 Meiling Cao1†
Meiling Cao1† Hongkun Jiang
Hongkun Jiang- 1Department of Neonatology, The First Hospital of China Medical University, Shenyang, Liaoning, China
- 2Department of Pediatrics, The First Hospital of China Medical University, Shenyang, Liaoning, China
- 3Department of Orthopaedic Surgery, Shengjing Hospital of China Medical University, Shenyang, Liaoning, China
Acute kidney injury (AKI) is a clinical syndrome associated with considerable morbidity and mortality. Despite therapeutic advancements, renal recovery and long-term outcomes remain suboptimal. Understanding the pathogenesis of AKI and identifying strategies to prevent its progression have become critical global health priorities. Mitochondrial dysfunction and changes in cellular energy metabolism play key roles in the pathophysiology of AKI. In patients with AKI, proximal tubular cells (PTCs) commonly exhibit impaired mitochondrial biogenesis, characterized by dysregulated mitochondrial dynamics, reduced fusion, and increased fission. Additionally, autophagy dysfunction may occur, contributing to compromised fatty acid β-oxidation (FAO) and subsequent energy deficits. To resolve this energy crisis, under the regulation of multiple signaling pathways, including AMP-activated protein kinase, mechanistic target of rapamycin complex 1, sirtuins, peroxisome proliferator-activated receptor alpha, peroxisome proliferator-activated receptor-γ coactivator 1α, and hypoxia-inducible factor-1 alpha, surviving PTCs may undergo a temporary shift toward glycolysis-dominant energy metabolism. This adaptive metabolic reprogramming is frequently associated with the activation of the pentose phosphate pathway and the suppression of gluconeogenesis. However, a sustained impairment of fatty acid oxidation (FAO) and continued reliance on glycolysis can result in the accumulation of lipids and glycolytic intermediates. This, in turn, may trigger inflammatory responses, promote epithelial-mesenchymal transition, impair tubular repair mechanisms, and contribute to the development of renal fibrosis. Collectively, these pathological processes facilitate the progression from acute kidney injury (AKI) to chronic kidney disease (CKD). Although interventions aimed at enhancing mitochondrial biogenesis, restoring mitochondrial and FAO homeostasis, and employing remote ischemic preconditioning have demonstrated potential in mitigating AKI progression, further investigation is required to address unresolved concerns related to their safety and clinical efficacy.
1 Introduction
Acute kidney injury (AKI) is a common and clinically significant syndrome characterized by an abrupt decline in renal function, typically indicated by elevated serum creatinine levels or reduced urine output (oliguria) (Hoste et al., 2018; Ronco et al., 2019). Epidemiological studies reported a high incidence of AKI across various populations. Despite the potential for renal function to return to baseline following appropriate treatment, long-term outcomes remain poor, with the condition contributing to considerable mortality and posing a substantial burden on global healthcare systems (Ronco et al., 2019; Gupta et al., 2024; Levey et al., 2020).
Mitochondria, as central organelles responsible for cellular energy production, play a key role in supporting diverse metabolic and physiological processes (Gewin, 2021). In the kidney, proximal tubular cells (PTCs) are particularly reliant on mitochondrial fatty acid β-oxidation (FAO) and oxidative phosphorylation (OXPHO) to meet their high energy demands (Han et al., 2016). These metabolic pathways generate significant amounts of adenosine triphosphate (ATP), which is essential for fueling the active transport and reabsorption functions of tubular epithelial cells (TECs) (Gewin, 2021; Houten et al., 2016).
In AKI, mitochondrial energy metabolic reprogramming plays a key role in disease pathogenesis. Mitochondrial dysfunction, characterized by impaired FAO and disrupted quality control mechanisms, has been frequently observed in affected patients (Freitas-Lima et al., 2020). Energy metabolic reprogramming in TECs is primarily driven by an imbalance in energy homeostasis, leading to a compensatory shift from FAO-dependent OXPHO to a glycolysis-dominant metabolic state. This adaptive shift resembles the Warburg effect commonly reported in tumor biology (Li Z. et al., 2021; Lan et al., 2016; Ma et al., 2022).
This metabolic transition is often accompanied by activation of the pentose phosphate pathway (PPP), which may contribute to cellular defense against oxidative stress induced by reactive oxygen species (ROS), as well as a concomitant suppression of gluconeogenesis (Scantlebery et al., 2021; van der Rijt et al., 2022). The enhanced glycolytic activity serves as a transient energy source, primarily sustaining the viability and limited functional capacity of the remaining TECs (van der Rijt et al., 2022). However, prolonged failure to reestablish normal mitochondrial metabolism may promote the progression of AKI to chronic kidney disease (CKD) (Shen et al., 2020).
Furthermore, mitochondrial dysfunction is closely implicated in AKI pathogenesis through the activation of inflammatory signaling cascades, which may lead to PTC apoptosis and necrosis (Jiang et al., 2020; Sun et al., 2019). These processes contribute to maladaptive repair, renal fibrosis, and ultimately the development of CKD.
Although advances in current treatment strategies have improved short-term outcomes, targeted therapies for AKI continue to face significant limitations. Changes in cellular metabolic pathways contribute to the progression of renal dysfunction. Early correction of energy metabolic reprogramming in TECs, along with the restoration of mitochondrial function, is considered a potentially valuable therapeutic approach to reduce the severity and long-term consequences of AKI (Ishimoto and Inagi, 2016).
This review outlines the metabolic transitions observed during AKI, emphasizing the underlying pathophysiological mechanisms and key regulatory pathways implicated in mitochondrial dysfunction and energy metabolism. In addition, current strategies for targeted therapeutic intervention are examined, along with their associated challenges and limitations.
2 AKI and mitochondrial energy metabolism
2.1 Basic structure and functions of mitochondria
Mitochondria are dynamic, double-membraned organelles typically exhibiting an ellipsoidal or rod-shaped morphology. Their structural components include the outer mitochondrial membrane (OMM), inner mitochondrial membrane (IMM), mitochondrial matrix, and intermembrane space (IMS) (Giacomello et al., 2020; Wang et al., 2024). The OMM serves as a selective barrier that regulates the diffusion of molecules and participates in mitochondrial signaling pathways. The IMM, consisting of the inner boundary membrane and cristae, is characterized by its inward folds that increase surface area for metabolic processes. This membrane provides the structural platform for OXPHO and the assembly of the, ETC (Trotta and Chipuk, 2017).
The mitochondrial matrix, enclosed by the IMM, supports several key metabolic processes, including the tricarboxylic acid (TCA) cycle and the later stages of glycolysis. The IMS plays a key role in numerous cellular functions, including protein and lipid biosynthesis, metal ion homeostasis, redox regulation, oxidative protein folding, initiation of apoptosis, and the maintenance of mitochondrial dynamics and structural integrity.
2.2 The key role of mitochondria in energy metabolism
The kidney exhibits a high resting metabolic rate, supported by a substantial mitochondrial content and elevated oxygen consumption. Different types of cells have different requirements for ATP, podocytes, endothelial cells and mesangial cells mainly use glucose for their energy supply, while renal tubular cells mainly use free fatty acids, glutamine, pyruvate, citrate and lactate as substrates for fuel supply through aerobic respiration (Scholz et al., 2021). Noteworthy primarily target the tubular epithelial cells especially the highly metabolically active proximal tubular segment (Isaka et al., 2011). PTCs are the most energy-demanding cells, which efficiently use FAO as a fuel source. This energy demand is primarily met through FAO and OXPHOS (Chung et al., 2019). FAO occurs predominantly within mitochondria and involves a sequence of cyclic reactions that convert fatty acids (FA) into acetyl-coenzyme A (acetyl-CoA). During this process, flavin adenine dinucleotide (FAD) is reduced to FADH2, and nicotinamide adenine dinucleotide (NAD+) is reduced to NADH (Foster, 2012). These reducing equivalents subsequently enter the mitochondrial electron transport chain, where their oxidation drives ATP production via OXPHOS. Species differences have been well documented for PPARα-regulated genes, such as those involved in peroxisome biogenesis and peroxisomal FAO (Lawrence et al., 2001; Sasaki et al., 2019).
2.3 Aberrant changes in mitochondrial structure and function during AKI
2.3.1 Mitochondrial dysfunction
Mitochondrial dysfunction in AKI is characterized by impairments in mitochondrial biogenesis, attenuation of mitophagy, disrupted mitochondrial dynamics, and reduced OXPHO activity (Figure 1) (Emma et al., 2016). Increased oxidative stress (OS) and excessive production of ROS are closely associated with the regulation of mitophagy (Zhu et al., 2023; Levine and Kroemer, 2019). This selective degradation of damaged mitochondria is mediated by key regulatory proteins, including PTEN-induced putative kinase 1, BCL2 interacting protein 3, and FUN14 domain-containing protein 1, and is essential for maintaining mitochondrial quality control (Quirós et al., 2016; Onishi et al., 2021; Lin et al., 2023). Upregulation of mitophagy represents an early cellular response aimed at promoting survival. However, excessive or sustained mitochondrial damage triggers pathological overactivation of mitophagy, contributing to cell death and tissue injury (Aggarwal et al., 2016).
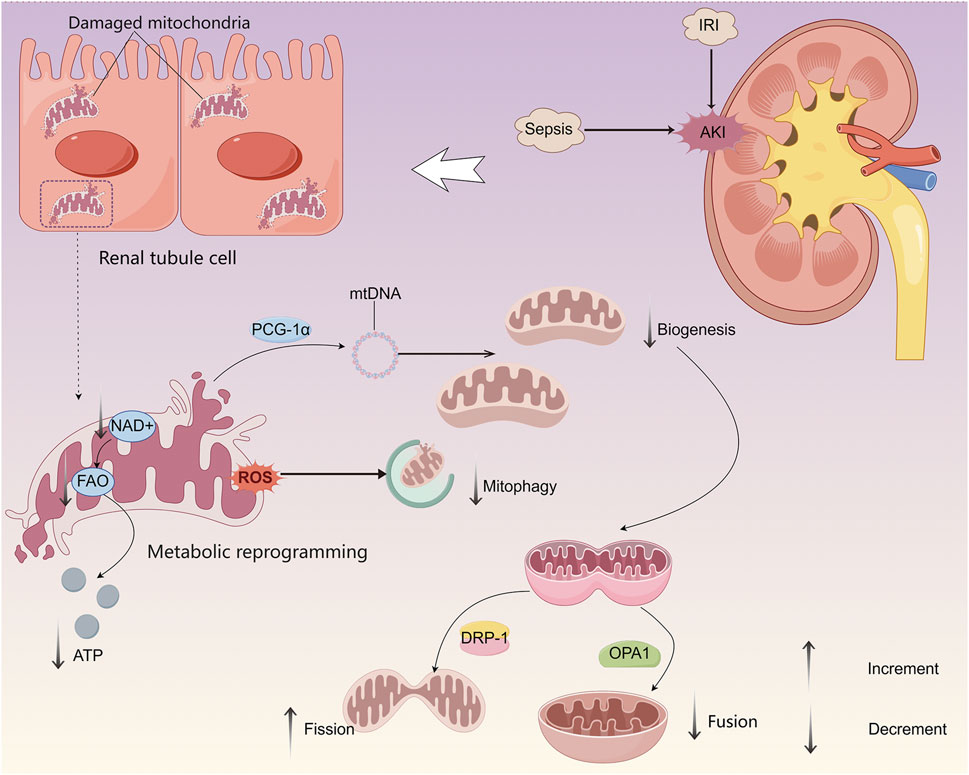
Figure 1. Mitochondrial dysfunction and its impact on energy metabolism in renal TECs during AKI. During AKI, mitochondrial dysfunction in TECs is characterized by impaired biogenesis, decreased mitochondrial fusion, increased fission, and dysregulated mitophagy, resulting in compromised mitochondrial quality control. These changes lead to a metabolic shift from FAO to glycolysis, serving as a transient compensatory mechanism to sustain minimal ATP production necessary for TEC survival.
During AKI, dysfunctional mitochondria can induce the expression of peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α), which facilitates mitochondrial biogenesis and supports the maintenance of mitochondrial structure, function, and quantity. This adaptive response may contribute to reduced cell death and attenuation of lipid accumulation (Cheng et al., 2018). In sepsis-induced AKI models, proximal tubular cell-specific knockdown of PGC-1α has been associated with exacerbated renal injury and disease progression (Tran et al., 2011).
As highly dynamic organelles, mitochondria depend on continuous cycles of fission and fusion to preserve network integrity, regulate organelle quantity and quality, and adapt to fluctuating energy demands. Mitochondrial dynamics is very nuanced, and a balance between fission and fusion is critical depending on the cellular environment/state (Fan et al., 2024; Wai and Langer, 2016). Mitochondrial fission is promoted by the activation and translocation of dynamin-related protein 1, whereas mitochondrial fusion is mediated by optic atrophy 1, whose function may be disrupted by proteolytic cleavage, OPA1 is repressed by proteolysis, leading to dysregulation of mitochondrial fusion. Dysregulation of these dynamic processes can result in mitochondrial fragmentation, further contributing to cellular dysfunction during AKI (Qin et al., 2019).
