 Toan Pham
Toan Pham Andrew Taberner
Andrew Taberner Anthony Hickey
Anthony Hickey June-Chiew Han
June-Chiew Han- 1Auckland Bioengineering Institute, The University of Auckland, Auckland, New Zealand
- 2Department of Engineering Science and Biomedical Engineering, The University of Auckland, Auckland, New Zealand
- 3School of Biological Sciences, The University of Auckland, Auckland, New Zealand
Introduction: Type 2 diabetes (T2D) is a global epidemic, and heart failure is the primary cause of premature death among T2D patients. Mitochondrial dysfunction has been linked to decreased contractile performance in diabetic heart, partly due to a disturbance in the mitochondrial capacity to supply adequate metabolic energy to contractile proteins. MOTS-c, a newly discovered mitochondrial-derived peptide, has shown promise as a therapeutic for restoring energy homeostasis and muscle function in metabolic diseases. However, whether MOTS-c therapy improves T2D heart function by increasing mitochondrial bioenergetic function remains unknown.
Methods: Here we studied the mitochondrial bioenergetic function of heart tissues isolated from a rat model mimicking type 2 diabetes induced by a high-fat diet and low-dose streptozotocin. Treated diabetic group received MOTS-c (15 mg/kg) daily injection for 3 weeks. We employed high-resolution respirometric and fluorometric techniques to simultaneously assess mitochondrial ATP production and hydrolysis capacity, reactive oxygen species (ROS) production, and oxygen flux in cardiac tissue homogenates.
Results: We found that untreated T2D rats had hyperglycemia, poor glucose control, and left ventricular hypertrophy relative to controls. T2D mitochondria showed decreased oxygen flux at the oxidative phosphorylation (OXP) while ROS production, ATP production and hydrolysis rates remained unchanged. Diabetic rats treated with MOTS-c showed decreased fasting glucose levels, improved glucose homeostasis, and decreased degree of cardiac hypertrophy. At the subcellular level, MOTS-c treated mitochondria showed increased OXPHOS respiration and ROS levels and decreased ATP hydrolysis rate during anoxic conditions.
Discussion: These findings demonstrate beneficial effects of MOTS-c treatment on glucose homeostasis and suggest a useful therapeutic option for diabetic-related cardiomyopathy and mitochondrial dysfunction.
Introduction
Diabetes is rapidly becoming one of the most common chronic diseases. Over 90% of diabetic patients are diagnosed with Type 2 diabetes (T2D). Cardiovascular complications are the leading cause of morbidity and mortality in diabetic patients, with heart failure accounting for approximately 80% of all deaths (Boudina and Abel, 2007). Diabetic cardiomyopathy is defined by left ventricular hypertrophy, altered substrate metabolism (Amaral and Okonko, 2015; Stanley et al., 1997; Sharma et al., 2004) and, eventually, decreased contractile function (Marciniak et al., 2014; Montaigne et al., 2014). Mitochondrial dysfunction may precede contractile dysfunction and could be a key contributor to the decreased myocardial contractility seen in T2D patients (Montaigne et al., 2014; Anderson et al., 2009), as well as in rodent models of diabetes (Marciniak et al., 2014; Li et al., 2022; Tang et al., 2023). Understanding the underlying processes of diabetes-induced heart dysfunction is critical for establishing effective therapeutic options, yet it also remains an ongoing challenge for healthcare and biomedical research.
The heart is a high-energy-demanding organ and requires a continuous supply of adenosine triphosphate (ATP) to support constant mechanical work. Mitochondria primarily supply ATP via oxidative phosphorylation and occupy approximately 35% of the total volume of mammalian cardiac myocytes (Barth et al., 1992). Mitochondrial dysfunction often coincides with heart failure (Zhou and Tian, 2018). Emerging evidence has reported mitochondrial structural abnormalities (Tang et al., 2023; Boudina et al., 2007) and decreased bioenergetic function (Marciniak et al., 2014; Boudina et al., 2007; Croston et al., 2014; Pham et al., 2014) in diabetic hearts. As diabetes progresses, the heart decreases glucose utilisation, eventually relying almost exclusively on fatty acid (FA) oxidation to sustain ATP production (Amaral and Okonko, 2015; Stanley et al., 1997; Sharma et al., 2004). FA oxidation is less efficient than glucose oxidation, as it consumes more oxygen per unit of ATP generated (Lopaschuk et al., 2010). It also appears that increased reliance on FA oxidation in diabetic heart mitochondria associates with increased reactive oxygen species (ROS) production (Pham et al., 2014; Ye et al., 2004) and increased mitochondrial uncoupling protein expression (Boudina et al., 2007; Cole et al., 2011), which results in a decrease in the proton motive force and decreased ATP synthesis capacities (Pham et al., 2014; Ye et al., 2004).
MOTS-c is a newly discovered mitochondria-derived peptide encoded by the open reading frame in mitochondrial 12S rRNA. This small 16-amino-acid protein is highly conserved among species (Lee et al., 2015). Emerging data suggest that MOTS-c regulates cellular metabolism through AMPK-dependent pathways, enhancing glucose utilisation and stress responses (Lee et al., 2015; Kim et al., 2017; Merry et al., 2020a). MOTS-c is found in blood and other mitochondria-containing tissues. Blood MOTS-c levels were reported to be lower in T2D individuals (Ramanjaneya et al., 2019), gestational diabetes (Yin et al., 2022), coronary endothelial dysfunction (Qin et al., 2018), obese children and adolescents (Du et al., 2018), and streptozotocin-induced diabetic mice (Xu et al., 2024). Exogenous MOTS-c therapy has emerged as a promising therapeutic strategy for enhancing muscle mitochondrial function in metabolic disorders. In cell culture studies, MOTS-c administration has been demonstrated to increase mitochondrial ATP content in doxorubicin-induced senescent human fibroblasts (Kim et al., 2018) and HEK293 cells (Lee et al., 2015), as well as protecting against inflammation and oxidative stress in H9C2 cells (Shen et al., 2022). In animal studies, MOTS-c therapy has been shown to have a variety of positive benefits, including the prevention of age-related metabolic dysfunction (Li et al., 2019), ovariectomy-induced metabolic disturbances (Lu et al., 2019), insulin resistance in high-fat diet-induced obesity (Lee et al., 2015), and pressure overload-induced cardiac hypertrophy (Zhong et al., 2022). Recent research has also shown that MOTS-c treatment improved heart function in diabetic rats by enhancing glucose metabolism (Li et al., 2022; Yin et al., 2022) and upregulating antioxidant defences (Tang et al., 2023). However, the direct effect of MOTS-c therapy in restoring mitochondrial bioenergetic dysfunction in diabetic hearts is unknown.
We hypothesise that MOTS-c therapy may alleviate diabetic myocardial injury by improving mitochondrial respiration, increasing ATP production, and lowering oxidative stress levels. This study examined how MOTS-c treatment changed cardiac mitochondrial function in a well-established T2D rat model. This model has been shown to closely resemble the heart metabolic abnormalities seen in diabetic patients (Mansor et al., 2013; Guo et al., 2018). High-resolution respirometry and fluorometry were employed to investigate the mitochondrial bioenergetic function in cardiac tissue homogenates.
Methods
Animals
All experiments were conducted following protocols approved by the University of Auckland Animal Ethics Committee (AEC22653). Male Wistar rats (6–7 weeks old, 150–200 g) were randomly assigned to three groups: control (n = 12), untreated diabetic (n = 7), and treated diabetic (n = 11). Diabetes was induced using a combination of dietary and pharmacological interventions. Rats were fed with a high-fat diet (HFD, 43% digestible energy from lipids, SF04-001, Specialty Feeds, Australia) for a total duration of 15 weeks and received a low-dose intraperitoneal injection of streptozotocin (STZ, 25 mg/kg, in citrate buffer pH 4) at week 8 post-HFD. Control rats received standard chow and an injection of citrate buffer. At week 12, the treated diabetic group received daily intraperitoneal injections of MOTS-c (15 mg/kg/day) for 3 weeks, while the untreated diabetic and control groups received saline injections. All rats had water and food access in a 12-h light/dark cycle. Body weight and blood glucose were monitored weekly. At week 15, each rat was subjected to a glucose tolerance test following a 6-h fast before receiving an intraperitoneal injection of glucose bolus (2 g/kg, in sterile saline) and blood samples were obtained from the tail puncture. Blood glucose was measured using an Accu-Chek blood glucose meter at various time points (before the glucose tolerance test, then 15, 30, 60, 90, and 120 min after glucose administration).
