 Gayathri K. Natarajan
Gayathri K. Natarajan Jyotsna Mishra
Jyotsna Mishra Amadou K. S. Camara
Amadou K. S. Camara Wai-Meng Kwok
Wai-Meng Kwok- 1Department of Anesthesiology, Medical College of Wisconsin, Milwaukee, WI, United States
- 2Department of Physiology, Medical College of Wisconsin, Milwaukee, WI, United States
- 3Cancer Center, Medical College of Wisconsin, Milwaukee, WI, United States
- 4Cardiovascular Center, Medical College of Wisconsin, Milwaukee, WI, United States
- 5Department of Pharmacology and Toxicology, Medical College of Wisconsin, Milwaukee, WI, United States
Nearly 2 decades since its discovery as one of the genes responsible for the Wolf-Hirschhorn Syndrome (WHS), the primary function of the leucine-zipper EF-hand containing transmembrane 1 (LETM1) protein in the inner mitochondrial membrane (IMM) or the mechanism by which it regulates mitochondrial Ca2+ handling is unresolved. Meanwhile, LETM1 has been associated with the regulation of fundamental cellular processes, such as development, cellular respiration and metabolism, and apoptosis. This mini-review summarizes the diversity of cellular functions impacted by LETM1 and highlights the multiple roles of LETM1 in health and disease.
Introduction
Mitochondria accumulate large amounts of Ca2+ via highly regulated mechanisms of Ca2+ transport across the mitochondrial membranes and Ca2+ handling in the matrix (Rizzuto et al., 2012) and thereby, attenuate deleterious increases in cytosolic Ca2+ (Hajnóczky et al., 2014). Ca2+ uptake into mitochondria by the mitochondrial Ca2+ uniporter (MCU) macromolecular complex (MCUC) and Ca2+ efflux through the electrogenic Na+/Li+/Ca2+ exchanger (NCLX) are among the well-studied mechanisms of mitochondrial Ca2+ transport (Palty et al., 2010; Blomeyer et al., 2013; Mishra et al., 2017). Recently, the Leucine zipper EF-hand containing transmembrane 1 (LETM1) protein, expressed in the inner mitochondrial membrane (IMM) has been implicated in mitochondrial Ca2+ regulation. However, the mechanism by which LETM1 regulates Ca2+ is unresolved and controversial.
The Letm1 gene was originally identified as one of the genes deleted in patients afflicted with the Wolf-Hirschhorn Syndrome (WHS), a contiguous gene deletion disorder marked by severe growth and intellectual disability, hypotonia, and seizures (Endele et al., 1999; Schlickum et al., 2004). It is evolutionarily conserved in all eukaryotes, plants, and animals (Zhang et al., 2012; Tang et al., 2018; Lin and Stathopulos, 2019) with ubiquitous expression in mammalian tissues (Fagerberg et al., 2014; Uhlén et al., 2015). Letm1 is characterized as an essential gene, such that haploinsufficiency leads to manifestation of disease phenotype (Endele et al., 1999; Hart et al., 2014), and homozygous deletion is embryonically lethal (McQuibban et al., 2010; Jiang et al., 2013).
The LETM1 subunit (83.4 kDa) has a short hydrophobic N-terminal, a conserved proline-rich transmembrane domain, a hydrophilic C-terminal in the matrix, with two to four coiled-coil domains (CCDs), and a characteristic leucine-zipper sequence (Endele et al., 1999). Depending on the species, the C-terminus may include either one or two non-canonical Ca2+-binding EF-Hand motifs, or none as in the orthologs from yeast and from the protozoan Trypanosome brucei (Nowikovsky et al., 2004; Hasegawa and van der Bliek, 2007). A TXXR motif, a consensus site for phosphorylation by PKC and Casein Kinase 2 (CK2), flanks the N-terminus of the conserved transmembrane domain (Figure 1). Electron microscopy data suggest that LETM1 subunits oligomerize into a putative hexameric configuration to form a functional exchanger with pH-sensitive conformation and conductance properties (Hasegawa and van der Bliek, 2007; Shao et al., 2016). The N-terminus of LETM1 was presumed to face the mitochondrial intermembrane space (IMS); however, recent predictions of a putative second transmembrane domain in the LETM1 protein has led to a reevaluation of the structure and orientation of the LETM1 subunit in the IMM (Lee et al., 2017; Austin and Nowikovsky, 2019).
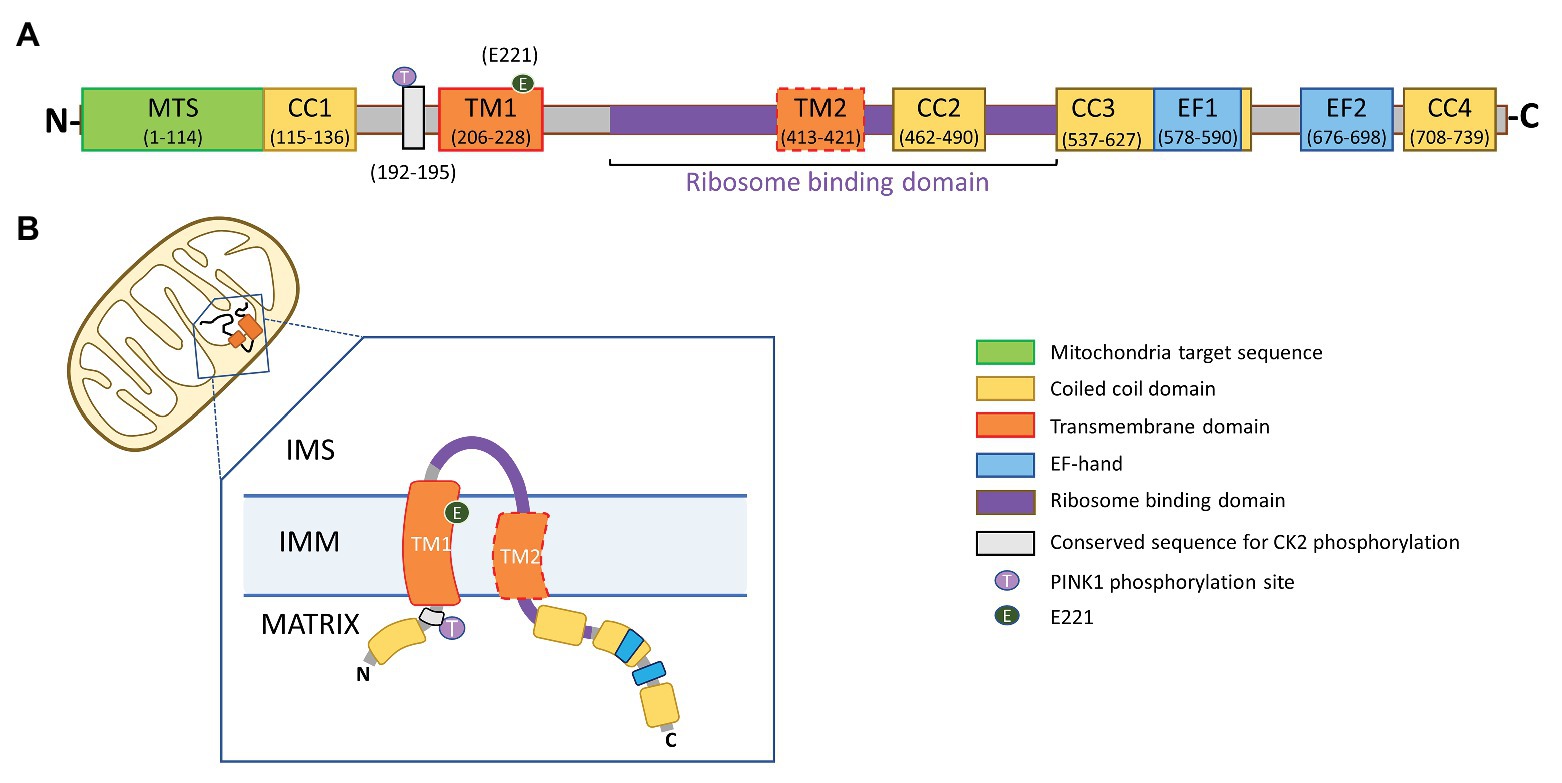
Figure 1. Proposed topology of leucine-zipper EF-hand containing transmembrane 1 (LETM1). (A) Domain architecture of LETM1. MTS, mitochondrial targeting sequence; CC1, CC2, CC3, and CC4, coiled-coil domains 1–4; TM1, conserved transmembrane domain 1; TM2, putative transmembrane domain 2; EF1, EF2, non-canonical Ca2+-binding EF-hand domains; and Gray Box – TXXR motif, a conserved consensus site for phosphorylation by protein kinase C and casein kinase 2 (CK2). The numbers in parentheses represent amino acid residue for human LETM1 sequence (Uniprot accession number: Q2VYF4). (B) Both the N- and C-termini of the LETM1 subunit are proposed to reside in the mitochondrial matrix. A portion of the ribosome binding domain is exposed to the inter-membrane space (IMS).
