 Samantha L. Gaytan1
Samantha L. Gaytan1 Ahmed Lawan2
Ahmed Lawan2 Jongwha Chang3
Jongwha Chang3 Md Nurunnabi4
Md Nurunnabi4 Sudip Bajpeyi1
Sudip Bajpeyi1 Jason B. Boyle1
Jason B. Boyle1 Sung Min Han5*
Sung Min Han5* Kisuk Min1*
Kisuk Min1*- 1Department of Kinesiology, College of Health Sciences, University of Texas at El Paso, El Paso, TX, United States
- 2Department of Biological Sciences, College of Science, University of Alabama in Huntsville, Huntsville, AL, United States
- 3Department of Pharmaceutical Sciences, Irma Lerma Rangel School of Pharmacy, Texas A&M University, College Station, TX, United States
- 4Department of Pharmaceutical Sciences, School of Pharmacy, University of Texas at El Paso, El Paso, TX, United States
- 5Department of Physiology and Aging, College of Medicine, Institute on Aging, University of Florida, Gainesville, FL, United States
Doxorubicin is a highly effective chemotherapeutic agent widely used to treat a variety of cancers. However, the clinical application of doxorubicin is limited due to its adverse effects on several tissues. One of the most serious side effects of doxorubicin is cardiotoxicity, which results in life-threatening heart damage, leading to reduced cancer treatment success and survival rate. Doxorubicin-induced cardiotoxicity results from cellular toxicity, including increased oxidative stress, apoptosis, and activated proteolytic systems. Exercise training has emerged as a non-pharmacological intervention to prevent cardiotoxicity during and after chemotherapy. Exercise training stimulates numerous physiological adaptations in the heart that promote cardioprotective effects against doxorubicin-induced cardiotoxicity. Understanding the mechanisms responsible for exercise-induced cardioprotection is important to develop therapeutic approaches for cancer patients and survivors. In this report, we review the cardiotoxic effects of doxorubicin and discuss the current understanding of exercise-induced cardioprotection in hearts from doxorubicin-treated animals.
Introduction
The number of cancer survivors living in the United States continues to increase annually due to the early detection of cancer and advances in treatment (Jiang et al., 2022). As of 2019, it was estimated that there are 16.9 million cancer survivors in the United States (Jiang et al., 2022). This represents approximately 5% of the population. The number is projected to reach 22.1 million by 2031 (Jiang et al., 2022). As the number of cancer survivals increases, their quality of life has become a critical issue. However, prolonged and combined cancer treatments, including cancer surgery, radiation therapy, and chemotherapy, are known to cause pain, chronic fatigue, muscle weakness, and physical dysfunction, impairing quality of life in cancer survivors (Galvao et al., 2009; Narayanan and Koshy, 2009). Approximately, 1 in 4 cancer survivors reported a decreased quality of life due to the side effects of cancer treatments (Weaver et al., 2012). Specifically, chemotherapy with anthracyclines (ANTs) has been shown to induce irreversible side effects (Tacar et al., 2013; Min et al., 2015). ANTs are a group of antineoplastic antibiotics that are highly effective chemotherapeutic agents used to treat a wide variety of cancers (Mukai et al., 2021). Unfortunately, the clinical use of ANTs is limited due to the development of cytotoxicity in the heart, resulting in cardiomyopathy and heart failure in cancer patients and survivors during cancer treatments or in even several years after cancer treatment (Kamphuis et al., 2020; Abdel-Qadir et al., 2021). Specifically, doxorubicin is among the ANTs used to manage and treat various types of malignancies and tumors (Min et al., 2015; Douedi and Carson, 2019; Lee et al., 2020; Wu et al., 2022). The primary therapeutic approach for preventing DOX-induced cardiotoxicity is to intervene with standard therapies for heart failure (Dragojevic-Simic et al., 2004; Yu et al., 2020). It has been well established that exercise training provides cardioprotective effects against DOX-induced adverse effects on the heart as a non-pharmacological cardioprotective strategy in cancer patients and survivors. This review provides an updated overview of cardiotoxicity associated with treatments of DOX and presents the current understanding of the exercise-induced protection against the cardiotoxicity.
Anthracycline therapy
ANTs are cytostatic antibiotics that are extracted from Streptomyces bacterium (Fujiwara et al., 1985; Octavia et al., 2012). ANTs include daunorubicin (DAU), doxorubicin (DOX), epirubicin (EPI), idarubicin (IDA), and valrubicin (VAL) (Figure 1) (Dimarco et al., 1963; Capelôa et al., 2020). DAU was the first ANT used to treat acute pediatric leukemia in 1964 (Dimarco et al., 1963). DOX was later isolated from a mutant of Streptomyces peucetius in 1969 (Arcamone et al., 1969). The structural formula of DAU and DOX is very similar, except for the substitution of a hydroxyl group at the carbon 14 position in DOX (Escherich et al., 2013). DAU is utilized against acute lymphoblastic and myeloblastic leukemias, whereas DOX is more effective in lymphomas, sarcomas, and a broad spectrum of solid tumors, such as breast, lung, bladder, and bone cancers (Minotti et al., 2004; Arcamone, 2009; Tacar et al., 2013). Later on, several newer ANTs have been developed to treat multiple types of cancers (Bonfante et al., 1980; McGowan et al., 2017). Although ANTs have been successful in treating a variety of cancers, they have been associated with both acute and chronic cardiotoxicity, depending on the cumulative dose of each agent (Table 1) (Venkatesh and Kasi, 2019; Rocca et al., 2020). For example, early adverse effects of DOX have been reported to reduce the left ventricular ejection fraction within months post-treatment with a cumulative dose ≥350 mg/g2 (Buzdar et al., 1985). Studies with 630 breast and lung cancer patients have revealed that 32 of those 630 patients (5.1%) had DOX-induced congestive heart failure (Swain et al., 2003). Most patients with congestive heart failure were treated with a cumulative dose of ≥500 mg/m2. The estimated cumulative percentages of patients with DOX-induced congestive heart failure were 5%, 16% and 48% at a cumulative doses of 400 mg/g2, 500 mg/m2, and 700 mg/m2, respectively (Swain et al., 2003). Therefore, the dosage adjustments of ANTs are required to prevent the effects of cardiotoxicity and maximize the therapeutic effects.
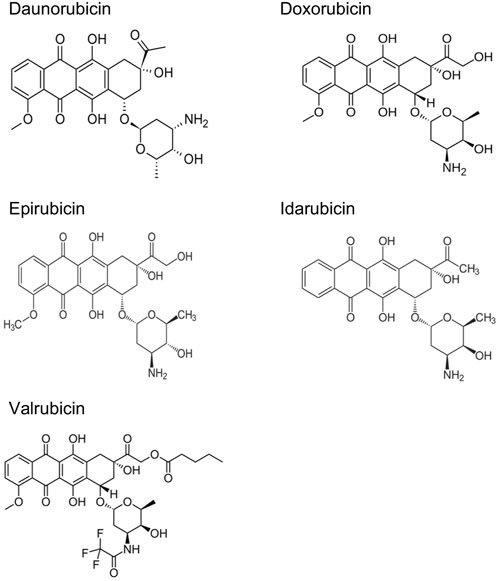
FIGURE 1. Chemical structures of main anthracyclines.
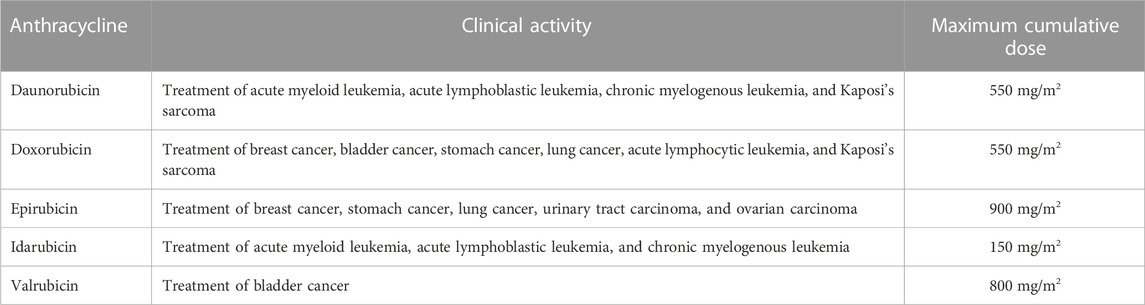
TABLE 1. Anthracyclines, therapeutic use and maximum recommended cumulative dose.
Mechanisms of ANT-induced cytotoxicity
It has been recognized that ANTs act through a combination of multiple mechanisms, including 1) intercalation into DNA, 2) poisons of topoisomerase II, and 3) production of reactive oxygen species.
DNA intercalation
The activity of ANTs results in strong inhibitory effects on nucleic acid synthesis (Gewirtz, 1999; Shandilya et al., 2020). Nuclear DNA has been recognized as the primary target of ANTs (Karadurmus et al., 2021). ANTs consist of flat aromatic moieties that intercalate between DNA base pairs (Figure 2) (Frederick et al., 1990). The intercalation inhibits DNA and RNA synthesis, subsequently blocking the transcription and replication in highly replicating cells (Gewirtz, 1999). The specificity, binding affinity, and the binding mode of each ANT depend on differences in the sequence of the DNA base (Shandilya et al., 2020). The intercalation of ANTs can distort DNA and interfere with the nuclear functions in cancer cells (Wang et al., 1987; Chaires, 1990). ANTs also intercalate mitochondrial DNA (Lebrecht and Walker) to induce single or double-strand mtDNA breaks and quantitative defects in mtDNA copy number (Tewey et al., 1984; Lawrence et al., 1993; Lebrecht and Walker, 2007). Both the mutation and deletion of mtDNA-lesion compromise the synthesis of mtDNA-encoded respiratory chain subunits in the mitochondrial inner membrane, contributing to the marked mitochondrial toxicity (Lebrecht and Walker, 2007; Ashley and Poulton, 2009). Consequently, ANT-induced mitochondrial toxicity causes increased production of mitochondrial reactive oxygen species (Priya et al., 2017), which is one of the mechanisms of ANT-induced cardiotoxicity (Lebrecht and Walker, 2007).
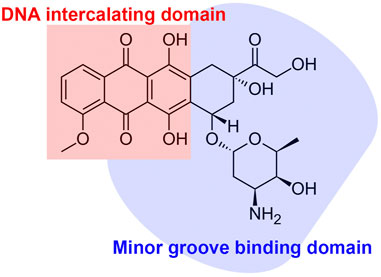
FIGURE 2. The schematic chemical structure and functional domain of anthracycline.
Topoisomerase II poisoning
Along with DNA intercalation, topoisomerase II is also considered as one of the primary targets of ANT-induced cytotoxic activity in cancer cells. Topoisomerase II is a nuclear enzyme that manages DNA tangles and supercoils by cutting both strands of the DNA helix during replication and transcription (Deweese and Osheroff, 2009). ANTs intercalated into DNA form a stable ANT-DNA-topoisomerase II ternary complex, thereby poisoning the enzyme activity. This ternary complex impends the relegation of breaks in the double-stranded DNA (Shandilya et al., 2020). As a result, ANTs induce irreversible DNA damage, leading to genomic instability and ultimately apoptotic cell death in rapidly dividing cancer cells (Li and Liu, 2001; Minotti et al., 2004; Minev, 2011). This ANT-induced topoisomerase II poisoning is also the molecular basis of cardiotoxicity. Since topoisomerase II β is present in cardiomyocytes (Capranico et al., 1992; Vejpongsa and Yeh, 2014), the inhibition of its isoform has been shown to induce long-term side effects of ANTs in cardiac muscle, resulting in cardiomyopathy (Cornarotti et al., 1996; Austin and Marsh, 1998).