Drug
Mitochondria-derived peptide MOTS-c (MRWQEMGYIFYPRKLR) was synthesised by GenScript Biotech Ltd, Singapore and kept in powder form at −80 °C. MOTS-c was freshly dissolved in sterile saline before injection and used within 30 min.
Tissue preparation
On the experiment day, rats were deeply anesthetised with isoflurane and injected with heparin (1,000 IU/kg). Following cervical dislocation, the heart was dissected, submerged in cold Tyrode solution, and then Langendorff-perfused with oxygenated Tyrode solution at room temperature. The Tyrode solution contained (in mmol/L): 130 NaCl, 6 KCl, 1 MgCl2, 0.3 CaCl2, 0.5 NaH2PO4, 10 HEPES, 10 glucose, and 20 2,3-butanedione monoxime (pH 7.4, adjusted with Tris). Ventricular free wall thickness, heart mass, and tibial length were measured.
Mitochondrial respiration was measured in fresh left ventricular samples. Fresh heart tissues (approximately 30 mg) were transferred to 750 µL cold MiRO5 buffer (without MgCl2), which contained (in mmol/L) 110 sucrose, 60 K-lactobionate, 20 HEPES, 20 taurine, 10 KH2PO4, 0.5 EGTA, 1 g/L BSA fatty acid free, and pH 7.1 adjusted at 30 °C. A small amount of MgCl2 (1 mmol/L) was used during the experiment to avoid signal interference with Magnesium Green fluorescence-based ATP measurement. Cardiac tissue samples were minced with small scissors, homogenised for 10 s at medium speed (Omni International, Hennesaw, GA) and transferred to oxygraph chambers for mitochondrial experiments. The remaining homogenate samples were stored at −80 °C for later enzymatic analysis.
Mitochondrial experiments
Mitochondrial respiration was measured using a high-resolution fluo-respirometer (O2k, Oroboros Instruments, Innsbruck, Austria). This device has two separate 2-mL chambers. Each chamber has a polarographic oxygen sensor and a sealed stopper for substrate-inhibitor titration. Two detachable fluorimeters (Oroboros Instruments) were inserted in front of each chamber window to simultaneously measure the fluorescence of specific fluorophores (as described below) and O2 consumption rate. O2 tension was calibrated at an air saturation level (at 101 kPa) before each experiment. All measurements were conducted at 37 °C while the stirring speed was set to 750 rpm. All substrates, uncouplers, and inhibitors were added manually using Hamilton syringes.
Protocol 1: ATP production and hydrolysis
Magnesium Green (MgG, cell impermeant, M3733 Thermo Fisher Scientific) fluorescence (excitation: 470 nm, emission: 520 nm) was used to measure ATP-ADP exchange (Pham et al., 2014; Power et al., 2014) in a modified MiRO5 buffer (without MgCl2). MgG (5 μmol/L) and MgCl2 (1 mmol/L) were added before adding homogenate samples (2 mg) and waiting for signal stability. Complex I (CI) substrates, including malate (2 mmol/L), glutamate (10 mmol/L), and pyruvate (5 mmol/L) were used to induce leak respiration, which reflects O2 consumption compensating ion leaks without ATP synthesis, despite the presence of a small amount of ADP (∼0.03 μmol/L) in homogenate samples might contribute to negligible oxidative phosphorylation (OXPHOS) -linked respiration. ADP (2.5 mmol/L) was added to measure NADH-linked OXPHOS respiration in the presence of CI substrates, followed by succinate (10 mmol/L) to activate both CI and CII OXPHOS. Tissues were exposed to anoxia for 20 min to measure the maximal rate of ATP hydrolysis, followed by opening the stoppers to equilibrate with air to restore O2 concentration. Oligomycin (2 μmol/L) was added to inhibit ATP synthase, to return the MgG signal level to baseline, where this fluorescence level corresponded to no ATP synthesis activity by the mitochondria, allowing estimation of ATP-independent MgG signal. Antimycin A (1 μmol/L) was used to inhibit complex III (CIII) and allowed the measurement of non-mitochondrial O2 consumption.
Protocol 2: ROS production
Amplex UltraRed (AUR, 25 μmol/L), together with horseradish peroxidase (HRP, 10 U) and superoxide dismutase (SOD, 10 U), were used to assess mitochondrial H2O2 production (Pham et al., 2020; Hedges et al., 2020). Exogenous SOD converts superoxide radicals into H2O2, which then reacts with HRP and AUR to create resorufin (excitation: 525 nm, emission: 550 nm). H2O2 signal was calibrated with three times the known amount of H2O2 (122 μmol/L). MgCl2 (1 mmol/L) and homogenate samples (2 mg) were added to each chamber. CI substrates (malate, glutamate, pyruvate) and CII substrate (succinate) were added to initiate leak respiration linked to CI and CII. ADP (2.5 mmol/L) stimulated OXPHOS respiration from both CI and CII, while multiple titrations of carbonyl cyanide m-chlorophenyl hydrazone (CCCP, 1 μmol/L) assessed uncoupled respiration, demonstrating the maximal capacity of electron transfer system. Antimycin A (1 μmol/L) inhibited complex III to determine residual OCR. Complex IV (CIV) respiration was measured using ascorbate (2 mmol/L) and N,N,N′,N′-tetramethyl-p-phenylenediamine dihydrochloride (TMPD, 0.5 mmol/L), followed by inhibition with azide (100 mmol/L).
Protocol 3: fatty acid oxidation
MgCl2 (1 mmol/L) and homogenate samples (2 mg) were added to each chamber. To assess fatty acid metabolism, malate (0.1 mmol/L) and palmitoyl-L-carnitine (40 μmol/L) were used to initiate leak respiration. ADP (2.5 mmol/L) stimulated fatty acid-mediated OXPHOS respiration, followed by succinate (10 mmol/L) for CII-mediated OXPHOS. Antimycin A (1 μmol/L) was added to determine non-mitochondrial OCR.
Citrate synthase activity
A protease inhibitor cocktail (25 μL, cOmplete Mini EDTA-free, Roche, Mannheim, Germany) was added to frozen homogenate samples. The samples were then thawed, vortexed, and centrifuged at 5,000 g for 10 min at 4 °C. Supernatants were collected and diluted 25-fold in phosphate-buffered saline to assess citrate synthase (CS) activity. Absorbance was measured at 412 nm every 30 s for a duration of 10 min with a microplate reader at room temperature in Tris-HCl buffer (50 mmol/L, pH 8) containing oxaloacetate (0.5 mmol/L), acetyl-CoA (0.1 mmol/L), and 5,5-dithiobis-(2-nitrobenzoic acid) (0.2 mmol/L). The CS activity was determined from the absorbance slope using an extinction coefficient value of 13,600 L/mol/cm and normalised for protein content. Total protein concentration was measured with a BCA protein kit (Thermo Fisher Scientific, Waltham, MA, USA).
Data analyses
Respirometric and fluorometric data were recorded and analysed offline using DatLab 7.1 (Oroboros Instruments). O2 flux data were corrected for residual O2 consumption following the addition of antimycin, except for CIV respiration, where O2 flux data derived from the autooxidation effect were subtracted for O2 flux after CIV inhibition with sodium azide addition. ATP flux signals were calibrated using separate assays without tissue samples, as previously described (Pham et al., 2014) and corrected for the background signal of ATP synthase inhibition by oligomycin. H2O2 measurements were corrected for background H2O2 levels, which were measured before adding homogenate samples. Data were normalised to tissue wet mass.
