 Jingli Gao1†
Jingli Gao1† Liuyifei Huang1†Yuzhan Zhang1
Liuyifei Huang1†Yuzhan Zhang1 Lei Wei1
Lei Wei1 Zhixiang Yu1
Zhixiang Yu1 Yan Xing1Jinguo Yuan1
Yan Xing1Jinguo Yuan1 Xiaoxuan Ning2*
Xiaoxuan Ning2* Shiren Sun1*
Shiren Sun1*- 1Department of Nephrology, Xijing Hospital, Fourth Military Medical University, Xi’an, China
- 2Department of Geriatric, Xijing Hospital, Air Force Medical University, Xi’an, China
Acute kidney injury (AKI) represents a clinical syndrome with a bleak short-term prognosis, posing a high risk for the development of chronic kidney diseases and end-stage kidney disease. The underlying mechanisms of AKI are still not fully understood, and effective intervention strategies remain elusive. Enormous energy is required to meet the functional activity in hypermetabolic tubular epithelial cells (TECs), the most vulnerable cell types during AKI. Recent evidence has shed light on the reprogramming of metabolic pathways and the shift in energy substrates under pathological conditions. The reprogrammed metabolic pathway initially serves to compensate for energy shortages and supply substrates for cell repair during the early stages of AKI. However, sustained metabolic dysregulation tend to become detrimental for tubular repair and regeneration. Intriguingly, dynamic alterations in specific metabolites extend beyond their conventional roles as metabolic byproducts, actively participating in pathophysiology through multifaceted regulatory mechanisms during AKI. As yet, clinical therapy for AKI has not yet incorporated the intervention of metabolic disorders, highlighting a vast potential for extensive application. This review aims to summarize recent studies on the role of metabolic pathway reprogramming and metabolites in AKI, while discussing promising therapeutic strategies targeting metabolic reprogramming.
1 Introduction
Acute kidney injury (AKI) is characterized by an abrupt deterioration of kidney function, manifested by elevated serum creatinine levels and decreased urinary output within a 7-day period. The etiologies of AKI are categorized into pre-renal, renal, and post-renal factors, primarily involving ischemia/reperfusion injury (IRI), toxins, and urinary obstruction (Kellum et al., 2021). Patients with AKI face an 8.8-fold higher risk of developing chronic kidney disease (CKD), a 3.1-fold higher risk of end stage renal disease (ESRD), and a 2.0-fold higher risk of mortality compared to non-AKI populations (Coca et al., 2012). Among the most abundant intrinsic renal cells, tubular epithelial cells (TECs) are extremely sensitive and susceptible to AKI. The fate of TECs and kidney tissue is influenced by various pathological processes, including but not limited to inflammation (Matsushita et al., 2020), oxidative stress (Aranda-Rivera et al., 2021), cell cycle arrest (De Chiara et al., 2021), epigenetic regulation (Guo et al., 2019), and gut microbiota dysbiosis (Saranya and Viswanathan, 2023).
In recent decades, there has been a growing focus on the role and function of cellular metabolism of organs in disease processes. Maintaining metabolic homeostasis is fundamental requirement for cell growth, proliferation, and the specialized physiological functions (O'Brien et al., 2020). TECs have high metabolic activity and primarily rely on fatty acid oxidation (FAO) rather than glucose oxidation for their energy supply under physiological conditions. Complementary energy substrates including amino acids (AAs), ketone bodies, pyruvate, and lactate further contribute as fuel for aerobic respiration to generate adenosine triphosphate (ATP) in tubular cells (Scholz et al., 2021). Pathological stress induces profound metabolic alterationscharacterized by mitochondrial dysfunction and an imbalance of 5′-adenosine monophosphate (AMP)-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) in TECs. This metabolic reprogramming recapitulates the embryonic development and differentiation patterns observed in nephron progenitor cells (NPCs) which initially serving as an adaptive response. Paradoxically, sustained metabolism reprogramming contributes to maladaptive repair of TECs and deterioration in kidney function (Zhu et al., 2022; Wang G. et al., 2022).
Apart from metabolic disorders, endogenous intermediate metabolites play a role not only as products of the metabolic process but also as influencers of the outcome of multiple tissues during nutrient stress. Endogenous intermediate metabolites exert multifaceted regulatory effects through two principal mechanisms, (1) transducing signaling pathways by binding with receptors, (2) covalently modification of amino acid residues via post-translational modifications (PTMs) which may affect protein localization, conformational properties, and intermolecular interaction networks by competing with other binding partners (Figlia et al., 2020; Wang and Lei, 2018).
Currently, there are few effective or potent therapies available for halting the progression of AKI. Given that the dysregulation of metabolic homeostasis considerably disrupts cells and tissues, exploring key metabolic pathways and metabolites may yield novel biomarkers and therapeutic strategies for AKI. In this review, we aim to elucidate how reprogrammed metabolic pathways, along with the differential metabolites, dedicate TECs fate and renal outcome in AKI, with particular emphasis on their translational potential for AKI management (Table 1).
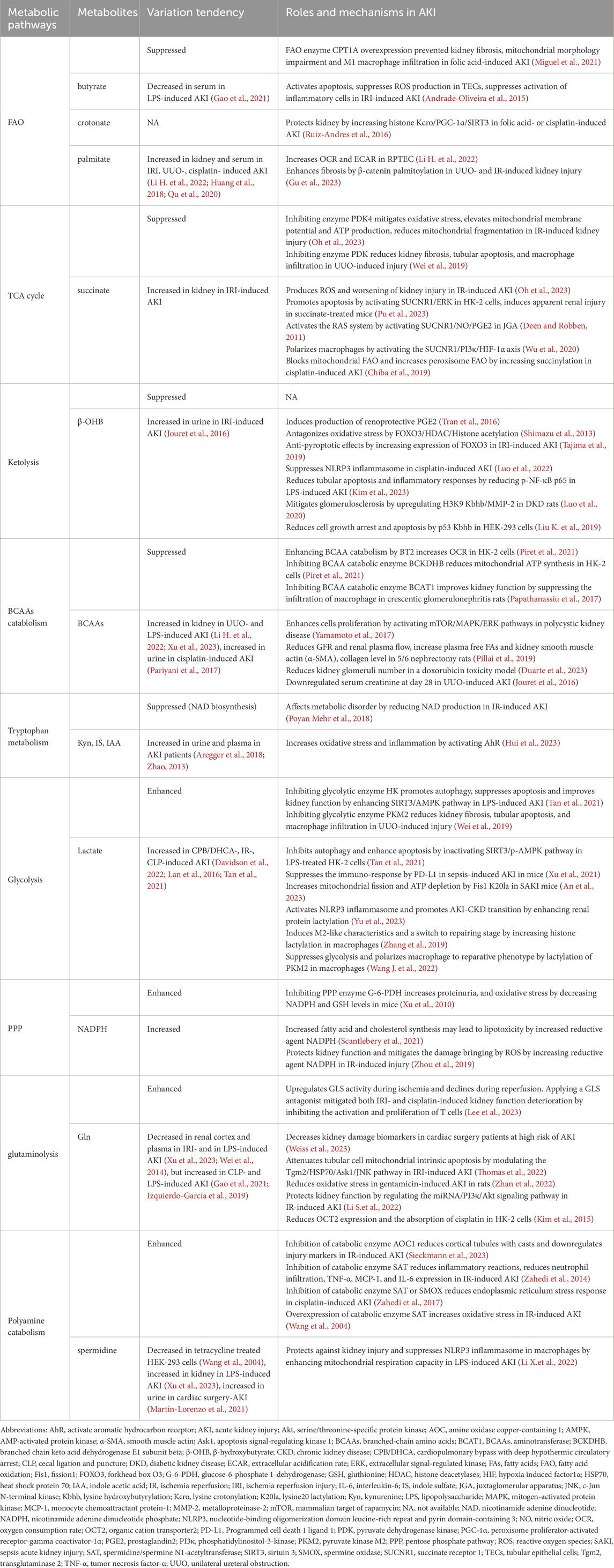
Table 1. Roles of metabolic pathways and metabolites in signal transduction, protein modifications, and the effects on AKI.
2 Downregulated metabolic pathways
2.1 Fatty acid oxidation
Fatty acids (FAs) are transported by the CD36 membrane glycoprotein, fatty acid binding protein (FABP), and fatty acid transport protein (FATP) in TECs. In the mitochondrial membrane, long-chain fatty acids (LCFAs, C18 ≤ C22), medium-chain fatty acids (MCFAs, C6≤C12), and short-chain fatty acids (SCFAs, ≤C6) are catalyzed by long/medium/short-chain acyl-CoA synthetase (ACSL, ACSM, ACSS) to produce acyl coenzyme A (acyl-CoA) (Figure 1). Carnitine palmitoyl-transferase 1A (CPT1A) acts as a crucial, speed-limiting enzyme on the inner membrane of mitochondria. It catalyzes the transformation of acyl-CoA from coenzyme A to L-carnitine, generating acyl-carnitine to facilitate the transfer of fatty acids from cytosol to the mitochondria. Then palmitoyl-transferase 2 (CPT2) reversely releases acyl-carnitine, reverting it to acyl-CoA and carnitine in mitochondrial matrix. Subsequently, acyl-CoA undergoes stepwise oxidation to acetyl coenzyme A (Ac-CoA), which then enters the tricarboxylic acid (TCA) cycle. This cycle produces nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2), both participating in oxidative phosphorylation to produce ATP and H2O. Additionally, FAO can also take place in peroxisomes, which mainly catalyze very long chain fatty acids (VLCFAs, ≥C22) into SCFAs. These degraded SCFAs can be transferred to mitochondria or cytosol to be utilized or secreted.
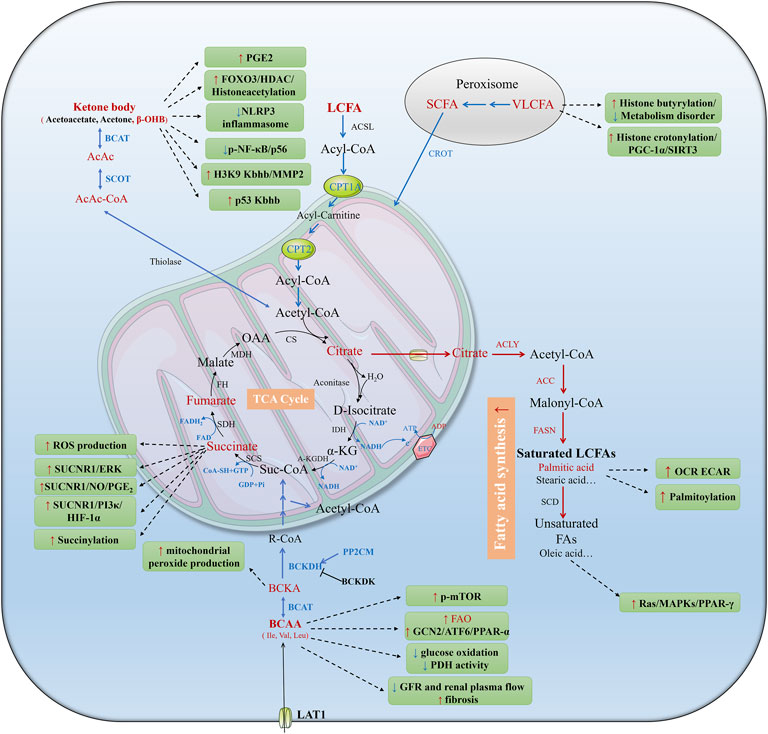
Figure 1. Metabolic reprogramming of tubular epithelial cells (TECs) during AKI. AKI can be induced by hypoxia, toxins, or urinary obstruction. AKI results in mitochondrial dysfunction and low oxygen availability which impedes the electron transport chain and aerobic respiration. The shift from FAO to glycolysis is widely studied in AKI. In addition, ketolysis, BCAA catabolism, glutaminolysis, polyamine metabolism and their metabolites play different roles in AKI. Red and blue fonts and arrows mean up- and downregulated pathways and metabolites in AKI, black fonts and arrows mean unidentified pathways and metabolites in previous studies. BCAAs, branched-chain amino acids; Suc-CoA, succinyl coenzyme; acetyl-CoA, acetyl coenzyme; acyl-CoA, acyl coenzyme; TCA, tricarboxylic acid; Gln, glutamine; Glu, glutamate; α-KG, α-ketoglutarate; NAD, nicotinamide adenine dinucleotide; FAD, flavin adenine dinucleotide; NADPH, nicotinamide adenine dinucleotide phosphate; G-6-P, glucose-6-phosphate; PPP, pentose phosphate pathway; FA, fatty acid; FAO, fatty acid oxidation; FAS, fatty acid synthesis; CPT1, carnitine palmitoyl-transferase 1; CPT2, carnitine palmitoyl-transferase 2; Try, tryptophan; QUIN, quinolinic acid; ATP, adenosine triphosphate; ADP, adenosine diphosphate; ETC, electron transport chain; e−, electron; GABA, γ-aminobutyric acid; Put, putrescine; Spd, spermidine; Spm, spermine. GSSG, oxidized glutathione; GSH, reduced glutathione; H2O2, hydrogen peroxide.