Production of reactive oxygen species
One of the mechanisms responsible for ANT-induced cytotoxicity is the generation of excessive reactive oxygen species (Priya et al., 2017). ROS, including superoxide radical (O2•−), hydrogen peroxide (H2O2), and hydroxyl radical (HO•), are byproducts of the normal metabolisms and play roles in homeostasis in normal cells (Rocca et al., 2020). However, excessively high and persistent levels of ROS result in an imbalance between the production of free radicals and antioxidant defense systems, triggering oxidative stress and cellular damage (Devasagayam et al., 2004). It has been demonstrated that mitochondria are one of the major sites for ANT-induced oxidative stress and cellular damage (Min et al., 2015). One electron is transferred from NADPH to the flavoprotein in the mitochondrial electron transport chain. The quinone ring of ANTs acts as an electron acceptor to form semiquinone, which produces superoxide anion (Berthiaume and Wallace, 2007). This reaction is catalyzed by NADH reductase at complex I in the inner mitochondrial membrane (Berthiaume and Wallace, 2007; Murabito et al., 2020). The superoxide dismutase neutralizes superoxide anion into hydrogen peroxide (Stěrba et al., 2013). The production of superoxide anion and hydrogen peroxide stimulates enzyme-mediated reduction-oxidation cycles, producing reactive and destructive hydroxyl radicals, which cause nucleic acid damage, protein alkylation, and lipid peroxidation, followed by apoptosis (Figure 3) (Simůnek et al., 2009; Angsutararux et al., 2015). In addition to mitochondrial ROS, other enzymes including NADPH oxidase (NOX) and nitric oxide synthase (NOS) also contribute to DOX-induced cardiotoxicity (Gilleron et al., 2009; Lin et al., 2019). NOX has been identified as one of the most important sources of ANT-induced ROS (Gilleron et al., 2009; Priya et al., 2017; Lin et al., 2019). NOX is a transmembrane enzyme that is located in intracellular organelles and consists of serval isoforms (Panday et al., 2015). Specifically, NOX2 has been shown to contribute to ANT-induced cardiotoxicity (Wojnowski et al., 2005; Zhao et al., 2010). Growing evidence shows that NOX2 deficiency attenuates superoxide production, preventing cardiomyocytes cell death, myocardial fibrosis, and leukocyte infiltration following DOX administration (Zhao et al., 2010; McLaughlin et al., 2017). NOS is also one of the contributing enzymes to oxidative stress and damage to cardiac muscle following DOX treatment. NOS catalyzes the conversion of L-arginine to nitric oxide (Mukai et al.) (Knowles and Moncada, 1994). Three NOS isoforms have been identified in mammals, including neuronal NOS (nNOS), cytokine-inducible NOS (iNOS) and endothelial NOS (eNOS) (Knowles and Moncada, 1994). DOX administration increases the levels of NO through the activation of eNOS and iNOS (Vásquez-Vivar et al., 1997). eNOS has been shown to catalyze NADPH-dependent superoxide formation following DOX treatment by directly binding the reductase domain of eNOS (Vásquez-Vivar et al., 1997). Eventually, the overproduction of ROS and NO generates reactive nitrogen species (RNS), which lead to cardiotoxicity following ANT treatment (Fogli et al., 2004; Štěrba et al., 2013).
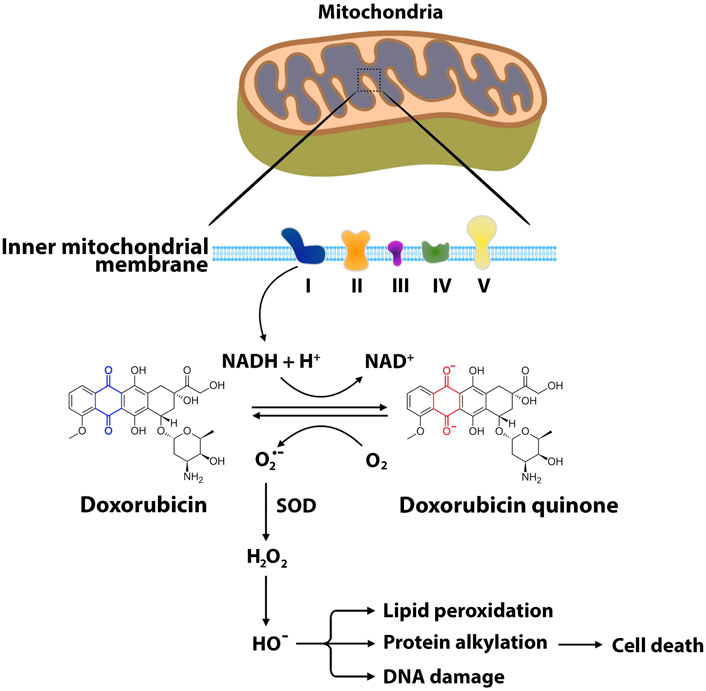
FIGURE 3. Main pathways of ANT-induced oxidative stress. The formation of reactive oxygen species begins with one-electron reduction of the quinone moiety through NADH reductase at complex I of the electron transport chain. In this reaction, the quinone ring of ANTs such as doxorubicin accepts the electron to form semiquinone, producing superoxide anion. Superoxide dismutase neutralizes the superoxide anion into hydrogen peroxide. Hydroxyl radical is produced from hydrogen peroxide through enzyme-mediated reduction-oxidation cycles. ROS interact with mitochondrial DNA, proteins, lipids, and other biomolecules, leading to cellular oxidative damage and eventually apoptosis. O2•−, superoxide radical; H2O2, hydrogen peroxide; HO•, hydroxyl radical; SOD, superoxide dismutase; NAD, nicotinamide adenine dinucleotide.
DOX-induced cardiomyopathy
DOX-induced cardiotoxicity can lead to the development of cardiomyopathy and, ultimately, congestive heart failure (Jeyaseelan et al., 1997; Singal et al., 2000). Echocardiographic analysis shows that ventricular ejection fraction, fractional shortening, and diastolic function can be reduced in the hearts treated with DOX (Lee et al., 1987; Willis et al., 2019). The dysfunction of cardiac contractility with DOX exposure results from a decrease in cardiac mass, which causes cardiac muscle atrophy and cardiac wall thinning (Min et al., 2015; Willis et al., 2019; Ye et al., 2021). The DOX-induced cardiac atrophy can be identified with an atrophic shift of myosin heavy chain isoform from alpha isoform to beta isoform and increased atrial natriuretic peptide (Willis et al., 2019). Additionally, DOX administration also causes cardiac morphological changes, such as dilated ventricles and increased myocardial fibrosis (Gyulkhasyan et al., 2019; Levick et al., 2019). DOX has also been shown to induce cardiac muscle damage through intracellular proteolytic systems (Min et al., 2015; Montalvo et al., 2020). Mammalian cells regulate the balance between protein synthesis and protein degradation, depending on the cellular demand (Rothman, 2010; Dasuri et al., 2013). Proteolytic systems stimulate protein degradation in response to cellular stresses. Although the process of protein breakdown is required for cell survival, the excessive activation of proteolytic systems in response to pathological stress can accelerate protein degradation, leading to muscular atrophy and dysfunction (Hu et al., 2008; Powers et al., 2012). Abundant evidence indicates that DOX-induced ROS production contributes to the activation of proteolytic systems. DOX-induced ROS production contributes to activation of four main proteolytic systems: 1) Ubiquitin proteasome system, 2) calpain, 3) caspase-3, and 4) autophagy.
DOX-induced activation of ubiquitin-proteasome system
The Ubiquitin proteasome system (UPS) is an ATP-dependent proteolytic system composed of numerous ubiquitin ligase enzymes and a large proteolytic complex called the proteasome (Grune et al., 2003; Liu et al., 2016; Hyatt et al., 2019). The UPS plays a role in the protein breakdown that occurs during muscle damage (Murton et al., 2008; Powers et al., 2011). The UPS requires polyubiquitination of proteins through ubiquitin ligase enzymes, including E1 (ubiquitin-activating enzyme), E2, and E3 (Ichimura et al., 2000; Bodine et al., 2001). The polyubiquitinated proteins that are damaged or deemed unnecessary are degraded by the proteasome. Specifically, two muscle-specific E3 ligases, Muscle Atrophy F-box (MAFbx)/atrogin-1 and Muscle-Ring Finger-1 (MuRF-1), contribute to the UPS-mediated protein degradation in cardiac muscle. Numerous studies have indicated that DOX treatment stimulates UPS in cardiac muscle, leading to cardiac muscle damage (Powers et al., 2011; Derouiche et al., 2014; Min et al., 2015; Montalvo et al., 2020). Specifically, DOX treatment activates UPS through mitochondrial ROS production in cardiac muscle (Montalvo et al., 2020). Indeed, DOX administration significantly increases both mitochondrial H2O2 production and MAFbx, whereas mitochondria-targeted antioxidant protects mitochondria against DOX-induced oxidative stress and attenuates the expression of atrogin-1/MAFbx in cardiac muscle (Montalvo et al., 2020). Another study also showed that exercise preconditioning improves mitochondrial biogenesis and prevents gene expression of MuRF-1 in DOX-administrated cardiac muscle (Kavazis et al., 2014). A recent study demonstrated that a DOX dose-dependent (1–25 mg/kg) increases MuRF-1 mRNA and protein levels in myocardial tissues, accompanied by decreases in cardiac mass and cardiomyocyte cross-sectional area (Willis et al., 2019). However, mice lacking MuRF-1 were protected against DOX-induced cardiac atrophy and contractile dysfunction (Willis et al., 2019). These findings suggest that DOX administration induces pathological protein degradation through the activation of UPS in cardiac muscle.
DOX-mediated calpain activity
Calpain is an intracellular calcium-dependent cysteine protease (Khorchid and Ikura, 2002; Vickers, 2017). Calpain exists as an inactive proenzyme in the cytosol. When intracellular calcium levels increase, the proenzyme form of calpain is converted to its active form, which cleaves cytoplasmic and nuclear substrates, leading to apoptosis (Momeni, 2011). Calpain activation has been implicated in myocardial injuries, including ischemia/reperfusion myocardial injury, pressure overload-induced cardiomyopathy, and heart failure (Li et al., 2009; Wang et al., 2018b; Wang et al., 2020). It has been demonstrated that DOX treatment causes calcium overload, which increases calpain activity (Szenczi et al., 2005; Emanuelov et al., 2010). Campos et al. established that the expression of active calpain increased in cardiomyocytes isolated from DOX-treated rats that showed dystrophin disruption in cardiac muscle (Campos et al., 2011). However, a calcium-blocking agent prevented calpain activation and preserved cardiac function. Another study also investigated the calpain-induced cardiomyopathy in rats injected with DOX (Min et al., 2015). Echocardiography analysis showed that DOX administration resulted in impaired cardiac function with decreased fractional shortening and thinning of the septal and left ventricular posterior wall. In contrast, rats treated with a calpain inhibitor prior to DOX injection attenuated cardiac dysfunction. In vitro experiments have also shown that DOX induces calpain activation in cardiomyocytes (Lim et al., 2004). Calpain activation in cardiomyocytes treated with DOX resulted in myofilament protein degradation and necrosis, while calpain inhibitors preserved the myofilament protein degradation (Lim et al., 2004). These studies indicate that calpain activation is one of the contributors that cause DOX-induced cardiomyopathy.