Statistical analyses
Statistical analyses were performed with GraphPad Prism 10.0. All data are presented as mean ± SEM unless otherwise stated. One-way analysis of variance test was used to examine group differences in all data, except that two-way analysis of variance was used to analyse blood glucose data in the glucose tolerance test. If a significant difference (p < 0.05) was detected, Fisher’s least significant difference post-hoc test was used to identify pairwise differences. All graphs were created using Prism software.
Results
Morphological characteristics of the rats
Untreated diabetic rats had notably higher fasting glucose levels than control and MOTS-c-treated diabetic rats, with no difference between the latter two groups (Figure 1A). We excluded rats with type 1 diabetic signs, including hyperglycaemia (>30 mmol/L), polyuria, and substantial weight loss. Untreated diabetic rats demonstrated decreased glucose clearance capability compared to controls, whereas MOTS-c-treated rats showed improved glucose handling, although blood glucose levels remained higher than the control group (Figure 1B).
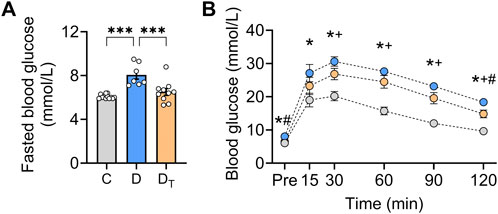
Figure 1. Glucose homeostasis in diabetic rats after MOTS-c treatment. (A) Fasted blood glucose levels. (B) Blood glucose levels were measured before and after a glucose bolus in control, untreated diabetic and treated diabetic rats. C, Control group (labelled grey), D, untreated diabetic group (labelled blue), DT: MOTS-c treated diabetic group (labelled orange). The colour of each of the three groups matches (A) Data are presented as means ± SEM. *p < 0.05 Control vs. untreated diabetic; +p < 0.05 control vs. treated diabetic; #p < 0.05 untreated diabetic vs. treated diabetic rats.
Compared to the controls, the untreated diabetic rats exhibited increases in body mass, tibial length, heart mass and left-ventricular thickness, mimicking T2D symptoms (Table 1). Three weeks of MOTS-c treatment alleviated these increases. No statistical differences in any parameters were detected between the control group and the treated diabetic group.
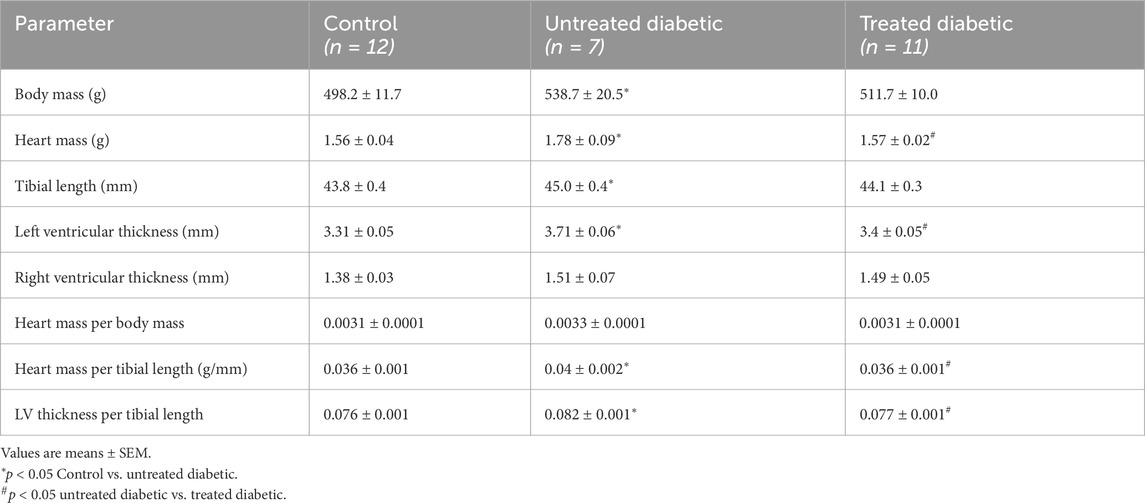
Table 1. Morphological characteristics of the rats at the time of sacrifice.
Mitochondrial respiration
Untreated diabetic heart mitochondria showed no significant differences in O2 consumption rate or O2 flux from carbohydrate-supported substrates (addition of malate, glutamate, and pyruvate) at CI and CII leak, uncoupled respiration, or CIV respiration states compared to other groups (Figure 2). However, diabetic mitochondria had a lower mitochondrial O2 flux in CI and CII OXPHOS state compared to the control and MOTS-c-treated diabetic groups, with no difference between the latter two groups (Figure 2D). The activity of CS normalised to protein content was significantly higher in the MOTS-c-treated diabetic group than in the control and untreated diabetic groups, indicating that the MOTS-c-treated group had increased mitochondrial content (Figure 2G).
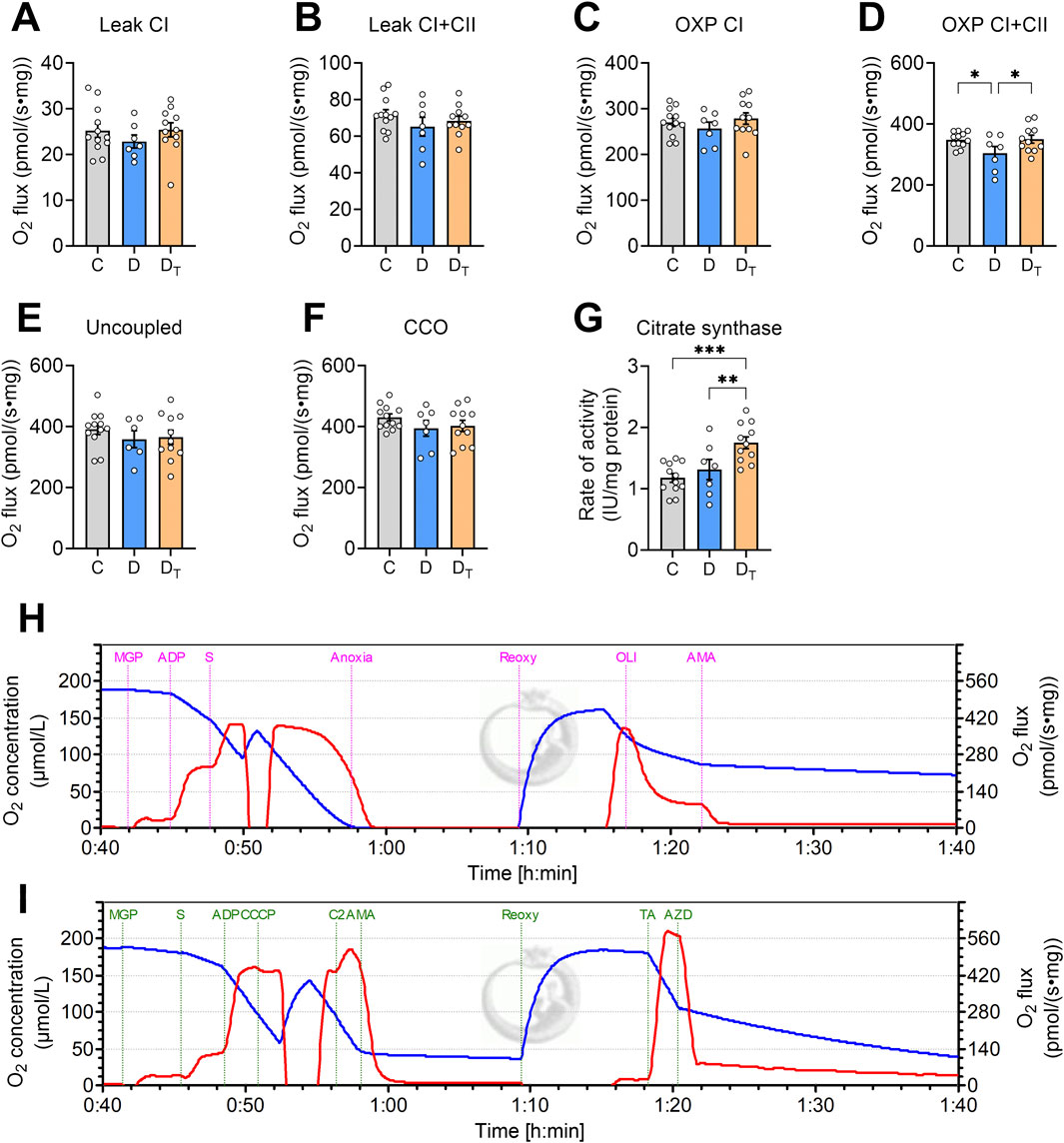
Figure 2. Mitochondrial O2 flux measured from left ventricular homogenates. O2 flux is normalised to tissue wet weight (A–F). (G) Citrate synthase activity as a measure of mitochondrial content, IU unit is in µmol/min. Representative traces of O2 concentration (blue) and O2 flux per tissue mass (red) from Protocol #1 (H) and Protocol #2 (I). CI: complex I, CII, complex II, OXP: oxidative phosphorylation, CCO: cytochrome C oxidase, CCCP: carbonyl cyanide m-chlorophenyl hydrazone, TA: N, N, N′, N′-tetramethyl-p-phenylenediamine dihydrochloride and ascorbate, C, Control group, D, untreated diabetic group, DT, MOTS-c treated diabetic group. All data are presented as mean ± SEM.