2.3.2 Dysfunction of mitochondrial energy metabolism
A reduction in the expression of peroxisome proliferator-activated receptor alpha (PPARα), a key regulator of FA homeostasis, has been associated with decreased activity of carnitine palmitoyltransferase 1 (CPT1), thereby impairing the transport of FAs into the mitochondrial matrix (Chung et al., 2018). This disruption may result in impaired oxidation of long-chain FAs and contribute to cellular necrosis, accompanied by downregulation of enzymes associated with FAO (Freitas-Lima et al., 2020; Erpicum et al., 2018).
Glycolysis may briefly enhance energy production and offer partial protective effects in the setting of AKI, but its prolonged activation has been linked to adverse renal outcomes. Although glycolysis can sustain basic cellular functions under stress conditions, persistent reliance on this pathway, along with the accumulation of lactate, promotes renal fibrosis (Hu et al., 2020). The combined effects of dysregulated FAO and sustained glycolytic activity contribute to the progression from AKI to CKD through multiple interrelated mechanisms (Li Z. et al., 2021). Enhanced glycolysis in a short period compensates for impaired energy production and exerts partial renal-protective effects. However, long-term shut down of FAO and enhanced glycolysis lead to inflammation, lipid accumulation, and fibrosis (Zhu et al., 2022).
2.3.3 Enhancement of OS
ROS in AKI primarily originate from mitochondrial activity and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complexes (Cristofano et al., 2021; Zorov et al., 2014). Hypoxia and subsequent reperfusion serve as key initiating factors for ROS overproduction. During AKI, dysregulation of the antioxidant defense system impairs the clearance of excessive ROS, thereby disrupting redox homeostasis and promoting OS.
Elevated ROS levels can oxidatively modify mitochondrial lipids and proteins, resulting in functional disruption of the electron transport chain (ETC.) and increased permeability of the mitochondrial membrane. These changes ultimately compromise mitochondrial bioenergetics and contribute to cellular injury during the course of AKI (Hall et al., 2013).
2.4 The role of mitochondrial dysfunction in the pathogenesis of AKI
Mitochondrial dysfunction plays a key role in the pathophysiological progression of AKI. During AKI, mitochondria frequently exhibit morphological abnormalities, including swelling and fragmentation, which can facilitate the release of pro-apoptotic mediators and trigger the opening of the mitochondrial permeability transition pore. These processes lead to apoptosis and necrosis of PTCs, thereby aggravating renal injury and promoting AKI progression (Sun et al., 2020). Damaged mitochondria release cardiolipin and other bioactive molecules that activate Nucleotide oligomerization domain (NOD)-like receptors, contributing to elevated levels of pro-inflammatory cytokines and chemokines and result in sustained renal inflammation and injury (Szeto et al., 2017). Mitochondrial damage persists beyond the acute ischemic phase, maintaining chronic inflammasome activation and ultimately leading to progressive renal fibrosis.
Under renal injury and OS conditions, excessive ROS generation and the release of pro-apoptotic factors such as apoptosis-inducing factor impairs mitochondrial membrane potential and promotes both caspase-dependent and caspase-independent apoptotic pathways. There are two main caspase-dependent apoptotic pathways of apoptosis: the intrinsic pathway and the extrinsic pathway. The intrinsic pathway is triggered by disruptions in the cellular microenvironment, demarcated by mitochondrial outer membrance permeabilization, and precipitated mainly by caspases 3 (Sanz et al., 2023). The extrinsic pathway is initiated by the binding of proapoptotic ligands to death receptors on the cell surface, leading to the formation of the death-inducing signaling complex (DISC) and the activation of caspases (Qiao et al., 2024). Besides that, caspase-independent apoptotic pathway, known as the AIF/Endo G pathway, induces mitochondrial AIF release (Luo et al., 2021). Endo G acts as a modulator. Forced BNIP-3 expression by plasmid transfection results in mitochondrial Endo G release and nuclear translocation. BH3 domain of BNIP-3 interacted with anti-apoptotic protein to form dimers, which was able to promote the apoptosis (Luo et al., 2021; Liu et al., 2016).
Mitochondrial dysfunction disrupts communication between mitochondria and the endoplasmic reticulum (ER) (Igwebuike et al., 2020; Hasegawa and Inagi, 2020), contributing to tubulointerstitial inflammation and fibrosis. Various forms of renal injury are associated with ER stress and activation of the unfolded protein response (UPR), which can initially support cell survival by restoring ER and mitochondrial homeostasis through distinct signaling pathways (Zhou et al., 2025). However, when adaptive UPR mechanisms fail, they may instead promote cell death (Bartoszewska and Collawn, 2020). Due to hypoxia, mitochondrial dysfunction, and disordered nutrient-sensing pathways, the metabolic process in the TECs shifts from FAO to glycolysis. Long-term shut down of FAO and enhanced glycolysis lead to inflammation, lipid accumulation, and fibrosis. In AKI, TECs undergo autophagy-mediated phenotypic conversion to a pro-fibrotic state, leading to the secretion of fibroblast growth factor 2, which activates fibroblasts and contributes to maladaptive renal repair and fibrosis (Livingston et al., 2023).
Mitochondrial dysfunction not only contributes to the onset of AKI but also accelerates its progression. Renal injury is often accompanied by an intense inflammatory response (Kong et al., 2024). Necrotic tubular cells release damage-associated molecular patterns (DeWolf et al., 2022), which activate toll-like receptors on renal parenchymal and resident immune cells, stimulating the production of pro-inflammatory cytokines and chemokines. This cascade further promotes the transition from AKI to CKD. Although renal cells retain a capacity for self-repair, incomplete restoration of PTCs results in cell cycle arrest at the G2/M phase, a phenomenon that has been associated with tubulointerstitial fibrosis and irreversible renal damage. Such progression increases the risk of CKD and may ultimately lead to end-stage renal disease (Gewin, 2021; Zhao et al., 2021).
3 Mechanisms of mitochondrial energy metabolic reprogramming
3.1 Transition of energy metabolism pathways
Under physiological conditions, TECs actively absorb circulating FAs through specific transporter proteins, including CD36 and FA-binding proteins. Following entry into the cytoplasm, FAs are converted to acyl-coenzyme A (acyl-CoA) and subsequently processed by CPT1, a key rate-limiting enzyme located on the outer mitochondrial membrane. CPT1 catalyzes the formation of acyl-carnitines, which are transported into mitochondria where they are reconverted to acyl-CoA by CPT2 on the inner mitochondrial membrane. These acyl-CoAs then undergo β-oxidation to produce acetyl-CoA, which enters the TCA cycle for complete oxidation. The reducing equivalents NADH and FADH2 generated during FAO enter the, ETC, facilitating ATP production through OXPHO (Li et al., 2020). Very long-chain FAs require preliminary processing in peroxisomes before entering mitochondria for further oxidation (Houten et al., 2020).
Under normal metabolic conditions, the uptake, intracellular transport, and oxidation of FAs are tightly regulated to maintain lipid homeostasis, preventing lipid accumulation in the cytoplasm. This FAO-OXPHO-driven energy supply mode enables rapid ATP generation but requires high levels of oxygen, rendering TECs particularly susceptible to hypoxic injury. As a result, proximal tubules are more vulnerable to damage during episodes of AKI.
Along with FA metabolism, TECs serve as key sites of renal gluconeogenesis. Enzymes of renal gluconeogenesis, including phosphoenolpyruvate carboxykinase (PCK) and fructose-1,6-bisphosphatase (FBP), are predominantly expressed in TECs.
Under fasting or stress conditions, TECs utilize lactate as primary substrate for gluconeogenesis, which maintains systemic glucose homeostasis by coordinating with glucose reabsorption. Under normal physiological conditions, the glycolytic activity of TECs remains low, and ATP generated through aerobic glycolysis accounts for only a minor proportion (∼4%) of total ATP production (Guder and Ross, 1984).
During AKI, mitochondrial FAO and the transport of FAs into mitochondria are significantly impaired due to mitochondrial dysfunction and disruption of quality control mechanisms (Kang et al., 2015). In rat models of AKI induced by cisplatin, lipopolysaccharide (LPS), or ischemia-reperfusion (IR), significant reductions in ATP levels and intracellular lipid accumulation have been observed (Bhargava and Schnellmann, 2017). These models indicate reduced FA uptake in TECs, along with downregulation of key FAO enzymes such as CPT1 and long-chain acyl-CoA dehydrogenase (ACADL), as well as reduced expression and activity of PPARα, a central transcriptional regulator of FAO.
During both early and late reperfusion phases following IR injury, there is a notable increase in glucose uptake and glycolytic activity in the renal cortex. This metabolic shift is characterized by upregulation of rate-limiting glycolytic enzymes such as pyruvate kinase M2 isoform (PKM2) and hexokinase (HK), leading to elevated production of glycolytic end-products, including pyruvate and lactate (Li et al., 2020). Similar metabolic changes have been observed in isolated proximal tubules subjected to hypoxia/reoxygenation injury ex vivo, and increased activities of HK, PKM2, and lactate dehydrogenase (LDH) have been reported in mouse models of AKI (Li et al., 2020).
Collectively, these findings indicate a metabolic shift from FAO to glycolysis in TECs during AKI. Additionally, glucose is redirected into the PPP, which supports cellular repair and regeneration by producing NADPH and nucleotide precursors, and by maintaining intracellular levels of glutathione, a key antioxidant (Scantlebery et al., 2021). However, this shift is accompanied by a significant downregulation of gluconeogenic enzyme expression, resulting in hypoglycemia and lactate accumulation, which have been associated with increased severity of AKI and higher patient mortality (Legouis et al., 2020; Verissimo et al., 2022).
3.2 Modulation of key molecules
The metabolic reprogramming of TECs during AKI is regulated by an intricate signaling network involving AMP-activated protein kinase (AMPK), mechanistic target of rapamycin complex 1 (mTORC1), sirtuins (SIRTs), PPARα, PGC-1α, and hypoxia-inducible factor-1 alpha (HIF-1α). The activation of these pathways is initiated by different molecular signals depending on the etiology of AKI, as presented in Figure 2.
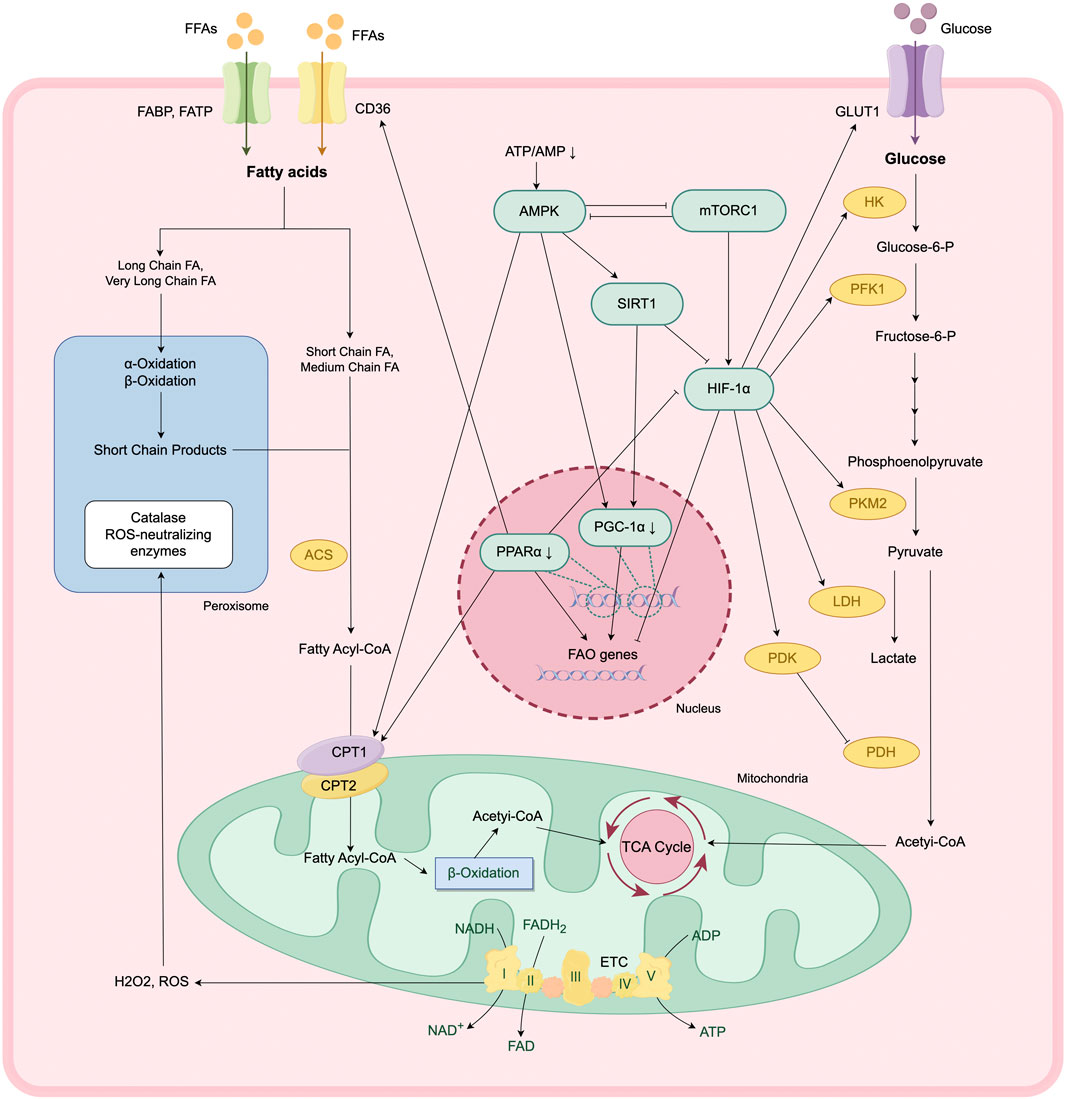
Figure 2. Metabolic reprogramming and regulation of key signaling pathways in AKI. In response to AKI, TECs undergo a metabolic transition from FAO- and OXPHO-dependent ATP production to a glycolysis-dominant state. This reprogramming is modulated by a complex signaling network, involving AMPK-SIRT1-PGC-1α axis, mTOR-HIF-1α pathway.