DOX-induced activation of caspase-3
Caspases are a family of cysteine protease enzymes that contribute to programmed cell death (Shi, 2004). In healthy cells, the caspases exist as dormant pro-enzymes. The caspases undergo cleavage events in response to a death-inducing signal to release a large subunit and a small subunit that heterodimerize into the active enzyme (Roy, 2000). It has been well known that caspase-3 is a crucial mediator of apoptosis by efficiently cleaving many key cellular proteins (Porter and Jänicke, 1999). In fact, caspase-3 is highly activated during the progression of multiple forms of heart diseases (Philipp et al., 2004; Putinski et al., 2013; Hashmi and Al-Salam, 2015; Min et al., 2015). Caspase-3 activation is capable of promoting the degradation of cardiac myofibrillar proteins, such as α-actin, α-actinin, and cTnT (Communal et al., 2002). Numerous studies have established that DOX treatment induces apoptosis through the activation of caspase-3 in cardiac muscle (Michihiko et al., 2006; Pointon et al., 2010; Min et al., 2015; Sun et al., 2016) and the inhibition of caspase-3 activity attenuates DOX-induced cardiotoxicity (Wang et al., 2001; Zhang et al., 2009a; Ma et al., 2013). Although the regulation of caspase-3 activity is complex and involves several interconnected signaling pathways, both extrinsic and intrinsic pathways have been postulated to activate caspase-3 in cardiac muscle treated with DOX. DOX treatment can induce the extrinsic apoptotic pathway via the upregulation of death receptors (Nakamura et al., 2000). Death ligands, including FasL and TNFα, bind to their receptors, leading to caspase-8 activation. Activated caspase-8 increases caspase-3 activity, resulting in cardiomyocyte apoptosis (Zhao and Zhang, 2017; Pan et al., 2021). It is also feasible that DOX contributes to caspase-3 activation through the intrinsic apoptotic pathways by stimulating ROS production in cardiac muscle. Indeed, increased cellular levels of ROS have been reported to activate caspase-3 in a variety of cell types, including cardiomyocytes (Powers et al., 2011; Yeh et al., 2019). DOX can activate the core apoptosis regulators, such as Bax and Bak in the cytosol. The activated Bax/Bak are translocated from the cytosol to the outer membrane of mitochondria, increasing mitochondrial membrane permeability. Cytochrome c in the inner membrane of mitochondria is released into the cytoplasm (An et al., 2009). Subsequently, cytochrome c activates caspase-9, resulting in the activation of caspase-3 in DOX-treated cardiomyocytes (Wei et al., 2022). These findings many explain how DOX treatment can induce cardiomyocyte apoptosis, which causes cardiotoxicity.
DOX-mediated autophagic signaling
Autophagy is a homeostatic process by which cytoplasmic components are degraded and recycled under normal and stress conditions through lysosomal pathways (Hansen et al., 2018). Autophagy has emerged as a major regulator of cardiac homeostasis and function. The level of autophagy in cardiac muscle is low under normal conditions, whereas it is upregulated in response to pathological stress (Nishida et al., 2009). Under physiological conditions, autophagy is essential for optimal cellular function and survival as it removes damaged or unwanted proteins and organelles. Under pathological conditions, autophagy may be stimulated to induce toxic effects (Smuder et al., 2013; Cui et al., 2021). Excessive autophagy activation can cause damage to organelles such as the mitochondria and endoplasmic reticulum, releasing compounds into the cytoplasm (e.g., cytochrome c and calcium) that can induce cell death (Nishida et al., 2008; Zhang et al., 2009b; Nishida et al., 2009). The activation of autophagy begins with the formation of a phagophore through a system of autophagy proteins (Atg proteins) (Smuder et al., 2013). The phagophore, also known as a double-membrane structure, sequesters bulk cytoplasmic components, such as abnormal intracellular proteins, excess or damaged organelles, and invading microorganisms. The phagophore expands to a sealed, double-membrane vesicle called the autophagosome (Hollenstein and Kraft, 2020). Beclin-1 plays an important role in the initial steps of autophagosome formation by mediating the localization of other Atg proteins to the phagophore (Gustafsson and Gottlieb, 2008). Elongation of the autophagosome requires the interaction of several Atg proteins (Mizushima et al., 1999; Smuder et al., 2013). Specifically, Atg4 is responsible for the cleavage of microtubule-associated protein 1A/1B-light chain 3 (LC3) to LC3-I (Yang et al., 2015). Cleaved LC3-I is conjugated by Atg7, Atg3, and Atg12-Atg5-Atg16L complex, leading to LC3-II synthesis, which is recruited to the autophagosomal membrane for the elongation (Ichimura et al., 2000). P62 is an autophagosome cargo protein that is used as a reporter of autophagy activity (Liu et al., 2016). After the formation of the autophagosome, cytoplasmic components are delivered to the lysosomes (Mizushima et al., 2002). The outer membrane of the autophagosome fuses with the lysosome to form an autolysosome (Mizushima et al., 2002). Hydrolases in lysosomes degrade the autophagosome-delivered components (Mizushima et al., 2002). Much evidence shows that the activation of autophagic signaling is associated with various forms of heart disease, including heart failure, ischemia-reperfusion injury, and metabolic cardiomyopathies (Shimomura et al., 2001; Kostin et al., 2003; Kanamori et al., 2015). This suggests that autophagy emerges as a new therapeutic target for heart disease. DOX treatment also induces autophagic signaling in cardiac muscle. Indeed, DOX-induced autophagosome and autolysosome accumulation were confirmed in vivo by using GFP-LC3 and mRFP-GFP-LC3 transgenic mice (Abdullah et al., 2019). In this study, both acute DOX treatment (20 mg/kg) and chronic DOX treatment (5 mg/kg every week for 4 weeks) exhibited time-dependent accumulation of LC3B II levels in cardiac muscle. Conversely, it has been reported that inhibition of autophagy via 3-methyladenine (3-MA) is sufficient to protect against DOX-induced autophagy, mitochondrial dysfunction, and cardiac contractile dysfunction (Lu et al., 2009; Xu et al., 2012). A proposed mechanism responsible for DOX-induced autophagy is that DOX administration results in damage to the mitochondria and induction of Beclin-1 expression, leading to accelerated autophagy and cardiomyopathy (Lu et al., 2009). Other groups also demonstrated that anti-apoptotic protein Bcl-2 can form a complex with Beclin-1 to inhibit autophagic apoptosis and autophagosome formation in cardiomyocyte (Wang et al., 2018a; Yu et al., 2022). However, DOX treatment increases the expression of Beclin-1 and decreases Bcl-2 protein expression, increasing the Beclin-1/Bcl-2 ratio, which indicates the activation of autophagic signaling and apoptosis (Smuder et al., 2013). These findings implicate that autophagy antagonists may represent a therapeutic approach for the preservation and/or maintenance of cardiac muscle function during and/or doxorubicin treatment.
Exercise protects against DOX-induced cardiotoxicity
Since DOX treatment causes cardiac toxicity and dysfunction, pharmacological cardioprotective strategies, including chemoprotective agents (Dexrazoxane, Mesna, and Amifostine) (Dragojevic-Simic et al., 2004; Aluise et al., 2011; Yu et al., 2020) and neurohormonal therapy (β-blockers, angiotensin receptor blockers, angiotensin-converting enzyme inhibitors) have been broadly explored (Ibrahim et al., 2009; Chang et al., 2015; Beheshti et al., 2016). Alternatively, exercise has been investigated as a non-pharmacological cardioprotective strategy in cancer patients. Several studies have demonstrated that exercise training or physical activity prevents or mitigates cardiac dysfunction from DOX-induced cardiotoxicity. The cardioprotective effects of exercise may improve the chemotherapy completion rate by managing dose-limiting toxicity. Many studies have consistently shown that exercise may result in the preservation of left ventricular contractile function through various mechanisms, including increased cardiac expression of antioxidant enzymes, mitochondrial function, and reduced proapoptotic signaling (Kim and Hwang, 2015; Morton et al., 2019). The following section will summarize the potential mechanisms of exercise-induced cardioprotection against cardiotoxicity following DOX administration.
Effect of exercise on antioxidant capacity in DOX-treated cardiac muscle
Antioxidant enzymes
Antioxidants are defined as substances that attenuate, delay, or prevent oxidation of another substance. Cellular antioxidants are compartmentalized in organelles and the cytoplasm to mitigate ROS and maintain redox balance (Powers and Jackson, 2008). ROS is known to decrease the activity of antioxidant enzymes, which are essential to maintain mitochondrial function by removing or neutralizing ROS produced from DOX. It has been established that exercise enhances antioxidant activities in cardiac and skeletal muscle by upregulating various cellular antioxidant systems (Ji et al., 1998; Muthusamy et al., 2012; Wang et al., 2016). Kim et al. reported that 2 weeks of aerobic exercise was sufficient to increase antioxidant enzyme activity, including superoxide dismutase (SOD) and catalase in the cardiac muscle of rats following DOX administration (Kim and Hwang, 2015). Similarly, 2 weeks of low-intensity treadmill exercise significantly increased glutathione peroxidase (GPx), attenuating left ventricular dysfunction in rats during DOX treatments. In contrast, sedentary rats treated with DOX displayed an increase in caspase-3 activity and consequently exhibited left ventricular dysfunction (Chicco et al., 2006). Exercise preconditioning also exhibited cardioprotective effects on antioxidant production in cardiac muscle. Animals subjected to 2 weeks of preconditioning prevented DOX accumulation in the mitochondria of cardiac muscle and attenuated mitochondrial ROS production, leading to the preservation of cardiac muscle contractility (Morton et al., 2019). Additionally, moderate treadmill exercise prior to DOX treatment increased the expression of the antioxidant enzymes GPx1, catalase, and manganese superoxide dismutase (Fasipe et al., 2021) in cardiomyocytes (Scott et al., 2011). Upregulation of the mentioned enzymes allows for the regulation of elevated ROS by neutralizing or removing the reactive forms and therefore, the preservation of mitochondrial function in DOX-treated cardiac muscle.
Non-enzymatic antioxidants
In addition to antioxidant enzymes, non-enzymatic antioxidants such as glutathione (GSH) have a critical role in the cardiac antioxidant defense system (Ascensao et al., 2007). The non-enzymatic antioxidants also interrupt and inactivate toxic free radical chain reactions. Ascensão et al. showed that DOX administration increases the amount of oxidized GSH (GSSG) in the cardiac tissue of mice, suggesting elevated oxidative byproduct release from cardiac tissue treated with DOX (Ascensão et al., 2005a). However, endurance swimming exercise reduced products of oxidative protein damage by 18.1% compared to the non-exercise group. This observation indicated that exercise induces cardiac redox adaptations that attenuate DOX-induced damage. Additionally, endurance-trained mice showed diminished levels of GSSH compared with non-trained mice. This indicates that exercise increases cardiac tissue GSH intake capacity and protects the myocardium from DOX-induced oxidative stress. Consistent with these findings, Demirel et al. found that exercise increased MnSOD and GSH concentrations, both of which remove oxidant precursors, providing antioxidant protection in cardiomyocytes (Demirel et al., 2001). In regard to mechanisms of exercise-induced non-enzymatic antioxidants, Wang et al. showed that acute exercise increased the expression of redox effector factor-1 (Ref1) and nuclear factor erythroid 2-related factor 2 (Nrf2) genes and proteins in skeletal muscle. The increased expression of these proteins was associated with mitochondrial H2O2 production and GSH and MnSOD activity (Wang et al., 2016). The authors suggest that the exercise-induced release of H2O2 stimulates the activation of the Ref1 signaling pathway. It is established that exercise-induced oxidative stress activates Nrf2, a redox-sensitive transcription factor that reduces the production of ROS by modulating the antioxidant defense systems (Muthusamy et al., 2012). Additionally, exercise increases the levels of ROS-generating NADPH oxidase-4 (Nox4) (Fasipe et al., 2021). An increase in Nox4 stimulates activation of Nrf2 which then increases the nuclear transcription of antioxidant genes, ultimately decreasing cardiomyocyte susceptibility to chemotoxic agents. These finding indicate that exercise produces adaptations in cardiac tissues that maintain the redox balance in cardiomyocytes. Specifically, exercise induces adaptations to the glutathione system and Ref1 signaling pathway, indicating the non-enzymatic antioxidants may serve as protective mechanisms against DOX toxicity.