Figure 3 presents data on the respiratory control ratio, which measures mitochondrial coupling efficiency, defined as the ratio of O2 flux in the OXPHOS state and O2 flux in the leak state. MOTS-c treated diabetic mitochondria exhibited a significantly higher respiratory control ratio than the untreated group (Figure 3A). Untreated diabetic mitochondria had a higher uncoupling control ratio than the control and MOTS-c treated groups (Figure 3B). There were no significant differences in cytochrome c-oxidase or CIV activity relative to the coupled OXPHOS state among the groups (Figure 3C).
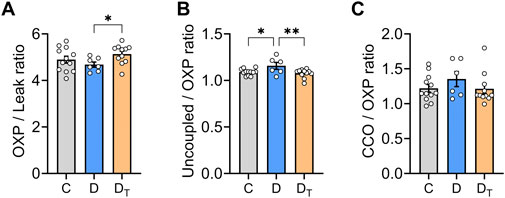
Figure 3. Mitochondrial respiratory flux control ratios. (A) The respiratory control ratio, a measure of mitochondrial coupling efficiency, was calculated using both Complex I and Complex II substrates with and without ADP. (B) Uncoupling control ratio was calculated as a ratio of O2 flux in uncoupled respiration state compared to oxidative phosphorylation (OXP) state. (C) Activity of cytochrome c-oxidase (CIV) relative to the coupled OXP state. C, Control group, D, untreated diabetic group, DT, MOTS-c treated diabetic group. Data are expressed as mean ± SEM.
Figure 4 shows data of mitochondrial respiration supported by a fatty acid-based substrate (palmitoyl-L-carnitine) and a low concentration of malate. When normalised to tissue wet mass, mitochondrial fatty acid oxidation-mediated respiration was similar among groups.
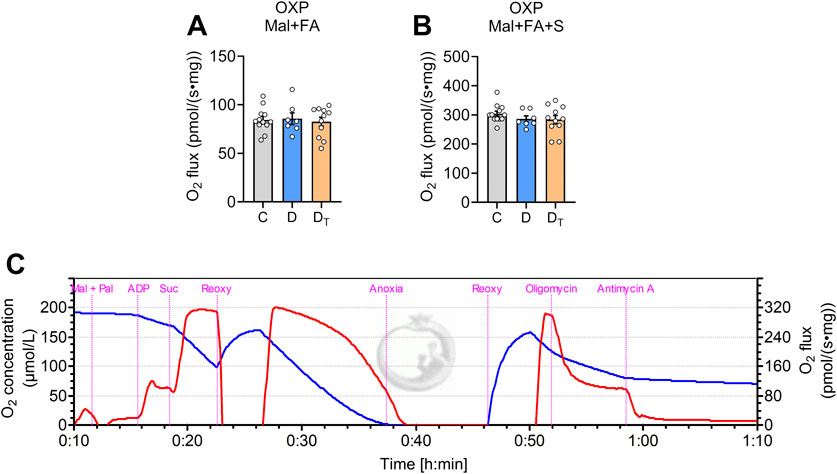
Figure 4. Respiration capacity of mitochondrial palmitoyl-carnitine oxidation. Mitochondrial O2 flux measured from left ventricular homogenates normalised to tissue wet weight (A,B) Representative traces of O2 concentration (blue) and O2 flux per tissue mass (red) from Protocol #3 (C) Mal, malate, Pal, palmitoyl-L-carnitine, Suc, succinate, Reoxy, reoxygenation, C, Control group, D, untreated diabetic group, DT, MOTS-c treated diabetic group. Data are expressed as mean ± SEM.
Steady-state ATP synthesis and hydrolysis measurements
When normalised to tissue wet mass, ATP production rate in the OXPHOS state did not differ among groups (Figure 5). Similarly, no significant differences were observed in the P/O ratio, a measure of mitochondrial energy efficiency, when expressed relative to steady-state O2 flux (Figure 5C). During anoxia, ATP synthase operates in reverse, consuming ATP and resulting in negative ATP flux, and no difference was observed between control and untreated diabetic groups (Figure 5B). However, MOTS-c treated diabetic mitochondria exhibited significantly lower ATP hydrolysis rates than untreated diabetic groups.
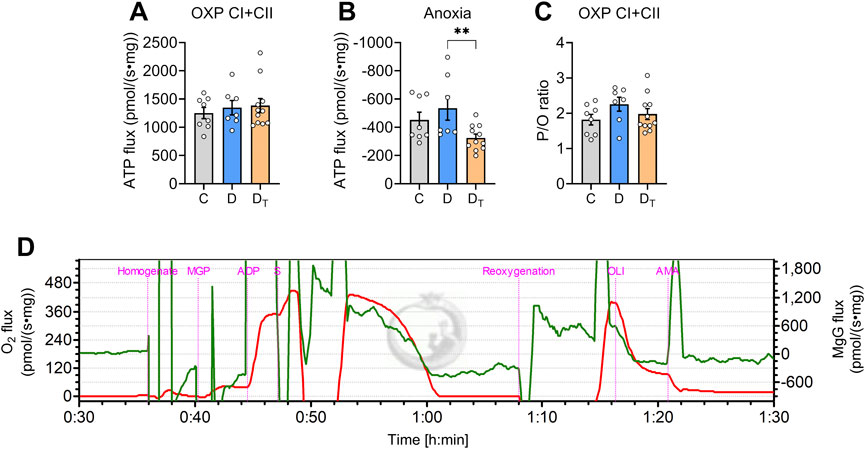
Figure 5. ATP production and hydrolysis capacity in left ventricular homogenates. Rates of ATP production per tissue mass in normoxic (A) and anoxic [(B) negative values] conditions. (C) Steady-state P/O ratio was calculated using the rate of ATP production relative to the oxygen flux in oxidative phosphorylation (OXP) respiration state. (D) Representative traces of O2 flux per tissue mass (red) and MgG signal flux (Green) from Protocol #1. C, Control group, D, untreated diabetic group, DT, MOTS-c treated diabetic group. Data are expressed as mean ± SEM.