3.2.1 HIF-1α
Renal hypoxia is a common feature across various AKI models, regardless of the initiating insult. The transcription factor HIF-1α plays a key role in mediating cellular responses to hypoxia and contributes to the metabolic reprogramming observed during AKI (Zhang et al., 2025). Under hypoxic conditions, the activity of cytoplasmic prolyl-hydroxylases is inhibited, preventing the hydroxylation and subsequent ubiquitination of HIF-1α, enhancing the stability of HIF-1α. Stabilized HIF-1α then translocates into the nucleus, where it forms a transcriptionally active heterodimer with HIF-1β. This complex binds to hypoxia response elements in the promoter regions of target genes, regulating the transcription of genes involved in metabolic adaptation and glycolysis (Lee S. H. et al., 2021).
HIF-1α inhibits the transcription of FA uptake genes (such as CD36) and β-oxidation rate-limiting enzyme genes (such as ACOX1, CPT1A), thereby suppress FAO activity in TECs (Li W. et al., 2021). While it enhances the transcription of glucose transport genes (such as GLUT1 and GLUT3) and key rate-limiting enzymes in glycolysis (such as PKM2 and HK2). Concurrently, HIF-1α upregulates the expression of LDH and pyruvate dehydrogenase kinase (PDK), promoting the conversion of pyruvate to lactate and inhibiting the entry of pyruvate into TCA cycle, further shifting energy metabolism toward glycolysis (Cai et al., 2020).
Along with its role in metabolic regulation, HIF-1α influences mitochondrial quality control. It mediates mitophagy, which provides cytoprotective effects by reducing apoptosis and ROS production during renal injury (Fu et al., 2020).
3.2.2 PPARα/PGC-1α
PPARα is a ligand-activated nuclear transcription factor that is highly expressed in proximal renal tubules. It plays a key role in maintaining intracellular FA homeostasis by regulating the transcription of FAO-related enzymes in mitochondria and peroxisomes, and modulates cellular FA uptake through regulating the expression of CD36 and CPT1 (Emma et al., 2016). Additionally, PPARα exert indirect regulation over glycolysis by attenuating the transcriptional activity of HIF-1α (Iwaki et al., 2019). In murine models of ischemia-reperfusion injury (IRI) and drug-induced AKI, PPARα expression is significantly downregulated. Restoration of PPARα activity ameliorates metabolic dysregulation in these models (Lakhia et al., 2018). Conversely, reduced PPARα expression contributes to increased lipid accumulation and the development of fibrotic phenotypes in TECs, as demonstrated in CKD models.
PGC-1α is a key regulator of mitochondrial biogenesis (Fontecha-Barriuso et al., 2020). By activating nuclear receptors such as PPARα and ERRα, PGC-1α upregulates the expression of FAO key enzymes (e.g., CPT1A, ACOX1) and mitochondrial respiratory chain complex genes, maintaining energy homeostasis in TECs (Li and Susztak, 2018). Reduced expression of PGC-1α is consistently observed in animal models of AKI. TEC-specific knockout of PGC-1α results in exacerbated renal injury, whereas PGC-1α overexpression in TECs enhances NAD+ levels, thereby promoting mitochondrial biogenesis and mitophagy. These effects collectively reduce lipid accumulation and tubular injury and improve survival following renal IRI (Li et al., 2023; Tran et al., 2016). PGC-1α contributes to peroxisomal remodeling, further supporting its role in lipid metabolism and cellular homeostasis.
3.2.3 mTOR/AMPK
The mechanistic target of rapamycin (mTOR) signaling pathway is a well-characterized nutrient-sensing pathway in the kidney. In response to external stimuli (e.g., hypoxia, ischemia, drug-induced injury), mTORC1 promotes the translation of HIF-1α, thereby driving a metabolic shift from oxidative phosphorylation to glycolysis (Cao et al., 2020; Cheng et al., 2014). Activation of mTOR pathway has been documented across multiple murine models of AKI. Pharmacological inhibition of mTORC1 suppresses excessive glycolysis and TEC proliferation, thereby ameliorating renal fibrosis (Cao et al., 2020; Cheng et al., 2014). In addition, mTORC1 inhibits mitophagy, further contributing to mitochondrial dysfunction (Wang et al., 2020).
AMPK, another key energy sensor in renal cells, negatively regulates mTOR signaling pathway. AMPK activation is triggered by a decrease in the ATP/AMP ratio, leading to SIRT1-mediated deacetylation and suppression of HIF-1α activity, thereby attenuating glycolysis (Clark and Parikh, 2021). AMPK enhances PGC-1α expression via SIRT1 activation, promoting mitochondrial biogenesis and mitophagy (Price et al., 2012). Furthermore, AMPK supports FAO by increasing CPT1 activity, which may contribute to improved ATP generation (Clark and Parikh, 2021).
Interestingly, the effects of AMPK activation appear to be time-dependent. Early-stage AMPK activation during AKI may fulfill elevated energy demands and support cellular repair mechanisms (Bao et al., 2019). However, during the later stages of injury, AMPK activity may be suppressed through mechanisms that remain to be fully elucidated, potentially exacerbating renal damage (Jin et al., 2020).
4 Specific role of mitochondrial energy metabolic reprogramming in AKI
During AKI, metabolic reprogramming in TECs may temporarily compensate for deficits in energy supply, support cellular survival, and partially mitigate tissue damage, thereby facilitating early repair processes. However, sustained suppression of FAO along with persistent enhancement of glycolysis can result in intracellular lipid accumulation, activation of inflammatory pathways, and fibrogenesis, collectively contributing to the progression of AKI (Figure 3).
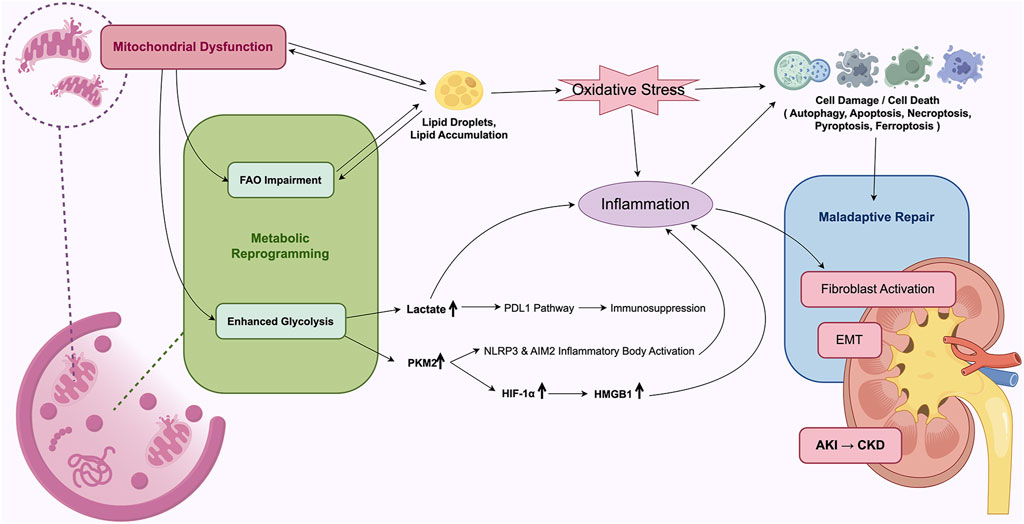
Figure 3. Mechanistic overview of mitochondrial energy metabolism reprogramming in AKI. Mitochondrial dysfunction and metabolic reprogramming leads to maladaptive repair of TECs, driving the development of AKI to CKD.
4.1 Exacerbation of renal TECs injury
Impairment of FAO results in the excessive accumulation of cytotoxic lipid species—such as triglycerides, free fatty acids (FFAs), cholesterol, and lysophosphatidylcholine—within TECs. This condition disrupts cellular homeostasis and contributes to cellular injury and death, a process commonly referred to as lipotoxicity (Jang et al., 2020; Todorović et al., 2021). Lipid accumulation adversely affects mitochondrial function, further impairing FAO, which reduces FFA utilization and promotes additional lipid deposition (Baek et al., 2022; Xu et al., 2022). This establishes a self-perpetuating cycle that may intensify renal injury. A similar bidirectional relationship is observed between mitochondrial dysfunction and lipid accumulation, whereby impaired mitochondria exhibit continuous electron leakage from complex I of the, ETC, resulting in the generation of ROS. These ROS trigger lipid peroxidation by stimulating the release of lipid radicals from membrane-associated triglycerides and phosphatidylcholine, thereby exacerbating lipid-induced cellular damage (Zhang et al., 2021).
The primary mechanism underlying renal injury associated with lipid overload appears to involve OS, but it is also closely linked to ER stress, defective autophagy, and inflammatory responses (Chen et al., 2018; Lee E. S. et al., 2021). Furthermore, metabolic reprogramming during AKI has been closely associated with multiple forms of programmed cell death in TECs, including apoptosis, necroptosis, pyroptosis, and ferroptosis.
Lactate accumulation may further amplify glycolytic activity by stabilizing HIF-1α, thereby creating a positive feedback loop that exacerbates mitochondrial dysfunction and cellular damage. Experimental studies have demonstrated that the glycolytic inhibitor 2-deoxyglucose can activate autophagy through the upregulation of SIRT3 and phosphorylated AMPK, reducing apoptosis in both sepsis-induced TEC injury models and LPS-stimulated HK-2 cells, ultimately improving cell viability (Tan et al., 2020; Tan et al., 2021). Additionally, pharmacologic agents such as shikonin (a PKM2 inhibitor) and dichloroacetate (a PDK1 inhibitor) have been shown to suppress TEC apoptosis and attenuate renal interstitial fibrosis (Wei et al., 2019).
4.2 Enhancing inflammatory response
Ischemic and toxic insults to the kidneys not only result in direct cellular injury but also elicit a robust inflammatory response. The dynamic balance between pro-inflammatory and anti-inflammatory pathways plays a critical role in determining the extent of subsequent renal damage and repair (Lee et al., 2024). TECs, while being primary targets of injury, also actively contribute to the regulation of the renal inflammatory microenvironment.
The glycolytic enzyme PKM2 exerts pro-inflammatory effects. Elevated PKM2 expression activates the transcription of HIF-1α and promotes the release of high mobility group box 1 (HMGB1) from activated macrophages, thereby amplifying sterile inflammation, particularly in sepsis-associated AKI, and enhancing TEC susceptibility to OS (Yang et al., 2014). In both IRI and sepsis-induced AKI models, HMGB1 release by TECs has been implicated in the promotion of renal inflammation (Zhao et al., 2023). Furthermore, PKM2 activation contributes to the intracellular accumulation of succinate, a signaling metabolite that stabilizes HIF-1α and promotes interleukin-1 beta (IL-1β) production during inflammatory responses (Tannahill et al., 2013). Inhibition of PKM2 within renal tissue has been associated with downregulation of HIF-1α, reduced TEC apoptosis, and amelioration of LPS-induced AKI (Yang et al., 2014). In sepsis-induced AKI models, PKM2 inhibition attenuates the activation of NLR family pyrin domain containing 3 (NLRP3) and absent in melanoma 2 (AIM2) inflammasomes, decreases the release of IL-1β, IL-18, and HMGB1 by macrophages, and ultimately improves survival outcomes (Xie et al., 2016).
