 Jingyi Yang
Jingyi Yang Yingdong Wang1
Yingdong Wang1 Fangping Lu
Fangping Lu- 1Department of Pathophysiology, Mudanjiang Medical University, Mudanjiang, China
- 2Department of General Surgery, Mudanjiang Medical University Affiliated Hongqi Hospital, Mudanjiang, China
Introduction: ERS-induced apoptosis may play a pivotal role in diabetic skeletal muscle atrophy. However, the specific mechanisms by which ERS regulates skeletal muscle atrophy in diabetes remain unclear. The research examines the impact of endoplasmic reticulum stress (ERS) on skeletal muscle atrophy in type 2 diabetes mellitus (T2DM) mice.
Methods: Leptin receptor-deficient Db/db mice (n = 7, 24-week-old, male) were employed as a type 2 diabetes model, while age-matched male C57BL/6J mice (n = 7) served as normal controls. Pathway enrichment analysis of differentially expressed genes was performed based on transcriptome sequencing data, focusing on apoptosis, ERS, and ubiquitin-proteasome pathways. Skeletal muscle morphology was assessed via anatomical observation, Laminin Staining, and immunoblotting analysis (WB). WB was used to detect ERS markers (ATF6, p-eIF2α, Bip, p-JNK, Chop), apoptosis-related proteins (Bcl2, Bax, Cleaved Caspase-3, CytC), p-Akt, and muscle atrophy marker Atrogin1.
Results: Transcriptomic enrichment analysis confirmed specific activation of apoptosis, ERS, and ubiquitin-proteasome pathways. WB revealed upregulated ERS-related proteins, increased apoptotic proteins, decreased p-Akt expression, elevated Atrogin1 levels, and enhanced proteolytic activity. Db/db mice exhibited significant skeletal muscle atrophy, with Laminin Staining demonstrating reduced cross-sectional area (CSA) of muscle fibers.
Discussion: These findings uncovers a dual regulatory mechanism underlying diabetic muscle atrophy. The diabetic skeletal muscle microenvironment exhibits elevated oxidative stress and significantly enhanced ER stress, which promotes direct muscle atrophy through ER stress sensor-mediated apoptosis. Concurrently, sustained ER stress suppresses Akt activity while upregulating the muscle-specific E3 ubiquitin ligase Atrogin1, thereby accelerating proteolysis and inducing indirect muscle wasting. These findings provide crucial mechanistic insights into diabetic skeletal myopathy, highlighting the ER stress signaling network as a promising therapeutic target for mitigating muscle atrophy in diabetes.
1 Introduction
Diabetes mellitus (DM) represents a global metabolic dysregulation syndrome, where T2DM is pathophysiologically characterized by insulin resistance. T2DM is often accompanied by various complications, including skeletal muscle atrophy. Skeletal muscle, serving as the principal regulator of systemic glucose homeostasis, functions as the primary insulin-target tissue that mediates postprandial glucose disposal through GLUT4-dependent uptake mechanisms (Sylow et al., 2021). Skeletal muscle atrophy is pathologically associated with diverse clinical contexts spanning: (1) age-related sarcopenia; (2) chronic diseases; and (3) malignancy-associated cachexia (Ishii et al., 2008; Mendes et al., 2015; Zhang H. et al., 2023; Zhang et al., 2024). Emerging evidence demonstrates that dysregulated mROS production and caspase-3 activation constitute pathophysiological mechanisms driving proteolytic degradation in atrophying muscle (Plant et al., 2009; Min et al., 2011; Zhang et al., 2024).
The endoplasmic reticulum (ER) serves as a central organelle for cellular proteostasis, coordinating protein synthesis, post-translational modification, secretory trafficking, and folding quality control (Yoshida, 2007). ERS can be triggered by various pathological and physiological factors, including Ca2+ dysregulation, redox imbalance, defective protein folding, and other stressors (Jiang et al., 2021). In patients with T2DM, obesity is frequently associated with enhanced adipokine secretion, which subsequently promotes excessive reactive oxygen species (ROS) generation and induces systemic oxidative stress (Fernandez-Sanchez et al., 2011). As the primary defense mechanism against oxidative damage, the endogenous antioxidant system mobilizes crucial enzymes including superoxide dismutase (SOD) and catalase (CAT) to establish the first-line protective barrier against oxidative assault (Jomova et al., 2024). This compensatory upregulation represents a critical adaptive response to maintain redox homeostasis. Furthermore, the persistent oxidative imbalance contributes to the accumulation of misfolded proteins, thereby triggering endoplasmic reticulum (ER) stress through impaired protein folding capacity (Ong and Logue, 2023). Physiological Unfolded Protein Response (UPR) activation under moderate ERS orchestrates proteostatic adaptation through three transmembrane sensors. Conversely, sustained ERS drives terminal UPR signaling that activates caspase-dependent apoptotic cascades, culminating in programmed cell death (Park et al., 2019; Guo et al., 2024). The UPR is mediated by three evolutionarily conserved sensors: inositol-requiring enzyme 1α (IRE1α); protein kinase R (PKR)-like ER kinase (PERK); and activating transcription factor 6 (ATF6) (Casas-Martinez et al., 2024). Under proteostatic equilibrium, these sensors remain sequestered by the ER chaperone BiP/GRP78. During ERS, misfolded protein overload competitively dissociates BiP/GRP78 from the sensors, enabling their oligomerization and activation. PERK activation catalyzes eIF2α phosphorylation, which transiently attenuates global translation while paradoxically enabling ATF4-mediated transcriptional upregulation of pro-apoptotic factor CHOP. ATF4 and CHOP synergistically stimulate the expression of genes related to apoptosis, autophagy, and antioxidant responses (Lu et al., 2024). IRE1α endoribonuclease activity executes XBP1 mRNA splicing, generating sXBP1 for UPR gene activation, while concomitantly activating apoptosis signal-regulating kinase 1 (ASK1)-JNK1 signaling (Brozzi et al., 2015; Lu et al., 2024). Activated ATF6 translocates to the Golgi for proteolytic cleavage, yielding a transcriptionally active fragment that represses anti-apoptotic Bcl-2 proteins while inducing pro-apoptotic effectors. In myocytes, ATF6 hyperactivation provokes caspase-9-mediated intrinsic apoptosis via mitochondrial permeabilization (Kapuy, 2024).
Akt (protein kinase B, PKB), a pivotal node in insulin receptor substrate (IRS) signaling, coordinates cellular proliferation, survival, and metabolic homeostasis through phosphorylation-dependent regulation of downstream effectors (Viglietto et al., 2002; Bao et al., 2004; Li et al., 2007). Following insulin receptor activation, phosphatidylinositol-3,4,5-trisphosphate (PIP3) recruits Akt to the plasma membrane via pleckstrin homology (PH) domain interaction, enabling PDK1-mediated phosphorylation at Thr308 within the activation loop (Stephens et al., 1998). Concomitant mTOR complex 2 (mTORC2)-dependent phosphorylation at Ser473 in the hydrophobic motif confers full catalytic competence (Zeng et al., 2007). Activated Akt modulates downstream targets, including FoxOs, cell cycle regulators, mTOR, and GSK3, to regulate diverse physiological functions (Manning and Cantley, 2007; Vantler et al., 2023).
ERS-induced apoptosis may play a pivotal role in diabetic skeletal muscle atrophy. However, the specific mechanisms by which ERS regulates skeletal muscle atrophy in diabetes remain unclear. This research aims to explore the molecular mechanisms of ERS-mediated skeletal muscle atrophy in diabetic mice. By comparing skeletal muscle alterations, ERS marker expression, and apoptosis between diabetic and control groups, we seek to provide new insights into the pathogenesis of diabetic muscle atrophy and theoretical support for developing ERS-targeted therapies.