H2O2 production measurements
H2O2 production at various respiratory states did not differ between control and untreated diabetic groups (Figure 6). MOTS-c-treated diabetic mitochondria exhibited higher H2O2 production per tissue mass at the CI and CII leak state than untreated diabetic mitochondria (Figure 6A). At the same CI and CII leak state, H2O2 production expressed as a percentage of single oxygen flux (H2O2/O) was significantly higher in treated diabetic mitochondria than in control and untreated diabetic groups (Figure 6E).
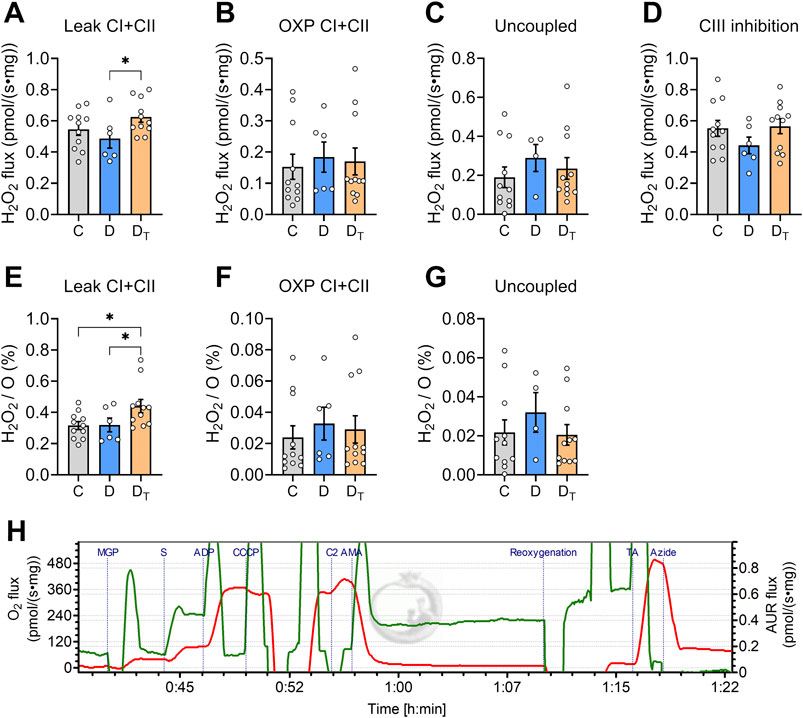
Figure 6. H2O2 production rates in left ventricular homogenates. (A–D) H2O2 production rate per tissue mass at various respiration states; (E–G) H2O2 production relative to oxygen flux (H2O2/O). (H) Representative traces of O2 flux per tissue mass (red) and AUR signal flux (Green) from Protocol #2. CI, complex I, CII, complex II, OXP, oxidative phosphorylation, C, Control group, D, untreated diabetic group, DT, MOTS-c treated diabetic group. Data are expressed as mean ± SEM.
Discussion
This study examined the effects of MOTS-c on mitochondrial bioenergetic function in Type 2 diabetic hearts. Our findings reveal that untreated diabetic rats gained more weight, had poorer glucose regulation, developed left-ventricular hypertrophy and had lower mitochondrial respiration at the OXPHOS respiratory state compared to the control group. MOTS-c treatment significantly lowered weight gain, improved glucose handling, and reversed cardiac hypertrophy. MOTS-c improved carbohydrate-supported mitochondrial respiration and increased citrate synthase activity. However, MOTS-c treated diabetic mitochondria decreased the capacity of ATP hydrolysis during anoxia while having no effect on FA-supported mitochondrial respiration, mitochondrial ATP production and ROS production in CI-CII OXP state.
Animal morphological phenotypes
In this study, we employed a T2D-like rat model induced by a combination of a high-fat diet and low-dose STZ injection, which has been shown to resemble cardiac metabolic abnormalities in diabetic individuals (Mansor et al., 2013; Guo et al., 2018). In this study, we confirmed that rats on this regimen increased body weight and had poor glucose homeostasis. Rats with substantial weight loss and hyperglycaemia, which resembled Type 1 diabetes, were excluded from this study. Our findings demonstrate that MOTS-c treatment effectively delayed weight gain in diabetic rats, which is consistent with recent research in HFD-induced obese mice (Lee et al., 2015) and T1D mice (Xu et al., 2024). These investigations also found that MOTS-c treatment did not affect food intake, indicating that MOTS-c may directly increase whole-body metabolic rate.
MOTS-c treatment has been demonstrated to lower blood glucose levels in T1D mice (Xu et al., 2024), T1D rats (Li et al., 2022), or gestational diabetic mice (Yin et al., 2022). Our findings extend these reports as MOTS-c treatment decreased fasted blood glucose levels and improved glucose handling in T2D rats. We also observed an 8% decrease in left ventricular wall thickness in the treated diabetic group, highlighting the potential beneficial effects of MOTS-c on restoring cardiac dysfunction, as consistently reported in previous studies on T1D model (Li et al., 2022; Wu N. et al., 2023). Previous studies reported that MOTS-c treatment induces activation of AMPK signalling pathway, which improves insulin sensitivity and upregulates GLUT4 expression in skeletal muscle, enabling efficient glucose uptake and metabolism (Lee et al., 2015; Yin et al., 2022).
Carbohydrate- and FA-supported mitochondrial respiration
In untreated diabetic rats, mass-specific oxygen flux from carbohydrate-supported substrates (including malate, glutamate and pyruvate) was 13% lower in CI + CII OXP respiratory state than in the control. This observation is consistent with decreased mitochondrial respiration found in our previous study in T1D rat model (Pham et al., 2014) and others in T2D mouse model (Boudina et al., 2007). Our data in this present work show that MOTS-c treatment restores mitochondrial respiration per tissue mass, aligning with previous findings of increased mitochondrial respiration in senescent cells (Kim et al., 2018). The increased respiration following MOTS-c treatment may be attributed to increased mitochondrial content rather than intrinsic functional gain. It is possible that MOTS-c treatment increased CS activity per each mitochondrion without increasing total mitochondrial volume, which warrants further investigation to confirm this assumption. Nevertheless, our findings are consistent with previous findings in T1D rats (Tang et al., 2023; Wang et al., 2022) reported increases in both CS activity and mitochondrial number, and preserved mitochondrial structure in the treated diabetic group. This is likely attributed to increased activation of the AMPK pathway to induce increased mitochondrial biogenesis biomarkers (Xu et al., 2024) in response to MOTS-c treatment.
Additionally, untreated diabetic mitochondria had a greater uncoupled respiratory ratio, suggesting impaired phosphorylation efficiency despite preserved electron transfer system (ETS) capacity. This apparent uncoupling was also restored by MOTS-c treatment. CIV oxygen flux, which represents the terminal step of the ETS, remained unchanged across groups. Given that CIV flux typically exceeds ETS flux in most mitochondria (Pham et al., 2020; Po et al., 2016), our findings indicate that neither T2D nor MOTS-c treatment altered maximal oxygen flux through CIV activity (Figure 2J).
Compared to glucose oxidation, fatty acid oxidation is less efficient as it generates less ATP per oxygen unit consumed (Barclay and Loiselle, 2020). Plasma levels of free fatty acids and triglycerides have been reported to be higher in diabetic patients (Anderson et al., 2009) and rats (Tang et al., 2023). Interestingly, despite increased fatty acid transport in diabetic hearts (Bonen et al., 2009; Chabowski et al., 2008; Ljubkovic et al., 2019), maximal fatty acid-supported mitochondrial respiration was found to be unchanged in left ventricular tissues from T2D patients (Ljubkovic et al., 2019), or decreased in atrial tissues from T2D patients (Anderson et al., 2009; Croston et al., 2014) and left ventricular tissue in a T1D mouse model (Boudina et al., 2007), indicating a maladaptive metabolic response to abundant FA availability. Our data reveal no differences in mitochondrial respiration from fatty acid-supported substrates among groups, consistent with the report in T2D patient ventricular tissues (Ljubkovic et al., 2019), suggesting that the effect of T2D on FA-mediated mitochondrial respiration could be tissue region-dependent. We further note that the homogenate samples used in this study may contain some levels of endogenous fatty acids, which could confound our findings. Future research would focus on changes in fatty acid transport proteins and substrate oxidation pathways, and the use of isolated mitochondria to better understand energy substrate metabolism in both ventricular and atrial tissues of diabetic hearts.