2.1.1 Suppressed FAO during AKI
During AKI, mitochondrial dysfunction can disrupt the electron transport chain (ETC). Ischemia-induced low oxygen availability hampers electron transport in the ETC, ultimately inhibiting substrate oxidation in the TCA cycle. Consequently, upstream FAO is also halted. Applying single-cell combinatorial indexing RNA sequencing in uIRI (unilateral ischemia reperfusion injury) and UUO (unilateral ureteral obstruction) mice, Li et al. found a negative correlation between the proportion of failed-repaired proximal tubular cells (FR-PTC) and FAO activity (Li H. et al., 2022). Lipid droplet accumulation significantly increased in uIRI at 6 h on Day (D)2 and decreased nearly to baseline at D7 and D14. By contrast, lipid droplets gradually accumulated gradually over time in UUO mice (Li H. et al., 2022). Kidney transplant recipients who underwent severe IRI were found to have enrichment of LCFAs in their urine (Rinaldi et al., 2022).
Researchers observed that kidney CD36 scavenger receptor significantly increased in patients with AKI and in mice with cisplatin-induced AKI (Ma et al., 2024). Tubule-specific CD36 overexpression exacerbated proteinuria and fibrosis in mice with folic acid-induced AKI (Jung et al., 2018), indicating that increased CD36 was associated with the progression of AKI. A study revealed that kidney CPT1A levels in patients with CKD, and kidney CPT2 levels in patients post-kidney transplantation were significantly correlated with eGFR (Miguel et al., 2021). In addition, serum levels of short- and middle-chain acylcarnitines were elevated in patients with CKD group compared to healthy controls (Miguel et al., 2021), indicating a reduction of fatty acid transportation into the mitochondria. Treatment with the CPT1 inhibitor, etomoxir, suppressed ATP production, elevated apoptosis, and led to dedifferentiation in TECs (Kang et al., 2015). In a study by Miguel et al., tubule-specific overexpression of CPT1A in mice prevented kidney fibrosis, mitochondrial morphology impairment, and M1 macrophage infiltration induced by folic acid nephropathy (Miguel et al., 2021). Furthermore, the speed-limiting enzyme for FAO, acyl-coenzyme A oxidase 1 (ACOX), was found to be decreased after IR-induced injury in TECs, showing a negative correlation with kidney function (Chen et al., 2023). Carnitine O-Octanoyltransferase (CROT), responsible for transferring fatty acids from peroxisomes to mitochondria for FAO, and alpha-methylacyl-CoA racemase (AMACR), involved in peroxisome β-oxidation, were also decreased in IR-induced AKI (Chen et al., 2023). These studies collectively suggest the suppression of both mitochondrial and peroxisomal FAO.
2.1.2 Metabolites of FAO
The intermediate carboxylic acids generate several acyl-CoAs, and these acyl-CoAs could emerge as “donors” transferred to AA residuals, such as lysine, by acyltransferase or removed by deacylase. Acylated proteins resulting from these processes can have an impact on various signaling functions. While enzymes like lysine acetyltransferase (KAT) and histone deacetylase (HDAC), responsible for adding and removing acetyl-CoA, have been extensively studied in the past, it is essential to recognize that the abundance of metabolites can also affect post-translational modification either enzymatically or non-enzymatically (Simithy et al., 2017;Sabari et al., 2017).
As a LCFA, palmitate accumulated in both kidney and serum during AKI induced by IRI, UUO, and cisplatin (Li H. et al., 2022; Huang et al., 2018; Qu et al., 2020). Palmitate treatment has been shown to increase both the oxygen consumption rate and extracellular acidification rate simultaneously (Li H. et al., 2022), indicating an enhancement of FAO and glycolysis at the same time in human renal proximal TECs. However, Gu et al. observed a decrease in palmitoyltransferase which significantly downregulated palmitoylation of β-catenin and delayed degradation of β-catenin, ultimately contributing to enhanced fibrosis in both UUO- and IR-induced AKI in mice, as well as in TGF-β1 stimulated TECs (Gu et al., 2023). Oleic acid, an unsaturated LCFA, could have a protective effect by mitigating inflammation and oxidative stress through the Ras/MAPKs/PPAR-γ signaling pathway in lipopolysaccharide (LPS)-induced AKI (Zhang B. et al., 2022).
SCFAs, including butyrate, crotonate, malonate, acetate, etc., are composed of 1-6 carbon atoms. SCFAs are mainly derived from the intestinal microbiota but can also originate from the β-oxidation of fatty acids and fatty acid synthesis in cytosol. They can bind with the G protein-coupled receptors and regulate pathophysiological processes (Pluznick, 2014). The levels of SCFAs, especially butyrate, were found to be significantly lower in patients with CKD compared to healthy individuals (Wang et al., 2019). Additionally, serum butyrate levels was significantly downregulated in LPS-induced AKI models. (Gao et al., 2021). Butyrate treatment has demonstrated protective effects on the kidneys during IR-induced injury by activating apoptosis, suppressing reactive oxygen species production in TECs, and suppressing the activation of inflammatory cells (Andrade-Oliveira et al., 2015). Systemic administration of sodium butyrate increased histone butyrylation, ameliorated lipid and glucose metabolic disorders, and attenuated renal inflammation and fibrosis, while the protective effect could be eliminated by histone modification enzyme p300 inhibitor A485 in diabetic kidney disease (DKD)mice (Zhou et al., 2022).However, the roles of butyrate and butyrylation need to be further elucidated in AKI.
As a byproduct of fatty acid β-oxidation, crotonate and histone crotonylation were found to coincide periodically with the expression of fatty acid β-oxidation genes in yeast (Gowans et al., 2019), and their levels could be regulated by FAO enzymes (Zhang Y. et al., 2022). Conversely, Ruiz-Andres et al. demonstrated an increase in histone crotonylation in folic acid- and cisplatin-induced AKI (Ruiz-Andres et al., 2016). The administration of crotonate was shown to upregulate histone crotonylation, prevent the reduction of peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC-1α), and maintain sirtuin 3 (SIRT3) expression, ultimately offering protection against AKI (Ruiz-Andres et al., 2016).
2.1.3 Metabolites in TCA cycle
Low oxygen levels impede oxidative phosphorylation, resulting in the accumulation of intermediate metabolites in the TCA cycle. Renal metabolites in the TCA cycle, including succinate and cis-aconitate, are increased in UUO-induced AKI (Zhao et al., 2016). Additionally, serum levels of citrate, succinate, and fumarate, along with kidney succinate, are increased in cisplatin-induced AKI (Qu et al., 2020).
Succinate that accumulated during hypoxia underwent oxidation during the reperfusion stage, leading to a rapid induction of reactive oxygen species in multiple tissues (Chouchani et al., 2014). Pharmacological inhibition of succinate accumulation has been shown to protect the heart, brain (Chouchani et al., 2014), and kidneys (Oh et al., 2023) from IRI. Succinate can also be secreted into the interstitium, activating succinate receptor 1 in various cell types and triggering several signaling pathways, including upregulating extracellular signal-regulated kinase and promoting apoptosis in HK-2 cells (Pu et al., 2023). This induces the secretion of nitric oxide and prostaglandin2 (PGE2), thereby activating the renin-angiotensin system in the juxtaglomerular apparatus (Deen and Robben, 2011), and affecting macrophage polarization through the phosphatidylinositol-3 kinase/hypoxia-induced factor-1α axis (Wu et al., 2020). Succinate and α-ketoglutaric acid can also serve as “donors” for lysine succinylation, which has been found to increase during IR-induced AKI. Sirtuin5 serves as a succinylation deacylase, and sirtuin5 knockout mice exhibited hypersuccinylation, protecting mice from cisplatin-induced AKI (Chiba et al., 2019). This protection was attributed to the blockage of mitochondrial FAO and the augmentation of peroxisome FAO, which reduced oxidative stress and compensated for the energy shortage in TECs.
Ac-CoA, acetate, and α-ketoglutaric acid are more than intermediate metabolites during aerobic respiration, also participating in covalent donors to regulate protein lysine acetylation (Sivanand et al., 2018). Acetylation is a transient and reversible protein modification, highly correlated with the concentration of these “donors” (Trefely et al., 2020). Kidney tissue acetate concentration is reduced in rats with UUO-induced AKI (Zhao et al., 2016). One study showed a decrease in histone acetylation in kidneys exposed to IRI and in HK-2 cells subjected to hypoxia/reoxygenation (Tajima et al., 2019). Transforming growth factor β1 stimulation induced a shift from aerobic respiratory to glycolysis, leading to a reduction in Ac-CoA concentration and histone 3 acetylation, thereby activating renal myofibroblasts and causing renal fibrosis (Smith et al., 2019). On the other hand, Hewitson et al. found that global acetylation of histone H3 at lysine 9 (H3K9) increased in TECs after UUO compared with a control group (Hewitson et al., 2017). Beyond histones, acetylation also took place in nonhistone proteins such as p53, with increased acetylation observed in sepsis-induced AKI mice. This acetylation of p53 suppressed autophagy and exacerbated tubular injury (Sun et al., 2021). These findings underscore that the availability of Ac-CoA, along with the activity and expression of histone acetyltransferases and histone deacetylases, are predominant factors influencing protein acetylation.
2.2 Ketone body oxidation
Ketone bodies comprise acetoacetate, acetone, and β-hydroxybutyrate (β-OHB), generated through the degradation of fatty acids in the liver and subsequently transported to peripheral tissues for utilization as a fuel source. In peripheral tissues, β-OHB dehydrogenase 1 (BDH1) oxidizes β-OHB to acetoacetate. Acetoacetate is then activated to acetoacetyl-CoA by succinyl-CoA:3 oxoacid-CoA transferase (SCOT), and further catalyzed by acetyl-CoA acetyltransferase (ACAT) to produce two molecules of Ac-CoA.
Some evidence suggests that ketolysis might be suppressed in AKI. Proteomic analysis revealed a significant decrease in the levels of ketone body oxidative enzymes, including BDH1, SCOT, and ACAT1 in the kidneys of mice with LPS-induced AKI (Xu et al., 2023). Single cell RNA sequencing showed an initial decrease followed by a gradual recovery in the gene expression of Bdh1, Acat1, and Oxct1 (SCOT) in the proximal tubule of uIRI mice (Kirita et al., 2020). In contrast, these genes exhibited a continuous decrease from the outset in UUO mice (Li H. et al., 2022) (single cell sequencing database: http://humphreyslab.com/SingleCell/). Consistent with these findings, urine β-OHB was significantly increased in IRI-induced AKI mice (Jouret et al., 2016), and urine acetoacetate levels were elevated in cisplatin-induced AKI rats (Pariyani et al., 2017).
In addition to its involvement in ketolysis, the metabolite β-OHB also interacted with AKI. Administration of β-OHB protects kidneys via various mechanisms. These include inducing renoprotective PGE2 production (Tran et al., 2016), inhibiting histone deacetylase and increasing global histone acetylation, further increasing forkhead box O3 (FOXO3) and antagonizing oxidative stress (Shimazu et al., 2013). β-OHB suppresses the mTOR signal pathway in DKD mice (Tomita et al., 2020), and exhibits anti-pyroptotic effects by increasing expression of FOXO3 in IR-induced AKI (Tajima et al., 2019). Moreover, β-OHB reduced the inflammatory response, oxidative stress, and tubular injury by decreasing Phospho-nuclear factor kappa-light-chain-enhancer of activated B cells subunit 65 (p-NF-κB p65) expression in LPS-induced AKI, and by suppressing the nucleotide-binding oligomerization domain leucine-rich repeat and pyrin domain-containing 3 (NLRP3) inflammasome in Cisplatin-induced AKI (Kim et al., 2023; Luo et al., 2022; Kim et al., 2024). β-OHB also plays signaling roles by inducing lysine hydroxybutyrylation (Kbhb) (Xie et al., 2016) in both histone and non-histone proteins. Administration of β-OHB increased H3K9 Kbhb, upregulated metalloproteinase-2 (MMP-2) expression and downregulated collagen Ⅳ levels, mitigating glomerulosclerosis in DKD rats (Luo et al., 2020). Additionally, β-OHB increased p53 Kbhb, inactivating p53 and resulting in the reduction of cell growth arrest and apoptosis in cultured cells (Liu K. et al., 2019). Although the protective effects of β-OHB in AKI are emerging, the characteristics and roles of ketolysis and Kbhb remain unclear in AKI and require further exploration.
2.3 BCAAs catabolism
Branched-chain amino acids (BCAAs) are essential AAs that cannot be synthesized in vivo and must be obtained from the diet. The three primary BCAAs are leucine, isoleucine, and valine. BCAA aminotransferase (BCAT1) initiates the catabolic metabolism of BCAAs, facilitating the transfer of amino from BCAAs to other keto acids and generating branched-chain keto acids (BCKAs). Subsequently, BCKA dehydrogenase (BCKD) catalyzes BCKAs. BCKD kinase (BCKDK) phosphorylates and inactivates BCKD, while the mitochondrion-localized protein phosphatase-2C (PP2Cm) specifically dephosphorylates and activates BCKD (Nie et al., 2018). Through a series of reactions, in vivo isotopic tracing shows that BCAAs produce Ac-CoA and succinyl-CoA, engaging in the TCA cycle (Neinast et al., 2019).