Lactic acid, acting both as a metabolic intermediate and a signaling molecule modulates immune and inflammatory responses. In sepsis-induced AKI, the accumulation of lactic acid in renal tubules activates the programmed death-1/programmed death-ligand 1 (PD-1/PD-L1) axis, leading to lymphocyte apoptosis and contributing to immunosuppression (Xu et al., 2021). Conversely, other studies have demonstrated that lactic acid may partially restore PGC-1α activity, reduce the expression of pro-inflammatory cytokines including TNF-α, IL-1β, and IL-6, alleviate renal injury, and improve survival in murine models of sepsis-induced AKI (Takakura and Zandi-Nejad, 2019).
In addition, lipid-derived mediators produced from the autoxidation of intracellular polyunsaturated FAs, such as α-linolenic acid and docosahexaenoic acid (DHA), exhibit anti-inflammatory effects (Lin et al., 2020). Exogenous DHA supplementation has been shown to significantly modulate oxylipin profiles in ischemia-induced AKI, thereby enhancing renal tubular function. However, excessive intake of unsaturated FAs may increase the production of pro-inflammatory lipid mediators and potentially worsen renal injury (Zhang et al., 2025; Sonnweber et al., 2018).
4.3 Facilitation of renal fibrosis
Renal fibrosis represents the final common pathological outcome in most CKD cases, with epithelial injury and aberrant repair processes serving as central events in its progression. In AKI, TECs may undergo cytoskeletal reorganization and extracellular matrix (ECM) accumulation, transitioning from an energy-demanding epithelial phenotype to a less metabolically active interstitial phenotype—a process known as epithelial-mesenchymal transition (EMT) (Grande et al., 2015). In addition, a subset of injured TECs may become arrested in the G2/M phase of the cell cycle and adopt a pro-fibrotic phenotype (Yang et al., 2010). Beyond the impact of endothelial damage, peritubular capillary rarefaction, pericyte detachment, nephron loss, interstitial hypoxia, and persistent inflammation, metabolic disturbances in TECs—particularly defective FAO and lipid accumulation—have been identified as key contributors to EMT and the progression of renal fibrosis through maladaptive repair mechanisms (Miguel et al., 2021).
In fibrotic kidneys, both the expression of FAO-associated enzymes and the transcriptional regulator PPARα are significantly downregulated. This reduction correlates positively with the extent of lipid accumulation. Restoration of FAO in experimental models of renal fibrosis have significantly reduced interstitial fibrotic changes (Kang et al., 2015). Evidence from folic acid-induced nephropathy in mice indicates that impaired FAO, rather than intracellular lipid droplet accumulation, serves as the principal determinant of tubulointerstitial fibrosis (Lan et al., 2016). Moreover, in a transgenic mouse model with kidney-specific overexpression of CD36, increased triglyceride and long-chain FA accumulation was insufficient to induce renal fibrosis, further supporting the notion that reduced FAO activity—rather than lipid storage alone—is more directly associated with fibrogenic progression in TECs (Kang et al., 2015).
Enhanced glycolytic activity has been implicated in the activation of fibroblasts, whereas pharmacological inhibition of glycolysis has been shown to significantly attenuate renal fibrosis (Ding et al., 2017). In a folic acid-induced AKI model, interstitial fibroblasts were activated and proliferated by metabolizing lactic acid produced by TECs during the later stages of injury. Early intervention using glycolysis inhibitors to limit tubular lactate production effectively suppressed fibroblast activation and mitigated renal fibrosis (Shen et al., 2020). Furthermore, pro-inflammatory microenvironments can promote fibroblast activation and ECM deposition. In a unilateral ureteral obstruction-induced model of renal interstitial fibrosis, upregulated glycolysis in TECs and elevated lactate levels contributed to a hypoxic and acidic microenvironment that inhibited podocyte proliferation and differentiation, thereby accelerating fibrotic remodeling (Li et al., 2018).
4.4 Effects on renal hemodynamics and systemic metabolism
The interaction between metabolic shifts and renal hemodynamics/tubuloglomerular feedback (TGF) is one of the core mechanisms of AKI, which involves imbalance of energy metabolism, accumulation of metabolites, oxidative stress, inflammation and regulation of signal pathways (Wada et al., 2024; Fogo and Harris, 2025). Glomerular filtration rate (GFR) affects the tubular reabsorption load, and TECs maintain ion transport by consuming ATP, which accounts for 80% of renal oxygen consumption. Energy status of TECs (e.g., ATP/AMP ratio) reversely regulates glomerular blood flow via the AMPK pathway. Lactate produced by TEC glycolysis dilates the afferent glomerular arteriole, while long-chain acyl-CoA released from impaired FAO enhances vasoconstriction.
Metabolic alterations during AKI also exert profound impacts on systemic metabolism. Impaired FAO and enhanced glycolysis in TECs lead to increased production of lactate and pyruvate, which serve as substrates for hepatic gluconeogenesis. In I/R models, lactate released by the kidney enters the liver through portal vein, where it activates pyruvate carboxylase (PC) and phosphoenolpyruvate carboxykinase (PEPCK), thereby promoting glucose synthesis (Yu et al., 2021). Concomitantly, reduced renal clearance of insulin and glucagon in AKI results in an elevated glucagon/insulin ratio, which triggers hepatic cAMP/PKA pathway and promotes gluconeogenesis (He et al., 2025).
5 Intervention and treatment of mitochondrial energy metabolic reprogramming in AKI
5.1 Mitochondrial-targeted peptide D-Arg-dimethylTyr-Lys-Phe-NH2 (SS-31)
The mitochondrial-targeted peptide SS-31 has demonstrated significant protective effects in renal tubular cells by attenuating OS, thereby reducing apoptosis and necrosis (Zuo et al., 2020; Cho et al., 2010; Lin et al., 2019). This agent contributes to the preservation of tubular function and provides protection against ischemia-induced renal injury. SS-31 stabilizes mitochondrial structure and function, prevents the collapse of mitochondrial membrane potential, and inhibits calcium-induced opening of the mitochondrial permeability transition (MPT) pore. These effects collectively promote ATP synthesis and reduce tubular cell loss (Table 1).
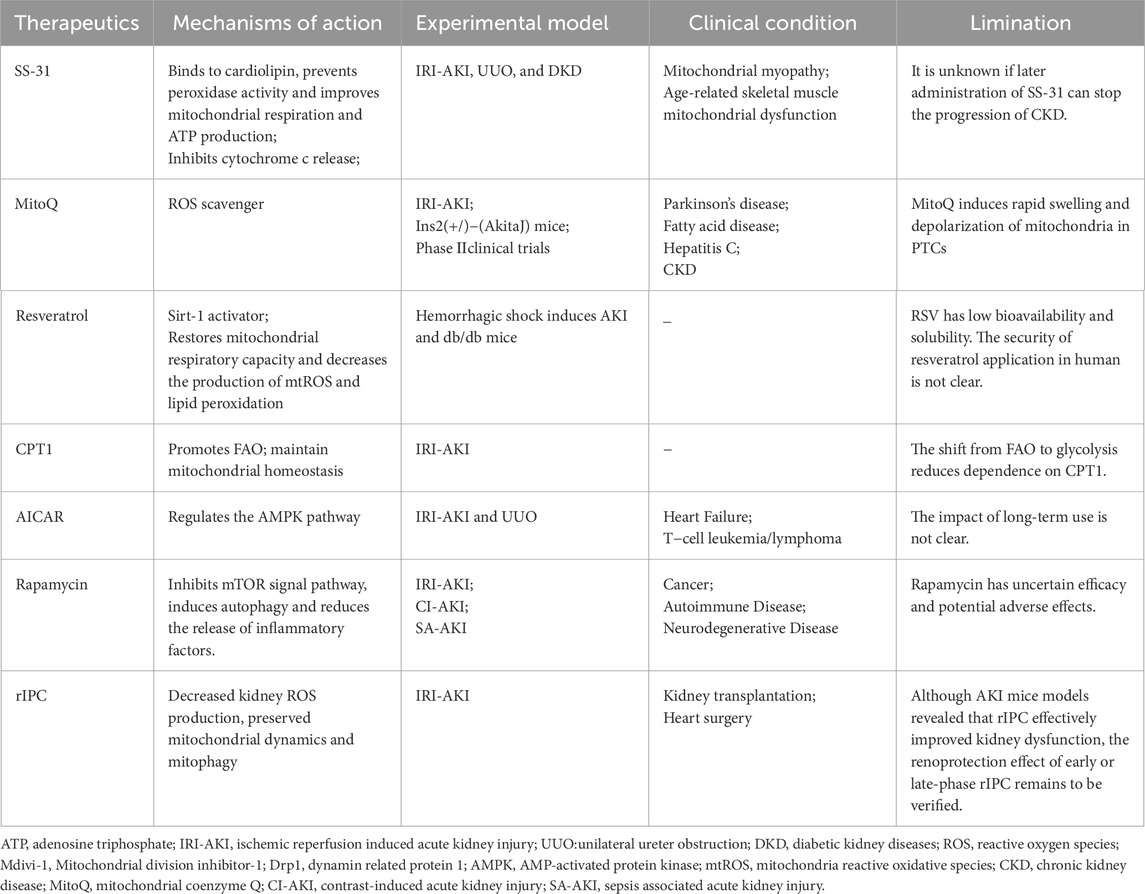
Table 1. Mitochondria-targeted therapeutics.
Moreover, SS-31 mitigates mitochondrial dysfunction and prevents subsequent cell death and inflammatory responses, which are implicated in the progression from AKI to CKD. By targeting upstream mitochondrial injury, SS-31 may serve as a therapeutic candidate for reducing long-term renal damage following AKI. It is not known if later administration of SS-31 can stop the progression of CKD (Szeto et al., 2017).
5.2 Mitochondria-targeted antioxidants
The targeted mitochondrial antioxidant mitoquinone (MitoQ) has demonstrated protective effects in AKI by attenuating OS and apoptosis. In models of renal IRI, pre-ischemic administration of MitoQ significantly reduces oxidative damage and the severity of renal injury (Dare et al., 2015). In db/db mice and angiotensin II-infused models, intraperitoneal injection of MitoQ alleviated excessive mitochondrial fission and restored mitochondrial autophagy, thereby mitigating tubular apoptosis and renal damage (Xiao et al., 2017; Zhu et al., 2021). Noteworthy recent in vitro data indicating that MitoQ causes mitochondrial swelling and depolarization in kidney tissue, but the doses were supraphysiological (Gottwald et al., 2018).
Resveratrol (RSV), a naturally occurring polyphenolic antioxidant, has indicated potential in improving mitochondrial function. RSV administration has been associated with increased mRNA and protein expression levels of PGC-1α and SIRT1, promoting mitochondrial homeostasis, restoring respiratory capacity, and attenuating cellular injury (Zhang et al., 2020). Unfortunately, this compound exhibits low bioavailability and solubility (Vesely et al., 2021). Clinical investigations of RSV in human populations remain in early stages, and further studies are needed to determine its efficacy and safety in patients with AKI.
5.3 FAO activators can improve mitochondrial energy metabolism in AKI
Impaired mitochondrial FAO in AKI represents a potential therapeutic target for mitochondrial-directed studies. Current strategies aimed at restoring FAO primarily involve compounds that activate the PPARα, PGC-1α, and AMPK signaling pathways (Console et al., 2020). Following renal IRI, FAO activity is significantly reduced. Modulation of CPT1 activity through pharmacological inhibition has been reported to act as a direct ligand for PPARα, thereby enhancing FAO levels and ameliorating fibrosis following IRI (Tanaka et al., 2011). Targeted modulation of CPT1 promotes PPARα-mediated FAO, protects TECs, and reduces renal injury in AKI models. Furthermore, restoration of energy homeostasis via PGC-1α activation in TECs attenuates both AKD and the progression to CKD.
Dysregulation of the mTOR and AMPK signaling pathways has been implicated in the metabolic reprogramming of TECs during AKI. Therapeutic modulation of this axis represents a promising approach to mitigating renal injury. Activation of AMPK using 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) enhances mitochondrial biogenesis, promotes mitochondrial FAO, and reduces renal damage in AKI models (Jin et al., 2020; McWilliam et al., 2021). In addition, both AICAR and RSV—a SIRT1 activator—have demonstrated efficacy in promoting mitochondrial adaptation and biogenesis, thereby reducing TEC injury and renal fibrosis in preclinical studies (Hong et al., 2017; Morigi et al., 2015; Xu et al., 2016). The impact of long-term use is not clear. Rapamycin, an mTOR inhibitor induces mitophagy and modulates cellular metabolism and proliferation. But rapamycin has uncertain efficacy and potential adverse effects (Moghadamnia et al., 2024).
Although FAO activators exhibit considerable therapeutic potential for AKI, the precise mechanisms by which FAO suppression promotes renal fibrogenesis remain incompletely understood. Moreover, the clinical translation of pharmacological agents targeting mitochondrial function and energy metabolism remains limited, highlighting the need for further investigation in both preclinical and clinical settings.