2 Materials and methods
2.1 Data collection and processing
The GSE22309 dataset was retrieved from the GEO database (https://www.ncbi.nlm.nih.gov/geo/) using the keyword “diabetes.” This dataset included human skeletal muscle samples from insulin-sensitive individuals and insulin-naïve diabetic patients. Differentially expressed genes (DEGs) were identified using GEO2R (|logFC| ≥ 1, FDR < 0.05). Gene Ontology (GO) analysis was conducted with DAVID and visualized with Cytoscape.
2.2 Animal experiments
All animal experiments were approved by the Animal Experimental Ethics Committee (IACUC-2303040). Leptin receptor-deficient Db/db mice (n = 7, 8-week-old, male) were employed as a type 2 diabetes model, while age-matched male C57BL/6J mice (n = 7) served as normal controls (Pandey and Dvorakova, 2020). Following 16 weeks of standardized housing under specific pathogen-free conditions, skeletal muscle tissues were collected for molecular characterization. All experimental animals were generously provided by Harbin Medical University.
2.3 Transcriptome data analysis
RNA from the hindlimb skeletal muscles of 5 control and 4 Db/db mice was sequenced on the Illumina platform by Metware Biotechnology Co., Ltd. (Wuhan, China).
2.4 Body weight and random blood glucose monitorin
Body weight was measured using an electronic balance (0.1 g resolution). Mice were briefly transferred to an empty cage for consistent weighing. Random blood glucose was assessed without fasting: a needle lightly punctured the tail vein, the first blood droplet was discarded, and subsequent droplets were analyzed via glucometer.
2.5 Intraperitoneal glucose tolerance test
Following 12-h fast. A sterile needle was used to puncture the tail tip, and blood droplets were collected for baseline (0-min) glucose measurement. Then glucose (2 g/kg) was administered i. p. Blood glucose was re-measured at 30, 60, and 120 min post-injection (same method) (Zhang Y. et al., 2023).
2.6 Antibodies
Anti-SOD (1:1,000), anti-p-eIF2α (1:1,000), anti-GAPDH (1:10,000), anti-ATF6 (1:1,000), anti-eIF2α (1:1,000), anti-p-JNK (1:1,000), anti-JNK (1:1,000), anti-CYTC (1:1,000), anti-Atrogin1 (1:1,000), anti-BiP (1:1,000), anti-Bax (1:1,000), anti-Caspase3 (1:1,000), anti-MYH4 (1:1,000), anti-CHOP (1:1,000), anti-MYOM1 (1:1,000) antibodies were purchased from Proteintech. Anti-TNNI2 (1:1,000), anti-CAT (1:1,000) antibodies were obtained from Affinity Biosciences (Qinke Biotechnology Co., Ltd.). Anti-Bcl2 (1:500) antibody was purchased from Wanlei Biotechnology Co., Ltd.
2.7 Immunoblotting analysis
Hind limb skeletal muscle tissues of mice were homogenized in pre-chilled RIPA buffer and centrifuged at low speed (3,000 rpm, 10 min, 4°C). The supernatant was collected and vortexed intermittently (20 s every 5 min for 30 min total). After centrifugation at 12,000 rpm for 30 min (4°C), the clarified supernatant was collected. Protein concentration was determined by BCA assay, and samples were adjusted to load 80 μg protein per well. The samples were mixed with 5× loading buffer at a 1:4 ratio, boiled at 100°C for 8 min, and separated by SDS-PAGE. Proteins were transferred to PVDF membranes (0.45 μm pore size) via wet transfer. Membranes were blocked for 90 min at room temperature, incubated with primary antibodies (4°C, 16 h) and HRP-conjugated secondary antibodies (25°C, 1 h). Protein bands were visualized by ECL and quantified using ImageJ software for normalization and statistical analysis.
2.8 Laminin staining
Immunofluorescence staining of hindlimb skeletal muscle (5 μm cryosections) was performed as follows: Tissues were fixed in 4% PFA, embedded in OCT compound, and sectioned using a cryostat. Antigen retrieval was performed by heating sections in citrate buffer (95°C, 20 min). After PBS washes, sections were blocked with blocking buffer (25°C, 30 min) and incubated with anti-laminin antibody (4°C, 16 h). Secondary antibody labeling was conducted under light-protected conditions (25°C, 1 h). Nuclei were counterstained with DAPI (5 min), and slides were mounted with antifade mounting medium. Images were acquired using confocal microscopy.
2.9 Statistical analysis
Data were analyzed using GraphPad Prism and expressed as mean ± SEM. Group comparisons were performed using Student’s t-test. The normality of data for all parametric tests (Student’s t-test) was verified using the Shapiro-Wilk test. Significance was set at *P < 0.05 and **P < 0.01.
3 Results
3.1 Differential gene expression in diabetic skeletal muscle
Differentially expressed genes (DEGs) were screened using GEO2R-normalized microarray data from the GEO database (GSE22309 dataset) with thresholds of |log2Fold Change| ≥1 and FDR < 0.05. A total of 227 diabetes mellitus (DM)-related DEGs were identified (Figure 1A). Protein-protein interaction network analysis conducted through the STRING database revealed gene clusters associated with ER homeostasis and apoptosis (Figure 1B). Gene Ontology (GO) enrichment analysis demonstrated significant enrichment in biological processes (BPs) including apoptosis regulation, proteostasis maintenance, and glycogen metabolism. Cellular components (CCs) were predominantly localized to the nucleus, cytoplasm, mitochondria, and ER. Molecular functions (MFs) involved protein binding, nucleic acid binding, ubiquitin-protein ligase interactions, and unfolded protein binding (Figure 1C). These bioinformatic analyses collectively indicate dysregulation of ER stress modulation, apoptotic damage, and proteostatic control in skeletal muscle of diabetic patients.
