 Xianying Zhu
Xianying Zhu Yuan Zhan
Yuan Zhan Yiya Gu1
Yiya Gu1 Ting Wang
Ting Wang Jungang Xie
Jungang Xie- 1Department of Respiratory and Critical Care Medicine, National Clinical Research Center for Respiratory Disease, Key Laboratory of Pulmonary Diseases of Health Ministry, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Department of Intensive Care Unit, Sun Yat-sen University Cancer Center, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Guangzhou, China
Chronic obstructive pulmonary disease (COPD), primarily attributed to cigarette smoke (CS), is characterized by multiple pathophysiological changes, including oxidative stress and inflammation. Stromal interaction molecule 1 (STIM1) is a Ca2+ sensor that regulates Ca2+ entry in different types of cells. The present study aimed to explore the relationship between CS-induced oxidative stress and inflammation, as well as the functional role of STIM1 thereinto. Our results showed that the reactive oxygen species (ROS)/STIM1/Ca2+ axis played a critical role in CS-induced secretion of interleukin (IL)-8 in human alveolar macrophages. Specifically, smokers with COPD (SC) showed higher levels of ROS in the lung tissues compared with healthy non-smokers (HN). STIM1 was upregulated in the lung tissues of COPD patients. The expression of STIM1 was positively associated with ROS levels and negatively correlated with pulmonary function. The expression of STIM1 was also increased in the bronchoalveolar lavage fluid (BALF) macrophages of COPD patients and PMA-differentiated THP-1 macrophages stimulated by cigarette smoke extract (CSE). Additionally, CSE-induced upregulation of STIM1 in PMA-differentiated THP-1 macrophages was inhibited by pretreatment with N-acetylcysteine (NAC), a ROS scavenger. Transfection with small interfering RNA (siRNA) targeting STIM1 and pretreatment with NAC alleviated CSE-induced increase in intracellular Ca2+ levels and IL-8 expression. Furthermore, pretreatment with SKF-96365 and 2-APB, the inhibitors of Ca2+ influx, suppressed CSE-induced secretion of IL-8. In conclusion, our study demonstrates that CSE-induced ROS production may increase the expression of STIM1 in macrophages, which further promotes the release of IL-8 by regulating Ca2+ entry. These data suggest that STIM1 may play a crucial role in CSE-induced ROS production and inflammation, and participate in the pathogenesis of COPD.
Introduction
Chronic obstructive pulmonary disease (COPD) is a major health problem that causes significant mortality and morbidity worldwide, and therefore places a substantial social and economic burden (Soriano et al., 2017). It is characterized by persistent respiratory symptoms, progressive airflow limitation associated with chronic inflammation and lung destruction, and ultimately irreversible lung function decline. Most cases of COPD are attributed to the exposure to noxious particles or gases, such as cigarette smoke (CS; Hogg and Senior, 2002; Mirza et al., 2018). The pathogenesis of COPD is extremely complicated and thus far remains unclear (Mizumura et al., 2014).
Oxidative stress occurs as a result of an imbalance between the production of free radicals and antioxidant defenses, which plays a crucial role in the pathogenesis of COPD (McGuinness and Sapey, 2017). Reactive oxygen species (ROS) is considered one of the most important by-products in oxygen metabolism and produced substantially during CS inhalation, resulting in a shift of balance to the oxidant side in COPD patients (Fischer et al., 2015). Specifically, oxidative stress promotes the release of proinflammatory mediators, including cytokines and peroxidation products of arachidonic acid (i.e., leukotrienes, prostanoids, and isoprostanes) (Khanna et al., 2013), and induce cell injury and apoptosis by activating and phosphorylating kinase cascades and transcription factors (Rahman, 2005). Oxidative stress is also involved in the genetic and epigenetic regulation of signaling pathways related to emphysema and chronic bronchitis phenotypes (Fischer et al., 2015). As oxidative stress plays a key role in the development of COPD, the therapeutic potential of many antioxidant agents has been evaluated, including thiols [e.g., N-acetylcysteine (NAC), carbocysteine] and antioxidant vitamins (e.g., vitamin C, D, and E) (Biswas et al., 2013; Tse et al., 2013). Chronic airway inflammation is recognized as one of the most critical pathophysiological mechanisms contributing to the pathogenesis of COPD, which causes structural alteration, lumen narrowing, and alveolar destruction (Lange et al., 2021). The relationships among CS, ROS, and inflammation have been under investigation (Lin et al., 2010; Tse et al., 2013). However, the role of stromal interaction molecule 1 (STIM1) in the alveolar macrophages of COPD patients has not been identified.
Stromal interaction molecule 1, a Ca2+ sensor located in the endoplasmic reticulum (ER), can promote multiple pathological processes, including inflammation and muscle metabolism, partly by regulating Ca2+ entry (Zhang et al., 2014; Kassan et al., 2016). A previous study showed that cigarette smoke extract (CSE) upregulated the protein expression of STIM1 in human airway smooth muscle (Wylam et al., 2015). In addition, lipopolysaccharide (LPS) promoted inflammatory cytokine secretion by increasing the protein expression of STIM1 in murine microglial cells and endothelial cells (Gandhirajan et al., 2013; Heo et al., 2015). STIM1 also acts as a ROS sensor to induce Ca2+ entry (Heo et al., 2015). However, the role of STIM1 in the alveolar macrophages of COPD patients has not been identified.
We hypothesized that STIM1 may participate in oxidative stress and pulmonary inflammation in alveolar macrophages and therefore contribute to the pathogenesis of COPD. In the current study, we measured the expression of STIM1 in the lung homogenates and bronchoalveolar lavage fluid (BALF) of COPD patients. The ROS levels and STIM1 expression in CSE-exposed THP-1 cells were examined. CSE-induced changes in cytokine secretion and intracellular Ca2+ levels were also explored by treating cells with antioxidant NAC and silencing STIM1.
Materials and Methods
Subjects
Normal lung specimens were collected from patients who underwent surgical resection for pulmonary lump in Tongji Hospital, Wuhan, China, between 2017 and 2021. BALF samples were intentionally obtained from patients who underwent bronchoscopy in Tongji Hospital between 2015 and 2017. COPD was diagnosed according to the Global Initiative for Chronic Obstructive Lung Disease (GOLD) criteria (Vestbo et al., 2013). Patients with a post-bronchodilator forced expiratory volume in 1 s (FEV1)/forced vital capacity ratio of less than 70% were enrolled. Age- and gender-matched non-smokers and smokers without COPD were also recruited as control subjects. Participants were excluded if they suffered from asthma, severe lung infections, or other obstructive lung diseases. This study was approved by the Ethics Committees of the Tongji Hospital (TJ-IRB 20140415 and TJ-IRB20210346) and written informed consent was obtained from all subjects.
Preparation of Alveolar Macrophages
Alveolar macrophages in BALF were obtained by performing bronchoscopy following the international guidelines (Meyer et al., 2012). A total of 120 ml sterile isotonic saline solution at 37°C were separated into four aliquots and flushed into the right middle lobe. After each flush, the fluid was aspirated immediately and gently, collected in sterile centrifuge tubes, and kept on ice. Approximately 50% BALF was recovered. Samples were then filtered through a 40-μm cell strainer and centrifuged at 1,000 × g for 10 min at 4°C. Cell pellets were resuspended in RPMI-1640 medium containing 20% fetal bovine serum, 200 U/ml penicillin, and 200 ug/ml streptomycin. Slides were prepared by cytocentrifugation at 1,200 rpm for 5 min and then fixed in 4% formaldehyde for immunofluorescence staining. Cells were cultured in 12-well culture plates in a 5% CO2 humidified incubator at 37°C for 2 h. Then, non-adherent cells were removed by washing the plates with RPMI-1640 medium, yielding monolayers that contained at least 95% macrophages. Cell morphology was analyzed and proteins were extracted.
Preparation of Cigarette Smoke Extract
Cigarette smoke extract was prepared by bubbling the smoke from two burning cigarettes (3R4F, University of Kentucky) at a rate of 1 cigarette/5 min to a 50 ml centrifuge tube containing 20 ml of RPMI-1640 medium. The pH was adjusted to 7.4. The solution was then filtered through a 0.22-μm filter to eliminate bacteria.
Cell Culture
Human-derived THP-1 cells (ATCC® TIB-202) were cultured in RPMI-1640 medium containing 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin in a humidified incubator with 5% CO2 at 37°C. Cells were treated with phorbol myristate acetate (PMA, 100 nM) for 48 h to induce macrophage differentiation. In transfection experiment, cells were transfected with 50 nM small interfering RNA (siRNA) targeting STIM1 (5′-GTGGTACAGTGGCTGATCA-3′) or negative control sequence (RiboBio, Guangzhou, China) using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, United States) according to the manufacturer’s instructions. In pharmacological experiment, cells were pretreated with 3 mM NAC (Sigma-Aldrich, St. Louis, MO, United States) for 1 h, with 10 μM SKF-96365 (MedChemExpress, United States) for 2 h, or with 10 μM 2-APB (MedChemExpress, United States) for 2 h before CSE stimulation.