5.4 The potential value of remote ischemic preconditioning (rIPC) in preventing AKI in renal tissue within a clinical practice
The rIPC exerts organ-protective effects through two distinct temporal phases: an acute phase and a delayed phase (Jia et al., 2024; Bolli, 2000; Bolli, 2007). The rIPC is induced by transient episodes of ischemia and reperfusion in distant tissues, which can subsequently confer protection against future ischemic injury in target organs, including the kidneys (Cho et al., 2010; Jang et al., 2012; Jang et al., 2008). The acute phase of rIPC is primarily mediated by rapid post-translational modifications of pre-existing proteins and the activation of signal transduction cascades (Pei and Zhou, 2017). In contrast, the delayed phase involves transcriptional upregulation and synthesis of cytoprotective proteins, such as superoxide dismutase, heat shock proteins, and inducible nitric oxide synthase, along with modulation of anti-apoptotic pathways. Direct and indirect effects on mitochondria by IPC can result in the activation of AMPK, a master regulator of cellular metabolism. Changes in the activity of the posttranslational modifiers, SIRT1 and SIRT5, also contribute to the overall adaptive processes in cellular metabolism and mitochondrial functioning (Jackson et al., 2018). RIPC maintains mitochondrial quality through mitochondrial autophagy to clear dysfunctional mitochondria (Livingston et al., 2019), which is essential for restoring organ integrity. Wei et al. found that rIPC may attenuate mitochondrial dysfunction in AKI by inhibiting NOX4-ROS signaling (Long et al., 2022), rIPC protected kidney function and pathological injury and mitigated NADPH oxidase 4 (NOX4) upregulation in different AKI models (cisplatin, LPS and IRI). Livingston, M.J., et al. found that the mitophagy regulator PINK1 was activated during IPC. Knockdown of PINK1 suppressed mitophagy and reduced the cytoprotective effect of IPC (Livingston et al., 2019). IPC also suppressed mitochondrial depolarization, improved ATP production, and inhibited the generation of reactive oxygen species.
The rIPC is regarded as a simple, non-invasive, cost-effective, and generally safe intervention with substantial potential for clinical application. Its acute phase has demonstrated efficacy in providing renal protection in various experimental models (Long et al., 2022). Although AKI mice models revealed that rIPC effectively improved kidney dysfunction, the renoprotection effect of early or late-phase rIPC remains to be verified (Wei et al., 2025). Besides that, the clinical evidence supporting delayed rIPC remains limited due to small sample sizes, and further large-scale studies are needed to evaluate its safety and therapeutic effectiveness.
Among the molecular mediators implicated in rIPC, heat shock protein 70 (HSP70) possesses cytoprotective properties, particularly through inhibition of apoptosis (Stankiewicz et al., 2005). Experimental studies have indicated that rIPC upregulates intracellular HSP70 expression, which is subsequently released into the extracellular space and systemic circulation during stress conditions. HSP70 contributes to the protective mechanisms associated with ischemic preconditioning. Therefore, further research is warranted to clarify the role of HSP70 in rIPC-induced renal protection, including its regulatory mechanisms and potential therapeutic implications (Figure 4).
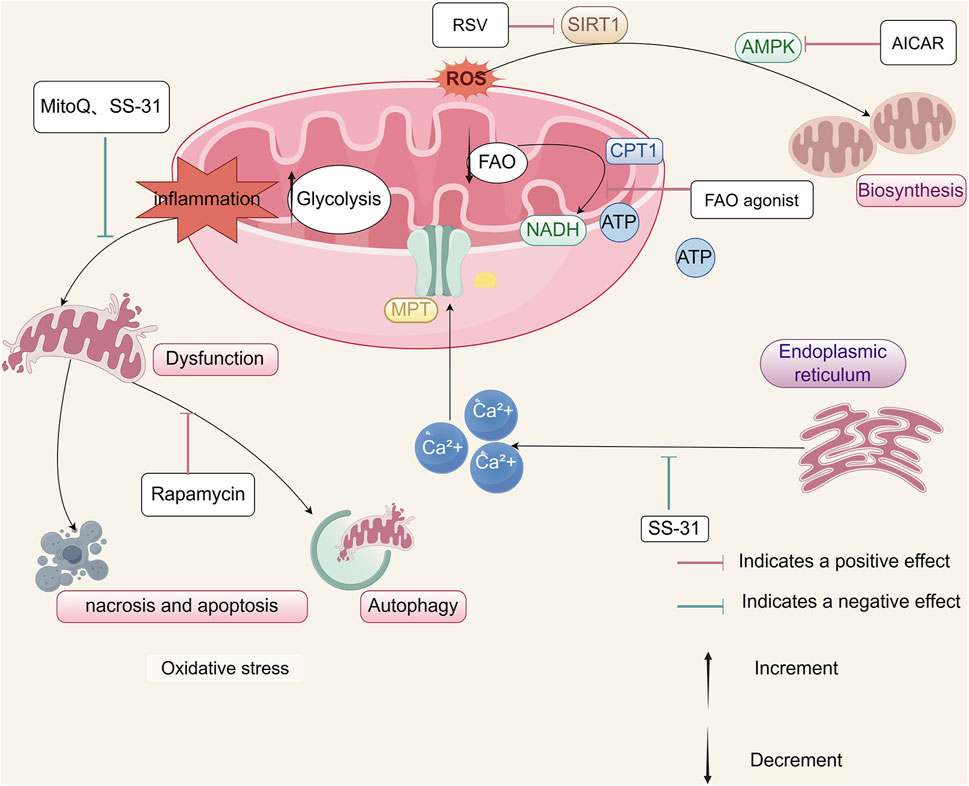
Figure 4. Therapeutic strategies targeting mitochondrial energy metabolism in AKI. Potential therapeutic approaches to mitigate mitochondrial dysfunction in AKI include: reduction of ROS using mitochondrial-targeted antioxidants such as SS-31 and MitoQ; inhibition of calcium-induced MPT pore opening by SS-31 to preserve ATP synthesis; enhancement of mitochondrial autophagy via rapamycin to maintain organelle quality; stimulation of mitochondrial biogenesis through RSV or AICAR; restoration of energy metabolism through FAO activation; and renal protection through rIPC induced by bilateral renal ischemia.
6 Conclusions and future prospects
The kidney, as the second most metabolically active organ in the human body, possesses a high mitochondrial density. These mitochondria serve not only as central sites for cellular energy metabolism but also as critical regulators of cell death pathways. Mitochondrial dysfunction and energy metabolic reprogramming are closely implicated in the onset and progression of AKI. This review has summarized current evidence regarding structural and functional changes in mitochondria, shifts in cellular energy metabolism, and recent advancements in the understanding of molecular mechanisms underlying these changes in AKI. Additionally, potential therapeutic strategies targeting mitochondrial dysfunction have been outlined, with emphasis on their relevance to AKI prevention and treatment.
Following AKI, TECs exhibit significant mitochondrial abnormalities, including disrupted dynamics of fission and fusion, impaired biogenesis, and altered morphology. These changes, regulated by a network of molecular pathways, lead to metabolic reprogramming characterized by downregulated FAO and enhanced glycolysis. This is accompanied by disturbances in membrane lipid and triglyceride metabolism, activation of the PPP, and suppression of gluconeogenesis. Although the upregulation of glycolysis offers transient metabolic compensation and oxidative stress resistance, sustained impairment of FAO results in lipid accumulation and progressive TEC injury, thereby promoting inflammation and fibrosis, ultimately resulting in poor renal outcomes. Early therapeutic interventions aimed at restoring mitochondrial function and enhancing FAO may prove to be more effective in halting disease progression rather than targeting downstream pathologies alone. Preclinical studies have indicated promising results using mitochondrial-targeted peptides, antioxidants, FAO activators, and rIPC in mitigating renal injury.
Despite the therapeutic potential of these strategies, several limitations and challenges remain. First, current knowledge of mitochondrial biology in the setting of AKI remains incomplete. The role of mitochondrial communication with other organelles, such as ER and lysosomes, requires further elucidation. Studies have shown that mitochondria-associated ER membranes (MAMs), the ER part directly connected to the mitochondria, participate in the pathogenesis of AKI by regulating calcium signaling, lipid metabolism and energy homeostasis (Igwebuike et al., 2020; Inagi, 2020). Second, existing research on metabolic reprogramming in AKI has predominantly focused on proximal tubular epithelial cells, while the metabolic responses of other renal cell types—including podocytes, endothelial cells, and interstitial cells—remain inadequately studied, to solve which single-cell metabolomics might be a promising method. Third, the precise mechanisms by which upstream and downstream regulatory molecules influence mitochondrial function and metabolic pathways are not fully understood. Circadian regulators such as brain and muscle arnt-like protein 1 (BMAL1) (Ye et al., 2022; Yang et al., 2024) may also interact with metabolic pathways in AKI. Additionally, the molecular basis through which FAO impairment contributes to renal fibrogenesis warrants further investigation.
Furthermore, although various mitochondrial-targeted interventions have demonstrated efficacy in preclinical models, their clinical translation remains limited. Challenges include inter-individual variability in metabolic responses due to heterogeneous AKI etiologies, dynamic shifts in metabolic states across AKI stages, and variability in inflammatory environments. Potential off-target effects and unforeseen adverse reactions also constrain the advancement of candidate therapeutics into clinical application. Identification of injury-specific therapeutic targets and optimization of intervention timing represent important areas for future research.
Despite these limitations, ongoing investigation is expected to yield deeper insights into the mitochondrial pathophysiology of AKI and support the development of more effective and clinically translatable therapeutic strategies.
Author contributions
MC: Writing – original draft, Funding acquisition, Writing – review and editing, Formal Analysis, Conceptualization. XZ: Formal Analysis, Writing – original draft, Software, Data curation. FX: Software, Writing – original draft, Formal Analysis, Data curation. MS: Writing – original draft, Formal Analysis, Software, Data curation. DZ: Data curation, Formal Analysis, Writing – review and editing, Software. LL: Software, Formal Analysis, Conceptualization, Project administration, Writing – review and editing, Data curation. HJ: Funding acquisition, Project administration, Writing – review and editing, Conceptualization.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Provincial Natural Science Foundation Joint Fund of Liaoning (2023-BSBA-363). Science and Technology Projects for People’s Livelihood of Liaoning Province (2021JH/10300008). Basic Research Projects of Liaoning Province (JYTMS20230073).
Acknowledgments
We would like to acknowledge the hard and dedicated work of all the staff that implemented the intervention and evaluation components of the study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AKI, Acute kidney injury; UPR, Unfolded Protein Response; ER, Endoplasmic reticulum; CKD, Chronic kidney disease; ESRD, End-stage renal disease; PTCs, proximal tubule cells; OXPHOS, oxidative phosphorylation; TECs, Tubular epithelial cells; FAO, Fatty acid β-oxidation; ATP, adenosine triphosphate; FAs, Fatty acids; PPARα, Peroxisome proliferator-activated receptor alpha; IR, Ischemia-reperfusion; CPT, Carnitine palmitoyltransferase; ACADL, Long-chain acyl-CoA dehydrogenase; HK, Hexokinase; PKM2, M2 isoform of pyruvate kinase; LDH, Lactate dehydrogenase; LPS, Lipopolysaccharides; PPP, Pentose phosphate pathway; GSH, Glutathione; 2-DG, 2-deoxyglucose; NAD+/NADH, Nicotinamide adenine dinucleotide; EMT, Epithelial-mesenchymal transition; HIF-1α, Hypoxia inducible factor-1; PHD, Prolyl-hydroxylases; VHL, Von-Hippel-Lindau; HRE, Hypoxia response elements; FADH2, Flavin adenine dinucleotide; PDK, Pyruvate dehydrogenase kinase; PGC-1α, Peroxisome proliferator-activated receptor-γ coactivator 1α; AMPK, AMP-activated protein kinase; AICAR, 5-Aminoimidazole-4-carboxamide ribonucleotide; PINK1, PTEN induced putative kinase 1; BNIP3, BCL2 interacting protein 3; FUNDC1, FUN14 domain-containing protein 1; HMGB1, High mobility group box 1; NLRP3, NLR family pyrin domain containing 3; AIM2, Absent in melanoma 2; TGF-β, Transforming growth factor-beta; SS-31, D-Arg-dimethylTyr-Lys-Phe-NH2; MitoQ, Mitoquinone; TNF-α, Tumor necrosis factor-α; OMM, mitochondrial membrane; IMM, inner mitochondrial membrane; IMS, mitochondrial intermembrane space; ETC, electron transport chain; ROS, reactive oxygen species; SIRT1, sirtuin1; FGF2, fibroblast growth factor 2; Glut1, Glucose transporter 1; FABP, fatty acid-binding protein; FBP, fructose-1,6-bisphosphatase; PCK, Phosphoenolpyruvate Carboxykinase; OS, oxidative stress; PD-L1, programmed cell death ligand 1; HSP70, the 70-kDa heat shock proteins; RSV, resveratrol; BRI, bilateral renal ischemia; DAMPs, damage associated molecular patterns; ADP, adenosine diphosphate; IL, interleukin; mtDNA, mitochondrial DNA; rIPC, remote ischemic preconditioning; PTEN, phosphatase and tensin homologue; FFA, free fatty acid; ECM, extracellular matrix; NOX4, Nicotinamide adenine dinucleotide phosphate oxidase 4; TGF, tubuloglomerular feedback; GFR, glomerular filtration rate; PC, pyruvate carboxylase; PEPCK, phosphoenolpyruvate carboxykinas.