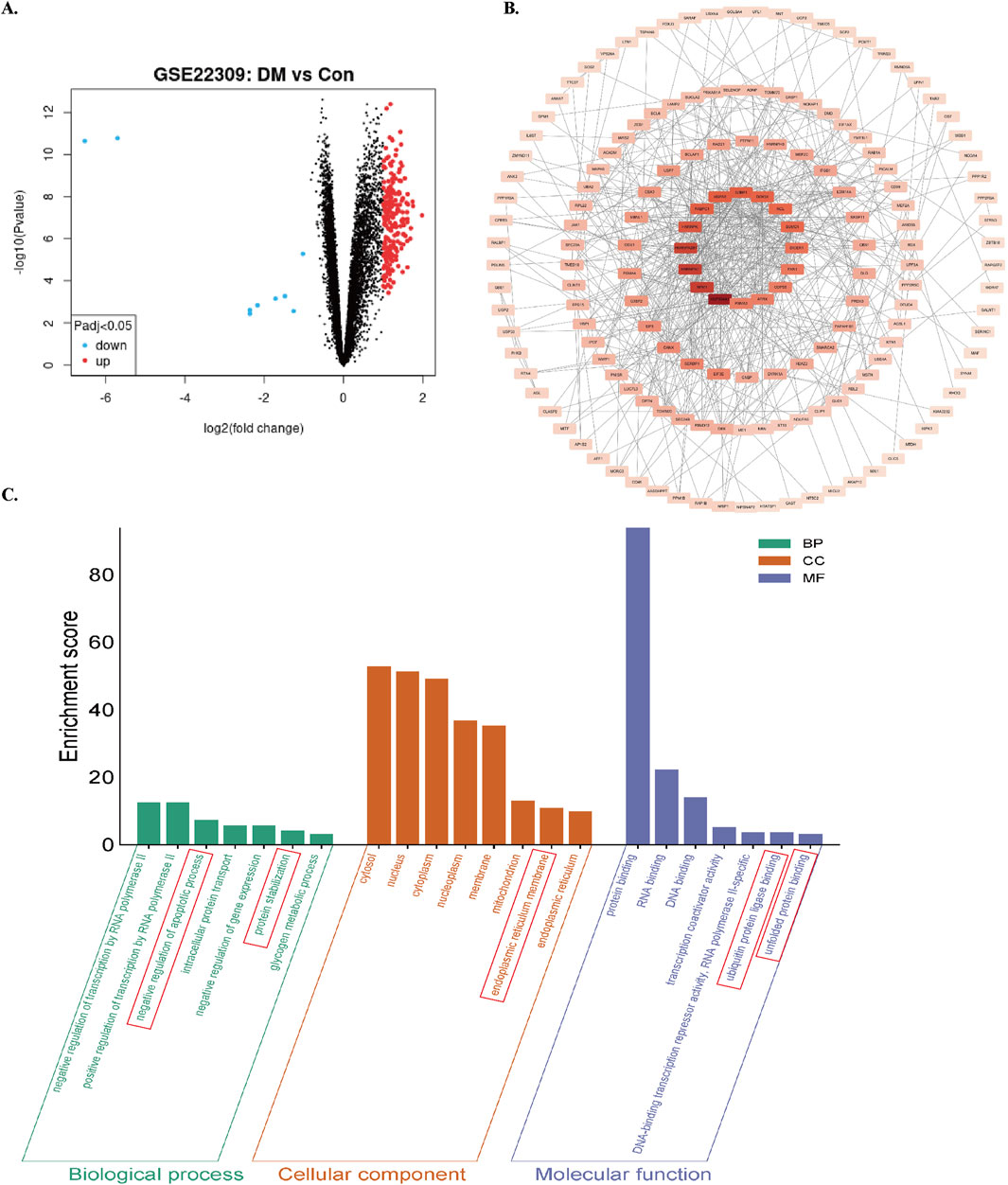
Figure 1. Screening and enrichment analysis of DEGs in the GSE22309 dataset. (A) Volcano plot of DEGs comparing skeletal muscle from insulin-sensitive individuals (Con) and untreated diabetic patients (DM). using red/blue color dots to denote upregulation and downregulation. (B) PPI network of DEGs. (C) GO enrichment analysis of DEGs. Enrichment score reflects the number of enriched genes; higher values indicate stronger enrichment.
3.2 Upregulated ER stress and apoptosis-associated genes in diabetic murine skeletal muscle
To further explore differential gene expression patterns, transcriptome sequencing was conducted on hindlimb skeletal muscle tissues from control and Db/db mice. DEGs were filtered using thresholds of |log2Fold Change| ≥ 1 and FDR <0.05, resulting in 1,764 DEGs (1,182 upregulated and 582 downregulated in Db/db mice) (Figure 2A). Cross-referencing analysis with 1,827 UPR-associated genes from the GeneCards database identified 106 overlapping genes (Figure 2B). Gene Ontology (GO) enrichment analysis demonstrated significant enrichment of BPs related to ER stress, insulin response, and apoptosis (Figure 2C). Furthermore, intersection analysis of 2,584 type 2 diabetes-associated genes curated from CTD, GeneCards, and DisGeNET databases revealed substantial overlap with ER stress and apoptosis-related genes (Figure 2D). Upregulated ER Stress and Apoptosis-Associated Genes in Diabetic Murine Skeletal Muscle: Integrated multi-omics analyses reveal a significant association between ER stress activation and apoptotic pathway enrichment in diabetic murine models.
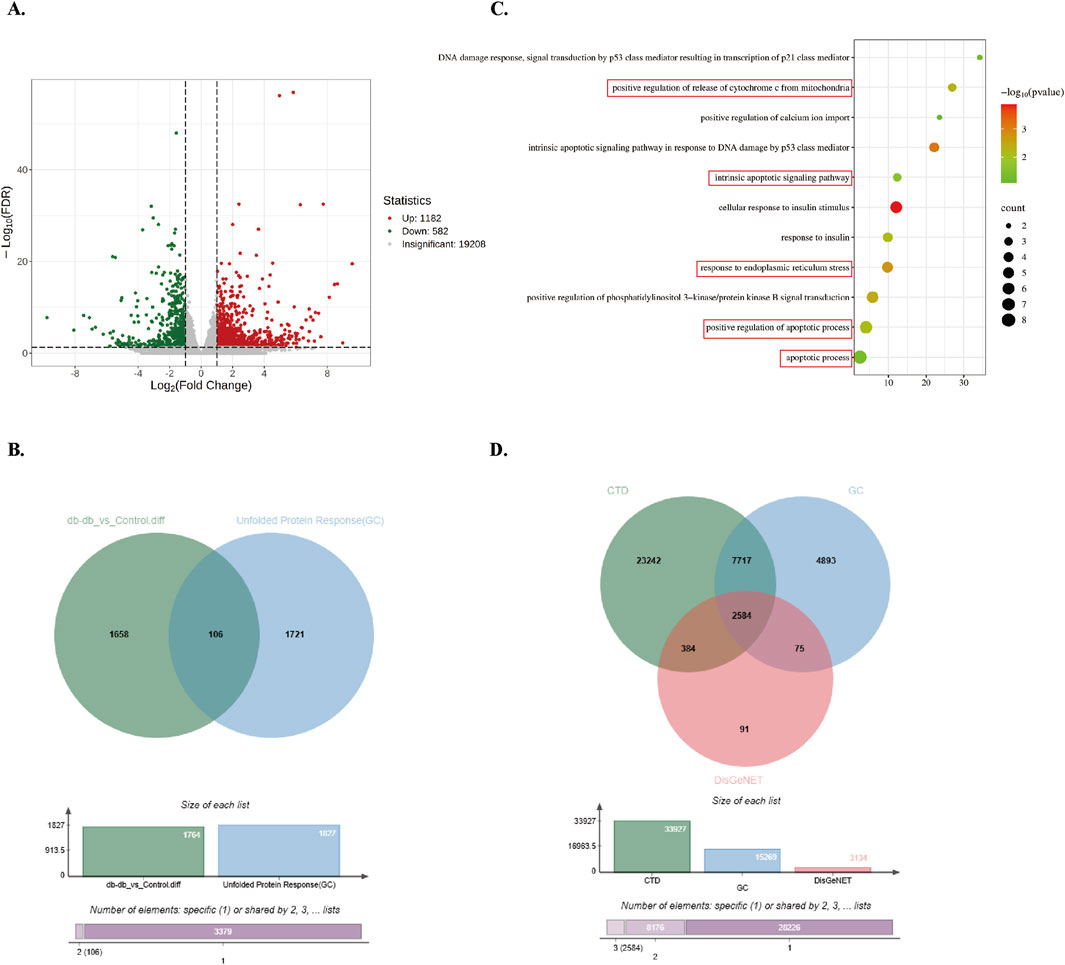
Figure 2. Screening and enrichment analysis of Db/db-associated DEGs. (A) Volcano plot of Db/db-related DEGs, using red/blue color scales to denote upregulation and downregulation. (B) Venn diagram showing integration between DEGs and UPR-related genes from GeneCards. (C) GO BP enrichment analysis of overlapping genes. Dot size reflects gene count; color intensity represents P-value (darker = lower P). (D) Venn diagram of T2DM-related genes from DisGeNET, GeneCards, and CTD.
3.3 Metabolic characterization of diabetic murine models
To validate the establishment of T2DM in murine models, bodyweight, random blood glucose levels, and glucose tolerance were systematically assessed in 8-week-old mice. Db/db mice demonstrated significantly greater bodyweight relative to controls (Figure 3A). The random blood glucose concentrations in diabetic mice were substantially elevated compared to controls (Figure 3B). Intraperitoneal glucose tolerance testing (IPGTT) further revealed impaired glycemic regulation in Db/db mice pared to control mice (Figure 3C). Quantification of the glucose tolerance curve area under the curve (AUC) showed significantly elevated values in diabetic versus controls (Figure 3D). These findings collectively demonstrate disrupted glucose homeostasis in Db/db mice, confirming successful T2DM model establishment.
