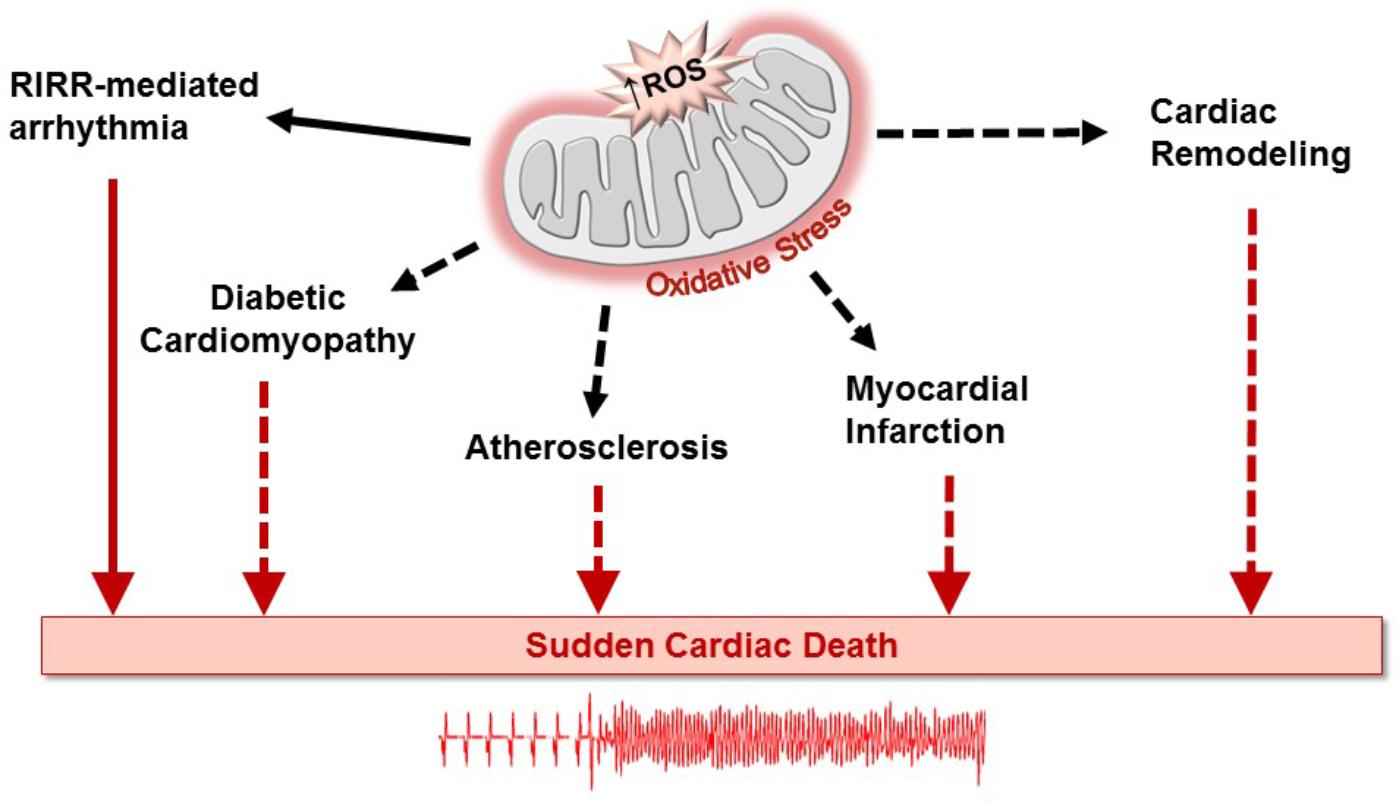
FIGURE 1. Oxidative stress created by excessive generation of mitochondrial reactive oxygen species (ROS) plays well-recognized roles in the development of an array of cardiovascular disorders leading to sudden cardiac death. ROS production can be amplified through the process of ROS-induced ROS release (RIRR), causing cellular dysfunction and death, directly creating a pro-arrhythmic substrate (solid arrows). In a relatively more indirect manner, ROS production can increase the risk of lethal arrhythmias by promoting adverse remodeling of various pathways in the diabetic heart (dashed arrows) (Wilson et al., 2018). Similarly, ROS production can contribute to the progression of atherosclerosis and myocardial infarction as well as to heart failure-associated cardiac remodeling, increasing the susceptibility of the heart to arrhythmias that can result in sudden cardiac death.
The Mitochondrial Translocator Protein
TSPO, formerly known as the peripheral benzodiazepine receptor (PBR) (Papadopoulos et al., 2006), is a structurally conserved molecule which is ubiquitously expressed in steroidogenic tissues, as well as brain, kidney, and heart cells (Rupprecht et al., 2010; Morin et al., 2016). It was discovered in 1977, and initially called PBR because of its ability to bind benzodiazepine drugs outside of the central nervous system (Braestrup and Squires, 1977). The 18-kDa molecule carries out a variety of essential roles such as cholesterol transport (Li and Papadopoulos, 1998), steroidogenesis (Besman et al., 1989; Papadopoulos et al., 1997), and programmed cell death (Caballero et al., 2013). In eukaryotes TSPO is mainly expressed on the outer mitochondrial membrane, in close physical association with other mitochondrial channels such as the voltage-dependent anion channel (VDAC) within the mitochondrial membrane transition pore (mPTP) complex, and the inner membrane anion channel (IMAC) (Veenman et al., 2007; Motloch et al., 2015) (Figure 2). Cryo-electron microscopy and image analyses of the TSPO molecule from Rhodobacter sphaeroides revealed a dimeric quaternary structure, whereby each TSPO monomer consists of five transmembrane domains (Korkhov et al., 2010; Li et al., 2015). Although monomeric and oligomeric forms have been reported, the functional implications of TSPO polymerization have not been fully elucidated (Lacapere et al., 2001; Delavoie et al., 2003; Jaremko et al., 2014). The revelation of the 3-dimensional high-resolution image of the mouse TSPO has provided the opportunity to study the molecular interactions between this mitochondrial protein and its ligands, such as the antagonist PK11195 (Jaremko et al., 2014). The functional role of TSPO in various organs and cell types has been investigated primarily using TSPO agonists and antagonists (Fulda et al., 2010; Rupprecht et al., 2010). In order to fully appreciate the role of TSPO as a mediator of cardiac pro-arrhythmic risk, we begin by reviewing the concept of RIRR which directly links mitochondrial instability to myocyte excitability.