ATP production
In this study, we assessed ATP production and mitochondrial respiration at the OXPHOS state to calculate the P/O ratio, the indicator of mitochondrial energy efficiency. Using various electron inputs from CI and CII substrates, the P/O ratio provides information about mitochondrial ATP synthesis and turnover capabilities. We found no differences in maximal ATP production rate or P/O ratio across groups, showing mitochondrial energy efficiency remained preserved both in diabetes and under treatment. This contradicts previous reports of lower mitochondrial ATP production in T1D rats (Pham et al., 2014), T2D mice (Boudina et al., 2007), and systemic hypertensive rats (Po et al., 2016). Interestingly, only T1D hearts (Pham et al., 2014) had a lower mitochondrial P/O ratio, highlighting disease-specific bioenergetic alterations. Previous research in senescent cell cultures (Lee et al., 2015; Kim et al., 2018) reported higher ATP content, most likely resulting from increased glucose uptake (Lee et al., 2015). However, our findings show no change in ATP production rate in MOTS-c treated diabetic mitochondria, suggesting that increased ATP content could arise from increased glycolysis pathway (Lee et al., 2015; Kim et al., 2018) instead of the oxidative phosphorylation process.
Real-time ATP measurements enabled us to determine ATP consumption during anoxia (Goo et al., 2013). Under anoxic circumstances, mitochondria halt electron transport and proton pumping while ATP synthase reverses to hydrolyse ATP to balance ionic homeostasis. Our data indicate that the mitochondrial ATP hydrolysis rate was significantly lower in MOTS-c treated diabetic group compared to untreated diabetic group. In other words, MOTS-c moderated lower ATP consumption during anoxia without compromising ATP production in normoxia. It has been demonstrated that diabetic hearts are more susceptible to ischemic insults (Paulson, 1997). Our findings suggest a protective role of MOTS-c in conserving ATP during ischemia while maintaining myocardial ATP levels during post-ischemic recovery.
H2O2 production
We simultaneously quantified mitochondrial H2O2 output and oxygen consumption rates at different respiratory states. Under conditions of high succinate and limited ADP, which simulate ischemic conditions (Chouchani et al., 2014), H2O2 production increased rapidly due to superoxide production at CI via reverse electron transport. In this respiratory state, the MOTS-c treatment group generated a higher H2O2 production, and this was more pronounced when expressed as a percentage of O2 flux (Figure 6I), compared to controls. Interestingly, transient bursts in H2O2 production may be important signals for cellular adaptation against stress (Ristow and Zarse, 2010; Yun and Finkel, 2014). The initial action of MOTS-c treatment could increase antioxidant responses by increasing protein expressions of superoxide dismutase, catalase, and glutathione peroxidase 4, as reported in a previous study on T1D hearts (Tang et al., 2023). This response is mediated through Nrf2 activation as a key regulator of antioxidant gene transcription (da Costa et al., 2019; Chen and Maltagliati, 2018; Merry et al., 2020b). This upregulated antioxidant defence system may protect cardiomyocytes from additional damage arising from increased oxidative stress, a known primary driver of diabetic cardiomyopathy pathogenesis (Anderson et al., 2009; Boudina et al., 2007; Pham et al., 2014; Ye et al., 2004).
MOTS-c doses and treatment duration
The biological activity and therapeutic effects of MOTS-c treatment are highly dose-dependent, resulting in significant variations in experimental results. Currently, no defined MOTS-c dosage ensures optimal treatment effects across different diseases. The literature describes various MOTS-c dosages, each generating diverse biological reactions. The reported doses include 0.5 mg/kg (Li et al., 2022; Yuan et al., 2021; Yang et al., 2024), 1 mg/kg (Xu et al., 2024), 5 mg/kg (Wu N. et al., 2023; Li et al., 2024; Jiang et al., 2023), 7.5 mg/kg (Zempo et al., 2021), 10 mg/kg (Yin et al., 2022), 15 mg/kg (Reynolds et al., 2021; Wu J. et al., 2023; Kumagai et al., 2024), or 50 mg/kg (Yin et al., 2020). Generally, lower doses (0.5–5 mg/kg) were usually taken over 8–12 weeks of treatment, whereas higher doses (10–15 mg/kg) were used in shorter periods, within 2–4 weeks. It is important to consider potential side effects associated with higher MOTS-c doses, including injection site responses and digestive disturbances. Our study, using the MOTS-c dose of 15 mg/kg daily over 3 weeks, was based on a previous study showing beneficial effects of MOTS-c in a similar timeframe and dose range (Reynolds et al., 2021). Future research is encouraged to explore the ideal MOTS-c dosage and treatment duration, all of which balance therapeutic efficacy and safety.
Conclusion
T2D rats exhibited impaired glucose-handling ability, gained more weight compared to the control group, and developed cardiac hypertrophy. T2D heart mitochondria showed decreased mitochondrial respiration without mitochondrial ATP production or ROS production changes. MOTS-c treatment improved glucose handling and reversed cardiac hypertrophy in diabetic rats. The MOTS-c treatment further restored mitochondrial respiration by increasing the mitochondrial content and decreasing the ATP hydrolysis rate. Our findings suggest that MOTS-c may be a promising treatment for mitochondrial dysfunction in diabetic settings.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by The University of Auckland Animal Ethics Committee. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
TP: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review and editing. AT: Conceptualization, Validation, Visualization, Writing – review and editing. AH: Conceptualization, Methodology, Validation, Visualization, Writing – review and editing. J-CH: Conceptualization, Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Heart Foundation of New Zealand Research Fellowship (1869, TP), Senior Research Fellowship (2027, J-CH) and Project Grant (1929, J-CH), Emerging Researcher First Grant (21/653, TP) from the Health Research Council of New Zealand, James Cook Research Fellowship (UOA1902, AT) and Marsden Project Grant (MFP-UOA2206, J-CH) from the Royal Society of New Zealand, and Lottery Health Shared Research Equipment grant from Lottery Grants Board of New Zealand (TP).