The functional role of LETM1 in mitochondria is controversial and extensively debated as to whether it is primarily a K+/H+ exchanger (KHE) as originally surmised or a Ca2+/H+ exchanger (CHE), as subsequent studies have shown (Nowikovsky et al., 2012; Austin and Nowikovsky, 2019; Li et al., 2019; Lin and Stathopulos, 2019). Regardless, LETM1 is implicated in mitochondrial and cellular functions, such that loss of LETM1 impacts mitochondrial ionic homeostasis, bioenergetics, morphology and biogenesis, cell viability, and development. Furthermore, LETM1 has been shown to interact with proteins that regulate varied signaling processes, such as import and assembly of critical respiratory supercomplexes in the IMM, cellular glucose metabolism, and neuronal function (Figure 2). Consequently, changes in LETM1 expression and function are associated with pathophysiologies, such as insulin resistance in obesity, tumorigenesis, and seizures (Hasegawa and van der Bliek, 2007; Dimmer et al., 2008; Tamai et al., 2008; Jiang et al., 2013; Hart et al., 2014; Park et al., 2014; Li et al., 2019). It is unclear as to how LETM1, localized to the IMM, regulates and impacts diverse cellular functions. It is expected that as a CHE and/or KHE in the IMM, LETM1 would regulate mitochondrial function. However, the studies revealing interactions between LETM1 and regulatory proteins suggest that LETM1 function may extend beyond its exchanger role in the IMM. This mini-review briefly summarizes the current status of the debate regarding LETM1 function as a CHE or KHE. It also highlights the diversity of cellular functions impacted by the loss or gain of LETM1 and the possible mechanisms involved in LETM1-mediated regulation of mitochondrial metabolism and cellular signaling.
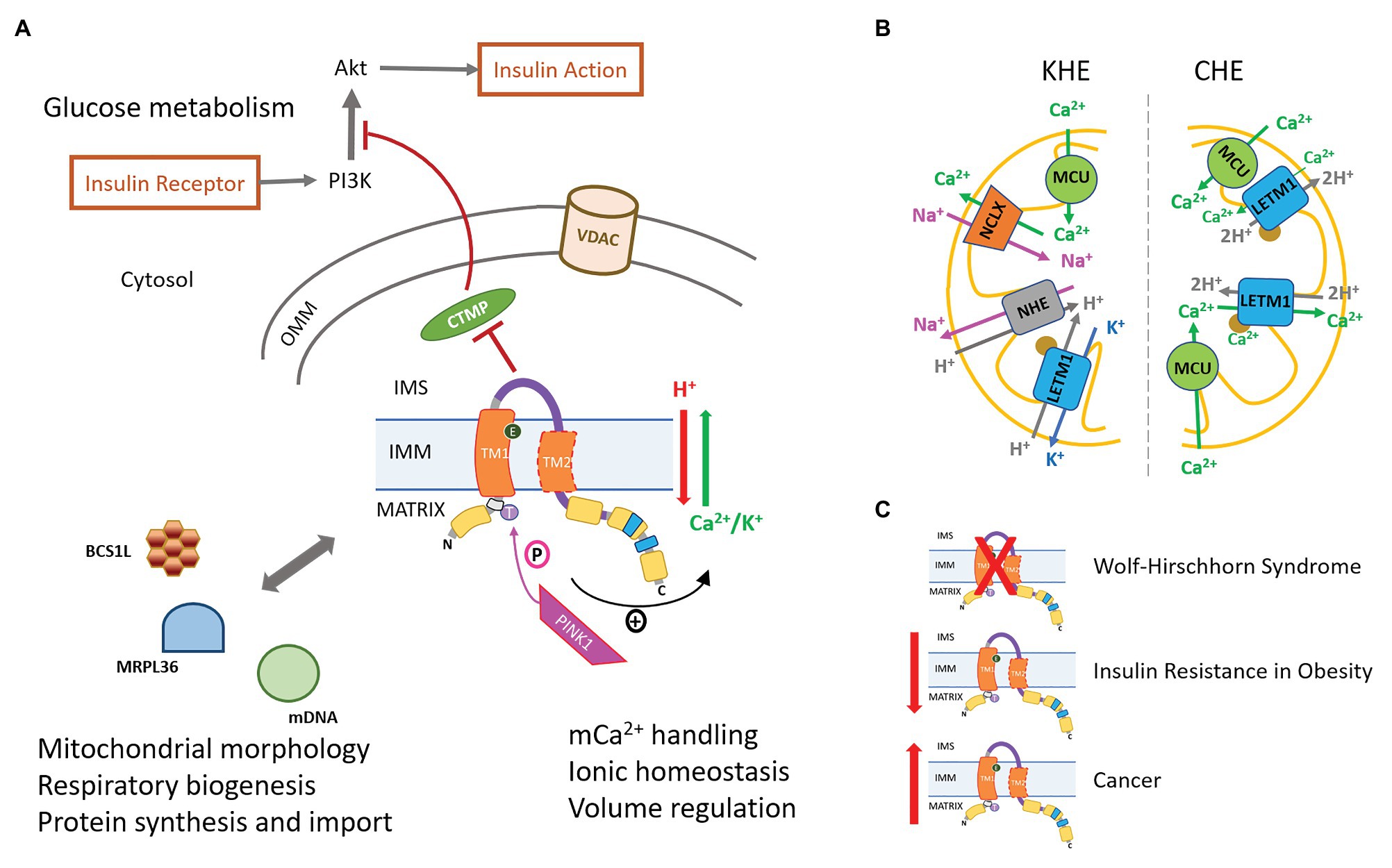
Figure 2. LETM1-mediated regulation of cellular physiology and pathophysiology. (A) LETM1-mediated Ca2+/H+ exchanger (CHE) or K+/H+ exchanger (KHE) activity regulates mitochondrial Ca2+ handling and bioenergetics. The serine-threonine kinase PINK1, associated with Parkinson’s Disease (PD), phosphorylates LETM1 at T192 and enhances exchanger activity (see text for more details). Interactions of LETM1 with signaling proteins, such as CTMP, BCS1L, and MRPL36, regulate glucose metabolism, mitochondrial morphology, and protein assembly in the inner mitochondrial membrane (IMM), respectively. VDAC, voltage-dependent anion channel (B) Proposed mechanisms of LETM1 as KHE (left) or CHE (right). The brown circle represents EF-hand in mammalian LETM1. (C) Disease phenotypes associated with either the absence [Wolf-Hirschhorn Syndrome (WHS)], decreased expression (Insulin resistance in obesity), or increased expression (cancer) of LETM1.
LETM1: Che or Khe?
Early evidence for LETM1 as a putative KHE (LETM1-KHE) came from studies in mitochondria expressing MDM38Δ, a mutant yeast ortholog of LETM1. MDM38Δ mitochondria exhibited higher matrix K+ content, decreased IMM potential (ΔΨ)m, loss of K+ acetate-induced swelling, and dissolution of cristae structure and mitochondrial morphology (Nowikovsky et al., 2004; Froschauer et al., 2005). These effects were replicated in mitochondria of other lower and higher organisms and cell cultures with knockout or knockdown of corresponding LETM1 orthologs, including Caenorhabditis elegans, HeLa cells, Drosophila melanogaster, and T. brucei (Hasegawa and van der Bliek, 2007; Dimmer et al., 2008; McQuibban et al., 2010; Hashimi et al., 2013). The K+ ionophore, nigericin, or the expression of human LETM1 reversed these effects, suggesting that the swelling and resultant effects on morphology and function were caused by the loss of an evolutionarily conserved KHE function in LETM1-deficient mitochondria (Nowikovsky et al., 2004).