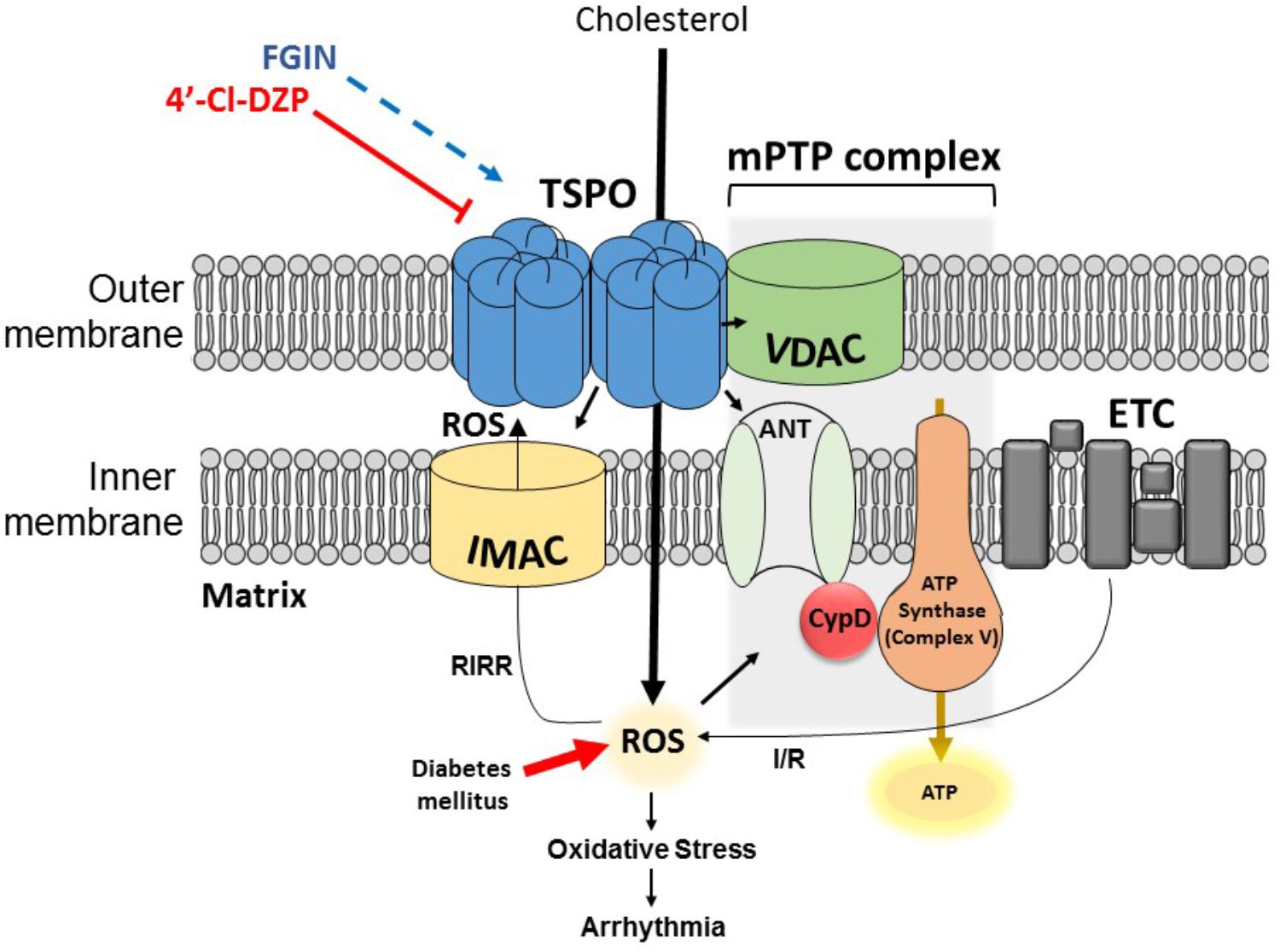
FIGURE 2. Simplified illustration of a TSPO dimer, IMAC, and the mPTP complex on the mitochondrial membrane. TSPO is located on the outer mitochondrial membrane and facilitates cholesterol transport into the matrix. The close apposition of TSPO with IMAC and mPTP allows it to carry out its modulatory role in processes such as RIRR. During normal mitochondrial respiration, electrons which escape the ETC can combine with oxygen forming O-2 anions. ROS-scavenging enzymes work toward removing the ROS and hence keeping the cells healthy. Excessive production and/or defective scavenging of ROS under pathological conditions such as I/R and/or diabetes mellitus can activate ROS-sensitive IMAC, which amplifies ROS levels via RIRR. Escalating oxidative stress then activates the mPTP complex which can result in ΔΨm depolarization, leading to mitochondrial dysfunction, ischemic injury, and electrical remodeling paving way to arrhythmia. Additionally, internalized cholesterol can become oxidized by accumulating ROS, generating oxysterols which further enhance oxidative stress. The most common TSPO ligands 4′-chlorodiazepam (4′-Cl-DZP) and FGIN-1-27 have been used by many studies to study the involvement of TSPO in these processes.
Ros-Induced Ros-Release and Mitochondrial Instability as a Mediator of Cardiac Arrhythmias
Mitochondria have long been recognized as indispensable sources of adenosine triphosphate (ATP) in energy-reliant organs such as the heart. Almost counterintuitively, it later became apparent that these specialized organelles can also control cell death in response to injury. In healthy mammalian cells, the preservation of ATP synthesis by complex V is achieved by maintaining a proton gradient across the inner mitochondrial membrane (Mitchell and Moyle, 1965a,b), which in turn, generates an electrochemical gradient that is responsible for maintaining a polarized mitochondrial membrane potential (Kroemer et al., 2007). Mitochondrial respiration is always accompanied with ROS production through leakage of electrons that subsequently react with oxygen to form superoxide anions (O-2) (Turrens, 2003). Under certain pathological conditions such as diabetes, the production of ROS can exceed the capacity with which protective antioxidant defense systems eliminate these toxic agents. Oxidative stress, as well as secondary factors such as mitochondrial Ca2+ overload can prime the formation of mPTP on the inner mitochondrial membrane (Zorov et al., 2000; Aon et al., 2003, 2006, 2007; Halestrap and Pasdois, 2009). This is responsible, at least in part, for mitochondrial membrane permeabilization (Green, 2005), which can be underpinned by the process of RIRR (Zorov et al., 2000; Aon et al., 2003, 2006; Brady et al., 2006; Yang et al., 2010). Traditionally, mPTP has been thought to exist as a complex of proteins comprising of VDAC, adenine nucleotide translocator (ANT), and cyclophilin D (CypD) (Figure 2). Nevertheless, genetic studies in recent years have challenged this traditionally accepted model of mPTP structure. For more details on this subject matter, we refer the reader to another review (Kwong and Molkentin, 2015).