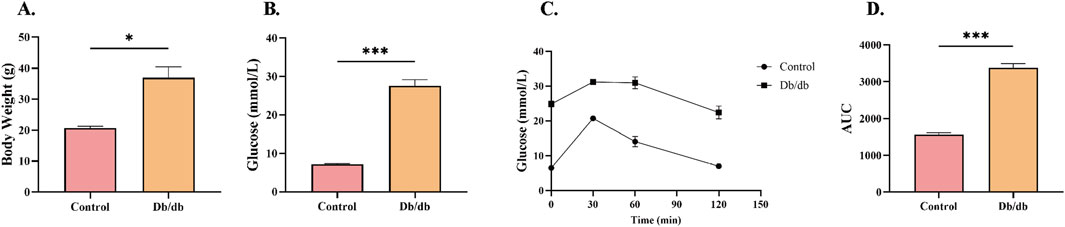
Figure 3. Metabolic Profile Characterization in Control and Db/db Murine models. (A) Body weight comparison at 8 weeks post-modeling. (B) Random blood glucose measurements between Control and Db/db cohorts. (C) Intraperitoneal glucose tolerance test (IPGTT) trajectories. (D) Quantitative area under the curve (AUC) analysis of glycemic responses. Data: mean ± SEM. *P < 0.05 was considered statistically significant. n = 3.
3.4 ERS activation in Db/db murine models
ERS has been mechanistically linked to oxidative stress, ubiquitin-proteasome pathway activation, and inflammatory signaling (Zhang H. et al., 2023). The quantity levels of critical antioxidant enzymes superoxide dismutase (SOD) and catalase (CAT), which counteract oxidative damage (Jomova et al., 2024), were significantly downregulated in Db/db mice compared to Control cohorts (Figures 4A–C), indicative of exacerbated oxidative stress. Furthermore, proteomic analysis revealed elevated expression of ERS markers including the ER transmembrane sensor ATF6, phosphorylated eukaryotic initiation factor 2α (p-eIF2α) as a downstream target of PERK signaling, and the ER chaperone BiP/GRP78 in Db/db mice compared to control mice (Figures 4D–G). These findings collectively demonstrate concomitant augmentation of oxidative stress and ERS pathways in diabetic murine models.
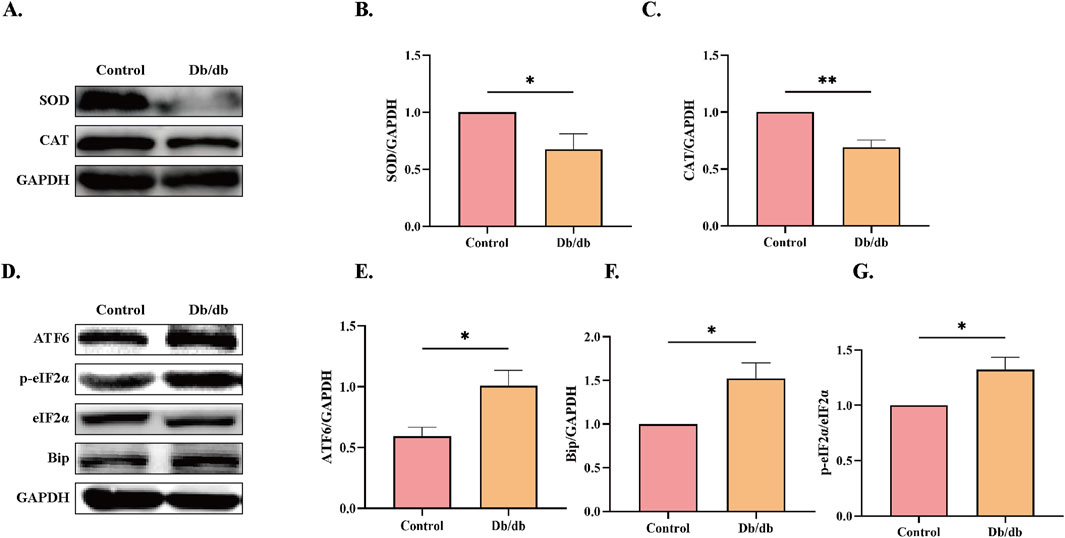
Figure 4. ERS Protein Levels in Control vs. Db/db Mice. (A) superoxide dismutase (SOD) and catalase (CAT) protein levels. Quantitative analysis of (B) SOD, n = 5, (C) CAT, n = 4. (D) Activating transcription factor 6 (ATF6), immunoglobulin heavy chain binding protein (Bip), and Phosphorylation of eukaryotic initiation factor-2α (p-eIF2α) protein levels. Quantitative analysis of (E) ATF6, n = 4, (F) Bip, n = 4, (G) p-eIF2α, n = 3. Data: mean ± SEM. *P < 0.05 was considered statistically significant.
3.5 Enhanced apoptotic activation in Db/db murine models
To further investigate whether ERS induces elevated apoptosis in Db/db mice, we performed protein-level analyses. CHOP and JNK, downstream targets of the PERK and IRE1α pathways, are well-known apoptosis-related proteins (Lakshmanan et al., 2011). CHOP-deficient mice exhibit reduced caspase-3 activation, increased Bcl-2/Bax ratios, and attenuated apoptosis in cardiac tissues (Fu et al., 2010). Additionally, colistin-induced JNK activation has been shown to significantly elevate the Bax/Bcl-2 ratio (Lu et al., 2017). Our results revealed increased expression of CHOP and phosphorylated JNK (p-JNK) in Db/db mice compared to control mice (Figures 5A–C). Furthermore, we assessed the levels of apoptosis-related protein markers and observed elevated expression of pro-apoptotic proteins, including cleaved caspase-3 (C-Caspase3), cytochrome C (CytC), and Bax, alongside reduced levels of the anti-apoptotic protein Bcl-2 in Db/db mice versus control mice (Figures 5D–H). These findings collectively indicate that ERS contributes to enhanced apoptosis in Db/db mice.
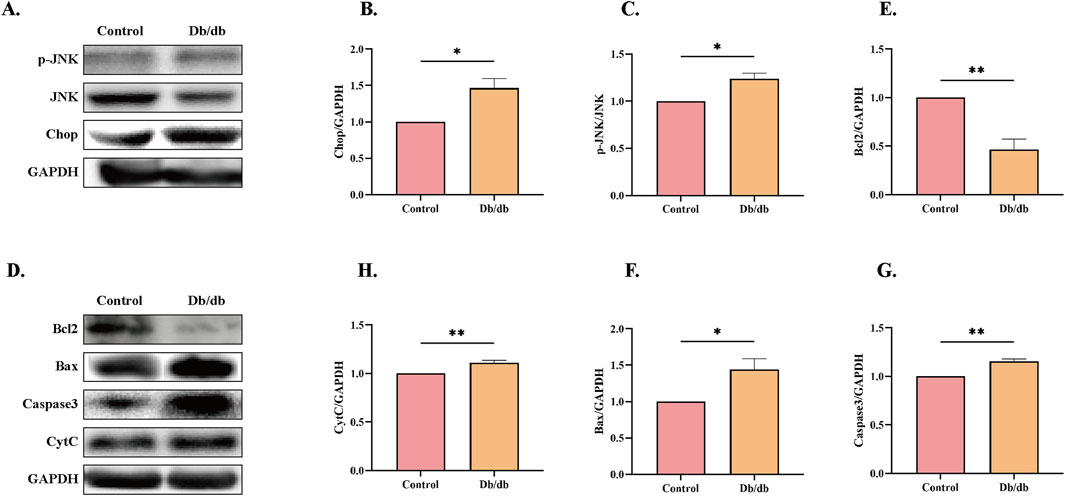
Figure 5. Apoptotic Pathway Activation Profile in Control and Db/db Murine models. (A) Immunoblot analysis of phosphorylated c-Jun N-terminal kinase (p-JNK) and C/EBP Homologous Protein (CHOP). Quantitative analysis of (B) p-JNK, n = 3, (C) CHOP, n = 3. (D) Protein levels of Cleaved caspase-3 (c-Casp3), Cytochrome c (Cyt c), B-cell lymphoma 2 (Bcl-2), and Bcl-2-associated X protein (Bax). Quantitative analysis of (E) Bcl-2, n = 3, (F) Bax, n = 3, (G) c-Casp3, n = 3, (H) Cyt c, n = 4. Data: mean ± SEM. *P < 0.05 was considered statistically significant.