With regard to the kidney, proteomics revealed a significant decrease in the protein levels of BCAA catabolic enzymes, including BCKD E1 subunit alpha (BCKDHA), BCKD E1 subunit beta (BCKHB), mitochondrial medium-chain specific acyl-CoA dehydrogenase (ACADM), isovaleryl-CoA dehydrogenase (IVD), 3-hydroxyisobutyryl-CoA hydrolase (HIBCH), and methylcrotonoyl-CoA carboxylase (MCCC1, MCCC2), in LPS-induced AKI in mice kidney (Xu et al., 2023). The gene expression of kidney BCAAs catabolism-related genes, such as Bckdha, Bckdhb, Acadm, Mut, Ivd, Hibch, Mccc1, and Mccc2, significantly decreased in Cisplatin, aristolochic acid I-induced and UUO-induced AKI (Piret et al., 2021). Similarly, the expression of Bckdha, Bckdhb, and Ppm1k genes was downregulated in injured proximal tubules (Li H. et al., 2022). In addition, the gene expression of BCAA catabolic enzymes positively correlated with eGFR in human species (Piret et al., 2021). Further experiments revealed that BCKDHB knock out reduced mitochondrial ATP synthesis, and that the BCAA catabolism enhancer BT2 improved tubular ferroptosis (Sone et al., 2023) and increased the oxygen consumption rate in HK-2 cells (Piret et al., 2021). Activation of BCAA catabolism improved renal fibrosis, epithelial-mesenchymal transition and inflammation in DKD mice (Deng et al., 2025). These findings demonstrated that BCAA catabolism was suppressed in AKI, potentially exacerbating the energy shortage and affecting cell death. In addition, the catabolism of BCAAs also affects macrophages; the BCAT1 inhibitor ERG240 downregulated oxygen consumption and glycolysis in LPS-treated macrophages, leading to a less pro-inflammatory phenotype (Papathanassiu et al., 2017). Inhibition of BCAT1 reduced glomerular crescents, serum creatinine, and proteinuria levels by suppressing macrophage infiltration in crescentic glomerulonephritis rats (Papathanassiu et al., 2017). Additionally, Shen J et al. found that cerebral BCAAs accumulated due to microbiota changes in ischemic stroke rats. BCAAs activated the protein kinase B/activator of transcription/NF-κB axis, exacerbating microglia-induced neuroinflammation (Shen et al., 2023).
Impaired BCAA catabolism leads to the accumulation of BCAAs and BCKAs, The concentration of BCAAs increased in mice kidney cortical tissue during UUO (Li H. et al., 2022), while kidney isoleucine and valine concentrations were elevated in LPS (Xu et al., 2023)- and cardiopulmonary bypass with deep hypothermic circulatory arrest (CPB/DHCA) (Davidson et al., 2022)-induced AKI. In rats with cisplatin-induced AKI, urine leucine and valine levels increased (Pariyani et al., 2017). In human, urinary leucine was proven to be positively correlated with the degree of contrast-induced acute kidney injury (Cheng et al., 2022), and another study showed increased urinary valine and leucine have a good prediction efficacy for pediatric AKI (Muhle-Goll et al., 2020). By contrast, another study reported a decrease in urine and kidney BCAAs during UUO-induced injury (Jouret et al., 2016), as well as a decrease in isoleucine and valine in plasma during UIRI injury (Shan et al., 2023). These discrepancies may arise from variations in disease stages, experimental methodologies, or tissue-specific metabolic responses.BCAAs, particularly leucine, mainly transmit signals by targeting mTOR pathway, participating in protein synthesis, cell proliferation, inflammation, and oxidative stress in various tissues and cell types (Zhenyukh et al., 2017; Yamamoto et al., 2017; Zhang et al., 2016). BCKAs, in a dosage-dependent manner, promoted mitochondrial peroxide production (Sun et al., 2016). Moreover, BCAAs impact pathological outcomes by regulating metabolic pathways. Accumulated BCAAs enhanced FAO by activating the general control nonderepresible-2 (GCN2)/activating transcription factor-6 (ATF6)/peroxisome proliferation-activated receptor alpha (PPAR-α) pathway (Li et al., 2020) and suppressed glucose oxidation by inhibiting pyruvate dehydrogenase activity (Li et al., 2017) in IR-induced myocardial injury. In the liver, BCAAs suppressed lipogenesis by blocking protein kinase B2/sterol regulatory element-binding protein/insulin-induced gene 2a signaling and enhanced glycogenesis by regulating protein kinase B 2/FOXO1 signaling (Zhao et al., 2020). In the kidney, BCAAs supplementation reduced GFR and renal plasma flow, increased plasma free fatty acids, and kidney smooth muscle actin collagen levels in 5/6 nephrectomy rats (Pillai et al., 2019). A L-Leucine-rich diet reduced kidney glomeruli numbers in a doxorubicin toxicity model (Duarte et al., 2023). However, another study demonstrated that the administration of BCAAs downregulated serum creatinine at D28 after UUO in rats, suggesting that BCAAs might partially prevent UUO-induced AKI (Jouret et al., 2016). Nonetheless, it remains unclear whether excessive BCAAs activate signaling transduction or enhance BCAA catabolism to compensate for the energy shortage in AKI.
2.4 Tryptophan metabolism
Tryptophan undergoes metabolism through three ways: the kynurenine pathway, the indole pathway, and the serotonin pathway (Hui et al., 2023). In the kynurenine pathway, tryptophan transforms into quinolinic acid which is then catalyzed by the speed-limiting enzyme quinolinate phosphoribosyltransferase, which is abundant in TECs. The process results in the de novo synthesis of nicotinamide adenine dinucleotide (NAD), an electron acceptor in TCA cycle. However, Mehr et al. discovered a downregulation of quinolinate phosphoribosyltransferase expression in mice, leading to increased renal and urine quinolinic acid levels and a decrease in NAD levels, and this shift in metabolism contributed to the adverse outcomes of IR-induced AKI (Poyan Mehr et al., 2018). Furthermore, other metabolites of tryptophan, such as kynurenine and indoxyl sulfate, activate the aromatic hydrocarbon receptor, initiating oxidative stress and inflammation that mediate kidney pathogenesis (Hui et al., 2023).
3 Upregulated metabolic pathways
3.1 Glycolysis
Glycolysis is a process that catabolizes glucose and supplies ATP without oxygen in the cytosol (Figure 2). Glucose uptake is mainly mediated by sodium glucose transporter 1/2 from the tubular lumen into proximal tubular cells, and glucose transporter type 1/2 which lies on the basolateral membrane and is responsible for transferring glucose intracellularly to the interstitium, or the reverse. Glucose is initially phosphorylated by hexokinase to glucose-6-phosphate (G-6-P). After a series of reactions, G-6-P produces phosphoenolpyruvate, which is then catalyzed by pyruvate kinase M2 to generate pyruvate. Pyruvate is transferred into mitochondria and catalyzed by pyruvate dehydrogenase to produce Ac-CoA, participating in the TCA cycle under normoxia conditions. By contrast, under hypoxic conditions, pyruvate is catalyzed by lactate dehydrogenase to form lactate. The early proximal TECs reabsorb glucose from the tubule lumen, but owning to scarce hexokinase, they hardly use glucose as energy substrate. The situation is reversed in medulla TECs due to low oxygen tension in healthy conditions (Klein et al., 1981; Uchida and Endou, 1988).
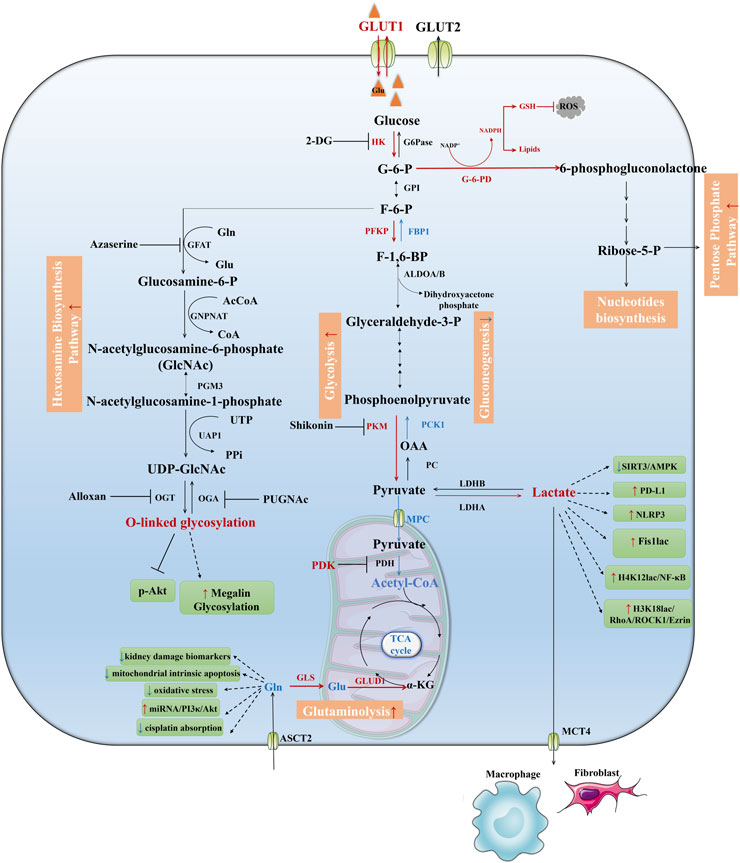
Figure 2. Downregulated pathways in TECs during AKI. Fatty acids are transported into the mitochondria for oxidation as acylcarnitines through the activity of carnitine CPT1 and CPT2, then acyl-CoA undergoes stepwise oxidation to Ac-CoA and enters the TCA cycle which coupling with oxidative phosphorylation to produce ATP. This process is shut down during AKI which leads to maladaptive tubule repair. Ketolysis, BCAA catabolism, NAD de novo biosynthesis from tryptophan were suppressed during AKI as well. IDH, Isocitrate dehydrogenase; A-KGDH, α-Ketoglutaric acid dehydrogenase; SCS, Succinyl-CoA synthetase; FH, Fumarase; SDH, Succinic dehydrogenase; MDH, Malate dehydrogenase; CS, Citrate synthase; SCD, Stearoyl-CoA Desaturase.
A metabolomic shift occurs in TECs during AKI to compensate for the energy shortage resulting from the mitochondrial dysfunction-induced FAO and oxidative phosphorylation deficiency. It was shown that glycolysis is upregulated in various models of AKI, including cecal ligation and puncture-, IRI-, and cisplatin-induced AKI (Lan et al., 2016; Gómez et al., 2017; Xie et al., 2023). Tubular hypoxia-induced factor-1α activation enhanced glucose transporter type 1 mRNA expression, which means hypoxia may facilitate glucose transporter type 1-mediated glucose uptake from the basolateral side for glycolysis (Farsijani et al., 2016). On one hand, IRI kidneys exhibited elevated levels of lactate, increased expression of glycolysis enzymes including hexokinase 2, phosphofructokinases, and pyruvate kinase M2 (Lan et al., 2016; Legouis et al., 2020), increased activity of hexokinase, and decreased expression of gluconeogenesis enzymes fructose-1,6-bisphosphatase 1 and phosphoenolpyruvate carboxykinase (Legouis et al., 2020). Lactate dehydrogenase has been widely used as a biomarker for predicting AKI (Heidari Beigvand et al., 2021; Popov et al., 2017; Bagshaw et al., 2007). 2-Deoxy-D-glucose (2-DG), the inhibitor of hexokinase, has been shown to promote autophagy, suppress apoptosis, and improve kidney function by enhancing SIRT3/AMPK pathway in LPS-induced AKI (Tan et al., 2021). Additionally, the pyruvate kinase M2 inhibitor shikonin rescued kidney fibrosis in UUO-induced injury, reducing tubular apoptosis, fibrosis, and fibroblast and macrophage infiltration, whereas it did not attenuate fibrosis in mouse proximal tubular cell line (Wei et al., 2019). On the other hand, mitochondrial pyruvate channel was downregulated during AKI, indicating a decrease in mitochondrial pyruvate uptake (Rauckhorst et al., 2023). Pyruvate dehydrogenase (PDH), the gateway enzyme linking glycolysis with TCA cycle, is inactivated when phosphorylated by pyruvate dehydrogenase kinase. Lan et al. showed that PDH phosphorylation was upregulated in the IRI mice kidney (Li et al., 2020). Additionally, Oh et al. found that the proximal tubule cell-specific knockout of pyruvate dehydrogenase kinase 4 (PDK4) or pharmacological inhibition of PDK alleviated IR-induced kidney injury by reducing succinate levels in tubules (Zhao et al., 2016). This concurrently mitigated oxidative stress, elevated mitochondrial membrane potential and ATP production, together with reduced mitochondrial fragmentation (Oh et al., 2023; Wei et al., 2019). These findings indicated that pyruvate tends to be directed toward anaerobic lactate production in TECs rather than incorporation into the aerobic TCA cycle. This metabolic shift contributes to worsening kidney function in AKI.