Sollot and colleagues (Zorov et al., 2000) pioneered the concept of RIRR to describe how ROS injuries confined to distinct areas of a cardiomyocyte are able to quickly spread through a wider network of mitochondria, culminating in oxidative stress at a cellular level (Zorov et al., 2006). RIRR is responsible for the autocatalytic amplification of ROS levels, eventually leading to cell death. Two modes of RIRR have been proposed based on the identity of the mitochondrial pathway that mediates the process, namely the mPTP or the IMAC (Brady et al., 2006; Yang et al., 2010). Initially, the connection between mPTP opening and oxidative stress-dependent destabilization of ΔΨm, leading to cell death was demonstrated by Zorov et al. (2000). This was followed by studies by Aon et al. (2003) who provided strong evidence for the involvement of IMAC as a mediator of RIRR in metabolic oscillations. Pharmacological studies confirmed that IMAC facilitates superoxide release (Takahashi and Asada, 1983; Paky et al., 1993), providing the mechanism by which this anion channel contributes to RIRR. TSPO antagonists such as 4′-chlorodiazepam and PK11195 inhibit anion transport by IMAC, consistent with a strong modulatory role of TSPO on this ROS-sensitive channel (Beavis, 1989; Kinnally et al., 1993). In response to stress, IMAC activation occurs first, ultimately followed by mPTP activation at higher stress levels (Aon et al., 2007; Motloch et al., 2015). Collectively, these are the key elements in the series of events leading up to ROS-induced cell death. Although brief perturbations in ΔΨm may not influence cell survival to a large extent, prolonged periods of ΔΨm instability are known to mediate mitochondrial dysfunction and cell death (Marchetti et al., 1996; Zamzami et al., 2005).
The relevance of metabolic oscillations to electrophysiological behavior was examined using photo-induced oxidation of cardiomyocytes. These seminal studies demonstrated that cyclical oscillations of the action potentials (AP) were generated in phase with ΔΨm oscillations. AP recovery was found to depend upon ΔΨm recovery, and this suggested a profound mitochondrial control of myocyte excitability, at least in vitro (Aon et al., 2003). More recently, we and others (Zhou et al., 2014; Alleman et al., 2016) examined the relationship between ΔΨm stability and arrhythmogenesis in response to oxidative stress. AP oscillations were generated by “out-of-phase” oscillations of sarcolemmal KATP channels during RIRR (Aon et al., 2003). Furthermore, the opening of sarcolemmal KATP channels may give rise to the phenomenon of “metabolic sink”, whereby conduction wavefronts are hindered when they encounter heterogeneous current sinks in the tissue. These current sinks are formed in regions having high open probability of sarcolemmal KATP channels (Akar et al., 2005; Akar and O’Rourke, 2011; Zhou et al., 2014). The testing of the anti-arrhythmic effects of KATP channel inhibition using glibenclamide resulted in conflicting results, including reports of adverse effects (del Valle et al., 2001), whilst the pro-arrhythmic effects of channel activation have been demonstrated by multiple groups (Fedorov et al., 2011; Xie et al., 2015). In our studies sarcolemmal KATP channel inhibition using glibenclamide did not prevent the initiation of reperfusion arrhythmias in the ex vivo perfused guinea pig heart (Akar et al., 2005). This highlighted the necessity for a better understanding of the upstream elements such as TSPO which could potentially modulate the deleterious opening of sarcolemmal KATP channels during RIRR.
Consistent with cellular studies of RIRR, we demonstrated that exposure of intact hearts to high doses of exogenous pro-oxidants such as H2O2 provoked two distinct ROS peaks. While the initial low amplitude peak coincided with the exogenous stressor, the second (large amplitude) peak (which we termed P2) occurred following not during the exogenous stress, consistent with a RIRR response (Biary et al., 2011). Functionally, hearts that exhibited P2 were prone to ventricular fibrillation, whereas those that did not were relatively more protected (Biary et al., 2011). In a subsequent study, we investigated the relationship between the stability of the mitochondrial membrane in response to oxidative stress and the pro-arrhythmic potential of guinea pig hearts (Xie et al., 2014). Specifically, we modulated the threshold and rate of decline of the mitochondrial membrane potential in response to exogenous pro-oxidant challenge using a variety of agents that affected the activity of key mitochondrial ion channels. Once again, hearts that exhibited rapid ΔΨm decline were associated with low thresholds for sustained arrhythmias (Xie et al., 2014). More recently, Alleman et al. (2016) elegantly demonstrated that the stabilization of the mitochondrial membrane potential may underpin exercise-mediated protection against reperfusion arrhythmias.
In light of studies showing that TSPO blockade was highly effective in abolishing ΔΨm instability, we and others examined the impact of TSPO inhibition on arrhythmia propensity. Indeed, TSPO inhibition protected against ischemia-induced AP duration (APD) shortening and inexcitability (Akar et al., 2005). In contrast, IMAC activation using the TSPO agonist FGIN-1-27 enhanced APD shortening and promoted conduction failure under ischemic conditions (Akar et al., 2005). In these hearts, high-resolution optical AP mapping revealed areas of conduction block, which gave rise to sustained re-entrant arrhythmias upon reperfusion. In contrast, TSPO inhibition protected against ischemia-induced conduction block and reperfusion-related arrhythmias. Highlighting the role of TSPO as a chief mediator of post-ischemic arrhythmias, Brown and colleagues observed similar anti-arrhythmic effects of TSPO blockade in a rabbit model of ischemia-reperfusion injury, which were not apparent in those hearts treated with the mPTP blocker, CsA (O’Rourke, 2000; Aon et al., 2003; Brown et al., 2008). In addition to pharmacological inhibition of TSPO, cardiac-specific knockdown of this gene also proved to be protective against reperfusion arrhythmias in spontaneously hypertensive rats (Ilkan et al., 2018). Ongoing studies will help determine if this novel cardiotropic TSPO gene silencing approach may have a role in combatting oxidative stress-related arrhythmias in the heart.
Tspo in Diabetic Pathophysiology
The use of TSPO ligands in a variety of experimental settings has led to their translation to clinical trials for treatment of neurological and psychiatric diseases (Rupprecht et al., 2010). The utility of these ligands in metabolic diseases, however, has been the subject of very few investigations. A notable exception is an elegant study by Gut et al. (2013) in which treatment of zebrafish larvae with 4′-chlorodiazepam or PK11195 caused a marked decrease in systemic glucose levels, suggesting a potential role for treatment of diabetic complications. The compounds were also found to be activators of a fasting-like energy state, protecting obese mice from the undesirable effects of metabolic dysregulation (Gut et al., 2013; Gut, 2015). Other groups studied the effects of pharmacological manipulation of TSPO on adipocyte functions. Since adipose tissue is a vital integrator of glucose homeostasis, it plays a major role in the pathophysiology of metabolic diseases including diabetes (Rosen and Spiegelman, 2006). The Papadopoulos laboratory postulated that TSPO in adipose tissues could serve as a pharmacological target in the treatment of type-2 diabetes mellitus (Li and Papadopoulos, 2015). To that end, they demonstrated the efficacy of two separate TSPO ligands in improving glucose uptake and adipogenesis through TSPO activation (Li and Papadopoulos, 2015). These authors argued that the anti-diabetic effects of these ligands are mediated via modulation of mitochondrial function, and in particular, cholesterol transport thereby improving biogenesis of the lipid bilayer (Li and Papadopoulos, 2015). Of note, TSPO expression is reduced in adipocytes from obese and diabetic mice and humans compared to those from their healthy non-diabetic counterparts (Arner et al., 2010; Thompson et al., 2013; Li and Papadopoulos, 2015). The significance of these observations was underscored by genetic knockdown studies. In particular, TSPO depletion in adipocytes led to impaired glucose uptake and adipogenesis. These findings are consistent with the notion that TSPO plays a critical role in the maintenance of normal adipocyte homeostasis (Li and Papadopoulos, 2015).