3.6 Reduced Akt activity and activation of E3 ubiquitin ligase in Db/db murine models
A significant decrease in phosphorylated Akt (p-Akt) levels was observed in Db/db mice compared to control mice (Figures 6A,B). Additionally, Atrogin-1 protein levels were markedly elevated compared to the Control (Figures 6C,D). These results indicate that ER stress in Db/db mice likely reduces Akt activity, leading to the activation of the E3 ligase Atrogin-1 and subsequent skeletal muscle atrophy.
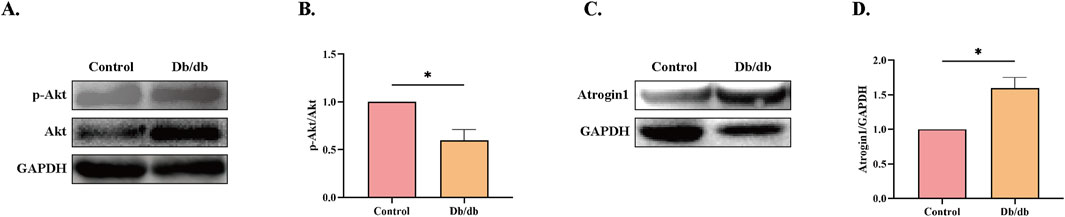
Figure 6. Protein Levels in Control vs. Db/db Mice. (A) Phosphorylated Protein Kinase B (p-Akt) protein levels. (B) Quantification of p-Akt. (C) Muscle Atrophy F-box protein 1 (Atrogin-1) protein levels. (D) Quantification of Atrogin-1. Statistical significance as above. Data: mean ± SEM. *P < 0.05 was considered statistically significant. n = 3.
3.7 Skeletal muscle atrophy in Db/db murine mice
To assess skeletal muscle atrophy in Db/db mice, we first examined gastrocnemius muscle morphology, which revealed a significant reduction in muscle mass compared to control mice (Figures 7A,B). Immunofluorescence staining of laminin further confirmed atrophy, demonstrating a marked decrease in the cross-sectional area (CSA) of muscle fibers in Db/db mice compared to control mice (Figures 7C,D). Consistent with these morphological changes, Western blot analysis showed significantly reduced expression levels of key skeletal muscle structural proteins—TNNI2, MYH4, and MYOM1—in Db/db mice compared to controls (Figures 7E–H). Together, these findings provide conclusive evidence that Db/db mouse exhibit skeletal muscle atrophy.
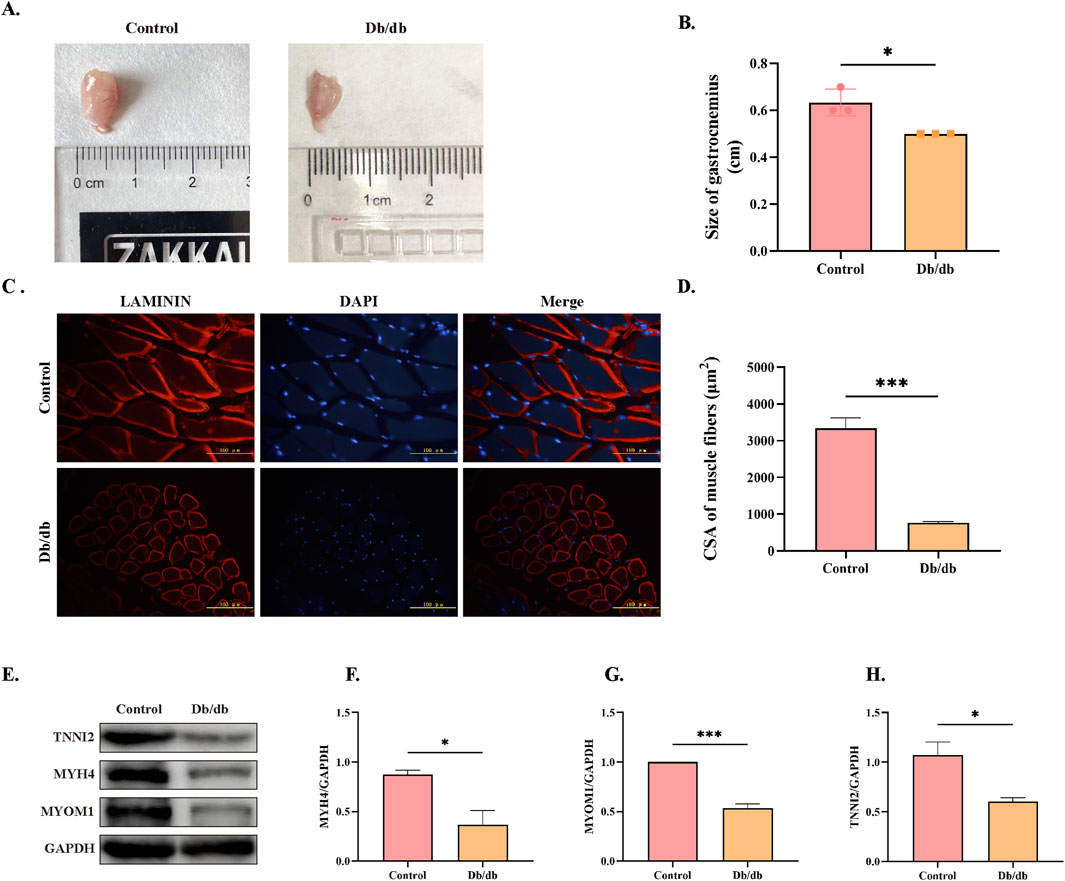
Figure 7. Morphological and Structural Protein Expression Differences in Skeletal Muscle Between Control and Db/db Murine Mice. (A) Gross anatomy of gastrocnemius muscles. (B) Quantification of gastrocnemius muscle mass. (C) Laminin immunofluorescence staining of skeletal muscle cross-sections, Scale bar = 100 μm. (D) Quantification of muscle fiber cross-sectional area (CSA). (E) Western blot analysis of skeletal muscle structural proteins: Troponin I, Fast Skeletal Muscle9 (TNNI2), Myosin Heavy Chain 4 (MYH4), and Myomesin 1 (MYOM1). Quantitative analysis of (F) MYH4, (G) MYOM1, (H) TNNI2. Data: mean ± SEM. *P < 0.05 was considered statistically significant. n = 3.
