 Leonardo Máximo Cardoso
Leonardo Máximo Cardoso Richard David Wainford
Richard David Wainford- 1Department of Biological Sciences, Institute of Exact and Biological Sciences, Federal University of Ouro Preto, Ouro Preto, Brazil
- 2Department of Cardiology, School of Medicine, Emory University, Atlanta, GA, United States
Systemic arterial hypertension (SAH) is a prevalent condition affecting humans and other mammals, with high salt intake recognized as a major risk factor for its development and progression. This review examines the intricate interplay between dietary salt, immune signaling, neural regulation and renal mechanisms in the pathophysiology of salt-sensitive hypertension (SSH). High salt consumption not only directly influences blood pressure but also induces low-grade inflammation by activating both innate and adaptive immune responses, particularly promoting pro-inflammatory T cell (TH17/IL-17) and macrophage phenotypes. These immune alterations impact key organs involved in blood pressure regulation, including the kidneys and central nervous system (CNS). In the CNS, salt-induced immune activation, especially microglial activation and cytokine production in regions such as the paraventricular nucleus, enhances sympathetic outflow and contributes to neurogenic hypertension. Disruption of the blood-brain barrier further facilitates immune cell infiltration and perpetuates neuroinflammation. Additionally, recent evidence shows that high salt intake alters the gut microbiome, reducing its diversity and favoring pro-inflammatory bacterial populations, which further amplify immune dysregulation via the gut-grain axis. The role of the kidneys in sodium handling is modulated by immune cell infiltration and cytokine-drive changes in sodium channel expression, reinforcing salt sensitivity. Aging and sex differences further modulate these pathways, increasing SSH risk in older individuals and postmenopausal women. Emerging therapeutic strategies targeting the gut microbiota, immune signaling, and neural pathways offer promise improvement for SAH management. However, further research is needed to clarify causal mechanisms and optimize interventions that address the neural-immune-microbiome axis in hypertension.
1 Introduction
Systemic arterial hypertension (SAH), characterized by a persistent elevation in blood pressure (BP), is a clinical condition that transcends the human species, affecting various mammalian species. Hypertension is the single largest risk factor for numerous diseases, including ischemic brain infarction, intracranial hemorrhage, myocardial infarction, renal diseases, and congestive heart failure (APA, 2020; WHO, 2012; Frame and Wainford, 2017). It has been widely documented that intrinsic factors like genetic background, sex and age (Nist et al., 2024; Kim et al., 2024; Demirci et al., 2025) as well as extrinsic factor like smoking (Groppelli et al., 1992), alcohol (Lima et al., 1999), sedentarism (Alvarez et al., 1999), and eating habits (Yamamoto-Kimura et al., 1996) are associated with the development of SAH. Among eating habits, salt intake plays a key role in the development of SAH (Grillo et al., 2019) and will be further discussed here.
High salt intake can fulfill physiological and homeostatic functions, particularly when the individual sodium balance is low and body sodium content must be replenished. Under this condition, the high salt intake is driven by an increase in sodium appetite, which results in a behavioral state in which the individual seeks and consumes higher than normal salt amounts, also called need-induced sodium appetite (Geerling and Loewy, 2008). Curiously, mammals also exhibit high sodium intake behavior which is not driven by physiological needs for body sodium replenishment but for what is understood as a hedonic stimulus termed need-free sodium appetite (Geerling and Loewy, 2008). For the purpose of this review, we will focus on the need-free sodium intake and its impact on the BP regulation and contributions to hypertension.
High salt intake has long been associated with “stiffen and harden pulse”, a condition first described by Chinese physician Huang Di´s Neijing Swen, about 2500 BCE (Bailey and Dhaun, 2024; Ha, 2014). However, it was only in the beginning of the 20th century that the association between high salt intake and hypertension was established by Ambard and Beaujard using modern scientific methodology and standards (Ambard and Beaujard, 1904). Since then, several experimental and trial studies have supported these observations and provided evidence for the impact that high salt intake has on the pathophysiology of hypertension. In the 1940´s and 1950´s, studies by Kempner showed that the BP in 2 of the 6 patients under a strict rice fruit diet declined to normal levels and rose again after 20 g of sodium chloride was added daily (Grollman et al., 1945). In essence, this observation gave birth to the idea that would be later elaborated into the concept of the salt-sensitivity of BP (SSBP). The SSBP can be currently defined as a trait where the individual BP (animal or human) displays a change parallel to the changes in salt intake (Eli et al., 2016; Weinberger, 1996a; Weinberger, 1996b; Weinberger, 2006). The SSBP is a trait normally distributed in humans and is present in both the normotensive and hypertensive individuals. An important and influential American study showed that after salt depletion and following sodium load, 26% of the normotensive patients were ascribed as salt sensitive while 51% of the hypertensive patients were salt sensitive (Weinberger et al., 1986). Despite limitations (Bailey and Dhaun, 2024) this study established that hypertensive patients have higher probability of displaying a SSBP phenotype and launched the basis for the understanding that salt sensitivity is an important cardiovascular risk factor “independent of and as powerful as BP” by itself (Bailey and Dhaun, 2024; Eli et al., 2016).
Over the last 100 years scientific investigations have repeatedly demonstrated that renal pressure-natriuresis, hormonal hydroelectrolytic balance and neural sympathetic mediated events are key to the SSBP (Bie and Evans, 2017a; Bie and Evans, 2017b). These studies have revealed intricate interactions among different systems to sustain high BP and other detrimental outcomes of high salt intake. Of particular interest in this context is a growing body of evidence that has linked the immune cellular and signaling mechanisms not only to hypertension but also to the SSBP (Bailey and Dhaun, 2024; Mattson, 2014; Mattson, 2019; Rucker et al., 2018; Maaliki et al., 2022) and experimental and clinical studies led to the idea that hypertension may be an “inflammatory” disease (Boos and Lip, 2006; Solak et al., 2016). Although the immune events that take place along hypertension development and maintenance hold some characteristics of an “inflammatory state” such as heightened immune-related signaling molecules in end-organs linked to BP control and hypertension, it is crucial to acknowledge that this concept may deviate from the conventional definition of inflammation. Classically, inflammation is defined as a protective process by which the body’s immune system responds to injury, infection, or harmful stimuli. The four typical signs of acute inflammation were first described by Celsus in ancient Rome (30–38 B.C.) (Chandrasoma and Taylor, 1998) and are defined as: Rubor (redness), Calor (heat), Tumor (swelling) and Dolor (pain). Those signs are not always present in end-organs associated with the development of hypertension and make one wonder whether “inflammation” truthy describes the role of the immune system in the development of hypertension. However, some authors have elaborated a new concept to describe a chronic, subclinical, sterile and systemic condition characterized by a persistent, mild elevation of inflammatory markers in the body (Minihane et al., 2015; Ronnback and Hansson, 2019; van de Vyver, 2023) that would better represent the role of the immune system in hypertension. This concept is termed “low-grade inflammation” (LGI) or “metabolic inflammation” or “metainflammation” (van de Vyver, 2023) and has been implicated in the pathogenesis of many non-communicable chronic diseases, including salt-sensitive hypertension (SSH) (Boos and Lip, 2006; Al-Nimer and Alhusseiny, 2016).
Based on the currently available experimental evidence, this review will explore some aspects of the intricate interplay between the immune system and its pro-inflammatory signaling molecules (PIM), the central nervous system (CNS) and the renal system in SSH.
2 The immune system and the salt-sensitivity of blood pressure: an overview
The role of the immune system in BP regulation and hypertension development became evident with pharmacological studies in rodents showing that, under certain conditions, the inhibition or lacking of cyclooxygenase-1, a key enzyme in the prostanoids synthesis pathway, can lower the BP of Ang II-salt hypertensive rats (Asirvatham-Jeyaraj et al., 2013; Athirakul et al., 2001). In addition, a 2007 study by Guzik and cols. Showed that activation of immune cells contributed to the hypertension induced by angiotensin II (Ang II) infusion and deoxycorticosterone acetate (DOCA) plus high-salt diet in Rag1−/− mutant mouse (Guzik et al., 2007). This particular study showed that the knockout mice for Rag1 lack mature T cells and B cells, which protected them from vascular dysfunction and remodeling in addition to reducing the hypertensive responses to Ang II and DOCA-salt (Guzik et al., 2007). Clinical studies also showed that patients with hypertension had a higher proportion of immunosenescent, proinflammatory, and cytotoxic CD8+ T cells in their blood. Furthermore, immunohistochemical assessment of renal biopsy specimens from patients with hypertensive nephrosclerosis revealed increased expression of the T-cell chemokine interferon (IFN)-inducible T-cell α chemoattractant in the proximal and distal tubules suggesting that T-cell-driven inflammation may also play a role in human hypertension, especially in the kidney (Youn et al., 2013).
One of the most important risk factors for hypertension, high salt intake, is found to alter the activation of inflammatory cells in both the innate and adaptive immune system (Afsar et al., 2018; Miyauchi et al., 2024; Li et al., 2022). Murine macrophages and T cells exposed to high salt have been found to express a pro-inflammatory state both, in vitro and in vivo (Binger et al., 2015; Jantsch et al., 2015; Kleinewietfeld et al., 2013; Wu et al., 2013; Zhang WC. et al., 2015). Additionally, a modest increase in extracellular salt concentration induces the serum glucocorticoid kinase 1 (SGK1) expression, a serine/threonine kinase 4 that promotes IL-23R expression by deactivating the transcription factor Forkhead box protein O1 (FOXO1) in mice. FOXO1 is a direct repressor of IL-23R expression and enhances the IL-17-producing T helper lymphocyte cells (TH17) differentiation in vitro and in vivo through IL-23 mechanisms (Wu et al., 2013). The TH17 polarization of naïve immune cells has been reported in both humans and mice (Miyauchi et al., 2024; Kleinewietfeld et al., 2013; Jorg et al., 2016; Wilck et al., 2017; Yakoub et al., 2024). Also, high salt concentrations in the medium can increase the production of PIM like TNF-α, IL-2 and granulocyte-macrophage colony-stimulating factor by cultured TH17 cells (Kleinewietfeld et al., 2013). Moreover, the SGK1 activation has also been shown to stimulate the mineralocorticoid-mediated expression of epithelial sodium channels (ENaC) in the kidneys of mice (Wulff et al., 2002) what indicates that the SGK1 pathway is shared by inflammatory and sodium homeostasis mechanisms.
High salt also impacts macrophage core functions both in vitro and in vivo, especially because macrophages exhibit chemotactic migration in response to salt gradients in vitro (Muller et al., 2024). The incubation of bone marrow-derived macrophages with high concentrations of sodium chloride (NaCl) elicited a strong pro-inflammatory phenotype characterized by enhanced pro-inflammatory cytokines (PIC) production, increased expression of immune-stimulatory molecules, and an antigen-independent boost of T cell proliferation through pathways that may involve the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and the mitogen-activated protein kinase (MAPK) signaling (Hucke et al., 2016). Additional evidence shows a direct action of NaCl in inducing pro-inflammatory phenotypes. Zhang and cols. Showed that a roughly 33% increase in medium NaCl concentration significantly induced interleukin 6 (IL-6) and MCP-1 (or CCL2) production by retinal pigment epithelium (ARPE-19) cells in culture (Zhang D. et al., 2015). This effect was not mediated by osmolarity, as an equivalent osmotic mannitol addition to the medium had no effect on IL-6 or MCP-1 production (Zhang D. et al., 2015). In addition to the NF-κB and MAPK pathways, evidence from humans and experimental animal studies have shown that myeloid-specific janus kinase 2 (JAK2) also contributes to LGI and SSH. The JAK2 signaling pathway plays crucial roles in many cellular processes and physiological functions such as cell growth and survival, hematopoiesis, immune regulation, inflammation, metabolism and gene transcription (Bader and Meyer, 2022). High salt upregulated gene expression of the JAK/STAT/SMAD pathway in human monocytes and the ablation of JAK2 signaling attenuated the SSH hypertension in mice (Saleem et al., 2024). The authors also found that JAK2 increased production of highly reactive isolevuglandins (IsoLG) and IL-6 by renal antigen presenting cells (APC). In addition, JAK2 also activates T cells and increases production of IL-6, interleukin 17A (IL-17A), and tumor necrosis factor-alpha (TNF-α) in mice (Saleem et al., 2024).
Although the evidence suggests that sodium can drive inflammation-related signaling pathways in immune cells, increased salt intake actually produces very small Cohen effect size in steady-state plasma sodium concentrations of certain models of SSH, such as the Dahl-salt-sensitive model (DSS) (Stocker, 2023; Nakamura and Cowley, 1989) or the HS12W model (Gomes et al., 2017). This poses some limitation to the general idea that high salt intake can affect immune cell physiology. It also may suggest that the activation of the immune system by high salt intake may include an indirect result of increased sodium “traffic” throughout the body. Indeed, assessment of serum sodium concentrations in humans showed only a modest (∼1.4%) increase in systemic serum sodium concentration after a high salt meal, lasting at least 2 h, and without changes in hormones involved with body fluid homeostasis or endothelial function, such as endothelin-1 (ET-1), vasopressin (AVP), and atrial natriuretic peptide (ANP) (Dickinson et al., 2014). One important question here yet to be answered is whether this transient increase in sodium concentration of the extracellular fluid could be enough to unleash important changes in immune cells phenotype toward a pro-inflammatory state and contribute to the LGI over time? Since sodium absorption by the intestine doesn´t distribute throughout body volume at once, the great gradient of sodium through the intestine wall could make these local immune cells more susceptible to sodium-driven changes in phenotype and cytokines profile production as described in in vitro studies. On the other hand, recent evidence has shown that high salt intake can result in non-osmotic sodium accumulation in the skin (Titze et al., 2003) and muscle tissue (Rossitto et al., 2020) pointing toward the possibility that immune cells in these regions could also be impacted by high sodium gradients and contribute to systemic LGI.
In addition to salt-induced immune cell differentiation and activation, sodium can enter into dendritic cells (DC) via the amiloride-sensitive epithelial sodium channel (ENaC) and activate the production of the PIM IL-1β and promote T cell production of IL-17A and IFN-γ (Barbaro et al., 2017). It has been proposed that higher sodium influx through ENaC in DC increases sodium/calcium exchange, thus activating phosphokinase C (PKC) and NADPH-oxidase (Barbaro et al., 2017). The increased reactive oxygen species production drives the production of IsoLG and the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome that activates caspase-1 and increases the production of IL-1β by the DC (Pitzer et al., 2022). Interesting, when such activated DC are adoptively transferred into naïve mice, they prime hypertension in response to a sub-pressor dose of Ang II (Barbaro et al., 2017). These experimental findings are further supported by a study showing that amiloride exerts anti-inflammatory effects by decreasing the PMI TNF and IL-6 plasma levels, but not IL-17A levels, in patients with resistant hypertension and type 2 diabetes (Thangaraj et al., 2024).
From a vascular perspective, transient high salt intake promotes T-cell-mediated hypertensive vascular injury in a mice model of high-salt intake drinking 1% NaCl solution (Yakoub et al., 2024). The authors showed that in transient high salt-treated hypertensive mice, the aortic injury was associated with increased inflammation, accumulation of neutrophils, monocytes, CD69+CD4+ T cells, as well as CD4+ and CD8+ memory T cells. Moreover, high salt intake sensitized the animals for angiotensin II-induced vascular lesions by increasing the expression of aortic RORγt as well as splenic CD4+TH17 and CD8+TC1 T cells in Ang II-treated mice (Yakoub et al., 2024). The authors suggested that transient high salt intake induces a subclinical T-cell-mediated aortic immune response, which is enhanced by Ang II and contributes to the vascular lesion in these animals.
Collectively, these data strongly indicate that sodium can deeply affect the physiology of immune cells toward a LGI state, which strongly correlates with SSH. Certain pieces of evidence suggest that sodium-induced modifications of immune cells physiology have a causal relationship with hypertension and further studies are needed to fully understand the role of the immune signaling system in the pathophysiology of hypertension.
2.1 Key points
1. The immune system influences BP regulation and the development of hypertension.
2. High salt intake triggers a pro-inflammatory state in both innate and adaptive immune systems affecting macrophages and T cells and promoting the production of pro-inflammatory mediators.
3. Sodium can modify macrophage physiology leading to pro-inflammatory phenotypes, increased production of PIM (like IL-6 and MCP-1), immune-stimulatory molecules, and activation of NF-κB and MAPK signaling pathways.
4. Sodium influx through ENaC in dendritic cells can induce production of IL-1β through inflammasome activation and promote the IL-17A and INF-γ production by T cells.
3 Salt-sensitive hypertension and the central nervous system immune system
The central nervous system (CNS) plays a pivotal role in the BP regulation, both in normotension and hypertension (Guyenet, 2006) and this concept has been grounded on several findings in the literature showing that neurogenic mechanisms are important for heightened BP and, especially, for SSH (Eli et al., 2016; Maaliki et al., 2022; Adams et al., 2008). The major findings in this section are summarized in Table 1.
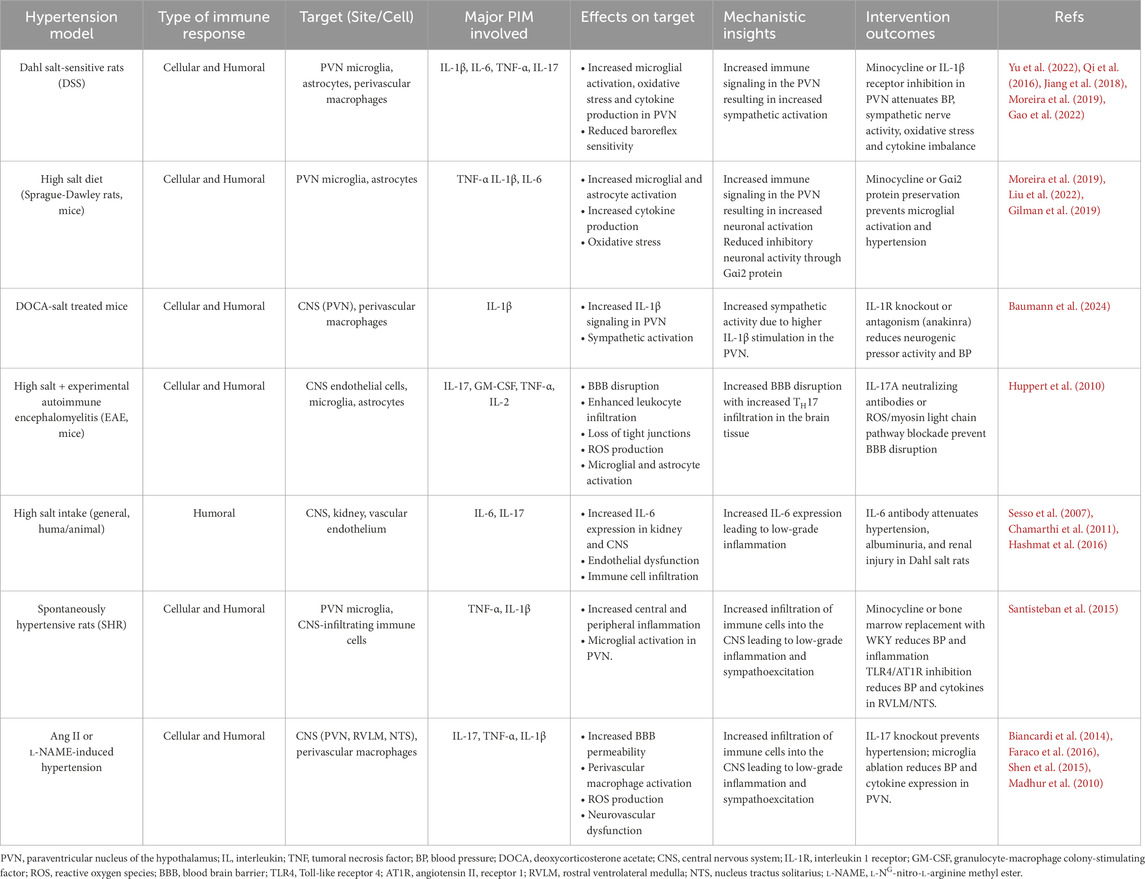
Table 1. Known effects of the immune system on neurogenic blood pressure control that contribute to salt sensitive hypertension and the outcomes of interventions.