As the final product of glycolysis, lactate levels significantly increased in AKI models, including the CPB/DHCA (Davidson et al., 2022), IRI (Lan et al., 2016), cisplatin (Pariyani et al., 2017), and sepsis (Tan et al., 2021) models. It has been repeatedly shown that lactate serves as a critical risk factor for AKI (Liu Z. et al., 2019; Zhao et al., 2022). Moreover, lactate plays various roles in contributing to the progression of AKI. Tan et al. found that lactate attenuated the expression of SIRT3 and phosphorylated AMPK, inhibited autophagy and enhanced apoptosis in LPS-treated HK-2 cells (Tan et al., 2021). Lactate also upregulated programmed cell death ligand 1 expression, suppressing the immune response in sepsis-induced AKI in mice (Xu et al., 2021). In addition, lactate can participate in post-translational modelling as the donor, with lactylation primarily determined by the concentration of lactate and the activity of glycolysis (Zhang et al., 2019). Lactate-dependent modification occurred both in histone proteins and non-histone proteins and exerting diverse effects during AKI. Renal protein lactylation activated the NLR family pyrin domain containing 3 inflammasome and promoted the transition from AKI to CKD (Yu et al., 2023). Wang et al. found that the key glycolytic enzyme, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) lead to the accumulation of lactate during ischemia-reperfusion injury which would increase histone 4 lysine 12 lactylation which further activate NF-κB signaling pathway and result in kidney fibrosis (Wang et al., 2024). Sheng et al. found that the downregulation of the deacetylase SIRT3, mediated hyperacetylation and inactivation of pyruvate dehydrogenase E1 subunit alpha 1 (PDHEα1), leading to increased lactate levels in TECs. The additional lactate led to elevated lactylation and increased levels of mitochondrial fission 1 protein, lysine 20, which subsequently led to excessive mitochondrial fission and ATP depletion in septic AKI mice (An et al., 2023).
Moreover, excessive lactate can be excreted into the interstitial microenvironment and is absorbed by monocarboxylate transporter, or activates G protein-coupled cell receptor 81 to interact with interstitial cells such as fibroblasts (Jin et al., 2021) and inflammatory cells (Yang et al., 2022), potentially affecting kidney function. Lactate derived from TECs is absorbed by fibroblasts via monocarboxylate transporter 1, promoting fibroblast activation and proliferation in folic-acid-induced AKI (Shen et al., 2020). The administration of lactate significantly reduced the extracellular acidification rate in LPS-treated macrophages. Increased lactylation of pyruvate kinase M2 pyruvate kinase M2 at the K62 site promoted pyruvate kinase M2 tetramerization, and inhibited pyruvate kinase M2 translocation into the nucleus. This suppression of glycolysis results in macrophage polarization toward a reparative phenotype (Wang J. et al., 2022).
To summarize, AKI enhances glycolysis in TECs, acting as an alternative source of energy supply during low oxygen availability. However, continuous glycolysis and the accumulation of lactate aggravates tubular injury, promoting AKI progression.
3.2 Pentose pyruvate pathway
The pentose phosphate pathway catabolizes glucose without ATP production. Glucose is catalyzed by G-6-PDH so as to biosynthesize 5-ribose phosphate for nucleotide production and generate nicotinamide adenine dinucleotide phosphate (NADPH) from the electron acceptor NADP+. Scantlebery et al. observed a significant upregulation of pentose phosphate pathway-related genes, including glucose-6-phosphate 1-dehydrogenase, transaldolase 1, transketolase, and ribose-phosphate diphosphokinase 1, following IR-AKI in mice kidneys. The upregulated NADPH levels may serve as a reductive agent in fatty acid and cholesterol synthesis, leading to lipotoxicity in TECs (Scantlebery et al., 2021). On the other hand, a Nature study revealed that inhibition of the glycolysis enzyme pyruvate kinase M2 compels glucose flux toward the pentose phosphate pathway in proximal tubular cells (Zhou et al., 2019), and upregulated NADPH could mitigate damage caused by reactive oxygen species, protecting against IR-induced kidney injury (Zhou et al., 2019). Moreover, G-6-PDH-deficient mice exhibited higher proteinuria, oxidative stress, and lower NADPH and glutathione levels in the kidney (Xu et al., 2010). These apparently dual functions of the pentose phosphate pathway necessitate further research to fully elucidate its role in AKI.
3.3 Hexosamine biosynthetic pathway
The hexosamine biosynthetic pathway is another branched process of glucose metabolism. Firstly, glucose is metabolized to G-6-P by hexokinase, then phosphohexose isomerase catalyzes G-6-P to fructose-6-phosphate. Glutamine fructose-6-phosphate amidotransferase (GFAT) deaminizes glutamine and produces glucosamine-6-phosphate, then acetyl-CoA and Uridine-5′-triphosphate (UTP) are introduced into this process to generate uridine 5′-diphospho-N-acetyl-D-glucosamine. Finally, O-linked N-acetylglucosaminyltransferase (OGT) and O-GlcNAcase serve as “writer” and “reader” to add or remove O-linked β-N-acetylglucosamine moieties (O-GlcNAc) on serine or threonine residues of proteins. This process is known as O-GlcNAcylation which also post-translationally modifies protein activity and function.
The hexosamine biosynthetic pathway and its metabolites are augmented and play cytoprotective roles during acute stress periods (Zachara et al., 2004). Hu et al. confirmed that enhanced O-GlcNAc signaling transduction reduced oxidative stress and cell apoptosis through inhibiting phosphorylation of protein kinase B. Inhibition of this process using alloxan, an OGT inhibitor, worsened kidney function in contrast-induced-AKI (Hu et al., 2017). Another study showed that remote ischemia preconditioning attenuated oxidative stress and tubular apoptosis through enhancing the hexosamine biosynthetic pathway and O-GlcNAc glycosylation levels in contrast-induced AKI, while pharmacological inhibition of GFAT abolished this protective effect (Hu et al., 2018). However, long-term augmentation of O-GlcNAcylation tended to harm kidney function. Spontaneously hypertensive rats exhibited that hyper-O-GlcNAcylation, GFAT and OGT level were positively correlated with proteinuria, while inhibiting GFAT reduced proteinuria. This could be attributed to reduced protein reabsorption in tubular cells due to O-GlcNAcylated megalin (Silva-Aguiar et al., 2018).
3.4 Glutaminolysis
Glutaminolysis is the catabolic process of glutamine. Glutamine is catalyzed by glutaminase, resulting in the generation of glutamate. Subsequently, glutamate is deaminated by glutamate dehydrogenase to produce α-ketoglutarate. The catabolism of glutamine also serves as a component of anaplerosis, that aims to maintain the homeostasis of intermediates within the TCA cycle. Glutaminolysis functions as an alternative pathway, providing the carbon skeleton for biosynthesis and energy expenditure during nutrition stress.
Metallo et al. found that MRC-5 cells (human lung fibroblasts) treated with isotope-labeled glutamine were heavily reliant on reductive glutamine metabolism to synthesize Ac-CoA for lipid synthesis in hypoxia environments (Metallo et al., 2011). In cardiac studies, researchers observed a significant increase in glutaminase-1, the speed-limiting enzyme in glutaminolysis, during angiogenesis II-induced hypertrophy and proliferation in cardiomyocytes and fibroblasts. Correspondingly, the inhibition of glutaminase-1 prevented pathological cardiac remodeling by suppressing anaplerosis from glutamine, thereby impeding the biosynthesis of nucleic acids and lipids in mice (Yoshikawa et al., 2022). However, other studies also showed that glutaminase-regulated glutaminolysis also elevated the NADPH/NADP+ ratio as well as glutathione levels against oxidative stress, which is beneficial for cell growth and proliferation (Tong et al., 2021; Yang et al., 2017).
Emerging evidence suggests metabolic rewiring involving enhanced glutaminolysis may occur during AKI progression. Single-cell RNA sequencing revealed an increase in the expression of the glutaminolysis enzyme Gls and Glud1 genes in proximal tubules in both uIRI and UUO mice (Li H. et al., 2022; Kirita et al., 2020), this transcriptional signature indicating enhanced glutaminolysis in kidney TECs (single-cell sequencing database: http://humphreyslab.com/SingleCell/). Nowik et al. demonstrated that the uptake and breakdown of glutamine increased to maintain an acid-base balance in acidosis-induced AKI (Nowik et al., 2008). Aligned with metabolomic profiling, Wei et al. found concentrations of glutamine decreased in the renal cortex and plasma in IRI- (Wei et al., 2014) and LPS-induced AKI (Xu et al., 2023), suggesting accelerated glutaminolytic flux in AKI. However, contradictory results have been reported in other studies, where some showed an increase in serum glutamine levels in cecal ligation and puncture-induced AKI in a pig model (Izquierdo-Garcia et al., 2019) and in LPS-induced AKI (Gao et al., 2021).
Glutaminolysis may also impact inflammatory cells; T cells exhibited metabolic reprogramming during IRI, with upregulated glutaminase activity during the ischemia period and a decline during reperfusion. Applying a glutaminase antagonist mitigated both IRI- and cisplatin-induced kidney function deterioration by inhibiting the activation and proliferation of T cells (Lee et al., 2023). Nevertheless, there is insufficient evidence to assert that glutaminolysis undergoes changes in TECs and affects the progression of AKI, making it an area worth exploring. Collectively, enhanced glutaminolysis appears detrimental to myocardial cells in pathological conditions, and renal glutaminolysis gene expression is upregulated in uIRI and UUO mice kidneys. However, the precise role of glutaminolysis in TECs and AKI remains unclear.
Numerous studies have demonstrated that the administration of glutamine can offer protection against AKI. In a randomized controlled trial, glutamine supplementation was found to significantly reduce kidney damage biomarkers in cardiac surgery patients at a high risk of AKI (Weiss et al., 2023). Mechanistically, glutamine attenuated IRI-induced AKI by modulating the glutamine gamma glutamyltransferase 2/heat shock protein 70/apoptosis signal-regulating kinase/c-Jun N-terminal kinase pathway and diminished mitochondrial intrinsic apoptosis in TECs (Thomas et al., 2022). Glutamine reduced oxidative stress in gentamicin-induced AKI in rats (Zhan et al., 2022), targeted microRNA/Notch and microRNA/phosphoinositide-3-kinase/protein kinase B signaling pathways in IR rats (Li S. et al., 2022), reduced organic cation transporter2 expression, and reduced the absorption of cisplatin HK-2 cells (Kim et al., 2015), among other mechanisms.
3.5 Polyamine metabolism in AKI
Polyamines, which include putrescine, spermidine, and spermine, are essential for cell proliferation, chromatin organization, gene regulation, cell death and immune system functions (Holbert et al., 2022). The synthesis of polyamine is initiated by the speed-limiting enzyme ornithine decarboxylase, which converts L-ornithine to putrescine. Subsequently, the addition of an aminopropyl group is carried out by spermidine synthase and spermine synthase, leading to the production of spermidine and spermine. The catabolism of polyamine involves the spermidine/spermine N1-acetyltransferase/N1-acetylpolyamine oxidase cascade. Additionally, spermine can be directly oxidized to spermidine by spermine oxidase, while amine oxidase copper-containing 1 (AOC1) is responsible for the breakdown of putrescine. Polyamines and their metabolites entering the circulation can be utilized by cells throughout the body, thereby affecting microenvironments.
Sieckmann et al. found that the key synthetic enzyme ODC1 was downregulated, while the catabolic enzyme AOC1 was upregulated in multiple kidney injury models, including IRI, UUO, kidney transplantation, rhabdomyolysis, and streptozocin-induced diabetes (Sieckmann et al., 2023). The increased AOC1 was secreted into the bloodstream, where it further catabolized putrescine into toxins. Interestingly, the knockout of AOC1 didn’t affect kidney function but led to a reduction in cortical tubules with casts and downregulation of the injury marker lipocalin-2 in IRI mice (Sieckmann et al., 2023). Another study demonstrated an elevation in the expression of the speed-limiting enzyme in polyamine and the activity of spermine/spermidine N1-acetyltransferase and spermine oxidase, two critical enzymes in polyamine catabolism, in the kidneys during IR (Zahedi et al., 2014), septic (Xu et al., 2023), and cisplatin- (Zahedi et al., 2017) induced AKI. Inhibiting either of these enzymes resulted in DNA injury, inflammatory reaction (Zahedi et al., 2014), oxidative stress (Wang et al., 2004), and endoplasmic reticulum stress/unfolded protein response (Zahedi et al., 2017). In summary, polyamine catabolism was heightened in AKI, and blocking polyamine catabolism alleviated kidney injury.
Accordingly, due to the enhanced catabolism of polyamine, the concentrations of putrescine increased, and spermidine and spermine decreased in tetracycline treated HEK-293 cells (Wang et al., 2004). Kidney putrescine levels increased in LPS (Xu et al., 2023)- and CPB/DHCA (Davidson et al., 2022)-induced AKI, while putrescine, spermidine, and spermine all increased in both kidney and serum (Gao et al., 2021; Sieckmann et al., 2023). Notably, urine spermidine showed a high concentration in cardiac surgery-associated AKI and displayed a strong association with the AKI outcome (Martin-Lorenzo et al., 2021).
4 Potential therapeutic target of metabolic pathways in treating AKI
Several studies have investigated drugs targeting these metabolic pathways to thwart the progression of AKI (Table 2).