Heat shock proteins
The effect of exercise-induced heat shock proteins (HSPs) on cardioprotection against DOX-induced cardiotoxicity has also been investigated. HSPs are a large family of molecular chaperones that play roles in cell survival and development by regulating protein maturation, refolding and degradation (Miller and Fort, 2018). HSPs acts as endogenous antioxidants against DOX-induced oxidative stress in the cardiac muscle. Exercise training has proven to increase the expression of HSPs, such as HSP-60, HSP-70, and HSP-72, in cardiac muscle, ameliorating the progression of DOX-induced cardiomyopathy (Ascensão et al., 2005a; Ascensão et al., 2005b; Chicco et al., 2005). Cardioprotection by HSPs may result from improved nuclear-encoded protein import into the mitochondrial matrix and protein folding ultimately reducing cellular proteolysis in cardiomyocytes (Ascensão et al., 2012). However, low-intensity exercise had no significant effect on HSPs or SOD isoforms (Chicco et al., 2006). Venkatakrishnan et al. revealed that high expression of HSP27 and its phosphorylation exhibited cardioprotective effects. This study found that phosphorylation at serine 15 and 85 of HSP 27 through p38 MAPK was a key mechanism in reduction of apoptosis in cardiac H9C2 cells treated DOX (Venkatakrishnan et al., 2006). Another study investigated the mechanisms of how HSPs prevent DOX-induced oxidative stress and cardiotoxicity (Fan et al., 2008). This study demonstrated that cardiac-specific overexpression of HSP20 attenuated acute DOX-triggered apoptosis in cardiomyocyte. This study found that HSP20 interacted with phosphorated Akt (serine 473), suggesting that the cardioprotective effect of HSP20 depends on the activity of Akt.
Together, these studies demonstrate that exercise training is an effective cardioprotective approach to prevent DOX-induced cardiotoxicity through the upregulation of antioxidant enzymes.
Effect of exercise on mitochondrial function in DOX-treated cardiac muscle
DOX accumulation in mitochondria
Negatively charged cardiolipin is located on the inner membrane of the mitochondria and is essential for the activation of enzymes in the electron transport chain (ETC) (Renu et al., 2018). Doxorubicin has a cationic charge giving it a strong affinity for cardiolipin and therefore binds in an irreversible reaction (Renu et al., 2018; Smuder, 2019). The resulting cardiolipin-DOX complex allows for DOX accumulation in the mitochondria of cardiomyocytes and reduces cardiolipin availability to activate enzymes essential to complex II and IV in the ETC. Additionally, the reduced availability of cardiolipin removes a crucial binding site for cytochrome c. Consequently, oxidative phosphorylation is reduced, and the mitochondrial membrane is compromised, further enabling cardiotoxicity (Schirone et al., 2022). These reactions make mitochondria one of the major targets of DOX and therefore its dysfunction is the hallmark of DOX-induced cardiotoxicity (Wu et al., 2022). Although the mechanisms are not clear, it is possible that the protective effect of exercise is through the preservation of ETC function.
DOX removal from mitochondria
To preserve mitochondrial function and increase oxidative capacity, the accumulation of DOX needs to be expelled from the mitochondria. Exercise has demonstrated mitochondrial protection against DOX-induced myotoxicity. Morton et al. reported that 2 weeks of endurance exercise significantly reduced the accumulation of DOX in cardiac mitochondria and conserved mitochondrial respiratory function (Morton et al., 2019). This study investigated the effect of exercise on the expression of ATP-binding cassette (ABC) transporters (Leonessa and Clarke, 2003). These transport proteins are the regulators of chemotherapeutic drugs in cells by ATP-dependent transmembrane efflux. The authors showed that endurance exercise significantly upregulated mitochondria-specific ABC transporters located in the inner and outer mitochondrial membranes in cardiac muscle following DOX treatment. This study suggested that exercise-induced increase in the expression of ABC transport proteins may be responsible for the protective effects of exercise on the heart against DOX-induced cardiotoxicity. It is possible that ABC transporters located in the mitochondria can export the DOX accumulated in the inner membrane (Cole, 2014; Renu et al., 2018). Specifically, the transporters ABCB6, ABCB7, ABCB8, and ABCB10 are found in the inner and outer membranes of the mitochondria (Morton et al., 2019). Morton et al. showed that 2 weeks of endurance exercise preconditioning upregulated the expression and activity of all four ABC transporters. In addition to their ability to export chemotherapeutics, ABC transporters have their own unique abilities. For example, ABCB8 is known to increase mitochondrial iron export therefore reducing ROS that forms when DOX interacts with iron (Morton et al., 2019; Wallace et al., 2020). Interestingly, it has been identified that the ABC transporter, multidrug resistance protein 1 (MRP1), releases the antioxidant GSH (Cole, 2014; Renu et al., 2018). As mentioned, GSH neutralizes free radicals such as ROS and therefore, plays a critical role in maintaining oxidative capacity through the antioxidant defense system (Octavia et al., 2012). Although the mechanisms have not been elucidated, it is established that ABC transport proteins are not limited to the direct export of DOX. In fact, in addition to DOX removal, ABC transport proteins preserve mitochondrial function and reduce ROS to preserve mitochondrial function. Together, these studies suggest that one of the mechanisms by which exercise provides its protective effect may be by reducing the overall DOX present in the mitochondria.
Mitochondrial permeability transition
A characteristic of DOX-induced toxicity is a reduction in the mitochondrial calcium loading capacity. DOX toxicity increases calcium and phosphate overload and oxidative stress, leading to mitochondrial swelling and damage to the outer mitochondrial membrane increasing the susceptibility to permeability transition pore opening (mPTP). (Zoratti et al., 2005; Ascensão et al., 2011). When stimulated, the mPTP is responsible for the release of calcium and pro-apoptotic proteins, worsening cytotoxicity. However, exercise has been shown to defend against myocardial injury through its effect on mPTP. Ascensão et al. investigated acute endurance exercise as an intervention and showed the attenuation of calcium-induced mPTP opening in DOX-treated cardiac muscle (Ascensão et al., 2011) and chronic endurance exercise improved mitochondrial calcium tolerance (Ascensão et al., 2005b). These observations demonstrate the amelioration of mitochondrial dysfunction during and after DOX treatment. Together, these reports indicate that exercise training protects the heart from DOX-induced cardiotoxicity by protecting cardiac mitochondria-drive mPTP opening and consequently interfering with the magnitude of apoptotic pathways.
Effect of exercise on DOX-induced proteolytic systems
FOXO signaling pathway
Exercise has been shown to regulate the activation of proteolytic systems. This protective effect of exercise on the attenuation of proteolytic systems is associated with the reduced Forkhead-Box O (FOXO) signaling pathway (Kavazis et al., 2014). The increased FOXO nuclear translocation causes the amplification of FOXO target genes, such atrogin-1 and MuRF-1, leading to cardiac muscle atrophy (Bodine et al., 2001; Gomes et al., 2001). DOX treatment has been shown to activate FOXO signaling through phosphorylation. Xia et al. demonstrated that DOX increases the phosphorylation of FOXO1 at Ser-249 through cyclin-dependent kinase 2 (CDK2) (Xia et al., 2020). The activated FOXO1 stimulates the transcription of proapoptotic target gene Bcl-2-interacting mediator of cell death (Bim) in cardiac muscle following DOX treatment. However, a FOXO1 inhibitor or FOXO1-specific siRNAs protected cardiomyocytes against DOX-induced apoptosis. As acute endurance exercise attenuated the activation of FOXO1 and FOXO3 in cardiac muscle following DOX administration, decreasing the activity of muscle-specific E3 ligases and ultimately apoptotic activity (Kavazis et al., 2014) could be one of the potential mechanisms involved in exercise-induced regulation of proteolytic systems, contributing to mitigating the toxicity caused by DOX treatment. It is also possible that the reduced expression of FOXO target proteins may be due to the exercise-induced upregulation of PGC-1 alpha (PGC-1α) (Sandri et al., 2006; Kavazis et al., 2014). Sandri et al. showed that overexpression of PGC-1α resulted in a reduction in atrogin-1 and MuRF-1, reducing the capacity of FOXO3 (Sandri et al., 2006). Further, downregulation of PGC-1 α has been associated with skeletal muscle atrophy (Sandri et al., 2006). Therefore, the upregulation of PGC-1 α expression may be partially responsible for the protective effect of exercise by reducing FOXO activity and inhibiting the target gene MuRF-1 (Kavazis et al., 2014).
Autophagic signaling
Exercise intervention also regulates autophagic signaling in hearts from DOX treated animals. Using transmission electron microscopy analysis, Wang F. et al. showed that 2 weeks of acute treadmill exercise ameliorates an increase in the number of autophagosomes and abnormal mitochondria in the heart following DOX treatment (Wang et al., 2021). Another study also revealed that exercise preconditioning inhibits DOX-induced cardiac autophagy/lysosomal system (Smuder et al., 2013). Acute preconditioning attenuated the expression of Beclin-1 and increased anti-apoptotic protein Bcl-2, thereby inhibiting autophagosome initiation. Additionally, acute preconditioning inhibited the conjugation of Atg12 to Atg5 production, which is required for the elongation of the autophagosome. Eventually, the acute preconditioning suppressed lysosomal proteases, including cathepsin B, D, and L in hearts from DOX treated animals (Smuder et al., 2013).
DOX-induced apoptosis
Exercise training also prevents DOX-induced apoptosis in the heart. Alihemmati et al. exercised male Wistar rats with alternating intervals of high and low training for 1 h a day, 5 days a week, for 6 weeks using a rodent treadmill. The interval training cycle lasted 7 min with high intensity exercise (85–90% VO2max) for 4 min and the low intensity exercise (65–75% VO2max) for 3 min. After the 6 weeks of interval exercise training, the rats received 20 mg/kg of DOX (Alihemmati et al., 2019). This study showed that the 6 weeks of interval training reduces Bax protein expression and increases Bcl-2 protein expression, leading to a decreased Bax/Bcl-2 ratio in hearts from DOX treated animals, thereby reducing apoptosis. The authors also confirmed the effect of interval training on cardiomyocyte apoptosis through TUNEL staining. The 6 weeks of interval training reduced TUNEL-positive apoptotic cells in hearts from DOX treated animals. The authors also examined the activity of microRNAs that modulate the damage pathways in cardiomyocytes in response to heart disease. The interval training attenuated the overexpression of microRNA-499 in hearts from DOX treated animals, which is a potential biomarker for apoptotic effects in cardiomyocytes. Exercise has been shown to inhibit both intrinsic and extrinsic apoptotic pathways in DOX-treated animals. In this regard, Magalhães et al. reported that exercise preconditioning prevents the expression of caspase-9 and caspase-3 proteins in animals treated with DOX, indicating that exercise attenuates the DOX-induced intrinsic apoptotic pathway (Magalhães et al., 2017). Another study revealed that treating animals with DOX significantly increased the activity of both caspase-8 and caspase-9 in cardiac muscle, whereas 12 weeks of endurance treadmill training prevented the increases in cardiac muscle following DOX treatment, resulting in decreased caspase-3 activity (Marques-Aleixo et al., 2018). Collectively, these studies suggest that exercise training can prevent cardiac muscle degradation by alleviating the DOX-induced proteolytic systems (Figure 4).
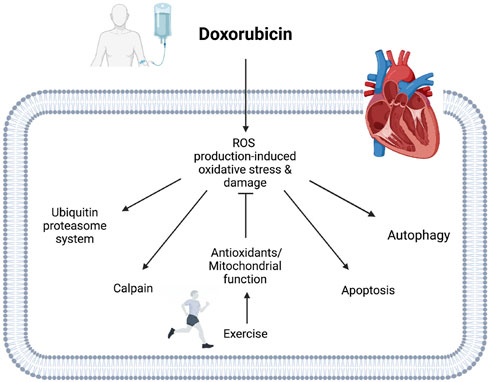
FIGURE 4. Potential mechanisms of exercise-induced cardioprotective effects against DOX cardiotoxicity. Exercise enhances cellular antioxidant defensive systems, including antioxidant enzyme, non-enzymatic antioxidant, and HSPs in hearts treated with DOX. Exercise also prevents DOX accumulation in mitochondria, improving mitochondrial function. Activated antioxidant systems and enhanced mitochondrial function mitigate DOX-induced oxidative stress and damage in cardiac muscle. The protective mechanisms of exercise can protect cardiac muscle by attenuating DOX-induced proteolytic systems.