The reflex control of BP regulation has been widely recognized for its ability to short-term buffer large BP fluctuations and prevent potentially harmful sudden increases or decreases in systemic BP (Guyenet, 2006; Wehrwein and Joyner, 2013). In SSH, salt-dependent increases in BP are usually accompanied by reduced baroreflex sensitivity (Ozaykan et al., 2017; Rosa et al., 2020; Wang et al., 2005). In addition to salt-related baroreflex impairments, excessive salt intake also leads to impairment in the reflex response to cardiopulmonary reflex activation. These reflexes are volume/chemical sensing mechanisms that regulate sympathetic and parasympathetic outflow to the cardiovascular system in response to volume expansion and/or chemical activation with metabolic byproducts and neurochemicals (Vasquez et al., 1997; Vasquez, 1994). In Dahl salt rats, a high salt diet sensitized the cardiopulmonary reflex-driven sympathetic response to hypervolemia in Dahl salt resistant rats but not in Dahl salt sensitive rats (Victor et al., 1986). Interestingly, Dahl salt sensitive rats had a smaller sympathetic inhibition response when challenged with volume expansion compared to Dahl salt resistant rats, even under low-salt diet (Ferrari A. et al., 1984; Ferrari AU. et al., 1984). These findings suggest that the salt-sensitivity trait by itself includes a reduced sensitivity of the cardiopulmonary reflex in Dahl salt sensitive rats regardless of high salt intake. Since these studies were conducted in urethane-anesthetized rats, the full understanding of the cardiopulmonary reflex role in SSH regulation remains limited. However, it provides important evidence of changes in short-term BP regulation due to the salt-sensitive trait. The plasticity of the cardiopulmonary reflex has also been investigated in hypertensive humans and, consistent with experimental findings, the results revealed that high salt intake potentiates the cardiopulmonary reflex gain and atrial natriuretic factor response only in salt resistant hypertensive patients (Trimarco et al., 1991). Interestingly, the reflex response to carotid baroreceptor unloading was unaffected by salt loading in none of the groups (Trimarco et al., 1991) suggesting that cardiopulmonary reflexes are more sensitive than baroreflex to high salt intake in salt resistant hypertensive patients.
The paraventricular nucleus of the hypothalamus (PVN) is a hypothalamic nucleus that orchestrates neural and hormonal responses to changes in blood sodium concentration, osmolality and BP through two distinct cell subtypes: the parvocellular neurons, which comprise autonomic regulatory neurons (Ferguson et al., 2008; Osborn et al., 2007), and the magnocellular neurons, which include oxytocin and vasopressin producing/containing neurons (Antunes-Rodrigues et al., 2004). Therefore, the PVN is a key forebrain region that plays important roles in neurogenic hypertension (Osborn et al., 2007). While the majority of descending sympathetic-related projections originating from PVN parvocellular neurons connect to the rostral ventral lateral medulla (RVLM) in the brainstem or to the intermedial lateral column in the spinal cord (Guyenet, 2006; Dampney et al., 2018), the majority of the projections from magnocellular PVN neurons project to the neurohypophysis and release vasopressin and oxytocin to the pituitary portal system when activated (Antunes-Rodrigues et al., 2004). The outcome is a multifaceted role of the PVN in regulating BP levels and sodium excretion by the kidney which contributes to the pathophysiology of SSH.
The immune signaling mechanisms in hypothalamic and brainstem regions controlling BP have received considerable attention in the recent years and the evidence gathered so far point toward important immune influence on the functional role of these regions in the SSH. The blockade of microglia activation with minocycline, a selective CNS-acting non-steroidal anti-inflammatory drug, in Dahl salt-sensitive rats can inhibit the augmented local production of IL-1β, IL-6, TNF-α and the oxidative stress in the PVN. Likewise, minocycline infusion in the PVN also attenuated the high BP, the ratio between resting RSNA/MaxRSNA, central prostaglandin E2 and plasm norepinephrine levels (Yu et al., 2022). The infusion of the IL-1β receptor inhibitor gevokizumab into the PVN of Dahl salt-sensitive rats also attenuated the BP, heart rate, and plasma norepinephrine levels (Qi et al., 2016) suggesting that this specific cytokine plays a role in the PVN-driven neurogenic pressor response. The authors also reported increased levels of NOX-2, NOX-4, IL-1β, NLRP3, Fra-LI and lower levels of IL-10 in Dahl salt-sensitive rats fed high salt diet and that gevokizumab restored the balance in the PVN (Qi et al., 2016). In addition, Jiang and cols. (2018) reported that Dahl salt-sensitive rats under high salt diet exhibited higher expression of PIC like TNF-α, IL-6, IL-1β and Fra1in the PVN and that intracerebroventricular infusion of highs sodium solution produced a marked increase in the expression of TNF-α, IL-6, IL-1β in Sprague-Dawley rats (Jiang et al., 2018). These studies highlight the role of high sodium concentrations in the PVN and its correlation to increased PIC production as well as the effect of the local anti-inflammatory agent minocycline or the IL-1β receptor inhibitor gevokizumab on SSH.
Although extensive research has advanced our understanding of immune signaling in the PVN regarding SSH, the role of immune signaling in the brainstem and its relationship to SSH is less advanced. It is known, however, that physiological reflex activation of the sympathetic circuit by bilateral carotid occlusion decreases the plasma levels of TNF and IL-1β, and increased the levels of IL-10 in endotoxemia induced by lipopolysaccharide in rats (Brognara et al., 2021a). Likewise, the activation of the Bezold-Jarisch reflex (the chemosensitive cardiopulmonary reflex) in endotoxemic rats reduced plasma levels of TNF and spleen levels of IL-6 (Brognara et al., 2021b). Although this evidence suggests that the autonomic activation of descending vagal pathways may influence peripheral PIM production, it does not provide further insights on how PIM can influence reflex regulation of the BP in the brainstem. On the other hand, toll-like receptor 4 and angiotensin II receptor 1 inhibition reduces BP as well as IL-6 and TNF-α protein density in the RVLM and nucleus tractus solitarius (NTS) of spontaneous hypertensive rats corroborating the idea that immune signaling in neuronal circuitries controlling BP is important for the hypertension development (Mowry et al., 2021).
3.1 Peripheral immune cell infiltration in the CNS and hypertension
Peripheral immune cell infiltration in the CNS has been associated with enhanced sympathetic drive, resulting in BP increase, whereas inhibition of this immune signaling in the CNS ameliorates hypertension (Nist et al., 2024; Moreira et al., 2019; Xue et al., 2016; Yu et al., 2010). This has been demonstrated by a study in which chimeric spontaneously hypertensive rats (SHR) reconstituted with Wistar-Kyoto (WKY) bone marrow resulted in significant BP reduction associated with attenuation of both central and peripheral immune activation (Santisteban et al., 2015). The authors also reported elevated BP and increased central and peripheral “inflammation” in chimeric WKY rats reconstituted with SHR bone marrow (Santisteban et al., 2015). When microglia activation was pharmacologically inhibited with minocycline, hypertension was attenuated in SHR (Santisteban et al., 2015). These findings strongly suggest that extravasation of bone-marrow-derived cells into the CNS, particularly into the PVN, is an important event in hypertension.
The infiltration of circulating immune cells into the CNS is believed to be associated with increased permeability of the blood-brain barrier (BBB) (Dinh et al., 2025). Hypertension has been shown to be linked to increased BBB permeability in the PVN, NTS and RVLM regions (Nist et al., 2024; Biancardi et al., 2014; Ueno et al., 2004). However, a causal relation between increased BBB permeability and hypertension is yet to be determined. Such an increase in the BBB permeability has been shown to allow systemic Ang II to enter into the nervous tissue parenchyma thus activating AT1 receptor on perivascular macrophages (PVM) within the brain (Biancardi et al., 2014; Faraco et al., 2016). The activation of PVM promotes pathogenic actions in key cardiovascular controlling regions of the brain to set neurovascular dysfunction through reactive oxygen species (ROS) production via the superoxide producing enzyme NOX2 during chronic hypertension (Faraco et al., 2016). Interestingly, the autonomic nervous system (SNA) also innervates bone marrow (Katayama et al., 2006) and spleen (Carnevale et al., 2016), and increased sympathetic outflow can stimulate mobilization and release of hematopoietic stem cells into blood stream through adrenergic neurotransmission stimulation (Hanoun et al., 2015; Sant et al., 2016). Sympathetic stimulation of bone marrow has been shown to favor enhanced proinflammatory responses in a mature innate immune system (Harwani et al., 2012). Selective ablation of splenic nerve prevents T cell egression from spleen and infiltration into renal and aorta tissue and protects against hypertension (Carnevale et al., 2016). This interplay between sympathetic drive and peripheral immune cell stimulation has the potential to contribute to a positive feedback loop in which more peripheral cells can infiltrate the CNS and stimulate local immune signaling allowing further sympathetic stimulation and, ultimately, aggravates the hypertension over time. However, increased sympathetic drive is a common finding in individuals with hypertension already established and raises the question of whether hypertension-related LGI is cause or consequence of high BP. For instance, unilateral renal sympathetic denervation, an approach which reduces BP, diminishes inflammatory cells activation in experimental models of hypertension (Xiao et al., 2015) and in human hypertensive patients (Zaldivia et al., 2017). One limitation here is that clinical studies could not discriminate between direct and indirect outcomes from renal denervation regarding direct nerve ablation effects on BP versus indirect effect resulting from attenuation of innate immunity on BP.
3.2 Microglia and salt-sensitive hypertension
The local production of pro- and anti-inflammatory molecules by microglia has also been reported as an important source of immune regulation of BP in SSH (Moreira et al., 2019; Shen et al., 2015; Shi et al., 2010). Increased activation of microglial has been implicated in cardiovascular disease and hypertension development. Microglial, characterized by their branched morphology, are tissue-resident macrophages of the CNS, and play pivotal role in monitoring the presence of pathogens in the CNS tissue and modulating synaptic and neuronal activities (Wang et al., 2022). Microglia activation states are based on the peripheral macrophages classification and include the M1 and M2 states in addition to the resting state M0 (Wang et al., 2022; Dubbelaar et al., 2018). Microglia activation is accompanied by morphological shifting from a small soma cell with ramified protrusions to an amoeboid-like morphology that enables microglia motility and phagocytic function (Dubbelaar et al., 2018; Kettenmann et al., 2011). This transition involves a complex interplay of different molecular and cellular mechanisms that include NF-κB signaling (Singh et al., 2022). Current literature on the molecular mechanisms leading to microglia activation in SSH primarily focuses on hypertension in a general manner and lacks specific aspects pertaining to the SSBP. In addition to the pattern recognition receptors expressed by microglial cells like the Toll-like receptors (TLR2, TLR3 TLR4 and TLR9) that can activate microglial cells (Olson and Miller, 2004), pro-inflammatory cytokines like IL-1β (Swaroop et al., 2016), IL-6 (Recasens et al., 2021), TNF-α (Bras et al., 2020), and INF-γ (Olson and Miller, 2004) can also activate microglial cells. Given the involvement of IL-1β, the factors that also activate NLRP3 inflammasome, as discussed above (Pitzer et al., 2022; Menu and Vince, 2011), may significantly contribute to microglia activation in response to high salt exposure. Interesting, the levels of specific immune signaling molecules can polarize resting microglia to the M1 or M2 states and, therefore, drive transition toward pro or anti-inflammatory phenotypes of active microglia. For instance, the signaling by LPS and INF-γ drive the microglia polarization toward the M1 phenotype which increases the expression of pro-inflammatory modulators like TNF-α, INF-γ, iNOS IL-1β, IL-2, IL-6, COX-2 CXCL9 and CXCL10 by microglia (Li et al., 2021). On the other hand, the signaling by IL-4 and IL-13 or LPS, IL-1β and TNF-α or IL-10 and TGF-β drive the polarization toward the M2 phenotype and increase the expression of mostly anti-inflammatory regulators (Li et al., 2021).
The role of microglia in hypertension has been demonstrated by a study in which selective pharmacological ablation of microglia in transgenic CD11b-DTR mice with either Ang II or L-NG-nitro-l-arginine methyl ester (l-NAME) induced hypertension reduced BP, attenuated the expression of TNF-α, IL-1β, and glutamate receptors in the PVN as well as the plasma levels of vasopressin, and kidney norepinephrine concentrations (Shen et al., 2015). These findings indicate that neuronal excitation of hypothalamic pathways involved with sympathetic (parvocellular cells in PVN) and hydric (vasopressin secretion) control is under influence of microglial cells and the local action of cytokines. Yet, the fact that kidney norepinephrine concentrations are also diminished in microglia-depleted animals indicate that sympathetic-mediated changes in renal function are also affected by local immune signaling within the PVN.
The mechanism by which high salt intake induces the activation of microglia in Sprague-Dawley rats may involve a Gαi2 signal transduction pathway. The downregulation of brain Gαi2 proteins by continuous i. c.v. Infusion of a phosphodiesterase ODN probe that selectively and specifically targets Gαi2 proteins not only produced a minocycline sensitive hypertension due to high salt intake but also increase the number of active microglia cells in the PVN of Sprague-Dawley rats (Moreira et al., 2019).
The activation of PVN-specific microglial cells in Sprague Dawley rats due to high salt intake (Moreira et al., 2019; Liu et al., 2022) is commonly associated with increased sympathetic-vascular coupling drive of the vascular tonus (Liu et al., 2022) and plasma noradrenaline (Moreira et al., 2019; Liu et al., 2022). In addition, high salt intake resulted in a more than double activation of microglia in the PVN of Dahl-salt-sensitive rats (DSS) and was also associated with elevated renal sympathetic nerve activity and production of central prostaglandin E2 as well as increased oxidative stress in the same region (Yu et al., 2022). In addition to rat models of SSH, mice under high salt diet also exhibited increased microglia activation and increased production of TNF-α in the PVN (Gilman et al., 2019). Most of these studies were associated with salt-related hypertension and indicate an important contribution of the microglia activation in the PVN to the SSH.
3.3 Central-acting interleukins/cytokines and salt sensitive hypertension
From a molecular perspective, several CNS-acting cytokines and interleukins, both peripherally and centrally generated, are involved with hypertension and include IL-1β, IL-6, IL-17, TNF-α and INF-γ while the major anti-inflammatory cytokine related with CNS immune signaling in hypertension is IL-10 (Yu et al., 2022; Moreira et al., 2019; Liu et al., 2022; Gilman et al., 2019; Yang et al., 2022; Konsman, 2022). The challenges imposed by high salt exposure have been shown to stimulate the production of IL-1β, IL-6, TNF-α (Afsar et al., 2018; Deng et al., 2017), IL-17 and differentiation of TH17 cells (Afsar et al., 2018; Balan et al., 2022), upregulation of MCP-1 and MIP-2 chemokines (Afsar et al., 2018; Yang et al., 2022), activation of microglia (Deng et al., 2017), activation of JAK2/STAT3 pathway in astrocytes (Deng et al., 2017) and reduce noninflammatory innate immune cell activation through reduction of IL-4 and IL-13-stimulated macrophage (Binger et al., 2015). The prevailing hypothesis suggests that higher production/action of immune signaling molecules in the CNS contributes to the heightened activity of neuronal pathways regulating BP through sympathetic activity and, therefore, contributes to hypertension. The immune signaling molecule production that is influenced by high salt consumption alters the immune balance within the CNS, favoring a pro-inflammatory profile. This shift in immune balance contributes to the development of sodium-induced neurogenic hypertension through brain-peripheral organs axis mechanisms, especially the gut-brain axis.
3.3.1 IL-17
A recent concept has linked the immune related change that occurs in the gut as a consequence of high salt intake with immune related events that occur in the brain to produce hypertension. This concept has been referred to as the gut-brain axis and has TH17 cells and IL-17 as their major focus of investigation (Carabotti et al., 2015; Dai et al., 2023; Palmu et al., 2021). As reported by Wang and cols. (2012), clinical findings support the idea that TH17/IL-17 play a role in the development of essential hypertension in humans as a positive correlation was found between hypertensive and non-hypertensive groups and TH17 cells count in the peripheral blood (Wang et al., 2012). Other findings indicate that IL-17 may be associated with hypertension development because knockout mice for IL-17 do not sustain hypertension produced by chronic infusion of Ang II (Madhur et al., 2010). The authors suggest that endothelial dysfunction driven by oxidative stress is the most likely molecular mechanism involved in this process and may be triggered by IL-17 (Madhur et al., 2010). On the other hand, clinical evidence indicates that neither IL-17-producing cells (Youn et al., 2013) nor circulating IL-17 levels (Al-Nimer and Alhusseiny, 2016) are positively correlated with increased levels of BP. However, one caveat must be considered here: most of the clinical studies searching for a correlation between circulating levels of IL-17 and hypertension were carried out in patients with stablished hypertension, many under pharmacological treatment for hypertension and no assessment of salt intake. assessment.
IL-17 is a PIM produced by the TH17 subset of CD4+ T cells and is an important signaling molecule in both acute and chronic inflammatory processes (Iwakura et al., 2011). It has been recently implicated in SSH (Wilck et al., 2017; Aguiar et al., 2017; Wenzel et al., 2019) and, since then, it has been ascribed as an important immune-derived player in the SSH. Experimental data have shown that high-salt intake increase the intestinal gut population of TH17 lymphocytes (Jorg et al., 2016; Wilck et al., 2017; Wenzel et al., 2019) and, consequently, augmented IL-17-mediated signaling throughout the body, especially when occurring in a pro-inflammatory environment (Matthias et al., 2020). Among the effects of IL-17 in the brain, it has been shown to disrupt the blood-brain-barrier (Kebir et al., 2007), drive local activation of glial cells as well as local production of IL-17 by glial cells (Deng et al., 2017; Waisman et al., 2015), and activate its receptor (IL17RA/RC) on microglia and astrocyte in the CNS (Zimmermann et al., 2013). In addition to the action on glial cells, IL-17 can directly modulate neuronal synaptic transmission by enhancing excitatory postsynaptic currents and suppress inhibitory postsynaptic synaptic currents and γ-aminobutyric acid (GABA) induced currents in lamina IIo somatostatin-expressing neurons in mouse spinal cord slices (Luo et al., 2019).
TH17 lymphocytes are shown to promote BBB disruption and favor a pro-inflammatory state of the CNS (Kebir et al., 2007). The mechanisms seem to involve the activation of IL-17 and IL-22 receptors on brain endothelial cells, and their activation permeabilizes the BBB (Kebir et al., 2007). This contributes to the migration of other immune cells into the brain parenchyma as well as the translocation of larger molecules (immune molecules included) from the blood to the brain tissue. In the CNS, IL-17 can activate microglia and further cause microvascular dysfunctions (Zimmermann et al., 2013). The authors state that their data argues against a direct role of IL-17A in producing neuronal tissue damage but acts as a modulating factor in a network of locally produced cytokines (Zimmermann et al., 2013). This interpretation is shared by other authors showing that IL-17 recruits other inflammatory cells to the CNS in hypertension (Harrison et al., 2012). Importantly, experimental evidence that direct microinjection of IL-17 into the lateral ventricle or PVN can elicit increases in BP, heart rate and renal sympathetic nerve activity strongly indicates that IL-17 can influence neuronal pathways controlling BP (Cao et al., 2021).
Although many studies imply that high polarization of TH17 cells is linked to a high production of IL-17, much of the evidence is, indeed, provided by in vitro experiments (Kleinewietfeld et al., 2013; Wu et al., 2013; Wenzel et al., 2019) and evidence of the interplay between high IL-17 production and neurogenic mechanisms leading to SSH are still fledgling. To the best of our knowledge, literature lack solid evidence that high salt intake increases the circulating levels of IL-17, what may suggest that infiltration of IL-17-producing cells may be the major source of this interleukin in end-organs like the brain and kidney. In this scenario, the potent pro-inflammatory properties of the IL-17 as well as its direct action on the central nervous system suggest it as an activator of neurogenic mechanisms that contribute to SSH.