Recent evidence also indicates that mitochondrial cholesterol buildup may be a key step in disease progression (Paradis et al., 2013; Musman et al., 2017). Paradis et al. (2013) reported that reperfusion of ischemic rat myocardium is linked with an accumulation of mitochondrial cholesterol, which in turn, causes the generation of oxysterols via oxidation of cholesterol by ROS. Interestingly, 4′-chlorodiazepam inhibited cholesterol accumulation and mitochondrial injury through oxysterol formation (Paradis et al., 2013). These findings revealed a novel mechanism of TSPO-related mitochondrial dysfunction that is distinct from RIRR. This alternative mechanism has been hypothesized to be of particular relevance to hypercholesterolemia, a hallmark of type-2 diabetes mellitus (Musman et al., 2017). Indeed, elevated cholesterol levels are well-known risk factors for various cardiovascular diseases including thrombosis and cardiac ischemia-reperfusion injury. Moreover, there is substantial evidence for exacerbation of cardiac injury (Hoshida et al., 1996; Scalia et al., 2001; Osipov et al., 2009), and defective cardioprotective pathways in hypercholesterolemic and diabetic conditions (Bouhidel et al., 2008; Peart and Headrick, 2009; Ferdinandy et al., 2014; Wu et al., 2014). In a follow-up study, Musman et al. (2017) demonstrated enhanced oxysterol formation in a standard rat model of type-2 diabetes mellitus. Remarkably, 4′-chlorodiazepam inhibited cholesterol transfer into mitochondria and reduced oxysterol buildup, reinstating oxidative phosphorylation and preventing mPTP opening (Musman et al., 2017). Therefore, the inhibition of cholesterol uptake by 4′-chlorodiazepam may represent a potential therapeutic strategy against ischemia-reperfusion injury in diabetes mellitus and other metabolic diseases. Preliminary work by our lab examined the role of TSPO ligands in post-ischemic arrhythmogenesis of the diabetic heart (Hu et al., 2016). In a rat model of obesity and type-2 diabetes mellitus, in which we and others found that classically cardioprotective pathways targeting mitochondria are generally impaired, we verified the effectiveness of TSPO inhibition by 4′-chlorodiazepam in protection against these arrhythmias. Future studies employing genetic knockdown and over-expression strategies are needed to better understand the role of TSPO in the electrophysiology of the diabetic heart both at baseline and in response to oxidative stress.
While the focus of this article is on the role of myocyte TSPO expression in arrhythmogenesis through the regenerative process of RIRR, mechanisms by which TSPO can contribute to sudden death is likely to be multi-factorial and not merely restricted to this phenomenon. Indeed, TSPO is expressed in numerous cell types and not just myocytes. In fact there is substantial evidence of robust TSPO expression in the endothelium, vascular smooth muscle cells, adipose tissue, platelets, and macrophages (Veenman and Gavish, 2006; Bird et al., 2010; Li and Papadopoulos, 2015; Hellberg et al., 2018). Of note, because TSPO expression in non-myocyte populations (namely macrophages) increases markedly during inflammation, TSPO serves as powerful biomarker in diabetes mellitus, atherosclerosis and other inflammatory diseases in an ever-growing number of PET studies (Pugliese et al., 2010; Hellberg et al., 2018; Ran et al., 2018). In addition to serving as a biomarker of inflammatory disease, TSPO actively participates in the regulation of non-myocyte cellular functions that likely influence arrhythmia vulnerability. For example, in macrophages, genetic and pharmacologic TSPO inhibition reduces cellular lipid content and prevents foam cell formation during atherogenesis (Taylor et al., 2014). Use of specific ligands also suggested an interesting role for TSPO in mediating white and brown adipose tissue homeostasis, pointing to its potential as a therapeutic target in the metabolic syndrome (Thompson et al., 2013). Interestingly, both epicardial adipose tissue and macrophages secrete inflammatory adipokines and cytokines which can induce structural and electrical remodeling of the myocardium (Mazurek et al., 2003; Melo et al., 2004; Iacobellis et al., 2005; Abed et al., 2013; Francis Stuart et al., 2016; Samanta et al., 2016). This provides a plausible link between non-myocyte TSPO activity and arrhythmogenesis, especially in the setting of diabetes mellitus.
Conclusion and Future Directions
A growing body of evidence highlights the role of oxidative stress as a major mediator of arrhythmias in the setting of metabolic diseases such as diabetes (Anderson et al., 2009; Duicu et al., 2016; Peiro et al., 2016; Zhang et al., 2017). Glucose fluctuations in diabetic patients promote excessive production of ROS (Saito et al., 2014; Wu et al., 2016), which can supersede the protective antioxidant defense systems that normally operate in healthy myocardium. This leads to oxidative stress and mitochondrial dysfunction, and is often regarded as a hallmark feature of the diabetic heart. Mitochondrial dysfunction gives rise to and exacerbates numerous cardiovascular complications including endothelial dysfunction (Higashi et al., 2009), atherosclerosis (Harrison et al., 2003), myocardial infarction (Misra et al., 2009), and diabetic cardiomyopathy (Jia et al., 2018), all of which can lead to sudden cardiac death (Zipes and Wellens, 1998). In addition, the pathological phenomenon of RIRR is a major mediator of oxidative stress-driven cellular electrical dysfunction and death (Zorov et al., 2000; Aon et al., 2003, 2006, 2007). This process generates ROS endogenously as a response to elevated ROS levels. Given its well-characterized links to ROS-releasing mitochondrial channels, TSPO has emerged as a key hub in the regulation of mitochondrial function and the cardiac response to oxidative stress. Our understanding of the role of TSPO in diabetes will expand by combining insights gained from pharmacological and genetic studies targeting this critical outer mitochondrial membrane protein in the diabetic heart.
Author Contributions
Both authors have contributed to the drafting, writing, and final editing of this article.