Conclusion
Cancer therapy has significantly improved, and as a result, the lifespan of cancer survivors has increased. Therefore, it is important to develop countermeasures to prevent chemotherapy-induced cardiotoxicity that impairs the quality of life for survivors. Various mechanisms are involved in DOX-induced cardiotoxicity. Given the abundance of reports indicating that exercise can result in a protective phenotype of the heart against the cardiotoxicity, exercise therapy as a non-pharmacological intervention can be an effective clinical approach to prevent or reverse the side effects of chemotherapy.
Investigations into the mechanisms responsible for exercise-induced cardioprotection against the cardiotoxicity from chemotherapy are still in the early stages. Thus, further research is required to provide comprehensive evidence considering various exercise type, intensity, and duration to develop exercise training protocols for cancer patients and survivors.
Author contributions
SG, AL, JC, MN, SB, JB, SH, and KM contributed to conception and design of the article. SG and KM wrote the manuscript. SG, AL, JC, SH, and KM edited the manuscript. SG and SH created figures. All authors read and approved the submitted version.
Funding
This report was supported by the American Federation for Aging Research (AGR DT 07-2502019 and AGR DTD 09-15-2021 to SH).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdel-Qadir, H., Bobrowski, D., Zhou, L., Austin, P. C., Calvillo-Argüelles, O., Amir, E., et al. (2021). Statin exposure and risk of heart failure after anthracycline-or trastuzumab-based chemotherapy for early breast cancer: A propensity score‒matched cohort study. J. Am. Heart Assoc. 10, e018393. doi:10.1161/JAHA.119.018393
Abdullah, C. S., Alam, S., Aishwarya, R., Miriyala, S., Bhuiyan, M. A. N., Panchatcharam, M., et al. (2019). Doxorubicin-induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration. Sci. Rep. 9, 2002–2020. doi:10.1038/s41598-018-37862-3
Alihemmati, A., Ebadi, F., Moghadaszadeh, M., Asadi, M., Zare, P., and Badalzadeh, R. (2019). Effects of high-intensity interval training on the expression of microRNA-499 and pro-and anti-apoptotic genes in doxorubicin-cardiotoxicity in rats. J. Electrocardiol. 55, 9–15. doi:10.1016/j.jelectrocard.2019.02.009
Aluise, C. D., Miriyala, S., Noel, T., Sultana, R., Jungsuwadee, P., Taylor, T. J., et al. (2011). 2-Mercaptoethane sulfonate prevents doxorubicin-induced plasma protein oxidation and TNF-α release: Implications for the reactive oxygen species-mediated mechanisms of chemobrain. Free Radic. Biol. Med. 50, 1630–1638. doi:10.1016/j.freeradbiomed.2011.03.009
An, J., Li, P., Li, J., Dietz, R., and Donath, S. (2009). ARC is a critical cardiomyocyte survival switch in doxorubicin cardiotoxicity. J. Mol. Med. 87, 401–410. doi:10.1007/s00109-008-0434-z
Angsutararux, P., Luanpitpong, S., and Issaragrisil, S. (2015). Chemotherapy-induced cardiotoxicity: Overview of the roles of oxidative stress. Oxid. Med. Cell Longev. 2015, 795602. doi:10.1155/2015/795602
Arcamone, F., Franceschi, G., Penco, S., and Selva, A. (1969). Adriamycin (14-hydroxydaunomycin), a novel antitumor antibiotic. Tetrahedron Lett. 10, 1007–1010. doi:10.1016/s0040-4039(01)97723-8
Arcamone, F. M. (2009). Fifty years of chemical research at Farmitalia. Chemistry–A Eur. J. 15, 7774–7791. doi:10.1002/chem.200900292
Ascensao, A., Ferreira, R., and Magalhaes, J. (2007). Exercise-induced cardioprotection--biochemical, morphological and functional evidence in whole tissue and isolated mitochondria. Int. J. Cardiol. 117, 16–30. doi:10.1016/j.ijcard.2006.04.076
Ascensão, A., Lumini-Oliveira, J., Machado, N. G., Ferreira, R. M., Gonçalves, I. O., Moreira, A. C., et al. (2011). Acute exercise protects against calcium-induced cardiac mitochondrial permeability transition pore opening in doxorubicin-treated rats. Clin. Sci. 120, 37–49. doi:10.1042/CS20100254
Ascensão, A., Magalhães, J., Soares, J., Ferreira, R., Neuparth, M., Marques, F., et al. (2005a). Endurance training attenuates doxorubicin-induced cardiac oxidative damage in mice. Int. J. Cardiol. 100, 451–460. doi:10.1016/j.ijcard.2004.11.004
Ascensão, A., Magalhães, J., Soares, J. M., Ferreira, R., Neuparth, M. J., Marques, F., et al. (2005b). Moderate endurance training prevents doxorubicin-induced in vivo mitochondriopathy and reduces the development of cardiac apoptosis. Am. J. Physiology-Heart Circulatory Physiology 289, H722–H731. doi:10.1152/ajpheart.01249.2004
Ascensão, A., Oliveira, P. J., and Magalhães, J. (2012). Exercise as a beneficial adjunct therapy during doxorubicin treatment—role of mitochondria in cardioprotection. Int. J. Cardiol. 156, 4–10. doi:10.1016/j.ijcard.2011.05.060
Ashley, N., and Poulton, J. (2009). Mitochondrial DNA is a direct target of anti-cancer anthracycline drugs. Biochem. Biophys. Res. Commun. 378, 450–455. doi:10.1016/j.bbrc.2008.11.059
Austin, C. A., and Marsh, K. L. (1998). Eukaryotic DNA topoisomerase II beta. Bioessays 20, 215–226. doi:10.1002/(SICI)1521-1878(199803)20:3<215::AID-BIES5>3.0.CO;2-Q
Beheshti, A. T., Toroghi, H. M., Hosseini, G., Zarifian, A., Shandiz, F. H., and Fazlinezhad, A. (2016). Carvedilol administration can prevent doxorubicin-induced cardiotoxicity: A double-blind randomized trial. Cardiology 134, 47–53. doi:10.1159/000442722
Berthiaume, J. M., and Wallace, K. B. (2007). Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell Biol. Toxicol. 23, 15–25. doi:10.1007/s10565-006-0140-y
Bodine, S. C., Latres, E., Baumhueter, S., Lai, V. K.-M., Nunez, L., Clarke, B. A., et al. (2001). Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294, 1704–1708. doi:10.1126/science.1065874
Bonfante, V., Bonadonna, G., Villani, F., and Martini, A. (1980). Preliminary clinical experience with 4’-epidoxorubicin in advanced human neoplasia. Cancer chemo-and immunopharmacology. Springer.
Buzdar, A. U., Marcus, C., Blumenschein, G. R., and Smith, T. L. (1985). Early and delayed clinical cardiotoxicity of doxorubicin. Cancer 55, 2761–2765. doi:10.1002/1097-0142(19850615)55:12<2761::aid-cncr2820551206>3.0.co;2-p
Campos, E. C., O'Connell, J. L., Malvestio, L. M., Romano, M. M. D., Ramos, S. G., Celes, M. R. N., et al. (2011). Calpain-mediated dystrophin disruption may be a potential structural culprit behind chronic doxorubicin-induced cardiomyopathy. Eur. J. Pharmacol. 670, 541–553. doi:10.1016/j.ejphar.2011.09.021
Capelôa, T., Benyahia, Z., Zampieri, L. X., Blackman, M. C., and Sonveaux, P. (2020). Metabolic and non-metabolic pathways that control cancer resistance to anthracyclines. Seminars in cell & developmental biology. Elsevier, 181–191.
Capranico, G., Tinelli, S., Austin, C. A., Fisher, M. L., and Zunino, F. (1992). Different patterns of gene expression of topoisomerase II isoforms in differentiated tissues during murine development. Biochimica Biophysica Acta (BBA)-Gene Struct. Expr. 1132, 43–48. doi:10.1016/0167-4781(92)90050-a
Chaires, J. B. (1990). Biophysical chemistry of the daunomycin-DNA interaction. Biophys. Chem. 35, 191–202. doi:10.1016/0301-4622(90)80008-u
Chang, S.-A., Lim, B.-K., Lee, Y. J., Hong, M.-K., Choi, J.-O., and Jeon, E.-S. (2015). A novel angiotensin type I receptor antagonist, fimasartan, prevents doxorubicin-induced cardiotoxicity in rats. J. Korean Med. Sci. 30, 559–568. doi:10.3346/jkms.2015.30.5.559
Chicco, A. J., Hydock, D. S., Schneider, C. M., and Hayward, R. (2006). Low-intensity exercise training during doxorubicin treatment protects against cardiotoxicity. J. Appl. physiology 100, 519–527. doi:10.1152/japplphysiol.00148.2005
Chicco, A. J., Schneider, C. M., and Hayward, R. (2005). Voluntary exercise protects against acute doxorubicin cardiotoxicity in the isolated perfused rat heart. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289, R424–r431. doi:10.1152/ajpregu.00636.2004
Cole, S. P. (2014). Multidrug resistance protein 1 (MRP1, ABCC1), a "multitasking" ATP-binding cassette (ABC) transporter. J. Biol. Chem. 289, 30880–30888. doi:10.1074/jbc.R114.609248
Communal, C., Sumandea, M., De Tombe, P., Narula, J., Solaro, R. J., and Hajjar, R. J. (2002). Functional consequences of caspase activation in cardiac myocytes. Proc. Natl. Acad. Sci. U. S. A. 99, 6252–6256. doi:10.1073/pnas.092022999
Cornarotti, M., Tinelli, S., Willmore, E., Zunino, F., Fisher, L. M., Austin, C. A., et al. (1996). Drug sensitivity and sequence specificity of human recombinant DNA topoisomerases IIalpha (p170) and IIbeta (p180). Mol. Pharmacol. 50, 1463–1471.
Cui, J., Zhao, S., Li, Y., Zhang, D., Wang, B., Xie, J., et al. (2021). Regulated cell death: Discovery, features and implications for neurodegenerative diseases. Cell Commun. Signal. 19, 120–129. doi:10.1186/s12964-021-00799-8
Dasuri, K., Zhang, L., and Keller, J. N. (2013). Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic. Biol. Med. 62, 170–185. doi:10.1016/j.freeradbiomed.2012.09.016
Demirel, H. A., Powers, S. K., Zergeroglu, M. A., Shanely, R. A., Hamilton, K., Coombes, J., et al. 2001. Short-term exercise improves myocardial tolerance to in vivo ischemia-reperfusion in the rat. J. Appl. Physiol., 91, 2205–2212. doi:10.1152/jappl.2001.91.5.2205
Derouiche, F., Bôle-Feysot, C., Naïmi, D., and Coëffier, M. (2014). Hyperhomocysteinemia-induced oxidative stress differentially alters proteasome composition and activities in heart and aorta. Biochem. Biophysical Res. Commun. 452, 740–745. doi:10.1016/j.bbrc.2014.08.141
Devasagayam, T. P., Tilak, J. C., Boloor, K. K., Sane, K. S., Ghaskadbi, S. S., and Lele, R. D. (2004). Free radicals and antioxidants in human health: Current status and future prospects. J. Assoc. Physicians India 52, 794–804.