4 Discussion
Maintaining redox homeostasis is crucial for normal physiological functions. Under pathological conditions, oxidative stress disrupts this balance and participates in the pathogenesis of multiple diseases including DM, inflammatory responses, and tumors (Martemucci et al., 2023). As a vital component of cellular stress responses, ERS interacts closely with oxidative stress, the UPS, and inflammatory signaling pathways through molecular mechanisms (Liu et al., 2023). SOD and CAT, serving as the first line of defense against oxidative stress, constitute the most robust protective barrier against oxidative damage (Jomova et al., 2024). Glutathione (GSH), a central antioxidant, maintains redox homeostasis in biological systems. Studies in the epididymis of T2DM rodents demonstrated significantly reduced glutathione peroxidase (GPx) activity and elevated glutathione reductase (GR) levels (Diniz et al., 2022). Another study further revealed that diminished GSH pools in diabetic systems exacerbate oxidative stress and inflammatory responses through redox imbalance (Dawi et al., 2024). These findings collectively imply that disrupted glutathione metabolism may underlie diabetes-associated tissue dysfunction. Research have revealed that oxidative stress disrupts nascent ER protein folding, triggering the UPR to handle misfolded proteins and activating downstream stress sensors that induce ER stress (Peserico et al., 2020). Our results demonstrated that diabetic mice exhibited elevated oxidative stress levels accompanied by reduced protein expression of antioxidant enzymes SOD and CAT. Concurrently, increased protein levels of ER chaperone Bip, UPR stress sensors ATF6 and IRE1α, along with their downstream regulators p-eIF2α, CHOP, and p-JNK were observed. Persistent ER stress ultimately leads to apoptosis-mediated cell death (Yang et al., 2008), while experimental evidence shows that knocking down CHOP or JNK protein levels attenuates apoptosis induction (Yang et al., 2021). The release of CytC into cytoplasm represents a critical event in the mitochondrial-dependent intrinsic apoptotic pathway, where cytosolic CytC combines with apoptotic protease-activating factors to initiate caspase cascade reactions, ultimately causing cellular damage (Wang et al., 2016). Our results showed upregulation of cleaved Caspase-3, Bax and CytC (pro-apoptotic) with concomitant downregulation of Bcl-2 (anti-apoptotic), indicating the occurrence of apoptosis that directly contributes to skeletal muscle atrophy.
Skeletal muscle atrophy primarily arises from three key mechanisms: increased proteolysis, reduced protein synthesis, and impaired myofiber regeneration. These processes are regulated by multiple molecular pathways, including the ubiquitin-proteasome system (UPS), autophagy-lysosomal pathway (ALP), calpain system, caspase pathway, IGF-1/Akt protein synthesis pathway, myostatin signaling, and muscle satellite cells (Ji et al., 2022). Among these, the PI3K/Akt/mTOR signaling pathway plays a central role in modulating protein synthesis in skeletal muscle. Activation of Akt inhibits FoxO-mediated transcription of muscle-specific E3 ubiquitin ligases, such as MuRF1 and Atrogin-1, while simultaneously promoting protein synthesis (Egerman and Glass, 2014). Chronic ER stress has been shown to impair the RTK/PI3K/AKT signaling pathway (Qin et al., 2010; Lu et al., 2017). For example, ER stress in aged rat livers suppresses insulin signaling, inhibits Akt, and upregulates FOXO expression (Kim et al., 2019). FoxOs are transcription factors that regulate E3 ubiquitin ligases. When inhibiting the Akt pathway, nuclear FoxOs promote the expression of MuRF1/atrogin-1, activating the UPS and driving protein degradation and skeletal muscle atrophy (Ji et al., 2022). Similarly, dietary selenium deficiency or excess in rainbow trout modulates the Akt/FoxO3a pathway, accelerating ubiquitin-mediated muscle protein degradation (Zhang et al., 2022).
In our research, we observed reduced Akt activity and elevated Atrogin-1 expression in the skeletal muscle of diabetic mice compared to control mice, accompanied by significant muscle atrophy. This muscular degeneration inevitably leads to impaired motor function, severely impacting the quality of life of affected individuals and imposing substantial personal, familial, and societal burdens.
While this study revealed significant dysregulation of ERS-related molecular markers in diabetic muscle atrophy, the causal relationships remain to be directly validated through functional interventions. Notably, previous investigations have demonstrated that 4-phenylbutyric acid (4-PBA), a chemical chaperone and ERS inhibitor, can ameliorate triptolide (TP)/lipopolysaccharide (LPS)-induced ERS-associated apoptosis and hepatic oxidative stress (Cheng et al., 2024). Furthermore, emerging evidence indicates that pharmacological inhibition of ERS with Salubrinal effectively attenuates ferroptosis and cellular damage in diabetic myocardial ischemia/reperfusion injury (Li et al., 2020). Based on these mechanistic insights, we recommend the following strategies to establish causal relationships: (1) Pharmacological validation using FDA-approved ERS inhibitors (e.g., 4-PBA or tauroursodeoxycholic acid [TUDCA]) in both Db/db mice and diabetic myotube models to assess potential reversal of muscle atrophy phenotypes; (2) Genetic validation through targeted knockdown of core ERS regulators (such as PERK, ATF6, or IRE1α) to systematically evaluate their regulatory effects on apoptotic pathways and ubiquitin-proteasome-mediated protein degradation. These investigations will advance the development of ERS-targeted therapies for diabetic sarcopenia.
5 Conclusion
As demonstrated in Figure 8, our research uncovers a dual regulatory mechanism underlying diabetic muscle atrophy. The diabetic skeletal muscle microenvironment exhibits elevated oxidative stress and significantly enhanced ER stress, which promotes direct muscle atrophy through ER stress sensor-mediated apoptosis. Concurrently, sustained ER stress suppresses Akt activity while upregulating the muscle-specific E3 ubiquitin ligase Atrogin-1, thereby accelerating proteolysis and inducing indirect muscle wasting. These findings provide crucial mechanistic insights into diabetic skeletal myopathy, highlighting the ER stress signaling network as a promising therapeutic target for mitigating muscle atrophy in diabetes. Future studies should elucidate the long-term consequences of ER stress modulation, identifying additional molecular players in this pathological cascade, and developing comprehensive intervention strategies to prevent and treat this debilitating complication.
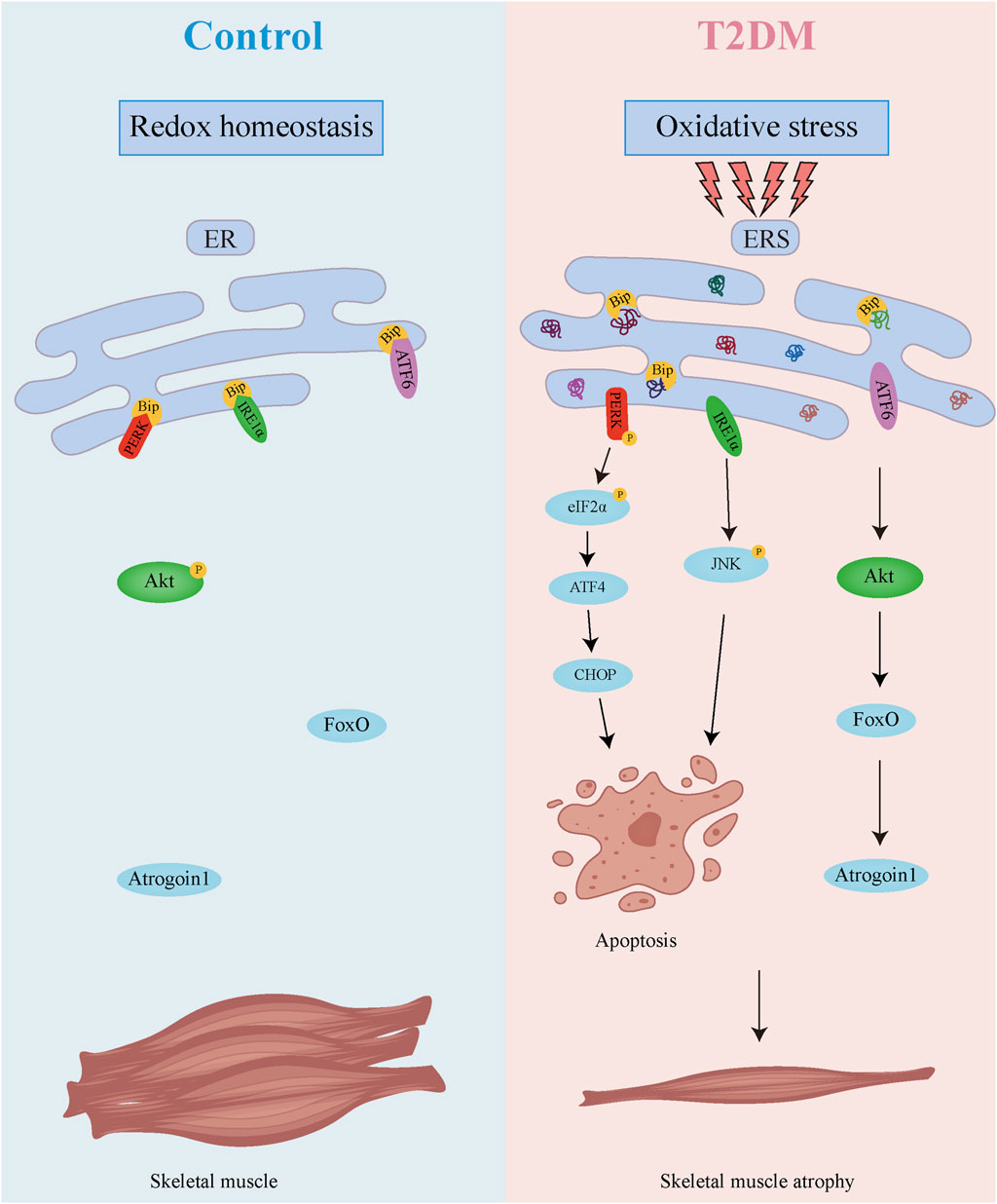
Figure 8. Mechanism diagram of skeletal muscle atrophy in T2DM.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by the Animal Experimental Ethics Committee (IACUC-2303040). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
JY: Writing – original draft, Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Resources, Software, Validation, Visualization. YW: Conceptualization, Data curation, Formal Analysis, Investigation, Writing – original draft. YX: Conceptualization, Data curation, Investigation, Methodology, Writing – original draft. XJ: Software, Supervision, Writing – review and editing. FL: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by Health Commission of Heilongjiang Provincial (Grant numbers: 20230202080190).
Acknowledgments
We are grateful to Wuhan Metware Biotechnology Co., Ltd. for assisting in sequencing and/or bioinformatics analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Bao S., Ouyang G., Bai X., Huang Z., Ma C., Liu M., et al. (2004). Periostin potently promotes metastatic growth of colon cancer by augmenting cell survival via the Akt/PKB pathway. Cancer Cell 5 (4), 329–339. doi:10.1016/s1535-6108(04)00081-9
Brozzi F., Nardelli T. R., Lopes M., Millard I., Barthson J., Igoillo-Esteve M., et al. (2015). Cytokines induce endoplasmic reticulum stress in human, rat and mouse beta cells via different mechanisms. Diabetologia 58 (10), 2307–2316. doi:10.1007/s00125-015-3669-6
Casas-Martinez J. C., Samali A., McDonagh B. (2024). Redox regulation of UPR signalling and mitochondrial ER contact sites. Cell. Mol. Life Sci. 81 (1), 250. doi:10.1007/s00018-024-05286-0
Cheng R., Jiang Y., Zhang Y., Ismail M., Zhang L., Jiang Z., et al. (2024). Proteasome activity inhibition mediates endoplasmic reticulum stress-apoptosis in triptolide/lipopolysaccharide-induced hepatotoxicity. Cell. Biol. Toxicol. 40 (1), 60. doi:10.1007/s10565-024-09903-3
Dawi J., Misakyan Y., Affa S., Kades S., Narasimhan A., Hajjar F., et al. (2024). Oxidative stress, glutathione insufficiency, and inflammatory pathways in type 2 diabetes mellitus: implications for therapeutic interventions. Biomedicines 13 (1), 18. doi:10.3390/biomedicines13010018
Diniz A., Alves M. G., Candeias E., Duarte A. I., Moreira P. I., Silva B. M., et al. (2022). Type 2 diabetes induces a pro-oxidative environment in rat epididymis by disrupting SIRT1/PGC-1α/SIRT3 pathway. Int. J. Mol. Sci. 23 (16), 8912. doi:10.3390/ijms23168912
Egerman M. A., Glass D. J. (2014). Signaling pathways controlling skeletal muscle mass. Crit. Rev. Biochem. Mol. Biol. 49 (1), 59–68. doi:10.3109/10409238.2013.857291
Fernandez-Sanchez A., Madrigal-Santillan E., Bautista M., Esquivel-Soto J., Morales-Gonzalez A., Esquivel-Chirino C., et al. (2011). Inflammation, oxidative stress, and obesity. Int. J. Mol. Sci. 12 (5), 3117–3132. doi:10.3390/ijms12053117
Fu H. Y., Okada K., Liao Y., Tsukamoto O., Isomura T., Asai M., et al. (2010). Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation 122 (4), 361–369. doi:10.1161/CIRCULATIONAHA.109.917914
Guo S., Tong Y., Li T., Yang K., Gao W., Peng F., et al. (2024). Endoplasmic reticulum Stress-Mediated cell death in renal fibrosis. Biomolecules 14 (8), 919. doi:10.3390/biom14080919
Ishii A., Ohkoshi N., Tamaoka A. (2008). G.P.9.13 Expressions of FOXO1 and FOXO3a in skeletal muscle atrophy associated with neuromuscular diseases. Neuromuscul. Disord. 9-10 (18), 789. doi:10.1016/j.nmd.2008.06.225
Ji Y., Li M., Chang M., Liu R., Qiu J., Wang K., et al. (2022). Inflammation: roles in skeletal muscle atrophy. Antioxidants 11 (9), 1686. doi:10.3390/antiox11091686
Jiang X., Yu W., Wu S., Tang L., Zhong G., Wan F., et al. (2021). Arsenic (III) and/or Antimony (III) induced disruption of calcium homeostasis and endoplasmic reticulum stress resulting in apoptosis in mice heart. Ecotoxicol. Environ. Saf. 220, 112394. doi:10.1016/j.ecoenv.2021.112394
Jomova K., Alomar S. Y., Alwasel S. H., Nepovimova E., Kuca K., Valko M. (2024). Several lines of antioxidant defense against oxidative stress: antioxidant enzymes, nanomaterials with multiple enzyme-mimicking activities, and low-molecular-weight antioxidants. Arch. Toxicol. 98 (5), 1323–1367. doi:10.1007/s00204-024-03696-4
Kapuy O. (2024). Mechanism of decision making between autophagy and apoptosis induction upon endoplasmic reticulum stress. Int. J. Mol. Sci. 25 (8), 4368. doi:10.3390/ijms25084368
Kim D. H., Ha S., Choi Y. J., Dong H. H., Yu B. P., Chung H. Y. (2019). Altered FoxO1 and PPARγ interaction in age-related ER stress-induced hepatic steatosis. Aging (Albany NY) 11 (12), 4125–4144. doi:10.18632/aging.102042
Lakshmanan A. P., Thandavarayan R. A., Palaniyandi S. S., Sari F. R., Meilei H., Giridharan V. V., et al. (2011). Modulation of AT-1R/CHOP-JNK-Caspase12 pathway by olmesartan treatment attenuates ER stress-induced renal apoptosis in streptozotocin-induced diabetic mice. Eur. J. Pharm. Sci. 44 (5), 627–634. doi:10.1016/j.ejps.2011.10.009
Li W., Li W., Leng Y., Xiong Y., Xia Z. (2020). Ferroptosis is involved in diabetes myocardial Ischemia/Reperfusion injury through endoplasmic reticulum stress. DNA. Cell. Biol. 39 (2), 210–225. doi:10.1089/dna.2019.5097
Li X., Monks B., Ge Q., Birnbaum M. J. (2007). Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature 447 (7147), 1012–1016. doi:10.1038/nature05861
Liu H., Lai W., Nie H., Shi Y., Zhu L., Yang L., et al. (2023). PM2.5 triggers autophagic degradation of Caveolin-1 via endoplasmic reticulum stress (ERS) to enhance the TGF-β1/Smad3 axis promoting pulmonary fibrosis. Environ. Int. 181, 108290. doi:10.1016/j.envint.2023.108290
Lu Y., Zhou J., Wang H., Gao H., Ning E., Shao Z., et al. (2024). Endoplasmic reticulum stress-mediated apoptosis and autophagy in osteoarthritis: from molecular mechanisms to therapeutic applications. Cell Stress Chaperones 29 (6), 805–830. doi:10.1016/j.cstres.2024.11.005
Lu Z., Miao Y., Muhammad I., Tian E., Hu W., Wang J., et al. (2017). Colistin-induced autophagy and apoptosis involves the JNK-Bcl2-Bax signaling pathway and JNK-p53-ROS positive feedback loop in PC-12 cells. Chem. Biol. Interact. 277, 62–73. doi:10.1016/j.cbi.2017.08.011