Acknowledgments
We want to acknowledge Linley Nisbet, Maryam Rahmani, and the Vernon Jansen Unit team for their practical assistance in animal husbandry. We also appreciate Troy Merry’s discussion during the early phase of the project design. Finally, we acknowledge the animals whose sacrifice made this research possible.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. Grammarly was used to polish the English.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Amaral N., Okonko D. O. (2015). Metabolic abnormalities of the heart in type II diabetes. Diab. Vasc. Dis. Res. 12 (4), 239–248. doi:10.1177/1479164115580936
Anderson E., Kypson A., Rodriguez E., Anderson C., Lehr E., Neufer P. (2009). Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 54 (20), 1891–1898. doi:10.1016/j.jacc.2009.07.031
Barclay C. J., Loiselle D. S. (2020). An equivocal final link - quantitative determination of the thermodynamic efficiency of ATP hydrolysis - sullies the chain of electric, ionic, mechanical and metabolic steps underlying cardiac contraction. Front. Physiol. 11, 183. doi:10.3389/fphys.2020.00183
Barth E., Stammler G., Speiser B., Schaper J. (1992). Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J. Mol. Cell Cardiol. 24 (7), 669–681. doi:10.1016/0022-2828(92)93381-s
Bonen A., Holloway G. P., Tandon N. N., Han X. X., McFarlan J., Glatz J. F., et al. (2009). Cardiac and skeletal muscle fatty acid transport and transporters and triacylglycerol and fatty acid oxidation in lean and Zucker diabetic fatty rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297 (4), R1202–R1212. doi:10.1152/ajpregu.90820.2008
Boudina S., Abel E. D. (2007). Diabetic cardiomyopathy revisited. Circulation 115 (25), 3213–3223. doi:10.1161/CIRCULATIONAHA.106.679597
Boudina S., Sena S., Theobald H., Sheng X., Wright J. J., Hu X. X., et al. (2007). Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 56 (10), 2457–2466. doi:10.2337/db07-0481
Chabowski A., Górski J., Glatz J. F., Jj P. L., Bonen A. (2008). Protein-mediated fatty acid uptake in the heart. Curr. Cardiol. Rev. 4 (1), 12–21. doi:10.2174/157340308783565429
Chen Q. M., Maltagliati A. J. (2018). Nrf2 at the heart of oxidative stress and cardiac protection. Physiol. Genomics 50 (2), 77–97. doi:10.1152/physiolgenomics.00041.2017
Chouchani E. T., Pell V. R., Gaude E., Aksentijević D., Sundier S. Y., Robb E. L., et al. (2014). Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515 (7527), 431–435. doi:10.1038/nature13909
Cole M. A., Murray A. J., Cochlin L. E., Heather L. C., McAleese S., Knight N. S., et al. (2011). A high fat diet increases mitochondrial fatty acid oxidation and uncoupling to decrease efficiency in rat heart. Basic Res. Cardiol. 106 (3), 447–457. doi:10.1007/s00395-011-0156-1
Croston T. L., Thapa D., Holden A. A., Tveter K. J., Lewis S. E., Shepherd D. L., et al. (2014). Functional deficiencies of subsarcolemmal mitochondria in the type 2 diabetic human heart. Am. J. Physiol. Heart Circ. Physiol. 307 (1), H54–H65. doi:10.1152/ajpheart.00845.2013
da Costa R. M., Rodrigues D., Pereira C. A., Silva J. F., Alves J. V., Lobato N. S., et al. (2019). Nrf2 as a potential mediator of cardiovascular risk in metabolic diseases. Front. Pharmacol. 10, 382. doi:10.3389/fphar.2019.00382
Du C., Zhang C., Wu W., Liang Y., Wang A., Wu S., et al. (2018). Circulating MOTS-c levels are decreased in obese male children and adolescents and associated with insulin resistance. Pediatr. Diabetes 19, 1058–1064. doi:10.1111/pedi.12685
Goo S., Pham T., Han J. C., Nielsen P., Taberner A., Hickey A., et al. (2013). Multiscale measurement of cardiac energetics. Clin. Exp. Pharmacol. Physiol. 40 (9), 671–681. doi:10.1111/1440-1681.12139
Guo X. X., Wang Y., Wang K., Ji B. P., Zhou F. (2018). Stability of a type 2 diabetes rat model induced by high-fat diet feeding with low-dose streptozotocin injection. J. Zhejiang Univ. Sci. B 19 (7), 559–569. doi:10.1631/jzus.B1700254
Hedges C. P., Pham T., Shetty B., Masson S. W. C., Hickey A. J. R., Shepherd P. R., et al. (2020). Prolonged treatment with a PI3K p110α inhibitor causes sex- and tissue-dependent changes in antioxidant content, but does not affect mitochondrial function. Biosci. Rep. 40 (10). doi:10.1042/BSR20201128
Jiang J., Chang X., Nie Y., Xu L., Yang L., Peng Y., et al. (2023). Orally administered MOTS-c analogue ameliorates dextran sulfate sodium-induced colitis by inhibiting inflammation and apoptosis. Eur. J. Pharmacol. 939, 175469. doi:10.1016/j.ejphar.2022.175469
Kim S. J., Mehta H. H., Wan J., Kuehnemann C., Chen J., Hu J. F., et al. (2018). Mitochondrial peptides modulate mitochondrial function during cellular senescence. Aging (Albany NY) 10 (6), 1239–1256. doi:10.18632/aging.101463
Kim S. J., Xiao J., Wan J., Cohen P., Yen K. (2017). Mitochondrially derived peptides as novel regulators of metabolism. J. Physiol. 595 (21), 6613–6621. doi:10.1113/JP274472
Kumagai H., Kim S. J., Miller B., Natsume T., Wan J., Kumagai M. E., et al. (2024). Mitochondrial-derived microprotein MOTS-c attenuates immobilization-induced skeletal muscle atrophy by suppressing lipid infiltration. Am. J. Physiol. Endocrinol. Metab. 326 (3), E207–E214. doi:10.1152/ajpendo.00285.2023
Lee C., Zeng J., Drew B. G., Sallam T., Martin-Montalvo A., Wan J., et al. (2015). The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 21 (3), 443–454. doi:10.1016/j.cmet.2015.02.009
Li F., Jia Y., Fang J., Gong L., Zhang Y., Wei S., et al. (2024). Neuroprotective mechanism of MOTS-c in TBI mice: insights from integrated transcriptomic and metabolomic analyses. Drug Des. Devel. Ther. 18, 2971–2987. doi:10.2147/DDDT.S460265
Li Q., Lu H., Hu G., Ye Z., Zhai D., Yan Z., et al. (2019). Earlier changes in mice after D-galactose treatment were improved by mitochondria derived small peptide MOTS-c. Biochem. Biophys. Res. Commun. 513 (2), 439–445. doi:10.1016/j.bbrc.2019.03.194
Li S., Wang M., Ma J., Pang X., Yuan J., Pan Y., et al. (2022). MOTS-C and exercise restore cardiac function by activating of NRG1-ErbB signaling in diabetic rats. Front. Endocrinol. (Lausanne) 13, 812032. doi:10.3389/fendo.2022.812032
Ljubkovic M., Gressette M., Bulat C., Cavar M., Bakovic D., Fabijanic D., et al. (2019). Disturbed fatty acid oxidation, endoplasmic reticulum stress, and apoptosis in left ventricle of patients with type 2 diabetes. Diabetes 68 (10), 1924–1933. doi:10.2337/db19-0423
Lopaschuk G. D., Ussher J. R., Folmes C. D., Jaswal J. S., Stanley W. C. (2010). Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 90 (1), 207–258. doi:10.1152/physrev.00015.2009
Lu H., Wei M., Zhai Y., Li Q., Ye Z., Wang L., et al. (2019). MOTS-c peptide regulates adipose homeostasis to prevent ovariectomy-induced metabolic dysfunction. J. Mol. Med. Berl. 97 (4), 473–485. doi:10.1007/s00109-018-01738-w
Mansor L. S., Gonzalez E. R., Cole M. A., Tyler D. J., Beeson J. H., Clarke K., et al. (2013). Cardiac metabolism in a new rat model of type 2 diabetes using high-fat diet with low dose streptozotocin. Cardiovasc. Diabetol. 12, 136. doi:10.1186/1475-2840-12-136
Marciniak C., Marechal X., Montaigne D., Neviere R., Lancel S. (2014). Cardiac contractile function and mitochondrial respiration in diabetes-related mouse models. Cardiovasc. Diabetol. 13 (1), 118. doi:10.1186/s12933-014-0118-7