Immunohistochemical Analysis
Formalin (10%)-fixed, paraffin-embedded lung tissue sections of healthy non-smokers (HN), smokers without COPD, and smokers with COPD (SC) were deparaffinized using xylene and rehydrated in a graded ethanol series. Heat-induced antigen retrieval was performed using a microwave. After cooling with running tap water, sections were incubated with 3% hydrogen peroxide to block endogenous peroxidase activity, followed by 1-h incubation in 5% BSA-phosphate-buffered saline (PBS) solution at room temperature to avoid non-specific background. Then, slides were incubated with polyclonal rabbit anti-STIM1 (1:500; Proteintech, United States) antibody at 4°C overnight in a humidified chamber. After washing, sections were incubated with a peroxidase-conjugated goat anti-rabbit secondary antibody for 1 h at room temperature. The reactions were developed using a DAB substrate kit with hematoxylin as a counterstain. A Nikon Spot image acquisition and processing system (United States) was used for image assessment.
Immunofluorescence Staining
Cells were fixed with 4% paraformaldehyde for 15 min and stored at −80°C. After washing, slides were incubated in 3% hydrogen peroxide solution in the dark for 10 min. After three washes with PBS for 5 min, slides were blocked in 5% BSA for 20 min. Then, slides were incubated with polyclonal rabbit anti-STIM1 antibody (1:50, Proteintech, United States), monoclonal mouse anti-CD68 antibody (Abcam, United Kingdom), or isotype controls (Becton Dickinson, Franklin Lakes, United States) overnight at 4°C. After washing, slides were incubated with goat anti-rabbit and goat anti-mouse secondary antibodies (Aspen, Wuhan, China) for 50 min at 37°C. DAPI was used to counterstain the nuclei.
Detection of Reactive Oxygen Species Levels
Lung tissues (1 g) were obtained from subjects who underwent surgical resection as aforementioned and flash frozen with liquid nitrogen. Samples were thawed, maintained at 2–8°C, and homogenized with 9 mL PBS by a grinder. Then, tissue homogenates were centrifuged at 3,000 rpm for 10 min and the supernatant was collected and examined for concentration. The supernatants were incubated with 1 mmol/L DCFH-DA solution (Elabscience, China) for 30 min at 37°C. The ROS levels were detected by a microplate reader (Molecular Devices, China) at 525 nm.
The levels of ROS in THP-1 cells were detected by a Fluorometric Intracellular Ros Kit (Sigma-Aldrich, Darmstadt, Germany). After pretreated with NAC for 1 h, PMA-differentiated THP-1 cells were treated with CSE for 3 h. Then, cells were washed and incubated with RPMI-1640 containing 1 μM dichlorofluorescein diacetate (H2DCF-DA) for 30 min. After three washes with RPMI-1640 medium for 5 min, cells were digested by trypsin, washed again, and resuspended in 200 μl PBS. Finally, flow cytometry (BD Biosciences, SanJose, CA, United States) was performed to detect the fluorescent signal intensity at 525 nm to indicate the intracellular levels of ROS.
Measurement of Intracellular Ca2+
Intracellular Ca2+ levels were measured using the Fluo-3, AM Kit (Solarbio, Beijing, China) in accordance with the manufacturer’s protocol. After treated with CSE or other indicated agents, cells were incubated with 5 μM molecular probe Fluo-3, AM for 20 min at 37°C. Next, cells were cultured in HBSS containing 1% fetal bovine serum for 40 min and then resuspended in HBSS buffer saline. The Ca2+ levels were determined by flow cytometry (BD Biosciences, SanJose, CA, United States) at 525 nm.
Cell Viability Assay
Approximately 5,000 cells were seeded in each well of a 96-well plate (Corning, MA, United States). After 48-h incubation with different concentrations of CSE, Cell Counting Kit-8 (CCK-8; Promoter Biotechnology, Wuhan, China) was used to detect cell viability according to the manufacturer’s instructions. Optical density (OD) values were obtained using a microplate reader (Molecular Devices, China).
ELISA
The levels of interleukin (IL)-8 and IL-1β in PMA-differentiated THP-1 macrophages were quantified using DuoSet ELISA kit (R&D Systems, Minneapolis, MN, United States) and RayBio ELISA kit (RayBiotech, United States) according to the manufacturers’ instructions. The minimum detectable dose was 31.3 pg/mL and 0.3 pg/ml for IL-8 and IL-1β, respectively.
Real-Time Quantitative Polymerase Chain Reaction
Total RNA was extracted using Trizol (Takara, Japan) and reverse transcribed to cDNA using PrimeScriptTM RT reagent Kit with gDNA Eraser (Takara, Japan). The mRNA expression was assessed by real-time quantitative polymerase chain reaction (RT-qPCR) using TB Green® Premix Ex TaqTM II (Takara, Japan) on BioRad CFX384 (Bio-Rad, CA, United States). The parameters were as follows: 40 cycles at 95°C for 10 s, 59°C for 20 s, and 72°C for 30 s. The relative mRNA expression was determined using the 2–ΔΔCt method with β-actin as the internal control. The following primers were used in this experiment: β-actin (F: 5′-GCGCGGCTACAGCTTCA-3′; R: 5′-CTTAATGTCACGCACGATTTCC-3′), STIM1 (F: 5′-TTG TCCATGCAGTCCCCTAG-3′; R: 5′-GGTAGTGGTGATGGTG GTGA-3′).
Western Blot
Total protein was extracted using RIPA buffer supplemented with protease inhibitor cocktail (Servicebio, Wuhan, China). Equal amounts of proteins were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes. After blocking with Tris-buffered saline and tween-20 (TBST) containing 5% non-fat milk for 1 h, the membranes were incubated with rabbit polyclonal anti-STIM1 antibody (1:1,000, Proteintech, Chicago, IL, United States) or rabbit polyclonal anti-β-tubulin antibody (1:4,000, Sungene biotech, Shanghai, China) overnight at 4°C. The membranes were washed with TBST buffer, followed by 1-h incubation with a horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (1:4,000; Sigma-Aldrich, Darmstadt, Germany) at room temperature. After washed with TBST, the protein expression was detected using an ECL chemiluminescence detection kit (Advansta, California, United States) and quantified by ImageJ.
Statistical Analysis
All data were normally distributed and expressed as the mean ± SEM. Statistical significance was determined by Student’s t-test or one-way ANOVA with Newman–Keuls multiple comparison test as appropriate. The correlations were analyzed by Pearson correlation. The GraphPad Prism 8 Software (GraphPad Software, San Diego, CA, United States) was used for all statistical analysis and graphic generation. The FlowJo 10 software was used for flow cytometry analysis. P < 0.05 was considered statistically significant.
Results
Characteristics of All Subjects
The clinical characteristics of all subjects are shown in Tables 1, 2. Lung tissues were collected from 12 HN, 12 smokers without COPD (healthy smokers, HS), and 16 SC. BALF samples were obtained from 13 HN, 14 HS, and 12 SC. There was no significant difference in age and gender among these groups. Compare with the HN group, the smoking index of HS and SC was markedly increased. The FEV1% predicted and FEV1/FVC of SC were significantly lower than those of the HN and HS groups.
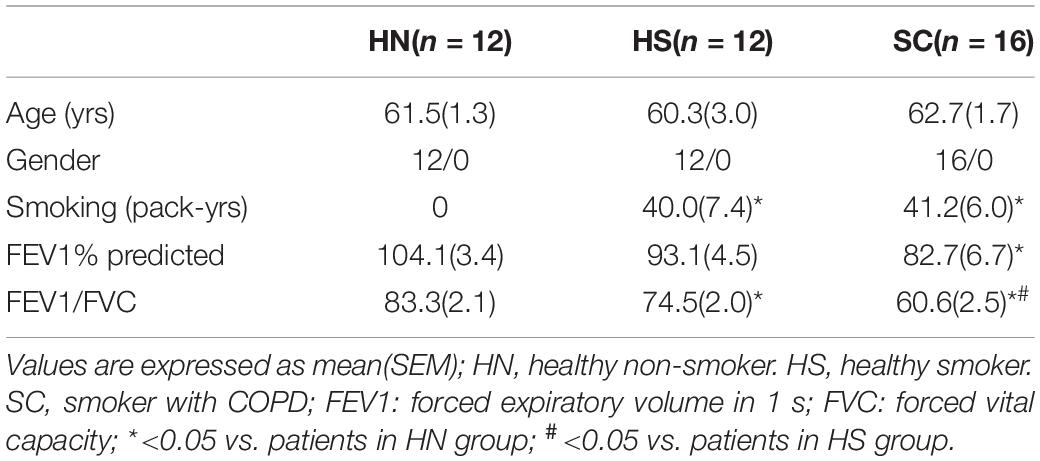
Table 1. Clinical characteristics for those subjects with collected lung tissues.