Manning B. D., Cantley L. C. (2007). AKT/PKB signaling: navigating downstream. Cell 129 (7), 1261–1274. doi:10.1016/j.cell.2007.06.009
Martemucci G., Portincasa P., Centonze V., Mariano M., Khalil M., D'Alessandro A. G. (2023). Prevention of oxidative stress and diseases by antioxidant supplementation. Med. Chem. 19 (6), 509–537. doi:10.2174/1573406419666221130162512
Mendes P., Wickerson L., Helm D., Janaudis-Ferreira T., Brooks D., Singer L. G., et al. (2015). Skeletal muscle atrophy in advanced interstitial lung disease. Respirology 20 (6), 953–959. doi:10.1111/resp.12571
Min K., Smuder A. J., Kwon O., Kavazis A. N., Szeto H. H., Powers S. K. (2011). Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J. Appl. Physiol. 111 (5), 1459–1466. doi:10.1152/japplphysiol.00591.2011
Ong G., Logue S. E. (2023). Unfolding the interactions between endoplasmic reticulum stress and oxidative stress. Antioxidants 12 (5), 981. doi:10.3390/antiox12050981
Pandey S., Dvorakova M. C. (2020). Future perspective of diabetic animal models. Endocr. Metab. Immune Disord.-Drug Targets 20 (1), 25–38. doi:10.2174/1871530319666190626143832
Park K., Lee S. E., Shin K., Uchida Y. (2019). Insights into the role of endoplasmic reticulum stress in skin function and associated diseases. FEBS J. 286 (2), 413–425. doi:10.1111/febs.14739
Peserico D., Stranieri C., Garbin U., Mozzini C C., Danese E., Cominacini L., et al. (2020). Ezetimibe prevents Ischemia/Reperfusion-Induced oxidative stress and Up-Regulates Nrf2/ARE and UPR signaling pathways. Antioxidants 9 (4), 349. doi:10.3390/antiox9040349
Plant P. J., Bain J. R., Correa J. E., Woo M., Batt J. (2009). Absence of caspase-3 protects against denervation-induced skeletal muscle atrophy. J. Appl. Physiol. 107 (1), 224–234. doi:10.1152/japplphysiol.90932.2008
Qin L., Wang Z., Tao L., Wang Y. (2010). ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 6 (2), 239–247. doi:10.4161/auto.6.2.11062
Stephens L., Anderson K., Stokoe D., Erdjument-Bromage H., Painter G. F., Holmes A. B., et al. (1998). Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science 279 (5351), 710–714. doi:10.1126/science.279.5351.710
Sylow L., Tokarz V. L., Richter E. A., Klip A. (2021). The many actions of insulin in skeletal muscle, the paramount tissue determining glycemia. Cell Metab. 33 (4), 758–780. doi:10.1016/j.cmet.2021.03.020
Vantler M., Schorscher M., Berghausen E. M., Wong D. W. L., Zhao X., Gnatzy-Feik L., et al. (2023). PI 3-kinase/AKT/FOXO1 signaling in smooth muscle cells protects against abdominal aortic aneurysm. Eur. Heart J. Supple1 (45). doi:10.1093/eurheartj/ehae666.2245
Viglietto G., Motti M. L., Bruni P., Melillo R. M., D'Alessio A., Califano D., et al. (2002). Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat. Med. 8 (10), 1136–1144. doi:10.1038/nm762
Wang Q., Lan X. F., Zhou L. N., Zhou J. P., Li N., Gu C. J., et al. (2016). Correlation between intermittent hypoxia-induced apoptosis of human umbilical vein endothelial cells and release of cytochrome C from mitochondria. Zhonghua Yi Xue Za Zhi 96 (30), 2385–2389. doi:10.3760/cma.j.issn.0376-2491.2016.30.005
Yang Y., Liu Y., Zhang Y., Ji W., Wang L., Lee S. C. (2021). Periplogenin activates ROS-ER stress pathway to trigger apoptosis via BIP-eIF2α- CHOP and ire1α-ASK1-JNK signaling routes. Anti-Cancer Agents Med. Chem. 21 (1), 61–70. doi:10.2174/1871520620666200708104559
Yang Z., Ke Y., Xu R., Peng P. (2008). Evaluation of peritumoral brain edema in intracranial meningiomas using CT perfusion imaging. Nan Fang. Yi Ke Da Xue Xue Bao 28 (8), 1460–1462.
Yoshida H. (2007). ER stress and diseases. FEBS J. 274 (3), 630–658. doi:10.1111/j.1742-4658.2007.05639.x
Zeng Z., Sarbassov D. D., Samudio I. J., Yee K. W. L., Munsell M. F., Ellen Jackson C., et al. (2007). Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood 109 (8), 3509–3512. doi:10.1182/blood-2006-06-030833
Zhang F., Teng Z., Wang L., Wang L., Huang T., Zhang X. (2022). Dietary selenium deficiency and excess accelerate ubiquitin-mediated protein degradation in the muscle of rainbow trout (Oncorhynchus mykiss) via akt/FoxO3a and NF-κB signaling pathways. Biol. Trace Elem. Res. 200 (3), 1361–1375. doi:10.1007/s12011-021-02726-x
Zhang H., Qi G., Wang K., Yang J., Shen Y., Yang X., et al. (2023). Oxidative stress: roles in skeletal muscle atrophy. Biochem. Pharmacol. 214, 115664. doi:10.1016/j.bcp.2023.115664
Zhang H., Wang B., Wang X., Huang C., Xu S., Wang J., et al. (2024). Handelin alleviates cachexia- and aging-induced skeletal muscle atrophy by improving protein homeostasis and inhibiting inflammation. J. Cachexia Sarcopenia Muscle 15 (1), 173–188. doi:10.1002/jcsm.13381
Keywords: type 2 diabetes mellitus, endoplasmic reticulum stress, apoptosis, skeletal muscle atrophy, proteostasis
Citation: Yang J, Wang Y, Xu Y, Jia X and Lu F (2025) Mechanisms of skeletal muscle atrophy in type 2 diabetes mellitus. Front. Physiol. 16:1607873. doi: 10.3389/fphys.2025.1607873
Received: 08 April 2025; Accepted: 04 June 2025;
Published: 25 June 2025.
Edited by:
Amilcare Barca, University of Salento, ItalyReviewed by:
Daniela Sayuri Mizobuti, Brazilian Biosciences National Laboratory (LNBio), BrazilLimin Shi, Johns Hopkins All Children’s Hospital, United States
Copyright © 2025 Yang, Wang, Xu, Jia and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fangping Lu, bHVmYW5ncGluZzA5MTBAMTYzLmNvbQ==; Xinqi Jia, amlheGlucWkwMTI5QDE2My5jb20=