References
Aggarwal S., Mannam P., Zhang J. (2016). Differential regulation of autophagy and mitophagy in pulmonary diseases. Am. J. Physiol. Lung Cell Mol. Physiol. 311 (2), L433–L452. doi:10.1152/ajplung.00128.2016
Baek J., Afshinnia F., Michailidis G., Pennathur S. (2022). Lipidomic approaches to dissect dysregulated lipid metabolism in kidney disease. Nat. Rev. Nephrol. 18 (1), 38–55. doi:10.1038/s41581-021-00488-2
Bao H., Zhang Q., Liu X., Song Y., Li X., Wang Z., et al. (2019). Lithium targeting of AMPK protects against cisplatin-induced acute kidney injury by enhancing autophagy in renal proximal tubular epithelial cells. Faseb J. 33 (12), 14370–14381. doi:10.1096/fj.201901712R
Bartoszewska S., Collawn J. F. (2020). Unfolded protein response (UPR) integrated signaling networks determine cell fate during hypoxia. Cell Mol. Biol. Lett. 25, 18. doi:10.1186/s11658-020-00212-1
Bhargava P., Schnellmann R. G. (2017). Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 13 (10), 629–646. doi:10.1038/nrneph.2017.107
Bolli R. (2000). The late phase of preconditioning. Circ. Res. 87 (11), 972–983. doi:10.1161/01.res.87.11.972
Bolli R. (2007). Preconditioning: a paradigm shift in the biology of myocardial ischemia. Am. J. Physiol. Heart Circ. Physiol. 292 (1), H19–H27. doi:10.1152/ajpheart.00712.2006
Cai T., Ke Q., Fang Y., Wen P., Chen H., Yuan Q., et al. (2020). Sodium-glucose cotransporter 2 inhibition suppresses HIF-1α-mediated metabolic switch from lipid oxidation to glycolysis in kidney tubule cells of diabetic mice. Cell Death Dis. 11 (5), 390. doi:10.1038/s41419-020-2544-7
Cao H., Luo J., Zhang Y., Mao X., Wen P., Ding H., et al. (2020). Tuberous sclerosis 1 (Tsc1) mediated mTORC1 activation promotes glycolysis in tubular epithelial cells in kidney fibrosis. Kidney Int. 98 (3), 686–698. doi:10.1016/j.kint.2020.03.035
Chen Q., Su Y., Ju Y., Ma K., Li W., Li W. (2018). Astragalosides IV protected the renal tubular epithelial cells from free fatty acids-induced injury by reducing oxidative stress and apoptosis. Biomed. Pharmacother. 108, 679–686. doi:10.1016/j.biopha.2018.09.049
Cheng C. F., Ku H. C., Lin H. (2018). PGC-1α as a pivotal factor in lipid and metabolic regulation. Int. J. Mol. Sci. 19 (11), 3447. doi:10.3390/ijms19113447
Cheng S. C., Quintin J., Cramer R. A., Shepardson K. M., Saeed S., Kumar V., et al. (2014). mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345 (6204), 1250684. doi:10.1126/science.1250684
Cho W. Y., Choi H. M., Lee S. Y., Kim M. G., Kim H. K., Jo S. K. (2010). The role of Tregs and CD11c(+) macrophages/dendritic cells in ischemic preconditioning of the kidney. Kidney Int. 78 (10), 981–992. doi:10.1038/ki.2010.266
Chung K. W., Dhillon P., Huang S., Sheng X., Shrestha R., Qiu C., et al. (2019). Mitochondrial damage and activation of the STING pathway lead to renal inflammation and fibrosis. Cell Metab. 30 (4), 784–799. doi:10.1016/j.cmet.2019.08.003
Chung K. W., Lee E. K., Lee M. K., Oh G. T., Yu B. P., Chung H. Y. (2018). Impairment of PPARα and the Fatty acid oxidation pathway aggravates renal fibrosis during aging. J. Am. Soc. Nephrol. 29 (4), 1223–1237. doi:10.1681/ASN.2017070802
Clark A. J., Parikh S. M. (2021). Targeting energy pathways in kidney disease: the roles of sirtuins, AMPK, and PGC1α. Kidney Int. 99 (4), 828–840. doi:10.1016/j.kint.2020.09.037
Console L., Scalise M., Giangregorio N., Tonazzi A., Barile M., Indiveri C. (2020). The link between the mitochondrial fatty acid oxidation derangement and kidney injury. Front. Physiol. 11, 794. doi:10.3389/fphys.2020.00794
Cristofano M. D., Giacomo D., F B. (2021). Mechanisms underlying the hormetic effect of conjugated linoleic acid: focus on Nrf2, mitochondria and NADPH oxidases. Free Radic. Biol. Med. 167, 276–286. doi:10.1016/j.freeradbiomed.2021.03.015
Dare A. J., Bolton E. A., Pettigrew G. J., Bradley J. A., Saeb-Parsy K., Murphy M. P. (2015). Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol. 5, 163–168. doi:10.1016/j.redox.2015.04.008
DeWolf S. E., Kasimsetty S. G., Hawkes A. A., Stocks L. M., Kurian S. M., McKay D. B. (2022). DAMPs released from injured renal tubular epithelial cells activate innate immune signals in healthy renal tubular epithelial cells. Transplantation 106 (8), 1589–1599. doi:10.1097/TP.0000000000004038
Ding H., Jiang L., Xu J., Bai F., Zhou Y., Yuan Q., et al. (2017). Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am. J. Physiol. Ren. Physiol. 313 (3), F561–F575. doi:10.1152/ajprenal.00036.2017
Emma F., Montini G., Parikh S. M., Salviati L. (2016). Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat. Rev. Nephrol. 12 (5), 267–280. doi:10.1038/nrneph.2015.214
Erpicum P., Rowart P., Defraigne J. O., Krzesinski J. M., Jouret F. (2018). What we need to know about lipid-associated injury in case of renal ischemia-reperfusion. Am. J. Physiol. Ren. Physiol. 315 (6), F1714–F1719. doi:10.1152/ajprenal.00322.2018
Fan X., Yang M., Lang Y., Lu S., Kong Z., Gao Y., et al. (2024). Mitochondrial metabolic reprogramming in diabetic kidney disease. Cell Death Dis. 15 (6), 442. doi:10.1038/s41419-024-06833-0
Fogo A. B., Harris R. C. (2025). Crosstalk between glomeruli and tubules. Nat. Rev. Nephrol. 21 (3), 189–199. doi:10.1038/s41581-024-00907-0
Fontecha-Barriuso M., Martin-Sanchez D., Martinez-Moreno J. M., Monsalve M., Ramos A. M., Sanchez-Niño M. D., et al. (2020). The role of PGC-1α and mitochondrial biogenesis in kidney diseases. Biomolecules 10 (2), 347. doi:10.3390/biom10020347
Foster D. W. (2012). Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. J. Clin. Invest 122 (6), 1958–1959. doi:10.1172/jci63967
Freitas-Lima L. C., Budu A., Arruda A. C., Perilhão M. S., Barrera-Chimal J., Araujo R. C., et al. (2020). PPAR-α deletion attenuates Cisplatin nephrotoxicity by modulating renal organic transporters MATE-1 and OCT-2. Int. J. Mol. Sci. 21 (19), 7416. doi:10.3390/ijms21197416
Fu Z. J., Wang Z. Y., Xu L., Chen X. H., Li X. X., Liao W. T., et al. (2020). HIF-1α-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol. 36, 101671. doi:10.1016/j.redox.2020.101671
Gewin L. S. (2021). Sugar or fat? Renal tubular metabolism reviewed in health and disease. Nutrients 13 (5), 1580. doi:10.3390/nu13051580
Giacomello M., Pyakurel A., Glytsou C., Scorrano L. (2020). The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 21 (4), 204–224. doi:10.1038/s41580-020-0210-7
Gottwald E. M., Duss M., Bugarski M., Haenni D., Schuh C. D., Landau E. M., et al. (2018). The targeted anti-oxidant MitoQ causes mitochondrial swelling and depolarization in kidney tissue. Physiol. Rep. 6 (7), e13667. doi:10.14814/phy2.13667
Grande M. T., Sánchez-Laorden B., López-Blau C., De Frutos C. A., Boutet A., Arévalo M., et al. (2015). Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 21 (9), 989–997. doi:10.1038/nm.3901
Guder W. G., Ross B. D. (1984). Enzyme distribution along the nephron. Kidney Int. 26 (2), 101–111. doi:10.1038/ki.1984.143
Gupta S., Glezerman I. G., Hirsch J. S., Chen K. L., Devaraj N., Wells S. L., et al. (2024). Derivation and external validation of a simple risk score for predicting severe acute kidney injury after intravenous cisplatin: cohort study. Bmj 384, e077169. doi:10.1136/bmj-2023-077169
Hall A. M., Rhodes G. J., Sandoval R. M., Corridon P. R., Molitoris B. A. (2013). In vivo multiphoton imaging of mitochondrial structure and function during acute kidney injury. Kidney Int. 83 (1), 72–83. doi:10.1038/ki.2012.328
Han S. H., Malaga-Dieguez L., Chinga F., Kang H. M., Tao J., Reidy K., et al. (2016). Deletion of Lkb1 in renal tubular epithelial cells leads to CKD by altering metabolism. J. Am. Soc. Nephrol. 27 (2), 439–453. doi:10.1681/ASN.2014121181
Hasegawa S., Inagi R. (2020). Organelle stress and crosstalk in Kidney disease. Kidney360 1 (10), 1157–1164. doi:10.34067/KID.0002442020
He B., Copps K. D., Stöhr O., Liu B., Hu S., Joshi S., et al. (2025). Spatial regulation of glucose and lipid metabolism by hepatic insulin signaling. Cell Metab. 37, 1568–1583.e7. doi:10.1016/j.cmet.2025.03.015
Hong Y. A., Bae S. Y., Ahn S. Y., Kim J., Kwon Y. J., Jung W. Y., et al. (2017). Resveratrol ameliorates contrast induced nephropathy through the activation of SIRT1-PGC-1α-Foxo1 signaling in mice. Kidney Blood Press Res. 42 (4), 641–653. doi:10.1159/000481804
Hoste E. A. J., Kellum J. A., Selby N. M., Zarbock A., Palevsky P. M., Bagshaw S. M., et al. (2018). Global epidemiology and outcomes of acute kidney injury. Nat. Rev. Nephrol. 14 (10), 607–625. doi:10.1038/s41581-018-0052-0
Houten S. M., Violante S., Ventura F. V., Wanders R. J. A. (2016). The biochemistry and physiology of mitochondrial fatty acid β-oxidation and its genetic disorders. Annu. Rev. Physiol. 78, 23–44. doi:10.1146/annurev-physiol-021115-105045
Houten S. M., Wanders R. J. A., Ranea-Robles P. (2020). Metabolic interactions between peroxisomes and mitochondria with a special focus on acylcarnitine metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 1866 (5), 165720. doi:10.1016/j.bbadis.2020.165720
Hu X., Xu Q., Wan H., Hu Y., Xing S., Yang H., et al. (2020). PI3K-Akt-mTOR/PFKFB3 pathway mediated lung fibroblast aerobic glycolysis and collagen synthesis in lipopolysaccharide-induced pulmonary fibrosis. Lab. Invest 100 (6), 801–811. doi:10.1038/s41374-020-0404-9
Igwebuike C., Yaglom J., Huiting L., Feng H., Campbell J. D., Wang Z., et al. (2020). Cross organelle stress response disruption promotes gentamicin-induced proteotoxicity. Cell Death Dis. 11 (4), 217. doi:10.1038/s41419-020-2382-7
Inagi R. (2020). The implication of organelle cross talk in AKI. Nephron 144 (12), 634–637. doi:10.1159/000508639
Isaka Y., Kimura T., Takabatake Y. (2011). The protective role of autophagy against aging and acute ischemic injury in kidney proximal tubular cells. Autophagy 7 (9), 1085–1087. doi:10.4161/auto.7.9.16465
Ishimoto Y., Inagi R. (2016). Mitochondria: a therapeutic target in acute kidney injury. Nephrol. Dial. Transpl. 31 (7), 1062–1069. doi:10.1093/ndt/gfv317
Iwaki T., Bennion B. G., Stenson E. K., Lynn J. C., Otinga C., Djukovic D., et al. (2019). PPARα contributes to protection against metabolic and inflammatory derangements associated with acute kidney injury in experimental sepsis. Physiol. Rep. 7 (10), e14078. doi:10.14814/phy2.14078
Jackson C. W., Escobar I., Xu J., Perez-Pinzon M. A. (2018). Effects of ischemic preconditioning on mitochondrial and metabolic neruoprotection: 5' adenosine monophosphate-activated protein kinase and sirtuins. Brain Circ. 4 (2), 54–61. doi:10.4103/bc.bc_7_18
Jang H. S., Kim J., Kim K. Y., Kim J. I., Cho M. H., Park K. M. (2012). Previous ischemia and reperfusion injury results in resistance of the kidney against subsequent ischemia and reperfusion insult in mice; a role for the Akt signal pathway. Nephrol. Dial. Transpl. 27 (10), 3762–3770. doi:10.1093/ndt/gfs097
Jang H. S., Kim J., Park Y. K., Park K. M. (2008). Infiltrated macrophages contribute to recovery after ischemic injury but not to ischemic preconditioning in kidneys. Transplantation 85 (3), 447–455. doi:10.1097/TP.0b013e318160f0d1
Jang H. S., Noh M. R., Jung E. M., Kim W. Y., Southekal S., Guda C., et al. (2020). Proximal tubule cyclophilin D regulates fatty acid oxidation in cisplatin-induced acute kidney injury. Kidney Int. 97 (2), 327–339. doi:10.1016/j.kint.2019.08.019
Jia P., Ji Q., Zou Z., Zeng Q., Ren T., Chen W., et al. (2024). Effect of delayed remote ischemic preconditioning on Acute kidney injury and outcomes in patients undergoing cardiac surgery: a randomized clinical trial. Circulation 150 (17), 1366–1376. doi:10.1161/CIRCULATIONAHA.124.071408
Jiang M., Bai M., Lei J., Xie Y., Xu S., Jia Z., et al. (2020). Mitochondrial dysfunction and the AKI-to-CKD transition. Am. J. Physiol. Ren. Physiol. 319 (6), F1105–f1116. doi:10.1152/ajprenal.00285.2020
Jin K., Ma Y., Manrique-Caballero C. L., Li H., Emlet D. R., Li S., et al. (2020). Activation of AMP-activated protein kinase during sepsis/inflammation improves survival by preserving cellular metabolic fitness. Faseb J. 34 (5), 7036–7057. doi:10.1096/fj.201901900R
Kang H. M., Ahn S. H., Choi P., Ko Y. A., Han S. H., Chinga F., et al. (2015). Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 21 (1), 37–46. doi:10.1038/nm.3762
Kong Y., Chen X., Liu F., Tang J., Zhang Y., Zhang X., et al. (2024). Ultrasmall Polyphenol-NAD(+) nanoparticle-mediated renal delivery for mitochondrial repair and anti-inflammatory treatment of AKI-to-CKD progression. Adv. Mater 36 (30), e2310731. doi:10.1002/adma.202310731
Lakhia R., Yheskel M., Flaten A., Quittner-Strom E. B., Holland W. L., Patel V. (2018). PPARα agonist fenofibrate enhances fatty acid β-oxidation and attenuates polycystic kidney and liver disease in mice. Am. J. Physiol. Ren. Physiol. 314 (1), F122–F131. doi:10.1152/ajprenal.00352.2017
Lan R., Geng H., Singha P. K., Saikumar P., Bottinger E. P., Weinberg J. M., et al. (2016). Mitochondrial pathology and glycolytic shift during proximal tubule Atrophy after Ischemic AKI. J. Am. Soc. Nephrol. 27 (11), 3356–3367. doi:10.1681/ASN.2015020177
Lawrence J. W., Li Y., Chen S., DeLuca J. G., Berger J. P., Umbenhauer D. R., et al. (2001). Differential gene regulation in human versus rodent hepatocytes by peroxisome proliferator-activated receptor (PPAR) alpha. PPAR alpha fails to induce peroxisome proliferation-associated genes in human cells independently of the level of receptor expresson. J. Biol. Chem. 276 (34), 31521–31527. doi:10.1074/jbc.M103306200
Lee E. S., Kang J. S., Kim H. M., Kim S. J., Kim N., Lee J. O., et al. (2021b). Dehydrozingerone inhibits renal lipotoxicity in high-fat diet-induced obese mice. J. Cell Mol. Med. 25 (18), 8725–8733. doi:10.1111/jcmm.16828
Lee K., Jang H. R., Rabb H. (2024). Lymphocytes and innate immune cells in acute kidney injury and repair. Nat. Rev. Nephrol. 20 (12), 789–805. doi:10.1038/s41581-024-00875-5
Lee S. H., Golinska M., Griffiths J. R. (2021a). HIF-1-Independent mechanisms regulating metabolic adaptation in hypoxic cancer cells. Cells 10 (9), 2371. doi:10.3390/cells10092371
Legouis D., Ricksten S. E., Faivre A., Verissimo T., Gariani K., Verney C., et al. (2020). Altered proximal tubular cell glucose metabolism during acute kidney injury is associated with mortality. Nat. Metab. 2 (8), 732–743. doi:10.1038/s42255-020-0238-1
Levey A. S., Eckardt K. U., Dorman N. M., Christiansen S. L., Hoorn E. J., Ingelfinger J. R., et al. (2020). Nomenclature for kidney function and disease: report of a kidney disease: improving global outcomes (KDIGO) consensus conference. Kidney Int. 97 (6), 1117–1129. doi:10.1016/j.kint.2020.02.010
Levine B., Kroemer G. (2019). Biological functions of autophagy genes: a disease perspective. Cell 176 (1-2), 11–42. doi:10.1016/j.cell.2018.09.048
Li J., Shi X., Chen Z., Xu J., Zhao R., Liu Y., et al. (2023). Aldehyde dehydrogenase 2 alleviates mitochondrial dysfunction by promoting PGC-1α-mediated biogenesis in acute kidney injury. Cell Death Dis. 14 (1), 45. doi:10.1038/s41419-023-05557-x
Li M., Jia F., Zhou H., Di J., Yang M. (2018). Elevated aerobic glycolysis in renal tubular epithelial cells influences the proliferation and differentiation of podocytes and promotes renal interstitial fibrosis. Eur. Rev. Med. Pharmacol. Sci. 22 (16), 5082–5090. doi:10.26355/eurrev_201808_15701
Li S. Y., Susztak K. (2018). The role of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) in kidney disease. Semin. Nephrol. 38 (2), 121–126. doi:10.1016/j.semnephrol.2018.01.003
Li W., Duan A., Xing Y., Xu L., Yang J. (2021b). Transcription-Based multidimensional regulation of Fatty acid metabolism by HIF1α in renal tubules. Front. Cell Dev. Biol. 9, 690079. doi:10.3389/fcell.2021.690079
Li Y., Nourbakhsh N., Pham H., Tham R., Zuckerman J. E., Singh P. (2020). Evolution of altered tubular metabolism and mitochondrial function in sepsis-associated acute kidney injury. Am. J. Physiol. Ren. Physiol. 319 (2), F229–F244. doi:10.1152/ajprenal.00390.2019
Li Z., Lu S., Li X. (2021a). The role of metabolic reprogramming in tubular epithelial cells during the progression of acute kidney injury. Cell Mol. Life Sci. 78 (15), 5731–5741. doi:10.1007/s00018-021-03892-w
Lin Q., Li S., Jiang N., Shao X., Zhang M., Jin H., et al. (2019). PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 26, 101254. doi:10.1016/j.redox.2019.101254
Lin Q., Li S., Jin H., Cai H., Zhu X., Yang Y., et al. (2023). Mitophagy alleviates cisplatin-induced renal tubular epithelial cell ferroptosis through ROS/HO-1/GPX4 axis. Int. J. Biol. Sci. 19 (4), 1192–1210. doi:10.7150/ijbs.80775
Lin S. H., Fan J., Zhu J., Zhao Y. S., Wang C. J., Zhang M., et al. (2020). Exploring plasma metabolomic changes in sepsis: a clinical matching study based on gas chromatography-mass spectrometry. Ann. Transl. Med. 8 (23), 1568. doi:10.21037/atm-20-3562
Liu G., Zou H., Luo T., Long M., Bian J., Liu X., et al. (2016). Caspase-Dependent and caspase-independent pathways are involved in cadmium-induced apoptosis in primary rat proximal tubular cell culture. PLoS One 11 (11), e0166823. doi:10.1371/journal.pone.0166823
Livingston M. J., Shu S., Fan Y., Li Z., Jiao Q., Yin X. M., et al. (2023). Tubular cells produce FGF2 via autophagy after acute kidney injury leading to fibroblast activation and renal fibrosis. Autophagy 19 (1), 256–277. doi:10.1080/15548627.2022.2072054
Livingston M. J., Wang J., Zhou J., Wu G., Ganley I. G., Hill J. A., et al. (2019). Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy 15 (12), 2142–2162. doi:10.1080/15548627.2019.1615822
Long Y. Q., Feng X. M., Shan X. S., Chen Q. C., Xia Z., Ji F. H., et al. (2022). Remote ischemic preconditioning reduces Acute Kidney injury after cardiac surgery: a systematic review and meta-analysis of randomized controlled trials. Anesth. Analg. 134 (3), 592–605. doi:10.1213/ANE.0000000000005804
Luo Q., Wu X., Zhao P., Nan Y., Chang W., Zhu X., et al. (2021). OTUD1 activates caspase-independent and caspase-dependent apoptosis by promoting AIF nuclear translocation and MCL1 degradation. Adv. Sci. (Weinh) 8 (8), 2002874. doi:10.1002/advs.202002874
Ma H., Guo X., Cui S., Wu Y., Zhang Y., Shen X., et al. (2022). Dephosphorylation of AMP-activated protein kinase exacerbates ischemia/reperfusion-induced acute kidney injury via mitochondrial dysfunction. Kidney Int. 101 (2), 315–330. doi:10.1016/j.kint.2021.10.028
McWilliam S. J., Wright R. D., Welsh G. I., Tuffin J., Budge K. L., Swan L., et al. (2021). The complex interplay between kidney injury and inflammation. Clin. Kidney J. 14 (3), 780–788. doi:10.1093/ckj/sfaa164
Miguel V., Tituaña J., Herrero J. I., Herrero L., Serra D., Cuevas P., et al. (2021). Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J. Clin. Invest 131 (5), e140695. doi:10.1172/JCI140695
Moghadamnia M., Dashti-Khavidaki S., Alimadadi H. (2024). Role of mTOR inhibitors in pediatric liver transplant recipients: a systematic review. Paediatr. Drugs 26 (6), 673–693. doi:10.1007/s40272-024-00648-4
Morigi M., Perico L., Rota C., Longaretti L., Conti S., Rottoli D., et al. (2015). Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Invest 125 (2), 715–726. doi:10.1172/JCI77632
Onishi M., Yamano K., Sato M., Matsuda N., Okamoto K. (2021). Molecular mechanisms and physiological functions of mitophagy. Embo J. 40 (3), e104705. doi:10.15252/embj.2020104705
Pei H., Zhou C. (2017). Cardiac or renal protection by delayed remote ischemic preconditioning in the clinical practice: potential additive effect from concurrent medications with pharmacological mimicking conditioning. Int. J. Cardiol. 234, 105–106. doi:10.1016/j.ijcard.2016.12.157
Price N. L., Gomes A. P., Ling A. J. Y., Duarte F. V., Martin-Montalvo A., North B. J., et al. (2012). SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 15 (5), 675–690. doi:10.1016/j.cmet.2012.04.003
Qiao S., Kang Y., Tan X., Zhou X., Zhang C., Lai S., et al. (2024). Nanomaterials-induced programmed cell death: focus on mitochondria. Toxicology 504, 153803. doi:10.1016/j.tox.2024.153803
Qin N., Cai T., Ke Q., Yuan Q., Luo J., Mao X., et al. (2019). UCP2-dependent improvement of mitochondrial dynamics protects against acute kidney injury. J. Pathol. 247 (3), 392–405. doi:10.1002/path.5198
Quirós P. M., Mottis A., Auwerx J. (2016). Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 17 (4), 213–226. doi:10.1038/nrm.2016.23
Ronco C., Bellomo R., Kellum J. A. (2019). Acute kidney injury. Lancet 394 (10212), 1949–1964. doi:10.1016/S0140-6736(19)32563-2
Sanz A. B., Sanchez-Niño M. D., Ramos A. M., Ortiz A. (2023). Regulated cell death pathways in kidney disease. Nat. Rev. Nephrol. 19 (5), 281–299. doi:10.1038/s41581-023-00694-0
Sasaki Y., Raza-Iqbal S., Tanaka T., Murakami K., Anai M., Osawa T., et al. (2019). Gene expression profiles induced by a novel selective peroxisome proliferator-activated receptor α modulator (SPPARMα) pemafibrate. Int. J. Mol. Sci. 20 (22), 5682. doi:10.3390/ijms20225682
Scantlebery A. M., Tammaro A., Mills J. D., Rampanelli E., Kors L., Teske G. J., et al. (2021). The dysregulation of metabolic pathways and induction of the pentose phosphate pathway in renal ischaemia-reperfusion injury. J. Pathol. 253 (4), 404–414. doi:10.1002/path.5605