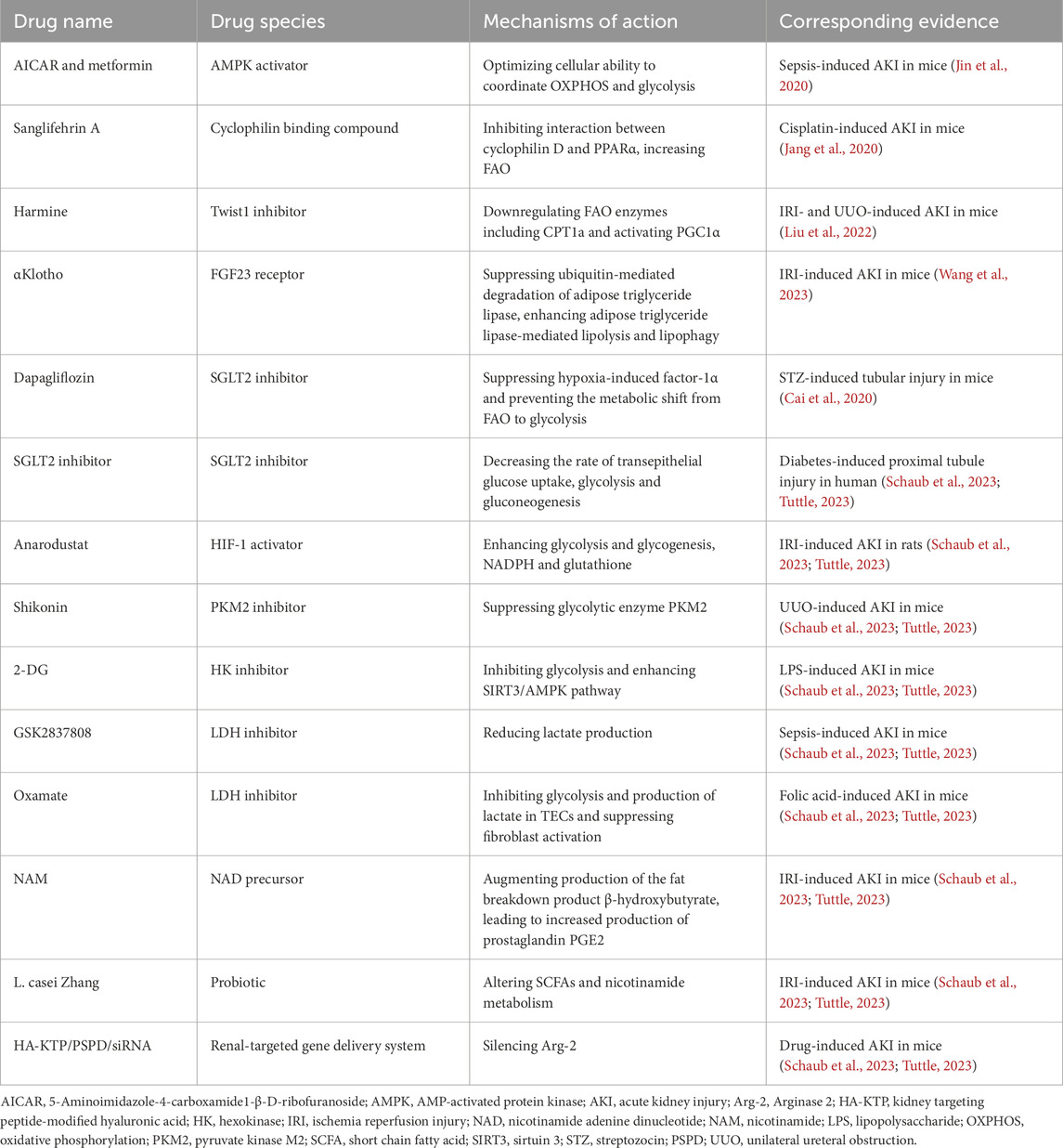
Table 2. Potential therapeutic targets of metabolic pathways in AKI.
Hypoxia-induced factor 1 (HIF-1) enhances glycolysis by increasing the expression of all glycolytic enzymes, including hexokinase 1, hexokinase 2, 6-phosphofructokinase, liver type, phosphofructokinase, platelet, aldolases (ALDA and ALDC), glyceraldehyde 3-phosphate dehydrogenase, and pyruvate kinase. Additionally, hypoxia-induced factor 1b suppresses FAO, reduces oxidative phosphorylation, and mitochondrial oxygen consumption. It also increases PDK1 and reduces cellular acetyl-CoA levels (Taylor and Scholz, 2022). In diabetic kidney disease, proximal tubules also exhibit metabolic shift from FAO to glycolysis which is associated with increased hypoxia-induced factor-1α. By suppressing HIF-1α by the sodium-glucose cotransporter-2 inhibitor (SGLT2i), dapagliflozin, prevented the metabolic shift from FAO to glycolysis in TECs (Cai et al., 2020). Indeed, applying the HIF-1 activator anarodustat, which inhibits the prolyl hydroxylase domain, rescued low-oxygen-treated HK-2 cells, though anarodustat enhanced glycolysis, which also increased glycogenesis, NADPH and glutathione, preserving sufficient glucose for energy supply and providing substrates to counteract oxidative stress (Ito et al., 2020). This underscores the importance of considering metabolic disorder as a complex, systemic, and interactive process when implementing interventions.
Employing single-cell RNA sequencing to explore differentially-expressed genes in kidney tissue from young patients with Type 2 diabetes and controls, Schaub et al. found that SGLT2i decreased the rate of transepithelial glucose uptake. This resulted in decreased glycolysis and gluconeogenesis in the proximal tubule, suppressing the activation of mTOR complex 1 and mitigating diabetes-induced proximal tubule injury (Schaub et al., 2023; Tuttle, 2023).
Liu L et al. found that Twist1, a transcription factor implicated in fibrotic pathogenesis across multiple organs, was upregulated in uIR- and UUO-induced kidney injury. Conditional knockout of twist1 in proximal tubular cells reversed the downregulation of FAO enzymes, including acyl-coenzyme A oxidase 1 and CPT1a, by activating peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC1-α), thereby alleviating fibrosis in AKI mice (Liu et al., 2022). Furthermore, Harmine, a Twist1 inhibitor, prevented fatty acid metabolic disorders and fibrogenesis, suggesting Twist1 could be a potential therapy target for AKI in the future.
αKlotho suppressed ubiquitin-mediated degradation of adipose triglyceride lipase, enhancing adipose triglyceride lipase-mediated lipolysis and lipophagy, subsequently protecting mice from IRI-induced AKI (Wang et al., 2023).
Probiotics were also been investigated as metabolic modulator in acute kidney injury. For instance, probiotic Lactobacillus casei Zhang (L. casei Zhang) could alter SCFAs and nicotinamide metabolism and protect kidney injury in IRI-induced AKI (Zhu et al., 2021).
Newly developed technology regulating the expression of metabolic enzyme was also applied in AKI. Utilizing spermine (SPD) as a monomer for siRNA delivery to downregulate Arg-2 expression, and assembling with kidney targeting peptide (KTP)-modified hyaluronic acid (HA) to improve the in vivo delivery and renal targeting of the gene vector. This system showed the alleviation of kidney injury through the mechanisms including promotion of mitochondrial autophagy, mitigation of oxidative stress, and inhibition of apoptosis in drug-induced AKI (Gu et al., 2024).
Numerous other interventions have attempted to target metabolic upstream molecules such as AMPK (Jin et al., 2020; Harley et al., 2022), peroxisome proliferator-activated receptor gamma, peroxisome proliferator-activated receptor-γ coactivator 1α (Tran et al., 2016), and others. Additionally, other interventions have focused on post-translational modifications of metabolites, including histone deacetylases (Guo et al., 2019), and SIRT (Huang et al., 2022).
5 Conclusion and perspectives
Accumulating evidence has recently unraveled that the metabolic pathway and energy substrate were switched during AKI. Catabolism of fatty acids, ketone bodies and BCAAs was shut down and replaced with enhanced glycolysis, glutaminolysis, pentose pyruvate pathway, and polyamine catabolism (Figure 3; Table 1). The reprogrammed metabolic pathway aims to compensate for energy shortages during the early stages of AKI, however, it tends to be detrimental for TEC repair. The differential metabolites triggered by metabolic rewriting also affect the kidney outcome mechanistically.

Figure 3. Upregulated pathways in TECs during AKI. Glycolysis, pentose phosphate pathway, hexokinase biosynthesis pathway, glutaminolysis pathway were enhanced during AKI. And their final products or intermediate metabolites played different roles during AKI. HK, hexokinase; G-6-P, glucose-6-phosphate; F-6-P, fructose-6-phosphate; GFAT, glutamine fructose-6-phosphate amidotransferase; phosphoenolpyruvate (PEP), then PEP is catalyzed by pyruvate kinase M2 (PKM2); fructose-1,6-bisphosphatase 1 (FBP1), phosphoenolpyruvate carboxykinase (PCK1/2), PC, pyruvate carboxylase.
In addition, metabolic heterogeneity across AKI etiologies still need to be verified, some evidence indeed suggests distinct energy utilization patterns among IRI, sepsis, nephrotoxin-induced AKI, driven by their unique pathophysiological mechanisms. IRI was characterized by abrupt ATP depletion due to hypoxia, proximal tubules shift to anaerobic glycolysis (increased PFK-1 and lactate accumulation). Mitochondrial dysfunction persists even post-reperfusion, with impaired fatty acid β-oxidation (CPT1 downregulation) and ROS-induced damage to ETC complexes. Sepsis-induced AKI was mainly mitochondrial dysfunction and systemic inflammatory-driven metabolic reprogramming. Despite preserved renal blood flow, microcirculatory shunting creates “cytopathic hypoxia.” Proinflammatory cytokines (e.g., TNF-α, IL-6) induce Warburg-like metabolic reprogramming–enhanced glycolysis (HIF-1α stabilization) but suppressed OXPHOS. Mitochondrial uncoupling (UCP2 upregulation) further reduces ATP yield. Lipolysis and amino acid catabolism are amplified to fuel gluconeogenesis. During nephrotoxin-induced AKI, ROS-mediated disruption of OXPHOS and substrate-specific toxicity. Direct tubular toxicity disrupts mitochondrial integrity (reduced cristae density, cytochrome c release). For instance, Cisplatin inhibits Complex I/IV and Krebs cycle enzymes (aconitase suppression), blocking both glucose and fatty acid metabolism. Persistent NADPH oxidase activation exacerbates oxidative stress, impairing redox-sensitive metabolic sensors (AMPK/PGC-1α axis). Despite distinct initiating insults, different etiologies of AKI ultimately converge on overlapping pathophysiological mechanisms in TECs, resulting in strikingly similar metabolic reprogramming during advanced injury stages.
Given that metabolic pathways are dynamic, reversible, systemic, and intricate processes, researchers commonly employ a combination of proteomics, metabolomics, and transcriptomics to analyze enzyme expression, metabolite concentration, and isotope-labeled metabolites for tracking outlets. Recently, single-cell RNA sequencing and spatial metabolomics have provided new insights into the transcriptome and metabolism of various cell types. However, even with the integration of different methodologies, accurate and consistent answers for metabolic pathways may not always be achieved. Therefore, additional studies are imperative to further elucidate the role of metabolic pathways that may underly disease processes in the kidney.
In conclusion, numerous studies have demonstrated that metabolic rewriting is inevitable in AKI. This review delved into the dual roles of adaptive metabolic shifts and their maladaptive consequences during AKI, aiming to inspire researchers to prioritize metabolic insights into clinically diagnostic/prognostic biomarkers and precision therapies.