However, in contrast with yeast or the protozoan T. brucei, LETM1 orthologs of higher organisms possess EF-hands, which may act as Ca2+ sensors and implicate LETM1 in Ca2+ regulation. Accordingly, LETM1 was identified as a putative CHE (LETM1-CHE) in a genome-wide RNAi screening in D. melanogaster cells, where mitochondrial Ca2+ influx/efflux was coupled to H+ extrusion/influx, respectively. LETM1-knockdown (LETM1-KD) decoupled the Ca2+/H+ antiport while LETM1 overexpression increased pH-induced Ca2+ transport 5-fold (Jiang et al., 2009). In subsequent studies, recombinant LETM1 reconstituted into liposomes exhibited LETM1-mediated Ca2+ transport that is bidirectional and insensitive to K+ and Na2+, indicating that LETM1 is a CHE (Jiang et al., 2009; Tsai et al., 2014; Shao et al., 2016). Both Ca2+ uptake and release were driven by changes in pH (Jiang et al., 2009; Tsai et al., 2014) and enhanced by the phosphomimetic T192E located in the N-terminus of the LETM1 subunit (Huang et al., 2017b).
In cells and isolated mitochondria, it is unclear whether LETM1-CHE mediates either mitochondrial Ca2+ uptake or efflux. In histamine-stimulated HeLa cells, LETM1-KD or a mutation of negative-charged glutamate residue in the transmembrane domain increased mitochondrial Ca2+, indicating a loss of LETM1-mediated Ca2+ efflux (Shao et al., 2016). In isolated rat heart mitochondria, Na+-independent Ca2+ efflux increased with extra-matrix acidification and with increasing Ca2+ load and LETM1-KD in permeabilized H9c2 cells modified Ca2+ efflux, indicating that, in these cells, LETM1-CHE mediates Ca2+ efflux (Natarajan et al., 2020). In contrast, in another study using histamine-stimulated HeLa cells, LETM1-KD significantly diminished Ca2+ uptake in the submicromolar Ca2+ range (Jiang et al., 2009). In endothelial cells, LETM1, along with MCU, may mediate mitochondrial Ca2+ uptake in response to slow and small Ca2+ elevations due to store-operated Ca2+ entry (SOCE) following Ca2+ depletion in the sarco/endo-plasmic reticulum (SR/ER) and/or failure of Ca2+ sequestration through the sarco/endo-plasmic reticular Ca2+ ATPase (SERCA; Waldeck-Weiermair et al., 2013). In this study, LETM1-KD significantly diminished mitochondrial Ca2+ uptake under conditions of slow Ca2+ increases during SOCE (Patergnani et al., 2011; Waldeck-Weiermair et al., 2011, 2013). Other studies have shown that LETM1-CHE may mediate both Ca2+-influx and efflux. For instance, Doonan et al. (2014) showed that in histamine-challenged HeLa cells, LETM1-KD or the expression of a LETM1-mutant with deleted EF-hands significantly diminished both Ca2+ influx and efflux rates in mitochondria. Similar changes in histamine-induced mitochondrial Ca2+ were observed in LETM1-silenced B lymphocytes, hematopoietic cells, neonatal rat ventricular myocytes, and in fibroblasts from WHS patients (Doonan et al., 2014). Since the expression of MCU remained unaffected in LETM1-KD cells, the decrease in mCa2+ uptake rate was associated with the loss of a LETM1-dependent Ca2+ influx. However, the decrease in Ca2+ uptake rate was also accompanied by an increase in total mCa2+ and decrease in Ca2+ efflux rate. This suggested that the decreased Ca2+ uptake rate may be a consequence of the loss of a LETM1-dependent Ca2+ efflux. Thus, it is yet to be established whether LETM1-CHE facilitates Ca2+ influx or efflux or both (Figure 2B).
Despite studies supporting a LETM1-CHE function, recent studies have provided further evidence that human LETM1 also is primarily a KHE. Studies in HeLa cells have shown that overexpression of LETM1 increases the K+-stimulated proton gradient, but has no effect on the mCa2+ efflux rate in response to a histamine challenge (Marchi et al., 2014). Another study in HeLa cells suggested that LETM1-based regulation of mitochondrial Ca2+ is achieved by a LETM1-KHE activity whereby it regulates the activity of the mitochondrial Na+/H+ exchanger (mNHE), ultimately affecting NCLX-mediated Ca2+ efflux (Figure 2B; Austin et al., 2017). Alternatively, it may be speculated that swelling in LETM1-deficient mitochondria is caused by changes in K+ transport due to accumulation of Ca2+ (Halestrap et al., 1986; Gogvadze et al., 2004; Kaasik et al., 2007), and nigericin-mediated KHE activity may alleviate the effects of Ca2+ imbalance to reverse swelling in these cells. This notion is supported by the observation that nigericin stimulated a K+ driven H+ flux in LETM1-KD Drosophila S2 cells without restoring pH-driven Ca2+ exchange (Jiang et al., 2009). Thus, the controversy over the primary function of LETM1 in cells persists. In the absence of LETM1-specific inhibitors, it is even more challenging to delineate the role of LETM1 in mitochondrial Ca2+ regulation.
The Diversity of LETM1-Induced Pathophysiology
LETM1 in Cellular Metabolic Dysregulation
Evidence indicates that LETM1 mediates several steps in the cellular metabolic signaling pathway, such that LETM1 deficiency is associated with impairment of both cellular and mitochondrial metabolism. Several studies document the role of LETM1 in regulating cellular metabolism through its impact on insulin secretion and glucose uptake by cells. In mitochondria of insulin-secreting pancreatic β-cells, MCU−/− as well as LETM1-KD prevented Ca2+-induced matrix acidification, decreased substrate-stimulated ATP production, and impaired glucose-stimulated insulin secretion (Quan et al., 2015). These observations suggest that LETM1 functions as CHE and mediates mitochondrial Ca2+ efflux in these cells.
However, LETM1 has also been shown to regulate cellular glucose metabolism through interactions with other signaling proteins (Figure 2A). In these examples, a LETM1-CHE/KHE function is not clear. For instance, LETM1 has been identified as a constitutive binding partner of carboxy-terminal-modulator-protein (CTMP; Piao et al., 2009b), a protein that localizes to the IMS in membrane-bound and free forms and regulates mitochondrial biogenesis by positively influencing mitochondrial fission (Parcellier et al., 2009). CTMP can bind to the hydrophobic motif of protein kinase B (Akt) and inhibit its phosphorylation (Maira et al., 2001; Parcellier et al., 2009). The nature of the interaction between LETM1 and CTMP is unknown, although, like CTMP, LETM1 has also been shown to regulate mitochondrial biogenesis. LETM1 overexpression has been correlated with increased cleavage of the mitochondrial fusion protein OPA1, leading to increased mitochondrial fragmentation (Piao et al., 2009b). It is suggested that, in obesity, LETM1 and CTMP may counter-regulate each other’s function since LETM1 expression is inversely correlated with the expression of CTMP and positively correlated with Akt phosphorylation and activity (Park et al., 2014). Thus, in liver cells from obese and high fat diet-fed mice, a decrease in LETM1 expression was associated with a reciprocal increase in CTMP expression and a decrease in Akt activity, suppression of insulin signaling and cellular glucose uptake, and development of insulin resistance (Park et al., 2014). The impact of LETM1 on insulin signaling was recently demonstrated in an epigenome-wide study of genes undergoing differential DNA methylation in obese individuals that identified one of nine differential CpG methylation sites in the LETM1 gene to be associated with fasting insulin levels (Liu et al., 2019). The association between LETM1 methylation and fasting insulin levels suggests that it can serve as an epigenetic marker to indicate future development of insulin resistance and Type II diabetes in obese individuals.
Another example of LETM1-dependent regulation of cellular metabolism, where its function as a CHE/KHE is not readily apparent is the association between LETM1 and pyruvate oxidation. Jiang et al. (2013) showed that loss of LETM1 impaired pyruvate oxidation during aerobic glycolysis, but did not affect fatty acid oxidation during fasting. This was confirmed from metabolic profiles of liver tissue, which showed significant decrease in β-hydroxybutyrate (BHB), a downstream metabolite of pyruvate, and fatty acid oxidation, in LETM1+/− mice compared to WT mice during glycolysis, with no difference in BHB levels during fasting (Jiang et al., 2013). LETM1-KD also led to an increase in phosphorylated pyruvate dehydrogenase, the inactive form of the enzyme, and a decreased oxidation of pyruvate to acetyl-CoA (Durigon et al., 2018).
LETM1 also regulates mitochondrial metabolism, whereby its deficiency can decrease ATP production by compromising the assembly of respiratory complexes. A conserved 14-3-3-like domain in the LETM1 subunit has been shown to bind to the mitochondrial ribosomal protein L36 (MRPL36) and to nucleoprotein complexes called mitochondrial nucleoids, which are mitochondrial DNA (mDNA) complexed with regulatory proteins (Chen and Butow, 2005; Camara et al., 2009; Piao et al., 2009a; Lupo et al., 2011; He et al., 2012; Gilkerson et al., 2013; Durigon et al., 2018). Consequently, LETM1-KD is correlated with abnormal clustering of mitochondrial nucleoids and improper import and formation of respiratory complexes, thereby compromising mitochondrial respiration and concomitant energy metabolism (Frazier et al., 2006; Tamai et al., 2008; Lupo et al., 2011; Durigon et al., 2018). In addition, the interaction of LETM1 with BCS1L, a mitochondrial chaperone protein and AAA-ATPase, is shown to be critical for respiratory complex formation and assembly of respiratory supercomplexes. Knockdown of either LETM1 or BCS1L resulted in non-formation of complexes I and III and decreased formation of complex IV, leading to disassembly of respiratory supercomplexes, decreased ATP production, and increased formation of mitochondrial reactive oxygen species (mROS; Tamai et al., 2008). In yeast mitochondria expressing a mutant LETM1 homolog, the expression of several mitochondrially encoded proteins, including cytochrome b, a subunit of the complex III of the respiratory chain, was found to be significantly downregulated (Frazier et al., 2006). Furthermore, yeast mitochondria transfected with LETM1 lacking the MRPL36 binding domain exhibited a selective lack of respiratory complexes III and IV, suggesting a downregulation of protein translation (Lupo et al., 2011). These observations suggest that LETM1 may impact mitochondrial respiration and cellular metabolism by regulating the translation, transport, and assembly of respiratory complexes of the IMM.
In contrast, in mammalian cells, LETM1-KD was not associated with a decrease in the expression levels of respiratory chain subunits or disassembly of respiratory supercomplexes (Hasegawa and van der Bliek, 2007; Dimmer et al., 2008). Studies also showed that LETM1-KD did not affect respiratory complex assembly, but significantly inhibited complex IV activity (Doonan et al., 2014) or decreased complex II-dependent oxygen consumption (Aral et al., 2020). In both cases, mROS production was increased. The effects of LETM1-KD were partially reversed by either LETM1 reconstitution or overexpression of mitochondria-targeted expression of manganese superoxide dismutase, and glutathione peroxidase (Doonan et al., 2014) or by supplementation with melatonin, a highly effective antioxidant and free-radical scavenger (Aral et al., 2020). LETM1-deficient mitochondria exhibit a 91% increase in the frequency of “mitoflashes,” which are stochastic and discrete bursts of superoxide anion production observed in respiring mitochondria, with concomitant matrix alkalinization (Hou et al., 2014; Schwarzländer et al., 2014; Li et al., 2016; Wang et al., 2016). In these studies, the reduced mitochondrial metabolism was instead attributed to LETM1-KD-induced changes in mitochondrial morphology. This is supported by the consistent observations of disordered and swollen mitochondria in LETM1-deficient cells. Restoration of mitochondrial morphology by reconstitution of LETM1 or stimulation of K+ transport using ionophores suggested that the absence of a LETM1-CHE/KHE activity may underlie the distorted morphology, lack of respiratory supercomplex formation, and decreased ATP production (Nowikovsky et al., 2004; Froschauer et al., 2005; Hasegawa and van der Bliek, 2007; Dimmer et al., 2008; Hashimi et al., 2013). However, a recent study demonstrated that just the insertion of LETM1 complexes into the bilayer is sufficient to form membrane invaginations and maintain cristae structure. The structural domain in the LETM1 subunit responsible for this property lies within the conserved ribosomal binding domain (Figure 1; Nakamura et al., 2020). Considering the importance of the ribosomal binding domain for protein translation and assembly, these observations suggest that LETM1 may influence mitochondrial morphology and formation of respiratory supercomplexes independent of mitochondrial ionic homeostasis.
Thus, it is possible that different LETM1-dependent mechanisms underlie the effects of LETM1-KD on respiratory supercomplex formation and compromised bioenergetics. Further studies are required to understand how a LETM1-CHE/KHE function also regulates its interactions with proteins, such as BCS1L, MRPL36, and CTMP and impacts both mitochondrial and cellular metabolism.
LETM1 in Neuronal Disorders
Parkinson’s disease (PD) is a progressive neurodegenerative disorder, associated with mitochondrial dysfunction (Bose and Beal, 2016). LETM1 is implicated in PD because of its putative association with the phosphatase and tensin homolog deleted on chromosome 10 (PTEN)-induced kinase 1 (PINK-1), mutations of which are found in PD models (Huang et al., 2017b). PINK-1, a mitochondria-targeted serine/threonine kinase, is imported into the IMM under normal conditions, and translocated to the OMM during mitophagy (Fallaize et al., 2015). In in vitro experiments, PINK-1 directly phosphorylated LETM1 at Thr192, which increased Ca2+ release through LETM1 in liposomes and LETM1-T192E protected PINK-1-deficient neurons by rescuing mitochondrial Ca2+ homeostasis (Huang et al., 2017b). The study suggests that, in neuronal mitochondria under physiological conditions, LETM1 is constitutively phosphorylated by PINK-1 and regulates Ca2+ handling via a LETM1-CHE activity (Figure 2A). Future studies elucidating the effects of PINK-1 mutations on LETM1-CHE activity will provide better insight into a possible role for LETM1 in the pathogenesis of PD.
LETM1-deficiency is linked to the pathogenesis of epileptic seizures, one of the major phenotypes in WHS patients (Endele et al., 1999; McQuibban et al., 2010; Hart et al., 2014). LETM1-deficient neurons in D. melanogaster exhibit motor defects, which correlates with reduced release of neurotransmitters (McQuibban et al., 2010). This is consistent with a reduced expression of LETM1 in patients with temporal lobe epilepsy and in a rat pilocarpine-induced epilepsy model, exhibiting mitochondrial swelling, early onset of the first seizure, and increased seizure frequency and duration (Zhang et al., 2014). Seizures are typically closely associated with excess ROS emission, altered metabolic pathways, and compromised mitochondrial bioenergetics and mitochondrial dysfunction (Kann et al., 2005; Folbergrová and Kunz, 2012; Khurana et al., 2013).
In patients with amyotrophic lateral sclerosis (ALS), LETM1 expression is implicated in mitochondrial dysfunction. For example, a common mouse model used to study ALS, the end-stage superoxide dismutase SOD1(G93A) transgenic mice, exhibited a 2-fold increase in LETM1 expression in motor neurons of the hypoglossal nucleus (hMNs). This correlated with a decrease in mitochondrial Ca2+ uptake and imbalance in mitochondrial Ca2+ homeostasis (Mühling et al., 2014).
The mechanism by which LETM1 expression regulates the progression of epilepsy or ALS is unknown. Nevertheless, the association of LETM1-KD-induced mitochondrial dysfunction and Ca2+ dysregulation suggests that the loss of a LETM1-CHE activity and resulting Ca2+ dysregulation may underlie pathophysiology. Interestingly, however, LETM1-KD in the epilepsy model also decreased the expression of mitochondrial cytochrome b, a component of complex III (Zhang et al., 2014). Given the role of LETM1 in the formation of respiratory supercomplexes and protein import via interactions with BCS1L and MRPL36, it is likely that this aspect of LETM1-depedent regulation also plays a role in the effects of LETM1-KD in epilepsy. Furthermore, LETM1+/− mice manifest both impaired glucose metabolism and seizure activity (Jiang et al., 2013). Given that LETM1 regulates mitochondrial and cellular metabolism (described in “LETM1 in cellular metabolic dysregulation” in this article), this indicates that LETM1-deficiency induced mitochondrial dysfunction underlies both PD and ALS.
LETM1 in Cancer
Cancer cells depend upon mitochondria for metabolic reprogramming in cancer initiation, progression, and resistance to therapy (Vyas et al., 2016). LETM1-mediated regulation of mitochondrial biogenesis, metabolism, and cell survival signaling has provided impetus to investigate LETM1 activity in various human cancers (Dimmer et al., 2008; Piao et al., 2009a; Li et al., 2015). Multiple human malignancies, including breast, colon, esophagus, lung, ovary, rectum, stomach, and uterine cervix, report high LETM1-expression profile correlating with poor prognosis, low survival rates, upregulation of cancer stemness genes, and enhanced angiogenesis (Piao et al., 2009a, 2019a,b, 2020; Li et al., 2015, 2020; Wang et al., 2015, 2020; Huang et al., 2017a; Yang et al., 2018). In thyroid, prostate, ovarian, and gastric cancers, LETM1 overexpression is correlated with increased cell survival via enhanced Akt signaling (Lee et al., 2016; Piao et al., 2020; Wang et al., 2020; Zhang et al., 2020). This is further corroborated by the observation in prostate cancer cells, that the PI3K inhibitor LY294002, which stopped the migration and invasion of tumor cells, also caused the downregulation of LETM1 (Piao et al., 2020). This suggests that LETM1 may mediate Akt signaling through its interaction with the Akt inhibitor, CTMP. In lung cancer, LETM1 overexpression promoted tumor formation by inhibiting 5'-adenosine monophosphate activated protein kinase (AMPK), a cellular bioenergetic sensor that activates autophagy at depleted ATP levels (Hwang et al., 2010). AMPK inhibition would lead to deregulation of the normal progression of the cell cycle. Accordingly, LETM1-KD reduced the expression of cancer stemness-related genes, increased AMPK activation, arrested cell cycle, lowered the number and size of tumor spheroids, and markedly inhibited cell proliferation (Che et al., 2020; Li et al., 2020; Piao et al., 2020).
In contrast with studies that associate LETM1-overexpression with promotion of tumorigenesis, other studies demonstrated that LETM1 over-expression impaired mitochondrial biogenesis, compromised ATP production, and elicited necrotic cancer cell death in lung (Hwang et al., 2010) and hepatocellular carcinoma (Shin et al., 2013). In these studies, LETM1 overexpression correlated with suppression of Akt activity and cell survival signaling while facilitating apoptosis (Hwang et al., 2010; Shin et al., 2013). However, it is to be noted that in Shin et al. (2013), co-delivery of LETM1 with CTMP to tumor cells as a treatment strategy in hepatocellular carcinoma, led to overexpression of both LETM1 and CTMP and correlated with distorted mitochondrial morphology and increased apoptosis. The authors surmised that the decrease in Akt phosphorylation was a result of increased apoptosis caused by mitochondrial dysfunction in LETM1-overexpressed cells.
Alterations in cellular and mitochondrial Ca2+ homeostasis and ensuing mitochondrial dysfunction are a hallmark of tumorigenesis (Romero-Garcia and Prado-Garcia, 2019). It remains to be determined how changes in LETM1 expression alter mitochondrial Ca2+ regulation. The correlation of LETM1 expression with Akt phosphorylation suggests that, in addition to LETM1-CHE/KHE activity, regulation of tumorigenesis by LETM1 may involve signaling through its constitutive binding partner CTMP in cancer cells.
LETM1 may also be involved in initiating resveratrol-induced cancer cell death (Madreiter-Sokolowski et al., 2016). In this study, resveratrol, a naturally occurring polyphenol, induced apoptosis in HeLa and human vascular endothelial (EA.hy926) cancer cells but not somatic cells. The authors proposed that the specific inhibitory action of resveratrol on the F1 subunit of ATP synthase may decrease ATP production and inhibit the activity of SERCA in the MAM region of cancer cells, triggering SOCE in these cells (Madreiter-Sokolowski et al., 2016). In the absence of SERCA-mediated Ca2+ sequestration in the ER, this would lead to increased MCU/LETM1-mediated Ca2+ uptake into mitochondria, resulting in mitochondrial Ca2+ overload and apoptosis (Waldeck-Weiermair et al., 2013). This hypothesis provides a possible mechanism by which the increased expression of LETM1 in cancer cells may be utilized as a means for delivering therapy. However, this hypothesis remains to be investigated. A comprehensive understanding of the mechanism by which LETM1 regulates mitochondrial Ca2+ handling, interactions with regulatory proteins such as CTMP and inter-organelle signaling is necessary to explore LETM1 as a potential reliable prognosticator and therapeutic target in cancer treatment.
Perspective
The diverse impact of LETM1 deficiency, as outlined above, suggests that LETM1 expression and function is critical for short-term dynamic processes such as cellular metabolism as well as long-term processes important for cell survival and development. Although the primary function of LETM1 as CHE or KHE is still being debated, it is evident that loss of LETM1 function adversely impacts mitochondrial Ca2+ handling in several cell types. Whether, this is due to the loss of a LETM1-CHE function or a consequence of the loss of a LETM1-KHE function on mitochondrial Ca2+ handling needs to be resolved. The association between changes in LETM1 expression and effects on mitochondrial Ca2+ handling as described in this review argues in favor of a CHE function for LETM1. It is to be noted, however, that evidence for LETM1-CHE activity has been largely obtained from observations in in vitro experimental conditions where the LETM1 protein is studied in isolation, devoid of other ionic activities that determine ionic homeostasis, and osmotic balance in vivo (Jiang et al., 2009; Tsai et al., 2014; Shao et al., 2016). Conversely, cell studies covering the spectrum from lower to higher-order eukaryotes, while providing compelling evidence for an evolutionarily conserved LETM1-KHE function (Nowikovsky et al., 2004; Dimmer et al., 2008; McQuibban et al., 2010; Hashimi et al., 2013), do not account for the role of EF-hands in mammalian LETM1 in a KHE function. Our recent study in rat cardiac mitochondria conducted in Na+-free conditions to render NCLX inactive, showed that the contribution of a Na+-independent Ca2+ efflux (CHE), presumably encoded by LETM1, increased with matrix Ca2+ load and that CHE may be able to sense matrix Ca2+ buffering (Natarajan et al., 2020). The studies favoring a LETM1-KHE function cannot explain how a LETM1-KHE may regulate Ca2+ handling in the absence of NCLX. Future studies will need to address these knowledge gaps as to how the LETM1-CHE function in in vitro conditions is modulated in the presence or absence of one or more EF-hands in the LETM1 protein or factors such as the presence of other ionic exchangers.
LETM1 interacts with proteins, such as CTMP, BCS1L, and MRPL36, and regulates processes like Akt signaling and protein import and assembly. Changes in LETM1 expression also affect these processes, implicating it in the pathophysiology of several disease phenotypes, including metabolic, neurodegenerative, and neoplastic diseases (Figure 2C). The link between the primary function of LETM1 as CHE/KHE and its interaction with these proteins is not apparent. Specifically, is the CHE or KHE function of LETM1 independent of its actions on signaling proteins and respiratory complex assembly? Some insight is obtained from a recent study, which showed in in vitro experiments that insertion of LETM1 complexes into membrane bilayers alone led to the formation of membrane invaginations (Nakamura et al., 2020). Four residues in the C-terminus of the LETM1 subunit were found to be critical for LETM1 complex formation and maintaining morphology of the IMM (Nakamura et al., 2020), which is crucial for the proper assembly and function of essential IMM proteins (Bulthuis et al., 2018). These residues are distinct from the transmembrane and EF-Hand domains of the LETM1 subunit, which are important for LETM1-CHE activity. Since the formation of LETM1 complexes is regulated by the chaperone protein BCS1L (Tamai et al., 2008), this suggests that the interaction of LETM1 with BCS1L may occur at sites independent of the domains that regulate the LETM1-CHE/KHE function. It is, however, possible that the domain(s) regulating LETM1-CHE/KHE would influence the interaction of LETM1 with proteins such as BCS1L and hence the regulation of mitochondrial morphology and vice versa. Given the increasing importance of inter-organelle communications and signaling for integrated cellular function in health and disease (Javadov et al., 2020), this aspect of LETM1-mediated signaling, in addition to its exchanger function needs further investigation.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
Funding for this work was provided by NIH R01 HL131673.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aral, C., Demirkesen, S., Bircan, R., and Sirin, D. Y. (2020). Melatonin reverses the oxidative stress and mitochondrial dysfunction caused by LETM1 silencing. Cell Biol. Int. 44, 795–807. doi: 10.1002/cbin.11274
Austin, S., and Nowikovsky, K. (2019). LETM1: essential for mitochondrial biology and cation homeostasis? Trends Biochem. Sci. 44, 648–658. doi: 10.1016/j.tibs.2019.04.002
Austin, S., Tavakoli, M., Pfeiffer, C., Seifert, J., Mattarei, A., De Stefani, D., et al. (2017). LETM1-mediated K+ and Na+ homeostasis regulates mitochondrial Ca2+ efflux. Front. Physiol. 8:839. doi: 10.3389/fphys.2017.00839
Blomeyer, C. A., Bazil, J. N., Stowe, D. F., Pradhan, R. K., Dash, R. K., and Camara, A. K. S. (2013). Dynamic buffering of mitochondrial Ca2+ during Ca2+ uptake and Na+-induced Ca2+ release. J. Bioenerg. Biomembr. 45, 189–202. doi: 10.1007/s10863-012-9483-7
Bose, A., and Beal, M. F. (2016). Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 139, 216–231. doi: 10.1111/jnc.13731
Bulthuis, E. P., Adjobo-Hermans, M. J. W., Willems, P. H. G. M., and Koopman, W. J. H. (2018). Mitochondrial morphofunction in mammalian cells. Antioxid. Redox Signal. 30, 2066–2109. doi: 10.1089/ars.2018.7534
Camara, A. K. S., Lesnefsky, E. J., and Stowe, D. F. (2009). Potential therapeutic benefits of strategies directed to mitochondria. Antioxid. Redox Signal. 13, 279–347. doi: 10.1089/ars.2009.2788
Che, N., Yang, Z., Liu, X., Li, M., Feng, Y., Zhang, C., et al. (2020). Suppression of LETM1 inhibits the proliferation and stemness of colorectal cancer cells through reactive oxygen species–induced autophagy. J. Cell. Mol. Med. 25, 2110–2120. doi: 10.1111/jcmm.16169
Chen, X. J., and Butow, R. A. (2005). The organization and inheritance of the mitochondrial genome. Nat. Rev. Genet. 6, 815–825. doi: 10.1038/nrg1708
Dimmer, K. S., Navoni, F., Casarin, A., Trevisson, E., Endele, S., Winterpacht, A., et al. (2008). LETM1, deleted in Wolf–Hirschhorn syndrome is required for normal mitochondrial morphology and cellular viability. Hum. Mol. Genet. 17, 201–214. doi: 10.1093/hmg/ddm297
Doonan, P. J., Chandramoorthy, H. C., Hoffman, N. E., Zhang, X., Cárdenas, C., Shanmughapriya, S., et al. (2014). LETM1-dependent mitochondrial Ca2+ flux modulates cellular bioenergetics and proliferation. FASEB J. 28, 4936–4949. doi: 10.1096/fj.14-256453
Durigon, R., Mitchell, A. L., Jones, A. W., Manole, A., Mennuni, M., Hirst, E. M., et al. (2018). LETM 1 couples mitochondrial DNA metabolism and nutrient preference. EMBO Mol. Med. 10:e8550. doi: 10.15252/emmm.201708550
Endele, S., Fuhry, M., Pak, S. -J., Zabel, B. U., and Winterpacht, A. (1999). LETM1, a novel gene encoding a putative ef-hand ca2+-binding protein, flanks the wolf–hirschhorn syndrome (WHS) critical region and is deleted in most WHS patients. Genomics 60, 218–225. doi: 10.1006/geno.1999.5881
Fagerberg, L., Hallström, B. M., Oksvold, P., Kampf, C., Djureinovic, D., Odeberg, J., et al. (2014). Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 13, 397–406. doi: 10.1074/mcp.M113.035600
Fallaize, D., Chin, L. -S., and Li, L. (2015). Differential submitochondrial localization of PINK1 as a molecular switch for mediating distinct mitochondrial signalling pathways. Cell. Signal. 27, 2543–2554. doi: 10.1016/j.cellsig.2015.09.020
Folbergrová, J., and Kunz, W. S. (2012). Mitochondrial dysfunction in epilepsy. Mitochondrion 12, 35–40. doi: 10.1016/j.mito.2011.04.004
Frazier, A. E., Taylor, R. D., Mick, D. U., Warscheid, B., Stoepel, N., Meyer, H. E., et al. (2006). Mdm38 interacts with ribosomes and is a component of the mitochondrial protein export machinery. J. Cell Biol. 172, 553–564. doi: 10.1083/jcb.200505060
Froschauer, E., Nowikovsky, K., and Schweyen, R. J. (2005). Electroneutral K+/H+ exchange in mitochondrial membrane vesicles involves Yol027/Letm1 proteins. Biochim. Biophys. Acta Rev. Cancer 1711, 41–48. doi: 10.1016/j.bbamem.2005.02.018
Gilkerson, R., Bravo, L., Garcia, I., Gaytan, N., Herrera, A., Maldonado, A., et al. (2013). The mitochondrial nucleoid: integrating mitochondrial DNA into cellular homeostasis. Cold Spring Harb. Perspect. Biol. 5:a011080. doi: 10.1101/cshperspect.a011080
Gogvadze, V., Robertson, J. D., Enoksson, M., Zhivotovsky, B., and Orrenius, S. (2004). Mitochondrial cytochrome c release may occur by volume-dependent mechanisms not involving permeability transition. Biochem. J. 378, 213–217. doi: 10.1042/bj20031193
Hajnóczky, G., Booth, D., Csordás, G., Debattisti, V., Golenár, T., Naghdi, S., et al. (2014). Reliance of ER-mitochondrial calcium signaling on mitochondrial EF-hand Ca2+ binding proteins: Miros, MICUs, LETM1 and solute carriers. Curr. Opin. Cell Biol. 29, 133–141. doi: 10.1016/j.ceb.2014.06.002
Halestrap, A. P., Quinlan, P. T., Whipps, D. E., and Armston, A. E. (1986). Regulation of the mitochondrial matrix volume in vivo and in vitro. Biochem. J. 236, 779–787. doi: 10.1042/bj2360779
Hart, L., Rauch, A., Carr, A. M., Vermeesch, J. R., and O’Driscoll, M. (2014). LETM1 haploinsufficiency causes mitochondrial defects in cells from humans with Wolf-Hirschhorn syndrome: implications for dissecting the underlying pathomechanisms in this condition. Dis. Model. Mech. 7, 535–545. doi: 10.1242/dmm.014464
Hasegawa, A., and van der Bliek, A. M. (2007). Inverse correlation between expression of the Wolfs Hirschhorn candidate gene Letm1 and mitochondrial volume in C. elegans and in mammalian cells. Hum. Mol. Genet. 16, 2061–2071. doi: 10.1093/hmg/ddm154
Hashimi, H., McDonald, L., Stříbrná, E., and Lukeš, J. (2013). Trypanosome Letm1 protein is essential for mitochondrial potassium homeostasis. J. Biol. Chem. 288, 26914–26925. doi: 10.1074/jbc.M113.495119
He, J., Cooper, H. M., Reyes, A., Di Re, M., Sembongi, H., Litwin, T. R., et al. (2012). Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res. 40, 6109–6121. doi: 10.1093/nar/gks266
Hou, T., Wang, X., Ma, Q., and Cheng, H. (2014). Mitochondrial flashes: new insights into mitochondrial ROS signalling and beyond. J. Physiol. 592, 3703–3713. doi: 10.1113/jphysiol.2014.275735
Huang, E., Qu, D., Huang, T., Rizzi, N., Boonying, W., Krolak, D., et al. (2017b). PINK1-mediated phosphorylation of LETM1 regulates mitochondrial calcium transport and protects neurons against mitochondrial stress. Nat. Commun. 8:1399. doi: 10.1038/s41467-017-01435-1
Huang, B., Zhang, J., Zhang, X., Huang, C., Hu, G., Li, S., et al. (2017a). Suppression of LETM1 by siRNA inhibits cell proliferation and invasion of bladder cancer cells. Oncol. Rep. 38, 2935–2940. doi: 10.3892/or.2017.5959
Hwang, S. -K., Piao, L., Lim, H. -T., Minai-Tehrani, A., Yu, K. -N., Ha, Y. -C., et al. (2010). Suppression of lung tumorigenesis by leucine zipper/EF hand–containing transmembrane-1. PLoS One 5:e12535. doi: 10.1371/journal.pone.0012535
Javadov, S., Kozlov, A. V., and Camara, A. K. S. (2020). Mitochondria in health and diseases. Cell 9:1177. doi: 10.3390/cells9051177
Jiang, D., Zhao, L., and Clapham, D. E. (2009). Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 326, 144–147. doi: 10.1126/science.1175145
Jiang, D., Zhao, L., Clish, C. B., and Clapham, D. E. (2013). Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf-Hirschhorn syndrome. Proc. Natl. Acad. Sci. U. S. A. 110:E2249–E2254. doi: 10.1073/pnas.1308558110
Kaasik, A., Safiulina, D., Zharkovsky, A., and Veksler, V. (2007). Regulation of mitochondrial matrix volume. Am. J. Physiol. Cell Physiol. 292, C157–C163. doi: 10.1152/ajpcell.00272.2006
Kann, O., Kovács, R., Njunting, M., Behrens, C. J., Otáhal, J., Lehmann, T. -N., et al. (2005). Metabolic dysfunction during neuronal activation in the ex vivo hippocampus from chronic epileptic rats and humans. Brain 128, 2396–2407. doi: 10.1093/brain/awh568
Khurana, D. S., Valencia, I., Goldenthal, M. J., and Legido, A. (2013). Mitochondrial dysfunction in epilepsy. Semin. Pediatr. Neurol. 20, 176–187. doi: 10.1016/j.spen.2013.10.001
Lee, S. -Y., Kang, M. -G., Shin, S., Kwak, C., Kwon, T., Seo, J. K., et al. (2017). Architecture mapping of the inner mitochondrial membrane proteome by chemical tools in live cells. J. Am. Chem. Soc. 139, 3651–3662. doi: 10.1021/jacs.6b10418
Lee, J., Lee, W. K., Seol, M. -Y., Lee, S. G., Kim, D., Kim, H., et al. (2016). Coupling of LETM1 up-regulation with oxidative phosphorylation and platelet-derived growth factor receptor signaling via YAP1 transactivation. Oncotarget 7, 66728–66739. doi: 10.18632/oncotarget.11456
Li, H., Piao, L., Xu, D., and Xuan, Y. (2020). LETM1 is a potential biomarker that predicts poor prognosis in gastric adenocarcinoma. Exp. Mol. Pathol. 112:104333. doi: 10.1016/j.yexmp.2019.104333
Li, W., Sun, T., Liu, B., Wu, D., Qi, W., Wang, X., et al. (2016). Regulation of mitoflash biogenesis and signaling by mitochondrial dynamics. Sci. Rep. 6:32933. doi: 10.1038/srep32933
Li, Y., Tran, Q., Shrestha, R., Piao, L., Park, S., Park, J., et al. (2019). LETM1 is required for mitochondrial homeostasis and cellular viability (Review). Mol. Med. Rep. 19, 3367–3375. doi: 10.3892/mmr.2019.10041
Li, N., Zheng, Y., Xuan, C., Lin, Z., Piao, L., and Liu, S. (2015). LETM1 overexpression is correlated with the clinical features and survival outcome of breast cancer. Int. J. Clin. Exp. Pathol. 8, 12893–12900.
Lin, Q. -T., and Stathopulos, P. B. (2019). Molecular mechanisms of leucine zipper ef-hand containing transmembrane protein-1 function in health and disease. Int. J. Mol. Sci. 20:286. doi: 10.3390/ijms20020286
Liu, J., Carnero-Montoro, E., van Dongen, J., Lent, S., Nedeljkovic, I., Ligthart, S., et al. (2019). An integrative cross-omics analysis of DNA methylation sites of glucose and insulin homeostasis. Nat. Commun. 10:2581. doi: 10.1038/s41467-019-10487-4
Lupo, D., Vollmer, C., Deckers, M., Mick, D. U., Tews, I., Sinning, I., et al. (2011). Mdm38 is a 14-3-3-like receptor and associates with the protein synthesis machinery at the inner mitochondrial membrane. Traffic 12, 1457–1466. doi: 10.1111/j.1600-0854.2011.01239.x
Madreiter-Sokolowski, C. T., Gottschalk, B., Parichatikanond, W., Eroglu, E., Klec, C., Waldeck-Weiermair, M., et al. (2016). Resveratrol specifically kills cancer cells by a devastating increase in the ca2+ coupling between the greatly tethered endoplasmic reticulum and mitochondria. Cell. Physiol. Biochem. 39, 1404–1420. doi: 10.1159/000447844
Maira, S. -M., Galetic, I., Brazil, D. P., Kaech, S., Ingley, E., Thelen, M., et al. (2001). Carboxyl-terminal modulator protein (CTMP), a negative regulator of PKB/Akt and v-Akt at the plasma membrane. Science 294, 374–380. doi: 10.1126/science.1062030
Marchi, U. D., Santo-Domingo, J., Castelbou, C., Sekler, I., Wiederkehr, A., and Demaurex, N. (2014). NCLX protein, but not letm1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J. Biol. Chem. 289, 20377–20385. doi: 10.1074/jbc.M113.540898
McQuibban, A. G., Joza, N., Megighian, A., Scorzeto, M., Zanini, D., Reipert, S., et al. (2010). A Drosophila mutant of LETM1, a candidate gene for seizures in Wolf-Hirschhorn syndrome. Hum. Mol. Genet. 19, 987–1000. doi: 10.1093/hmg/ddp563
Mishra, J., Jhun, B. S., Hurst, S., O-Uchi, J., Csordás, G., and Sheu, S. -S. (2017). The mitochondrial Ca2+ uniporter: structure, function, and pharmacology. Handb. Exp. Pharmacol. 240, 129–156. doi: 10.1007/164_2017_1
Mühling, T., Duda, J., Weishaupt, J. H., Ludolph, A. C., and Liss, B. (2014). Elevated mRNA-levels of distinct mitochondrial and plasma membrane Ca2+ transporters in individual hypoglossal motor neurons of endstage SOD1 transgenic mice. Front. Cell. Neurosci. 8:353. doi: 10.3389/fncel.2014.00353
Nakamura, S., Matsui, A., Akabane, S., Tamura, Y., Hatano, A., Miyano, Y., et al. (2020). The mitochondrial inner membrane protein LETM1 modulates cristae organization through its LETM domain. Commun. Biol. 3, 99–11. doi: 10.1038/s42003-020-0832-5
Natarajan, G. K., Glait, L., Mishra, J., Stowe, D. F., Camara, A. K. S., and Kwok, W. -M. (2020). Total matrix Ca2+ modulates Ca2+ efflux via the Ca2+/H+ exchanger in cardiac mitochondria. Front. Physiol. 11:510600. doi: 10.3389/fphys.2020.510600
Nowikovsky, K., Froschauer, E. M., Zsurka, G., Samaj, J., Reipert, S., Kolisek, M., et al. (2004). The LETM1/YOL027 gene family encodes a factor of the mitochondrial K+ homeostasis with a potential role in the wolf-hirschhorn syndrome. J. Biol. Chem. 279, 30307–30315. doi: 10.1074/jbc.M403607200
Nowikovsky, K., Pozzan, T., Rizzuto, R., Scorrano, L., and Bernardi, P. (2012). The pathophysiology of LETM1. J. Gen. Physiol. 139, 445–454. doi: 10.1085/jgp.201110757
Palty, R., Silverman, W. F., Hershfinkel, M., Caporale, T., Sensi, S. L., Parnis, J., et al. (2010). NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. U. S. A. 107, 436–441. doi: 10.1073/pnas.0908099107
Parcellier, A., Tintignac, L. A., Zhuravleva, E., Dummler, B., Brazil, D. P., Hynx, D., et al. (2009). The carboxy-terminal modulator protein (CTMP) regulates mitochondrial dynamics. PLoS One 4:e5471. doi: 10.1371/journal.pone.0005471