Scholz H., Boivin F. J., Schmidt-Ott K. M., Bachmann S., Eckardt K. U., Scholl U. I., et al. (2021). Kidney physiology and susceptibility to acute kidney injury: implications for renoprotection. Nat. Rev. Nephrol. 17 (5), 335–349. doi:10.1038/s41581-021-00394-7
Shen Y., Jiang L., Wen P., Ye Y., Zhang Y., Ding H., et al. (2020). Tubule-derived lactate is required for fibroblast activation in acute kidney injury. Am. J. Physiol. Ren. Physiol. 318 (3), F689–F701. doi:10.1152/ajprenal.00229.2019
Sonnweber T., Pizzini A., Nairz M., Weiss G., Tancevski I. (2018). Arachidonic acid metabolites in cardiovascular and metabolic diseases. Int. J. Mol. Sci. 19 (11), 3285. doi:10.3390/ijms19113285
Stankiewicz A. R., Lachapelle G., Foo C. P. Z., Radicioni S. M., Mosser D. D. (2005). Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J. Biol. Chem. 280 (46), 38729–38739. doi:10.1074/jbc.M509497200
Sun J., Zhang J., Tian J., Virzì G. M., Digvijay K., Cueto L., et al. (2019). Mitochondria in sepsis-induced AKI. J. Am. Soc. Nephrol. 30 (7), 1151–1161. doi:10.1681/ASN.2018111126
Sun Y., Ge X., Li X., He J., Wei X., Du J., et al. (2020). High-fat diet promotes renal injury by inducing oxidative stress and mitochondrial dysfunction. Cell Death Dis. 11 (10), 914. doi:10.1038/s41419-020-03122-4
Szeto H. H., Liu S., Soong Y., Seshan S. V., Cohen-Gould L., Manichev V., et al. (2017). Mitochondria protection after Acute ischemia prevents prolonged upregulation of IL-1β and IL-18 and arrests CKD. J. Am. Soc. Nephrol. 28 (5), 1437–1449. doi:10.1681/ASN.2016070761
Takakura A., Zandi-Nejad K. (2019). Lactate-induced activation of HCA2 improves survival in mice with sepsis. Faseb J. 33 (6), 7625–7634. doi:10.1096/fj.201801982R
Tan C., Gu J., Chen H., Li T., Deng H., Liu K., et al. (2020). Inhibition of aerobic glycolysis promotes neutrophil to influx to the infectious site via CXCR2 in sepsis. Shock 53 (1), 114–123. doi:10.1097/SHK.0000000000001334
Tan C., Gu J., Li T., Chen H., Liu K., Liu M., et al. (2021). Inhibition of aerobic glycolysis alleviates sepsis-induced acute kidney injury by promoting lactate/Sirtuin 3/AMPK-regulated autophagy. Int. J. Mol. Med. 47 (3), 19. doi:10.3892/ijmm.2021.4852
Tanaka Y., Kume S., Araki S. i., Isshiki K., Chin-Kanasaki M., Sakaguchi M., et al. (2011). Fenofibrate, a PPARα agonist, has renoprotective effects in mice by enhancing renal lipolysis. Kidney Int. 79 (8), 871–882. doi:10.1038/ki.2010.530
Tannahill G. M., Curtis A. M., Adamik J., Palsson-McDermott E. M., McGettrick A. F., Goel G., et al. (2013). Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496 (7444), 238–242. doi:10.1038/nature11986
Todorović Z., Đurašević S., Stojković M., Grigorov I., Pavlović S., Jasnić N., et al. (2021). Lipidomics provides new Insight into pathogenesis and therapeutic targets of the ischemia-reperfusion injury. Int. J. Mol. Sci. 22 (6), 2798. doi:10.3390/ijms22062798
Tran M., Tam D., Bardia A., Bhasin M., Rowe G. C., Kher A., et al. (2011). PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Invest 121 (10), 4003–4014. doi:10.1172/JCI58662
Tran M. T., Zsengeller Z. K., Berg A. H., Khankin E. V., Bhasin M. K., Kim W., et al. (2016). PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531 (7595), 528–532. doi:10.1038/nature17184
Trotta A. P., Chipuk J. E. (2017). Mitochondrial dynamics as regulators of cancer biology. Cell Mol. Life Sci. 74 (11), 1999–2017. doi:10.1007/s00018-016-2451-3
van der Rijt S., Leemans J. C., Florquin S., Houtkooper R. H., Tammaro A. (2022). Immunometabolic rewiring of tubular epithelial cells in kidney disease. Nat. Rev. Nephrol. 18 (9), 588–603. doi:10.1038/s41581-022-00592-x
Verissimo T., Faivre A., Rinaldi A., Lindenmeyer M., Delitsikou V., Veyrat-Durebex C., et al. (2022). Decreased renal gluconeogenesis is a hallmark of chronic kidney disease. J. Am. Soc. Nephrol. 33 (4), 810–827. doi:10.1681/ASN.2021050680
Vesely O., Baldovska S., Kolesarova A. (2021). Enhancing bioavailability of nutraceutically used resveratrol and other stilbenoids. Nutrients 13 (9), 3095. doi:10.3390/nu13093095
Wada Y., Kidokoro K., Kondo M., Tokuyama A., Kadoya H., Nagasu H., et al. (2024). Evaluation of glomerular hemodynamic changes by sodium-glucose-transporter 2 inhibition in type 2 diabetic rats using in vivo imaging. Kidney Int. 106 (3), 408–418. doi:10.1016/j.kint.2024.05.006
Wai T., Langer T. (2016). Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab. 27 (2), 105–117. doi:10.1016/j.tem.2015.12.001
Wang H., Luo W., Chen H., Cai Z., Xu G. (2024). Mitochondrial dynamics and mitochondrial autophagy: molecular structure, orchestrating mechanism and related disorders. Mitochondrion 75, 101847. doi:10.1016/j.mito.2024.101847
Wang Y., Liu Z., Shu S., Cai J., Tang C., Dong Z. (2020). AMPK/mTOR signaling in autophagy regulation during cisplatin-induced acute kidney injury. Front. Physiol. 11, 619730. doi:10.3389/fphys.2020.619730
Wei Q., Su J., Dong G., Zhang M., Huo Y., Dong Z. (2019). Glycolysis inhibitors suppress renal interstitial fibrosis via divergent effects on fibroblasts and tubular cells. Am. J. Physiol. Ren. Physiol. 316 (6), F1162–F1172. doi:10.1152/ajprenal.00422.2018
Wei W., Yang L., Wang B., Tang L., Li J., Liu C., et al. (2025). Remote ischemic preconditioning attenuates mitochondrial dysfunction and ferroptosis of tubular epithelial cells by inhibiting NOX4-ROS signaling in Acute kidney injury. Int. J. Biol. Sci. 21 (5), 2313–2329. doi:10.7150/ijbs.105667
Xiao L., Xu X., Zhang F., Wang M., Xu Y., Tang D., et al. (2017). The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 11, 297–311. doi:10.1016/j.redox.2016.12.022
Xie M., Yu Y., Kang R., Zhu S., Yang L., Zeng L., et al. (2016). PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat. Commun. 7, 13280. doi:10.1038/ncomms13280
Xu J., Ma X., Yu K., Wang R., Wang S., Liu R., et al. (2021). Lactate up-regulates the expression of PD-L1 in kidney and causes immunosuppression in septic Acute Renal Injury. J. Microbiol. Immunol. Infect. 54 (3), 404–410. doi:10.1016/j.jmii.2019.10.006
Xu S., Gao Y., Zhang Q., Wei S., Chen Z., Dai X., et al. (2016). SIRT1/3 activation by resveratrol Attenuates Acute Kidney Injury in a septic rat model. Oxid. Med. Cell Longev. 2016, 7296092. doi:10.1155/2016/7296092
Xu S., Jia P., Fang Y., Jin J., Sun Z., Zhou W., et al. (2022). Nuclear farnesoid X receptor attenuates acute kidney injury through fatty acid oxidation. Kidney Int. 101 (5), 987–1002. doi:10.1016/j.kint.2022.01.029
Yang L., Besschetnova T. Y., Brooks C. R., Shah J. V., Bonventre J. V. (2010). Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 16 (5), 535–543. doi:10.1038/nm.2144
Yang L., Xie M., Yang M., Yu Y., Zhu S., Hou W., et al. (2014). PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat. Commun. 5, 4436. doi:10.1038/ncomms5436
Yang S., Ye Z., Chen W., Wang P., Zhao S., Zhou X., et al. (2024). BMAL1 alleviates sepsis-induced AKI by inhibiting ferroptosis. Int. Immunopharmacol. 142 (Pt B), 113159. doi:10.1016/j.intimp.2024.113159
Ye P., Li W., Huang X., Zhao S., Chen W., Xia Y., et al. (2022). BMAL1 regulates mitochondrial homeostasis in renal ischaemia-reperfusion injury by mediating the SIRT1/PGC-1α axis. J. Cell Mol. Med. 26 (7), 1994–2009. doi:10.1111/jcmm.17223
Yu S., Meng S., Xiang M., Ma H. (2021). Phosphoenolpyruvate carboxykinase in cell metabolism: roles and mechanisms beyond gluconeogenesis. Mol. Metab. 53, 101257. doi:10.1016/j.molmet.2021.101257
Zhang W., Li X., Wang S., Chen Y., Liu H. (2020). Regulation of TFEB activity and its potential as a therapeutic target against kidney diseases. Cell Death Discov. 6, 32. doi:10.1038/s41420-020-0265-4
Zhang X., Agborbesong E., Li X. (2021). The role of Mitochondria in Acute Kidney Injury and chronic Kidney disease and its therapeutic potential. Int. J. Mol. Sci. 22 (20), 11253. doi:10.3390/ijms222011253
Zhang Y., Zhu J. H., Zhou Y., Li Z. T., Liu H., Ma R. X., et al. (2025). Activation of HIF-1α C-terminal transactivation domain promotes tubulointerstitial fibrosis through hexokinase 2-mediated metabolic reprogramming. Cell Signal 127, 111531. doi:10.1016/j.cellsig.2024.111531
Zhao M., Wang Y., Li L., Liu S., Wang C., Yuan Y., et al. (2021). Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 11 (4), 1845–1863. doi:10.7150/thno.50905
Zhao Z. B., Marschner J. A., Iwakura T., Motrapu M., Kuang M., et al. (2023). Tubular epithelial cell HMGB1 promotes AKI-CKD transition by sensitizing cycling tubular cells to oxidative stress: a rationale for targeting HMGB1 during AKI recovery. J. Am. Soc. Nephrol. 34 (3), 394–411. doi:10.1681/ASN.0000000000000024
Zhou X., Li Z., Ren F., Deng H., Wen J., Xiang Q., et al. (2025). Endoplasmic reticulum stress and unfolded protein response in renal lipid metabolism. Exp. Cell Res. 446 (1), 114463. doi:10.1016/j.yexcr.2025.114463
Zhu D., Zhong J., Gong X., Wu X. (2023). Augmenter of liver regeneration reduces mitochondria-derived ROS and NLRP3 inflammasome activation through PINK1/Parkin-mediated mitophagy in ischemia-reperfusion-induced renal tubular injury. Apoptosis 28 (3-4), 335–347. doi:10.1007/s10495-022-01794-1
Zhu Z., Hu J., Chen Z., Feng J., Yang X., Liang W., et al. (2022). Transition of acute kidney injury to chronic kidney disease: role of metabolic reprogramming. Metabolism 131, 155194. doi:10.1016/j.metabol.2022.155194
Zhu Z., Liang W., Chen Z., Hu J., Feng J., Cao Y., et al. (2021). Mitoquinone protects podocytes from Angiotensin II-Induced mitochondrial dysfunction and injury via the Keap1-Nrf2 signaling pathway. Oxid. Med. Cell Longev. 2021, 1394486. doi:10.1155/2021/1394486
Zorov D. B., Juhaszova M., Sollott S. J. (2014). Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 94 (3), 909–950. doi:10.1152/physrev.00026.2013
Keywords: acute kidney injury (AKI), energy metabolic reprogramming, mitochondrial dysfunction, proximal tubular cells (PTC), therapeutic implications
Citation: Cao M, Zhao X, Xia F, Shi M, Zhao D, Li L and Jiang H (2025) Mitochondrial dysfunction and metabolic reprogramming in acute kidney injury: mechanisms, therapeutic advances, and clinical challenges. Front. Physiol. 16:1623500. doi: 10.3389/fphys.2025.1623500
Received: 06 May 2025; Accepted: 18 July 2025;
Published: 06 August 2025.
Edited by:
David Sebastián, University of Barcelona, SpainReviewed by:
Mi Liu, Southern Medical University, ChinaChidozie Okoye, University of Nigeria, Nsukka, Nigeria
Copyright © 2025 Cao, Zhao, Xia, Shi, Zhao, Li and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongkun Jiang, aGtqaWFuZ0BjbXUuZWR1LmNu; Lei Li, bGVpbGllaWVpbEAxMjYuY29t
†These authors have contributed equally to this work