Merry T. L., Chan A., Woodhead J. S. T., Reynolds J. C., Kumagai H., Kim S. J., et al. (2020a). Mitochondrial-derived peptides in energy metabolism. Am. J. Physiol. Endocrinol. Metab. 319 (4), E659–E666. doi:10.1152/ajpendo.00249.2020
Merry T. L., MacRae C., Pham T., Hedges C. P., Ristow M. (2020b). Deficiency in ROS-sensing nuclear factor erythroid 2-like 2 causes altered glucose and lipid homeostasis following exercise training. Am. J. Physiol. Cell Physiol. 318 (2), C337–C345. doi:10.1152/ajpcell.00426.2019
Montaigne D., Marechal X., Coisne A., Debry N., Modine T., Fayad G., et al. (2014). Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 130 (7), 554–564. doi:10.1161/CIRCULATIONAHA.113.008476
Paulson D. J. (1997). The diabetic heart is more sensitive to ischemic injury. Cardiovasc. Res. 34, 104–112. doi:10.1016/s0008-6363(97)00018-7
Pham T., Loiselle D., Power A., Hickey A. (2014). Mitochondrial inefficiencies and anoxic ATP hydrolysis capacities in diabetic rat heart. Am. J. Physiol. Cell Physiol. 307, C499–C507. doi:10.1152/ajpcell.00006.2014
Pham T., MacRae C. L., Broome S. C., D'Souza R F., Narang R., Wang H. W., et al. (2020). MitoQ and CoQ10 supplementation mildly suppresses skeletal muscle mitochondrial hydrogen peroxide levels without impacting mitochondrial function in middle-aged men. Eur. J. Appl. Physiol. 120 (7), 1657–1669. doi:10.1007/s00421-020-04396-4
Power A., Pearson N., Pham T., Cheung C., Phillips A., Hickey A. (2014). Uncoupling of oxidative phosphorylation and ATP synthase reversal within the hyperthermic heart. Physiol. Rep. 2 (9), e12138. doi:10.14814/phy2.12138
Power A., Pham T., Loiselle D., Crossman D., Ward M. L., Hickey A. (2016). Impaired ADP channeling to mitochondria and elevated reactive oxygen species in hypertensive hearts. Am. J. Physiol. Heart Circ. Physiol. 310, H1649–H1657. doi:10.1152/ajpheart.00050.2016
Qin Q., Delrio S., Wan J., Jay Widmer R., Cohen P., Lerman L. O., et al. (2018). Downregulation of circulating MOTS-c levels in patients with coronary endothelial dysfunction. Int. J. Cardiol. 254, 23–27. doi:10.1016/j.ijcard.2017.12.001
Ramanjaneya M., Bettahi I., Jerobin J., Chandra P., Abi Khalil C., Skarulis M., et al. (2019). Mitochondrial-derived peptides are down regulated in diabetes subjects. Front. Endocrinol. (Lausanne) 10, 331. doi:10.3389/fendo.2019.00331
Reynolds J. C., Lai R. W., Woodhead J. S. T., Joly J. H., Mitchell C. J., Cameron-Smith D., et al. (2021). MOTS-c is an exercise-induced mitochondrial-encoded regulator of age-dependent physical decline and muscle homeostasis. Nat. Commun. 12 (1), 470. doi:10.1038/s41467-020-20790-0
Ristow M., Zarse K. (2010). How increased oxidative stress promotes longevity and metabolic health: the concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 45 (6), 410–418. doi:10.1016/j.exger.2010.03.014
Sharma S., Adrogue J. V., Golfman L., Uray I., Lemm J., Youker K., et al. (2004). Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 18 (14), 1692–1700. doi:10.1096/fj.04-2263com
Shen C., Wang J., Feng M., Peng J., Du X., Chu H., et al. (2022). The mitochondrial-derived peptide MOTS-c attenuates oxidative stress injury and the inflammatory response of H9c2 cells through the Nrf2/ARE and NF-κB pathways. Cardiovasc. Eng. Technol. 13 (5), 651–661. doi:10.1007/s13239-021-00589-w
Stanley W. C., Lopaschuk G. D., McCormack J. G. (1997). Regulation of energy substrate metabolism in the diabetic heart. Cardiovasc. Res. 34 (1), 25–33. doi:10.1016/s0008-6363(97)00047-3
Tang M., Su Q., Duan Y., Fu Y., Liang M., Pan Y., et al. (2023). The role of MOTS-c-mediated antioxidant defense in aerobic exercise alleviating diabetic myocardial injury. Sci. Rep. 13 (1), 19781. doi:10.1038/s41598-023-47073-0
Wang M., Wang G., Pang X., Ma J., Yuan J., Pan Y., et al. (2022). MOTS-c repairs myocardial damage by inhibiting the CCN1/ERK1/2/EGR1 pathway in diabetic rats. Front. Nutr. 9, 1060684. doi:10.3389/fnut.2022.1060684
Wu J., Xiao D., Yu K., Shalamu K., He B., Zhang M. (2023b). The protective effect of the mitochondrial-derived peptide MOTS-c on LPS-induced septic cardiomyopathy. Acta Biochim. Biophys. Sin. (Shanghai) 55 (2), 285–294. doi:10.3724/abbs.2023006
Wu N., Shen C., Wang J., Chen X., Zhong P. (2023a). MOTS-C peptide attenuated diabetic cardiomyopathy in STZ-induced type 1 diabetic mouse model. Cardiovasc. Drugs Ther. 39, 491–498. doi:10.1007/s10557-023-07540-2
Xu L., Tang X., Yang L., Chang M., Xu Y., Chen Q., et al. (2024). Mitochondria-derived peptide is an effective target for treating streptozotocin induced painful diabetic neuropathy through induction of activated protein kinase/peroxisome proliferator-activated receptor gamma coactivator 1alpha -mediated mitochondrial biogenesis. Mol. Pain 20, 17448069241252654. doi:10.1177/17448069241252654
Yang L., Li M., Liu Y., Bai Y., Yin T., Chen Y., et al. (2024). MOTS-c is an effective target for treating cancer-induced bone pain through the induction of AMPK-mediated mitochondrial biogenesis. Acta Biochim. Biophys. Sin. (Shanghai) 56 (9), 1323–1339. doi:10.3724/abbs.2024048
Ye G., Metreveli N., Donthi R., Xia S., Xu M., Carlson E., et al. (2004). Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes 53, 1336–1343. doi:10.2337/diabetes.53.5.1336
Yin X., Jing Y., Chen Q., Abbas A. B., Hu J., Xu H. (2020). The intraperitoneal administration of MOTS-c produces antinociceptive and anti-inflammatory effects through the activation of AMPK pathway in the mouse formalin test. Eur. J. Pharmacol. 870, 172909. doi:10.1016/j.ejphar.2020.172909
Yin Y., Pan Y., He J., Zhong H., Wu Y., Ji C., et al. (2022). The mitochondrial-derived peptide MOTS-c relieves hyperglycemia and insulin resistance in gestational diabetes mellitus. Pharmacol. Res. 175, 105987. doi:10.1016/j.phrs.2021.105987
Yuan J., Wang M., Pan Y., Liang M., Fu Y., Duan Y., et al. (2021). The mitochondrial signaling peptide MOTS-c improves myocardial performance during exercise training in rats. Sci. Rep. 11 (1), 20077. doi:10.1038/s41598-021-99568-3
Zempo H., Kim S. J., Fuku N., Nishida Y., Higaki Y., Wan J., et al. (2021). A pro-diabetogenic mtDNA polymorphism in the mitochondrial-derived peptide, MOTS-c. Aging (Albany NY) 13 (2), 1692–1717. doi:10.18632/aging.202529
Zhong P., Peng J., Hu Y., Zhang J., Shen C. (2022). Mitochondrial derived peptide MOTS-c prevents the development of heart failure under pressure overload conditions in mice. J. Cell Mol. Med. 26 (21), 5369–5378. doi:10.1111/jcmm.17551
Keywords: MOTS-c, diabetic heart, mitochondrial respiration, ATP, reactive oxygen species
Citation: Pham T, Taberner A, Hickey A and Han J-C (2025) Mitochondria-derived peptide MOTS-c restores mitochondrial respiration in type 2 diabetic heart. Front. Physiol. 16:1602271. doi: 10.3389/fphys.2025.1602271
Received: 29 March 2025; Accepted: 02 June 2025;
Published: 30 June 2025.
Edited by:
Nazzareno Capitanio, University of Foggia, ItalyReviewed by:
Junxiao Ren, University of Florida, United StatesKasja Pavlović, University of Belgrade, Serbia
Copyright © 2025 Pham, Taberner, Hickey and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Toan Pham, dG9hbi5waGFtQGF1Y2tsYW5kLmFjLm56