3.3.2 IL-1β
IL-1β is a potent pro-inflammatory cytokine identified in 1984 (Auron et al., 1984) and primarily produced by activated macrophages. It mediates inflammatory responses, fever, and immune system activation. IL-1β is synthesized as an inactive precursor and is activated by caspase 1-mediated proteolytic cleavage, after what it can act on IL-1 receptors (Boraschi, 2022). Experimental findings have implicated IL-1β signaling with neurogenic mechanisms of the hypertension. Intracerebroventricular and PVN microinjections of IL-1β produce an increase in BP in normotensive Sprague-Dawley rats (Shi et al., 2011), indicating that pathways activated by IL-1β can play a role in hypertension. This signaling seems to include activation of perivascular macrophages in the brain tissue and increased expression/activation of type 2 cyclooxygenase (COX-2) thus leading to increased production of prostaglandin E2 (PGE2) (Yu et al., 2010). Heightened secretion of PGE2 by perivascular macrophages is believed to act in the PVN to increase the sympathetic drive to cardiovascular organs like the heart and vasculature (Yu et al., 2010). In a study carried out by Qi and cols., high salt diet (8% NaCl) increased BP, the biomarkers of inflammation within the PVN (including IL-1β expression) and norepinephrine plasma levels of DSS rats. Further, the bilateral microinjection of gevokizumab, an IL-1β inhibitor, in the PVN decreased the BP, the norepinephrine plasma levels, attenuated the levels of oxidative stress and restored the balance of cytokines within the PVN of DSS rats under high-salt diet (Qi et al., 2016). Corroborating these findings, both knockout of the IL-1 receptors (IL-1R) and pharmacological antagonism of IL-1R with anakinra (i.p.) reduced neurogenic pressor activity in deoxycorticosterone acetate (DOCA)-salt treated mice (Baumann et al., 2024). These findings indicate that central actions of IL-1β may be activating neuronal pathways controlling sympathetic drive to the cardiovascular system, thereby contributing to the neurogenic component of SSH.
3.3.3 TNF-α
In addition to these findings, high salt intake has been shown to drive central action/production of tumor necrosis factor alpha (TNF-α) in animals with SSBP, particularly in the PVN. A study by Gao and cols. (2022) showed that TNF-α injection into the PVN trigged a dose and time-dependent increase in mRNA expression of PIM including IL-1β and IL-6, chemokines (C–C Motif Chemokine Ligand 5 (CCL5) and C–C Motif Chemokine Ligand 12 (CCL12)) as well as inducible nitric oxide synthase (iNOS), and NF-kB in cultured brain neurons from neonatal SD rats (Gao et al., 2022). The authors also assessed and compared mRNA expression of these genes at a basal level as well as in response to TNF-α challenge between SD rats and Dahl salt-sensitive rats. They found that cultured neurons presented higher baseline levels and greater response to TNF-α challenge in mRNA expression of CCL5, iNOS and IL-1β (Gao et al., 2022). The central administration of TNF-α caused higher mRNA expression response for CCL12 in the PVN of Dahl-S rats compared to SD rats (Gao et al., 2022), strongly suggesting high sensitivity of specific downstream TNF-α-activated PIM signaling pathways in the PVN of Dahl-salt rats. In addition, TNF-α mRNA expression in the PVN has been shown to increase in Dahl-salt sensitive rats under high salt diet (4% NaCl) and chronic intracerebroventricular infusions of oligodeoxynucleotide (ODN), an inhibitor of the inhibitory G protein family alpha subunit (Gαi2), indicating that Gαi2 proteins mediate endogenous production of TNF-α in the PVN (Moreira et al., 2019), what may contribute to the development of SSH. The pieced evidence indicates that TNF-α signaling in the PVN of different hypertension models, including SSH models, is contributing to increased sympathetic drive (Moreira et al., 2019; Winklewski et al., 2015; Xiao and Harrison, 2020; Yu et al., 2019; Zera et al., 2009) further supporting the link between PIM signaling and neurogenic hypertension.
3.3.4 IL-6
Heightened IL-6 levels are reported as one of the most consistent PIM changes associated with human hypertension (Afsar et al., 2018; Sesso et al., 2007). For instance, hypertensive patients have higher baseline IL-6 levels when compared to normotensives regardless of the salt intake (Chamarthi et al., 2011). Officially named in 1988 (Sehgal et al., 1989), IL-6 is an interleukin encoded by a pleiotropic gene and is involved with immune regulation, B cell maturation, T cell differentiation (especially TH17), inflammation, hematopoiesis, BBB permeability, and acute phase response (Grebenciucova and VanHaerents, 2023). IL-6 is produced by a wide array of cell types and is rapidly synthesized in response to infections, tissue injuries, and other inflammatory stimuli (Grebenciucova and VanHaerents, 2023). Interesting, IL-6 can produce effects reminiscent of a hormone, which include the normal homeostatic control of vascular function, lipid metabolism, insulin resistance, iron transport, mitochondrial activities. IL-6 acts as a neurotrophic factor, supporting differentiation, maturation and survival of neuronal subtypes like dopaminergic and cholinergic neurons (Bader and Meyer, 2022). In addition, IL-6 can affect neuronal excitability by regulating the expression and function of ion channels such as voltage-gated sodium and calcium channels, receptor/ligand-gated ion channels, and synaptic function and plasticity by influencing neurotransmitter release and synaptic strength (Grebenciucova and VanHaerents, 2023; Gruol, 2015). It is noteworthy that neuronal activity itself can stimulate the production of IL-6 in neurons (Sallmann et al., 2000). Further, IL-6 also acts as a mediator in neuroimmune interactions between neurons and glial cells, allowing communication during inflammatory states or infection (Grebenciucova and VanHaerents, 2023).
High salt intake is a stimulator of IL-6 production in PVN of Dahl salt-sensitive rats, and its increase has been correlated with hypertension development in this model (Du et al., 2020). In Sprague-Dawley rats under high salt intake, the upstream mechanism that stimulates not only IL-6, but IL-1β and TNF-α as well, in the PVN involves the suppression of the activation of the alpha subunit of the inhibitory G protein family (Gαi2). Since PVN is an important forebrain region involved with sympathetic drive to the cardiovascular system, the authors proposed that increased inflammatory state in the PVN, indexed by the increased expression of pro-inflammatory cytokines, is associated with increased sympathetic drive thus contributing to the hypertension in Dahl salt-sensitive rats. Two different studies showed that knockout mice for IL-6 were protected from the Ang II-induced hypertension (Huang et al., 2006; Hat et al., 2024) and one study showed that IL-6 knockout mice were protected from Ang II plus salt induced hypertension as well (Hat et al., 2024). Although some advances have been made for the understanding of the IL-6 role in the BP regulatory brain pathways, the underlying mechanisms remain largely unknown and require further investigation.
From the current knowledge in the field, high salt intake has been shown to increase the expression of important PIM like TNF-α, IL-1β and IL-6 in the PVN of Dahl-salt sensitive rats but not of Sprague Dawley rats (Jiang et al., 2018). This finding may indicate that a) the genetic background of Dahl salt sensitive rats prompt higher PIM production in key regions of the brain controlling BP and b) Dahl salt sensitive rats are more susceptible to diet-sodium driven PIM production in the central nervous system. However, another study showed that direct injection of high-sodium solution into the lateral ventricle of Sprague Dawley rats stimulated increased expression of TNF-α, IL-1β and IL-6 in the PVN and the exposure of cultured hypothalamic neurons to high extracellular levels of NaCl also increased TNF-α and IL-6 production (Jiang et al., 2018). Interesting, Deng and cols. also showed that high salt treatment caused activation of astrocytes both in vivo and in vitro in Sprague Dawley rats (Deng et al., 2017). Together, the findings seem to lead to the conclusion that, indeed, Dahl salt sensitive rats may develop an inflammatory state in brain that is more intense than the one found in Sprague-Dawley rats. Additionally, TNF-α, IL-1β and IL-6 expressions by astrocytes were increased in cell culture assays and the authors suggest that JAK/STAT3 plays an important role in this process (Deng et al., 2017). Further studies confirmed those findings and suggested that the TNF-α, IL-1β and IL-6 increase in the PVN is associated with significant microglia activation and oxidative stress in Dahl salt sensitive rats (Yu et al., 2022).
Many aspects and details of such mechanisms remain unclear, mostly because some effects of the cytokines in key sites of the brain are not fully understood. Taking together, the pieced evidence indicates that high salt intake stimulates pro-inflammatory cytokines in cardioregulatory centers of the brain to produce a neurogenic-mediated increase in BP suggesting that brain LGI may be an important factor leading to hypertension. However, studies have not addressed the question whether brain cytokines increase before or after hypertension develop in experimental models and in hypertensive patients, what might establish (or not) the causal relationship between LGI and SSH.
3.4 Blood-brain barrier disruption, low-grade inflammation and salt-sensitive hypertension
The BBB is a highly selective semi-permeable barrier that separates the brain from the bloodstream. It is composed of endothelial cells, astrocyte end-feet, and pericytes embedded in the capillary basement membrane (Abbott et al., 2010). The BBB regulates the transfer of solutes and chemicals between the circulatory system and the CNS, protecting the brain from harmful substances and preserving the tight ionic composition of the extracellular fluid while allowing essential nutrients to pass through (Abbott et al., 2010). Disruptions in the BBB have been linked to the development of autonomic and cognitive impairments (Katsi et al., 2020), which could contribute to hypertension. On the other hand, hypertension itself might also contribute to BBB disruption and to cognitive impairments development (Katsi et al., 2020; Faraco, 2024; Pelisch et al., 2011), which raises some questions about cause-consequence relationship between BBB disruption and hypertension development.
High salt intake has been shown to disrupt the BBB through different mechanisms which involve changes in the tight junction proteins, inflammatory responses, activation of sodium-sensitive signaling pathways in brain microvascular endothelial cells, and the renin-angiotensin system. In a model of permanent cerebral ischemia, mice under high salt intake displayed significant enhanced ischemic brain damage which was associated with enhanced BBB disruption, increased leukocytes infiltration and loss of tight junction (TJ) proteins (Zhang T. et al., 2015). The authors also found that high sodium concentrations downregulated tight-junction proteins expression by endothelial cells through a p38/MAPK/SGK1 pathway in immortalized murine brain microvascular endothelial cell line bEnd.3 (Zhang T. et al., 2015). These findings suggest that exposure of brain endothelial cells to high sodium chloride concentrations increase BBB permeability through such mechanisms. Interestingly, high salt intake has been shown to specifically increase sodium concentration in the CSF of the HS12W model (Gomes et al., 2017), Dahl salt sensitive rats (Nakamura and Cowley, 1989; Huang et al., 2004), and one-kidney, one-wrap renal hypertension model (Haywood et al., 1984; Leenen et al., 2002), without parallel changes in plasma sodium concentration. That would indicate that high sodium concentration in the brain can be achieved in vivo. Whether that level of sodium concentration is sufficient to drive the p38/MAPK/SGK1 pathway activation is still to be determined. In addition, Zhang and cols. (2015) also reported a positive correlation between urinary sodium levels and ischemic lesion size in stroke patients (Zhang T. et al., 2015), suggesting that BBB disruption by high-sodium intake may occur in humans as well.
The disruption of the BBB also involves a complex interplay between peripheral and central-generated PIM. One of the key players in this process is the high salt intake-related cytokine IL-17, which has been demonstrated to be involved with BBB disruption in an experimental autoimmune encephalomyelitis (EAE) model induced in C57BL/6 mice. In this model, IL-17A induced the production of reactive oxygen species (ROS), which activated the endothelial contractile machinery. This activation was accompanied by a downregulation of the tight junction molecule occludin (Huppert et al., 2010), an important tight-junction protein in endothelial cells. Blocking either ROS formation or myosin light chain phosphorylation or sequestering IL-17 with IL-17A-neutralizing antibodies prevented IL-17A-induced BBB disruption (Huppert et al., 2010). A study by Kleinewietfeld also reported that high infiltration of TH17 cells into the CNS is present in a mouse model of experimental autoimmune encephalomyelitis (EAE) under high salt diet with co-involvement of the pro-inflammatory cytokines GM-CSF, TNF-α and IL-2 (Kleinewietfeld et al., 2013). In addition, cytokines like IL-1β, IL-6, IL-9, IL-17, INF-γ, TNF-α and CCL2 can impact tight-junction production and allocation, damage endothelial cells integrity, activate astrocytes and microglial cells and induce the penetration of peripheral immune cells into the CNS (Huang et al., 2021) through the disrupted BBB.
In addition to the role of immune-mediated mechanisms, disruption of the BBB has also been associated with high BP by itself (González-Marrero et al., 2012; Mueller and Heistad, 1980) and in some cases like the Ang II-induced high BP, BBB disruptions are reported to precede hypertension development in experimental models (Capone et al., 2011). Conversely, the majority of studies demonstrate disruption of the BBB after hypertension has been established, making it challenging to establish a causal relationship between BBB disruption, inflammation, and hypertension based on the current available knowledge. For instance, it has been demonstrated that capillary permeability of brain regions classically associated with sympathetic drive and BP control like PVN, NTS and RVLM are increased in hypertensive SHR but normal in pre-hypertensive SHR (Buttler et al., 2017). The injury process in vascular tissue stimulates the infiltration of inflammatory cells into the brain, especially in the perivascular tissue (Yu et al., 2010), reinforcing the LGI process. Once in the brain tissue, those cells are responsible for secreting other PIM such as IL-6, IL-1β and TNF-α locally (Hashmat et al., 2016; Winklewski et al., 2015) thus affecting local neurotransmission and neuronal activity of cardioregulatory centers in the brain. Consequently, it is plausible to speculate that salt-driven lesions in the BBB at critical sites for BP regulation can trigger the production of PIM within the brain in a positive feedback loop, thereby further activating neurogenic mechanisms associated with the development of SSH.
3.4.1 Key points
1. CNS is important for BP regulation in both normotension and SSH, with neurogenic mechanisms contributing to elevating BP.
2. Cardiovascular reflexes like baroreflex and cardiopulmonary reflex are impaired or altered in SSH, with Dahl salt-sensitive rats showing reduced reflex sensitivity even under low salt intake.
3. The hypothalamic paraventricular nucleus (PVN) plays a key role in SSH and the immune signaling within the PVN, including microglial activation and proinflammatory cytokine production, are shown to largely contribute to high BP.
4. Though less studied, immune signaling in brainstem areas like RVLM and NTS also contribute to hypertension, although its role in SSH is still to be further explored.
5. High salt intake can disrupt the BBB and allow immune cells to infiltrate CNS. Higher immune cell infiltration in the CNS is associated with high BP, especially in the SSH.
4 The gut microbiome and the gut-brain axis in salt-sensitive hypertension
4.1 Gut microbiome
Over the past decade, a growing number of studies have allocated substantial resources to comprehending the impact of alterations in the gut microbiome on BP regulation (Palmu et al., 2021; Calderon-Perez et al., 2020; Canale et al., 2021; Cardoso, 2024; Marques et al., 2018; Pluznick, 2016). A portion of these studies have specifically focused on the effects of excessive salt consumption on the changes in gut microbiome and its subsequent influence on BP regulatory mechanisms (Palmu et al., 2021; Canale et al., 2021; Abais-Battad et al., 2021; Chrysant, 2024; Elijovich et al., 2020; Mu et al., 2024). A prevailing consensus among various authors is that excessive salt consumption adversely affects the host’s health by diminishing the α and β diversity of the gut microbiome and leverage the balance toward the predominance of bacterial species that promote an inflammatory state and the production of detrimental metabolites (Wilck et al., 2017; Calderon-Perez et al., 2020; Elijovich et al., 2020; Mu et al., 2024; Mell et al., 2015; Xu et al., 2022; Yin et al., 2024). The imbalance between health-promoting and disease-causing gut bacteria, known as dysbiosis, is theorized to have a substantial impact on BP regulation through the gut-brain axis signaling system thus contributing to SSH in experimental models and humans (Elijovich et al., 2020; Nagase et al., 2020).
The gut microbiome produces several metabolites with bioactive properties that influence brain chemistry, stress response, cognitive and autonomic functions of the host. In this context, gut microbiome dysbiosis has been associated with pathophysiological mechanisms that may cause cardiovascular diseases. Indeed, high salt intake results in dysbiosis in the gut microbiome. This has been shown to cause the host’s differentiation of naïve T cells into pro-inflammatory TH17 cells, shifting the immune system towards a pro-inflammatory state, and promote the production of pro-hypertensive short-chain fatty acids (SCFA) by specific groups of gut bacteria (Wilck et al., 2017; Wang et al., 2022; Abais-Battad et al., 2021; Elijovich et al., 2020; Xu et al., 2022; Yin et al., 2024; Chakraborty et al., 2018; Chen et al., 2020). Such events in the gut affect the inflammatory signaling mechanisms within different organs controlling BP, which include the brain and the kidneys. These mechanisms have been actively discussed within the concept of signaling axis that involve mostly the gut, brain and kidneys. The gut-brain axis has received considerable attention recently due to its ability to influencing BP control, especially in SSH. By the current definition, the gut-brain axis refers to the bidirectional communication network between the gastrointestinal tract (GI tract) and the CNS. This complex system involves multiple key pathways, including neural, endocrine, immune, and humoral links which influence each other’s functions and BP levels (Carabotti et al., 2015; Mell et al., 2015). Evidence has shown that the gut-brain axis affects neural mechanisms controlling BP. Although much has been investigated on the correlation between hypertension and gut microbiome dysbiosis, the pathophysiological mechanism through which it occurs remains mostly unknown. Whether high salt intake can cause neurogenic hypertension through increased sympathetic outflow due to gut microbiome dysbiosis is yet to be further investigated. However, evidence shows that IL-17, a product of TH17 cells that proliferate under gut microbiome dysbiosis (Wilck et al., 2017), can act in the PVN to increase sympathetic outflow (Cao et al., 2021), and corroborates the theory that dysbiosis may be causally related to neurogenic SSH.
The study by Wilck et al. (2017) has shown that a 4% NaCl diet for 14 days caused a reduction in the L. murinus population of mice intestinal microbiota (Wilck et al., 2017). Such change was accompanied by increased BP and increased population of CD4 TH17 pro-inflammatory cells in the intestinal mucosa (Wilck et al., 2017). When high-salt-fed mice were switched back to a regular salt diet, the intestinal population of L. murinus as well as BP returned to normal, strongly suggesting that the impact of high salt intake on Lactobacillus murinus population: a) is correlated with hypertension in these animals and b) are reversible (Wilck et al., 2017). A study conducted by Bier and colleagues (2018) also showed that excessive salt consumption elevated the relative abundance of the taxa belonging to the Erwinia genus, Christensenellaceae, and Corynebacteriaceae families in Dahl salt-sensitive rats (Bier et al., 2018) suggesting the existence of “halophilic” bacteria in the gut microbiome. Conversely, it also reduced the abundance of the Anaerostipes genus in this model (Bier et al., 2018). These findings suggest that, beyond the Lactobacillus genus, the abundancy of other genera and even entire families of the gut microbiome can be impacted by high salt intake. In addition, previous (Linz et al., 2012; Pacha, 1998) and recent (de Assis Braga et al., 2025) studies also showed that high salt intake can, indeed, increase the content of sodium in the feces in vivo further supporting the hypothesis that, despite the vast majority of sodium intake is absorbed by the small intestine, a significant amount of sodium reaches the large intestine and function as selective pressure mechanism of the environment on gut microbiome. Such changes appear to be correlated with a shift in the relative abundance of specific families and genera that favor specific pro-hypertensive bacterial groups over rather than a general and unspecific impact on the gut microbiome of SSH models (de Assis Braga et al., 2025).