Author contributions
JG: Conceptualization, Writing – original draft, Writing – review and editing. LH: Writing – original draft, Writing – review and editing. YZ: Writing – review and editing. LW: Writing – review and editing. ZY: Writing – review and editing. YX: Writing – review and editing. JY: Writing – review and editing. XN: Conceptualization, Funding acquisition, Supervision, Writing – review and editing. SS: Conceptualization, Funding acquisition, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. National Natural Science Foundation of China grants (Reference number: 82170722, 82270715) supported this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
An S., Yao Y., Hu H., Wu J., Li J., Li L., et al. (2023). PDHA1 hyperacetylation-mediated lactate overproduction promotes sepsis-induced acute kidney injury via Fis1 lactylation. Cell death and Dis. 14, 457. doi:10.1038/s41419-023-05952-4
Andrade-Oliveira V., Amano M. T., Correa-Costa M., Castoldi A., Felizardo R. J. F., de Almeida D. C., et al. (2015). Gut bacteria products prevent AKI induced by ischemia-reperfusion. J. Am. Soc. Nephrol. JASN 26, 1877–1888. doi:10.1681/asn.2014030288
Aranda-Rivera A. K., Cruz-Gregorio A., Aparicio-Trejo O. E., Pedraza-Chaverri J. (2021). Mitochondrial redox signaling and oxidative stress in kidney diseases. Biomolecules 11, 1144. doi:10.3390/biom11081144
Aregger F., Uehlinger D. E., Fusch G., Bahonjic A., Pschowski R., Walter M., et al. (2018). Increased urinary excretion of kynurenic acid is associated with non-recovery from acute kidney injury in critically ill patients. BMC Nephrol. 19, 44. doi:10.1186/s12882-018-0841-5
Bagshaw S. M., Langenberg C., Haase M., Wan L., May C. N., Bellomo R. (2007). Urinary biomarkers in septic acute kidney injury. Intensive care Med. 33, 1285–1296. doi:10.1007/s00134-007-0656-5
Cai T., Ke Q., Fang Y., Wen P., Chen H., Yuan Q., et al. (2020). Sodium-glucose cotransporter 2 inhibition suppresses HIF-1α-mediated metabolic switch from lipid oxidation to glycolysis in kidney tubule cells of diabetic mice. Cell death and Dis. 11, 390. doi:10.1038/s41419-020-2544-7
Chen J., Zheng Q. Y., Wang L. M., Luo J., Chen K. H., He Y. N. (2023). Proteomics reveals defective peroxisomal fatty acid oxidation during the progression of acute kidney injury and repair. Heliyon 9, e18134. doi:10.1016/j.heliyon.2023.e18134
Cheng L., Wang L., Chen B., Wang C., Wang M., Li J., et al. (2022). A multiple-metabolites model to predict preliminary renal injury induced by iodixanol based on UHPLC/Q-Orbitrap-MS and (1)H-NMR. Metabolomics Official J. Metabolomic Soc. 18, 85. doi:10.1007/s11306-022-01942-3
Chiba T., Peasley K. D., Cargill K. R., Maringer K. V., Bharathi S. S., Mukherjee E., et al. (2019). Sirtuin 5 regulates proximal tubule fatty acid oxidation to protect against AKI. J. Am. Soc. Nephrol. 30, 2384–2398. doi:10.1681/ASN.2019020163
Chouchani E. T., Pell V. R., Gaude E., Aksentijević D., Sundier S. Y., Robb E. L., et al. (2014). Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435. doi:10.1038/nature13909
Coca S. G., Singanamala S., Parikh C. R. (2012). Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int. 81, 442–448. doi:10.1038/ki.2011.379
Davidson J. A., Robison J., Khailova L., Frank B. S., Jaggers J., Ing R. J., et al. (2022). Metabolomic profiling demonstrates evidence for kidney and urine metabolic dysregulation in a piglet model of cardiac surgery-induced acute kidney injury. Am. J. physiology. Ren. physiology 323, F20–f32. doi:10.1152/ajprenal.00039.2022
De Chiara L., Conte C., Antonelli G., Lazzeri E. (2021). Tubular cell cycle response upon AKI: revising old and new paradigms to identify novel targets for CKD prevention. Int. J. Mol. Sci. 22, 11093. doi:10.3390/ijms222011093
Deen P. M., Robben J. H. (2011). Succinate receptors in the kidney. J. Am. Soc. Nephrol. JASN 22, 1416–1422. doi:10.1681/asn.2010050481
Deng X., Tang C., Fang T., Li T., Li X., Liu Y., et al. (2025). Disruption of branched-chain amino acid homeostasis promotes the progression of DKD via enhancing inflammation and fibrosis-associated epithelial-mesenchymal transition. Metabolism Clin. Exp. 162, 156037. doi:10.1016/j.metabol.2024.156037
Duarte P. R. A., Franco R. R., Vilela D. D., Caixeta D. C., de Souza A. V., Deconte S. R., et al. (2023). Effects of an L-leucine-rich diet on liver and kidneys in a doxorubicin toxicity model. Life (Basel, Switz.) 13, 1823. doi:10.3390/life13091823
Farsijani N. M., Liu Q., Kobayashi H., Davidoff O., Sha F., Fandrey J., et al. (2016). Renal epithelium regulates erythropoiesis via HIF-dependent suppression of erythropoietin. J. Clin. investigation 126, 1425–1437. doi:10.1172/jci74997
Figlia G., Willnow P., Teleman A. A. (2020). Metabolites regulate cell signaling and growth via covalent modification of proteins. Dev. cell 54, 156–170. doi:10.1016/j.devcel.2020.06.036
Gao H., Yang T., Chen X., Song Y. (2021). Changes of lipopolysaccharide-induced acute kidney and liver injuries in rats based on metabolomics analysis. J. Inflamm. Res. 14, 1807–1825. doi:10.2147/jir.S306789
Gómez H., Kellum J. A., Ronco C. (2017). Metabolic reprogramming and tolerance during sepsis-induced AKI. Nat. Rev. Nephrol. 13, 143–151. doi:10.1038/nrneph.2016.186
Gowans G. J., Bridgers J. B., Zhang J., Dronamraju R., Burnetti A., King D. A., et al. (2019). Recognition of histone crotonylation by Taf14 links metabolic state to gene expression. Mol. cell 76, 909–921.e3. doi:10.1016/j.molcel.2019.09.029
Gu M., Jiang H., Tan M., Yu L., Xu N., Li Y., et al. (2023). Palmitoyltransferase DHHC9 and acyl protein thioesterase APT1 modulate renal fibrosis through regulating β-catenin palmitoylation. Nat. Commun. 14, 6682. doi:10.1038/s41467-023-42476-z
Gu X.-R., Liu K., Deng Y.-X., Xiang B. X., Zhou L. Y., Yin W. J., et al. (2024). A renal-targeted gene delivery system derived from spermidine for arginase-2 silencing and synergistic attenuation of drug-induced acute kidney injury. Chem. Eng. J. 486, 150125. doi:10.1016/j.cej.2024.150125
Guo C., Dong G., Liang X., Dong Z. (2019). Epigenetic regulation in AKI and kidney repair: mechanisms and therapeutic implications. Nat. Rev. Nephrol. 15, 220–239. doi:10.1038/s41581-018-0103-6
Harley G., Katerelos M., Gleich K., de Souza D. P., Narayana V. K., Kemp B. E., et al. (2022). Blocking AMPK signalling to acetyl-CoA carboxylase increases cisplatin-induced acute kidney injury and suppresses the benefit of metformin. Biomed. and Pharmacother. = Biomedecine and Pharmacother. 153, 113377. doi:10.1016/j.biopha.2022.113377
Heidari Beigvand H., Heidari K., Hashemi B., Saberinia A. (2021). The value of lactate dehydrogenase in predicting rhabdomyolysis-induced acute renal failure; a narrative review. Archives Acad. Emerg. Med. 9, e24. doi:10.22037/aaem.v9i1.1096
Hewitson T. D., Holt S. G., Tan S. J., Wigg B., Samuel C. S., Smith E. R. (2017). Epigenetic modifications to H3K9 in renal tubulointerstitial cells after unilateral ureteric obstruction and TGF-β1 stimulation. Front. Pharmacol. 8, 307. doi:10.3389/fphar.2017.00307
Holbert C. E., Cullen M. T., Casero R. A., Stewart T. M. (2022). Polyamines in cancer: integrating organismal metabolism and antitumour immunity. Nat. Rev. Cancer 22, 467–480. doi:10.1038/s41568-022-00473-2
Hu J., Chen R., Jia P., Fang Y., Liu T., Song N., et al. (2017). Augmented O-GlcNAc signaling via glucosamine attenuates oxidative stress and apoptosis following contrast-induced acute kidney injury in rats. Free Radic. Biol. and Med. 103, 121–132. doi:10.1016/j.freeradbiomed.2016.12.032
Hu J., Wang Y., Zhao S., Chen J., Jin S., Jia P., et al. (2018). Remote ischemic preconditioning ameliorates acute kidney injury due to contrast exposure in rats through augmented O-GlcNAcylation. Oxidative Med. Cell. Longev. 2018, 4895913. doi:10.1155/2018/4895913
Huang C., Jiang S., Gao S., Wang Y., Cai X., Fang J., et al. (2022). Sirtuins: research advances on the therapeutic role in acute kidney injury. Phytomedicine Int. J. phytotherapy Phytopharm. 101, 154122. doi:10.1016/j.phymed.2022.154122
Huang H., van Dullemen L. F. A., Akhtar M. Z., Faro M. L. L., Yu Z., Valli A., et al. (2018). Proteo-metabolomics reveals compensation between ischemic and non-injured contralateral kidneys after reperfusion. Sci. Rep. 8, 8539. doi:10.1038/s41598-018-26804-8
Hui Y., Zhao J., Yu Z., Wang Y., Qin Y., Zhang Y., et al. (2023). The role of tryptophan metabolism in the occurrence and progression of acute and chronic kidney diseases. Mol. Nutr. and food Res. 67, e2300218. doi:10.1002/mnfr.202300218
Ito M., Tanaka T., Ishii T., Wakashima T., Fukui K., Nangaku M. (2020). Prolyl hydroxylase inhibition protects the kidneys from ischemia via upregulation of glycogen storage. Kidney Int. 97, 687–701. doi:10.1016/j.kint.2019.10.020
Izquierdo-Garcia J. L., Nin N., Cardinal-Fernandez P., Rojas Y., de Paula M., Granados R., et al. (2019). Identification of novel metabolomic biomarkers in an experimental model of septic acute kidney injury. Am. J. physiology. Ren. physiology 316, F54–f62. doi:10.1152/ajprenal.00315.2018
Jang H. S., Noh M. R., Jung E. M., Kim W. Y., Southekal S., Guda C., et al. (2020). Proximal tubule cyclophilin D regulates fatty acid oxidation in cisplatin-induced acute kidney injury. Kidney Int. 97, 327–339. doi:10.1016/j.kint.2019.08.019
Jin K., Ma Y., Manrique-Caballero C. L., Li H., Emlet D. R., Li S., et al. (2020). Activation of AMP-activated protein kinase during sepsis/inflammation improves survival by preserving cellular metabolic fitness. FASEB J. official Publ. Fed. Am. Soc. Exp. Biol. 34, 7036–7057. doi:10.1096/fj.201901900R
Jin Z., Lu Y., Wu X., Pan T., Yu Z., Hou J., et al. (2021). The cross-talk between tumor cells and activated fibroblasts mediated by lactate/BDNF/TrkB signaling promotes acquired resistance to anlotinib in human gastric cancer. Redox Biol. 46, 102076. doi:10.1016/j.redox.2021.102076
Jouret F., Leenders J., Poma L., Defraigne J. O., Krzesinski J. M., de Tullio P. (2016). Nuclear magnetic resonance metabolomic profiling of mouse kidney, urine and serum following renal ischemia/reperfusion injury. PloS one 11, e0163021. doi:10.1371/journal.pone.0163021
Jung J. H., Choi J. E., Song J. H., Ahn S. H. (2018). Human CD36 overexpression in renal tubules accelerates the progression of renal diseases in a mouse model of folic acid-induced acute kidney injury. Kidney Res. Clin. Pract. 37, 30–40. doi:10.23876/j.krcp.2018.37.1.30
Kang H. M., Ahn S. H., Choi P., Ko Y. A., Han S. H., Chinga F., et al. (2015). Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 21, 37–46. doi:10.1038/nm.3762
Kellum J. A., Romagnani P., Ashuntantang G., Ronco C., Zarbock A., Anders H. J. (2021). Acute kidney injury. Nat. Rev. Dis. Prim. 7, 52. doi:10.1038/s41572-021-00284-z
Kim H. J., Park D. J., Kim J. H., Jeong E. Y., Jung M. H., Kim T. H., et al. (2015). Glutamine protects against cisplatin-induced nephrotoxicity by decreasing cisplatin accumulation. J. Pharmacol. Sci. 127, 117–126. doi:10.1016/j.jphs.2014.11.009
Kim M. J., Kim Y. S., Kim S. R., Lee D. W., Lee S. B., Kim I. Y. (2023). β-hydroxybutyrate ameliorates sepsis-induced acute kidney injury. Mol. Biol. Rep. 50, 8915–8923. doi:10.1007/s11033-023-08713-w
Kim M. J., Kim Y. S., Kim S. R., Lee D. W., Lee S. B., Kim I. Y. (2024). Pre-treatment with β-hydroxybutyrate mitigates cisplatin-induced acute kidney injury. Biochem. biophysical Res. Commun. 695, 149482. doi:10.1016/j.bbrc.2024.149482
Kirita Y., Wu H., Uchimura K., Wilson P. C., Humphreys B. D. (2020). Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc. Natl. Acad. Sci. U. S. A. 117, 15874–15883. doi:10.1073/pnas.2005477117
Klein K. L., Wang M. S., Torikai S., Davidson W. D., Kurokawa K. (1981). Substrate oxidation by isolated single nephron segments of the rat. Kidney Int. 20, 29–35. doi:10.1038/ki.1981.100
Lan R., Geng H., Singha P. K., Saikumar P., Bottinger E. P., Weinberg J. M., et al. (2016). Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J. Am. Soc. Nephrol. JASN 27, 3356–3367. doi:10.1681/asn.2015020177