Deweese, J. E., and Osheroff, N. (2009). The DNA cleavage reaction of topoisomerase II: Wolf in sheep's clothing. Nucleic Acids Res. 37, 738–748. doi:10.1093/nar/gkn937
Dimarco, A., Soldati, M., Fioretti, A., Dasdia, T., and Fioretti, A. (1963). The activity of daunomycin, a new antitumor antibiotic, on normal and tumor cells growing « in vitro ». Tumori 49, 235–251. doi:10.1177/030089166304900402
Dragojevic-Simic, V. M., Dobric, S. L., Bokonjic, D. R., Vucinic, Z. M., Sinovec, S. M., Jacevic, V. M., et al. (2004). Amifostine protection against doxorubicin cardiotoxicity in rats. Anti-cancer drugs 15, 169–178. doi:10.1097/00001813-200402000-00011
Emanuelov, A. K., Shainberg, A., Chepurko, Y., Kaplan, D., Sagie, A., Porat, E., et al. (2010). Adenosine A3 receptor-mediated cardioprotection against doxorubicin-induced mitochondrial damage. Biochem. Pharmacol. 79, 180–187. doi:10.1016/j.bcp.2009.08.010
Escherich, G., Zimmermann, M., Janka-Schaub, G., and Group, C. S. (2013). Doxorubicin or daunorubicin given upfront in a therapeutic window are equally effective in children with newly diagnosed acute lymphoblastic leukemia. A randomized comparison in trial CoALL 07-03. Pediatr. blood cancer 60, 254–257. doi:10.1002/pbc.24273
Fan, G.-C., Zhou, X., Wang, X., Song, G., Qian, J., Nicolaou, P., et al. (2008). Heat shock protein 20 interacting with phosphorylated Akt reduces doxorubicin-triggered oxidative stress and cardiotoxicity. Circulation Res. 103, 1270–1279. doi:10.1161/CIRCRESAHA.108.182832
Fasipe, B., Li, S., and Laher, I. (2021). Harnessing the cardiovascular benefits of exercise: Are Nrf2 activators useful? Sports Med. Health Sci. 3, 70–79. doi:10.1016/j.smhs.2021.04.002
Fogli, S., Nieri, P., and Cristina Breschi, M. (2004). The role of nitric oxide in anthracycline toxicity and prospects for pharmacologic prevention of cardiac damage. FASEB J. 18, 664–675. doi:10.1096/fj.03-0724rev
Frederick, C. A., Williams, L. D., Ughetto, G., Van Der Marel, G. A., Van Boom, J. H., Rich, A., et al. (1990). Structural comparison of anticancer drug-DNA complexes: Adriamycin and daunomycin. Biochemistry 29, 2538–2549. doi:10.1021/bi00462a016
Fujiwara, A., Hoshino, T., and Westley, J. W. (1985). Anthracycline antibiotics. Crit. Rev. Biotechnol. 3, 133–157. doi:10.3109/07388558509150782
Galvao, D., Taaffe, D., Spry, N., Joseph, D., Turner, D., and Newton, R. (2009). Reduced muscle strength and functional performance in men with prostate cancer undergoing androgen suppression: A comprehensive cross-sectional investigation. Prostate cancer prostatic Dis. 12, 198–203. doi:10.1038/pcan.2008.51
Gewirtz, D. A. (1999). A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem. Pharmacol. 57, 727–741. doi:10.1016/s0006-2952(98)00307-4
Gilleron, M., Marechal, X., Montaigne, D., Franczak, J., Neviere, R., and Lancel, S. (2009). NADPH oxidases participate to doxorubicin-induced cardiac myocyte apoptosis. Biochem. Biophys. Res. Commun. 388, 727–731. doi:10.1016/j.bbrc.2009.08.085
Gomes, M. D., Lecker, S. H., Jagoe, R. T., Navon, A., and Goldberg, A. L. (2001). Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. 98, 14440–14445. doi:10.1073/pnas.251541198
Grune, T., Merker, K., Sandig, G., and Davies, K. J. (2003). Selective degradation of oxidatively modified protein substrates by the proteasome. Biochem. biophysical Res. Commun. 305, 709–718. doi:10.1016/s0006-291x(03)00809-x
Gustafsson, Å. B., and Gottlieb, R. A. (2008). Recycle or die: The role of autophagy in cardioprotection. J. Mol. Cell. Cardiol. 44, 654–661. doi:10.1016/j.yjmcc.2008.01.010
Gyulkhasyan, T., Hakobyan, T., Parikh, A., Peketi, P., Enrick, M., Shockling, L., et al. (2019). Doxorubicin-induced cardiomyopathy: Prevention and treatment by a coronary specific vasodilator. FASEB J. 33, 685. doi:10.1096/fasebj.2019.33.1_supplement.685.14
Hansen, M., Rubinsztein, D. C., and Walker, D. W. (2018). Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 19, 579–593. doi:10.1038/s41580-018-0033-y
Hashmi, S., and Al-Salam, S. (2015). Acute myocardial infarction and myocardial ischemia-reperfusion injury: A comparison. Int. J. Clin. Exp. Pathol. 8, 8786–8796.
Hollenstein, D. M., and Kraft, C. (2020). Autophagosomes are formed at a distinct cellular structure. Curr. Opin. Cell Biol. 65, 50–57. doi:10.1016/j.ceb.2020.02.012
Hu, J., Klein, J. D., Du, J., and Wang, X. H. (2008). Cardiac muscle protein catabolism in diabetes mellitus: Activation of the ubiquitin-proteasome system by insulin deficiency. Endocrinology 149, 5384–5390. doi:10.1210/en.2008-0132
Hyatt, H., Deminice, R., Yoshihara, T., and Powers, S. K. (2019). Mitochondrial dysfunction induces muscle atrophy during prolonged inactivity: A review of the causes and effects. Archives Biochem. biophysics 662, 49–60. doi:10.1016/j.abb.2018.11.005
Ibrahim, M. A., Ashour, O. M., Ibrahim, Y. F., El-Bitar, H. I., Gomaa, W., and Abdel-Rahim, S. R. (2009). Angiotensin-converting enzyme inhibition and angiotensin AT1-receptor antagonism equally improve doxorubicin-induced cardiotoxicity and nephrotoxicity. Pharmacol. Res. 60, 373–381. doi:10.1016/j.phrs.2009.05.007
Ichimura, Y., Kirisako, T., Takao, T., Satomi, Y., Shimonishi, Y., Ishihara, N., et al. (2000). A ubiquitin-like system mediates protein lipidation. Nature 408, 488–492. doi:10.1038/35044114
Jeyaseelan, R., Poizat, C., Wu, H.-Y., and Kedes, L. (1997). Molecular mechanisms of doxorubicin-induced cardiomyopathy: Selective suppression of reiske iron-sulfur protein, ADP/ATP translocase, and phosphofructokinase genes is associated with ATP depletion in rat cardiomyocytes. J. Biol. Chem. 272, 5828–5832. doi:10.1074/jbc.272.9.5828
Ji, L. L., Leeuwenburgh, C., Leichtweis, S., Gore, M., Fiebig, R., Hollander, J., et al. (1998). Oxidative stress and aging. Role of exercise and its influences on antioxidant systems. Ann. N. Y. Acad. Sci. 854, 102–117. doi:10.1111/j.1749-6632.1998.tb09896.x
Jiang, C., Deng, L., Karr, M. A., Wen, Y., Wang, Q., Perimbeti, S., et al. (2022). Chronic comorbid conditions among adult cancer survivors in the United States: Results from the National Health Interview Survey, 2002-2018. Cancer 128, 828–838. doi:10.1002/cncr.33981
Kamphuis, J. A., Linschoten, M., Cramer, M. J., Doevendans, P. A., Asselbergs, F. W., and Teske, A. J. (2020). Early-and late anthracycline-induced cardiac dysfunction: Echocardiographic characterization and response to heart failure therapy. Cardio-oncology 6, 23–13. doi:10.1186/s40959-020-00079-3
Kanamori, H., Takemura, G., Goto, K., Tsujimoto, A., Mikami, A., Ogino, A., et al. (2015). Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy 11, 1146–1160. doi:10.1080/15548627.2015.1051295
Karadurmus, L., Dogan-Topal, B., Kurbanoglu, S., Shah, A., and Ozkan, S. A. (2021). The interaction between DNA and three intercalating anthracyclines using electrochemical DNA nanobiosensor based on metal nanoparticles modified screen-printed electrode. Micromachines (Basel) 12, 1337. doi:10.3390/mi12111337
Kavazis, A. N., Smuder, A. J., and Powers, S. K. (2014). Effects of short-term endurance exercise training on acute doxorubicin-induced FoxO transcription in cardiac and skeletal muscle. J. Appl. physiology 117, 223–230. doi:10.1152/japplphysiol.00210.2014
Khorchid, A., and Ikura, M. (2002). How calpain is activated by calcium. Nat. Struct. Biol. 9, 239–241. doi:10.1038/nsb0402-239
Kim, E.-J., and Hwang, S. (2015). Effects of aerobic exercise on antioxidants in rat models with cardiomyopathy. Phys. Ther. rehabilitation Sci. 4, 17–21. doi:10.14474/ptrs.2015.4.1.17
Knowles, R. G., and Moncada, S. (1994). Nitric oxide synthases in mammals. Biochem. J. 298 (2), 249–258. doi:10.1042/bj2980249
Kostin, S., Pool, L., Elsässer, A., Hein, S., Drexler, H. C., Arnon, E., et al. (2003). Myocytes die by multiple mechanisms in failing human hearts. Circ. Res. 92, 715–724. doi:10.1161/01.RES.0000067471.95890.5C
Lawrence, J. W., Darkin-Rattray, S., Xie, F., Neims, A. H., and Rowe, T. C. (1993). 4-Quinolones cause a selective loss of mitochondrial DNA from mouse L1210 leukemia cells. J. Cell. Biochem. 51, 165–174. doi:10.1002/jcb.240510208
Lebrecht, D., and Walker, U. A. (2007). Role of mtDNA lesions in anthracycline cardiotoxicity. Cardiovasc Toxicol. 7, 108–113. doi:10.1007/s12012-007-0009-1
Lee, B. H., Goodenday, L. S., Muswick, G. J., Yasnoff, W. A., Leighton, R. F., and Skell, R. T. (1987). Alterations in left ventricular diastolic function with doxorubicin therapy. J. Am. Coll. Cardiol. 9, 184–188. doi:10.1016/s0735-1097(87)80099-2
Lee, Y., Kwon, I., Jang, Y., Cosio-Lima, L., and Barrington, P. (2020). Endurance exercise attenuates doxorubicin-induced cardiotoxicity. Med. Sci. Sports Exerc. 52, 25–36. doi:10.1249/MSS.0000000000002094
Leonessa, F., and Clarke, R. (2003). ATP binding cassette transporters and drug resistance in breast cancer. Endocrine-related cancer 10, 43–73. doi:10.1677/erc.0.0100043
Levick, S. P., Soto-Pantoja, D. R., Bi, J., Hundley, W. G., Widiapradja, A., Manteufel, E. J., et al. (2019). Doxorubicin-induced myocardial fibrosis involves the neurokinin-1 receptor and direct effects on cardiac fibroblasts. Heart Lung Circ. 28, 1598–1605. doi:10.1016/j.hlc.2018.08.003
Li, T.-K., and Liu, L. F. (2001). Tumor cell death induced by topoisomerase-targeting drugs. Annu. Rev. Pharmacol. Toxicol. 41, 53–77. doi:10.1146/annurev.pharmtox.41.1.53
Li, Y., Arnold, J. M. O., Pampillo, M., Babwah, A. V., and Peng, T. (2009). Taurine prevents cardiomyocyte death by inhibiting NADPH oxidase-mediated calpain activation. Free Radic. Biol. Med. 46, 51–61. doi:10.1016/j.freeradbiomed.2008.09.025
Lim, C. C., Zuppinger, C., Guo, X., Kuster, G. M., Helmes, M., Eppenberger, H. M., et al. (2004). Anthracyclines induce calpain-dependent titin proteolysis and necrosis in cardiomyocytes. J. Biol. Chem. 279, 8290–8299. doi:10.1074/jbc.M308033200
Lin, J., Fang, L., Li, H., Li, Z., Lyu, L., Wang, H., et al. (2019). Astragaloside IV alleviates doxorubicin induced cardiomyopathy by inhibiting NADPH oxidase derived oxidative stress. Eur. J. Pharmacol. 859, 172490. doi:10.1016/j.ejphar.2019.172490
Liu, W. J., Ye, L., Huang, W. F., Guo, L. J., Xu, Z. G., Wu, H. L., et al. (2016). p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol. Biol. Lett. 21, 29. doi:10.1186/s11658-016-0031-z
Lu, L., Wu, W., Yan, J., Li, X., Yu, H., and Yu, X. (2009). Adriamycin-induced autophagic cardiomyocyte death plays a pathogenic role in a rat model of heart failure. Int. J. Cardiol. 134, 82–90. doi:10.1016/j.ijcard.2008.01.043
Ma, J., Wang, Y., Zheng, D., Wei, M., Xu, H., and Peng, T. (2013). Rac1 signalling mediates doxorubicin-induced cardiotoxicity through both reactive oxygen species-dependent and-independent pathways. Cardiovasc. Res. 97, 77–87. doi:10.1093/cvr/cvs309
Magalhães, J., Ascensão, A., Padrão, A. I., Aleixo, I. M., Santos-Alves, E., Rocha-Rodrigues, S., et al. (2017). Can exercise training counteract doxorubicin-induced oxidative damage of testis proteome? Toxicol. Lett. 280, 57–69. doi:10.1016/j.toxlet.2017.08.010
Marques-Aleixo, I., Santos-Alves, E., Torrella, J., Oliveira, P., Magalhães, J., and Ascensão, A. (2018). Exercise and doxorubicin treatment modulate cardiac mitochondrial quality control signaling. Cardiovasc. Toxicol. 18, 43–55. doi:10.1007/s12012-017-9412-4
Mcgowan, J. V., Chung, R., Maulik, A., Piotrowska, I., Walker, J. M., and Yellon, D. M. (2017). Anthracycline chemotherapy and cardiotoxicity. Cardiovasc. drugs Ther. 31, 63–75. doi:10.1007/s10557-016-6711-0
Mclaughlin, D., Zhao, Y., O'Neill, K. M., Edgar, K. S., Dunne, P. D., Kearney, A. M., et al. (2017). Signalling mechanisms underlying doxorubicin and Nox2 NADPH oxidase-induced cardiomyopathy: Involvement of mitofusin-2. Br. J. Pharmacol. 174, 3677–3695. doi:10.1111/bph.13773
Michihiko, U., Yoshihiko, K., Koh-Ichi, Y., Nobuyuki, M., Motoyuki, I., Takashi, M., et al. (2006). Doxorubicin induces apoptosis by activation of caspase-3 in cultured cardiomyocytes in vitro and rat cardiac ventricles in vivo. J. Pharmacol. Sci. 101, 151–158. doi:10.1254/jphs.fp0050980
Miller, D. J., and Fort, P. E. (2018). Heat shock proteins regulatory role in neurodevelopment. Front. Neurosci. 12, 821. doi:10.3389/fnins.2018.00821
Min, K., Kwon, O. S., Smuder, A. J., Wiggs, M. P., Sollanek, K. J., Christou, D. D., et al. (2015). Increased mitochondrial emission of reactive oxygen species and calpain activation are required for doxorubicin-induced cardiac and skeletal muscle myopathy. J. physiology 593, 2017–2036. doi:10.1113/jphysiol.2014.286518
Minev, B. (2011). Cancer management in man: Chemotherapy, biological therapy, hyperthermia and supporting measures. Springer Science & Business Media.