Corroborating experimental data, a recent study has described dysbiosis of the intestinal microbiota in hypertensive patients. This dysbiosis was characterized by reduced biodiversity and distinct bacterial signatures compared to the normotensive counterpart (Silveira-Nunes et al., 2020). Along with a reduction in Bacteroidetes members, hypertensive individuals displayed increased proportions of Lactobacillus and Akkermansia and decreased relative abundances Roseburia and Faecalibacterium within the Lachnospiraceae and Ruminococcaceae families (Silveira-Nunes et al., 2020). The increased proportion of Lactobacillus genera in hypertensive patients may indicate that, different of experimental findings in mice, the reduction in abundance of the genus Lactobacillus is not essential for the hypertension in humans. This study also reported a pro-inflammatory immune profile in hypertensive individuals with an increase in TNF/IFN-γ ratio, and in TNF and IL-6 production in peripheral blood when compared to normotensive subjects (Silveira-Nunes et al., 2020). Despite lending support for experimental data, this study did not address important questions like whether the changes in gut microbiota are, indeed, a cause for the hypertension in humans or if dietary salt consumption was correlated with the reported changes in microbiota. This question was addressed in a paper by Nagase et al. (2020) which concluded that consumption of low-salt diet was ineffective in regulating hypertension in individuals with a specific gut microbiome composition (Nagase et al., 2020). Therefore, the authors defend the idea that restoration of the gut microbiome should be considered a new better approach for controlling BP and preventing hypertension in humans (Nagase et al., 2020).
4.2 Short-chain fatty acids
Short-chain fatty acids (SCFA) have been recognized as important molecules from gut microbiome metabolism that play a significant role in the gut-brain axis signaling system. By definition, SCFA are a group of saturated aliphatic organic acids with a chain length of one to six carbon atoms. The most common SCFA produced by gut microbiome are acetate (C2), propionate (C3), and butyrate (C4), which are produced primarily through the anaerobic fermentation of indigestible polysaccharides, such as dietary fiber and resistant starch (Silva et al., 2020). The ratio of acetate:propionate:butyrate in the colon has been estimated in 3:1:1, meaning that acetate is the most abundant SCFA produced (He et al., 2020; Liu et al., 2021). The SCFA, produced by luminal large intestine bacteria, can enter the body through monocarboxylate transporters (Coady et al., 2004) or through simple diffusion (Charney et al., 1998). The SCFA plasma concentration in the host is, however, considerably smaller than colonic concentration. For instance, acetate has a typical plasma concentration about 1,000 to 2,000 times smaller (50–100 μmol/L) than colonic concentration (100 mmol/L), albeit it is still substantial (Xu et al., 2022). Acetate (produced by most of the Enterococcus species) and propionate (produced by Bacteroidetes, Acidaminococcus and Salmonella) are easily absorbed and transported to the liver. In the liver, propionate is known to promote the intrahepatic gluconeogenesis (Xu et al., 2022). In addition, bacteria from the genera Clostridium, Eubacterium and Roseburia are known as the major sources of butyrate (Liu et al., 2021; Wong et al., 2006; Nicholson et al., 2012; Louis and Flint, 2017; Sasaki et al., 2020).
The role of SCFA and mechanisms in the SSH is yet to be further explored. However, different studies have pointed out some beneficial functions for SCFA in the host´s health. For instance, Wu and cols. (2021) showed that oral administration of the SCFA sodium butyrate attenuated the hypertension and renal damage in DOCA/salt rats by a mechanism that involves inhibition of the MR/SGK1 pathway (Wu et al., 2021). Additionally, dietary sodium reduction has been shown to increase circulating SCFA which are associated with decreased BP and improved arterial compliance (Chen et al., 2020). On the other hand, high salt intake has been shown to reduce the concentrations of SCFA such as acetate, propionate, and butyrate in fecal samples of mice fed 8% salt diet, what was associated with cognitive dysfunction and gut dysbiosis (Hu et al., 2020). However, Dahl salt sensitive rats displayed increased levels of fecal acetate, propionic acid and isobutyrate, but not butyrate, when fed a 4% salt diet (Bier et al., 2018), contrasting with the high-salt intake findings in mice. A study by Bruning and cols. (2020) also raised some issues regarding the benefits of SCFA on BP regulation. The authors reported that microinjection of the acetate into the PVN of anesthetized rats produced an increase in splanchnic sympathetic nerve activity and BP (Bruning et al., 2020). Also, the pre-injection of kynurenic acid, an ionotropic EAA receptor antagonist, attenuated the sympathoexcitation and BP increase produced by acetate (Bruning et al., 2020), strongly suggesting that the acetate-induced excitation in the PVN could involve EAA neurotransmission.
SCFA produce their biological effects in the host by activating specific G protein-coupled receptors expressed throughout the body such as GPR41 and Olfr78, which are expressed in blood vessels and other tissues. Evidence from whole body knockout mice showed that the lack of GPR41 receptors led to hypertension in these animals while Olfr78 knockout mice are normotensive (Xu et al., 2022). Also, the SCFA GPR109A receptor in the rostral ventrolateral medulla (RVLM) plays a role in central BP control, where activation by nicotinic acid leads to l-glutamate release, subsequently increasing sympathetic activity and BP (Rezq and Abdel-Rahman, 2016). In addition, the EC50 values for acetate, propionate and butyrate are largely different for the GRP41, GRP43 and Olfr78 receptors (Xu et al., 2022; Lu et al., 2022; Pluznick, 2014). For instance, acetate has the lower affinity while propionate and butyrate have higher affinity for SCFA receptors (Table 2). This pharmacological aspect of the SCFA may also, at least in part, contribute to different and, sometimes, conflicting effects of SCFA on BP. Together, these findings picture a complex interaction between SCFA production and host’s response to it, especially concerning SSH. However, studies in humans have shown that dietary sodium reduction increases circulating levels of 2-methylbutyrate, butyrate, hexanoate, isobutyrate, and valerate SCFA and that such an increase in circulating SCFA was associated with BP reduction and improvement of arterial compliance (Chen et al., 2020; Ríos-Covian et al., 2016). Further studies are needed to establish whether SCFA effects on autonomic control of BP are specific to each individual molecule, the combination of them or if it is dependent on the major action site in the brain. A summary of the major findings is shown in Table 2.
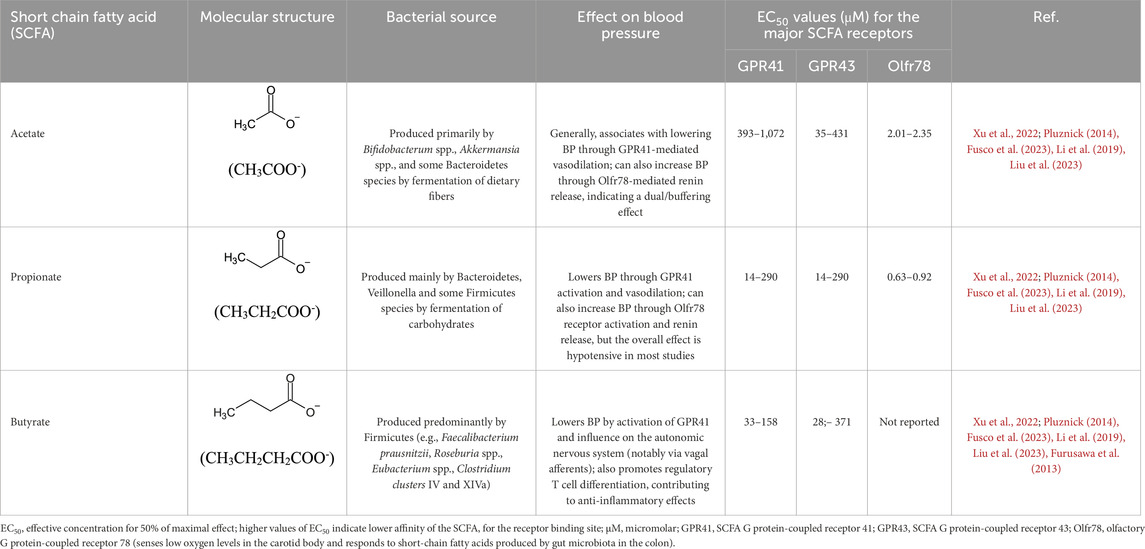
Table 2. Major short chain fatty acids produced by the gut microbiome and the effects on blood pressure regulation.
4.2.1 Key points
1. High salt intake can influence gut microbiome composition.
2. Composition of the gut microbiome can affect BP regulation by mechanisms involving inflammatory signaling and detrimental metabolite production.
3. The gut-brain axis, a bidirectional signaling network between the gut and the CNS, plays a role in BP control.
4. Dysbiosis in the gut microbiome can lead to increased sympathetic outflow and neurogenic hypertension.
5. Experimental data shows that high salt intake can cause dysbiosis shifting the Firmicutes/Bacteroidetes ration which is correlated with high BP and pro-inflammatory state.
6. In humans, dysbiosis in hypertensive patients is characterized by reduced biodiversity and distinct microbiome composition signature compared to normotensive individuals.
7. Short-chain fatty acids (SCFA), produced by gut microbiome, play a role in BP regulation. While some studies show beneficial effect of SCFA on BP, others indicate potential adverse effects, highlighting the complexity of their role in hypertension.
5 Kidney, salt-sensitive hypertension and inflammation
Sodium balance is tightly regulated by kidneys under a wide range of sodium intake (Bagordo et al., 2024). Although recent evidence indicates that both human and rodents display nonosmolar sodium retention in skin and skeletal muscle under high salt intake (Thowsen et al., 2022; Titze, 2014), the kidneys still have a dominant role in the sodium handling by the body. The traditional view that high salt intake can lead to hypertension because of increased water intake followed by plasma volume expansion, increased cardiac output and autoregulatory vasoconstriction has been challenged by current evidence showing that plasma volume does not increase under high salt intake that mimics the human setting in a rodent model of SSH (Gomes et al., 2017; de Souza et al., 2021). The results of these studies suggest that hypertension is initially driven by neurogenic mechanisms with renal histological and functional disfunctions after the establishment of the hypertension (de Souza et al., 2021).
The accumulation of sodium in tissues has been demonstrated to contribute to systemic LGI (Sahinoz et al., 2021) which can impact renal functions through a variety of mechanisms (Frame and Wainford, 2017). For instance, the infiltration of immune cells in the kidneys has been shown to play an important role in the renal mechanisms that contribute to SSH in experimental models (Mattson, 2014). The genetic deletion of the recombination-activating gen 1 (Rag1) in Dahl-salt sensitive rats resulted in a significant reduction in T and B cells in the blood and spleen under normal salt diet (Mattson et al., 2013). Also, the exposure of these animals to high salt diet produced a blunted infiltration of T cells into the kidney that is typically found in wild type Dahl salt sensitive rats following high salt intake (Mattson et al., 2013). The reduction of T cells infiltration into the kidneys of Rag1-null Dahl salt sensitive rats was accompanied by a significant reduction in the SSH of these animals (Mattson et al., 2013). Furthermore, the infiltration of immune cells into the kidneys of Wistar rats under high salt diet following l-NAME administration was described as responsible for the impaired pressure natriuresis that contributes to SSH (Franco et al., 2013). The progression of renal injury in obese Dahl salt sensitive leptin mutant rats was associated with increased expression of the macrophage inflammatory protein 3-α (MIP3α) and, consequently, increased immune cell recruitment and infiltration in the renal tissue, thus producing renal low-grade inflammation (Ekperikpe et al., 2023). Interestingly, high salt, but not high BP, induces immune cell activation, renal infiltration, high expression of Na+/K+-ATPase and SSH in ovariectomized rats (Vlachovsky et al., 2024) showing that high salt intake has a more prominent effect on immune kidney system than high BP alone.
As immune cells infiltrate the kidneys, PIM drive important changes in the expression and activity of sodium channels in the nephron affecting sodium handling by kidney. This was be demonstrated in an AngII-driven hypertension mouse model in which i) RAS-mediated hypertension was accompanied by elevated levels of the macrophage cytokine IL-1 in the kidney (Crowley et al., 2010) and ii) the IL-1R deficiency or blockade limits BP elevation in this model by mitigating sodium reabsorption via the NKCC2 co-transporter in the nephron through a mechanism dependent on nitric oxide production by intra-renal macrophages (Zhang et al., 2016). The authors also showed that sodium balance in the IL-1R knockout mice became negative under Ang-II infusion while sodium balance in wild type mice increased, and that pre-treatment with furosemide abrogated the difference. Given that sodium balance is a reliable indicator of natriuresis relative to sodium intake, the findings indicate that IL-1R may contribute to the Ang-II-induced sodium retention, as proposed by the authors (Crowley et al., 2010). However, plasma sodium concentrations were not addressed in this work, leaving questions on whether IL-1 cytokine is actually influencing extracellular fluid concentration of sodium and/or affecting sodium retention in the skin and muscle (Titze, 2014), extra or intracellularly (Thowsen et al., 2022) following high sodium intake. Further evidence indicates that IL-1β plays a direct role in SSH and this is supported by findings that inflammasome components NLRP3, ASC, and caspase-1 were not only present in distal tubules and collecting ducts of Dahl-salt sensitive rats but also increased under high salt intake (Zhu et al., 2016). Additionally, the infusion of the caspase-1 inhibitor Ac-YVAD-cmk as well as the transplantation of mesenchymal stem cells into the renal medulla of Dahl-salt sensitive rats reduced the NLRP3 inflammasome activation and the salt-induced hypertension (Zhu et al., 2016), linking the inflammasome-derived mature IL-1β signaling system in the kidney to the SSH. Further supporting the role of IL-1β on renal sodium handling, diabetic db/db mice harboring an IL-1 receptor type 1 knockout bone marrow (db/dbIL1RKO) phenotype displayed lower expression and activity of epithelial sodium channel (ENaC) expression and activity compared with db/db transplanted with a wild-type bone marrow (Veiras et al., 2022). The db/db mice features high levels of kidney cortical IL-1β expression and, alongside with diabetes, have progressive increase in BP levels when exposed to high salt diet which involves an impaired downregulation of ENaC in the kidney epithelial cells in response to high sodium (Veiras et al., 2021). When exposed to high salt intake, the db/dbIL1RKO displayed a salt resistant BP as well as lower renal inflammation with reduced IL-1β-induced production of IL-6 by macrophages in the kidneys (Veiras et al., 2022). These findings indicate that kidney cortical IL-1β overproduction increases renal ENaC expression and activity through a mechanism that involves macrophages polarization toward a proinflammatory phenotype and renal IL-6 accumulation, with concurrent salt sensitivity of the BP (Veiras et al., 2022).
Chronic infusion of Ang II is well known for its ability to increase BP levels and significantly decrease natriuresis when experimental animals are challenged with saline solutions. Also, Ang II regulates the synthesis of pro-inflammatory cytokines and chemokines in the kidneys of female Wistar rats (Ruiz-Ortega et al., 2002) and can increase abundance of the phosphorylated forms of the Na-K-2Cl cotransporter (NKCC), Na-Cl cotransporter, Ste20/SPS-1-related proline-alanine-rich kinase in the distal tubule as well as ENaC expression in the collecting duct in mice (Kamat et al., 2015). Parallel to its direct effects on renal expression of sodium handling ion channels, Ang II, DOCA-salt and norepinephrine cause T cells and monocytes/macrophages to accumulate in the kidney and increase the local production of pro-inflammatory cytokines like INF-γ and IL-17 (Itani and Harrison, 2015). In this context, knockout mice for INF-γ (IFN-γ(−/−)) or IL-17A (IL-17A (−/−)) displayed a blunted response to Ang II-induced hypertension (Kamat et al., 2015) indicating that Ang II-driven hypertensin also involves downstream pathways activated by INF-γ and IL-17 cytokines. However, INF-γ(−/−) and IL-17 (−/−) did not affected the Ang II-induced increase in abundance of the phosphorylated forms of the Na-K-2Cl cotransporter, Na-Cl cotransporter, Ste20/SPS-1-related proline-alanine-rich kinase in the distal tubule but significantly decreased the abundance of Na/H-exchanger isoform 3, sodium phosphate transporter isoform 2 (NaPi2) and the motor myosin VI in the proximal tubule (Kamat et al., 2015). These findings suggest that both INF-γ and IL-17 can interfere with the renal sodium handling by altering the expression of sodium transporters within the initial portion of the nephron. Given the total amount of filtered sodium reabsorbed in the portion of the nephron, even small changes in ENaC expression could have a significant impact on sodium retention and its impact on BP.
The exposure of ENaC in M-1 cortical collecting duct cells to IL-6 increased the protein expression of α-ENaC, β-ENaC, γ-ENaC and prostasin as well as the amiloride-sensitive sodium current in a study by Li and cols. (2010) (Li et al., 2010). The SSBP is also strongly correlated with the T-lymphocytes levels of mRNA for IL-6 isolated from kidneys when compared to circulating T-lymphocytes in Dahl salt sensitive rats (Rudemiller et al., 2014). By treating these animals with an IL-6 neutralizing antibody, Hashmat and cols. (2016) showed that a decreased renal cortical level of IL-6 was correlated with reduced glomerular and tubular damaged as well as with BP levels in Dahl salt sensitive rats under high salt intake (Hashmat et al., 2016) suggesting the IL-6 may exacerbate may be involved in the salt sensitivity of Dahl salt sensitive rats.
The role of TNF-α in the renal changes that occur in response to high salt intake challenge are particularly interesting, especially because of the poor understanding of the exact mechanisms underlying the role of TNF-α in the SSH. While TNF-α antagonism has attenuates the hypertensive responses in many hypertensive animal models, contrasting findings demonstrate that the direct systemic administration of TNF-α usually induces hypotensive as well as natriuretic responses (Mehaffey and Majid, 2017) indicating that the solo increase in the circulating TNF-α levels may oppose the SSH. Such contrasting outcomes are speculated to result, at least in part, from differential activities of the two TNF-α cell surface receptors expressed in the kidney tissue. The TNFR1 is usually found in the proximal tubule, collecting duct, vascular endothelium and vascular smooth muscle while TNFR2 are usually found in the proximal tubule, collecting duct, vascular endothelium but not in the vascular smooth muscle (Mehaffey and Majid, 2017; Castillo et al., 2012). Studies conducted in TNFR1 knockout (TNFR1KO) and TNFR2 knockout (TNFR2KO) mice have elucidated the differential effects of both receptors on renal sodium handling and renal injury induced by TNF-α. The TNF-α infusion in mice triggers a surge in urinary volume output and fractional excretion of sodium, while the absence of TNFR1 attenuates this response (Castillo et al., 2012). Also, the TNF-α natriuretic activity is amiloride-sensitive suggesting that such effect may be mediated by ENaC activity (Majid, 2011; Shahid et al., 2008). Corroborating the role of TNFR1 role in renal sodium handling, recent findings showed that TNFR1 activity is downregulated in eNOSKO mice, which facilitates salt retention and contribute to SSH under high salt intake (Majid et al., 2024). On the other hand, mice lacking TNFR2 displayed less Ang II-induced fibrotic changes and macrophage extravasation indicating that TNF-α action on TNFR2 promotes renal tissue damage through proinflammatory pathway (Mehaffey and Majid, 2017; Singh et al., 2013). In summary, the actions of TNF-α in the kidney strongly depend on the receptor type it acts on and may oppose the sodium retention in mice under high-salt intake, probably counteracting its pro-hypertensive effects. That would strongly indicate that the role of TNF-α may depend on the production of other cytokines and chemokines in SSH.
The immune events that originate in the kidneys not only affect local renal functions but also influence immune events that occur in the brain. The renal and central nervous system signal each other through afferent (sensory) and efferent (motor) sympathetic autonomic nerves and recent findings suggest that signaling system has an important role in the SSH. Using the rat DOCA-salt model, Banek and cols. Showed that afferent renal nerve activity is augmented and that both, total and selective afferent renal nerves ablation, attenuated the DOCA-salt induced hypertension by about 50% (Banek et al., 2016). In addition, the authors also found that total, but not selective, afferent renal denervation attenuated the DOCA-salt induced renal inflammation assessed by CD3+, CD4+ and CD8+ cell counting and inflammatory markers (GRO/KC, MCP-1, IL-1β, IL-2, IL-6, IL-17a, and TNF-α) in the kidney (Banek et al., 2016). The findings suggest that while afferent renal nerves contribute to hypertension in the rat DOCA-salt model, the efferent renal nerves contribute to renal inflammation. The authors also proposed that renal inflammation may drive afferent renal activity to the central nervous system thus contribute to a positive feedback loop to the renal inflammation and to hypertension. Further studies on DOCA-salt model showed that the IL-1R activation is partially responsible for the increased afferents renal nerve activity and that IL-1β is increased in the kidneys and urine of DOCA-salt mice (Baumann et al., 2024). As in the DOCA-salt model of hypertension, both total renal denervation and afferent renal denervation attenuated the hypertension in the 2 kidneys, one clip (2K1C) model (Lauar et al., 2023). In addition to increased pro-inflammatory cytokines (TNF-α and IL-1β) in the clipped kidney and urine, the authors found increased expression of these cytokines in the hypothalamus of 2K1C rats. Curiously, both, total and afferent renal denervation decreased the levels of TNF-α, but only afferent renal denervation decreased the levels of IL-1β in the hypothalamus of 2K1C rats (Lauar et al., 2023), suggesting that renal inflammation can trigger an immune response in the central nervous system of this hypertension model. Together, the findings indicate a complex reinforcing interplay between the kidneys and the central nervous system to sustain SSH through immune-mediated mechanisms.