Funding
This work was supported by NIH grants to FGA: R01 HL137259, R21 AG054211, R21 HL114378, R01 HL113497.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abed, H. S., Samuel, C. S., Lau, D. H., Kelly, D. J., Royce, S. G., Alasady, M., et al. (2013). Obesity results in progressive atrial structural and electrical remodeling: implications for atrial fibrillation. Heart Rhythm 10, 90–100. doi: 10.1016/j.hrthm.2012.08.043
Agarwal, G., and Singh, S. K. (2017). Arrhythmias in type 2 diabetes mellitus. Indian J. Endocrinol. Metab. 21, 715–718. doi: 10.4103/ijem.IJEM_448_16
Akar, F. G., Aon, M. A., Tomaselli, G. F., and O’Rourke, B. (2005). The mitochondrial origin of postischemic arrhythmias. J. Clin. Invest. 115,3527–3535. doi: 10.1172/JCI25371
Akar, F. G., and O’Rourke, B. (2011). Mitochondria are sources of metabolic sink and arrhythmias. Pharmacol. Ther. 131, 287–294. doi: 10.1016/j.pharmthera.2011.04.005
Alleman, R. J., Tsang, A. M., Ryan, T. E., Patteson, D. J., McClung, J. M., Spangenburg, E. E., et al. (2016). Exercise-induced protection against reperfusion arrhythmia involves stabilization of mitochondrial energetics. Am. J. Physiol. Heart Circ. Physiol. 310, H1360–H1370. doi: 10.1152/ajpheart.00858.2015
Anderson, E. J., Kypson, A. P., Rodriguez, E., Anderson, C. A., Lehr, E. J., and Neufer, P. D. (2009). Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 54, 1891–1898. doi: 10.1016/j.jacc.2009.07.031
Aon, M. A., Cortassa, S., Maack, C., and O’Rourke, B. (2007). Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J. Biol. Chem. 282, 21889–21900. doi: 10.1074/jbc.M702841200
Aon, M. A., Cortassa, S., Marban, E., and O’Rourke, B. (2003). Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J. Biol. Chem. 278, 44735–44744. doi: 10.1074/jbc.M302673200
Aon, M. A., Cortassa, S., and O’Rourke, B. (2006). The fundamental organization of cardiac mitochondria as a network of coupled oscillators. Biophys. J. 91, 4317–4327. doi: 10.1529/biophysj.106.087817
Arner, E., Westermark, P. O., Spalding, K. L., Britton, T., Ryden, M., Frisen, J., et al. (2010). Adipocyte turnover: relevance to human adipose tissue morphology. Diabetes Metab. Res. Rev. 59, 105–109. doi: 10.2337/db09-0942
Aune, D., Schlesinger, S., Norat, T., and Riboli, E. (2018). Diabetes mellitus and the risk of sudden cardiac death: a systematic review and meta-analysis of prospective studies. Nutr. Metab. Cardiovasc. Dis. 28, 543–556. doi: 10.1016/j.numecd.2018.02.011
Bajaj, S., and Khan, A. (2012). Antioxidants and diabetes. Indian J. Endocrinol. Metab. 16, S267–S271.
Bashan, N., Kovsan, J., Kachko, I., Ovadia, H., and Rudich, A. (2009). Positive and negative regulation of insulin signaling by reactive oxygen and nitrogen species. Physiol. Rev. 89, 27–71. doi: 10.1152/physrev.00014.2008
Beavis, A. D. (1989). On the inhibition of the mitochondrial inner membrane anion uniporter by cationic amphiphiles and other drugs. J. Biol. Chem. 264, 1508–1515.
Besman, M. J., Yanagibashi, K., Lee, T. D., Kawamura, M., Hall, P. F., and Shively, J. E. (1989). Identification of des-(Gly-Ile)-endozepine as an effector of corticotropin-dependent adrenal steroidogenesis: stimulation of cholesterol delivery is mediated by the peripheral benzodiazepine receptor. Proc. Natl. Acad. Sci. U.S.A. 86, 4897–4901. doi: 10.1073/pnas.86.13.4897
Biary, N., Xie, C., Kauffman, J., and Akar, F. G. (2011). Biophysical properties and functional consequences of reactive oxygen species (ROS)-induced ROS release in intact myocardium. J. Physiol. 589, 5167–5179. doi: 10.1113/jphysiol.2011.214239
Bird, J. L., Izquierdo-Garcia, D., Davies, J. R., Rudd, J. H., Probst, K. C., Figg, N., et al. (2010). Evaluation of translocator protein quantification as a tool for characterising macrophage burden in human carotid atherosclerosis. Atherosclerosis 210, 388–391. doi: 10.1016/j.atherosclerosis.2009.11.047
Bouhidel, O., Pons, S., Souktani, R., Zini, R., Berdeaux, A., and Ghaleh, B. (2008). Myocardial ischemic postconditioning against ischemia-reperfusion is impaired in ob/ob mice. Am. J. Physiol. Heart Circ. Physiol. 295, H1580–H1586. doi: 10.1152/ajpheart.00379.2008
Brady, N. R., Hamacher-Brady, A., Westerhoff, H. V., and Gottlieb, R. A. (2006). A wave of reactive oxygen species (ROS)-induced ROS release in a sea of excitable mitochondria. Antioxid. Redox Signal. 8, 1651–1665. doi: 10.1089/ars.2006.8.1651
Braestrup, C., and Squires, R. F. (1977). Specific benzodiazepine receptors in rat brain characterized by high-affinity (3H)diazepam binding. Proc. Natl. Acad. Sci. U.S.A. 74, 3805–3809. doi: 10.1073/pnas.74.9.3805
Brown, D. A., Aon, M. A., Akar, F. G., Liu, T., Sorarrain, N., and O’Rourke, B. (2008). Effects of 4’-chlorodiazepam on cellular excitation-contraction coupling and ischaemia-reperfusion injury in rabbit heart. Cardiovasc. Res. 79, 141–149. doi: 10.1093/cvr/cvn053
Caballero, B., Veenman, L., and Gavish, M. (2013). Role of mitochondrial translocator protein (18 kDa) on mitochondrial- related cell death processes. Recent Pat. Endocr. Metab. Immune Drug Discov. 7, 86–101. doi: 10.2174/1872214811307020002
del Valle, H. F., Lascano, E. C., Negroni, J. A., and Crottogini, A. J. (2001). Glibenclamide effects on reperfusion-induced malignant arrhythmias and left ventricular mechanical recovery from stunning in conscious sheep. Cardiovasc. Res. 50, 474–485. doi: 10.1016/S0008-6363(01)00209-7
Delavoie, F., Li, H., Hardwick, M., Robert, J. C., Giatzakis, C., Peranzi, G., et al. (2003). In vivo and in vitro peripheral-type benzodiazepine receptor polymerization: functional significance in drug ligand and cholesterol binding. Biochemistry 42, 4506–4519. doi: 10.1021/bi0267487
Duicu, O. M., Lighezan, R., Sturza, A., Balica, R., Vaduva, A., Feier, H., et al. (2016). Assessment of mitochondrial dysfunction and monoamine oxidase contribution to oxidative stress in human diabetic hearts. Oxid. Med. Cell. Longev. 2016:8470394. doi: 10.1155/2016/8470394
Fedorov, V. V., Glukhov, A. V., Ambrosi, C. M., Kostecki, G., Chang, R., Janks, D., et al. (2011). Effects of KATP channel openers diazoxide and pinacidil in coronary-perfused atria and ventricles from failing and non-failing human hearts. J. Mol. Cell Cardiol. 51, 215–225. doi: 10.1016/j.yjmcc.2011.04.016
Ferdinandy, P., Hausenloy, D. J., Heusch, G., Baxter, G. F., and Schulz, R. (2014). Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol. Rev. 66, 1142–1174. doi: 10.1124/pr.113.008300