Lee K., Thompson E. A., Gharaie S., Patel C. H., Kurzhagen J. T., Pierorazio P. M., et al. (2023). T cell metabolic reprogramming in acute kidney injury and protection by glutamine blockade. JCI insight 8, e160345. doi:10.1172/jci.insight.160345
Legouis D., Ricksten S. E., Faivre A., Verissimo T., Gariani K., Verney C., et al. (2020). Altered proximal tubular cell glucose metabolism during acute kidney injury is associated with mortality. Nat. Metab. 2, 732–743. doi:10.1038/s42255-020-0238-1
Li H., Dixon E. E., Wu H., Humphreys B. D. (2022). Comprehensive single-cell transcriptional profiling defines shared and unique epithelial injury responses during kidney fibrosis. Cell metab. 34, 1977–1998.e9. doi:10.1016/j.cmet.2022.09.026
Li S., Huang X., Wang S., Chu X., Aierken M. (2022). Analysis of microRNA expression after glutamine intervention in acute renal ischemia-reperfusion injury. J. Healthc. Eng. 2022, 2401152. doi:10.1155/2022/2401152
Li T., Zhang Z., Kolwicz S. C., Abell L., Roe N. D., Kim M., et al. (2017). Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cell metab. 25, 374–385. doi:10.1016/j.cmet.2016.11.005
Li X., Zhou X., Liu X., Jiang X., Shi B., et al. (2022). Spermidine protects against acute kidney injury by modulating macrophage NLRP3 inflammasome activation and mitochondrial respiration in an eIF5A hypusination-related pathway. Mol. Med. (Camb. Mass.) 28, 103. doi:10.1186/s10020-022-00533-1
Li Y., Xiong Z., Yan W., Gao E., Cheng H., Wu G., et al. (2020). Branched chain amino acids exacerbate myocardial ischemia/reperfusion vulnerability via enhancing GCN2/ATF6/PPAR-α pathway-dependent fatty acid oxidation. Theranostics 10, 5623–5640. doi:10.7150/thno.44836
Liu K., Li F., Sun Q., Lin N., Han H., You K., et al. (2019). p53 β-hydroxybutyrylation attenuates p53 activity. Cell death and Dis. 10, 243. doi:10.1038/s41419-019-1463-y
Liu L., Ning X., Wei L., Zhou Y., Zhao L., Ma F., et al. (2022). Twist1 downregulation of PGC-1α decreases fatty acid oxidation in tubular epithelial cells, leading to kidney fibrosis. Theranostics 12 (8), 3758–3775. doi:10.7150/thno.71722
Liu Z., Meng Z., Li Y., Zhao J., Wu S., Gou S., et al. (2019). Prognostic accuracy of the serum lactate level, the SOFA score and the qSOFA score for mortality among adults with Sepsis. Scand. J. trauma, Resusc. Emerg. Med. 27, 51. doi:10.1186/s13049-019-0609-3
Luo S., Yang M., Han Y., Zhao H., Jiang N., Li L., et al. (2022). β-Hydroxybutyrate against Cisplatin-Induced acute kidney injury via inhibiting NLRP3 inflammasome and oxidative stress. Int. Immunopharmacol. 111, 109101. doi:10.1016/j.intimp.2022.109101
Luo W., Yu Y., Wang H., Liu K., Wang Y., Huang M., et al. (2020). Up-regulation of MMP-2 by histone H3K9 β-hydroxybutyrylation to antagonize glomerulosclerosis in diabetic rat. Acta diabetol. 57, 1501–1509. doi:10.1007/s00592-020-01552-2
Ma Y., Huang L., Zhang Z., Yang P., Chen Q., Zeng X., et al. (2024). CD36 promotes tubular ferroptosis by regulating the ubiquitination of FSP1 in acute kidney injury. Genes and Dis. 11, 449–463. doi:10.1016/j.gendis.2022.12.003
Martin-Lorenzo M., Ramos-Barron A., Gutierrez-Garcia P., Martin-Blazquez A., Santiago-Hernandez A., Rodrigo Calabia E., et al. (2021). Urinary spermidine predicts and associates with in-hospital acute kidney injury after cardiac surgery. Antioxidants (Basel, Switz.) 10, 896. doi:10.3390/antiox10060896
Matsushita K., Saritas T., Eiwaz M. B., McClellan N., Coe I., Zhu W., et al. (2020). The acute kidney injury to chronic kidney disease transition in a mouse model of acute cardiorenal syndrome emphasizes the role of inflammation. Kidney Int. 97, 95–105. doi:10.1016/j.kint.2019.06.022
Metallo C. M., Gameiro P. A., Bell E. L., Mattaini K. R., Yang J., Hiller K., et al. (2011). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384. doi:10.1038/nature10602
Miguel V., Tituaña J., Herrero J. I., Herrero L., Serra D., Cuevas P., et al. (2021). Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J. Clin. investigation 131, e140695. doi:10.1172/jci140695
Muhle-Goll C., Eisenmann P., Luy B., Kölker S., Tönshoff B., Fichtner A., et al. (2020). Urinary NMR profiling in pediatric acute kidney injury-A pilot study. Int. J. Mol. Sci. 21, 1187. doi:10.3390/ijms21041187
Neinast M. D., Jang C., Hui S., Murashige D. S., Chu Q., Morscher R. J., et al. (2019). Quantitative analysis of the whole-body metabolic fate of branched-chain amino acids. Cell metab. 29, 417–429.e4. doi:10.1016/j.cmet.2018.10.013
Nie C., He T., Zhang W., Zhang G., Ma X. (2018). Branched chain amino acids: beyond nutrition metabolism. Int. J. Mol. Sci. 19, 954. doi:10.3390/ijms19040954
Nowik M., Lecca M. R., Velic A., Rehrauer H., Brändli A. W., Wagner C. A. (2008). Genome-wide gene expression profiling reveals renal genes regulated during metabolic acidosis. Physiol. Genomics 32, 322–334. doi:10.1152/physiolgenomics.00160.2007
O'Brien C. M., Mulukutla B. C., Mashek D. G., Hu W. S. (2020). Regulation of metabolic homeostasis in cell culture bioprocesses. Trends Biotechnol. 38, 1113–1127. doi:10.1016/j.tibtech.2020.02.005
Oh C. J., Kim M. J., Lee J. M., Kim D. H., Kim I. Y., Park S., et al. (2023). Inhibition of pyruvate dehydrogenase kinase 4 ameliorates kidney ischemia-reperfusion injury by reducing succinate accumulation during ischemia and preserving mitochondrial function during reperfusion. Kidney Int. 104, 724–739. doi:10.1016/j.kint.2023.06.022
Papathanassiu A. E., Ko J. H., Imprialou M., Bagnati M., Srivastava P. K., Vu H. A., et al. (2017). BCAT1 controls metabolic reprogramming in activated human macrophages and is associated with inflammatory diseases. Nat. Commun. 8, 16040. doi:10.1038/ncomms16040
Pariyani R., Ismail I. S., Azam A., Khatib A., Abas F., Shaari K., et al. (2017). Urinary metabolic profiling of cisplatin nephrotoxicity and nephroprotective effects of Orthosiphon stamineus leaves elucidated by (1)H NMR spectroscopy. J. Pharm. Biomed. analysis 135, 20–30. doi:10.1016/j.jpba.2016.12.010
Pillai S. M., Herzog B., Seebeck P., Pellegrini G., Roth E., Verrey F. (2019). Differential impact of dietary branched chain and aromatic amino acids on chronic kidney disease progression in rats. Front. physiology 10, 1460. doi:10.3389/fphys.2019.01460
Piret S. E., Guo Y., Attallah A. A., Horne S. J., Zollman A., Owusu D., et al. (2021). Krüppel-like factor 6-mediated loss of BCAA catabolism contributes to kidney injury in mice and humans. Proc. Natl. Acad. Sci. U. S. A. 118, e2024414118. doi:10.1073/pnas.2024414118
Pluznick J. (2014). A novel SCFA receptor, the microbiota, and blood pressure regulation. Gut microbes 5, 202–207. doi:10.4161/gmic.27492
Popov S. V., Guseinov R. G., Martov A. G., Muratov T. M., Tabynbaev N. B. (2017). Biomarkers of acute hypoxia-reoxygenation injury to nercycites during laparoscopic resection of renal parenchyma. Urol. (Mosc. Russ. 1999), 120–125.
Poyan Mehr A., Tran M. T., Ralto K. M., Leaf D. E., Washco V., Messmer J., et al. (2018). De novo NAD(+) biosynthetic impairment in acute kidney injury in humans. Nat. Med. 24, 1351–1359. doi:10.1038/s41591-018-0138-z
Pu M., Zhang J., Zeng Y., Hong F., Qi W., Yang X., et al. (2023). Succinate-SUCNR1 induces renal tubular cell apoptosis. Am. J. physiology. Cell physiology 324, C467–c476. doi:10.1152/ajpcell.00327.2022
Qu X., Gao H., Sun J., Tao L., Zhang Y., Zhai J., et al. (2020). Identification of key metabolites during cisplatin-induced acute kidney injury using an HPLC-TOF/MS-based non-targeted urine and kidney metabolomics approach in rats. Toxicology 431, 152366. doi:10.1016/j.tox.2020.152366
Rauckhorst A. J., Martinez G. V., Andrade G. M., Wen H., Kim J. Y., Simoni A., et al. (2023). Tubular mitochondrial pyruvate carrier disruption elicits redox adaptations that protect from acute kidney injury. bioRxiv : the preprint server for biology, 526492. doi:10.1101/2023.01.31.526492
Rinaldi A., Lazareth H., Poindessous V., Nemazanyy I., Sampaio J. L., Malpetti D., et al. (2022). Impaired fatty acid metabolism perpetuates lipotoxicity along the transition to chronic kidney injury. JCI insight 7, e161783. doi:10.1172/jci.insight.161783
Ruiz-Andres O., Sanchez-Niño M. D., Cannata-Ortiz P., Ruiz-Ortega M., Egido J., Ortiz A., et al. (2016). Histone lysine crotonylation during acute kidney injury in mice. Dis. models and Mech. 9, 633–645. doi:10.1242/dmm.024455
Sabari B. R., Zhang D., Allis C. D., Zhao Y. (2017). Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. cell Biol. 18, 90–101. doi:10.1038/nrm.2016.140
Saranya G. R., Viswanathan P. (2023). Gut microbiota dysbiosis in AKI to CKD transition. Biomed. and Pharmacother. = Biomedecine and Pharmacother. 161, 114447. doi:10.1016/j.biopha.2023.114447
Scantlebery A. M., Tammaro A., Mills J. D., Rampanelli E., Kors L., Teske G. J., et al. (2021). The dysregulation of metabolic pathways and induction of the pentose phosphate pathway in renal ischaemia-reperfusion injury. J. pathology 253, 404–414. doi:10.1002/path.5605
Schaub J. A., AlAkwaa F. M., McCown P. J., Naik A. S., Nair V., Eddy S., et al. (2023). SGLT2 inhibitors mitigate kidney tubular metabolic and mTORC1 perturbations in youth-onset type 2 diabetes. J. Clin. investigation 133, e164486. doi:10.1172/jci164486
Scholz H., Boivin F. J., Schmidt-Ott K. M., Bachmann S., Eckardt K. U., Scholl U. I., et al. (2021). Kidney physiology and susceptibility to acute kidney injury: implications for renoprotection. Nat. Rev. Nephrol. 17, 335–349. doi:10.1038/s41581-021-00394-7
Shan D., Wang Y. Y., Chang Y., Cui H., Tao M., Sheng Y., et al. (2023). Dynamic cellular changes in acute kidney injury caused by different ischemia time. iScience 26, 106646. doi:10.1016/j.isci.2023.106646
Shen J., Guo H., Liu S., Jin W., Zhang Z. W., Zhang Y., et al. (2023). Aberrant branched-chain amino acid accumulation along the microbiota-gut-brain axis: crucial targets affecting the occurrence and treatment of ischaemic stroke. Br. J. Pharmacol. 180, 347–368. doi:10.1111/bph.15965
Shen Y., Jiang L., Wen P., Ye Y., Zhang Y., Ding H., et al. (2020). Tubule-derived lactate is required for fibroblast activation in acute kidney injury. Am. J. physiology. Ren. physiology 318, F689–f701. doi:10.1152/ajprenal.00229.2019
Shimazu T., Hirschey M. D., Newman J., He W., Shirakawa K., Le Moan N., et al. (2013). Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Sci. (New York, N.Y.) 339, 211–214. doi:10.1126/science.1227166
Sieckmann T., Schley G., Ögel N., Kelterborn S., Boivin F. J., Fähling M., et al. (2023). Strikingly conserved gene expression changes of polyamine regulating enzymes among various forms of acute and chronic kidney injury. Kidney Int. 104, 90–107. doi:10.1016/j.kint.2023.04.005
Silva-Aguiar R. P., Bezerra N. C. F., Lucena M. C., Sirtoli G. M., Sudo R. T., Zapata-Sudo G., et al. (2018). O-GlcNAcylation reduces proximal tubule protein reabsorption and promotes proteinuria in spontaneously hypertensive rats. J. Biol. Chem. 293, 12749–12758. doi:10.1074/jbc.RA118.001746
Simithy J., Sidoli S., Yuan Z. F., Coradin M., Bhanu N. V., Marchione D. M., et al. (2017). Characterization of histone acylations links chromatin modifications with metabolism. Nat. Commun. 8, 1141. doi:10.1038/s41467-017-01384-9
Sivanand S., Viney I., Wellen K. E. (2018). Spatiotemporal control of acetyl-CoA metabolism in chromatin regulation. Trends Biochem. Sci. 43, 61–74. doi:10.1016/j.tibs.2017.11.004
Smith E. R., Wigg B., Holt S., Hewitson T. D. (2019). TGF-β1 modifies histone acetylation and acetyl-coenzyme A metabolism in renal myofibroblasts. Am. J. Physiol. Ren. Physiol. 316, F517–F529. doi:10.1152/ajprenal.00513.2018
Sone H., Lee T. J., Lee B. R., Heo D., Oh S., Kwon S. H. (2023). MicroRNA-mediated attenuation of branched-chain amino acid catabolism promotes ferroptosis in chronic kidney disease. Nat. Commun. 14, 7814. doi:10.1038/s41467-023-43529-z
Sun H., Olson K. C., Gao C., Prosdocimo D. A., Zhou M., Wang Z., et al. (2016). Catabolic defect of branched-chain amino acids promotes heart failure. Circulation 133, 2038–2049. doi:10.1161/circulationaha.115.020226