5.1 Key points
1. The kidneys play a dominant role in regulating sodium balance in the body, even under high salt intake. Recent evidence challenges the traditional view that high salt intake led to hypertension through increased plasma volume and suggests that hypertension due to high salt intake is initiated by neurogenic mechanisms.
2. Sodium accumulation in tissues contribute to systemic low-grade inflammation which may impact renal function. Genetic studies implicate increased immune cell infiltration in the kidney as a mechanism that contribute to SSH specially because hypertension is reduced when immune cell infiltration is blunted.
3. IL-1 influences sodium retention and hypertension since IL-1R deficiency or blockade limits BP elevation by reducing sodium reabsorption in the nephron.
4. Ang II regulates pro-inflammatory cytokines production in the kidneys leading to renal damage and altering sodium transporters expression.
5. Renal and central nervous systems communicate to each other through afferent and efferent sympathetic renal nerves contributing to SSH. Inflammatory signaling in the kidneys trigger immune responses in the brain that further contribute to sympathetic stimulation, creating a positive feedback loop that contributes to SSH.
6 Age and low-grade inflammation in the salt-sensitive hypertension
Aging is an inevitable ongoing process that is part of life and is also an important risk factor for cardiovascular disease (North and Sinclair, 2012). Despite its importance for cardiovascular diseases, the underlying mechanisms behind the processes that build up age-related impairments to the cardiovascular system are not fully understood. However, it is important to point out that the SSBP is a trait that becomes more pronounced as individuals age (Osanai et al., 1993) and, therefore, can be regarded as an additional risk factor for those carrying this trait. The association between age and SSBP has been documented and has been linked to vascular dysfunction and diminished kidney function. Factors as epigenetic modifications, diet, gut microbiome abundancy and diversity, intrinsic immune system setting, mitochondrial dysfunctions, increased levels of cortisol, immunosenescence and, regarding women, menopause are the prevailing elements that largely contribute to age-related cardiovascular disease (Demirci et al., 2025).
Recent experimental studies have shown that the aging process in Sprague-Dawley (SD) rats from 3 to 16 months is accompanied by increased BP, enhanced neurogenic pressor activity, BBB disruption, increased PIM (IL-6 and TNF-α) production, and microglia activation in the PVN of male Sprague-Dawley rats (Nist et al., 2024). These findings indicate that aging is associated with increased brain production of pro-inflammatory molecules in the PVN, a key brain region controlling sympathetic nerve activity and BP of male rats. The authors also found that treatment with losartan, an AT1R antagonist used as first-line treatment of hypertension, improved BP levels as well as reduced microglia activation and IL-6 and TNF-α production in the PVN of 16 months old male rats (Nist et al., 2024). Interestingly, females SD rats lack the increased BP and increased PIM production in the PVN during the same aging period indicating that female SD rats do not undergo the same cardiovascular issues that male SD rats due to aging. On the other hand, earlier studies showed that Dahl salt sensitive female rats, even under low-sodium diet, displayed an increase in BP over time, from the age of 3 months to the age of 12 months (Hinojosa-Laborde et al., 2004) indicating that the salt sensitive trait in these animals may overcome the protective effects of sexual hormones during the evaluated aging period. Ovariectomy accelerated the development of hypertension in Dahl salt rats, while the ovariectomy accompanied by estrogen replacement attenuated its development (Hinojosa-Laborde et al., 2004). These findings are aligned with results from studies showing an increase in salt sensitivity over the aging process is also reported in humans (Sander, 2002; Schulman et al., 2006) and further highlight the importance of the steroid hormones in the SSBP protection as salt-resistance women pre-menopause become salt-sensitive after menopause (Schulman et al., 2006).
A growing number of studies have showed that the decline in expression of the Klotho protein is linked to aging in humans and experimental animals (Abraham and Li, 2022). Klotho is a membrane/soluble protein expressed in the choroid plexus of the brain, a structure primarily linked to the CSF production, and the convoluted tubules in the kidney (Abraham and Li, 2022). The circulating levels of the soluble form of Klotho (α-Klotho) have been shown to directly correlate with glomerular filtration rate suggesting its diagnostic importance in kidney function decline, especially during aging (John et al., 2011). Interesting, the missense single nucleotide polymorphism in the Klotho gene, rs9536314, do humans is also associated with SSH (Citterio et al., 2020). In addition, experimental findings showed that high sodium intake led to SSH in mice Klotho heterozygous knockout mice as wells as in aged mice and that Klotho supplemented reversed SSH in these animals (Kawarazaki et al., 2020). Also, Klotho deficiency in heterozygous knockout mice is associated with renal damage and SSH through CCR2-mediated inflammation (Zhou et al., 2015), linking the Klotho deficiency with immune regulation of SSBP. Whether Klotho blood levels can function as a reliable marker of SSBP, especially in aged people, is yet to be determined.
The ongoing findings point toward the important role of immune signaling in key brain regions controlling autonomic regulation of BP in experimental models and may represent an important step for the understanding of its role in the human set of SSBP.
6.1 Key points
1. Aging is an important risk factor for cardiovascular diseases that encompass factors like epigenetic modifications, diet, gut microbiome changes, immune system setting, mitochondrial dysfunctions, hormones changes and other factors that contribute to age-related BP elevation over time.
2. Experimental data have shown that aging is associated with hypertension development along with increased neurogenic pressor activity, BBB disruption and low-grade neuroinflammation.
3. Interesting, female experimental animals did not display the same increased BP associated with PIM production as male during aging. However, the salt-sensitive trait found in Dahl-salt sensitive rats resulted in increased BP during aging suggesting this trait may overcome the protective effects of female sexual hormones.
4. The decline in Klotho protein expression is linked to aging and SSH. The mechanisms seem to involve renal function decline and inflammation.
7 Therapeutic implications and future directions
High salt intake is important risk factor for cardiovascular and kidney disease and yet reductions in salt intake have proved a challenging task to be achieved in the modern western society. Although the daily salt consumption has, indeed, reduced from the 18th century to current days (Ha, 2014), it still far from the recommendations by worldwide health authorities. Therefore, new strategies and approaches must be considered to mitigate the effects of the high salt intake, especially when considering the aging and longer lifespan our society conquer over the past century. The gut microbiota plays a crucial role in regulating BP and kidney function through its interaction with the immune and nervous systems. Therapeutic strategies such as dietary interventions, probiotics, prebiotics, symbiotic, and fecal microbiota transplantation have shown promise, under experimental and controlled conditions, in modulating gut microbiota to manage hypertension and chronic kidney disease (Al-Habsi et al., 2024; Baldi et al., 2021). These interventions aim to restore microbial balance, enhance gut barrier function, and reduce systemic inflammation, which are critical for lowering BP and protecting kidney and brain health. Targeting neural pathways that regulate immunity, and inflammation offers a novel approach to treating kidney diseases and hypertension. The use of anti-inflammatory agents that specifically target the CNS like minocycline or drugs with specific action on cytokine receptors like IL-1R and TNFR antagonists may be proven effective in counteract the pro-inflammatory effects of high salt intake and work alongside conventional anti-hypertensive therapies. SCFA, produced by gut microbiota, have anti-inflammatory effects and play a role in BP regulation. Enhancing SCFA production through dietary modifications or supplementation could be a therapeutic strategy to mitigate hypertension and its associated kidney damage.
However, further research is needed to elucidate the precise mechanisms by which the gut microbiota influences hypertension and kidney diseases. Understanding these pathways will aid in the development of targeted therapies that can more effectively modulate the neuron-immune-microbiome axis and may mitigate the detrimental effects of high salt intake on the cardiovascular and renal systems. As our understanding of the microbiome’s role in hypertension and kidney diseases grows, there is potential for developing personalized treatment strategies. These could involve tailoring dietary interventions and microbiota-targeted therapies based on individual microbiome profiles. Conducting well-designed clinical trials to test the efficacy of microbiota-targeted interventions in humans is crucial. These trials will help validate the therapeutic potential of probiotics, prebiotics, and other microbiome-related strategies in managing hypertension and salt-related kidney diseases. In addition, utilizing metagenomics, metabolomics, and other omics technologies can provide comprehensive insights into the microbiome’s role in disease pathogenesis. This integration will facilitate the identification of novel biomarkers and therapeutic targets.
8 Conclusion
In this review, we focused on key findings that substantiate the role of the immune signaling pathway in the pathophysiology of SSH, particularly emphasizing certain aspects of the neural and renal mechanisms. The salt-induced challenges that prompt the immune system to corroborate pro-hypertensive mechanisms also entail significant alterations in the gut microbiome. Consequently, the trafficking of large amounts of salt through the body not only impairs autonomic and volume-related regulation of the cardiovascular system, but also adversely affects the commensal bacterial community in the gut, which subsequently reverberates in the immune system, contributing to a low-grade, chronic inflammatory state associated with important neural and renal pathophysiological mechanisms. The multiplicity of signaling mechanisms involved and the overlapping effects of aging and sexual dimorphism underscore the complexity of SSH, necessitating novel approaches and further investigation in this field to develop effective management strategies for hypertension. A chart of our working hypothesis is summarized in Figure 1.
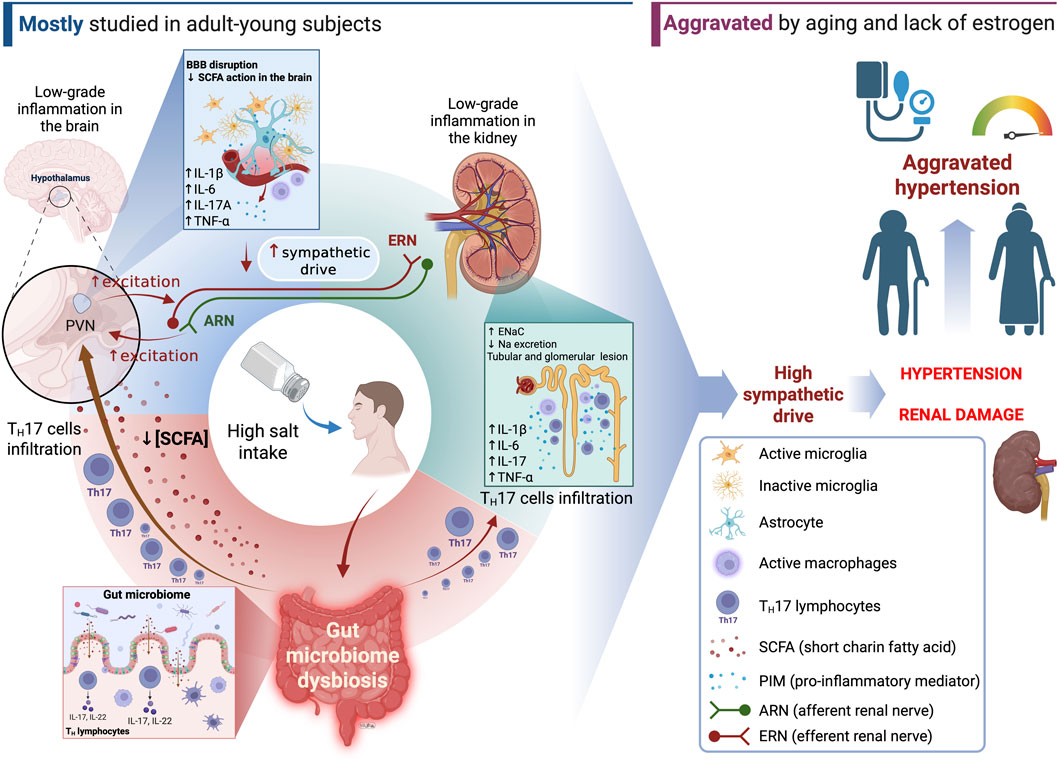
Figure 1. Summary of the major findings and working hypothesis for some of the mechanisms underlying the SSH. Gut microbiome dysbiosis can influence TH cell differentiation and the chemical diversity/quantity of the short chain fatty acids produced by bacterial metabolisms. These changes can promote immune responses in the central nervous system (particularly in the paraventricular nucleus of the hypothalamus–PVN) that shift the autonomic control of the BP toward a more active sympathetic drive. In addition, high salt intake and gut dysbiosis can induce immune signaling and immune cell infiltration in the kidney, affecting sodium handling and renal function. Together, those changes can favor an increase in total peripheral resistance and sodium/volume retention over time, largely contributing the hypertension development. It is noteworthy that age and sex differences can influence the ways such changes take place and aggravate hypertension under these low-grade inflammation state, further leading the higher levels of BP. SCFA, short chain fatty acids; PIM, pro-inflammatory cytokines; ARN, afferent renal nerve; ERN, efferent renal nerve; IL-1β, interleukine 1β; IL-6, interleukine 6; IL-17A, interleukine 17A; TNF-α, tumor necrosis factor α.
Author contributions
LC: Writing – review and editing, Conceptualization, Writing – original draft. RW: Supervision, Writing – review and editing, Conceptualization, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Institutes of Health (NIH) grants R01AG062515, and R01 AG075963 and Hevolution Foundation grants HF-GRO-23–1199246–43 to RDW; Minas Gerais State Research Foundation (FAPEMIG; Grant# APQ-02105-24); Federal University of Ouro Preto; Higher Education Personnel Improvement Coordination (CAPES); National Council for Scientific and Technological Development (CNPq) to LMC.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abais-Battad J. M., Saravia F. L., Lund H., Dasinger J. H., Fehrenbach D. J., Alsheikh A. J., et al. (2021). Dietary influences on the dahl SS rat gut microbiota and its effects on salt-sensitive hypertension and renal damage. Acta Physiol. (Oxf) 232 (4), e13662. doi:10.1111/apha.13662
Abbott N. J., Patabendige A. A., Dolman D. E., Yusof S. R., Begley D. J. (2010). Structure and function of the blood-brain barrier. Neurobiol. Dis. 37 (1), 13–25. doi:10.1016/j.nbd.2009.07.030
Abraham C. R., Li A. (2022). Aging-suppressor Klotho: prospects in diagnostics and therapeutics. Ageing Res. Rev. 82, 101766. doi:10.1016/j.arr.2022.101766
Adams J. M., McCarthy J. J., Stocker S. D. (2008). Excess dietary salt alters angiotensinergic regulation of neurons in the rostral ventrolateral medulla. Hypert 52 (5), 932–937. doi:10.1161/HYPERTENSIONAHA.108.118935
Afsar B., Kuwabara M., Ortiz A., Yerlikaya A., Siriopol D., Covic A., et al. (2018). Salt intake and immunity. Hypert 72 (1), 19–23. doi:10.1161/HYPERTENSIONAHA.118.11128
Aguiar S. L. F., Miranda M. C. G., Guimaraes M. A. F., Santiago H. C., Queiroz C. P., Cunha P. D. S., et al. (2017). High-salt diet induces IL-17-Dependent gut inflammation and exacerbates colitis in mice. Front. Immunol. 8, 1969. doi:10.3389/fimmu.2017.01969
Al-Habsi N., Al-Khalili M., Haque S. A., Elias M., Olqi N. A., Al Uraimi T. (2024). Health benefits of prebiotics, probiotics, synbiotics, and postbiotics. Nutrients 16 (22), 3955. doi:10.3390/nu16223955
Al-Nimer M. M., Alhusseiny A. (2016). Assessment of serum interleukins-17 and 1β levels in hypertensive patients treated with angiotensin converting enzyme And/Or angiotensin II receptor blockers. Saudi Journ Healt Sci. 5 (3), 125–129. doi:10.4103/2278-0521.195814
Alvarez Li F. C., Espinosa Brito A. D., Ordunez Garcia P. O., Silva Aycaguer L. C. (1999). Risk markers and high blood pressure. The Cienfuegos global project. Longitudinal study 1992-1994. Rev. Investig. clinica; organo del Hosp. Enfermedades la Nutr. 51 (3), 151–158.
Antunes-Rodrigues J., de Castro M., Elias L. L., Valenca M. M., McCann S. M. (2004). Neuroendocrine control of body fluid metabolism. Physiol. Rev. 84 (1), 169–208. doi:10.1152/physrev.00017.2003
Asirvatham-Jeyaraj N., King A. J., Northcott C. A., Madan S., Fink G. D. (2013). Cyclooxygenase-1 inhibition attenuates angiotensin II-salt hypertension and neurogenic pressor activity in the rat. Am. J. physiology Heart circulatory physiology 305 (10), H1462–H1470. doi:10.1152/ajpheart.00245.2013
Athirakul K., Kim H. S., Audoly L. P., Smithies O., Coffman T. M. (2001). Deficiency of COX-1 causes natriuresis and enhanced sensitivity to ACE inhibition. Kidney Int. 60 (6), 2324–2329. doi:10.1046/j.1523-1755.2001.00072.x
Auron P. E., Webb A. C., Rosenwasser L. J., Mucci S. F., Rich A., Wolff S. M., et al. (1984). Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proc. Natl. Acad. Sci. U. S. A. 81 (24), 7907–7911. doi:10.1073/pnas.81.24.7907
Bader M. S., Meyer S. C. (2022). JAK2 in myeloproliferative neoplasms: still a protagonist. Pharm. (Basel) 15 (2), 160. doi:10.3390/ph15020160
Bagordo D., Rossi G. P., Delles C., Wiig H., Rossitto G. (2024). Tangram of sodium and fluid balance. Hypertension 81 (3), 490–500. doi:10.1161/HYPERTENSIONAHA.123.19569
Bailey M. A., Dhaun N. (2024). Salt sensitivity: causes, consequences, and recent advances. Hypertension 81 (3), 476–489. doi:10.1161/HYPERTENSIONAHA.123.17959
Balan Y., Packirisamy R. M., Mohanraj P. S. (2022). High dietary salt intake activates inflammatory cascades via Th17 immune cells: impact on health and diseases. Arch. Med. Sci. 18 (2), 459–465. doi:10.5114/aoms.2020.96344
Baldi S., Mundula T., Nannini G., Amedei A. (2021). Microbiota shaping - the effects of probiotics, prebiotics, and fecal microbiota transplant on cognitive functions: a systematic review. World J. Gastroenterol. 27 (39), 6715–6732. doi:10.3748/wjg.v27.i39.6715
Banek C. T., Knuepfer M. M., Foss J. D., Fiege J. K., Asirvatham-Jeyaraj N., Van Helden D., et al. (2016). Resting afferent renal nerve discharge and renal inflammation: elucidating the role of afferent and efferent renal nerves in deoxycorticosterone acetate salt hypertension. Hypertension 68 (6), 1415–1423. doi:10.1161/HYPERTENSIONAHA.116.07850
Barbaro N. R., Foss J. D., Kryshtal D. O., Tsyba N., Kumaresan S., Xiao L., et al. (2017). Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. 21 (4), 1009–1020. doi:10.1016/j.celrep.2017.10.002
Baumann D., Van Helden D., Evans L. C., Vulchanova L., Dayton A., Osborn J. W. (2024). IL-1R mediated activation of renal sensory nerves in DOCA-salt hypertension. Hypertension 81 (8), 1811–1821. doi:10.1161/HYPERTENSIONAHA.123.22620
Biancardi V. C., Son S. J., Ahmadi S., Filosa J. A., Stern J. E. (2014). Circulating angiotensin II gains access to the hypothalamus and brain stem during hypertension via breakdown of the blood-brain barrier. Hypertension 63 (3), 572–579. doi:10.1161/HYPERTENSIONAHA.113.01743
Bie P., Evans R. G. (2017a). Neurohumoral-renal interactions in blood pressure control. Acta Physiol. (Oxf) 219 (1), 239–240. doi:10.1111/apha.12748
Bie P., Evans R. G. (2017b). Normotension, hypertension and body fluid regulation: brain and kidney. Acta Physiol. (Oxf) 219 (1), 288–304. doi:10.1111/apha.12718
Bier A., Braun T., Khasbab R., Di Segni A., Grossman E., Haberman Y., et al. (2018). A high salt diet modulates the gut microbiota and short chain fatty acids production in a salt-sensitive hypertension rat model. Nutrients 10 (9), 1154. doi:10.3390/nu10091154
Binger K. J., Gebhardt M., Heinig M., Rintisch C., Schroeder A., Neuhofer W., et al. (2015). High salt reduces the activation of IL-4- and IL-13-stimulated macrophages. J. Clin. investigation 125 (11), 4223–4238. doi:10.1172/JCI80919
Boos C. J., Lip G. Y. (2006). Is hypertension an inflammatory process? Curr. Pharm. Des. 12 (13), 1623–1635. doi:10.2174/138161206776843313
Boraschi D. (2022). What is IL-1 for? The functions of Interleukin-1 across evolution. Front. Immunol. 13, 872155. doi:10.3389/fimmu.2022.872155
Bras J. P., Bravo J., Freitas J., Barbosa M. A., Santos S. G., Summavielle T., et al. (2020). TNF-alpha-induced microglia activation requires miR-342: impact on NF-kB signaling and neurotoxicity. Cell Death Dis. 11 (6), 415. doi:10.1038/s41419-020-2626-6
Brognara F., Castania J. A., Kanashiro A., Dias D. P. M., Salgado H. C. (2021a). Physiological sympathetic activation reduces systemic inflammation: role of baroreflex and chemoreflex. Front. Immunol. 12, 637845. doi:10.3389/fimmu.2021.637845
Brognara F., Castania J. A., Ribeiro A. B., Santos-Junior N. N., Salgado H. C. (2021b). The bezold-jarisch Reflex and the inflammatory response modulation in unanesthetized endotoxemic rats. Front. Physiol. 12, 745285. doi:10.3389/fphys.2021.745285
Bruning J. R., Ghannam R. B., Chen Q.-h., Shan Z., Miodonski G., Techtmann S., et al. (2020). Microbial derived short chain fatty-acids and autonomic regulation of cardiovascular function. FASEB J. 34 (S1), 1. doi:10.1096/fasebj.2020.34.s1.06549
Buttler L., Jordao M. T., Fragas M. G., Ruggeri A., Ceroni A., Michelini L. C. (2017). Maintenance of blood-brain barrier integrity in hypertension: a novel benefit of exercise training for autonomic control. Front. Physiol. 8, 1048. doi:10.3389/fphys.2017.01048
Calderon-Perez L., Gosalbes M. J., Yuste S., Valls R. M., Pedret A., Llaurado E., et al. (2020). Gut metagenomic and short chain fatty acids signature in hypertension: a cross-sectional study. Sci. Rep. 10 (1), 6436. doi:10.1038/s41598-020-63475-w
Canale M. P., Noce A., Di Lauro M., Marrone G., Cantelmo M., Cardillo C., et al. (2021). Gut dysbiosis and Western diet in the pathogenesis of essential arterial hypertension: a narrative review. Nutrients 13 (4), 1162. doi:10.3390/nu13041162
Cao Y., Yu Y., Xue B., Wang Y., Chen X., Beltz T. G., et al. (2021). IL (Interleukin)-17A acts in the brain to drive neuroinflammation, sympathetic activation, and hypertension. Hypertension 78 (5), 1450–1462. doi:10.1161/HYPERTENSIONAHA.121.18219
Capone C., Faraco G., Park L., Cao X., Davisson R. L., Iadecola C. (2011). The cerebrovascular dysfunction induced by slow pressor doses of angiotensin II precedes the development of hypertension. Am. J. physiology Heart circulatory physiology 300 (1), H397–H407. doi:10.1152/ajpheart.00679.2010
Carabotti M., Scirocco A., Maselli M. A., Severi C. (2015). The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 28 (2), 203–209.
Cardoso A. M. (2024). Microbial influence on blood pressure: unraveling the complex relationship for health insights. Microbiome Res. Rep. 3 (2), 22. doi:10.20517/mrr.2023.73
Carnevale D., Perrotta M., Pallante F., Fardella V., Iacobucci R., Fardella S., et al. (2016). A cholinergic-sympathetic pathway primes immunity in hypertension and mediates brain-to-spleen communication. Nat. Commun. 7 (1), 13035. doi:10.1038/ncomms13035
Castillo A., Islam M. T., Prieto M. C., Majid D. S. (2012). Tumor necrosis factor-alpha receptor type 1, not type 2, mediates its acute responses in the kidney. Am. J. physiology Ren. physiology 302 (12), F1650–F1657. doi:10.1152/ajprenal.00426.2011
Chakraborty S., Galla S., Cheng X., Yeo J. Y., Mell B., Singh V., et al. (2018). Salt-Responsive metabolite, beta-Hydroxybutyrate, attenuates hypertension. Cell Rep. 25 (3), 677–689. doi:10.1016/j.celrep.2018.09.058
Chamarthi B., Williams G. H., Ricchiuti V., Srikumar N., Hopkins P. N., Luther J. M., et al. (2011). Inflammation and hypertension: the interplay of interleukin-6, dietary sodium, and the renin-angiotensin system in humans. Am. J. Hypertens. 24 (10), 1143–1148. doi:10.1038/ajh.2011.113
Chandrasoma P., Taylor C. R. (1998) “Chapter 3. The acute inflammatory response,” in Concise pathology. 3. New York, NY: The McGraw-Hill Companies.
Charney A. N., Micic L., Egnor R. W. (1998). Nonionic diffusion of short-chain fatty acids across rat Colon. Am. J. physiology 274 (3), G518–G524. doi:10.1152/ajpgi.1998.274.3.G518
Chen L., He F. J., Dong Y., Huang Y., Wang C., Harshfield G. A., et al. (2020). Modest sodium reduction increases circulating short-chain fatty acids in untreated hypertensives: a randomized, double-blind, placebo-controlled trial. Hypertension 76 (1), 73–79. doi:10.1161/HYPERTENSIONAHA.120.14800
Chrysant S. G. (2024). The role of gut microbiota in the development of salt-sensitive hypertension and the possible preventive effect of exercise. Expert Rev. Cardiovasc Ther. 22 (6), 265–271. doi:10.1080/14779072.2024.2364031
Citterio L., Delli Carpini S., Lupoli S., Brioni E., Simonini M., Fontana S., et al. (2020). Klotho gene in human salt-sensitive hypertension. Clin. J. Am. Soc. Nephrol. 15 (3), 375–383. doi:10.2215/CJN.08620719
Coady M. J., Chang M. H., Charron F. M., Plata C., Wallendorff B., Sah J. F., et al. (2004). The human tumour suppressor gene SLC5A8 expresses a na+-Monocarboxylate cotransporter. J. physiology 557 (Pt 3), 719–731. doi:10.1113/jphysiol.2004.063859
Crowley S. D., Song Y. S., Sprung G., Griffiths R., Sparks M., Yan M., et al. (2010). A role for angiotensin II type 1 receptors on bone marrow-derived cells in the pathogenesis of angiotensin II-dependent hypertension. Hypertension 55 (1), 99–108. doi:10.1161/HYPERTENSIONAHA.109.144964
Dai Y., Shen Z., Khachatryan L. G., Vadiyan D. E., Karampoor S., Mirzaei R. (2023). Unraveling mechanistic insights into the role of microbiome in neurogenic hypertension: a comprehensive review. Pathol. Res. Pract. 249, 154740. doi:10.1016/j.prp.2023.154740
Dampney R. A., Michelini L. C., Li D. P., Pan H. L. (2018). Regulation of sympathetic vasomotor activity by the hypothalamic paraventricular nucleus in normotensive and hypertensive states. Am. J. physiology Heart circulatory physiology 315 (5), H1200-H1214–H14. doi:10.1152/ajpheart.00216.2018
de Assis Braga D. C., Carlos Batista M. A., Guerra-Sa R., Alves da Silva T. C., Carneiro M. A. A., da Silva Lanna M. C., et al. (2025). Psidium guajava leaves extract alters colonic microbiome composition and reduces intestinal sodium absorption in rats exposed to a high-sodium diet. J. Pharm. Pharmacol. 77 (1), 111–126. doi:10.1093/jpp/rgae137
de Souza J. A., Becker L. K., Batista M. A. C., de Assis Braga D. C., Gomes P. M., Alzamora A. C., et al. (2021). Swimming training improves cardiovascular autonomic dysfunctions and prevents renal damage in rats fed a high-sodium diet from weaning. Exp. Physiol. 106 (2), 412–426. doi:10.1113/EP088892
Demirci M., Afolabi J. M., Kirabo A. (2025). Aging and sex differences in salt sensitivity of blood pressure. Clin. Sci. (Lond). 139 (2), 199–212. doi:10.1042/CS20240788
Deng Z., Wang Y., Zhou L., Shan Y., Tan S., Cai W., et al. (2017). High salt-induced activation and expression of inflammatory cytokines in cultured astrocytes. Cell Cycle 16 (8), 785–794. doi:10.1080/15384101.2017.1301330
Dickinson K. M., Clifton P. M., Burrell L. M., Barrett P. H., Keogh J. B. (2014). Postprandial effects of a high salt meal on serum sodium, arterial stiffness, markers of nitric oxide production and markers of endothelial function. Atherosclerosis 232 (1), 211–216. doi:10.1016/j.atherosclerosis.2013.10.032
Dinh Q. N., Vinh A., Lo C., Wong Zhang D. E., Figueiredo Galvao H. B., Selvaraji S., et al. (2025). Hypertension promotes neuroinflammation, brain injury and cognitive impairment. Brain Behav. Immun. Health 48, 101059. doi:10.1016/j.bbih.2025.101059
Du C. P., Wang M., Geng C., Hu B., Meng L., Xu Y., et al. (2020). Activity-induced SUMOylation of neuronal nitric oxide synthase is associated with plasticity of synaptic transmission and extracellular signal-regulated kinase 1/2 signaling. Antioxidants and redox Signal. 32 (1), 18–34. doi:10.1089/ars.2018.7669
Dubbelaar M. L., Kracht L., Eggen B. J. L., Boddeke E. (2018). The kaleidoscope of microglial phenotypes. Front. Immunol. 9, 1753. doi:10.3389/fimmu.2018.01753
Ekperikpe U. S., Poudel B., Shields C. A., Mandal S., Cornelius D. C., Williams J. M. (2023). Neutralizing MIP3α reduces renal immune cell infiltration and progressive renal injury in young obese dahl salt-sensitive rats. J. Pharmacol. Exp. Ther. 384 (3), 445–454. doi:10.1124/jpet.122.001298
Elijovich F., Weinberger M. H., Anderson C. A., Appel L. J., Bursztyn M., Cook N. R., et al. (2016). Salt sensitivity of blood pressure: a scientific statement from the American heart association. Hypertension 68 (3), e7–e46. doi:10.1161/HYP.0000000000000047
Elijovich F., Laffer C. L., Sahinoz M., Pitzer A., Ferguson J. F., Kirabo A. (2020). The gut microbiome, inflammation, and salt-sensitive hypertension. Curr. Hypertens. Rep. 22 (10), 79. doi:10.1007/s11906-020-01091-9
Faraco G. (2024). Dietary salt, vascular dysfunction, and cognitive impairment. Cardiovasc Res. 120, 2349–2359. doi:10.1093/cvr/cvae229
Faraco G., Sugiyama Y., Lane D., Garcia-Bonilla L., Chang H., Santisteban M. M., et al. (2016). Perivascular macrophages mediate the neurovascular and cognitive dysfunction associated with hypertension. J. Clin. investigation 126 (12), 4674–4689. doi:10.1172/JCI86950
Ferguson A. V., Latchford K. J., Samson W. K. (2008). The paraventricular nucleus of the hypothalamus - a potential target for integrative treatment of autonomic dysfunction. Expert Opin. Ther. Targets 12 (6), 717–727. doi:10.1517/14728222.12.6.717
Ferrari A., Gordon F. J., Mark A. L. (1984a). Impairment of cardiopulmonary baroreflexes in dahl salt-sensitive rats fed low salt. Am. J. physiology 247 (1 Pt 2), H119–H123. doi:10.1152/ajpheart.1984.247.1.H119
Ferrari A. U., Gordon F. J., Mark A. L. (1984b). Primary impairment of cardiopulmonary baroreflexes in dahl salt-sensitive rats. J. Hypertens. Suppl. 2 (3), S401–S403.
Frame A. A., Wainford R. D. (2017). Renal sodium handling and sodium sensitivity. Kidney Res. Clin. Pract. 36 (2), 117–131. doi:10.23876/j.krcp.2017.36.2.117
Franco M., Tapia E., Bautista R., Pacheco U., Santamaria J., Quiroz Y., et al. (2013). Impaired pressure natriuresis resulting in salt-sensitive hypertension is caused by tubulointerstitial immune cell infiltration in the kidney. Am. J. physiology Ren. physiology 304 (7), F982–F990. doi:10.1152/ajprenal.00463.2012
Furusawa Y., Obata Y., Fukuda S., Endo T. A., Nakato G., Takahashi D., et al. (2013). Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504 (7480), 446–450. doi:10.1038/nature12721
Fusco W., Lorenzo M. B., Cintoni M., Porcari S., Rinninella E., Kaitsas F., et al. (2023). Short-chain fatty-acid-producing bacteria: key components of the human gut microbiota. Nutrients 15 (9), 2211. doi:10.3390/nu15092211
Gao H., Bigalke J., Jiang E., Fan Y., Chen B., Chen Q. H., et al. (2022). TNFα triggers an augmented inflammatory response in brain neurons from dahl salt-sensitive rats compared with normal sprague dawley rats. Cell Mol. Neurobiol. 42 (6), 1787–1800. doi:10.1007/s10571-021-01056-9
Geerling J. C., Loewy A. D. (2008). Central regulation of sodium appetite. Exp. Physiol. 93 (2), 177–209. doi:10.1113/expphysiol.2007.039891
Gilman T. L., Mitchell N. C., Daws L. C., Toney G. M. (2019). Neuroinflammation contributes to high salt intake-augmented neuronal activation and active coping responses to acute stress. Int. J. Neuropsychopharmacol. 22 (2), 137–142. doi:10.1093/ijnp/pyy099
Gomes P. M., Sa R. W. M., Aguiar G. L., Paes M. H. S., Alzamora A. C., Lima W. G., et al. (2017). Chronic high-sodium diet intake after weaning lead to neurogenic hypertension in adult wistar rats. Sci. Rep. 7 (1), 5655. doi:10.1038/s41598-017-05984-9
González-Marrero I., Castañeyra-Ruiz L., González-Toledo J M., Castañeyra-Ruiz A., de Paz-Carmona H., Ruiz-Mayor L., et al. (2012). High blood pressure effects on the brain barriers and choroid plexus secretion. Neurosci. Med. 03 (01), 60–64. doi:10.4236/nm.2012.31009
Grebenciucova E., VanHaerents S. (2023). Interleukin 6: at the interface of human health and disease. Front. Immunol. 14, 1255533. doi:10.3389/fimmu.2023.1255533
Grillo A., Salvi L., Coruzzi P., Salvi P., Parati G. (2019). Sodium intake and hypertension. Nutrients 11 (9), 1970–1986. doi:10.3390/nu11091970
Grollman A., Harrison T. R., Mason M. F., Baxter J., Crampton J., Reichsman F. (1945). Sodium restriction in the diet for hypertension. Jama-J Am. Med. Assoc. 129 (8), 533–537.
Groppelli A., Giorgi D. M., Omboni S., Parati G., Mancia G. (1992). Persistent blood pressure increase induced by heavy smoking. J. Hypertens. 10 (5), 495–499. doi:10.1097/00004872-199205000-00014
Gruol D. L. (2015). IL-6 regulation of synaptic function in the CNS. Neuropharmacology 96 (Pt A), 42–54. doi:10.1016/j.neuropharm.2014.10.023
Guyenet P. G. (2006). The sympathetic control of blood pressure. Nat. Rev. Neurosci. 7 (5), 335–346. doi:10.1038/nrn1902
Guzik T. J., Hoch N. E., Brown K. A., McCann L. A., Rahman A., Dikalov S., et al. (2007). Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 204 (10), 2449–2460. doi:10.1084/jem.20070657
Ha S. K. (2014). Dietary salt intake and hypertension. Electrolyte Blood Press 12 (1), 7–18. doi:10.5049/EBP.2014.12.1.7
Hanoun M., Maryanovich M., Arnal-Estape A., Frenette P. S. (2015). Neural regulation of hematopoiesis, inflammation, and cancer. Neuron 86 (2), 360–373. doi:10.1016/j.neuron.2015.01.026
Harrison D. G., Marvar P. J., Titze J. M. (2012). Vascular inflammatory cells in hypertension. Front. Physiol. 3 (128), 128. doi:10.3389/fphys.2012.00128
Harwani S. C., Chapleau M. W., Legge K. L., Ballas Z. K., Abboud F. M. (2012). Neurohormonal modulation of the innate immune system is proinflammatory in the prehypertensive spontaneously hypertensive rat, a genetic model of essential hypertension. Circulation Res. 111 (9), 1190–1197. doi:10.1161/CIRCRESAHA.112.277475
Hashmat S., Rudemiller N., Lund H., Abais-Battad J. M., Van Why S., Mattson D. L. (2016). Interleukin-6 inhibition attenuates hypertension and associated renal damage in dahl salt-sensitive rats. Am. J. physiology Ren. physiology 311 (3), F555–F561. doi:10.1152/ajprenal.00594.2015
Hatcher M. D., Duan R., Lee D. L. (2024). 544 Interleukin-6 protects renal dysfunction in mouse models of hypertension and salt-sensitive hypertension. J. Clin. Transl. Sci. 8 (s1), 162. doi:10.1017/cts.2024.464
Haywood J. R., Buggy J., Fink G. D., DiBona G. F., Johnson A. K., Brody M. J. (1984). Alterations in cerebrospinal fluid sodium and osmolality in rats during one-kidney, one-wrap renal hypertension. Clin. Exp. Pharmacol. and physiology 11 (5), 545–549. doi:10.1111/j.1440-1681.1984.tb00865.x
He J., Zhang P., Shen L., Niu L., Tan Y., Chen L., et al. (2020). Short-chain fatty acids and their association with signalling pathways in inflammation, glucose and lipid metabolism. Int. J. Mol. Sci. 21 (17), 6356. doi:10.3390/ijms21176356
Hinojosa-Laborde C., Craig T., Zheng W., Ji H., Haywood J. R., Sandberg K. (2004). Ovariectomy augments hypertension in aging female dahl salt-sensitive rats. Hypertension 44 (4), 405–409. doi:10.1161/01.HYP.0000142893.08655.96
Hu L., Zhu S., Peng X., Li K., Peng W., Zhong Y., et al. (2020). High salt elicits brain inflammation and cognitive dysfunction, accompanied by alternations in the gut microbiota and decreased SCFA production. J. Alzheimer's Dis. JAD. 77 (2), 629–640. doi:10.3233/JAD-200035
Huang B. S., Van Vliet B. N., Leenen F. H. (2004). Increases in CSF [na+] precede the increases in blood pressure in dahl S rats and SHR on a high-salt diet. Am. J. physiology Heart circulatory physiology 287 (3), H1160–H1166. doi:10.1152/ajpheart.00126.2004
Huang B. S., Amin M. S., Leenen F. H. (2006). The central role of the brain in salt-sensitive hypertension. Curr. Opin. Cardiol. 21 (4), 295–304. doi:10.1097/01.hco.0000231398.64362.94
Huang X., Hussain B., Chang J. (2021). Peripheral inflammation and blood-brain barrier disruption: effects and mechanisms. CNS Neurosci. Ther. 27 (1), 36–47. doi:10.1111/cns.13569
Hucke S., Eschborn M., Liebmann M., Herold M., Freise N., Engbers A., et al. (2016). Sodium chloride promotes pro-inflammatory macrophage polarization thereby aggravating CNS autoimmunity. J. Autoimmun. 67, 90–101. doi:10.1016/j.jaut.2015.11.001
Huppert J., Closhen D., Croxford A., White R., Kulig P., Pietrowski E., et al. (2010). Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. 24 (4), 1023–1034. doi:10.1096/fj.09-141978
Itani H. A., Harrison D. G. (2015). Memories that last in hypertension. Am. J. physiology Ren. physiology 308 (11), F1197–F1199. doi:10.1152/ajprenal.00633.2014
Iwakura Y., Ishigame H., Saijo S., Nakae S. (2011). Functional specialization of interleukin-17 family members. Immunity 34 (2), 149–162. doi:10.1016/j.immuni.2011.02.012
Jantsch J., Schatz V., Friedrich D., Schroder A., Kopp C., Siegert I., et al. (2015). Cutaneous na+ storage strengthens the antimicrobial barrier function of the skin and boosts macrophage-driven host defense. Cell Metab. 21 (3), 493–501. doi:10.1016/j.cmet.2015.02.003
Jiang E., Chapp A. D., Fan Y., Larson R. A., Hahka T., Huber M. J., et al. (2018). Expression of proinflammatory cytokines is upregulated in the hypothalamic paraventricular nucleus of dahl salt-sensitive hypertensive rats. Front. Physiol. 9, 104. doi:10.3389/fphys.2018.00104
John G. B., Cheng C. Y., Kuro-o M. (2011). Role of klotho in aging, phosphate metabolism, and CKD. Am. J. Kidney Dis. 58 (1), 127–134. doi:10.1053/j.ajkd.2010.12.027
Jorg S., Kissel J., Manzel A., Kleinewietfeld M., Haghikia A., Gold R., et al. (2016). High salt drives Th17 responses in experimental autoimmune encephalomyelitis without impacting myeloid dendritic cells. Exp. Neurol. 279, 212–222. doi:10.1016/j.expneurol.2016.03.010
Kamat N. V., Thabet S. R., Xiao L., Saleh M. A., Kirabo A., Madhur M. S., et al. (2015). Renal transporter activation during angiotensin-II hypertension is blunted in interferon-gamma-/- and interleukin-17A-/- mice. Hypertension 65 (3), 569–576. doi:10.1161/HYPERTENSIONAHA.114.04975
Katayama Y., Battista M., Kao W. M., Hidalgo A., Peired A. J., Thomas S. A., et al. (2006). Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 124 (2), 407–421. doi:10.1016/j.cell.2005.10.041
Katsi V., Marketou M., Maragkoudakis S., Didagelos M., Charalambous G., Parthenakis F., et al. (2020). Blood-brain barrier dysfunction: the undervalued frontier of hypertension. J. Hum. Hypertens. 34 (10), 682–691. doi:10.1038/s41371-020-0352-2
Kawarazaki W., Mizuno R., Nishimoto M., Ayuzawa N., Hirohama D., Ueda K., et al. (2020). Salt causes aging-associated hypertension via vascular Wnt5a under klotho deficiency. J. Clin. investigation 130 (8), 4152–4166. doi:10.1172/JCI134431
Kebir H., Kreymborg K., Ifergan I., Dodelet-Devillers A., Cayrol R., Bernard M., et al. (2007). Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 13 (10), 1173–1175. doi:10.1038/nm1651
Kettenmann H., Hanisch U. K., Noda M., Verkhratsky A. (2011). Physiology of microglia. Physiol. Rev. 91 (2), 461–553. doi:10.1152/physrev.00011.2010
Kim K., Nist K. M., Puleo F., McKenna J., Wainford R. D. (2024). Sex differences in dietary sodium evoked NCC regulation and blood pressure in Male and female sprague-dawley, dahl salt-resistant, and dahl salt-sensitive rats. Am. J. physiology Ren. physiology 327 (2), F277–F289. doi:10.1152/ajprenal.00150.2023
Kleinewietfeld M., Manzel A., Titze J., Kvakan H., Yosef N., Linker R. A., et al. (2013). Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496 (7446), 518–522. doi:10.1038/nature11868
Konsman J. P. (2022). Cytokines in the brain and neuroinflammation: we didn't starve the fire. Pharm. (Basel) 15 (2), 140. doi:10.3390/ph15020140
Lauar M. R., Evans L. C., Van Helden D., Fink G. D., Banek C. T., Menani J. V., et al. (2023). Renal and hypothalamic inflammation in renovascular hypertension: role of afferent renal nerves. Am. J. physiology Regul. Integr. Comp. physiology 325 (4), R411–R422. doi:10.1152/ajpregu.00072.2023
Leenen F. H., Ruzicka M., Huang B. S. (2002). The brain and salt-sensitive hypertension. Curr. Hypertens. Rep. 4 (2), 129–135. doi:10.1007/s11906-002-0037-y
Li K., Guo D., Zhu H., Hering-Smith K. S., Hamm L. L., Ouyang J., et al. (2010). Interleukin-6 stimulates epithelial sodium channels in mouse cortical collecting duct cells. Am. J. physiology Regul. Integr. Comp. physiology 299 (2), R590–R595. doi:10.1152/ajpregu.00207.2009
Li L. Z., Tao S. B., Ma L., Fu P. (2019). Roles of short-chain fatty acids in kidney diseases. Chin. Med. J. Engl. 132 (10), 1228–1232. doi:10.1097/CM9.0000000000000228
Li J., Shui X., Sun R., Wan L., Zhang B., Xiao B., et al. (2021). Microglial phenotypic transition: signaling pathways and influencing modulators involved in regulation in central nervous system diseases. Front. Cell Neurosci. 15, 736310. doi:10.3389/fncel.2021.736310
Li X., Alu A., Wei Y., Wei X., Luo M. (2022). The modulatory effect of high salt on immune cells and related diseases. Cell Prolif. 55 (9), e13250. doi:10.1111/cpr.13250
Lima C. T., Carvalho F. M., Quadros Cde A., Goncalves H. R., Silva Junior J. A., Peres M. F., et al. (1999). Arterial hypertension and alcoholism among workers in an oil refinery. Revista Panam. de salud publica = Pan Am. J. public health 6 (3), 185–191. doi:10.1590/s1020-49891999000800006
Linz D., Wirth K., Linz W., Heuer H. O., Frick W., Hofmeister A., et al. (2012). Antihypertensive and laxative effects by pharmacological inhibition of sodium-proton-exchanger subtype 3-mediated sodium absorption in the gut. Hypertension 60 (6), 1560–1567. doi:10.1161/HYPERTENSIONAHA.112.201590
Liu P., Wang Y., Yang G., Zhang Q., Meng L., Xin Y., et al. (2021). The role of short-chain fatty acids in intestinal barrier function, inflammation, oxidative stress, and colonic carcinogenesis. Pharmacol. Res. 165, 105420. doi:10.1016/j.phrs.2021.105420
Liu G., Cheng J., Zhang T., Shao Y., Chen X., Han L., et al. (2022). Inhibition of microbiota-dependent trimethylamine N-Oxide production ameliorates high salt diet-induced sympathetic excitation and hypertension in rats by attenuating central neuroinflammation and oxidative stress. Front. Pharmacol. 13, 856914. doi:10.3389/fphar.2022.856914
Liu X. F., Shao J. H., Liao Y. T., Wang L. N., Jia Y., Dong P. J., et al. (2023). Regulation of short-chain fatty acids in the immune system. Front. Immunol. 14, 1186892. doi:10.3389/fimmu.2023.1186892
Louis P., Flint H. J. (2017). Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 19 (1), 29–41. doi:10.1111/1462-2920.13589
Lu Y., Zhang Y., Zhao X., Shang C., Xiang M., Li L., et al. (2022). Microbiota-derived short-chain fatty acids: implications for cardiovascular and metabolic disease. Front. Cardiovasc. Med. 9, 900381–902022. doi:10.3389/fcvm.2022.900381
Luo H., Liu H. Z., Zhang W. W., Matsuda M., Lv N., Chen G., et al. (2019). Interleukin-17 regulates neuron-glial communications, synaptic transmission, and neuropathic pain after chemotherapy. Cell Rep. 29 (8), 2384–2397. doi:10.1016/j.celrep.2019.10.085
Maaliki D., Itani M. M., Itani H. A. (2022). Pathophysiology and genetics of salt-sensitive hypertension. Front. Physiol. 13, 1001434. doi:10.3389/fphys.2022.1001434
Madhur M. S., Lob H. E., McCann L. A., Iwakura Y., Blinder Y., Guzik T. J., et al. (2010). Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 55 (2), 500–507. doi:10.1161/HYPERTENSIONAHA.109.145094
D. S. A. Majid (2011). “Tumor necrosis Factor-α and kidney function: experimental findings in mice,” Advances in TNF family research (New York, NY: Springer New York).
Majid D. S. A., Prieto M. C., Castillo A., Chamberlain C., Navar L. G. (2024). Augmentation of nitric oxide deficient hypertension by high salt diet is associated with reduced TNF-Alpha receptor type 1 expression in the kidneys. Am. J. Hypertens. 37 (9), 717–725. doi:10.1093/ajh/hpae066
Marques F. Z., Mackay C. R., Kaye D. M. (2018). Beyond gut feelings: how the gut microbiota regulates blood pressure. Nat. Rev. Cardiol. 15 (1), 20–32. doi:10.1038/nrcardio.2017.120
Matthias J., Heink S., Picard F., Zeitrag J., Kolz A., Chao Y. Y., et al. (2020). Salt generates antiinflammatory Th17 cells but amplifies pathogenicity in proinflammatory cytokine microenvironments. J. Clin. investigation 130 (9), 4587–4600. doi:10.1172/JCI137786
Mattson D. L. (2014). Infiltrating immune cells in the kidney in salt-sensitive hypertension and renal injury. Am. J. physiology Ren. physiology 307 (5), F499–F508. doi:10.1152/ajprenal.00258.2014
Mattson D. L. (2019). Immune mechanisms of salt-sensitive hypertension and renal end-organ damage. Nat. Rev. Nephrol. 15 (5), 290–300. doi:10.1038/s41581-019-0121-z
Mattson D. L., Lund H., Guo C., Rudemiller N., Geurts A. M., Jacob H. (2013). Genetic mutation of recombination activating gene 1 in dahl salt-sensitive rats attenuates hypertension and renal damage. Am. J. physiology Regul. Integr. Comp. physiology 304 (6), R407–R414. doi:10.1152/ajpregu.00304.2012
Mehaffey E., Majid D. S. A. (2017). Tumor necrosis factor-alpha, kidney function, and hypertension. Am. J. physiology Ren. physiology 313 (4), F1005-F1008–F8. doi:10.1152/ajprenal.00535.2016
Mell B., Jala V. R., Mathew A. V., Byun J., Waghulde H., Zhang Y., et al. (2015). Evidence for a link between gut microbiota and hypertension in the dahl rat. Physiol. Genomics 47 (6), 187–197. doi:10.1152/physiolgenomics.00136.2014
Menu P., Vince J. E. (2011). The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin. Exp. Immunol. 166 (1), 1–15. doi:10.1111/j.1365-2249.2011.04440.x
Minihane A. M., Vinoy S., Russell W. R., Baka A., Roche H. M., Tuohy K. M., et al. (2015). Low-grade inflammation, diet composition and health: current research evidence and its translation. Br. J. Nutr. 114 (7), 999–1012. doi:10.1017/S0007114515002093
Miyauchi H., Geisberger S., Luft F. C., Wilck N., Stegbauer J., Wiig H., et al. (2024). Sodium as an important regulator of immunometabolism. Hypertension 81 (3), 426–435. doi:10.1161/HYPERTENSIONAHA.123.19489
Moreira J. D., Chaudhary P., Frame A. A., Puleo F., Nist K. M., Abkin E. A., et al. (2019). Inhibition of microglial activation in rats attenuates paraventricular nucleus inflammation in Gαi2 protein-dependent, salt-sensitive hypertension. Exp. Physiol. 104 (12), 1892–1910. doi:10.1113/EP087924
Mowry F. E., Peaden S. C., Stern J. E., Biancardi V. C. (2021). TLR4 and AT1R mediate blood-brain barrier disruption, neuroinflammation, and autonomic dysfunction in spontaneously hypertensive rats. Pharmacol. Res. 174, 105877. doi:10.1016/j.phrs.2021.105877
Mu Y. F., Gao Z. X., Mao Z. H., Pan S. K., Liu D. W., Liu Z. S., et al. (2024). Perspectives on the involvement of the gut microbiota in salt-sensitive hypertension. Hypertens. Res. 47 (9), 2351–2362. doi:10.1038/s41440-024-01747-y
Mueller S. M., Heistad D. D. (1980). Effect of chronic hypertension on the blood-brain barrier. Hypertension 2 (6), 809–812. doi:10.1161/01.hyp.2.6.809
Muller L., Nasr A. R., Jux B., Makdissi N., Trowbridge J. W., Schmidt S. V., et al. (2024). Differential impact of high-salt levels in vitro and in vivo on macrophage core functions. Mol. Biol. Rep. 51 (1), 343. doi:10.1007/s11033-024-09295-x
Nagase S., Karashima S., Tsujiguchi H., Tsuboi H., Miyagi S., Kometani M., et al. (2020). Impact of gut microbiome on hypertensive patients with low-salt intake: Shika study results. Front. Med. 7, 475. doi:10.3389/fmed.2020.00475
Nakamura K., Cowley A. W. (1989). Sequential changes of cerebrospinal fluid sodium during the development of hypertension in dahl rats. Hypertension 13 (3), 243–249. doi:10.1161/01.hyp.13.3.243
Nicholson J. K., Holmes E., Kinross J., Burcelin R., Gibson G., Jia W., et al. (2012). Host-gut microbiota metabolic interactions. Science. 336 (6086), 1262–1267. doi:10.1126/science.1223813
Nist K. M., Bard H., McBride B., Capriglione A. L., Moreira J. D., Farb D. H., et al. (2024). Losartan attenuates sex-dependent hypertension, neuroinflammation, and cognitive impairment in the aging male sprague-dawley rat. Geroscience 47, 3007–3026. doi:10.1007/s11357-024-01409-4
North B. J., Sinclair D. A. (2012). The intersection between aging and cardiovascular disease. Circulation Res. 110 (8), 1097–1108. doi:10.1161/CIRCRESAHA.111.246876
Olson J. K., Miller S. D. (2004). Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 173 (6), 3916–3924. doi:10.4049/jimmunol.173.6.3916
Osanai T., Kanazawa T., Yokono Y., Uemura T., Okuguchi T., Onodera K. (1993). Effect of aging on sensitivity of blood pressure to salt. Nihon Ronen Igakkai Zasshi 30 (1), 30–34. doi:10.3143/geriatrics.30.30
Osborn J. W., Fink G. D., Sved A. F., Toney G. M., Raizada M. K. (2007). Circulating angiotensin II and dietary salt: converging signals for neurogenic hypertension. Curr. Hypertens. Rep. 9 (3), 228–235. doi:10.1007/s11906-007-0041-3
Ozaykan B., Taskin E., Magemizoglu A. (2017). Effect of salt loading on baroreflex sensitivity in reduced renal mass hypertension. Clin. Exp. Hypertens. 39 (7), 592–600. doi:10.1080/10641963.2017.1299748
Pacha J. (1998). Sodium balance and jejunal ion and water absorption in dahl salt-sensitive and salt-resistant rats. Clin. Exp. Pharmacol. and physiology 25 (3-4), 220–224. doi:10.1111/j.1440-1681.1998.t01-9-.x
Palmu J., Lahti L., Niiranen T. (2021). Targeting gut microbiota to treat hypertension: a systematic review. Int. J. Environ. Res. Public Health 18 (3), 1248. doi:10.3390/ijerph18031248
Pelisch N., Hosomi N., Ueno M., Nakano D., Hitomi H., Mogi M., et al. (2011). Blockade of AT1 receptors protects the blood-brain barrier and improves cognition in dahl salt-sensitive hypertensive rats. Am. J. Hypertens. 24 (3), 362–368. doi:10.1038/ajh.2010.241
Pitzer A., Elijovich F., Laffer C. L., Ertuglu L. A., Sahinoz M., Saleem M., et al. (2022). DC ENaC-Dependent inflammasome activation contributes to salt-sensitive hypertension. Circulation Res. 131 (4), 328–344. doi:10.1161/CIRCRESAHA.122.320818
Pluznick J. (2014). A novel SCFA receptor, the microbiota, and blood pressure regulation. Gut Microbes 5 (2), 202–207. doi:10.4161/gmic.27492
Pluznick J. L. (2016). Gut microbiota in renal physiology: focus on short-chain fatty acids and their receptors. Kidney Int. 90 (6), 1191–1198. doi:10.1016/j.kint.2016.06.033
Qi J., Zhao X. F., Yu X. J., Yi Q. Y., Shi X. L., Tan H., et al. (2016). Targeting Interleukin-1 beta to suppress sympathoexcitation in hypothalamic paraventricular nucleus in dahl salt-sensitive hypertensive rats. Cardiovasc Toxicol. 16 (3), 298–306. doi:10.1007/s12012-015-9338-7
Recasens M., Almolda B., Perez-Clausell J., Campbell I. L., Gonzalez B., Castellano B. (2021). Chronic exposure to IL-6 induces a desensitized phenotype of the microglia. J. Neuroinflammation 18 (1), 31. doi:10.1186/s12974-020-02063-1
Rezq S., Abdel-Rahman A. A. (2016). Central GPR109A activation mediates glutamate-dependent pressor response in conscious rats. J. Pharmacol. Exp. Ther. 356 (2), 456–465. doi:10.1124/jpet.115.229146
Ríos-Covian D., Ruas-Madiedo P., Margolles A., Gueimonde M., De Los Reyes-Gavilán C., Salazar N. (2016). Intestinal short chain fatty acids and their link with diet and human health. Front. Microbiol. 7, 185. doi:10.3389/fmicb.2016.00185
Ronnback C., Hansson E. (2019). The importance and control of low-grade inflammation due to damage of cellular barrier systems that may lead to systemic inflammation. Front. Neurol. 10, 533. doi:10.3389/fneur.2019.00533
Rosa G. B., Cavalet L. C., de Melo A. B. S., Junior M. D. F., Lopes P. R., Cardoso R. A., et al. (2020). High salt intake during puberty leads to cardiac remodelling and baroreflex impairment in lean and obese Male wistar rats. Br. J. Nutr. 123 (6), 642–651. doi:10.1017/S0007114519003283
Rossitto G., Mary S., Chen J. Y., Boder P., Chew K. S., Neves K. B., et al. (2020). Tissue sodium excess is not hypertonic and reflects extracellular volume expansion. Nat. Commun. 11 (1), 4222. doi:10.1038/s41467-020-17820-2
Rucker A. J., Rudemiller N. P., Crowley S. D. (2018). Salt, hypertension, and immunity. Annu. Rev. Physiol. 80, 283–307. doi:10.1146/annurev-physiol-021317-121134
Rudemiller N., Lund H., Jacob H. J., Geurts A. M., Mattson D. L., PhysGen Knockout P. (2014). CD247 modulates blood pressure by altering T-lymphocyte infiltration in the kidney. Hypertension 63 (3), 559–564. doi:10.1161/HYPERTENSIONAHA.113.02191
Ruiz-Ortega M., Ruperez M., Lorenzo O., Esteban V., Blanco J., Mezzano S., et al. (2002). Angiotensin II regulates the synthesis of proinflammatory cytokines and chemokines in the kidney. Kidney Int. Suppl. 62 (82), S12–S22. doi:10.1046/j.1523-1755.62.s82.4.x
Sahinoz M., Elijovich F., Ertuglu L. A., Ishimwe J., Pitzer A., Saleem M., et al. (2021). Salt sensitivity of blood pressure in blacks and women: a role of inflammation, oxidative stress, and epithelial na(+) channel. Antioxidants and redox Signal. 35 (18), 1477–1493. doi:10.1089/ars.2021.0212
Saleem M., Aden L. A., Mutchler A. L., Basu C., Ertuglu L. A., Sheng Q., et al. (2024). Myeloid-specific JAK2 contributes to inflammation and salt sensitivity of blood pressure. Circulation Res. 135 (9), 890–909. doi:10.1161/CIRCRESAHA.124.323595
Sallmann S., Juttler E., Prinz S., Petersen N., Knopf U., Weiser T., et al. (2000). Induction of interleukin-6 by depolarization of neurons. J. Neurosci. official J. Soc. Neurosci. 20 (23), 8637–8642. doi:10.1523/JNEUROSCI.20-23-08637.2000
Sander G. E. (2002). High blood pressure in the geriatric population: treatment considerations. Am. J. Geriatr. Cardiol. 11 (4), 223–232. doi:10.1111/j.1076-7460.2002.00032.x
Santisteban M. M., Kim S., Pepine C. J., Raizada M. K. (2016). Brain-gut-bone marrow axis: implications for hypertension and related therapeutics. Circulation Res. 118 (8), 1327–1336. doi:10.1161/CIRCRESAHA.116.307709
Santisteban M. M., Ahmari N., Carvajal J. M., Zingler M. B., Qi Y., Kim S., et al. (2015). Involvement of bone marrow cells and neuroinflammation in hypertension. Circ. Res. 117 (2), 178–191. doi:10.1161/CIRCRESAHA.117.305853
Sasaki K., Sasaki D., Hannya A., Tsubota J., Kondo A. (2020). In vitro human colonic microbiota utilises D-beta-hydroxybutyrate to increase butyrogenesis. Sci. Rep. 10 (1), 8516. doi:10.1038/s41598-020-65561-5
Schulman I. H., Aranda P., Raij L., Veronesi M., Aranda F. J., Martin R. (2006). Surgical menopause increases salt sensitivity of blood pressure. Hypertension 47 (6), 1168–1174. doi:10.1161/01.HYP.0000218857.67880.75
Sehgal P. B., Grieninger G., Tosato G. (1989). Regulation of the acute phase and immune responses: Interleukin-6. Ann. N. Y. Acad. Sci. 557, 1–583. doi:10.1111/j.1749-6632.1989.tb23993.x
Sesso H. D., Wang L., Buring J. E., Ridker P. M., Gaziano J. M. (2007). Comparison of interleukin-6 and C-reactive protein for the risk of developing hypertension in women. Hypertension 49 (2), 304–310. doi:10.1161/01.HYP.0000252664.24294.ff
Shahid M., Francis J., Majid D. S. (2008). Tumor necrosis factor-alpha induces renal vasoconstriction as well as natriuresis in mice. Am. J. physiology Ren. physiology 295 (6), F1836–F1844. doi:10.1152/ajprenal.90297.2008
Shen X. Z., Li Y., Li L., Shah K. H., Bernstein K. E., Lyden P., et al. (2015). Microglia participate in neurogenic regulation of hypertension. Hypertension 66 (2), 309–316. doi:10.1161/HYPERTENSIONAHA.115.05333
Shi P., Diez-Freire C., Jun J. Y., Qi Y., Katovich M. J., Li Q., et al. (2010). Brain microglial cytokines in neurogenic hypertension. Hypertension 56 (2), 297–303. doi:10.1161/HYPERTENSIONAHA.110.150409
Shi Z., Gan X. B., Fan Z. D., Zhang F., Zhou Y. B., Gao X. Y., et al. (2011). Inflammatory cytokines in paraventricular nucleus modulate sympathetic activity and cardiac sympathetic afferent reflex in rats. Acta Physiol. (Oxf) 203 (2), 289–297. doi:10.1111/j.1748-1716.2011.02313.x
Silva Y. P., Bernardi A., Frozza R. L. (2020). The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front. Endocrinol. (Lausanne) 11, 25. doi:10.3389/fendo.2020.00025
Silveira-Nunes G., Durso D. F., Jr L., Cunha E. H. M., Maioli T. U., Vieira A. T., et al. (2020). Hypertension is associated with intestinal microbiota dysbiosis and inflammation in a Brazilian population. Front. Pharmacol. 11, 258. doi:10.3389/fphar.2020.00258
Singh P., Bahrami L., Castillo A., Majid D. S. (2013). TNF-Alpha type 2 receptor mediates renal inflammatory response to chronic angiotensin II administration with high salt intake in mice. Am. J. physiology Ren. physiology 304 (7), F991–F999. doi:10.1152/ajprenal.00525.2012
Singh A., Ansari V. A., Mahmood T., Ahsan F., Wasim R. (2022). Neurodegeneration: microglia: nf-kappab signaling pathways. Drug Res. (Stuttg) 72 (9), 496–499. doi:10.1055/a-1915-4861
Solak Y., Afsar B., Vaziri N. D., Aslan G., Yalcin C. E., Covic A., et al. (2016). Hypertension as an autoimmune and inflammatory disease. Hypertens. Res. 39 (8), 567–573. doi:10.1038/hr.2016.35
Stocker S. D. (2023). Altered neuronal discharge in the organum vasculosum of the Lamina terminalis contributes to dahl salt-sensitive hypertension. Hypertension 80 (4), 872–881. doi:10.1161/HYPERTENSIONAHA.122.20798
Swaroop S., Sengupta N., Suryawanshi A. R., Adlakha Y. K., Basu A. (2016). HSP60 plays a regulatory role in IL-1β-induced microglial inflammation via TLR4-p38 MAPK axis. J. Neuroinflammation 13 (1), 27. doi:10.1186/s12974-016-0486-x
Thangaraj S. S., Oxlund C. S., Andersen H., Svenningsen P., Stubbe J., Palarasah Y., et al. (2024). Amiloride lowers plasma TNF and interleukin-6 but not interleukin-17A in patients with hypertension and type 2 diabetes. Am. J. physiology Ren. physiology 327 (1), F37–F48. doi:10.1152/ajprenal.00268.2023
Thowsen I. M., Karlsen T. V., Nikpey E., Haslene-Hox H., Skogstrand T., Randolph G. J., et al. (2022). Na(+) is shifted from the extracellular to the intracellular compartment and is not inactivated by glycosaminoglycans during high salt conditions in rats. J. physiology 600 (10), 2293–2309. doi:10.1113/JP282715
Titze J. (2014). Sodium balance is not just a renal affair. Curr. Opin. Nephrol. Hypertens. 23 (2), 101–105. doi:10.1097/01.mnh.0000441151.55320.c3
Titze J., Lang R., Ilies C., Schwind K. H., Kirsch K. A., Dietsch P., et al. (2003). Osmotically inactive skin na+ storage in rats. Am. J. physiology Ren. physiology 285 (6), F1108–F1117. doi:10.1152/ajprenal.00200.2003
Trimarco B., Lembo G., Ricciardelli B., De Luca N., Rendina V., Condorelli G., et al. (1991). Salt-induced plasticity in cardiopulmonary baroreceptor reflexes in salt-resistant hypertensive patients. Hypertension 18 (4), 483–493. doi:10.1161/01.hyp.18.4.483
Ueno M., Sakamoto H., Liao Y. J., Onodera M., Huang C. L., Miyanaka H., et al. (2004). Blood-brain barrier disruption in the hypothalamus of young adult spontaneously hypertensive rats. Histochem Cell Biol. 122 (2), 131–137. doi:10.1007/s00418-004-0684-y
van de Vyver M. (2023). Immunology of chronic low-grade inflammation: relationship with metabolic function. J. Endocrinol. 257 (1), e220271. doi:10.1530/JOE-22-0271
Vasquez E. C. (1994). Contribution of the cardiopulmonary reflex to the cardiovascular regulation in normal and pathophysiological states. Braz. J. Med. Biol. Res. 27 (4), 1049–1064.
Vasquez E., Meyrelles S., Mauad H., Cabral A. (1997). Neural reflex regulation of arterial pressure in pathophysiological conditions: interplay among the baroreflex, the cardiopulmonary reflexes and the chemoreflex. Braz. J. Med. Biol. Res. 30 (4), 521–532. doi:10.1590/s0100-879x1997000400014
Veiras L. C., Shen J. Z. Y., Bernstein E. A., Regis G. C., Cao D., Okwan-Duodu D., et al. (2021). Renal inflammation induces salt sensitivity in Male Db/Db mice through dysregulation of ENaC. J Am Soc Nephrol 32 (5), 1131–1149. doi:10.1681/ASN.2020081112
Veiras L. C., Bernstein E. A., Cao D., Okwan-Duodu D., Khan Z., Gibb D. R., et al. (2022). Tubular IL-1β induces salt sensitivity in diabetes by activating renal macrophages. Circulation research 131 (1), 59–73. doi:10.1161/CIRCRESAHA.121.320239
Victor R. G., Morgan D. A., Thoren P., Mark A. L. (1986). High salt diet sensitizes cardiopulmonary baroreflexes in dahl salt-resistant rats. Hypertension 8 (6 Pt 2), II21–II27. doi:10.1161/01.hyp.8.6_pt_2.ii21
Vlachovsky S. G., Azurmendi P. J., Oddo E. M., Rodriguez R. S., Di Ciano L. A., Goette N. P., et al. (2024). High sodium, rather than high blood pressure, induces immune cell activation and renal infiltration in ovariectomized adult wistar rats. Biochem. Biophys Res Commun 722, 150147. doi:10.1016/j.bbrc.2024.150147
Waisman A., Hauptmann J., Regen T. (2015). The role of IL-17 in CNS diseases. Acta Neuropathol 129 (5), 625–637. doi:10.1007/s00401-015-1402-7
Wang D. S., Xie H. H., Shen F. M., Cai G. J., Su D. F. (2005). Blood pressure variability, cardiac baroreflex sensitivity and organ damage in experimentally hypertensive rats. Clinical and experimental pharmacology and physiology 32 (7), 545–552. doi:10.1111/j.1440-1681.2005.04229.x
Wang J., Chen J., Chen C., Huang S., Rao X., Zhong J. (2012). Elevated Th17 and IL-23 in hypertensive patients with acutly increased blood pressure. American Journal of Immunology 8, 27–32. doi:10.3844/ajisp.2012.27.32
Wang M., Pan W., Xu Y., Zhang J., Wan J., Jiang H. (2022). Microglia-mediated neuroinflammation: a potential target for the treatment of cardiovascular diseases. J Inflamm Res 15, 3083–3094. doi:10.2147/JIR.S350109
Wehrwein E. A., Joyner M. J. (2013). Regulation of blood pressure by the arterial baroreflex and autonomic nervous system. Handb Clin Neurol 117, 89–102. doi:10.1016/B978-0-444-53491-0.00008-0
Weinberger M. H. (1996a). Salt sensitivity: does it play an important role in the pathogenesis and treatment of hypertension? Current opinion in nephrology and hypertension 5 (3), 205–208. doi:10.1097/00041552-199605000-00003
Weinberger M. H. (1996b). Salt sensitivity of blood pressure in humans. Hypertension 27 (3 Pt 2), 481–490. doi:10.1161/01.hyp.27.3.481
Weinberger M. H. (2006). Pathogenesis of salt sensitivity of blood pressure. Current hypertension reports 8 (2), 166–170. doi:10.1007/s11906-006-0014-y
Weinberger M. H., Miller J. Z., Luft F. C., Grim C. E., Fineberg N. S. (1986). Definitions and characteristics of sodium sensitivity and blood pressure resistance. Hypertension 8 (6 Pt 2), II127–II134. doi:10.1161/01.hyp.8.6_pt_2.ii127
Wenzel U. O., Bode M., Kurts C., Ehmke H. (2019). Salt, inflammation, IL-17 and hypertension. Br J Pharmacol 176 (12), 1853–1863. doi:10.1111/bph.14359
Wilck N., Matus M. G., Kearney S. M., Olesen S. W., Forslund K., Bartolomaeus H., et al. (2017). Salt-responsive gut commensal modulates TH17 axis and disease. Nature 551 (7682), 585–589. doi:10.1038/nature24628
Winklewski P. J., Radkowski M., Wszedybyl-Winklewska M., Demkow U. (2015). Brain inflammation and hypertension: the chicken or the egg? J Neuroinflammation 12, 85. doi:10.1186/s12974-015-0306-8
Wong J. M., de Souza R., Kendall C. W., Emam A., Jenkins D. J. (2006). Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol 40 (3), 235–243. doi:10.1097/00004836-200603000-00015
Wu C., Yosef N., Thalhamer T., Zhu C., Xiao S., Kishi Y., et al. (2013). Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 496 (7446), 513–517. doi:10.1038/nature11984
Wu C., Chen Z., Zhang L., Zhu Y., Deng M., Huang C., et al. (2021). Sodium butyrate ameliorates deoxycorticosterone acetate/salt-induced hypertension and renal damage by inhibiting the MR/SGK1 pathway. Hypertens Res 44 (2), 168–178. doi:10.1038/s41440-020-00548-3
Wulff P., Vallon V., Huang D. Y., Volkl H., Yu F., Richter K., et al. (2002). Impaired renal na(+) retention in the sgk1-knockout mouse. The Journal of clinical investigation 110 (9), 1263–1268. doi:10.1172/JCI15696
Xiao L., Harrison D. G. (2020). Inflammation in hypertension. Canadian Journal of Cardiology 36 (5), 635–647. doi:10.1016/j.cjca.2020.01.013
Xiao L., Kirabo A., Wu J., Saleh M. A., Zhu L., Wang F., et al. (2015). Renal denervation prevents immune cell activation and renal inflammation in angiotensin II-Induced hypertension. Circulation research 117 (6), 547–557. doi:10.1161/CIRCRESAHA.115.306010
Xu J., Moore B. N., Pluznick J. L. (2022). Short-chain fatty acid receptors and blood pressure regulation: council on hypertension mid-career award for research excellence 2021. Hypertension 79 (10), 2127–2137. doi:10.1161/HYPERTENSIONAHA.122.18558
Xue B., Thunhorst R. L., Yu Y., Guo F., Beltz T. G., Felder R. B., et al. (2016). Central renin-angiotensin system activation and inflammation induced by high-fat diet sensitize angiotensin II-Elicited hypertension. Hypertension 67 (1), 163–170. doi:10.1161/HYPERTENSIONAHA.115.06263
Yakoub M., Rahman M., Kleimann P., Hoffe J., Feige M., Bouvain P., et al. (2024). Transient high salt intake promotes T-Cell-Mediated hypertensive vascular injury. Hypertension 81 (12), 2415–2429. doi:10.1161/HYPERTENSIONAHA.124.23115
Yamamoto-Kimura L., Zamora-Gonzalez J., Huerta-Alvarado S., Fajardo-Gutierrez A., Cardoso-Saldana G., Posadas-Romero C. (1996). High blood pressure and cardiovascular risk factors in an adult population of Mexico City. Characteristics of the studied population. Archives of medical research 27 (2), 213–222.
Yang J., Ran M., Li H., Lin Y., Ma K., Yang Y., et al. (2022). New insight into neurological degeneration: inflammatory cytokines and blood-brain barrier. Front Mol Neurosci 15, 1013933. doi:10.3389/fnmol.2022.1013933
Yin X., Duan C., Zhang L., Zhu Y., Qiu Y., Shi K., et al. (2024). Microbiota-derived acetate attenuates neuroinflammation in rostral ventrolateral medulla of spontaneously hypertensive rats. J Neuroinflammation 21 (1), 101. doi:10.1186/s12974-024-03061-3
Youn J. C., Yu H. T., Lim B. J., Koh M. J., Lee J., Chang D. Y., et al. (2013). Immunosenescent CD8+ T cells and C-X-C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension 62 (1), 126–133. doi:10.1161/HYPERTENSIONAHA.113.00689
Yu Y., Zhang Z. H., Wei S. G., Serrats J., Weiss R. M., Felder R. B. (2010). Brain perivascular macrophages and the sympathetic response to inflammation in rats after myocardial infarction. Hypertension 55 (3), 652–659. doi:10.1161/HYPERTENSIONAHA.109.142836
Yu Y., Cao Y., Bell B., Chen X., Weiss R. M., Felder R. B., et al. (2019). Brain TACE (tumor necrosis factor-alpha-converting enzyme) contributes to sympathetic excitation in heart failure rats. Hypertension 74 (1), 63–72. doi:10.1161/HYPERTENSIONAHA.119.12651
Yu X. J., Liu X. J., Guo J., Su Y. K., Zhang N., Qi J., et al. (2022). Blockade of microglial activation in hypothalamic paraventricular nucleus improves high salt-induced hypertension. Am J Hypertens 35 (9), 820–827. doi:10.1093/ajh/hpac052
Zaldivia M. T., Rivera J., Hering D., Marusic P., Sata Y., Lim B., et al. (2017). Renal denervation reduces monocyte activation and monocyte-platelet aggregate formation: an anti-inflammatory effect relevant for cardiovascular risk. Hypertension 69 (2), 323–331. doi:10.1161/HYPERTENSIONAHA.116.08373
Zera T., Ufnal M., Szczepanska-Sadowska E. (2009). Central TNF-α elevates blood pressure and sensitizes to central pressor action of angiotensin II in the infarcted rats. J. Physiol. Pharmacol. 59 (Suppl. 8), 117–121.
Zhang W. C., Zheng X. J., Du L. J., Sun J. Y., Shen Z. X., Shi C., et al. (2015a). High salt primes a specific activation state of macrophages, M(Na). Cell Res 25 (8), 893–910. doi:10.1038/cr.2015.87
Zhang D., Wang C., Cao S., Ye Z., Deng B., Kijlstra A., et al. (2015b). High-salt enhances the inflammatory response by retina pigment epithelium cells following lipopolysaccharide stimulation. Mediators Inflamm 2015, 197521. doi:10.1155/2015/197521
Zhang T., Fang S., Wan C., Kong Q., Wang G., Wang S., et al. (2015c). Excess salt exacerbates blood-brain barrier disruption via a p38/MAPK/SGK1-dependent pathway in permanent cerebral ischemia. Sci Rep 5, 16548. doi:10.1038/srep16548
Zhang J., Rudemiller N. P., Patel M. B., Karlovich N. S., Wu M., McDonough A. A., et al. (2016). Interleukin-1 receptor activation potentiates salt reabsorption in angiotensin II-Induced hypertension via the NKCC2 Co-transporter in the nephron. Cell Metab. 23 (2), 360–368. doi:10.1016/j.cmet.2015.11.013
Zhou X., Chen K., Lei H., Sun Z. (2015). Klotho gene deficiency causes salt-sensitive hypertension via monocyte chemotactic protein-1/CC chemokine receptor 2-mediated inflammation. J Am Soc Nephrol 26 (1), 121–132. doi:10.1681/ASN.2013101033
Zhu Q., Li X. X., Wang W., Hu J., Li P. L., Conley S., et al. (2016). Mesenchymal stem cell transplantation inhibited high salt-induced activation of the NLRP3 inflammasome in the renal medulla in dahl S rats. Am. J. Physiol. Renal Physiol. 310 (7), F621-F627–F7. doi:10.1152/ajprenal.00344.2015
Zimmermann J., Krauthausen M., Hofer M. J., Heneka M. T., Campbell I. L., Muller M. (2013). CNS-Targeted production of IL-17A induces glial activation, microvascular pathology and enhances the neuroinflammatory response to systemic endotoxemia. PloS one 8 (2), e57307. doi:10.1371/journal.pone.0057307
Keywords: hypertension, salt, inflammation, microbiome, short chain fatty acids, brain, kidney, salt sensitivity
Citation: Cardoso LM and Wainford RD (2025) The immune-microbiome axis in salt-sensitive hypertension: a focus on renal and neural mechanisms. Front. Physiol. 16:1653387. doi: 10.3389/fphys.2025.1653387
Received: 24 June 2025; Accepted: 28 August 2025;
Published: 08 October 2025.
Edited by:
Erika E Nishi, Federal Universtity of São Paulo, BrazilReviewed by:
Zhi-Lin Luan, Dalian Medical University, ChinaMarta Kuczeriszka, Polish Academy of Sciences, Poland
Copyright © 2025 Cardoso and Wainford. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard David Wainford, cmljaGFyZC5kYXZpZC53YWluZm9yZEBlbW9yeS5lZHU=
†ORCID: Leonardo Máximo Cardoso, orcid.org/0000-0001-9821-0484; Richard David Wainford, orcid.org/0000-0003-2830-5618