Park, J., Li, Y., Kim, S. -H., Yang, K. -J., Kong, G., Shrestha, R., et al. (2014). New players in high fat diet-induced obesity: LETM1 and CTMP. Metabolism 63, 318–327. doi: 10.1016/j.metabol.2013.10.012
Patergnani, S., Suski, J. M., Agnoletto, C., Bononi, A., Bonora, M., De Marchi, E., et al. (2011). Calcium signaling around mitochondria associated membranes (MAMs). Cell Commun. Signal 9:19. doi: 10.1186/1478-811X-9-19
Piao, L., Feng, Y., Yang, Z., Qi, W., Li, H., Han, H., et al. (2019a). LETM1 is a potential cancer stem-like cell marker and predicts poor prognosis in colorectal adenocarcinoma. Pathol. Res. Pract. 215:152437. doi: 10.1016/j.prp.2019.152437
Piao, L., Li, H., Feng, Y., Li, X., Cui, Y., and Xuan, Y. (2020). Leucine zipper-ef-hand containing transmembrane protein 1 is a potential prognostic biomarker and promotes cell progression in prostate cancer. Cancer Manag. Res. 12, 1649–1660. doi: 10.2147/CMAR.S236482
Piao, L., Li, Y., Kim, S. J., Byun, H. S., Huang, S. M., Hwang, S. -K., et al. (2009a). Association of LETM1 and MRPL36 contributes to the regulation of mitochondrial atp production and necrotic cell death. Cancer Res. 69, 3397–3404. doi: 10.1158/0008-5472.CAN-08-3235
Piao, L., Li, Y., Kim, S. J., Sohn, K. -C., Yang, K. -J., Park, K. A., et al. (2009b). Regulation of OPA1-mediated mitochondrial fusion by leucine zipper/EF-hand-containing transmembrane protein-1 plays a role in apoptosis. Cell. Signal. 21, 767–777. doi: 10.1016/j.cellsig.2009.01.020
Piao, L., Yang, Z., Feng, Y., Zhang, C., Cui, C., and Xuan, Y. (2019b). LETM1 is a potential biomarker of prognosis in lung non-small cell carcinoma. BMC Cancer 19, 898–899. doi: 10.1186/s12885-019-6128-9
Quan, X., Nguyen, T. T., Choi, S. -K., Xu, S., Das, R., Cha, S. -K., et al. (2015). Essential role of mitochondrial Ca2+ uniporter in the generation of mitochondrial pH gradient and metabolism-secretion coupling in insulin-releasing cells. J. Biol. Chem. 290, 4086–4096. doi: 10.1074/jbc.M114.632547
Rizzuto, R., De Stefani, D., Raffaello, A., and Mammucari, C. (2012). Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13, 566–578. doi: 10.1038/nrm3412
Romero-Garcia, S., and Prado-Garcia, H. (2019). Mitochondrial calcium: transport and modulation of cellular processes in homeostasis and cancer (Review). Int. J. Oncol. 54, 1155–1167. doi: 10.3892/ijo.2019.4696
Schlickum, S., Moghekar, A., Simpson, J. C., Steglich, C., O’Brien, R. J., Winterpacht, A., et al. (2004). LETM1, a gene deleted in Wolf–Hirschhorn syndrome, encodes an evolutionarily conserved mitochondrial protein. Genomics 83, 254–261. doi: 10.1016/j.ygeno.2003.08.013
Schwarzländer, M., Wagner, S., Ermakova, Y. G., Belousov, V. V., Radi, R., Beckman, J. S., et al. (2014). The “mitoflash” probe cpYFP does not respond to superoxide. Nature 514, E12–E14. doi: 10.1038/nature13858
Shao, J., Fu, Z., Ji, Y., Guan, X., Guo, S., Ding, Z., et al. (2016). Leucine zipper-EF-hand containing transmembrane protein 1 (LETM1) forms a Ca2+/H+ antiporter. Sci. Rep. 6:34174. doi: 10.1038/srep34174
Shin, J. -Y., Chung, Y. -S., Kang, B., Jiang, H. -L., Yu, D. -Y., Han, K., et al. (2013). Co-delivery of LETM1 and CTMP synergistically inhibits tumor growth in H-ras12V liver cancer model mice. Cancer Gene Ther. 20, 186–194. doi: 10.1038/cgt.2013.6
Tamai, S., Iida, H., Yokota, S., Sayano, T., Kiguchiya, S., Ishihara, N., et al. (2008). Characterization of the mitochondrial protein LETM1, which maintains the mitochondrial tubular shapes and interacts with the AAA-ATPase BCS1L. J. Cell Sci. 121, 2588–2600. doi: 10.1242/jcs.026625
Tang, G., Zhang, C., Ju, Z., Zheng, S., Wen, Z., Xu, S., et al. (2018). The mitochondrial membrane protein FgLetm1 regulates mitochondrial integrity, production of endogenous reactive oxygen species and mycotoxin biosynthesis in Fusarium graminearum. Mol. Plant Pathol. 19, 1595–1611. doi: 10.1111/mpp.12633
Tsai, M. -F., Jiang, D., Zhao, L., Clapham, D., and Miller, C. (2014). Functional reconstitution of the mitochondrial Ca2+/H+ antiporter Letm1. J. Gen. Physiol. 143, 67–73. doi: 10.1085/jgp.201311096
Uhlén, M., Fagerberg, L., Hallström, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., et al. (2015). Tissue-based map of the human proteome. Science 347:1260419. doi: 10.1126/science.1260419
Vyas, S., Zaganjor, E., and Haigis, M. C. (2016). Mitochondria and cancer. Cell 166, 555–566. doi: 10.1016/j.cell.2016.07.002
Waldeck-Weiermair, M., Deak, A. T., Groschner, L. N., Alam, M. R., Jean-Quartier, C., Malli, R., et al. (2013). Molecularly distinct routes of mitochondrial Ca2+ uptake are activated depending on the activity of the Sarco/endoplasmic reticulum Ca2+ ATPase (SERCA). J. Biol. Chem. 288, 15367–15379. doi: 10.1074/jbc.M113.462259
Waldeck-Weiermair, M., Jean-Quartier, C., Rost, R., Khan, M. J., Vishnu, N., Bondarenko, A. I., et al. (2011). Leucine zipper EF hand-containing transmembrane protein 1 (Letm1) and uncoupling proteins 2 and 3 (UCP2/3) contribute to two distinct mitochondrial Ca2+ uptake pathways. J. Biol. Chem. 286, 28444–28455. doi: 10.1074/jbc.M111.244517
Wang, J., Ding, W., Xu, Y., Tao, E., Mo, M., Xu, W., et al. (2020). Long non-coding RNA RHPN1-AS1 promotes tumorigenesis and metastasis of ovarian cancer by acting as a ceRNA against miR-596 and upregulating LETM1. Aging 12, 4558–4572. doi: 10.18632/aging.102911
Wang, C., Liu, Q., Chen, Y., Liu, S., Xu, J., Cui, X., et al. (2015). Clinical implication of leucine zipper/EF hand-containing transmembrane-1 overexpression in the prognosis of triple-negative breast cancer. Exp. Mol. Pathol. 98, 254–259. doi: 10.1016/j.yexmp.2014.12.012
Wang, X., Zhang, X., Huang, Z., Wu, D., Liu, B., Zhang, R., et al. (2016). Protons trigger mitochondrial flashes. Biophys. J. 111, 386–394. doi: 10.1016/j.bpj.2016.05.052
Yang, Z., Ni, W., Cui, C., Qi, W., Piao, L., and Xuan, Y. (2018). Identification of LETM1 as a marker of cancer stem-like cells and predictor of poor prognosis in esophageal squamous cell carcinoma. Hum. Pathol. 81, 148–156. doi: 10.1016/j.humpath.2018.07.001
Zhang, B., Carrie, C., Ivanova, A., Narsai, R., Murcha, M. W., Duncan, O., et al. (2012). LETM proteins play a role in the accumulation of mitochondrially encoded proteins in arabidopsis thaliana and AtLETM2 displays parent of origin effects. J. Biol. Chem. 287, 41757–41773. doi: 10.1074/jbc.M112.383836
Zhang, Y., Chen, L., Cao, Y., Chen, S., Xu, C., Xing, J., et al. (2020). LETM1 promotes gastric cancer cell proliferation, migration, and invasion via the PI3K/Akt signaling pathway. J. Gastric Cancer 20, 139–151. doi: 10.5230/jgc.2020.20.e12
Keywords: leucine-zipper EF-hand containing transmembrane 1, mitochondrial calcium hydrogen exchanger, mitochondrial potassium hydrogen exchanger, mitochondrial calcium handling, carboxy-terminal-modulator-protein, Wolf-Hirschhorn syndrome, cancer biology, cell metabolism, bioenergetics
Citation: Natarajan GK, Mishra J, Camara AKS and Kwok W-M (2021) LETM1: A Single Entity With Diverse Impact on Mitochondrial Metabolism and Cellular Signaling. Front. Physiol. 12:637852. doi: 10.3389/fphys.2021.637852
Edited by:
Ildikò Szabò, University of Padua, ItalyReviewed by:
Wolfgang F. Graier, Medical University of Graz, AustriaShane Austin, The University of the West Indies, Barbados
Copyright © 2021 Natarajan, Mishra, Camara and Kwok. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gayathri K. Natarajan, Z25hdGFyYWphbkBtY3cuZWR1