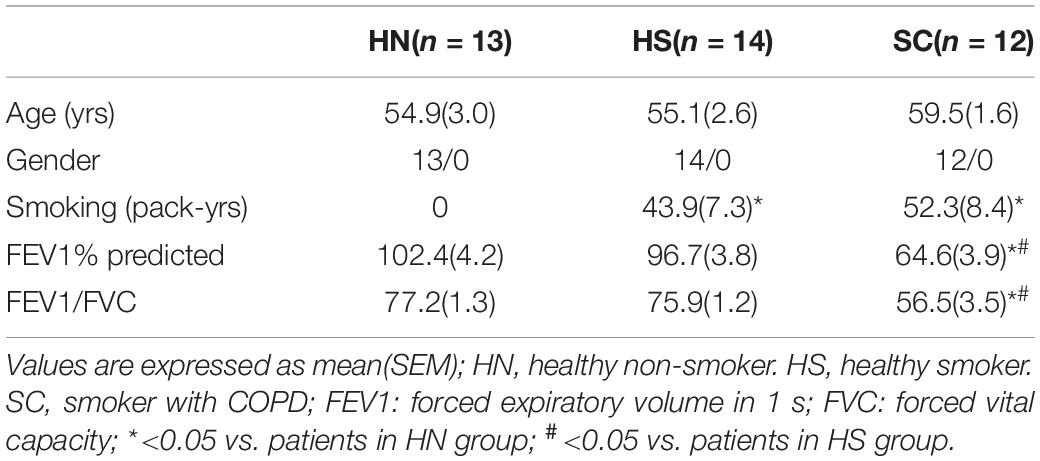
Table 2. Clinical characteristics for those subjects with collected alveolar macrophages.
Intrapulmonary Expression of Stromal Interaction Molecule 1 Is Increased in Chronic Obstructive Pulmonary Disease Patients
Immunohistochemistry was performed to detect the expression of STIM1 in the lung tissue of HN, HS, and SC (Figures 1A–C). STIM1 was mainly expressed in alveolar macrophages. Compared with the HN and HS groups, the expression of STIM1 in SC was notably increased. RT-qPCR and Western blot were performed to validate the expression of STIM1. Consistent with the results of immunohistochemistry, intrapulmonary expression of STIM1 in SC was prominently increased compared with the HN and HS group at both mRNA (Figure 1D) and protein (Figures 1E,F) levels. To explore the association between STIM1 expression and pulmonary function, the correlation between STIM1 protein expression and FEV1% predicted in the SC group was analyzed. We found that intrapulmonary STIM1 expression was negatively correlated with the pulmonary function of COPD patients (Figure 1G), indicating the potential role of STIM1 in the development of COPD.
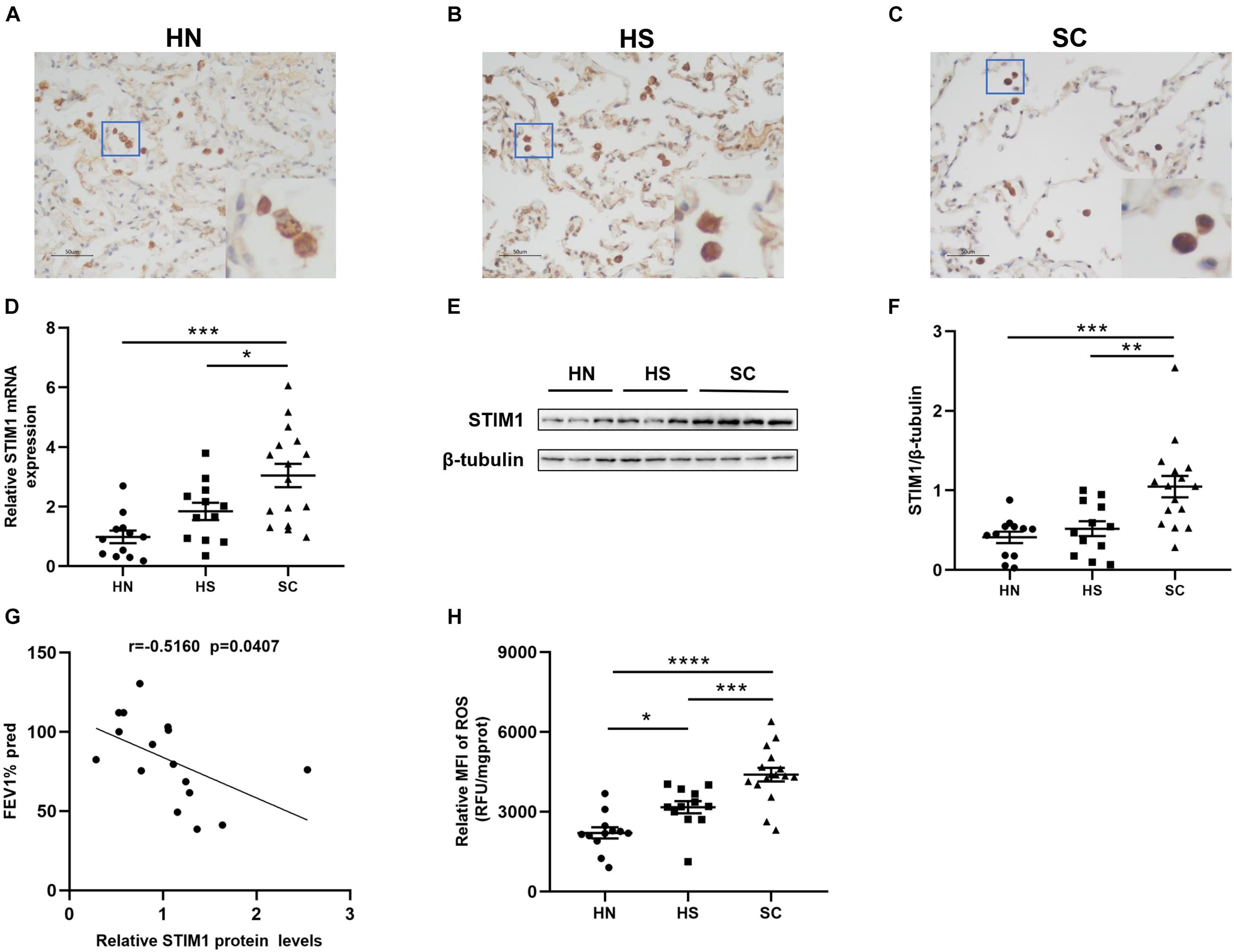
Figure 1. Intrapulmonary stromal interaction molecule 1 (STIM1) expression and reactive oxygen species (ROS) levels are enhanced in the lung of chronic obstructive pulmonary disease (COPD) patients. Representative immunohistochemical images of human lung tissues against STIM1 are shown. Compared with healthy non-smokers (HN) (A) and healthy smokers (HS) (B), STIM1 expression was markedly increased in the alveolar macrophages of smokers with COPD (SC) (C). Magnification = × 200. The red-boxed area indicates a region of higher magnification. The STIM1 mRNA expression was increased in SC relative to HN and HS (D). Representative STIM1 expression in whole-lung tissue homogenates was detected by Western blot (E) and quantified using ImageJ (F). Increased STIM1 is negatively associated with pulmonary function in COPD patients (G). The ROS levels of lung tissue in SC patients were significantly increased relative to HS and HN (H). Data are displayed as mean ± SEM, n = 12 for HN, n = 12 for HS, and n = 16 for SC. P-values were calculated using one-way ANOVA followed by Newman–Keuls test. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 represent significant differences. STIM1, stromal interaction molecule 1; FEV1%pred, forced expiratory volume in 1 s (FEV1)% predicted; ROS, reactive oxygen species; MFI, Mean fluorescent intensity; HN, healthy non-smoker; HS, healthy smoker; and SC, smoker with COPD.
Increased Reactive Oxygen Species Levels in Smokers With or Without Chronic Obstructive Pulmonary Disease
The levels of ROS in the lung tissue supernatants of SC and HS were significantly increased compared with the HN group (Figure 1H). HS showed higher ROS levels than HN, indicating that CS may directly lead to the elevation of ROS levels in the lungs. Moreover, significantly higher levels of ROS were observed in the lung tissues of SC compared with HS, suggesting enhanced oxidative stress in the lungs of SC. To determine the association between STIM1 expression and ROS production, we performed correlation analysis in all subjects. The results showed that pulmonary ROS levels were positively associated with STIM1 expression at both mRNA (Supplementary Figure 1A) and protein (Supplementary Figure 1B) levels.
Stromal Interaction Molecule 1 Is Expressed in Human Alveolar Macrophages and Upregulated in Macrophages Isolated From the Bronchoalveolar Lavage Fluid of Chronic Obstructive Pulmonary Disease Patients
Previous immunohistochemistry analysis showed that STIM1 was mainly expressed in alveolar macrophages. To further define the location and expression level of STIM1, immunofluorescence staining and Western blot were performed using alveolar macrophages isolated from the BALF samples. Immunofluorescence staining against CD68 (a marker of macrophages), STIM1, and DAPI revealed that STIM1 was predominantly located in the cytoplasm rather than the nucleus of macrophages (Figure 2A). Moreover, alveolar macrophages from different individuals were cultured for 24 h and the protein expression of STIM1 was detected by Western blot. The level of STIM1 in SC was significantly higher than that of the HS and HN groups, and there was no significant difference between the HN and HS groups (Figures 2B,C). These results indicated that STIM1 may play a key role in the development of COPD. We therewith treated human macrophages with 5% CSE to investigate the effect of CSE on STIM1 expression. We found that CSE significantly upregulated the expression of STIM1 in alveolar macrophages in vitro (Supplementary Figures 2A,B).
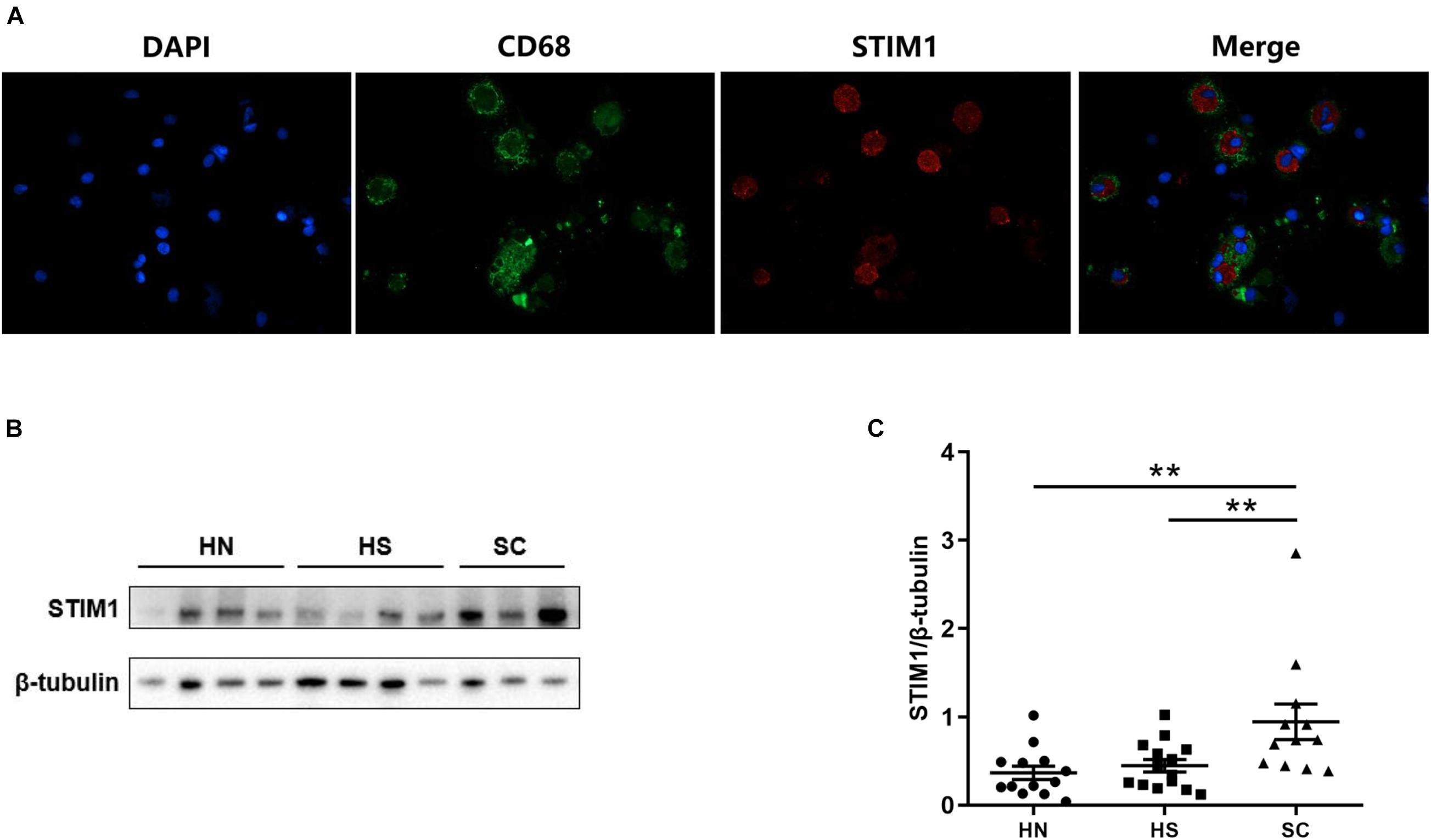
Figure 2. Stromal Interaction Molecule 1 is located in the human alveolar macrophages and up-regulated in macrophages isolated from bronchoalveolar lavage fluid (BALF) of COPD patients. Representative immunofluorescent images against STIM1 in macrophages isolated from BALF are shown (A). Representative western blot images of STIM1 expression in the macrophages of BALF are shown (B) and quantified using ImageJ (C). Data are displayed as mean ± SEM, n = 13 for HN, n = 14 for HS, and n = 12 for SC. P-values were calculated using one-way ANOVA followed by Newman–Keuls test. **P < 0.01 represent significant differences. STIM1, stromal interaction molecule 1; BALF: bronchoalveolar lavage fluid; HN, healthy non-smoker; HS, healthy smoker; and SC, smoker with COPD.
N-Acetylcysteine Alleviates Cigarette Smoke Extract-Induced Reactive Oxygen Species Production and Stromal Interaction Molecule 1 Expression in Phorbol Myristate Acetate-Differentiated THP-1 Cells
Cell viability was not affected by CSE at a concentration lower than or equal to 10% (Supplementary Figure 3). To assess the potential effects of CSE and NAC on intracellular ROS production, PMA-differentiated THP-1 cells were incubated with or without CSE at different concentrations for 3 h. Cells were also pretreated with 3 mM NAC or control medium for 1 h before CSE treatment. Flow cytometry analysis showed that 5 and 10% CSE significantly induced ROS production in cells (Figures 3A,B). CSE at a concentration of 5% was used to investigate the effect of NAC on ROS production. Compared with the control groups, cells treated with 5% CSE showed increased intracellular ROS production, while pretreatment with 3 mM NAC markedly reduced CSE-induced ROS production (Figures 3C,D).

Figure 3. N-acetylcysteine (NAC) alleviates cigarette smoke extract (CSE)-induced ROS production and STIM1 expression in PMA-differentiated THP-1 cells. The representative images of flow cytometry for intracellular ROS levels are shown (A,C) and quantified using FlowJo (B,D). The intracellular level of ROS was up-regulated as CSE concentration increased in PMA-differentiated THP-1 cells. Pretreatment with 3 mM NAC for 1 h alleviated CSE-induced ROS increase in PMA-differentiated THP-1 cells. The representative images of western blot of STIM1 expression are shown (E,G) and quantified using ImageJ (F,H). 5%CSE stimulation for 48 h enhanced STIM1 expression in PMA-differentiated THP-1 cells. Pretreatment with 3 mM NAC for 1 h alleviated CSE-induced STIM1 increase in PMA-differentiated THP-1 cells. Data are displayed as mean ± SEM of at least three independent experiments. P-values were calculated using one-way ANOVA followed by Newman–Keuls test. *P < 0.05, **P < 0.01, and ***P < 0.001 represent significant differences. MFI, Mean fluorescent intensity; ROS, reactive oxygen species; CSE, cigarette smoke extract; NAC, N-acetylcysteine; STIM1, stromal interaction molecule 1; and PMA, phorbol myristate acetate.
To further examine whether the expression of STIM1 was regulated by CSE-induced ROS, we measured the expression of STIM1 in PMA-differentiated THP-1 cells treated with different concentrations of CSE. We found that 5% CSE significantly increased the expression of STIM1 (Figures 3E,F). However, 1-h pretreatment with 3 mM NAC markedly suppressed CSE-induced upregulation of STIM1 in cells (Figures 3G,H). These results support the hypothesis that CSE-induced ROS may be involved in the regulation of STIM1 expression in macrophages.
Interleukin-8 Induction Is Reduced by N-Acetylcysteine and Stromal Interaction Molecule 1 Knockdown in Cigarette Smoke Extract-Stimulated Phorbol Myristate Acetate-Differentiated THP-1 Cells
The above data showed that intracellular ROS levels were upregulated by CSE treatment and NAC alleviated CSE-induced ROS production. CSE-induced upregulation of STIM1 in PMA-differentiated THP-1 cells was also inhibited by NAC. Thus, we speculated that CSE might increase the expression of STIM1 by promoting the production of ROS. To further investigate the effects of CSE-induced ROS on inflammatory cytokines and to determine whether it was subjected to the regulation of STIM1, we examined the effects of STIM1 knockdown and NAC pretreatment on IL-8 and IL-1β, two important cytokines in the pathogenesis of COPD, in PMA-differentiated THP-1 cells (Pauwels et al., 2011). Western blot was used to confirm STIM1 knockdown in cells (Figures 4A,B). NAC pretreatment suppressed CSE-induced IL-8 release in cells transfected with si-NC or si-STIM1. Knockdown of STIM1 further decreased the level of IL-8 in CSE-stimulated cells (Figure 4C). These results indicated a potential regulation of IL-8 production by ROS and STIM1 in CSE-challenged cells. Combined with previous findings that ROS regulated the expression of STIM1, we hypothesized that the regulation of IL-8 by ROS and STIM1 was initiated by CSE. Neither STIM1 knockdown nor NAC pretreatment significantly altered the production of IL-1β (Figure 4D).
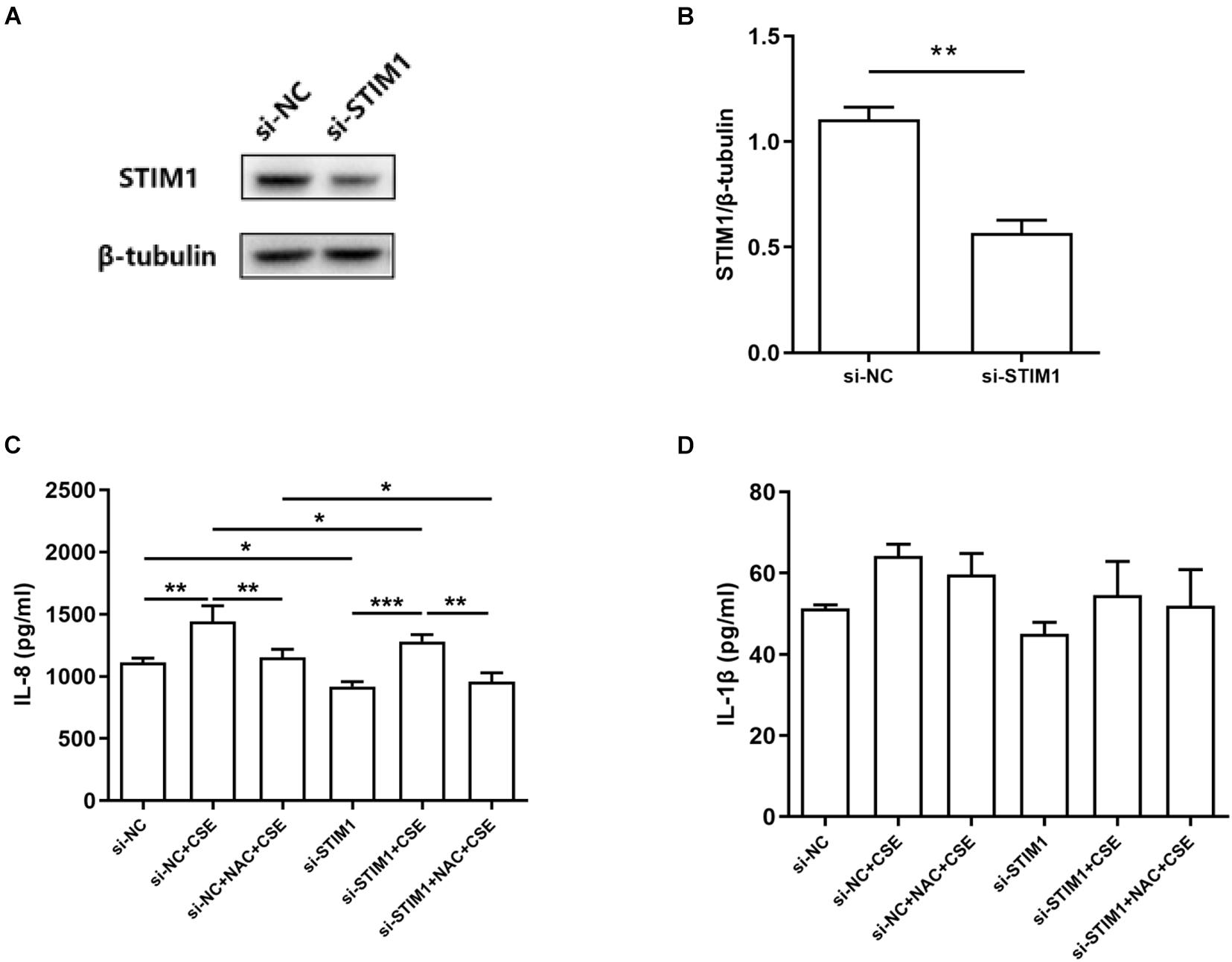
Figure 4. Interleukin-8 (IL-8) induction is mitigated by NAC and STIM1 knockdown in CSE-stimulated PMA-differentiated THP-1 cells. Relative STIM1 expression was detected by western blot in PMA-differentiated THP-1 cells after transfected with small interfering RNA (siRNA)-STIM1 (A) and quantified using ImageJ (B). P-values were calculated using Student’s t-test of at least three independent experiments. si-NC transfected and si-STIM1 transfected THP-1 cells were pretreated with 3 mM NAC for 1 h, and then the supernatant was collected 48 h after 5% CSE stimulation. The levels of IL-8 and IL-1β released from the cells are shown (C,D). Data are expressed as mean ± SEM of at least three independent experiments. P-values were calculated using Student t-test or one-way ANOVA followed by Newman–Keuls test as appropriate. *P < 0.05, **P < 0.01, and ***P < 0.001 represent significant differences. NC, negative control; STIM1, stromal interaction molecule 1; NAC, N-acetylcysteine; CSE, cigarette smoke extract; and PMA, phorbol myristate acetate.
Reactive Oxygen Species/Stromal Interaction Molecule 1 Regulates Interleukin-8 Production Through Intracellular Ca2+ Levels in Cigarette Smoke Extract-Stimulated Phorbol Myristate Acetate-Differentiated THP-1 Cells
To study whether intracellular Ca2+ is involved in the regulation of IL-8 by ROS/STIM1 in CSE-stimulated cells, we performed flow cytometry to detect intracellular Ca2+ levels. The results showed that pretreatment with NAC suppressed CSE-induced increase in intracellular Ca2+ levels, and STIM1 knockdown further decreased intracellular Ca2+ levels in CSE-stimulated cells (Figures 5A,B). Moreover, treatment with SKF-96365 and 2-APB, two pharmacological inhibitors targeting Ca2+ channels, significantly inhibited CSE-induced increase in IL-8 levels (Figure 5C). Taken together, it could be concluded that CSE promoted the release of IL-8 in PMA-differentiated THP-1 cells possibly through the ROS/STIM1/Ca2+ axis.
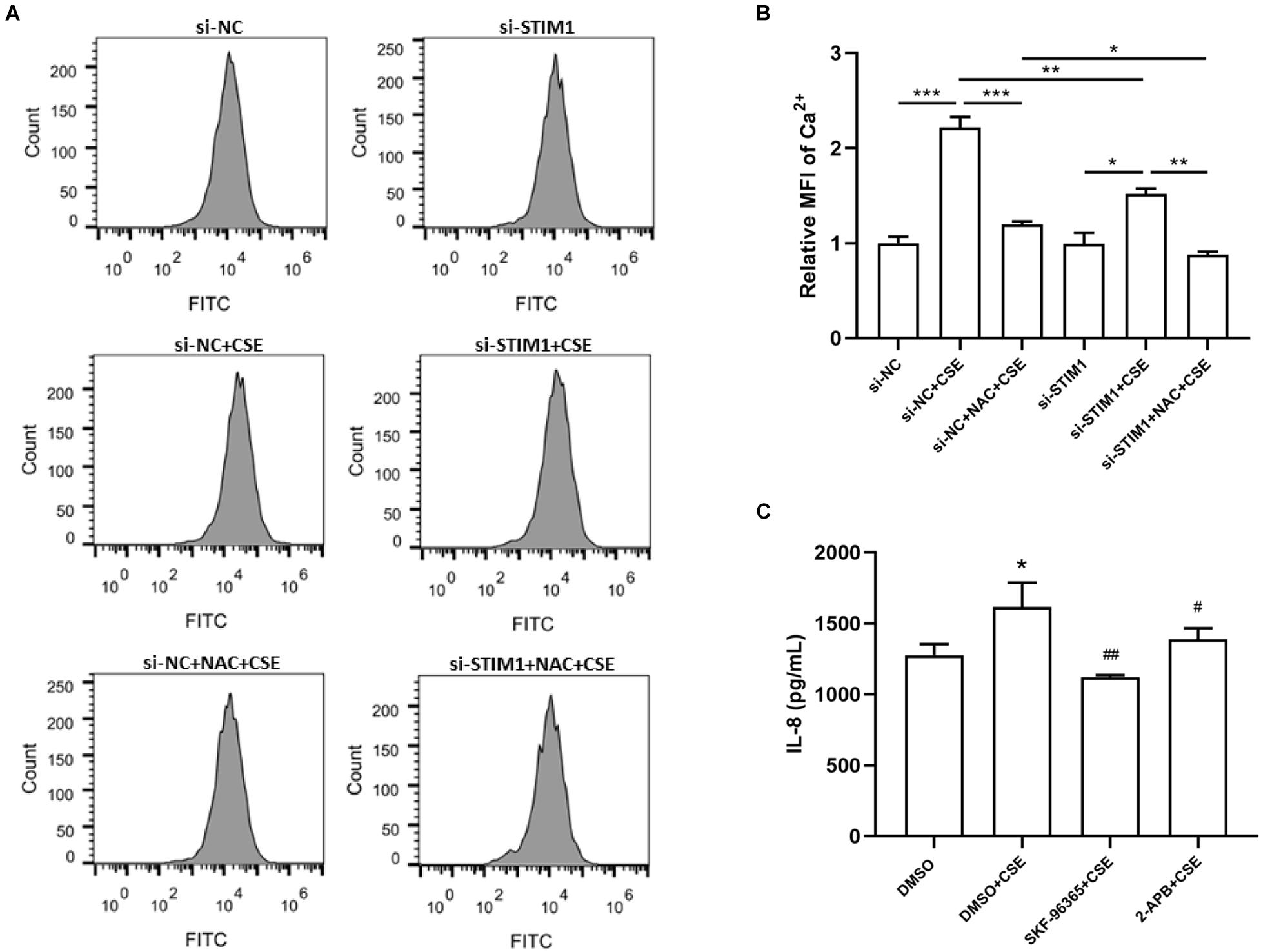
Figure 5. Reactive oxygen species-stromal interaction molecule 1-regulated IL-8 increase is mediated by intracellular Ca2+ level in CSE-stimulated PMA-differentiated THP-1 cells. The representative images of flow cytometry for detection of intracellular Ca2 + are shown (A) and quantified by FlowJo (B). Knockdown of STIM1 and pretreatment of 3 mM NAC in PMA-differentiated THP-1 cells alleviated CSE-induced intracellular Ca2 + increase. 2-h Pretreatments by SKF-96365 and 2-APB, respectively, the Ca2 + channel inhibitors, mitigated IL-8 increase 48 h after 5%CSE stimulation (C). Data are expressed as mean ± SEM of at least three independent experiments. P-values were calculated using one-way ANOVA followed by Newman–Keuls test. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. si-NC or DMSO. #P < 0.05, ##P < 0.01 vs. DMSO + CSE. NC, negative control; STIM1, stromal interaction molecule 1; MFI, Mean fluorescent intensity; CSE, cigarette smoke extract; NAC, N-acetylcysteine; DMSO, dimethyl sulfoxide; and PMA, phorbol myristate acetate.
Discussion
Although the major characteristic of COPD, oxidative stress and pulmonary inflammation, have been reported extensively (McGuinness and Sapey, 2017; Lange et al., 2021), the link between the two pathological processes remains poorly understood. The current study investigated the potential interplay between ROS production and inflammatory cytokine expression, as well as the role of STIM1, in alveolar macrophages in response to CS. We first demonstrated that the expression level of STIM1 in the lung tissue homogenates and alveolar macrophages isolated from the BALF of COPD patients was significantly higher than that of HN and HS. Also, CSE upregulated the expression of STIM1 in vitro. Moreover, intrapulmonary ROS production was markedly enhanced in COPD patients. Correlation analysis indicated a possible link between ROS production and STIM1 expression. In macrophages differentiated from THP-1 cells, administration of NAC, an inhibitor of ROS, effectively suppressed CSE-induced upregulation of STIM1, implying the regulation of STIM1 by ROS in response to CS. Importantly, both NAC pretreatment and STIM1 knockdown inhibited CSE-induced increase in intracellular Ca2+ levels and IL-8 release. Collectively, our study uncovered that CS promoted IL-8 secretion in human alveolar macrophages potentially through the ROS/STIM1/Ca2+ axis (Figure 6). These findings may contribute to a better understanding of the pathogenesis of COPD in terms of oxidative stress and inflammation.
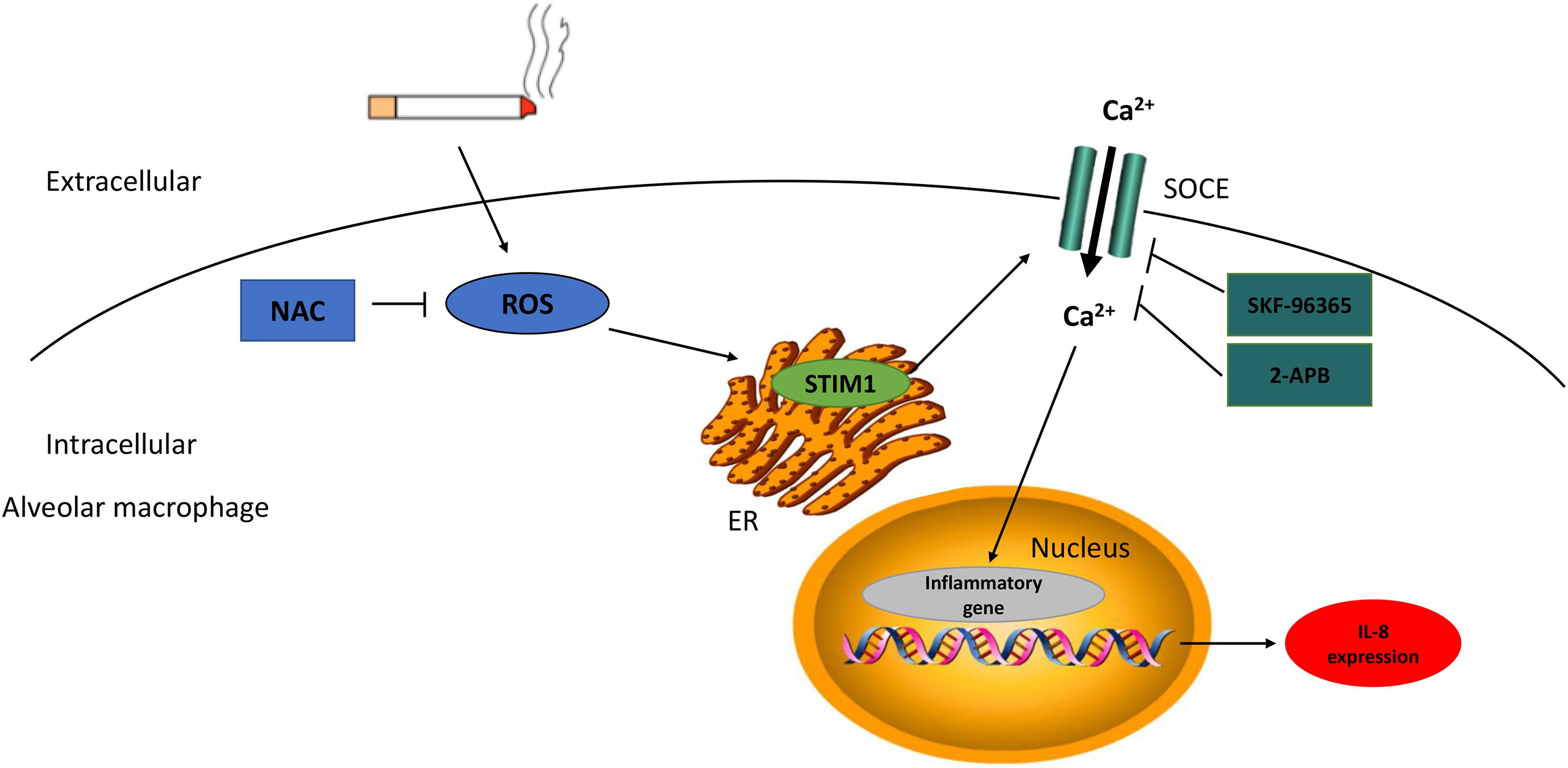
Figure 6. The scheme diagram of STIM1 in the regulation of cigarette smoke (CS)-stimulated alveolar macrophage. CS-induced intracellular ROS production enhanced STIM1 expression in alveolar macrophage, and further promoted inflammatory cytokine secretion through the regulation of Ca2 + entry. NAC can alleviate intracellular ROS levels and SKF-96365/2-APB can dampen Ca2 + influx, both of which can mitigate the IL-8 expression. NAC, N-acetylcysteine; ROS, reactive oxygen species; STIM1, stromal interaction molecule 1; SOCE, store-operated Ca2 + entry; and ER, endoplasmic reticulum.
Cigarette smoke contains approximately 4,700 chemical compounds and is implicated in many diseases, including cancer, cardiovascular diseases, and COPD (Stedman, 1968). Previous studies have demonstrated that CS increases ROS production in lung tissues and BALF both in vivo (Toledo et al., 2012; Li et al., 2013) and in vitro (Kayyali et al., 2003; Cipollina et al., 2014; Campos et al., 2017; Li et al., 2018). It has also been reported that CSE may provoke ROS production by enhancing the transcription and activity of ROS-generating enzymes (Kayyali et al., 2003). ROS may promote inflammation by activating stress kinases (e.g., c-Jun activated kinase, extracellular signal-regulated kinase, and p38) and redox-sensitive transcription factors (e.g., NF-κB and activator protein-1), which further induce the expression of multiple inflammatory genes, such as IL-8 and TNF-α (Rahman and Adcock, 2006; Zuo and Wijegunawardana, 2021). ROS also affect Ca2+ homeostasis by regulating Ca2+ transport proteins located in the plasma membrane, ER, and mitochondria. The link between ROS and inflammation in the lung has been explored (Madreiter-Sokolowski et al., 2020). Lin et al. (2015) found that CSE increased the levels of ROS in lung epithelial cells, which activated transient receptor potential ankyrin 1, increased Ca2+ influx, activated the MAPKs/NF-κB signaling pathway, and ultimately induced the expression of IL-8. Intracellular Ca2+ response can be completed inhibited by NAC (Lin et al., 2015). Previous studies have identified STIM1 as an important oxidative stress sensor (Hawkins et al., 2010) and an ER transmembrane protein (Soboloff et al., 2012), which senses the reduction of Ca2+ in the ER and promote inflammatory responses (Liou et al., 2005; Chen et al., 2013). That is to say, STIM1 translocates to the regions of the ER close to the plasma membrane, where it couples with and activates the plasma membrane Ca2+ channel protein Orai1 to increase Ca2+ influx (Park et al., 2009).
In this study, we found that STIM1 was mainly located in the cytoplasm of alveolar macrophages and the level of STIM1 in the lung tissues and alveolar macrophages of COPD patients was upregulated compared to other groups, which was consistent with the functional characteristics of STIM1. Moreover, correlation analysis demonstrated a significant negative association between STIM1 expression and lung function, indicating that STIM1 plays a vital role in the development of COPD. We further demonstrated that CSE upregulated STIM1 in human alveolar macrophages isolated from BALF and PMA-differentiated THP-1 macrophages in vitro. STIM1, as a critical oxidative stress sensor, is regulated by ROS through residue modification (Hawkins et al., 2010; Gandhirajan et al., 2013). However, whether ROS would affect the protein expression of STIM1 has not been reported. Here, we reported that NAC, a ROS inhibitor, suppressed CSE-induced upregulation of STIM1 protein in macrophages, suggesting the regulation of STIM1 by ROS.
Changes in intracellular Ca2+ levels are essential for proinflammatory cascade in macrophages, the predominant type of inflammatory cells in COPD (Zhou et al., 2006). Elevated IL-8 and IL-1β levels have been observed in the BALF and lung tissues of smokers and patients with COPD (Culpitt et al., 2003; Tomaki et al., 2007). Previous studies revealed that STIM1 played a role in IL-8 production in response to various stimuli in different types of cells (Zhou et al., 2014; Hong et al., 2015). In the current study, we speculated that CSE, a main resource of ROS, might promote Ca2+ influx by regulating STIM1 (a Ca2+ sensor) and further increase the production of IL-1β and IL-8 in human alveolar macrophages. Here, we unexpectedly found that CSE enhanced intracellular Ca2+ levels, but this effect was alleviated by STIM1 knockdown and NAC pretreatment, suggesting the regulation of intracellular Ca2+ levels by ROS and STIM1. Consistent with previous studies (Sarir et al., 2010), NAC inhibited CSE-induced upregulation of IL-8 in human macrophages. Knockdown of STIM1 by siRNA also decreased CSE-induced IL-8 release. The expression of IL-1β, however, was not affected by NAC pretreatment or STIM1 knockdown. Subsequently, we treated cells with Ca2+ channel inhibitors (SKF-96365 and 2-APB) before CSE stimulation to explore the role of intracellular Ca2+ on the expression of inflammatory cytokine IL-8. The results showed that Ca2+ channel inhibitors suppressed CSE-induced upregulation of IL-8, indicating that Ca2+ plays a critical role in CSE-stimulated inflammatory responses. These findings imply that STIM1 may regulate oxidative-stress-related inflammation by acting as a link between ROS and inflammatory cytokines, such as IL-8.
The present study has some limitations. Firstly, we did not validate the role of the ROS/STIM1/Ca2+ axis in COPD in genetic knockout animal models. In addition, further studies are needed to elucidate the exact mechanisms of ROS-mediated upregulation of STIM1 in CSE-stimulated human alveolar macrophages.
Conclusion
In conclusion, our study demonstrated that STIM1 was mainly expressed in alveolar macrophages and the level of STIM1 was increased in patients with COPD. Mechanically, CSE promoted the expression of inflammatory cytokine IL-8 in PMA-differentiated THP-1 macrophages possibly through the ROS/STIM1/Ca2+ axis. These findings revealed a critical role of STIM1 in ROS-related pulmonary inflammation in COPD and provided new insights into the pathogenic mechanism of COPD.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by the Tongji Hospital Ethics Committees. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
JX: conceptualization, methodology, supervision, and funding acquisition. XZ and YZ: project administration, investigation, formal analysis, software, and writing – original draft. YG and ZD: investigation. QH and TW: software. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (Nos. 81973986, 82170049, 82070032, and 81800041), the Health Research Fund of Wuhan (No. WX21Q07), and Health and family planning research project of Hubei (No. WJ2019M116).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2021.733650/full#supplementary-material
Supplementary Figure 1 | Intrapulmonary ROS levels are positively associated with STIM1 expression. The correlation between ROS levels and STIM1 mRNA expression in the lung specimens of all patients are shown (A). The correlation between ROS levels and STIM1 protein expression are shown (B). n = 12 for HN, n = 12 for HS, and n = 16 for SC. STIM1, stromal interaction molecule 1; ROS, reactive oxygen species; MFI, Mean fluorescent intensity; HN, healthy non-smoker; HS, healthy smoker; SC, smoker with COPD.
Supplementary Figure 2 | Cigarette smoke extract stimulation increased STIM1 expression in human macrophages isolated from BALF. Representative western blot image regarding STIM1 protein expression in macrophages treated with or without 5% CSE is shown (A) and quantified using ImageJ (B). Data are expressed as mean ± SEM of at least three independent experiments. P-values were calculated using Student t-test. ∗∗∗P < 0.001 represents significantly different relative to control. CSE concn, concentration of cigarette smoke extract; STIM1, stromal interaction molecule 1.
Supplementary Figure 3 | Cigarette smoke extract stimulation lower than 10% conducted no distinct influence on cell viability in PMA-differentiated THP-1 macrophage. The effects of CSE on HBE cell activity was shown above. Data are expressed as mean ± SEM of at least three independent experiments. P-values were calculated using one-way ANOVA followed by Newman–Keuls test. ∗∗P < 0.01 represents significant difference relative to control. CSE, cigarette smoke extract.
Abbreviations
BALF, bronchoalveolar lavage fluid; Ca2+, calcium; COPD, chronic obstructive pulmonary disease; CRAC, Ca2+ release-activated Ca2+; CSE, cigarette smoke extract; ER, endoplasmic reticulum; IL-8, interleukin-8; IL-1 β, interleukin-1 β; NAC, N-acetylcysteine; NF- κ B, nuclear factor kappa-light-chain-enhancer of activated B cell; Nrf2, nuclear factor erythroid 2-related factor2; PMA, phorbol myristate acetate; ROS, reactive oxygen species; STIM, stromal interaction molecule; SOCE, store-operated Ca2+-entry.
References
Biswas, S., Hwang, J. W., Kirkham, P. A., and Rahman, I. (2013). Pharmacological and dietary antioxidant therapies for chronic obstructive pulmonary disease. Curr. Med. Chem. 20, 1496–1530. doi: 10.2174/0929867311320120004
Campos, K. K. D., Araujo, G. R., Martins, T. L., Bandeira, A. C. B., Costa, G. P., Talvani, A., et al. (2017). The antioxidant and anti-inflammatory properties of lycopene in mice lungs exposed to cigarette smoke. J. Nutr. Biochem. 48, 9–20. doi: 10.1016/j.jnutbio.2017.06.004
Chen, G., Panicker, S., Lau, K. Y., Apparsundaram, S., Patel, V. A., Chen, S. L., et al. (2013). Characterization of a novel CRAC inhibitor that potently blocks human T cell activation and effector functions. Mol. Immunol. 54, 355–367. doi: 10.1016/j.molimm.2012.12.011
Cipollina, C., Di Vincenzo, S., Gerbino, S., Siena, L., Gjomarkaj, M., and Pace, E. (2014). Dual anti-oxidant and anti-inflammatory actions of the electrophilic cyclooxygenase-2-derived 17-oxo-DHA in lipopolysaccharide- and cigarette smoke-induced inflammation. Biochim. Biophys. Acta 1840, 2299–2309. doi: 10.1016/j.bbagen.2014.02.024
Culpitt, S. V., Rogers, D. F., Shah, P., De Matos, C., Russell, R. E., Donnelly, L. E., et al. (2003). Impaired inhibition by dexamethasone of cytokine release by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 167, 24–31. doi: 10.1164/rccm.200204-298oc
Fischer, B. M., Voynow, J. A., and Ghio, A. J. (2015). COPD: balancing oxidants and antioxidants. Int. J. Chron. Obstruct. Pulmon. Dis. 10, 261–276. doi: 10.2147/copd.s42414
Gandhirajan, R. K., Meng, S., Chandramoorthy, H. C., Mallilankaraman, K., Mancarella, S., Gao, H., et al. (2013). Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J. Clin. Invest. 123, 887–902.
Hawkins, B. J., Irrinki, K. M., Mallilankaraman, K., Lien, Y. C., Wang, Y., Bhanumathy, C. D., et al. (2010). S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J. Cell Biol. 190, 391–405. doi: 10.1083/jcb.201004152
Heo, D. K., Lim, H. M., Nam, J. H., Lee, M. G., and Kim, J. Y. (2015). Regulation of phagocytosis and cytokine secretion by store-operated calcium entry in primary isolated murine microglia. Cell Signal. 27, 177–186. doi: 10.1016/j.cellsig.2014.11.003
Hogg, J. C., and Senior, R. M. (2002). Chronic obstructive pulmonary disease – part 2: pathology and biochemistry of emphysema. Thorax 57, 830–834. doi: 10.1136/thorax.57.9.830
Hong, C. H., Chang, K. L., Wang, H. J., Yu, H. S., and Lee, C. H. (2015). IL-9 induces IL-8 production. J. Dermatol. Sci. 78, 206–214.
Kassan, M., Ait-Aissa, K., Radwan, E., Mali, V., Haddox, S., Gabani, M., et al. (2016). Essential role of smooth muscle STIM1 in hypertension and cardiovascular dysfunction. Arterioscler. Thromb Vasc. Biol. 36, 1900–1909. doi: 10.1161/atvbaha.116.307869
Kayyali, U. S., Budhiraja, R., Pennella, C. M., Cooray, S., Lanzillo, J. J., Chalkley, R., et al. (2003). Upregulation of xanthine oxidase by tobacco smoke condensate in pulmonary endothelial cells. Toxicol. Appl. Pharmacol. 188, 59–68. doi: 10.1016/s0041-008x(02)00076-5
Khanna, A., Guo, M., Mehra, M., and Royal, W. III (2013). Inflammation and oxidative stress induced by cigarette smoke in Lewis rat brains. J. Neuroimmunol. 254, 69–75. doi: 10.1016/j.jneuroim.2012.09.006
Lange, P., Ahmed, E., Lahmar, Z. M., Martinez, F. J., and Bourdin, A. (2021). Natural history and mechanisms of COPD. Respirology 26, 298–321. doi: 10.1111/resp.14007
Li, P. C., Tsai, W. H., and Chien, C. T. (2013). Dietary Monascus adlay supplements facilitate suppression of cigarette smoke-induced pulmonary endoplasmic reticulum stress, autophagy, apoptosis and emphysema-related PLGF in the rat. Food Chem. 136, 765–774. doi: 10.1016/j.foodchem.2012.08.007
Li, X., Michaeloudes, C., Zhang, Y., Wiegman, C. H., Adcock, I. M., Lian, Q., et al. (2018). Mesenchymal stem cells alleviate oxidative stress-induced mitochondrial dysfunction in the airways. J. Allergy Clin. Immunol. 141, 1634–1645.e5.
Lin, A. H., Liu, M. H., Ko, H. K., Perng, D. W., Lee, T. S., and Kou, Y. R. (2015). Lung epithelial TRPA1 transduces the extracellular ROS into transcriptional regulation of lung inflammation induced by cigarette smoke: the role of influxed Ca(2)(+). Mediators Inflamm. 2015:148367.
Lin, C. C., Lee, I. T., Yang, Y. L., Lee, C. W., Kou, Y. R., and Yang, C. M. (2010). Induction of COX-2/PGE(2)/IL-6 is crucial for cigarette smoke extract-induced airway inflammation: role of TLR4-dependent NADPH oxidase activation. Free Radic. Biol. Med. 48, 240–254. doi: 10.1016/j.freeradbiomed.2009.10.047
Liou, J., Kim, M. L., Heo, W. D., Jones, J. T., Myers, J. W., Ferrell, J. E. Jr., et al. (2005). STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15, 1235–1241. doi: 10.1016/j.cub.2005.05.055
Madreiter-Sokolowski, C. T., Thomas, C., and Ristow, M. (2020). Interrelation between ROS and Ca(2+) in aging and age-related diseases. Redox Biol. 36:101678. doi: 10.1016/j.redox.2020.101678
McGuinness, A. J., and Sapey, E. (2017). Oxidative stress in COPD: sources, markers, and potential mechanisms. J. Clin. Med. 6:21. doi: 10.3390/jcm6020021
Meyer, K. C., Raghu, G., Baughman, R. P., Brown, K. K., Costabel, U., du Bois, R. M., et al. (2012). American thoracic society committee on, an official American thoracic society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am. J. Respir. Crit. Care Med. 185, 1004–1014. doi: 10.1164/rccm.201202-0320st
Mirza, S., Clay, R. D., Koslow, M. A., and Scanlon, P. D. (2018). COPD guidelines: a review of the 2018 GOLD report. Mayo Clin. Proc. 93, 1488–1502. doi: 10.1016/j.mayocp.2018.05.026
Mizumura, K., Cloonan, S. M., Nakahira, K., Bhashyam, A. R., Cervo, M., Kitada, T., et al. (2014). Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Invest. 124, 3987–4003. doi: 10.1172/jci74985
Park, C. Y., Hoover, P. J., Mullins, F. M., Bachhawat, P., Covington, E. D., Raunser, S., et al. (2009). STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136, 876–890. doi: 10.1016/j.cell.2009.02.014
Pauwels, N. S., Bracke, K. R., Dupont, L. L., Van Pottelberge, G. R., Provoost, S., Vanden Berghe, T., et al. (2011). Role of IL-1alpha and the Nlrp3/caspase-1/IL-1beta axis in cigarette smoke-induced pulmonary inflammation and COPD. Eur. Respir J. 38, 1019–1028. doi: 10.1183/09031936.00158110
Rahman, I. (2005). Oxidative stress in pathogenesis of chronic obstructive pulmonary disease: cellular and molecular mechanisms. Cell Biochem. Biophys. 43, 167–188. doi: 10.1385/cbb:43:1:167
Rahman, I., and Adcock, I. M. (2006). Oxidative stress and redox regulation of lung inflammation in COPD. Eur. Respir. J. 28, 219–242. doi: 10.1183/09031936.06.00053805
Sarir, H., Mortaz, E., Janse, W. T., Givi, M. E., Nijkamp, F. P., and Folkerts, G. (2010). IL-8 production by macrophages is synergistically enhanced when cigarette smoke is combined with TNF-alpha. Biochem. Pharmacol. 79, 698–705. doi: 10.1016/j.bcp.2009.10.001
Soboloff, J., Rothberg, B. S., Madesh, M., and Gill, D. L. (2012). STIM proteins: dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 13, 549–565. doi: 10.1038/nrm3414
Soriano, J. B., Abajobir, A. A., Abate, K. H., Abera, S. F., Agrawal, A., Ahmed, M. B., et al. (2017). Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir. Med. 5, 691–706.
Stedman, R. L. (1968). The chemical composition of tobacco and tobacco smoke. Chem. Rev. 68, 153–207. doi: 10.1021/cr60252a002
Toledo, A. C., Magalhaes, R. M., Hizume, D. C., Vieira, R. P., Biselli, P. J., Moriya, H. T., et al. (2012). Aerobic exercise attenuates pulmonary injury induced by exposure to cigarette smoke. Eur. Respir. J. 39, 254–264. doi: 10.1183/09031936.00003411
Tomaki, M., Sugiura, H., Koarai, A., Komaki, Y., Akita, T., Matsumoto, T., et al. (2007). Decreased expression of antioxidant enzymes and increased expression of chemokines in COPD lung. Pulm Pharmacol. Ther. 20, 596–605. doi: 10.1016/j.pupt.2006.06.006
Tse, H. N., Raiteri, L., Wong, K. Y., Yee, K. S., Ng, L. Y., Wai, K. Y., et al. (2013). High-dose N-acetylcysteine in stable COPD: the 1-year, double-blind, randomized, placebo-controlled HIACE study. Chest 144, 106–118.
Vestbo, J., Hurd, S. S., Agusti, A. G., Jones, P. W., Vogelmeier, C., Anzueto, A., et al. (2013). Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am. J. Respir. Crit. Care Med. 187, 347–365. doi: 10.1164/rccm.201204-0596pp
Wylam, M. E., Sathish, V., VanOosten, S. K., Freeman, M., Burkholder, D., Thompson, M. A., et al. (2015). Mechanisms of cigarette smoke effects on human airway smooth muscle. PLoS One 10:e0128778.
Zhang, H., Clemens, R. A., Liu, F., Hu, Y., Baba, Y., Theodore, P., et al. (2014). STIM1 calcium sensor is required for activation of the phagocyte oxidase during inflammation and host defense. Blood 123, 2238–2249. doi: 10.1182/blood-2012-08-450403
Zhou, M. H., Zheng, H., Si, H., Jin, Y., Peng, J. M., He, L., et al. (2014). Stromal interaction molecule 1 (STIM1) and Orai1 mediate histamine-evoked calcium entry and nuclear factor of activated T-cells (NFAT) signaling in human umbilical vein endothelial cells. J. Biol. Chem. 289, 29446–29456. doi: 10.1074/jbc.m114.578492
Zhou, X., Yang, W., and Li, J. (2006). Ca2+- and protein kinase C-dependent signaling pathway for nuclear factor-kappaB activation, inducible nitric-oxide synthase expression, and tumor necrosis factor-alpha production in lipopolysaccharide-stimulated rat peritoneal macrophages. J. Biol. Chem. 281, 31337–31347. doi: 10.1074/jbc.m602739200
Keywords: oxidative stress, inflammation, stromal interaction molecule 1, macrophage, chronic obstructive pulmonary disease
Citation: Zhu X, Zhan Y, Gu Y, Huang Q, Wang T, Deng Z and Xie J (2021) Cigarette Smoke Promotes Interleukin-8 Production in Alveolar Macrophages Through the Reactive Oxygen Species/Stromal Interaction Molecule 1/Ca2+ Axis. Front. Physiol. 12:733650. doi: 10.3389/fphys.2021.733650
Received: 16 July 2021; Accepted: 22 September 2021;
Published: 08 October 2021.
Edited by:
Tzong-Shyuan Lee, National Taiwan University, TaiwanReviewed by:
Hsin-Kuo Bruce Ko, Taipei Veterans General Hospital, TaiwanMark Edward Wylam, Mayo Clinic, United States
Copyright © 2021 Zhu, Zhan, Gu, Huang, Wang, Deng and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jungang Xie, eGllampnZ0Bob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work