Sun M., Li J., Mao L., Wu J., Deng Z., He M., et al. (2021). p53 deacetylation alleviates sepsis-induced acute kidney injury by promoting autophagy. Front. Immunol. 12, 685523. doi:10.3389/fimmu.2021.685523
Tajima T., Yoshifuji A., Matsui A., Itoh T., Uchiyama K., Kanda T., et al. (2019). β-hydroxybutyrate attenuates renal ischemia-reperfusion injury through its anti-pyroptotic effects. Kidney Int. 95, 1120–1137. doi:10.1016/j.kint.2018.11.034
Tan C., Gu J., Li T., Chen H., Liu K., Liu M., et al. (2021). Inhibition of aerobic glycolysis alleviates sepsis-induced acute kidney injury by promoting lactate/Sirtuin 3/AMPK-regulated autophagy. Int. J. Mol. Med. 47, 19. doi:10.3892/ijmm.2021.4852
Taylor C. T., Scholz C. C. (2022). The effect of HIF on metabolism and immunity. Nat. Rev. Nephrol. 18, 573–587. doi:10.1038/s41581-022-00587-8
Thomas K., Zondler L., Ludwig N., Kardell M., Lüneburg C., Henke K., et al. (2022). Glutamine prevents acute kidney injury by modulating oxidative stress and apoptosis in tubular epithelial cells. JCI insight 7, e163161. doi:10.1172/jci.insight.163161
Tomita I., Kume S., Sugahara S., Osawa N., Yamahara K., Yasuda-Yamahara M., et al. (2020). SGLT2 inhibition mediates protection from diabetic kidney disease by promoting ketone body-induced mTORC1 inhibition. Cell metab. 32, 404–419.e6. doi:10.1016/j.cmet.2020.06.020
Tong Y., Guo D., Lin S. H., Liang J., Yang D., Ma C., et al. (2021). SUCLA2-coupled regulation of GLS succinylation and activity counteracts oxidative stress in tumor cells. Mol. cell 81, 2303–2316.e8. doi:10.1016/j.molcel.2021.04.002
Tran M. T., Zsengeller Z. K., Berg A. H., Khankin E. V., Bhasin M. K., Kim W., et al. (2016). PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531, 528–532. doi:10.1038/nature17184
Trefely S., Lovell C. D., Snyder N. W., Wellen K. E. (2020). Compartmentalised acyl-CoA metabolism and roles in chromatin regulation. Mol. Metab. 38, 100941. doi:10.1016/j.molmet.2020.01.005
Tuttle K. R. (2023). Digging deep into cells to find mechanisms of kidney protection by SGLT2 inhibitors. J. Clin. investigation 133, e167700. doi:10.1172/jci167700
Uchida S., Endou H. (1988). Substrate specificity to maintain cellular ATP along the mouse nephron. Am. J. physiology 255, F977–F983. doi:10.1152/ajprenal.1988.255.5.F977
Wang G., Heijs B., Kostidis S., Rietjens R. G. J., Koning M., Yuan L., et al. (2022). Spatial dynamic metabolomics identifies metabolic cell fate trajectories in human kidney differentiation. Cell stem cell 29, 1580–1593.e7. doi:10.1016/j.stem.2022.10.008
Wang J., Yang P., Yu T., Gao M., Liu D., Zhang J., et al. (2022). Lactylation of PKM2 suppresses inflammatory metabolic adaptation in pro-inflammatory macrophages. Int. J. Biol. Sci. 18, 6210–6225. doi:10.7150/ijbs.75434
Wang S., Lv D., Jiang S., Jiang J., Liang M., Hou F., et al. (2019). Quantitative reduction in short-chain fatty acids, especially butyrate, contributes to the progression of chronic kidney disease. Clin. Sci. (Lond. Engl. 1979) 133, 1857–1870. doi:10.1042/cs20190171
Wang Y., Li H., Jiang S., Fu D., Lu X., Lu M., et al. (2024). The glycolytic enzyme PFKFB3 drives kidney fibrosis through promoting histone lactylation-mediated NF-κB family activation. Kidney Int. 106, 226–240. doi:10.1016/j.kint.2024.04.016
Wang Y., Ran L., Lan Q., Liao W., Wang L., Wang Y., et al. (2023). Imbalanced lipid homeostasis caused by membrane αKlotho deficiency contributes to the acute kidney injury to chronic kidney disease transition. Kidney Int. 104, 956–974. doi:10.1016/j.kint.2023.08.016
Wang Y. P., Lei Q. Y. (2018). Metabolite sensing and signaling in cell metabolism. Signal Transduct. Target Ther. 3, 30. doi:10.1038/s41392-018-0024-7
Wang Z., Zahedi K., Barone S., Tehrani K., Rabb H., Matlin K., et al. (2004). Overexpression of SSAT in kidney cells recapitulates various phenotypic aspects of kidney ischemia-reperfusion injury. J. Am. Soc. Nephrol. JASN 15, 1844–1852. doi:10.1097/01.asn.0000131525.77636.d5
Wei Q., Su J., Dong G., Zhang M., Huo Y., Dong Z. (2019). Glycolysis inhibitors suppress renal interstitial fibrosis via divergent effects on fibroblasts and tubular cells. Am. J. physiology. Ren. physiology 316, F1162–f1172. doi:10.1152/ajprenal.00422.2018
Wei Q., Xiao X., Fogle P., Dong Z. (2014). Changes in metabolic profiles during acute kidney injury and recovery following ischemia/reperfusion. PloS one 9, e106647. doi:10.1371/journal.pone.0106647
Weiss R., Meersch M., Gerke M., Wempe C., Schäfers M., Kellum J. A., et al. (2023). Effect of glutamine administration after cardiac surgery on kidney damage in patients at high risk for acute kidney injury: a randomized controlled trial. Anesth. analgesia 137, 1029–1038. doi:10.1213/ane.0000000000006288
Wu J. Y., Huang T. W., Hsieh Y. T., Wang Y. F., Yen C. C., Lee G. L., et al. (2020). Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol. cell 77, 213–227.e5. doi:10.1016/j.molcel.2019.10.023
Xie W., He Q., Zhang Y., Xu X., Wen P., Cao H., et al. (2023). Pyruvate kinase M2 regulates mitochondrial homeostasis in cisplatin-induced acute kidney injury. Cell death and Dis. 14, 663. doi:10.1038/s41419-023-06195-z
Xie Z., Zhang D., Chung D., Tang Z., Huang H., Dai L., et al. (2016). Metabolic regulation of gene expression by histone lysine β-hydroxybutyrylation. Mol. cell 62, 194–206. doi:10.1016/j.molcel.2016.03.036
Xu J., Li J., Li Y., Shi X., Zhu H., Chen L. (2023). Multidimensional landscape of SA-AKI revealed by integrated proteomics and metabolomics analysis. Biomolecules 13, 1329. doi:10.3390/biom13091329
Xu J., Ma X., Yu K., Wang R., Wang S., Liu R., et al. (2021). Lactate up-regulates the expression of PD-L1 in kidney and causes immunosuppression in septic Acute Renal Injury. J. Microbiol. Immunol. Infect. = Wei mian yu gan ran za zhi 54, 404–410. doi:10.1016/j.jmii.2019.10.006
Xu Y., Zhang Z., Hu J., Stillman I. E., Leopold J. A., Handy D. E., et al. (2010). Glucose-6-phosphate dehydrogenase-deficient mice have increased renal oxidative stress and increased albuminuria. FASEB J. official Publ. Fed. Am. Soc. Exp. Biol. 24, 609–616. doi:10.1096/fj.09-135731
Yamamoto J., Nishio S., Hattanda F., Nakazawa D., Kimura T., Sata M., et al. (2017). Branched-chain amino acids enhance cyst development in autosomal dominant polycystic kidney disease. Kidney Int. 92, 377–387. doi:10.1016/j.kint.2017.01.021
Yang K., Fan M., Wang X., Xu J., Wang Y., Tu F., et al. (2022). Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell death Differ. 29, 133–146. doi:10.1038/s41418-021-00841-9
Yang L., Venneti S., Nagrath D. (2017). Glutaminolysis: a hallmark of cancer metabolism. Annu. Rev. Biomed. Eng. 19, 163–194. doi:10.1146/annurev-bioeng-071516-044546
Yoshikawa S., Nagao M., Toh R., Shinohara M., Iino T., Irino Y., et al. (2022). Inhibition of glutaminase 1-mediated glutaminolysis improves pathological cardiac remodeling. Am. J. physiology. Heart circulatory physiology 322, H749–h761. doi:10.1152/ajpheart.00692.2021
Yu F., Meng Y., Chen K., He Y. (2023). Wcn23-0490 protein lactylation leads to the chronic transition of acute kidney injury by activating inflammasomes. Kidney Int. Rep. 8, S32. doi:10.1016/j.ekir.2023.02.071
Zachara N. E., O'Donnell N., Cheung W. D., Mercer J. J., Marth J. D., Hart G. W. (2004). Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J. Biol. Chem. 279, 30133–30142. doi:10.1074/jbc.M403773200
Zahedi K., Barone S., Destefano-Shields C., Brooks M., Murray-Stewart T., Dunworth M., et al. (2017). Activation of endoplasmic reticulum stress response by enhanced polyamine catabolism is important in the mediation of cisplatin-induced acute kidney injury. PloS one 12, e0184570. doi:10.1371/journal.pone.0184570
Zahedi K., Barone S., Wang Y., Murray-Stewart T., Roy-Chaudhury P., Smith R. D., et al. (2014). Proximal tubule epithelial cell specific ablation of the spermidine/spermine N1-acetyltransferase gene reduces the severity of renal ischemia/reperfusion injury. PloS one 9, e110161. doi:10.1371/journal.pone.0110161
Zhan F., Wang X., Zhang J., Yi S., He P. (2022). Glutamine alleviates the renal dysfunction associated with gentamicin-induced acute kidney injury in Sprague-Dawley rats. Biotechnol. Appl. Biochem. 69, 323–329. doi:10.1002/bab.2111
Zhang B., Zeng M., Wang Y., Li M., Wu Y., Xu R., et al. (2022). Oleic acid alleviates LPS-induced acute kidney injury by restraining inflammation and oxidative stress via the Ras/MAPKs/PPAR-γ signaling pathway. Phytomedicine Int. J. phytotherapy Phytopharm. 94, 153818. doi:10.1016/j.phymed.2021.153818
Zhang D., Tang Z., Huang H., Zhou G., Cui C., Weng Y., et al. (2019). Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580. doi:10.1038/s41586-019-1678-1
Zhang F., Zhao S., Yan W., Xia Y., Chen X., Wang W., et al. (2016). Branched chain amino acids cause liver injury in obese/diabetic mice by promoting adipocyte lipolysis and inhibiting hepatic autophagy. EBioMedicine 13, 157–167. doi:10.1016/j.ebiom.2016.10.013
Zhang Y., Chen Y., Zhang Z., Tao X., Xu S., Zhang X., et al. (2022). Acox2 is a regulator of lysine crotonylation that mediates hepatic metabolic homeostasis in mice. Cell death and Dis. 13, 279. doi:10.1038/s41419-022-04725-9
Zhao H., Zhang F., Sun D., Wang X., Zhang X., Zhang J., et al. (2020). Branched-chain amino acids exacerbate obesity-related hepatic glucose and lipid metabolic disorders via attenuating Akt2 signaling. Diabetes 69, 1164–1177. doi:10.2337/db19-0920
Zhao J. (2013). Plasma kynurenic acid/tryptophan ratio: a sensitive and reliable biomarker for the assessment of renal function. Ren. Fail. 35, 648–653. doi:10.3109/0886022x.2013.790301
Zhao L., Dong M., Liao S., Du Y., Zhou Q., Zheng H., et al. (2016). Identification of key metabolic changes in renal interstitial fibrosis rats using metabonomics and pharmacology. Sci. Rep. 6, 27194. doi:10.1038/srep27194
Zhao X., Lu Y., Li S., Guo F., Xue H., Jiang L., et al. (2022). Predicting renal function recovery and short-term reversibility among acute kidney injury patients in the ICU: comparison of machine learning methods and conventional regression. Ren. Fail. 44, 1326–1337. doi:10.1080/0886022x.2022.2107542
Zhenyukh O., Civantos E., Ruiz-Ortega M., Sánchez M. S., Vázquez C., Peiró C., et al. (2017). High concentration of branched-chain amino acids promotes oxidative stress, inflammation and migration of human peripheral blood mononuclear cells via mTORC1 activation. Free Radic. Biol. and Med. 104, 165–177. doi:10.1016/j.freeradbiomed.2017.01.009
Zhou H. L., Zhang R., Anand P., Stomberski C. T., Qian Z., Hausladen A., et al. (2019). Metabolic reprogramming by the S-nitroso-CoA reductase system protects against kidney injury. Nature 565, 96–100. doi:10.1038/s41586-018-0749-z
Zhou T., Xu H., Cheng X., He Y., Ren Q., Li D., et al. (2022). Sodium butyrate attenuates diabetic kidney disease partially via histone butyrylation modification. Mediat. Inflamm. 2022, 7643322. doi:10.1155/2022/7643322
Zhu H., Cao C., Wu Z., Zhang H., Sun Z., Wang M., et al. (2021). The probiotic L. casei Zhang slows the progression of acute and chronic kidney disease. Cell metab. 33, 1926–1942.e8. doi:10.1016/j.cmet.2021.06.014
Keywords: acute kidney injury, fatty acid oxidation, glycolysis, BCAAs, ketolysis, glutaminolysis
Citation: Gao J, Huang L, Zhang Y, Wei L, Yu Z, Xing Y, Yuan J, Ning X and Sun S (2025) Acute kidney injury through a metabolic lens: pathological reprogramming mechanisms and clinical translation potential. Front. Physiol. 16:1602865. doi: 10.3389/fphys.2025.1602865
Received: 30 March 2025; Accepted: 16 May 2025;
Published: 06 June 2025.
Edited by:
Yue Zhang, Nanjing Medical University, ChinaCopyright © 2025 Gao, Huang, Zhang, Wei, Yu, Xing, Yuan, Ning and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shiren Sun, c3Vuc2hpcmVuQG1lZG1haWwuY29tLmNu; Xiaoxuan Ning, bmluZ3h4MDFAZm1tdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship