 Guopeng You
Guopeng You Jinwen Xie1
Jinwen Xie1 Shaocong Zhao
Shaocong Zhao- 1Department of Physical Education, Xiamen University of Technology, Xiamen, China
- 2Department of Physical Education, Changzhi Medical College, Changzhi, China
Cardiovascular diseases (CVDs) are the world’s leading cause of death, but there’s a gap between scientific research and real-world treatment. Exercise is a safe and effective way to prevent and manage CVDs, yet putting it into practice faces many challenges. This review shows how exercise protects the heart by improving metabolism, reducing inflammation and cell damage, and strengthening connections between heart cells and blood vessels. Exercise establishes a multi-organ defense network involving remote organs including the brain, skeletal muscle, adipose tissue, liver, and kidneys. To bridge the gap between research and clinical use, future efforts should focus on developing exercise-like drugs, personalized workout plans, and remote rehabilitation programs.
1 Introduction
Driven by global population aging and the widespread prevalence of risk factors, cardiovascular diseases (CVD) continue to exhibit rising incidence and mortality rates (Krishnan et al., 2025). Recent epidemiological studies reveal that while age-standardized CVD mortality decreased by 18.6% compared to 1990, over 20.5 million CVD-related deaths occurred globally in 2021, with ischemic heart disease (IHD) accounting for 48.3% of cases—a 72% absolute increase since 1990 (Martin et al., 2024). Data from the 2023 Report on Cardiovascular Health and Diseases in China underscore CVD as the leading cause of death nationwide, with coronary heart disease mortality demonstrating a persistent upward trajectory over the past decade (Center For Cardiovascular Diseases The Writing Committee Of The ReportOn Cardiovascular and Diseases In China, 2024). These findings highlight the urgent public health challenge posed by CVD.
Exercise intervention stands as a cornerstone strategy for CVD prevention and management. Clinical evidence confirms that regular, moderate physical activity significantly reduces CVD morbidity and mortality (Tucker et al., 2022). Animal experiments also shows that exercise improve the cardiac function of mice with myocardial infarction by inhibiting inflammation, oxidative stress and apoptosis (Bo et al., 2024; Wang et al., 2024; Hou et al., 2019). However, the 2023 WHO Global Report on Physical Activity indicates that 27.5% of adults fail to meet recommended exercise guidelines (150–300 min of moderate or 75–150 min of vigorous activity weekly) (Bull et al., 2020). This disparity between scientific evidence and clinical implementation reflects systemic barriers to cardiac exercise rehabilitation, including inadequate insurance coverage for long-term exercise prescriptions (only 31% of U.S. insurance plans include cardiac rehabilitation), patient misconceptions regarding exercise safety, and a critical shortage of specialized rehabilitation teams (Balady et al., 2011; Schopfer et al., 2020). Addressing these challenges requires a deeper understanding of the systemic biological mechanisms underlying exercise-induced cardioprotection and the development of translational pathways bridging basic research to clinical practice. This study, based on the perspective of integrative physiology, systematically sorts out the mechanism by which the heart benefits from exercise, promoting the integration of basic research and clinical application, and has significant theoretical and practical significance.
2 Molecular regulatory mechanisms of exercise-induced cardiac functional improvement
2.1 Metabolic reprogramming
The heart, as a high-energy-demand organ, relies on metabolic homeostasis to maintain functional integrity. Under physiological conditions, 60%–90% of adenosine triphosphate (ATP) in adult cardiomyocytes is derived from fatty acid β-oxidation, while glucose metabolism contributes 10%–30%, with alternative substrates such as ketones and lactate becoming critical under specific physiological or pathological conditions (Lopaschuk et al., 2021; Kolwicz et al., 2013). Mitochondria, the primary ATP producers in cardiomyocytes, also serve as major sources of reactive oxygen species (ROS). Dysregulated mitochondrial dynamics contribute to myocardial injury and disease progression across multiple pathological models (Forte et al., 2021). The heart exhibits remarkable metabolic plasticity, dynamically adjusting substrate preferences in response to physiological demands or pathological states. Exercise-induced physiological remodeling is often accompanied by enhanced mitochondrial function. Long-term regular exercise can increase myocardial oxygen consumption by 3–10-fold, which elevates ADP concentration to enhance oxidative phosphorylation efficiency, ultimately leading to adaptive alterations characterized by enlarged mitochondrial volume and increased cristae density (Ritterhoff and Tian, 2023; Qiu et al., 2022; Abel and Doenst, 2011). This metabolic remodeling is closely associated with exercise modality and duration, with its molecular basis involving AMPK-PGC-1α signaling axis-mediated regulation of mitochondrial biogenesis (Qiu et al., 2022; Abel and Doenst, 2011; Li et al., 2018; White et al., 1987). Collectively, exercise training not only promotes metabolic adaptation toward higher efficiency in healthy hearts but also rehabilitates impaired myocardial mitochondrial energy production capacity and efficiency, thereby facilitating functional recovery in diseased hearts.
2.2 Regulation of oxidative stress
The core pathological mechanism underlying myocardial oxidative injury stems from disrupted redox homeostasis of ROS. Under physiological conditions, approximately 90% of ROS (e.g., superoxide anion O2•- and hydrogen peroxide H2O2) derive from the mitochondrial electron transport chain (ETC), with the remainder originating from enzymatic systems such as NADPH oxidase (NOX) and xanthine oxidase (Zhou and Tian, 2018). In pathological states (e.g., ischemia-reperfusion injury, hypertensive cardiac hypertrophy) or during aging, mitochondrial complex I/III dysfunction amplifies electron leakage from the, ETC, coupled with upregulated NOX2/4 activity. These synergistic effects drive ROS production rates that substantially exceed the scavenging capacity of endogenous antioxidant systems (e.g., superoxide dismutase SOD and glutathione peroxidase GPx), culminating in oxidative overload (Burgoyne et al., 2012). This redox imbalance triggers lipid peroxidation (e.g., elevated malondialdehyde MDA), protein carbonylation (e.g., 3-nitrotyrosine accumulation), and mitochondrial DNA oxidative damage (e.g., increased 8-hydroxy-2′-deoxyguanosine 8-OHdG) via Fenton reactions, ultimately promoting cardiomyocyte apoptosis and contractile dysfunction (Tsutsui et al., 2011).
Distinct exercise modalities differentially regulate myocardial antioxidant systems. Moderate-intensity continuous training (MICT) significantly enhances SOD1/2 activity and the GSH/GSSG (Glutathione and glutathione disulfide) ratio, mediated by exercise-induced Sirt3 (Sirtuin 3) deacetyation (Sundaresan et al., 2009). High-intensity interval training activates the HIF-1α/Nrf2 (Hypoxia-inducible factor 1α/Nuclear factor erythroid 2-like 2) synergistic pathway through transient hypoxia, substantially improving Nrf2 nuclear translocation efficiency (Gliemann et al., 2016). Resistance exercise promotes FoxO transcription factor phosphorylation via IGF-1/Akt signaling, driving SOD2 and CAT gene expression, though with minimal effects on GPx regulation (Konopka and Harber, 2014). A randomized controlled trial in coronary artery disease patients revealed that 12-week aerobic exercise (150 min/week, it is the minimum recommended by the ACSM in reference to exercise) increased Smyd1 (SET And MYND Domain Containing 1) expression by 1.8-fold in myocardial biopsy samples, accompanied by a 37% reduction in plasma malondialdehyde (MDA) levels and a 4.2% improvement in left ventricular ejection fraction (LVEF) (Hambrecht et al., 1993).
2.3 Regulation of programmed cell death
Programmed cell death (PCD) in cardiomyocytes—encompassing apoptosis, ferroptosis, and pyroptosis—constitutes a critical regulatory mechanism for maintaining cardiac homeostasis through distinct molecular cascades. Apoptosis, a caspase-dependent process, physiologically eliminates senescent or damaged cardiomyocytes but is pathologically activated by stimuli that elevate the Bax/Bcl-2 ratio, triggering mitochondrial permeability transition pore (mPTP) opening, cytochrome C release, and caspase-3 activation, ultimately causing irreversible contractile unit loss (Krijnen et al., 2002). Ferroptosis, an iron-dependent, lipid peroxidation-driven death modality, involves glutathione peroxidase 4 (GPX4) inactivation and mitochondrial cristae collapse (Stockwell et al., 2017). Disrupted myocardial iron homeostasis (e.g., free iron overload) catalyzes polyunsaturated fatty acid (PUFA) peroxidation via the Fenton reaction, compromising plasma membrane integrity. Exercise preconditioning suppresses ferroptosis in doxorubicin-induced cardiotoxicity by activating mitochondrial superoxide-dependent AMPKα2, which inhibits the p53-SLC7A11 axis and enhances ROS scavenging, concurrently downregulating ferroptosis markers (ACSL4, PTGS2) (Tadokoro et al., 2020). Chronic exercise further bolsters antioxidant defenses via the Nrf2/GPX4 pathway (Wang et al., 2022a). Pyroptosis, a caspase-1-dependent inflammatory death initiated by NLRP3 (NOD-like receptor family, pyrin domain containing-3) inflammasome activation, features gasdermin D (GSDMD)-mediated pore formation and IL-1β release (Jankowska et al., 2008). The ROS-NLRP3-IL-18 axis amplifies inflammation during myocardial ischemia-reperfusion injury, impairing contractility (Gopalan et al., 2021). Combined curcumin and exercise intervention in hyperlipidemic rats downregulates pyroptosis genes (NLRP3, ASC (Apoptosis-associated speck-like protein containing a CARD), caspase-1) by inhibiting TLR4/MyD88/NF-κB (Toll-like receptor 4/Myeloid differentiation factor 88/Nuclear factor kappa-B) signaling and reducing serum IL-1β (Ramos et al., 2016). While exercise alone partially mitigates pyroptosis (Yang et al., 2025), its direct mechanisms (e.g., inflammasome regulation) require validation via conditional knockout models.
2.4 Epigenetic regulation
Epigenetic modifications—including dynamic alterations in DNA methylation, non-coding RNAs, and histone modifications—orchestrate exercise-induced cardioprotection by remodeling myocardial gene expression in response to pathological stress. Exercise modulates cardiac DNA methylation through regulation of DNMTs and TET demethylases (Benito et al., 2021; Haeusler et al., 2013). In hypertensive cardiac hypertrophy, hypomethylation of the ACE promoter elevates ACE mRNA expression, augmenting angiotensin II (Ang II)-mediated fibrosis (Benito et al., 2021); conversely, 12-week aerobic exercise increases ACE promoter methylation and reduces plasma Ang II (Haeusler et al., 2013). Furthermore, aerobic exercise ameliorates myocardial ischemia-reperfusion injury by suppressing METTL3-mediated m6A methylation, thereby stabilizing cell death-related mRNAs (Zhang et al., 2025). MiRNA is currently regarded as a potential therapeutic target and biomarker in the study of various physiological and pathological processes in cardiovascular diseases (van Rooij and Olson, 2007). Exercise also remodels myocardial miRNA profiles (Soci et al., 2011). Aerobic training increases the expression of miR-29, reduces the expression and concentration of collagen genes in the heart, and promotes physiological myocardial hypertrophy (Soci et al., 2011). Where in upregulated miR-29b directly targets collagen genes (COL1A1/COL3A1/ELN) to attenuate post-infarction fibrosis (Melo et al., 2014), while exercise-induced HIF-1α activates miR-126, promoting angiogenesis via PI3K/AKT/eNOS and MAPK pathways in infarcted hearts (Song et al., 2020). Intermittent aerobic exercise can inhibit the TGFβ pathway by up-regulating the expression of miR-101a, ultimately leading to a reduction in cardiac tissue fibrosis and scar formation (Xiao et al., 2017). Systemically, exercise-stimulated skeletal muscle releases exosomal miR-126-3p that suppresses cardiomyocyte VCAM-1 expression, mitigating endothelial inflammation (Bei et al., 2017). In addition, physical exercise is increasingly recognized for its ability to regulate cardiac function by regulating histone modifications (Zheng et al., 2025). Lehmann et al. found that compared with healthy controls, failing hearts showed reduced levels of HDAC4 N-terminal fragment (HDAC4-NT), and exercise was proven to increase HDAC4-NT levels to protect cardiac function (Lehmann et al., 2018). Meanwhile, during exercise, AMPK in the myocardium is activated and phosphorylates HDAC4. This phosphorylation reduces the inhibitory effect of HDAC4 on MEF2a, and this change helps improve cardiac function and glucose metabolism in mice with heart failure (Jiang et al., 2020).
3 Cellular crosstalk
3.1 Cardiomyocyte-endothelial dialogue
Cardiomyocytes and cardiac microvascular endothelial cells (CMECs) form dynamic functional units through paracrine signaling, mechanical coupling, and metabolic interactions, collectively maintaining cardiac homeostasis. This bidirectional communication (cardiomyocyte-endothelial dialogue) critically regulates energy metabolism, redox balance, and pathological remodeling.
Nitric oxide (NO) suppresses excessive L-type calcium channel activation in cardiomyocytes via the cGMP/PKG pathway, reducing diastolic Ca2+ concentration and alleviating calcium overload-induced systolic dysfunction (Ramos et al., 2016). Pathological conditions (e.g., hypertension) induce endothelial endothelin-1 (ET-1) overexpression, which activates the cardiomyocyte CaMKII/NFATc3 pathway through ETA receptors, driving pathological hypertrophy (Al-Khatib et al., 2018).
Exercise enhances cardiomyocyte-endothelial communication by improving paracrine signaling. Exercise upregulates NRG-1 expression in endothelial progenitor cells (EPCs). Post-myocardial infarction exercise increases eNOS expression and promotes angiogenesis through the NRG-1/ErbB4/PI3K/AKT signaling pathway (Huang et al., 2025). Additionally, the mechanisms by which exercise strengthens cardiomyocyte-endothelial communication involve enhanced mechanical coupling. Exercise-induced cyclic shear stress upregulates endothelial Piezo1 expression and inhibits ET-1 production via AMPKα activation (Clarkson et al., 1999). In conclusion, exercise improves cardiac function by reinforcing cardiac-endothelial communication, promoting endothelial proliferation, and enhancing angiogenesis.
3.2 Immune cell regulation
Immune cells serve as pivotal regulators of cardiac homeostasis and disease progression through dynamic orchestration of inflammatory responses and repair processes. During acute myocardial injury, vascular endothelial upregulation of adhesion molecules and resident macrophage-derived chemokines recruit neutrophils and monocytes for necrotic clearance and inflammation resolution. Subacutely, amplified pro-inflammatory signaling induces mast cell release of pro-fibrotic factors, driving maladaptive fibrosis and dysfunction. Exercise exerts potent immunomodulatory effects by mobilizing circulatory lymphocytes/neutrophils and reprogramming cardiac immunity: (1) Post-ischemic DAMP-activated TLR4/MyD88/NF-κB signaling promotes M1 macrophage polarization and pro-apoptotic cytokine secretion (IL-1β, TNF-α) (Nahrendorf and Swirski, 2016), which exercise counteracts via PPARα-mediated inhibition of NF-κB nuclear translocation to reduce M1 dominance (Santos et al., 2016); (2) Exercise-induced IL-4/IL-13 activates STAT6 to drive M2 macrophage polarization, enhancing TGF-β1/VEGF-A-dependent collagen remodeling and angiogenesis (Lavine et al., 2014). Single-cell sequencing confirms significant enrichment of pro-repair genes (Arg1, Ym1) in cardiac macrophages of exercised subjects (Epelman et al., 2014). Collectively, exercise mitigates pathological remodeling by suppressing excessive immune activation while promoting macrophage phenotypic switching toward reparative M2 polarization.
4 Exercise-mediated organ dialogue
Exercise has been substantiated as an effective approach for primary prevention and adjunctive therapy of cardiovascular diseases, with its clinical value strongly supported by evidence-based medicine. Recent breakthroughs in molecular mechanism studies further elucidate its therapeutic potential. From a systems biology perspective, exercise intervention establishes multidimensional regulatory networks that induce cascade adaptive changes spanning subcellular structures to tissue and organ levels. Importantly, these biological effects exhibit remarkable inter-organ crosstalk. Mechanistic investigations reveal that exercise-mediated cardioprotection operates through multiple organ axes, including but not limited to the brain-heart axis, skeletal muscle-heart axis, liver-heart axis, adipose-heart axis, kidney-heart axis, and gut-heart axis (Figure 1).
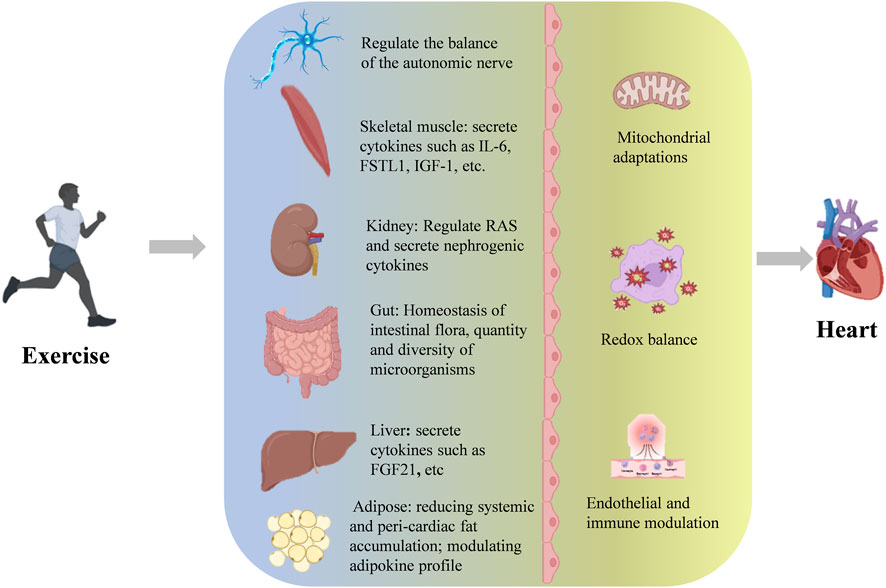
Figure 1. The multi-layered mechanisms by which exercise confers cardioprotection.
4.1 Autonomic nerve remodeling
4.1.1 Autonomic balance regulation
Cardiac autonomic regulation—sympathetic and parasympathetic balance—critically governs myocardial function, with its dysfunction being a primary driver of cardiac injury and heart failure (Floras, 2009). Pathological states (e.g., pressure overload) trigger hypothalamic paraventricular nucleus (PVN) glutamatergic neuron hyperexcitability, leading to chronic sympathetic overactivation characterized by excessive norepinephrine (NE) release and impaired reuptake (Florea and Cohn, 2014). While acutely compensatory, sustained NE excess exacerbates myocardial damage via β1-AR/cAMP/PKA-induced calcium overload and mitochondrial oxidative stress, accelerating ventricular remodeling (Floras, 2009). Exercise restores autonomic homeostasis through multi-tiered mechanisms: (1) Downregulating PVN angiotensin II type 1 receptor (AT1R) to suppress glutamatergic hyperactivity, improving ischemic cardiac function (Patel and Zheng, 2012); (2) Enhancing baroreceptor sensitivity while reducing β2-adrenergic receptor responsiveness to catecholamines (Fraga et al., 2007); (3) Restoring the adrenal GRK2-α2-AR-catecholamine axis to normalize sympathetic tone (Rengo et al., 2010). Conversely, impaired vagal activity reduces heart rate variability (HRV)—a biomarker of cardiac autonomic integrity (Billman et al., 2015)—which correlates with myocardial infarction and heart failure risk (Sessa et al., 2018; Hillebrand et al., 2013). Regular exercise elevates cardiac vagal tone, concurrently attenuating sympathetic activity and β2-AR sensitivity, thereby augmenting HRV and conferring cardioprotection in both healthy and diseased hearts (Routledge et al., 2010; Fiuza-Luces et al., 2018). In conclusion, exercise preserves and restores cardiac autonomic homeostasis, modulates HRV, and thereby confers cardioprotection.
4.1.2 Optimization of cardiovascular reflex regulation
The neural regulation of cardiac activity involves three primary reflex pathways: baroreflex, chemoreflex, and volume reflex. The baroreceptor reflex monitors blood pressure changes through the carotid sinus and aortic arch baroreceptors. When blood pressure rises, it reduces heart rate and cardiac output by enhancing vagal nerve activity, constituting an important mechanism for maintaining blood pressure homeostasis. Hypertensive patients generally have reduced baroreceptor sensitivity, and regular exercise not only lowers blood pressure but also effectively restores baroreflex sensitivity (Brum et al., 2000; Laterza et al., 2007), which has significant clinical implications for improving cardiac load. The chemoreceptor reflex is mainly activated by the carotid body and aortic body chemoreceptors when blood oxygen partial pressure drops, carbon dioxide partial pressure rises, and pH decreases, enhancing sympathetic nerve activity to increase heart rate and cardiac output. Notably, in chronic heart failure, chemoreceptor reflex sensitivity abnormally increases, leading to excessive sympathetic nerve activation and accelerating myocardial remodeling (Sun et al., 1999). Animal experiments have confirmed that exercise intervention can effectively inhibit the sensitivity of peripheral chemoreceptors in heart failure models (Li et al., 2008), providing a mechanistic explanation for the improvement of cardiac function through exercise rehabilitation. The volume receptor reflex monitors the circulatory volume status through atrial stretch receptors and cardiopulmonary pressure receptors. When blood volume increases, it reduces sympathetic nerve tension and enhances vagal nerve activity by inhibiting the renin-angiotensin-aldosterone system and increasing atrial natriuretic peptide secretion, thereby reducing cardiac output. In pathological conditions, weakened myocardial contractility leads to increased residual blood volume in the ventricle, and abnormally activated volume reflex may form a vicious cycle of “increased preload - reflexive cardiac function inhibition.” Exercise effectively regulate preload and afterload by promoting blood redistribution in skeletal muscles and improving venous return, possibly interrupting this pathological process. Existing evidence indicates that the multi-target regulation of cardiovascular reflexes by exercise is an important mechanism for its cardioprotective effect. However, the coordinated action mechanism of the three reflex pathways during exercise intervention, their temporal regulatory characteristics, and their dynamic balance relationship under pathological conditions still require systematic research. In particular, the differences in the effects of different exercise modes (intensity, duration) on each reflex pathway and the central integration mechanism are worthy of in-depth exploration.
4.1.3 Central neural remodeling mechanisms
The cardiac neural regulatory network exhibits multi-level characteristics, involving not only autonomic nerves and cardiovascular reflexes but also the “brain-heart” regulatory axis formed by the higher cortical-hypothalamic-brainstem pathways. Functional magnetic resonance imaging (fMRI) studies confirm that limbic systems (e.g., lateral prefrontal cortex, insular cortex, amygdala) form neural circuit connections with the heart via the hypothalamic PVN (Hu et al., 2023). Notably, the primary motor cortex (M1 region), traditionally associated with motor execution and cognitive functions (Levy et al., 2020; Shenoy et al., 2013; Bhattacharjee et al., 2021), has recently been found to exhibit anatomical connectivity with the heart (Li et al., 2021), Optogenetic experiments demonstrate that activating M1 glutamatergic neurons bidirectionally modulates heart rate and contractile function in both healthy and myocardial infarction (MI) mice (Bo et al., 2024).
Myocardial infarction-induced neural remodeling involves dual central and peripheral pathological alterations. MI enhances sympathetic excitatory input from the PVN to the rostral ventrolateral medulla (RVLM), forming a hyperactive “PVN-RVLM-sympathetic nerve” axis that directly causes abnormal β-adrenergic receptor density elevation and myocardial fibrosis (Koba et al., 2020; Wang et al., 2022b). Exercise intervention effectively suppresses sympathetic sprouting and catecholamine secretion by downregulating NADPH oxidase activity and inhibiting oxidative stress in the RVLM, demonstrating clear cardioprotective effects in MI animal models (Koba et al., 2014; Chen et al., 2014). Current evidence reveals that exercise improves cardiac autonomic balance by modulating multi-level central nodes in the “cortex-hypothalamus-brainstem” axis. However, critical questions remain unresolved: (1) hierarchical regulatory relationships among distinct brain regions in exercise-mediated cardioprotection; (2) temporal window characteristics of neural plasticity; (3) dose-response relationships between exercise intensity and neural remodeling effects. Particularly, the regulatory roles of non-motor cortical regions (e.g., insula, anterior cingulate cortex) in the “brain-heart” axis warrant further investigation.
4.2 Skeletal muscle-cardiac crosstalk
As the largest metabolic organ, skeletal muscle exerts its endocrine function as a pivotal mediator in exercise-induced cardioprotection. During exercise, skeletal muscle secretes over 650 bioactive myokines (Bay and Pedersen, 2020), which establish bidirectional communication with multiple organs (e.g., brain, cardiovascular system) via endocrine pathways, forming the systemic biological basis of exercise benefits (Severinsen and Pedersen, 2020). Notably, clinical experiment have shown that skeletal muscle-derived IL-6 exhibits unique anti-inflammatory properties in exercise physiology: while circulating IL-6 levels rise markedly during exercise, it induces monocytes to produce anti-inflammatory mediators (e.g., IL-1 receptor antagonist, IL-10) while suppressing the bioactivity of pro-inflammatory cytokines like TNF-α, creating a systemic anti-inflammatory milieu (Fiuza-Luces et al., 2018; Steensberg et al., 2003; Starkie et al., 2003). Animal experiments revealed that exercise-induced follistatin-like protein 1 (FSTL1) improve cardiac function after myocardial infarction through a comprehensive protective effect of inhibiting apoptosis, reducing fibrosis and promoting angiogenesis (Ouchi et al., 2008; Xi et al., 2021). Myokines such as irisin and insulin-like growth factor 1 (IGF-1) show significant cardioprotective effects in animal models of cardiac ischemia-reperfusion injury and pressure overload by regulating myocardial energy metabolism, enhancing antioxidant capacity and improving mitochondrial function (Ma et al., 2021; Li et al., 2024; Tan et al., 2023). Current evidence confirms that exercise regulates multiple pathophysiological processes (cardiac inflammatory microenvironment, angiogenesis, cell survival) through myokine cascades along the skeletal muscle-heart axis. However, critical gaps remain: (1) spatiotemporal secretion patterns of distinct myokines; (2) tissue-specific receptor distribution; (3) crosstalk between signaling pathways; and particularly; (4) dose-response relationships between exercise intensity and myokine secretion profiles require systematic investigation.
4.3 Kidney-heart crosstalk
The cardiorenal interaction plays a pivotal role in cardiovascular pathophysiology, classically exemplified by cardiorenal syndrome (CRS), where dysfunction of either the heart or kidneys triggers secondary injury in the other organ through neurohumoral regulation, hemodynamic alterations, and inflammatory responses (Ronco et al., 2010). Epidemiological studies reveal a significantly elevated cardiovascular mortality rate in chronic kidney disease (CKD) patients, with global CKD-related cardiovascular deaths reaching 1.8 million in 2021 (Martin et al., 2024). Pathologically, CKD-induced cardiac injury arises from a triad of mechanisms: (1) increased left ventricular end-diastolic pressure due to volume overload; (2) enhanced oxidative stress from uremic toxin accumulation; and (3) overactivation of the renin-angiotensin-aldosterone system (RAAS) (Chuasuwan and Kellum, 2012). Exercise ameliorates cardiorenal interactions through multiple pathways. Regular exercise reduces intraglomerular pressure and suppresses tubulointerstitial fibrosis progression, thereby decelerating CKD advancement (Shlipak et al., 2022; Yamakoshi et al., 2022). Exercise-induced enhanced sodium excretion and modified antidiuretic hormone sensitivity alleviate volume overload (Beetham et al., 2022). Studies demonstrate that exercise stimulates renal synthesis of ELABELA (ELA), a dual-organ protective peptide hormone. ELA activates the YAP-Akt-mTOR-P70S6K signaling network in cardiomyocytes, enhancing contractile reserve, promoting microvascular angiogenesis, and suppressing Ang II-induced pathological remodeling to preserve cardiac function (Zheng et al., 2021; Xi et al., 2023). Current evidence confirms that exercise intervention establishes a multi-target protective network against CRS by: (1) maintaining glomerular filtration function; (2) modulating RAAS activity; and (3) augmenting renoprotective factor secretion. However, critical gaps persist regarding: (1) temporal effects of exercise on cardiorenal crosstalk; (2) differential cardiorenal benefits across exercise modalities (endurance/resistance/high-intensity interval training); and (3) roles of other renally derived cytokines beyond ELA in exercise-mediated cardioprotection.
4.4 Gut-heart cross-talk
The gut microbiota, as the largest exogenous metabolic organ, dynamically regulates systemic homeostasis through microbiota-host co-metabolic networks. Microbial metabolites serve as chemical mediators of gut-organ axis communication and play vital roles in maintaining cardiovascular health (Nicholson et al., 2005; Dai et al., 2023). Gut dysbiosis impairs cardiac function via: (1) systemic inflammation triggered by pathogen-associated molecular pattern (PAMP) translocation; (2) pro-atherogenic metabolite production (e.g., trimethylamine N-oxide, TMAO); and (3) cholesterol homeostasis disruption caused by bile acid metabolism dysregulation. Exercise exerts significant regulatory effects on gut microbiota. It optimizes microbial composition (Kim and Kang, 2019) and enhances microbiome richness/diversity (Clarke et al., 2014). Notably, myocardial ischemia itself alters gut microbiota diversity. Both human and animal studies demonstrate post-myocardial infarction microbiome shifts (Tang et al., 2019; Liu et al., 2017), while exercise increases Butyricimonas and Akkermansia abundance, improving cardiac function in infarcted hearts (Liu et al., 2017). Mechanistically, exercise-induced microbial metabolic reprogramming generates cardioprotective molecules: 3-hydroxypyridinecarboxylic acid (3-HPA) and 4-hydroxybenzoic acid (4-HBA) activate the Nrf2-ARE pathway, reducing cardiomyocyte apoptosis by 42% and suppressing TGF-β/Smad3-mediated collagen deposition, ultimately limiting infarct size (Zhou et al., 2022). These findings suggest exercise protects ischemic hearts through dual mechanisms—microbiota structural optimization and functional metabolite production. However, critical gaps remain regarding: (1) exercise intensity-metabolite concentration gradients; (2) causal contributions of specific bacterial strains; and (3) tissue-specific delivery mechanisms of microbial metabolites.
4.5 Liver-heart crosstalk
Hepato-cardiac interactions play significant roles in interorgan pathophysiological communication. Common liver diseases may induce cardiac dysfunction (Correale et al., 2018). Exercise intervention disrupts this vicious cycle through multiple mechanisms. Exercise alleviates cirrhosis-associated cardiac remodeling and diastolic dysfunction (de Souza et al., 2021). At the molecular mechanism level, the core mediators of heart-liver interaction include: (1) Inflammatory signal cascade (Correale et al., 2018):Myocardial infarction induces inflammatory responses in the liver and leads to liver injury, while exercise-induced myogenic factor Irisin inhibits liver inflammatory responses and improves liver injury caused by myocardial infarction (Wang et al., 2023a). (2) Hepatogenic inducible factor: Hepatogenic Protein coagulation Factor XI (FXI) can activate the Bone Morphogenetic Protein (BMP)-Smad1/5 pathway in the heart, thereby inhibiting the genes involved in inflammation and fibrosis and protecting the cardiac function in heart failure (Cao et al., 2022). FGF21 is a cytokine mainly expressed by the liver and also an exercise-inducing factor. Exercise can maintain mitochondrial integrity through the FGF21-Sirtuin3 axis to protect cardiac function under pathological conditions (Jin et al., 2022). Exercise bidirectionally regulates hepato-cardiac crosstalk, attenuating cardiac injury through interorgan anti-inflammatory effects and direct myocardial protection via hepatokines. In heart failure, the ratio of phosphocreatine to ATP in the myocardium significantly decreases, leading to insufficient energy supply to the heart (Jullig et al., 2008). Exercise can induce the expression of myocardial FGF21 coreceptor β-klotho, promote the phosphorylation of FOXO3 through AMPK signaling, induce the expression of mitochondrial deacetylase Sirt3, promote the deacetylation of myocardial mitochondrial enzyme clusters to maintain mitochondrial integrity and function, and improve the efficiency of mitochondrial oxidative phosphorylation (Jin et al., 2022). Restore myocardial ATP levels and improve impaired cardiac function. During the rational remodeling process of heart disease, the preference for energy metabolism substrates shifts from fatty acid oxidation to glucose oxidation, resulting in a decrease in energy productivity (Gibb and Hill, 2018). Exercise can promote the expression of medium-chain acyl-coA dehydrogenase and 2, 4-dienyl-CoA reductase one in mitochondria, enhance the β -oxidation capacity of myocardial fatty acids, and improve the energy supply efficiency of the heart (Risikesan et al., 2023). However, unresolved questions include: (1) tissue-specific FGF21 regulation (liver vs. adipose) in response to exercise; (2) time-dose relationships between exercise intensity and FGF21 effects; and (3) roles of exercise-induced hepatic metabolites (e.g., bile acid derivatives) in the liver-heart axis.
4.6 Adipose-heart crosstalk
Obesity and metabolic syndrome are major cardiovascular risk factors, with adipose tissue playing a central role. Obesity induces systemic inflammation (Ghigliotti et al., 2014; Berg and Scherer, 2005), which paradoxically drives adipogenesis as an adaptive mechanism to prevent ectopic fatty acid deposition (Wernstedt Asterholm et al., 2014). Epicardial adipose tissue exhibits heightened adipogenic sensitivity compared to other visceral fat depots (Marchington and Pond, 1990). In obesity, epicardial fat acts as a sensor, mediating systemic inflammatory effects on myocardium akin to its adverse coronary impacts (Packer, 2018). Exercise reduces epicardial fat accumulation, mitigates oxidative stress/inflammation, and confers cardioprotection (Nyawo et al., 2021). Adipokines exhibit context-dependent cardiac effects: Leptin protects against cardiomyocyte hypertrophy/apoptosis under physiological conditions (Unger, 2005), but promotes adverse cardiovascular outcomes in obesity-related hyperleptinemia (Zhao et al., 2021). Exercise lowers hyperleptinemia and restores leptin’s cardioprotective effects (Lowndes et al., 2014). In summary, exercise combats adipose-mediated cardiac injury by: (1) reducing systemic and peri-cardiac fat accumulation; and (2) modulating adipokine profiles. However, mechanistic details of exercise-regulated leptin signaling and tissue-specific adipokine interactions require further elucidation.
5 Clinical translation opportunities and challenges
5.1 Exercise mimetics
Recent years have witnessed growing interest in bioactive oral compounds that mimic or amplify exercise benefits, termed “exercise mimetics” or “exercise pills.” These compounds aim to stimulate muscle adaptations akin to exercise, yet their capacity to fully replicate exercise effects remains contentious. Scholars argue that exercise-induced responses involve multifactorial, often redundant interactions among signaling kinases, downstream pathways, and spatiotemporal coordination, generating integrated adaptations to physiological challenges (Hsieh et al., 2025). Consequently, single-target interventions are unlikely to recapitulate the full exercise phenotype. Nevertheless, key regulatory nodes retain therapeutic potential. Over 100 myokines have been identified (Chow et al., 2022), though most remain functionally uncharacterized. Beyond myokines, muscle-derived metabolites during contraction also contribute to metabolic regulation, suggesting “exercise mimetics” may partially mimic metabolic benefits while neglecting multisystem adaptations (Hsieh et al., 2025; Hoffmann and Weigert, 2017). Future research should employ multi-omics technologies to delineate cardioprotective exercise factors, establish drug screening platforms based on exercise factor interactions, and develop targeted delivery systems for exercise-limited patients. Breakthroughs in these areas may bridge basic discoveries to clinical translation, advancing exercise mimetics from concept to application.
5.2 Personalized exercise prescription
Scientific exercise prescription adheres to the FITT-VP framework (Frequency, Intensity, Time, Type, Volume, Progression), which synergistically modulates energy metabolism, hemodynamics, and molecular signaling to determine pathophysiological outcomes. Given the stringent safety and efficacy requirements for cardiac exercise rehabilitation, careful consideration must be given to differential cardiac impacts associated with varying exercise parameters. Pandey et al. believe that there is a clear dose-response relationship between exercise and cardiovascular health benefits (Pandey et al., 2015). Zheng et al. demonstrated that individuals engaging in moderate and large exercise volumes exhibited superior cardiac structural parameters compared to those performing high-intensity exercise (Zheng et al., 2024). Zhang et al. found that the greatest benefits were gained when exercise was initiated in the acute phase after myocardial infarction, while the later the exercise intervention after myocardial infarction, the worse the exercise effect (Zhang et al., 2016). It is noteworthy that High-Intensity Interval Training (HIIT) demonstrates superior efficacy in suppressing pathological cardiac remodeling in patients with heart failure compared to aerobic exercise, resistance training, and combined training modalities (Wang et al., 2023b; Cornelis et al., 2016). Meanwhile, a higher training frequency (for example, more than twice a week) can better improve the endothelial function of patients with heart failure than a lower training frequency (for example, twice a week) (Fuertes-Kenneally et al., 2023). Pandey et al. observed that when the PA (Physical Activity) levels were 250 and 500 MET-min/wk, the risk of HF decreased by only 5% and 10%, respectively. However, individuals who engaged in physical activity at 1000 MET-min/wk and 4 times 2000 MET-min/wk had a 19% and 35% reduced risk of heart failure (Pandey et al., 2015). From this, it can be seen that exercise parameters are of vital importance to the effect of cardiac exercise rehabilitation. Although some basic research has compared the differences in the protective effects of different exercise parameters on the heart, in clinical practice, patients’ conditions, disease courses, and physical constitutions all vary. Therefore, the setting of exercise parameters needs to be considered appropriately based on the actual situation (Pei et al., 2023; Pei et al., 2021; Zhang et al., 2024; Sylviana et al., 2022). Future work should establish multidimensional parameter matrices encompassing exercise modalities and intervention windows, integrated with single-cell sequencing and metabolomics to map exercise parameter-molecular network-clinical outcome pathways. This paradigm may transcend empirical prescription, enabling precision cardiac rehabilitation.
Although exercise is regarded as a safe and effective remedy for preventing and treating cardiovascular diseases, its potential risks, contraindications and scientific avoidance strategies must be taken seriously. Patients with coronary heart disease who have not been evaluated may experience plaque rupture, acute myocardial infarction or malignant arrhythmias (such as ventricular fibrillation) during intense exercise. Long-term overexertion can lead to an increase in myocardial fibrosis markers, promote pathological myocardial hypertrophy, and at the same time, high-intensity endurance exercise increases the risk of atrial fibrillation (Patel and Link, 2025; Gerardin et al., 2021; Pandey et al., 2015). For patients with absolute contraindications such as unstable angina pectoris, severe aortic stenosis, acute myocarditis/pericarditis, etc., exercise should be strictly restricted. Patients with relative contraindications such as hypertension and severe arrhythmia need to undergo medical assessment before engaging in exercise. When experiencing chest pain/a feeling of oppression, dizziness, arrhythmia or shortness of breath during exercise, one must stop exercising immediately. In conclusion, only through scientific exercise can one maximize the benefits for the heart while avoiding risks.
5.3 Telemedicine-enabled cardiac rehabilitation
Digital transformation is driving a fourth medical revolution in cardiac rehabilitation, featuring wearable biosensors (e.g., Einthoven-style patch ECG), 5G telemedicine platforms, and deep learning-based early warning systems. Studies confirm that Internet of Things (IoT)-enabled home-based cardiac rehabilitation (HBCR) matches center-based programs in reducing major cardiovascular events and improving 6-min walk distance (Anderson et al., 2017; McDonagh et al., 2023). Multimodal data fusion enables real-time monitoring of exercise intensity, myocardial oxygen consumption, and arrhythmias, allowing dynamic prescription optimization. However, safety concerns persist for high-risk populations (Thomas et al., 2019), necessitating biomarker-based pre-event warning systems. Future efforts should integrate three tiers: (1) foundational research on predictive biomarkers; (2) technological innovation in remote monitoring; and (3) clinical implementation strategies. This “trinity” framework may shift cardiac rehabilitation from facility-dependent to intelligent, personalized paradigms (Figure 2).
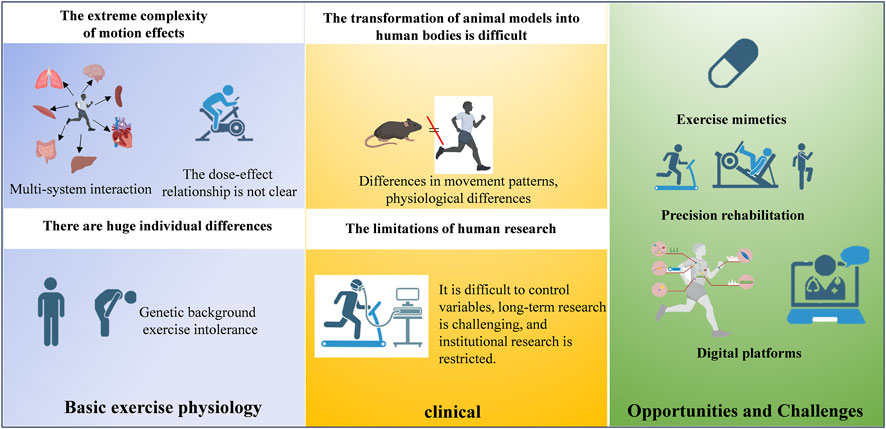
Figure 2. A diagram outlining the translational pipeline from basic exercise physiology to clinical implementation.
6 Conclusion
Exercise serves as a cornerstone intervention strategy for cardiovascular disease prevention and treatment, with its cardioprotective effects arising from the integration of multidimensional molecular regulatory networks and interorgan synergistic interactions. Although challenges persist in developing exercise mimetics due to target selectivity limitations and systemic complexity, multi-omics analysis of exercise factor networks provides new directions for precision drug design. Concurrently, intelligent upgrades in tele-rehabilitation technologies–particularly the integration of wearable devices with AI-based early warning systems–are facilitating the transition from empirical interventions to data-driven decision-making in cardiac care management.
Current research limitations primarily involve: (1) Insufficient quantitative characterization of dose-effect relationships between exercise parameters and molecular responses; (2) Unclear temporal window characteristics of interorgan communication signals and their interaction mechanisms within pathological microenvironments; (3) Incomplete understanding of maintenance and resolution mechanisms underlying exercise-induced epigenetic memory. Future studies should integrate single-cell spatiotemporal omics, optogenetic modulation, and organoid models to systematically elucidate the hierarchical architecture of exercise-activated cardioprotective networks, thereby establishing theoretical foundations for personalized cardiac rehabilitation strategies. Although the benefits of chronic exercise on physiology and molecular pathways have been established, there is still much to be discovered to establish better-designed clinical protocols and approaches (Figure 3).
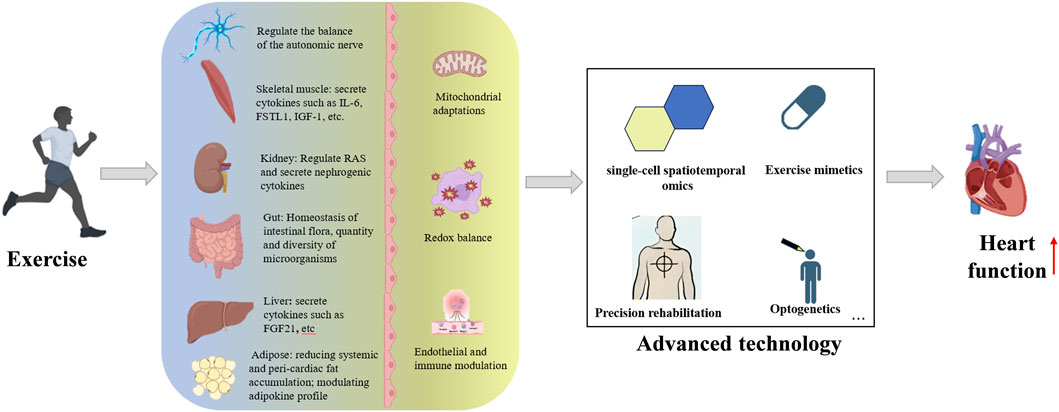
Figure 3. An integrative view of exercise promotes technological progress and heart health.
Author contributions
GY: Conceptualization, Funding acquisition, Investigation, Methodology, Resources, Writing – original draft, Writing – review and editing. JX: Conceptualization, Validation, Writing – original draft, Writing – review and editing. WT: Methodology, Writing – original draft, Writing – review and editing. SZ: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the social science fund of Fujian province Grants FJ2024A016 and FJ2025BF039 awarded to SZ and GY, respectively.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abel E. D., Doenst T. (2011). Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc Res. 90, 234–242. doi:10.1093/cvr/cvr015
AL-Khatib S. M., Stevenson W. G., Ackerman M. J., Bryant W. J., Callans D. J., Curtis A. B., et al. (2018). 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: executive summary: a report of the American college of cardiology/American Heart Association task force on clinical practice guidelines and the heart rhythm society. Circulation 138, e210–e271. doi:10.1161/CIR.0000000000000548
Anderson L., Sharp G. A., Norton R. J., Dalal H., Dean S. G., Jolly K., et al. (2017). Home-based versus centre-based cardiac rehabilitation. Cochrane Database Syst. Rev. 6, Cd007130. doi:10.1002/14651858.CD007130.pub4
Balady G. J., Ades P. A., Bittner V. A., Franklin B. A., Gordon N. F., Thomas R. J., et al. (2011). Referral, enrollment, and delivery of cardiac rehabilitation/secondary prevention programs at clinical centers and beyond: a presidential advisory from the American Heart Association. Circulation 124, 2951–2960. doi:10.1161/CIR.0b013e31823b21e2
Bay M. L., Pedersen B. K. (2020). Muscle-organ crosstalk: focus on immunometabolism. Front. Physiol. 11, 567881. doi:10.3389/fphys.2020.567881
Beetham K. S., Krishnasamy R., Stanton T., Sacre J. W., Douglas B., Isbel N. M., et al. (2022). Effect of a 3-Year lifestyle intervention in patients with chronic kidney disease: a randomized clinical trial. J. Am. Soc. Nephrol. 33, 431–441. doi:10.1681/asn.2021050668
Bei Y., Xu T., Lv D., Yu P., Xu J., Che L., et al. (2017). Exercise-induced circulating extracellular vesicles protect against cardiac ischemia-reperfusion injury. Basic Res. Cardiol. 112, 38. doi:10.1007/s00395-017-0628-z
Benito B., Martinez-Cordoba P. J., Guillamon M. D. (2021). Measurement and determinants of efficiency in the municipal police service. Eval. Program Plann 85, 101904. doi:10.1016/j.evalprogplan.2020.101904
Berg A. H., Scherer P. E. (2005). Adipose tissue, inflammation, and cardiovascular disease. Circ. Res. 96, 939–949. doi:10.1161/01.Res.0000163635.62927.34
Bhattacharjee S., Kashyap R., Abualait T., Annabel Chen S. H., Yoo W. K., Bashir S. (2021). The role of primary motor cortex: more than movement execution. J. Mot. Behav. 53, 258–274. doi:10.1080/00222895.2020.1738992
Billman G. E., Cagnoli K. L., Csepe T., Li N., Wright P., Mohler P. J., et al. (2015). Exercise training-induced bradycardia: evidence for enhanced parasympathetic regulation without changes in intrinsic sinoatrial node function. J. Appl. Physiol. 118, 1344–1355. doi:10.1152/japplphysiol.01111.2014
Bo W., Cai M., Ma Y., Di L., Geng Y., Li H., et al. (2024). Manipulation of glutamatergic neuronal activity in the primary motor cortex regulates cardiac function in normal and myocardial infarction mice. Adv. Sci. (Weinh) 11, e2305581. doi:10.1002/advs.202305581
Brum P. C., Da Silva G. J., Moreira E. D., Ida F., NegrãO C. E., Krieger E. M. (2000). Exercise training increases baroreceptor gain sensitivity in normal and hypertensive rats. Hypertension 36, 1018–1022. doi:10.1161/01.hyp.36.6.1018
Bull F. C., AL-Ansari S. S., Biddle S., Borodulin K., Buman M. P., Cardon G., et al. (2020). World Health organization 2020 guidelines on physical activity and sedentary behaviour. Br. J. Sports Med. 54, 1451–1462. doi:10.1136/bjsports-2020-102955
Burgoyne J. R., Mongue-Din H., Eaton P., Shah A. M. (2012). Redox signaling in cardiac physiology and pathology. Circ. Res. 111, 1091–1106. doi:10.1161/CIRCRESAHA.111.255216
Cao Y., Wang Y., Zhou Z., Pan C., Jiang L., Zhou Z., et al. (2022). Liver-heart cross-talk mediated by coagulation factor XI protects against heart failure. Science 377, 1399–1406. doi:10.1126/science.abn0910
Center for Cardiovascular Diseases the Writing Committee of the Report on Cardiovascular, H. and Diseases In CHINA, N (2024). Report on cardiovascular health and diseases in China 2023: an updated summary. Biomed. Environ. Sci. 37, 949–992. doi:10.3967/bes2024.162
Chen T., Cai M. X., Li Y. Y., He Z. X., Shi X. C., Song W., et al. (2014). Aerobic exercise inhibits sympathetic nerve sprouting and restores β-adrenergic receptor balance in rats with myocardial infarction. PLoS One 9, e97810. doi:10.1371/journal.pone.0097810
Chow L. S., Gerszten R. E., Taylor J. M., Pedersen B. K., VAN Praag H., Trappe S., et al. (2022). Exerkines in health, resilience and disease. Nat. Rev. Endocrinol. 18, 273–289. doi:10.1038/s41574-022-00641-2
Chuasuwan A., Kellum J. A. (2012). Cardio-renal syndrome type 3: epidemiology, pathophysiology, and treatment. Semin. Nephrol. 32, 31–39. doi:10.1016/j.semnephrol.2011.11.005
Clarke S. F., Murphy E. F., O'Sullivan O., Lucey A. J., Humphreys M., Hogan A., et al. (2014). Exercise and associated dietary extremes impact on gut microbial diversity. Gut 63, 1913–1920. doi:10.1136/gutjnl-2013-306541
Clarkson P., Montgomery H. E., Mullen M. J., Donald A. E., Powe A. J., Bull T., et al. (1999). Exercise training enhances endothelial function in young men. J. Am. Coll. Cardiol. 33, 1379–1385. doi:10.1016/s0735-1097(99)00036-4
Cornelis J., Beckers P., Taeymans J., Vrints C., Vissers D. (2016). Comparing exercise training modalities in heart failure: a systematic review and meta-analysis. Int. J. Cardiol. 221, 867–876. doi:10.1016/j.ijcard.2016.07.105
Correale M., Tarantino N., Petrucci R., Tricarico L., Laonigro I., Di Biase M., et al. (2018). Liver disease and heart failure: back and forth. Eur. J. Intern Med. 48, 25–34. doi:10.1016/j.ejim.2017.10.016
Dai Y., Sun Z., Zheng Y., Ge J. (2023). Recent advances in the gut microbiome and microbial metabolites alterations of coronary artery disease. Sci. Bull. (Beijing) 68, 549–552. doi:10.1016/j.scib.2023.03.009
De Souza S. L. B., Mota G. A. F., Gregolin C. S., Do Nascimento M., Luvizotto R. A. M., Bazan S. G. Z., et al. (2021). Exercise training attenuates cirrhotic cardiomyopathy. J. Cardiovasc Transl. Res. 14, 674–684. doi:10.1007/s12265-020-09997-0
Epelman S., Lavine K. J., Beaudin A. E., Sojka D. K., Carrero J. A., Calderon B., et al. (2014). Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40, 91–104. doi:10.1016/j.immuni.2013.11.019
Fiuza-Luces C., Santos-Lozano A., Joyner M., Carrera-Bastos P., Picazo O., Zugaza J. L., et al. (2018). Exercise benefits in cardiovascular disease: beyond attenuation of traditional risk factors. Nat. Rev. Cardiol. 15, 731–743. doi:10.1038/s41569-018-0065-1
Floras J. S. (2009). Sympathetic nervous system activation in human heart failure: clinical implications of an updated model. J. Am. Coll. Cardiol. 54, 375–385. doi:10.1016/j.jacc.2009.03.061
Florea V. G., Cohn J. N. (2014). The autonomic nervous system and heart failure. Circ. Res. 114, 1815–1826. doi:10.1161/circresaha.114.302589
Forte M., Schirone L., Ameri P., Basso C., Catalucci D., Modica J., et al. (2021). The role of mitochondrial dynamics in cardiovascular diseases. Br. J. Pharmacol. 178, 2060–2076. doi:10.1111/bph.15068
Fraga R., Franco F. G., Roveda F., De Matos L. N., Braga A. M., Rondon M. U., et al. (2007). Exercise training reduces sympathetic nerve activity in heart failure patients treated with carvedilol. Eur. J. Heart Fail 9, 630–636. doi:10.1016/j.ejheart.2007.03.003
Fuertes-Kenneally L., Manresa-Rocamora A., Blasco-Peris C., Ribeiro F., Sempere-Ruiz N., Sarabia J. M., et al. (2023). Effects and optimal dose of exercise on endothelial function in patients with heart failure: a systematic review and meta-analysis. Sports Med. Open 9, 8. doi:10.1186/s40798-023-00553-z
Gerardin B., Guedeney P., Bellemain-Appaix A., Levasseur T., Mustafic H., Benamer H., et al. (2021). Life-threatening and major cardiac events during long-distance races: updates from the prospective RACE PARIS registry with a systematic review and meta-analysis. Eur. J. Prev. Cardiol. 28, 679–686. doi:10.1177/2047487320943001
Ghigliotti G., Barisione C., Garibaldi S., Fabbi P., Brunelli C., Spallarossa P., et al. (2014). Adipose tissue immune response: novel triggers and consequences for chronic inflammatory conditions. Inflammation 37, 1337–1353. doi:10.1007/s10753-014-9914-1
Gibb A. A., Hill B. G. (2018). Metabolic coordination of physiological and pathological cardiac remodeling. Circ. Res. 123, 107–128. doi:10.1161/CIRCRESAHA.118.312017
Gliemann L., Nyberg M., Hellsten Y. (2016). Effects of exercise training and resveratrol on vascular health in aging. Free Radic. Biol. Med. 98, 165–176. doi:10.1016/j.freeradbiomed.2016.03.037
Gopalan V., Yaligar J., Michael N., Kaur K., Anantharaj R., Verma S. K., et al. (2021). A 12-week aerobic exercise intervention results in improved metabolic function and lower adipose tissue and ectopic fat in high-fat diet fed rats. Biosci. Rep. 41. doi:10.1042/BSR20201707
Haeusler R. A., Astiarraga B., Camastra S., Accili D., Ferrannini E. (2013). Human insulin resistance is associated with increased plasma levels of 12α-hydroxylated bile acids. Diabetes 62, 4184–4191. doi:10.2337/db13-0639
Hambrecht R., Niebauer J., Marburger C., Grunze M., Kalberer B., Hauer K., et al. (1993). Various intensities of leisure time physical activity in patients with coronary artery disease: effects on cardiorespiratory fitness and progression of coronary atherosclerotic lesions. J. Am. Coll. Cardiol. 22, 468–477. doi:10.1016/0735-1097(93)90051-2
Hillebrand S., Gast K. B., De Mutsert R., Swenne C. A., Jukema J. W., Middeldorp S., et al. (2013). Heart rate variability and first cardiovascular event in populations without known cardiovascular disease: meta-analysis and dose-response meta-regression. Europace 15, 742–749. doi:10.1093/europace/eus341
Hoffmann C., Weigert C. (2017). Skeletal muscle as an endocrine organ: the role of myokines in exercise adaptations. Cold Spring Harb. Perspect. Med. 7, a029793. doi:10.1101/cshperspect.a029793
Hou Z., Qin X., Hu Y., Zhang X., Li G., Wu J., et al. (2019). Longterm exercise-derived exosomal miR-342-5p: a novel exerkine for cardioprotection. Circ. Res. 124, 1386–1400. doi:10.1161/CIRCRESAHA.118.314635
Hsieh P. N., Shen S., Chukwurah M. I., Churchill T. W., Stewart K. M., Chung E. H., et al. (2025). Athlete's heart revisited: historical, clinical, and molecular perspectives. Circ. Res. 137, 231–254. doi:10.1161/CIRCRESAHA.125.325638
Hu J. R., Abdullah A., Nanna M. G., Soufer R. (2023). The brain-heart axis: neuroinflammatory interactions in cardiovascular disease. Curr. Cardiol. Rep. 25, 1745–1758. doi:10.1007/s11886-023-01990-8
Huang H., Huang G., Li R., Wei L., Yuan Z., Huang W. (2025). Exercise training after myocardial infarction enhances endothelial progenitor cells function via NRG-1 signaling. Cardiovasc Toxicol. 25, 411–426. doi:10.1007/s12012-025-09967-5
Jankowska E. A., Wegrzynowska K., Superlak M., Nowakowska K., Lazorczyk M., Biel B., et al. (2008). The 12-week progressive quadriceps resistance training improves muscle strength, exercise capacity and quality of life in patients with stable chronic heart failure. Int. J. Cardiol. 130, 36–43. doi:10.1016/j.ijcard.2007.07.158
Jiang H., Jia D., Zhang B., Yang W., Dong Z., Sun X., et al. (2020). Exercise improves cardiac function and glucose metabolism in mice with experimental myocardial infarction through inhibiting HDAC4 and upregulating GLUT1 expression. Basic Res. Cardiol. 115, 28. doi:10.1007/s00395-020-0787-1
Jin L., Geng L., Ying L., Shu L., Ye K., Yang R., et al. (2022). FGF21-Sirtuin 3 axis confers the protective effects of exercise against diabetic cardiomyopathy by governing mitochondrial integrity. Circulation 146, 1537–1557. doi:10.1161/circulationaha.122.059631
Jullig M., Hickey A. J., Chai C. C., Skea G. L., Middleditch M. J., Costa S., et al. (2008). Is the failing heart out of fuel or a worn engine running rich? A study of mitochondria in old spontaneously hypertensive rats. Proteomics 8, 2556–2572. doi:10.1002/pmic.200700977
Kim D., Kang H. (2019). Exercise training modifies gut microbiota with attenuated host responses to sepsis in wild-type mice. Faseb J. 33, 5772–5781. doi:10.1096/fj.201802481R
Koba S., Hisatome I., Watanabe T. (2014). Central command dysfunction in rats with heart failure is mediated by brain oxidative stress and normalized by exercise training. J. Physiol. 592, 3917–3931. doi:10.1113/jphysiol.2014.272377
Koba S., Hanai E., Kumada N., Watanabe T. (2020). Sympathoexcitatory input from hypothalamic paraventricular nucleus neurons projecting to rostral ventrolateral medulla is enhanced after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 319, H1197–h1207. doi:10.1152/ajpheart.00273.2020
Kolwicz S. C., Purohit S., Tian R. (2013). Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ. Res. 113, 603–616. doi:10.1161/circresaha.113.302095
Konopka A. R., Harber M. P. (2014). Skeletal muscle hypertrophy after aerobic exercise training. Exerc Sport Sci. Rev. 42, 53–61. doi:10.1249/JES.0000000000000007
Krijnen P. A., Nijmeijer R., Meijer C. J., Visser C. A., Hack C. E., Niessen H. W. (2002). Apoptosis in myocardial ischaemia and infarction. J. Clin. Pathol. 55, 801–811. doi:10.1136/jcp.55.11.801
Krishnan V., Huang X., Perak A. M., Coresh J., Ndumele C. E., Greenland P., et al. (2025). Discordance of 10- and 30-Year predicted risk for cardiovascular disease in US adults. JAMA 333, 1828–1831. doi:10.1001/jama.2025.2868
Laterza M. C., De Matos L. D., Trombetta I. C., Braga A. M., Roveda F., Alves M. J., et al. (2007). Exercise training restores baroreflex sensitivity in never-treated hypertensive patients. Hypertension 49, 1298–1306. doi:10.1161/hypertensionaha.106.085548
Lavine K. J., Epelman S., Uchida K., Weber K. J., Nichols C. G., Schilling J. D., et al. (2014). Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. U. S. A. 111, 16029–16034. doi:10.1073/pnas.1406508111
Lehmann L. H., Jebessa Z. H., Kreusser M. M., Horsch A., He T., Kronlage M., et al. (2018). A proteolytic fragment of histone deacetylase 4 protects the heart from failure by regulating the hexosamine biosynthetic pathway. Nat. Med. 24, 62–72. doi:10.1038/nm.4452
Levy S., Lavzin M., Benisty H., Ghanayim A., Dubin U., Achvat S., et al. (2020). Cell-type-specific outcome representation in the primary motor cortex. Neuron 107, 954–971.e9. doi:10.1016/j.neuron.2020.06.006
Li Y. L., Ding Y., Agnew C., Schultz H. D. (2008). Exercise training improves peripheral chemoreflex function in heart failure rabbits. J. Appl. Physiol. 105, 782–790. doi:10.1152/japplphysiol.90533.2008
Li F. H., Li T., Su Y. M., Ai J. Y., Duan R., Liu T. C. (2018). Cardiac basal autophagic activity and increased exercise capacity. J. Physiol. Sci. 68, 729–742. doi:10.1007/s12576-018-0592-x
Li Z., Li Z., Xu W., Li Y., Wang Q., Xu H., et al. (2021). The connectome from the cerebral cortex to the viscera using viral transneuronal tracers. Am. J. Transl. Res. 13, 12152–12167.
Li H., Qin S., Tang J., Wang T., Ren W., Di L., et al. (2024). Resistance exercise upregulates Irisin expression and suppresses myocardial fibrosis following myocardial infarction via activating AMPK-Sirt1 and inactivating TGFβ1-Smad2/3. Acta Physiol. (Oxf) 240, e14163. doi:10.1111/apha.14163
Liu Z., Liu H. Y., Zhou H., Zhan Q., Lai W., Zeng Q., et al. (2017). Moderate-intensity exercise affects gut microbiome composition and influences cardiac function in myocardial infarction mice. Front. Microbiol. 8, 1687. doi:10.3389/fmicb.2017.01687
Lopaschuk G. D., Karwi Q. G., Tian R., Wende A. R., Abel E. D. (2021). Cardiac energy metabolism in heart failure. Circ. Res. 128, 1487–1513. doi:10.1161/circresaha.121.318241
Lowndes J., Zoeller R. F., Kyriazis G. E., Miles M. P., Seip R. L., Moyna N. M., et al. (2014). Hyperleptinemia is associated with CRP, but not apolipoprotein E, and is reduced by exercise training. Int. J. Sport Nutr. Exerc Metab. 24, 524–531. doi:10.1123/ijsnem.2013-0200
Ma Y., Kuang Y., Bo W., Liang Q., Zhu W., Cai M., et al. (2021). Exercise training alleviates cardiac fibrosis through increasing fibroblast growth factor 21 and regulating TGF-β1-Smad2/3-MMP2/9 signaling in mice with myocardial infarction. Int. J. Mol. Sci. 22, 12341. doi:10.3390/ijms222212341
Marchington J. M., Pond C. M. (1990). Site-specific properties of pericardial and epicardial adipose tissue: the effects of insulin and high-fat feeding on lipogenesis and the incorporation of fatty acids in vitro. Int. J. Obes. 14, 1013–1022.
Martin S. S., Aday A. W., Almarzooq Z. I., Anderson C. A. M., Arora P., Avery C. L., et al. (2024). 2024 heart disease and stroke statistics: a report of US and global data from the American heart association. Circulation 149, e347–e913. doi:10.1161/CIR.0000000000001209
Mcdonagh S. T., Dalal H., Moore S., Clark C. E., Dean S. G., Jolly K., et al. (2023). Home-based versus centre-based cardiac rehabilitation. Cochrane Database Syst. Rev. 10, Cd007130. doi:10.1002/14651858.CD007130.pub5
Melo S. F., Fernandes T., Barauna V. G., Matos K. C., Santos A. A., Tucci P. J., et al. (2014). Expression of MicroRNA-29 and collagen in cardiac muscle after swimming training in myocardial-infarcted rats. Cell Physiol. Biochem. 33, 657–669. doi:10.1159/000358642
Nahrendorf M., Swirski F. K. (2016). Abandoning M1/M2 for a network model of macrophage function. Circ. Res. 119, 414–417. doi:10.1161/CIRCRESAHA.116.309194
Nicholson J. K., Holmes E., Wilson I. D. (2005). Gut microorganisms, Mammalian metabolism and personalized health care. Nat. Rev. Microbiol. 3, 431–438. doi:10.1038/nrmicro1152
Nyawo T. A., Pheiffer C., Mazibuko-Mbeje S. E., Mthembu S. X. H., Nyambuya T. M., Nkambule B. B., et al. (2021). Physical exercise potentially targets epicardial adipose tissue to reduce cardiovascular disease risk in patients with metabolic diseases: oxidative stress and inflammation emerge as major therapeutic targets. Antioxidants (Basel) 10, 1758. doi:10.3390/antiox10111758
Ouchi N., Oshima Y., Ohashi K., Higuchi A., Ikegami C., Izumiya Y., et al. (2008). Follistatin-like 1, a secreted muscle protein, promotes endothelial cell function and revascularization in ischemic tissue through a nitric-oxide synthase-dependent mechanism. J. Biol. Chem. 283, 32802–32811. doi:10.1074/jbc.M803440200
Packer M. (2018). Epicardial adipose tissue may mediate deleterious effects of obesity and inflammation on the myocardium. J. Am. Coll. Cardiol. 71, 2360–2372. doi:10.1016/j.jacc.2018.03.509
Pandey A., Garg S., Khunger M., Darden D., Ayers C., Kumbhani D. J., et al. (2015). Dose-response relationship between physical activity and risk of heart failure: a meta-analysis. Circulation 132, 1786–1794. doi:10.1161/CIRCULATIONAHA.115.015853
Patel N. S., Link M. S. (2025). Risk of Running-A contemporary look at cardiac arrest in long-distance races. JAMA 333, 1674–1675. doi:10.1001/jama.2025.3646
Patel K. P., Zheng H. (2012). Central neural control of sympathetic nerve activity in heart failure following exercise training. Am. J. Physiol. Heart Circ. Physiol. 302, H527–H537. doi:10.1152/ajpheart.00676.2011
Pei Z., Yang C., Guo Y., Dong M., Wang F. (2021). Effect of different exercise training intensities on age-related cardiac damage in male mice. Aging (Albany NY) 13, 21700–21711. doi:10.18632/aging.203513
Pei Z., Zhou R., Yao W., Dong S., Liu Y., Gao Z. (2023). Different exercise training intensities prevent type 2 diabetes mellitus-induced myocardial injury in male mice. iScience 26, 107080. doi:10.1016/j.isci.2023.107080
Qiu Y., Pan X., Chen Y., Xiao J. (2022). Hallmarks of exercised heart. J. Mol. Cell Cardiol. 164, 126–135. doi:10.1016/j.yjmcc.2021.12.004
Ramos J. S., Dalleck L. C., Ramos M. V., Borrani F., Roberts L., Gomersall S., et al. (2016). 12 min/week of high-intensity interval training reduces aortic reservoir pressure in individuals with metabolic syndrome: a randomized trial. J. Hypertens. 34, 1977–1987. doi:10.1097/HJH.0000000000001034
Rengo G., Leosco D., Zincarelli C., Marchese M., Corbi G., Liccardo D., et al. (2010). Adrenal GRK2 lowering is an underlying mechanism for the beneficial sympathetic effects of exercise training in heart failure. Am. J. Physiol. Heart Circ. Physiol. 298, H2032–H2038. doi:10.1152/ajpheart.00702.2009
Risikesan J., Heeboll S., Kumarathas I., Funck K. L., Sondergaard E., Johansen R. F., et al. (2023). Exercise increases myocardial free fatty acid oxidation in subjects with metabolic dysfunction-associated fatty liver disease. Atherosclerosis 372, 10–18. doi:10.1016/j.atherosclerosis.2023.03.015
Ritterhoff J., Tian R. (2023). Metabolic mechanisms in physiological and pathological cardiac hypertrophy: new paradigms and challenges. Nat. Rev. Cardiol. 20, 812–829. doi:10.1038/s41569-023-00887-x
Ronco C., Mccullough P., Anker S. D., Anand I., Aspromonte N., Bagshaw S. M., et al. (2010). Cardio-renal syndromes: report from the consensus conference of the acute dialysis quality initiative. Eur. Heart J. 31, 703–711. doi:10.1093/eurheartj/ehp507
Routledge F. S., Campbell T. S., Mcfetridge-Durdle J. A., Bacon S. L. (2010). Improvements in heart rate variability with exercise therapy. Can. J. Cardiol. 26, 303–312. doi:10.1016/s0828-282x(10)70395-0
Santos M. H., Higuchi Mde L., Tucci P. J., Garavelo S. M., Reis M. M., Antonio E. L., et al. (2016). Previous exercise training increases levels of PPAR-alpha in long-term post-myocardial infarction in rats, which is correlated with better inflammatory response. Clin. (Sao Paulo) 71, 163–168. doi:10.6061/clinics/2016(03)08
Schopfer D. W., Whooley M. A., Allsup K., Pabst M., Shen H., Tarasovsky G., et al. (2020). Effects of home-based cardiac rehabilitation on time to enrollment and functional status in patients with ischemic heart disease. J. Am. Heart Assoc. 9, e016456. doi:10.1161/JAHA.120.016456
Sessa F., Anna V., Messina G., Cibelli G., Monda V., Marsala G., et al. (2018). Heart rate variability as predictive factor for sudden cardiac death. Aging (Albany NY) 10, 166–177. doi:10.18632/aging.101386
Severinsen M. C. K., Pedersen B. K. (2020). Muscle-organ crosstalk: the emerging roles of myokines. Endocr. Rev. 41, 594–609. doi:10.1210/endrev/bnaa016
Shenoy K. V., Sahani M., Churchland M. M. (2013). Cortical control of arm movements: a dynamical systems perspective. Annu. Rev. Neurosci. 36, 337–359. doi:10.1146/annurev-neuro-062111-150509
Shlipak M. G., Sheshadri A., Hsu F. C., Chen S. H., Jotwani V., Tranah G., et al. (2022). Effect of structured, moderate exercise on kidney function decline in sedentary older adults: an ancillary analysis of the LIFE study randomized clinical trial. JAMA Intern Med. 182, 650–659. doi:10.1001/jamainternmed.2022.1449
Soci U. P., Fernandes T., Hashimoto N. Y., Mota G. F., Amadeu M. A., Rosa K. T., et al. (2011). MicroRNAs 29 are involved in the improvement of ventricular compliance promoted by aerobic exercise training in rats. Physiol. Genomics 43, 665–673. doi:10.1152/physiolgenomics.00145.2010
Song W., Liang Q., Cai M., Tian Z. (2020). HIF-1α-induced up-regulation of microRNA-126 contributes to the effectiveness of exercise training on myocardial angiogenesis in myocardial infarction rats. J. Cell Mol. Med. 24, 12970–12979. doi:10.1111/jcmm.15892
Starkie R., Ostrowski S. R., Jauffred S., Febbraio M., Pedersen B. K. (2003). Exercise and IL-6 infusion inhibit endotoxin-induced TNF-alpha production in humans. Faseb J. 17, 884–886. doi:10.1096/fj.02-0670fje
Steensberg A., Fischer C. P., Keller C., MøLLER K., Pedersen B. K. (2003). IL-6 enhances plasma IL-1ra, IL-10, and cortisol in humans. Am. J. Physiol. Endocrinol. Metab. 285, E433–E437. doi:10.1152/ajpendo.00074.2003
Stockwell B. R., Friedmann Angeli J. P., Bayir H., Bush A. I., Conrad M., Dixon S. J., et al. (2017). Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. doi:10.1016/j.cell.2017.09.021
Sun S. Y., Wang W., Zucker I. H., Schultz H. D. (1999). Enhanced peripheral chemoreflex function in conscious rabbits with pacing-induced heart failure. J. Appl. Physiol. 86, 1264–1272. doi:10.1152/jappl.1999.86.4.1264
Sundaresan N. R., Gupta M., Kim G., Rajamohan S. B., Isbatan A., Gupta M. P. (2009). Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Invest 119, 2758–2771. doi:10.1172/JCI39162
Sylviana N., Goenawan H., Susanti Y., Lesmana R., Megantara I., Setiawan S. (2022). Effect of different intensities aerobic exercise to cardiac angiogenesis regulation on Wistar rats. Pol. J. Vet. Sci. 25, 119–128. doi:10.24425/pjvs.2022.140848
Tadokoro T., Ikeda M., Ide T., Deguchi H., Ikeda S., Okabe K., et al. (2020). Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 5, e132747. doi:10.1172/jci.insight.132747
Tan Y., Feng P., Feng L., Shi L., Song Y., Yang J., et al. (2023). Low-dose exercise protects the heart against established myocardial infarction via IGF-1-upregulated CTRP9 in male mice. MedComm 4, e411. doi:10.1002/mco2.411
Tang T. W. H., Chen H. C., Chen C. Y., Yen C. Y. T., Lin C. J., Prajnamitra R. P., et al. (2019). Loss of gut microbiota alters immune system composition and cripples Postinfarction cardiac repair. Circulation 139, 647–659. doi:10.1161/circulationaha.118.035235
Thomas R. J., Beatty A. L., Beckie T. M., Brewer L. C., Brown T. M., Forman D. E., et al. (2019). Home-based cardiac rehabilitation: a scientific statement from the American association of cardiovascular and pulmonary rehabilitation, the American heart association, and the American college of cardiology. Circulation 140, e69–e89. doi:10.1161/cir.0000000000000663
Tsutsui H., Kinugawa S., Matsushima S. (2011). Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 301, H2181–H2190. doi:10.1152/ajpheart.00554.2011
Tucker W. J., Fegers-Wustrow I., Halle M., Haykowsky M. J., Chung E. H., Kovacic J. C. (2022). Exercise for primary and secondary prevention of cardiovascular disease: JACC focus seminar 1/4. J. Am. Coll. Cardiol. 80, 1091–1106. doi:10.1016/j.jacc.2022.07.004
Unger R. H. (2005). Hyperleptinemia: protecting the heart from lipid overload. Hypertension 45, 1031–1034. doi:10.1161/01.Hyp.0000165683.09053.02
VAN Rooij E., Olson E. N. (2007). MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J. Clin. Invest 117, 2369–2376. doi:10.1172/JCI33099
Wang H. T., Yang W. Q., Liu Y. Q. (2022a). Effects of aerobic exercise on Nrf2/GPX4/Ferroptosis pathway in myocardial injury in high-fat diet mice. Zhongguo Ying Yong Sheng Li Xue Za Zhi 38, 143–148. doi:10.12047/j.cjap.6234.2022.026
Wang K., You S., Hu H., Li X., Yin J., Shi Y., et al. (2022b). Effect of TLR4/MyD88/NF-kB axis in paraventricular nucleus on ventricular arrhythmias induced by sympathetic hyperexcitation in post-myocardial infarction rats. J. Cell Mol. Med. 26, 2959–2971. doi:10.1111/jcmm.17309
Wang T., Yu M., Li H., Qin S., Ren W., Ma Y., et al. (2023a). FNDC5/Irisin inhibits the inflammatory response and mediates the aerobic exercise-induced improvement of liver injury after myocardial infarction. Int. J. Mol. Sci. 24, 4159. doi:10.3390/ijms24044159
Wang T., Zhang L., Cai M., Tian Z. (2023b). Effects of different exercise modalities on inhibiting left ventricular pathological remodeling in patients with heart failure with reduced ejection fraction: a systematic review and network meta-analysis. Life Sci. 319, 121511. doi:10.1016/j.lfs.2023.121511
Wang J., Liu S., Meng X., Zhao X., Wang T., Lei Z., et al. (2024). Exercise inhibits doxorubicin-induced cardiotoxicity via regulating B cells. Circ. Res. 134, 550–568. doi:10.1161/CIRCRESAHA.123.323346
Wernstedt Asterholm I., Tao C., Morley T. S., Wang Q. A., Delgado-Lopez F., Wang Z. V., et al. (2014). Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 20, 103–118. doi:10.1016/j.cmet.2014.05.005
White F. C., Mckirnan M. D., Breisch E. A., Guth B. D., Liu Y. M., Bloor C. M. (1987). Adaptation of the left ventricle to exercise-induced hypertrophy. J. Appl. Physiol. 62, 1097–1110. doi:10.1152/jappl.1987.62.3.1097
XI Y., Hao M., Liang Q., Li Y., Gong D. W., Tian Z. (2021). Dynamic resistance exercise increases skeletal muscle-derived FSTL1 inducing cardiac angiogenesis via DIP2A-Smad2/3 in rats following myocardial infarction. J. Sport Health Sci. 10, 594–603. doi:10.1016/j.jshs.2020.11.010
XI Y., Li Y., Ren W., Bo W., Ma Y., Pan S., et al. (2023). ELABELA-APJ-Akt/YAP signaling axis: a novel mechanism of aerobic exercise in cardioprotection of myocardial infarction rats. Med. Sci. Sports Exerc 55, 1172–1183. doi:10.1249/mss.0000000000003143
Xiao L., He H., Ma L., Da M., Cheng S., Duan Y., et al. (2017). Effects of miR-29a and miR-101a expression on myocardial interstitial collagen generation after aerobic exercise in myocardial-infarcted rats. Arch. Med. Res. 48, 27–34. doi:10.1016/j.arcmed.2017.01.006
Yamakoshi S., Nakamura T., Xu L., Kohzuki M., Ito O. (2022). Exercise training ameliorates renal oxidative stress in rats with chronic renal failure. Metabolites 12, 836. doi:10.3390/metabo12090836
Yang B., Xu J., Dao X., Huang Y., Liang J., Huang J., et al. (2025). Aerobic exercise and PI3K inhibitor ameliorate obesity cardiomyopathy by alleviating pyroptosis in middle-aged mice. Int. J. Mol. Sci. 26, 4935. doi:10.3390/IJMS26104935
Zhang Y. M., Lu Y., Tang Y., Yang D., Wu H. F., Bian Z. P., et al. (2016). The effects of different initiation time of exercise training on left ventricular remodeling and cardiopulmonary rehabilitation in patients with left ventricular dysfunction after myocardial infarction. Disabil. Rehabil. 38, 268–276. doi:10.3109/09638288.2015.1036174
Zhang Y., Zhao Y., Song R., Tai W. (2024). Effects of different exercise intensities on the rat model of heart failure. Int. Heart J. 65, 713–722. doi:10.1536/ihj.24-154
Zhang X., Huang D. X., Xuan C., Li Y., Jiang Y., Wu X., et al. (2025). Aerobic exercise training attenuates ischemia-reperfusion injury in mice by decreasing the methylation level of METTL3-associated m6A RNA in cardiomyocytes. Life Sci. 361, 123294. doi:10.1016/j.lfs.2024.123294
Zhao S., Kusminski C. M., Scherer P. E. (2021). Adiponectin, leptin and cardiovascular disorders. Circ. Res. 128, 136–149. doi:10.1161/circresaha.120.314458
Zheng Q., Tian G., Xu F., Ci X., Luan R., Wu L., et al. (2021). The role of elabela in kidney disease. Int. Urol. Nephrol. 53, 1851–1857. doi:10.1007/s11255-021-02790-1
Zheng P., Li H., Kong N. (2024). The effect of different forms and amounts of exercise on cardiac structureand function (in Chinese). Chin. J. Ultrasound Med. 40, 44–47. doi:10.3969/j.issn.1002-0101.2024.01.014
Zheng X., Liu X., Guo Y., Lv Y., Lin C., Wang D., et al. (2025). Physical exercise and epigenetic modifications in skeletal muscle, brain, and heart. Epigenetics Chromatin 18, 12. doi:10.1186/s13072-025-00576-8
Zhou B., Tian R. (2018). Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Invest 128, 3716–3726. doi:10.1172/JCI120849
Keywords: cardiac rehabilitation, integrative physiology, exercise rehabilitation, cardiovascular, aerobic exercise
Citation: You G, Xie J, Tong W and Zhao S (2025) The new perspective of cardiac exercise rehabilitation: based on integrative physiology. Front. Physiol. 16:1651589. doi: 10.3389/fphys.2025.1651589
Received: 22 June 2025; Accepted: 12 August 2025;
Published: 21 August 2025.
Edited by:
Alex Cleber Improta-Caria, University of São Paulo, BrazilReviewed by:
Ursula Soci, School of Physical Education and Sports of Sao Paulo University, BrazilGuilherme Da Silva Rodrigues, Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo, Brazil
Leôncio Soares, Universidade Federal de Viçosa, Brazil
Copyright © 2025 You, Xie, Tong and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shaocong Zhao, c2hjemhhb0B4bXV0LmVkdS5jbg==