Minotti, G., Menna, P., Salvatorelli, E., Cairo, G., and Gianni, L. (2004). Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 56, 185–229. doi:10.1124/pr.56.2.6
Mizushima, N., Noda, T., and Ohsumi, Y. (1999). Apg16p is required for the function of the Apg12p–Apg5p conjugate in the yeast autophagy pathway. EMBO J. 18, 3888–3896. doi:10.1093/emboj/18.14.3888
Mizushima, N., Ohsumi, Y., and Yoshimori, T. (2002). Autophagosome formation in mammalian cells. Cell Struct. Funct. 27, 421–429. doi:10.1247/csf.27.421
Montalvo, R. N., Doerr, V., Min, K., Szeto, H. H., and Smuder, A. J. (2020). Doxorubicin-induced oxidative stress differentially regulates proteolytic signaling in cardiac and skeletal muscle. Am. J. Physiology-Regulatory, Integr. Comp. Physiology 318, R227–R233. doi:10.1152/ajpregu.00299.2019
Morton, A. B., Mor Huertas, A., Hinkley, J. M., Ichinoseki-Sekine, N., Christou, D. D., and Smuder, A. J. (2019). Mitochondrial accumulation of doxorubicin in cardiac and diaphragm muscle following exercise preconditioning. Mitochondrion 45, 52–62. doi:10.1016/j.mito.2018.02.005
Mukai, H., Uemura, Y., Akabane, H., Watanabe, T., Park, Y., Takahashi, M., et al. (2021). Anthracycline-containing regimens or taxane versus S-1 as first-line chemotherapy for metastatic breast cancer. Br. J. Cancer 125, 1217–1225. doi:10.1038/s41416-021-01531-6
Murabito, A., Hirsch, E., and Ghigo, A. (2020). Mechanisms of anthracycline-induced cardiotoxicity: Is mitochondrial dysfunction the answer? Front. Cardiovasc Med. 7, 35. doi:10.3389/fcvm.2020.00035
Murton, A., Constantin, D., and Greenhaff, P. (2008). The involvement of the ubiquitin proteasome system in human skeletal muscle remodelling and atrophy. Biochimica Biophysica Acta (BBA)-Molecular Basis Dis. 1782, 730–743. doi:10.1016/j.bbadis.2008.10.011
Muthusamy, V. R., Kannan, S., Sadhaasivam, K., Gounder, S. S., Davidson, C. J., Boeheme, C., et al. (2012). Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic. Biol. Med. 52, 366–376. doi:10.1016/j.freeradbiomed.2011.10.440
Nakamura, T., Ueda, Y., Juan, Y., Katsuda, S., Takahashi, H., and Koh, E. (2000). Fas-mediated apoptosis in adriamycin-induced cardiomyopathy in rats: In vivo study. Circulation 102, 572–578. doi:10.1161/01.cir.102.5.572
Narayanan, V., and Koshy, C. (2009). Fatigue in cancer: A review of literature. Indian J. Palliat. Care 15, 19–25. doi:10.4103/0973-1075.53507
Nishida, K., Kyoi, S., Yamaguchi, O., Sadoshima, J., and Otsu, K. (2009). The role of autophagy in the heart. Cell Death Differ. 16, 31–38. doi:10.1038/cdd.2008.163
Nishida, K., Yamaguchi, O., and Otsu, K. (2008). Crosstalk between autophagy and apoptosis in heart disease. Circulation Res. 103, 343–351. doi:10.1161/CIRCRESAHA.108.175448
Octavia, Y., Tocchetti, C. G., Gabrielson, K. L., Janssens, S., Crijns, H. J., and Moens, A. L. (2012). Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell. Cardiol. 52, 1213–1225. doi:10.1016/j.yjmcc.2012.03.006
Pan, Y., Pan, Y.-M., Liu, F.-T., Xu, S.-L., Gu, J.-T., Hang, P.-Z., et al. (2021). MicroRNA-98 ameliorates doxorubicin-induced cardiotoxicity via regulating caspase-8 dependent Fas/RIP3 pathway. Environ. Toxicol. Pharmacol. 85, 103624. doi:10.1016/j.etap.2021.103624
Panday, A., Sahoo, M. K., Osorio, D., and Batra, S. (2015). NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell Mol. Immunol. 12, 5–23. doi:10.1038/cmi.2014.89
Philipp, S., Pagel, I., Höhnel, K., Lutz, J., Buttgereit, J., Langenickel, T., et al. (2004). Regulation of caspase 3 and Fas in pressure overload-induced left ventricular dysfunction. Eur. J. heart Fail. 6, 845–851. doi:10.1016/j.ejheart.2004.01.014
Pointon, A. V., Walker, T. M., Phillips, K. M., Luo, J., Riley, J., Zhang, S.-D., et al. (2010). Doxorubicin in vivo rapidly alters expression and translation of myocardial electron transport chain genes, leads to ATP loss and caspase 3 activation. PloS one 5, e12733. doi:10.1371/journal.pone.0012733
Porter, A. G., and Jänicke, R. U. (1999). Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 6, 99–104. doi:10.1038/sj.cdd.4400476
Powers, S. K., Hudson, M. B., Nelson, W. B., Talbert, E. E., Min, K., Szeto, H. H., et al. (2011). Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit. Care Med. 39, 1749–1759. doi:10.1097/CCM.0b013e3182190b62
Powers, S. K., and Jackson, M. J. (2008). Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 88, 1243–1276. doi:10.1152/physrev.00031.2007
Powers, S. K., Wiggs, M. P., Duarte, J. A., Zergeroglu, A. M., and Demirel, H. A. (2012). Mitochondrial signaling contributes to disuse muscle atrophy. Am. J. Physiology-Endocrinology Metabolism 303, E31–E39. doi:10.1152/ajpendo.00609.2011
Priya, L. B., Baskaran, R., Huang, C.-Y., and Padma, V. V. (2017). Neferine ameliorates cardiomyoblast apoptosis induced by doxorubicin: Possible role in modulating NADPH oxidase/ROS-mediated NFκB redox signaling cascade. Sci. Rep. 7, 12283–12313. doi:10.1038/s41598-017-12060-9
Putinski, C., Abdul-Ghani, M., Stiles, R., Brunette, S., Dick, S. A., Fernando, P., et al. (2013). Intrinsic-mediated caspase activation is essential for cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. 110, E4079–E4087. doi:10.1073/pnas.1315587110
Renu, K., Abilash, V. G., Tirupathi Pichiah, B. T., and Arunachalam, S. (2018). Molecular mechanism of doxorubicin-induced cardiomyopathy - an update. Eur. J. Pharmacol. 818, 241–253. doi:10.1016/j.ejphar.2017.10.043
Rocca, C., Pasqua, T., Cerra, M. C., and Angelone, T. (2020). Cardiac damage in anthracyclines therapy: Focus on oxidative stress and inflammation. Antioxidants Redox Signal. 32, 1081–1097. doi:10.1089/ars.2020.8016
Rothman, S. (2010). How is the balance between protein synthesis and degradation achieved? Theor. Biol. Med. Model 7, 25. doi:10.1186/1742-4682-7-25
Roy, S. (2000). Caspases at the heart of the apoptotic cell death pathway. Chem. Res. Toxicol. 13, 961–962. doi:10.1021/tx000109k
Sandri, M., Lin, J., Handschin, C., Yang, W., Arany, Z. P., Lecker, S. H., et al. (2006). PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. U. S. A. 103, 16260–16265. doi:10.1073/pnas.0607795103
Schirone, L., D'Ambrosio, L., Forte, M., Genovese, R., Schiavon, S., Spinosa, G., et al. (2022). Mitochondria and doxorubicin-induced cardiomyopathy: A complex interplay. Cells, 11.
Scott, J. M., Khakoo, A., Mackey, J. R., Haykowsky, M. J., Douglas, P. S., and Jones, L. W. (2011). Modulation of anthracycline-induced cardiotoxicity by aerobic exercise in breast cancer: Current evidence and underlying mechanisms. Circulation 124, 642–650. doi:10.1161/CIRCULATIONAHA.111.021774
Shandilya, M., Sharma, S., Das, P. P., and Charak, S. (2020). Molecular-level understanding of the anticancer action mechanism of anthracyclines. Advances in precision medicine oncology. IntechOpen.
Shi, Y. (2004). Caspase activation, inhibition, and reactivation: A mechanistic view. Protein Sci. 13, 1979–1987. doi:10.1110/ps.04789804
Shimomura, H., Terasaki, F., Hayashi, T., Kitaura, Y., Isomura, T., and Suma, H. (2001). Autophagic degeneration as a possible mechanism of myocardial cell death in dilated cardiomyopathy. Jpn. Circ. J. 65, 965–968. doi:10.1253/jcj.65.965
Simůnek, T., Stérba, M., Popelová, O., Adamcová, M., Hrdina, R., and Gersl, V. (2009). Anthracycline-induced cardiotoxicity: Overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol. Rep. 61, 154–171. doi:10.1016/s1734-1140(09)70018-0
Singal, P. K., Li, T., Kumar, D., Danelisen, I., and Iliskovic, N. (2000). Adriamycin-induced heart failure: Mechanism and modulation. Mol. Cell Biochem. 207, 77–86. doi:10.1023/a:1007094214460
Smuder, A. J. (2019). Exercise stimulates beneficial adaptations to diminish doxorubicin-induced cellular toxicity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 317, R662–R672. doi:10.1152/ajpregu.00161.2019
Smuder, A. J., Kavazis, A. N., Min, K., and Powers, S. K. (2013). Doxorubicin-induced markers of myocardial autophagic signaling in sedentary and exercise trained animals. J. Appl. physiology 115, 176–185. doi:10.1152/japplphysiol.00924.2012
Stěrba, M., Popelová, O., Vávrová, A., Jirkovský, E., Kovaříková, P., Geršl, V., et al. (2013). Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid. Redox Signal 18, 899–929. doi:10.1089/ars.2012.4795
Štěrba, M., Popelová, O., Vávrová, A., Jirkovský, E., Kovaříková, P., Geršl, V., et al. (2013). Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxidants redox Signal. 18, 899–929. doi:10.1089/ars.2012.4795
Sun, Z., Yan, B., Yu, W. Y., Yao, X., Ma, X., Sheng, G., et al. (2016). Vitexin attenuates acute doxorubicin cardiotoxicity in rats via the suppression of oxidative stress, inflammation and apoptosis and the activation of FOXO3a. Exp. Ther. Med. 12, 1879–1884. doi:10.3892/etm.2016.3518
Swain, S. M., Whaley, F. S., and Ewer, M. S. (2003). Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer Interdiscip. Int. J. Am. Cancer Soc. 97, 2869–2879. doi:10.1002/cncr.11407
Szenczi, O., Kemecsei, P., Holthuijsen, M. F., Van Riel, N. A., Van Der Vusse, G. J., Pacher, P., et al. (2005). Poly (ADP-ribose) polymerase regulates myocardial calcium handling in doxorubicin-induced heart failure. Biochem. Pharmacol. 69, 725–732. doi:10.1016/j.bcp.2004.11.023
Tacar, O., Sriamornsak, P., and Dass, C. R. (2013). Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 65, 157–170. doi:10.1111/j.2042-7158.2012.01567.x
Tewey, K., Rowe, T., Yang, L., Halligan, B., and Liu, L.-F. (1984). Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 226, 466–468. doi:10.1126/science.6093249
Vásquez-Vivar, J., Martasek, P., Hogg, N., Masters, B. S., Pritchard, K. A., and Kalyanaraman, B. (1997). Endothelial nitric oxide synthase-dependent superoxide generation from adriamycin. Biochemistry 36, 11293–11297. doi:10.1021/bi971475e
Vejpongsa, P., and Yeh, E. (2014). Topoisomerase 2β: A promising molecular target for primary prevention of anthracycline-induced cardiotoxicity. Clin. Pharmacol. Ther. 95, 45–52. doi:10.1038/clpt.2013.201
Venkatakrishnan, C. D., Tewari, A. K., Moldovan, L., Cardounel, A. J., Zweier, J. L., Kuppusamy, P., et al. (2006). Heat shock protects cardiac cells from doxorubicin-induced toxicity by activating p38 MAPK and phosphorylation of small heat shock protein 27. Am. J. Physiol. Heart Circ. Physiol. 291, H2680–H2691. doi:10.1152/ajpheart.00395.2006
Vickers, N. J. (2017). Animal communication: When i’m calling you, will you answer too? Curr. Biol. 27, R713–R715. doi:10.1016/j.cub.2017.05.064
Wallace, K. B., Sardão, V. A., and Oliveira, P. J. (2020). Mitochondrial determinants of doxorubicin-induced cardiomyopathy. Circ. Res. 126, 926–941. doi:10.1161/CIRCRESAHA.119.314681
Wang, A. H., Ughetto, G., Quigley, G. J., and Rich, A. (1987). Interactions between an anthracycline antibiotic and DNA: Molecular structure of daunomycin complexed to d(CpGpTpApCpG) at 1.2-A resolution. Biochemistry 26, 1152–1163. doi:10.1021/bi00378a025
Wang, F., Chandra, J., and Kleinerman, E. S. (2021). Exercise intervention decreases acute and late doxorubicin-induced cardiotoxicity. Cancer Med. 10, 7572–7584. doi:10.1002/cam4.4283
Wang, G.-W., Klein, J. B., and Kang, Y. J. (2001). Metallothionein inhibits doxorubicin-induced mitochondrial cytochrome c release and caspase-3 activation in cardiomyocytes. J. Pharmacol. Exp. Ther. 298, 461–468.
Wang, H., Wang, H., Liang, E. Y., Zhou, L. X., Dong, Z. L., Liang, P., et al. (2018a). Thrombopoietin protects H9C2 cells from excessive autophagy and apoptosis in doxorubicin-induced cardiotoxicity. Oncol. Lett. 15, 839–848. doi:10.3892/ol.2017.7410
Wang, P., Li, C. G., Qi, Z., Cui, D., and Ding, S. (2016). Acute exercise stress promotes Ref1/Nrf2 signalling and increases mitochondrial antioxidant activity in skeletal muscle. Exp. Physiol. 101, 410–420. doi:10.1113/EP085493
Wang, S., Wang, H., Su, X., Liu, B., Wang, L., Yan, H., et al. (2020). β-Adrenergic activation may promote myosin light chain kinase degradation through calpain in pressure overload-induced cardiac hypertrophy: β-Adrenergic activation results in MLCK degradation. Biomed. Pharmacother. 129, 110438. doi:10.1016/j.biopha.2020.110438
Wang, Y., Chen, B., Huang, C.-K., Guo, A., Wu, J., Zhang, X., et al. (2018b). Targeting calpain for heart failure therapy: Implications from multiple murine models. JACC Basic Transl. Sci. 3, 503–517. doi:10.1016/j.jacbts.2018.05.004
Weaver, K. E., Forsythe, L. P., Reeve, B. B., Alfano, C. M., Rodriguez, J. L., Sabatino, S. A., et al. (2012). Mental and physical health–related quality of life among US cancer survivors: Population estimates from the 2010 national health interview survey. Cancer Epidemiol. Prev. Biomarkers 21, 2108–2117. doi:10.1158/1055-9965.EPI-12-0740
Wei, Y., Zhao, J., Xiong, J., Chai, J., Yang, X., Wang, J., et al. (2022). Wogonin reduces cardiomyocyte apoptosis from mitochondrial release of cytochrome c to improve doxorubicin-induced cardiotoxicity. Exp. Ther. Med. 23, 205–208. doi:10.3892/etm.2022.11128
Willis, M. S., Parry, T. L., Brown, D. I., Mota, R. I., Huang, W., Beak, J. Y., et al. (2019). Doxorubicin exposure causes subacute cardiac atrophy dependent on the striated muscle–specific ubiquitin ligase MuRF1. Circ. Heart Fail. 12, e005234. doi:10.1161/CIRCHEARTFAILURE.118.005234
Wojnowski, L., Kulle, B., Schirmer, M., Schlüter, G., Schmidt, A., Rosenberger, A., et al. (2005). NAD(P)H oxidase and multidrug resistance protein genetic polymorphisms are associated with doxorubicin-induced cardiotoxicity. Circulation 112, 3754–3762. doi:10.1161/CIRCULATIONAHA.105.576850
Wu, B. B., Leung, K. T., and Poon, E. N. (2022). Mitochondrial-targeted therapy for doxorubicin-induced cardiotoxicity. Int. J. Mol. Sci. 23, 1912. doi:10.3390/ijms23031912
Xia, P., Chen, J., Liu, Y., Fletcher, M., Jensen, B. C., and Cheng, Z. (2020). Doxorubicin induces cardiomyocyte apoptosis and atrophy through cyclin-dependent kinase 2–mediated activation of forkhead box O1. J. Biol. Chem. 295, 4265–4276. doi:10.1074/jbc.RA119.011571
Xu, X., Chen, K., Kobayashi, S., Timm, D., and Liang, Q. (2012). Resveratrol attenuates doxorubicin-induced cardiomyocyte death via inhibition of p70 S6 kinase 1-mediated autophagy. J. Pharmacol. Exp. Ther. 341, 183–195. doi:10.1124/jpet.111.189589
Yang, Z., Wilkie-Grantham, R. P., Yanagi, T., Shu, C. W., Matsuzawa, S., and Reed, J. C. (2015). ATG4B (Autophagin-1) phosphorylation modulates autophagy. J. Biol. Chem. 290, 26549–26561. doi:10.1074/jbc.M115.658088
Ye, S., Su, L., Shan, P., Ye, B., Wu, S., Liang, G., et al. (2021). LCZ696 attenuated doxorubicin-induced chronic cardiomyopathy through the TLR2-MyD88 complex formation. Front. Cell Dev. Biol. 9, 654051. doi:10.3389/fcell.2021.654051
Yeh, Y. H., Liang, C. Y., Chen, M. L., Tsai, F. M., Lin, Y. Y., Lee, M. C., et al. (2019). Apoptotic effects of hsian-tsao (Mesona procumbens Hemsley) on hepatic stellate cells mediated by reactive oxygen species and ERK, JNK, and caspase-3 pathways. Food Sci. Nutr. 7, 1891–1898. doi:10.1002/fsn3.1046
Yu, X., Ruan, Y., Huang, X., Dou, L., Lan, M., Cui, J., et al. (2020). Dexrazoxane ameliorates doxorubicin-induced cardiotoxicity by inhibiting both apoptosis and necroptosis in cardiomyocytes. Biochem. Biophysical Res. Commun. 523, 140–146. doi:10.1016/j.bbrc.2019.12.027
Yu, Y., Guo, D., and Zhao, L. 2022. MiR-199 aggravates doxorubicin-induced cardiotoxicity by targeting TAF9b. Evidence-Based Complementary Altern. Med., 2022, 1, 13. doi:10.1155/2022/4364779
Zhang, S.-H., Wang, W.-Q., and Wang, J.-L. (2009a). Protective effect of tetrahydroxystilbene glucoside on cardiotoxicity induced by doxorubicin in vitro and in vivo. Acta Pharmacol. Sin. 30, 1479–1487. doi:10.1038/aps.2009.144
Zhang, Y.-W., Shi, J., Li, Y.-J., and Wei, L. (2009b). Cardiomyocyte death in doxorubicin-induced cardiotoxicity. Archivum Immunol. Ther. Exp. 57, 435–445. doi:10.1007/s00005-009-0051-8
Zhao, L., and Zhang, B. (2017). Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci. Rep. 7, 44735–44811. doi:10.1038/srep44735
Zhao, Y., Mclaughlin, D., Robinson, E., Harvey, A. P., Hookham, M. B., Shah, A. M., et al. (2010). Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with Doxorubicin chemotherapy. Cancer Res. 70, 9287–9297. doi:10.1158/0008-5472.CAN-10-2664
Keywords: anthracycline, doxorubicin, cardiotoxicity, exercise training, oxidative stress, proteolytic systems
Citation: Gaytan SL, Lawan A, Chang J, Nurunnabi M, Bajpeyi S, Boyle JB, Han SM and Min K (2023) The beneficial role of exercise in preventing doxorubicin-induced cardiotoxicity. Front. Physiol. 14:1133423. doi: 10.3389/fphys.2023.1133423
Received: 28 December 2022; Accepted: 27 February 2023;
Published: 09 March 2023.
Edited by:
José Magalhães, University of Porto, PortugalReviewed by:
Antonio Alexandre Ascensao, University of Porto, PortugalEylem Taskin, Adiyaman University, Türkiye
Copyright © 2023 Gaytan, Lawan, Chang, Nurunnabi, Bajpeyi, Boyle, Han and Min. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kisuk Min, a21pbkB1dGVwLmVkdQ==; Sung Min Han, aGFuLnNAdWZsLmVkdQ==