Francis Stuart, S. D., De Jesus, N. M., Lindsey, M. L., and Ripplinger, C. M. (2016). The crossroads of inflammation, fibrosis, and arrhythmia following myocardial infarction. J. Mol. Cell Cardiol. 91, 114–122. doi: 10.1016/j.yjmcc.2015.12.024
Fulda, S., Galluzzi, L., and Kroemer, G. (2010). Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 9, 447–464. doi: 10.1038/nrd3137
Green, D. R. (2005). Apoptotic pathways: ten minutes to dead. Cell 121, 671–674. doi: 10.1016/j.cell.2005.05.019
Gut, P. (2015). Targeting mitochondrial energy metabolism with TSPO ligands. Biochem. Soc. Trans. 43, 537–542. doi: 10.1042/BST20150019
Gut, P., Baeza-Raja, B., Andersson, O., Hasenkamp, L., Hsiao, J., Hesselson, D., et al. (2013). Whole-organism screening for gluconeogenesis identifies activators of fasting metabolism. Nat. Chem. Biol. 9, 97–104. doi: 10.1038/nchembio.1136
Gutierrez, A., and Van Wagoner, D. R. (2015). Oxidant and inflammatory mechanisms and targeted therapy in atrial fibrillation: an update. J. Cardiovasc. Pharmacol. 66, 523–529. doi: 10.1097/FJC.0000000000000313
Halestrap, A. P., and Pasdois, P. (2009). The role of the mitochondrial permeability transition pore in heart disease. Biochim. Biophys. Acta 1787, 1402–1415. doi: 10.1016/j.bbabio.2008.12.017
Harrison, D., Griendling, K. K., Landmesser, U., Hornig, B., and Drexler, H. (2003). Role of oxidative stress in atherosclerosis. Am. J. Cardiol. 91, 7A–11A. doi: 10.1016/S0002-9149(02)03144-2
Hellberg, S., Liljenback, H., Eskola, O., Morisson-Iveson, V., Morrison, M., Trigg, W., et al. (2018). Positron emission tomography imaging of macrophages in atherosclerosis with (18)F-GE-180, a radiotracer for translocator protein (TSPO). Contrast Media Mol. Imaging 2018:9186902. doi: 10.1155/2018/9186902
Higashi, Y., Noma, K., Yoshizumi, M., and Kihara, Y. (2009). Endothelial function and oxidative stress in cardiovascular diseases. Circ. J. 73, 411–418. doi: 10.1253/circj.CJ-08-1102
Hoshida, S., Nishida, M., Yamashita, N., Igarashi, J., Hori, M., Kamada, T., et al. (1996). Amelioration of severity of myocardial injury by a nitric oxide donor in rabbits fed a cholesterol-rich diet. J. Am. Coll. Cardiol. 27, 902–909. doi: 10.1016/0735-1097(95)00538-2
Hu, J., Koh, W.-J., Chaoqin, X., and Akar, F. G. (2016). Role of the mitochondrial translocator protein (TSPO) in the proarrhythmic vulnerability of the diabetic heart. Paper Presented at the Circulation Research, Philadelphia, PA.
Huxley, R. R., Alonso, A., Lopez, F. L., Filion, K. B., Agarwal, S. K., Loehr, L. R., et al. (2012). Type 2 diabetes, glucose homeostasis and incident atrial fibrillation: the atherosclerosis risk in communities study. Heart 98, 133–138. doi: 10.1136/heartjnl-2011-300503
Huxley, R. R., Filion, K. B., Konety, S., and Alonso, A. (2011). Meta-analysis of cohort and case-control studies of type 2 diabetes mellitus and risk of atrial fibrillation. Am. J. Cardiol. 108, 56–62. doi: 10.1016/j.amjcard.2011.03.004
Iacobellis, G., Corradi, D., and Sharma, A. M. (2005). Epicardial adipose tissue: anatomic, biomolecular and clinical relationships with the heart. Nat. Clin. Pract. Cardiovasc. Med. 2, 536–543. doi: 10.1038/ncpcardio0319
Ilkan, Z., Strauss, B., Campana, C., and Akar, F. G. (2018). Optical action potential mapping in acute models of ischemia-reperfusion injury: probing the arrhythmogenic role of the mitochondrial translocator protein. Methods Mol. Biol. 1816, 133–143. doi: 10.1007/978-1-4939-8597-5_10
Jaremko, L., Jaremko, M., Giller, K., Becker, S., and Zweckstetter, M. (2014). Structure of the mitochondrial translocator protein in complex with a diagnostic ligand. Science 343, 1363–1366. doi: 10.1126/science.1248725
Jia, G., Hill, M. A., and Sowers, J. R. (2018). Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ. Res. 122, 624–638. doi: 10.1161/CIRCRESAHA.117.311586
Kinnally, K. W., Zorov, D. B., Antonenko, Y. N., Snyder, S. H., McEnery, M. W., and Tedeschi, H. (1993). Mitochondrial benzodiazepine receptor linked to inner membrane ion channels by nanomolar actions of ligands. Proc. Natl. Acad. Sci. U.S.A. 90, 1374–1378. doi: 10.1073/pnas.90.4.1374
Korkhov, V. M., Sachse, C., Short, J. M., and Tate, C. G. (2010). Three-dimensional structure of TspO by electron cryomicroscopy of helical crystals. Structure 18, 677–687. doi: 10.1016/j.str.2010.03.001
Kroemer, G., Galluzzi, L., and Brenner, C. (2007). Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87, 99–163. doi: 10.1152/physrev.00013.2006
Kwong, J. Q., and Molkentin, J. D. (2015). Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 21, 206–214. doi: 10.1016/j.cmet.2014.12.001
Lacapere, J. J., Delavoie, F., Li, H., Peranzi, G., Maccario, J., Papadopoulos, V., et al. (2001). Structural and functional study of reconstituted peripheral benzodiazepine receptor. Biochem. Biophys. Res. Commun. 284, 536–541. doi: 10.1006/bbrc.2001.4975
Lau, D. H., Nattel, S., Kalman, J. M., and Sanders, P. (2017). Modifiable risk factors and atrial fibrillation. Circulation 136, 583–596. doi: 10.1161/CIRCULATIONAHA.116.023163
Li, F., Liu, J., Zheng, Y., Garavito, R. M., and Ferguson-Miller, S. (2015). Protein structure. Crystal structures of translocator protein (TSPO) and mutant mimic of a human polymorphism. Science 347, 555–558. doi: 10.1126/science.1260590
Li, H., and Papadopoulos, V. (1998). Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology 139, 4991–4997. doi: 10.1210/endo.139.12.6390
Li, J., and Papadopoulos, V. (2015). Translocator protein (18 kDa) as a pharmacological target in adipocytes to regulate glucose homeostasis. Biochem. Pharmacol. 97, 99–110. doi: 10.1016/j.bcp.2015.06.020
Marchetti, P., Castedo, M., Susin, S. A., Zamzami, N., Hirsch, T., Macho, A., et al. (1996). Mitochondrial permeability transition is a central coordinating event of apoptosis. J. Exp. Med. 184, 1155–1160. doi: 10.1084/jem.184.3.1155
Mazurek, T., Zhang, L., Zalewski, A., Mannion, J. D., Diehl, J. T., Arafat, H., et al. (2003). Human epicardial adipose tissue is a source of inflammatory mediators. Circulation 108, 2460–2466. doi: 10.1161/01.CIR.0000099542.57313.C5
Melo, J., Voigt, P., Sonmez, B., Ferreira, M., Abecasis, M., Rebocho, M., et al. (2004). Ventral cardiac denervation reduces the incidence of atrial fibrillation after coronary artery bypass grafting. J. Thorac. Cardiovasc. Surg. 127, 511–516. doi: 10.1016/S0022-5223(03)01283-2
Misra, M. K., Sarwat, M., Bhakuni, P., Tuteja, R., and Tuteja, N. (2009). Oxidative stress and ischemic myocardial syndromes. Med. Sci. Monit. 15, Ra209–Ra219.
Mitchell, P., and Moyle, J. (1965a). Evidence discriminating between the chemical and the chemiosmotic mechanisms of electron transport phosphorylation. Nature 208, 1205–1206. doi: 10.1038/2081205a0
Mitchell, P., and Moyle, J. (1965b). Stoichiometry of proton translocation through the respiratory chain and adenosine triphosphatase systems of rat liver mitochondria. Nature 208, 147–151. doi: 10.1038/208147a0
Morin, D., Musman, J., Pons, S., Berdeaux, A., and Ghaleh, B. (2016). Mitochondrial translocator protein (TSPO): from physiology to cardioprotection. Biochem. Pharmacol. 105, 1–13. doi: 10.1016/j.bcp.2015.12.003
Motloch, L. J., Hu, J., and Akar, F. G. (2015). The mitochondrial translocator protein and arrhythmogenesis in ischemic heart disease. Oxid. Med. Cell. Longev. 2015:234104. doi: 10.1155/2015/234104
Movahed, M. R., Hashemzadeh, M., and Jamal, M. M. (2005). Diabetes mellitus is a strong, independent risk for atrial fibrillation and flutter in addition to other cardiovascular disease. Int. J. Cardiol. 105, 315–318. doi: 10.1016/j.ijcard.2005.02.050
Musman, J., Paradis, S., Panel, M., Pons, S., Barau, C., Caccia, C., et al. (2017). A TSPO ligand prevents mitochondrial sterol accumulation and dysfunction during myocardial ischemia-reperfusion in hypercholesterolemic rats. Biochem. Pharmacol. 142, 87–95. doi: 10.1016/j.bcp.2017.06.125
O’Rourke, B. (2000). Pathophysiological and protective roles of mitochondrial ion channels. J. Physiol. 529(Pt 1), 23–36. doi: 10.1111/j.1469-7793.2000.00023.x
Osipov, R. M., Bianchi, C., Feng, J., Clements, R. T., Liu, Y., Robich, M. P., et al. (2009). Effect of hypercholesterolemia on myocardial necrosis and apoptosis in the setting of ischemia-reperfusion. Circulation 120, S22–S30. doi: 10.1161/CIRCULATIONAHA.108.842724
Paky, A., Michael, J. R., Burke-Wolin, T. M., Wolin, M. S., and Gurtner, G. H. (1993). Endogenous production of superoxide by rabbit lungs: effects of hypoxia or metabolic inhibitors. J. Appl. Physiol. 74, 2868–2874. doi: 10.1152/jappl.1993.74.6.2868
Papadopoulos, V., Amri, H., Boujrad, N., Cascio, C., Culty, M., Garnier, M., et al. (1997). Peripheral benzodiazepine receptor in cholesterol transport and steroidogenesis. Steroids 62, 21–28. doi: 10.1016/S0039-128X(96)00154-7
Papadopoulos, V., Baraldi, M., Guilarte, T. R., Knudsen, T. B., Lacapere, J. J., Lindemann, P., et al. (2006). Translocator protein (18kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 27, 402–409. doi: 10.1016/j.tips.2006.06.005
Paradis, S., Leoni, V., Caccia, C., Berdeaux, A., and Morin, D. (2013). Cardioprotection by the TSPO ligand 4’-chlorodiazepam is associated with inhibition of mitochondrial accumulation of cholesterol at reperfusion. Cardiovasc. Res. 98, 420–427. doi: 10.1093/cvr/cvt079
Peart, J. N., and Headrick, J. P. (2009). Clinical cardioprotection and the value of conditioning responses. Am. J. Physiol. Heart Circ. Physiol. 296, H1705–H1720. doi: 10.1152/ajpheart.00162.2009
Peiro, C., Romacho, T., Azcutia, V., Villalobos, L., Fernandez, E., Bolanos, J. P., et al. (2016). Inflammation, glucose, and vascular cell damage: the role of the pentose phosphate pathway. Cardiovasc. Diabetol. 15:82. doi: 10.1186/s12933-016-0397-2
Pistrosch, F., Ganz, X., Bornstein, S. R., Birkenfeld, A. L., Henkel, E., and Hanefeld, M. (2015). Risk of and risk factors for hypoglycemia and associated arrhythmias in patients with type 2 diabetes and cardiovascular disease: a cohort study under real-world conditions. Acta Diabetol. 52, 889–895. doi: 10.1007/s00592-015-0727-y
Pugliese, F., Gaemperli, O., Kinderlerer, A. R., Lamare, F., Shalhoub, J., Davies, A. H., et al. (2010). Imaging of vascular inflammation with [11C]-PK11195 and positron emission tomography/computed tomography angiography. J. Am. Coll. Cardiol. 56, 653–661. doi: 10.1016/j.jacc.2010.02.063
Ran, C., Albrecht, D. S., Bredella, M. A., Yang, J., Yang, J., Liang, S. H., et al. (2018). PET imaging of human brown adipose tissue with the TSPO tracer [(11)C]PBR28. Mol. Imaging Biol 20, 188–193. doi: 10.1007/s11307-017-1129-z
Rosen, E. D., and Spiegelman, B. M. (2006). Adipocytes as regulators of energy balance and glucose homeostasis. Nature 444, 847–853. doi: 10.1038/nature05483
Rupprecht, R., Papadopoulos, V., Rammes, G., Baghai, T. C., Fan, J., Akula, N., et al. (2010). Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat. Rev. Drug Discov. 9, 971–988. doi: 10.1038/nrd3295
Saito, S., Teshima, Y., Fukui, A., Kondo, H., Nishio, S., Nakagawa, M., et al. (2014). Glucose fluctuations increase the incidence of atrial fibrillation in diabetic rats. Cardiovasc. Res. 104, 5–14. doi: 10.1093/cvr/cvu176
Samanta, R., Pouliopoulos, J., Thiagalingam, A., and Kovoor, P. (2016). Role of adipose tissue in the pathogenesis of cardiac arrhythmias. Heart Rhythm 13, 311–320. doi: 10.1016/j.hrthm.2015.08.016
Scalia, R., Gooszen, M. E., Jones, S. P., Hoffmeyer, M., Rimmer, D. M. III, Trocha, S. D., et al. (2001). Simvastatin exerts both anti-inflammatory and cardioprotective effects in apolipoprotein E-deficient mice. Circulation 103, 2598–2603. doi: 10.1161/01.CIR.103.21.2598
Stahn, A., Pistrosch, F., Ganz, X., Teige, M., Koehler, C., Bornstein, S., et al. (2014). Relationship between hypoglycemic episodes and ventricular arrhythmias in patients with type 2 diabetes and cardiovascular diseases: silent hypoglycemias and silent arrhythmias. Diabetes Care 37, 516–520. doi: 10.2337/dc13-0600
Takahashi, M. A., and Asada, K. (1983). Superoxide anion permeability of phospholipid membranes and chloroplast thylakoids. Arch. Biochem. Biophys. 226, 558–566. doi: 10.1016/0003-9861(83)90325-9
Taylor, J. M., Allen, A. M., and Graham, A. (2014). Targeting mitochondrial 18 kDa translocator protein (TSPO) regulates macrophage cholesterol efflux and lipid phenotype. Clin Sci 127, 603–613. doi: 10.1042/CS20140047
Thompson, M. M., Manning, H. C., and Ellacott, K. L. (2013). Translocator protein 18 kDa (TSPO) is regulated in white and brown adipose tissue by obesity. PLoS One 8:e79980. doi: 10.1371/journal.pone.0079980
Turrens, J. F. (2003). Mitochondrial formation of reactive oxygen species. J. Physiol. 552, 335–344. doi: 10.1113/jphysiol.2003.049478
Van Wagoner, D. R. (2008). Oxidative stress and inflammation in atrial fibrillation: role in pathogenesis and potential as a therapeutic target. J. Cardiovasc. Pharmacol. 52, 306–313. doi: 10.1097/FJC.0b013e31817f9398
Veenman, L., and Gavish, M. (2006). The peripheral-type benzodiazepine receptor and the cardiovascular system. Implications for drug development. Pharmacol. Ther. 110, 503–524. doi: 10.1016/j.pharmthera.2005.09.007
Veenman, L., Papadopoulos, V., and Gavish, M. (2007). Channel-like functions of the 18-kDa translocator protein (TSPO): regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr. Pharm. Des. 13, 2385–2405. doi: 10.2174/138161207781368710
Wilson, A. J., Gill, E. K., Abudalo, R. A., Edgar, K. S., Watson, C. J., and Grieve, D. J. (2018). Reactive oxygen species signalling in the diabetic heart: emerging prospect for therapeutic targeting. Heart 104, 293–299. doi: 10.1136/heartjnl-2017-311448
Wu, N., Shen, H., Liu, H., Wang, Y., Bai, Y., and Han, P. (2016). Acute blood glucose fluctuation enhances rat aorta endothelial cell apoptosis, oxidative stress and pro-inflammatory cytokine expression in vivo. Cardiovasc. Diabetol. 15:109. doi: 10.1186/s12933-016-0427-0
Wu, N., Zhang, X., Guan, Y., Shu, W., Jia, P., and Jia, D. (2014). Hypercholesterolemia abrogates the cardioprotection of ischemic postconditioning in isolated rat heart: roles of glycogen synthase kinase-3beta and the mitochondrial permeability transition pore. Cell Biochem. Biophys. 69, 123–130. doi: 10.1007/s12013-013-9778-2
Xie, C., Hu, J., Motloch, L. J., Karam, B. S., and Akar, F. G. (2015). The classically cardioprotective agent diazoxide elicits arrhythmias in type 2 diabetes mellitus. J. Am. Coll. Cardiol. 66, 1144–1156. doi: 10.1016/j.jacc.2015.06.1329
Xie, C., Kauffman, J., and Akar, F. G. (2014). Functional crosstalk between the mitochondrial PTP and KATP channels determine arrhythmic vulnerability to oxidative stress. Front. Physiol. 5:264. doi: 10.3389/fphys.2014.00264
Yang, L., Korge, P., Weiss, J. N., and Qu, Z. (2010). Mitochondrial oscillations and waves in cardiac myocytes: insights from computational models. Biophys. J. 98, 1428–1438. doi: 10.1016/j.bpj.2009.12.4300
Yang, W., Dall, T. M., Beronjia, K., Lin, J., Semilla, A. P., Chakrabarti, R., et al. (2018). Economic costs of diabetes in the U.S. in 2017. Diabetes Care 41, 917–928. doi: 10.2337/dci18-0007
Zamzami, N., Larochette, N., and Kroemer, G. (2005). Mitochondrial permeability transition in apoptosis and necrosis. Cell Death. Differ. 12(Suppl. 2), 1478–1480. doi: 10.1038/sj.cdd.4401682
Zhang, X., Zhang, Z., Zhao, Y., Jiang, N., Qiu, J., Yang, Y., et al. (2017). Alogliptin, a Dipeptidyl Peptidase-4 inhibitor, alleviates atrial remodeling and improves mitochondrial function and biogenesis in diabetic rabbits. J. Am. Heart Assoc. 6:e005945. doi: 10.1161/JAHA.117.005945
Zhou, L., Solhjoo, S., Millare, B., Plank, G., Abraham, M. R., Cortassa, S., et al. (2014). Effects of regional mitochondrial depolarization on electrical propagation: implications for arrhythmogenesis. Circ. Arrhythm. Electrophysiol. 7, 143–151. doi: 10.1161/CIRCEP.113.000600
Zipes, D. P., and Wellens, H. J. (1998). Sudden cardiac death. Circulation 98, 2334–2351. doi: 10.1161/01.CIR.98.21.2334
Zorov, D. B., Filburn, C. R., Klotz, L. O., Zweier, J. L., and Sollott, S. J. (2000). Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 192, 1001–1014. doi: 10.1084/jem.192.7.1001
Keywords: arrhythmias, reactive oxygen species, oxidative stress, mitochondria, diabetes
Citation: Ilkan Z and Akar FG (2018) The Mitochondrial Translocator Protein and the Emerging Link Between Oxidative Stress and Arrhythmias in the Diabetic Heart. Front. Physiol. 9:1518. doi: 10.3389/fphys.2018.01518
Received: 27 July 2018; Accepted: 09 October 2018;
Published: 26 October 2018.
Edited by:
Gaetano Santulli, Columbia University, United StatesReviewed by:
Jin O-Uchi, University of Minnesota Twin Cities, United StatesCrystal M. Ripplinger, University of California, Davis, United States
Copyright © 2018 Ilkan and Akar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fadi G. Akar, ZmFkaS5ha2FyQG1zc20uZWR1